
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystallization of smooth amorphous calcium phosphate microspheres to core–shell hydroxyapatite microspheres†
Mei-li Qi ab,
Zhaoxuan Longa,
Xiao-Cun Liuab,
Haijun Zhangc,
Jin Li*ab and
Shengkun Yao*de
ab,
Zhaoxuan Longa,
Xiao-Cun Liuab,
Haijun Zhangc,
Jin Li*ab and
Shengkun Yao*de
aSchool of Civil Engineering, Shandong Jiaotong University, Ji'nan 250357, China. E-mail: sdzblijin@163.com
bJinan Key Laboratory for Low-Carbon and Eco-Friendly Road Materials, Shandong Jiaotong University, Ji'nan 250357, China
cShanghai Tenth People's Hospital, School of Medicine, Tongji University, Shanghai 200092, China
dShandong Provincial Engineering and Technical Center of Light Manipulations and Shandong Provincial Key Laboratory of Optics and Photonic Device, School of Physics and Electronics, Shandong Normal University, Ji'nan 250014, China. E-mail: yaoshk@sdnu.edu.cn
eCollaborative Innovation Center of Light Manipulation and Applications, Shandong Normal University, Ji'nan 250358, China
First published on 13th August 2024
Abstract
Calcium phosphates (Ca-P) represent a significant class of biological minerals found in natural hard tissues. Crystallization through phase transformation of a metastable precursor is an effective strategy to guide the growth of crystalline Ca-P with exceptional functionality. Despite extensive research on Ca-P, the exact process during the crystallization of amorphous particles to hydroxyapatite (HA) remains elusive. Herein, pure HA microspheres with a core–shell structure are crystallized via dissolution and re-crystallization of smooth amorphous calcium phosphate (ACP) microspheres. The transformation is initiated with the increase of the hydrothermal treatment time in the presence of sodium trimetaphosphate and L-glutamic. The underlying mechanisms along with the kinetics of such transformation are explored. Nanocrystalline areas are formed on the smooth ACP microspheres and crystallization advances via nanometre-sized clusters formed by directional arrangement of nanocrystalline whiskers. Our findings shed light on a crucial but unclear stage in the genesis of HA crystals, specifically under the conditions of hydrothermal synthesis.
1. Introduction
Calcium phosphates (Ca-P), notably hydroxyapatite (HA), are essential biological minerals in natural hard tissues and are integral to bone tissue engineering applications.1,2 Composite biomaterials for bone tissue engineering often include HA or a Ca-P precursor that can transform into HA under physiological conditions. However, developing a straightforward and scalable method to produce pure HA under mild conditions remains challenging. One promising solution is the “biomimetics” approach, which involves non-classical crystallization through amorphous intermediates. Understanding the formation mechanisms of these apatitic minerals can enhance biomaterials development and pave the way for synthesizing nonbiogenic minerals.3–5Despite extensive research on Ca-P, the precise crystallization pathway of amorphous particles into HA, the mechanisms behind this transformation, and its kinetics are still under investigation. Studies on the nucleation of biological HA concur that HA does not form directly from solution but through an amorphous calcium phosphate (ACP) precursor.6,7 There is ongoing debate about the factors driving this ACP-mediated process, from ACP formation to its transition to HA. Tang's group suggested a solution-mediated surface nucleation process, where HA forms heterogeneously on ACP, with the transformation rate dependent on the initial ACP amount and calcium activity in solution.8–13 In contrast, Xie et al. and Wang et al. proposed that HA nucleation occurs within the ACP through a solid–solid transformation.14,15
Different proposed mechanisms for ACP-mediated HA formation suggest varying roles for additives. Additives can stabilize or destabilize ACP by altering its dissolution rate, affect the HA nucleation rate, or influence the chemistry and solubility of the ACP phase, impacting the solution's supersaturation. The presence of biomolecules, such as amino acids, can induce specific polymorph nucleation and affect crystal morphology.16–19 Sodium trimetaphosphate (STMP), a phosphorylating agent used in the food industry, can also introduce phosphate groups onto collagen surfaces, promoting mineralization.20,21 Therefore, the choice of biomolecules and phosphorus sources is crucial for creating Ca-P particles with desired structures.
This study introduces a one-step hydrothermal synthesis strategy for Ca-P microspheres, utilizing STMP as the phosphorus source and L-glutamic acid as an additive. ACP serves as an intermediate phase that readily transitions as a precursor for HA microsphere growth. By adjusting the reaction time, smooth ACP microspheres can be transformed into porous core–shell HA microspheres. The study investigates the phase evolution, morphology, particle size distribution, BET surface area, and pore size of these core–shell structured HA microspheres over time. The formation mechanism and crystallization process of HA under hydrothermal treatment is discussed, informed by the observed evolution.
2. Experimental
2.1. Materials
The anhydrous calcium chloride (CaCl2, AR), sodium trimetaphosphate ((NaPO3)3, AR), nitric acid (HNO3, AR), L-glutamic acid and urea (CO(NH2)2) were purchased from the Sinopharm Chemical Reagent Company Limited of China (Shanghai, China). All chemical reagents were utilized as received without additional purification.2.2. Preparation of the samples
Calcium phosphate microspheres were obtained through a hydrothermal route. In a typical step, 0.1 mol per L CaCl2, 0.02 mol per L (NaPO3)3 and 1 mol per L urea aqueous solutions were first mixed under magnetic stirring. A dilute nitric acid (HNO3) solution is then used to adjust the pH of the mixture to 3.5 to obtain a clear solution. When the pH of the solution is stable, 0.05 mol per L L-glutamic acid was added to it. The final mixture was hydrothermally treated at 180 °C in a high-pressure reactor lined with Teflon for 0.5, 1, 2, 3 and 8 h, respectively. After the treatment, the products are washed with deionized water and ethanol, centrifuged, and dried.2.3. Characterizations
The phase composition and functional groups of the samples were examined by powder X-ray diffraction (XRD, Bruker D8 Advance) in the 2θ range of 10–60° and Fourier transform infrared spectroscopy (FTIR, Nicolet IS50) collected between 4000 and 500 cm−1. The crystallite sizes (Dhkl) of the three strongest diffraction peak in as-prepared HA products were determined using the Debye–Scherrer approach.22,23 The degree of crystallinity of the HA samples was calculated by the Jade software. The surface morphologies were characterized using field scanning electron microscope (FE-SEM, Zeiss JSM-7610F) at an acceleration voltage of 5 kV equipped with an energy dispersive spectrometer (EDS) by dropping suspensions onto aluminum foils. The particle size distributions and physical gas adsorption performance were evaluated by laser particle size analyzer (Mastersizer 2000) and Micromeritics ASAP 2460 instrument. Prior to the FE-SEM and PSD tests, the samples were dispersed ultrasonically in anhydrous ethanol for 5 min.3. Results and discussion
3.1. Phase analysis
The phase composition and functional groups of the synthesized samples were characterized under varying hydrothermal durations using XRD and FTIR spectroscopy, as depicted in Fig. 1. At a hydrothermal duration of 0.5 h, the XRD pattern exhibited a broad and amorphous profile with a prominent peak at 2θ = 29° (Fig. 1a, black line), which is characteristic of the ACP phase.24 Upon increasing the hydrothermal time to 1 h, the predominant phase remained ACP, albeit with the emergence of several weak diffraction peaks (Fig. 1a, red line). This suggests the onset of crystallization and an impending change in the sample's morphology. Upon further extending the hydrothermal treatment to 2 h, the sample underwent a phase transition to a crystalline HA phase, as evidenced by the XRD pattern which aligns with the standard HA profile (JCPDS 09-0432, shown as the bottom bar chart in Fig. 1a). The three high-intensity diffraction peaks, observed at 2θ values of 31.7°, 32.1°, and 32.9°, are attributed to the crystallographic planes (211), (112), and (300) of the HA phase, respectively. The crystallinity of the HA sample significantly enhanced after 3 h of hydrothermal treatment and exhibited minimal further change when the hydrothermal duration was extended to 8 h (Fig. 1a, blue and purple lines). Noticeably, extending the hydrothermal treatment to 8 h, a longer time intervals, aims to explore the stability of the HA sample over an extended period, ensuring that the crystalline phase remains stable and does not undergo further significant changes. This investigation is crucial for understanding the optimal conditions for HA synthesis and its potential applications in various fields. | ||
| Fig. 1 XRD patterns (a) and FTIR spectra (b) of the samples prepared under different hydrothermal times. | ||
The crystallographic characteristics for the samples, such as crystallite sizes and crystallinity, are crucial parameters to delve deeper into the crystal growth dynamics.25 Herein, the Dhkl and the degree of crystallinity of HA subjected to varying durations of hydrothermal treatment were measured and are presented in Table 1. The crystallinity, along with the general trend in the growth of crystallite sizes, shows a significant increase, especially when the hydrothermal treatment time is extended from 2 h to 3 h. This suggests that the crystallites grow larger as the treatment time increases, which can be attributed to the prolonged exposure to the hydrothermal conditions that facilitate further crystallization. It is important to note that the calculations for these parameters, as performed using the Jade software, are approximate and should be taken as a reference.
| Hydrothermal treatment time (h) | D211 (nm) | D112 (nm) | D300 (nm) | Crystallinity (%) |
|---|---|---|---|---|
| 2 | 8.20 | 19.36 | 17.52 | 50.88 |
| 3 | 17.27 | 22.04 | 15.67 | 92.06 |
| 8 | 22.44 | 15.16 | 18.66 | 93.47 |
The functional groups of all samples, as revealed by FTIR spectroscopy, substantiate the phase transformation described earlier (refer to Fig. 1b). A distinct peak at 560 cm−1 emerges with hydrothermal treatments lasting up to 1 h, indicating the presence of ACP, while the broad absorption peak at 1115 cm−1 is a hallmark of ACP (as shown by the black and red lines in Fig. 2b).26 The peaks at 918 cm−1 and 560 cm−1 correspond to the stretching and bending vibrational modes of the P–O bonds in the PO43− group, respectively. As the hydrothermal duration extends beyond 2 h, the spectral patterns of the samples remain consistent (as indicated by the blue, green, and purple lines in Fig. 2b). The broad peaks at 3430 cm−1, 3370 cm−1, and 1650 cm−1 are characteristic of the hydroxyl (OH−) group present in water molecules. The peak at 3570 cm−1 is associated with the stretching vibrations of OH− within the HA structure, which suggests that the HA crystal lattice remains intact within the sample. The solitary peak at 1445 cm−1 is indicative of the characteristic vibration of CO32− ions, a product of urea hydrolysis. The broad and weakly defined doublet bands at 1460 cm−1 and 1420 cm−1, along with the peak at 876 cm−1, are aligned with the characteristic vibrations of the CO32− group. This suggests that CO32− ions are substituting for the B-site in the PO43− group within HA.27 It is a well-documented phenomenon for CO32− to replace PO43− in HA during urea hydrolysis, particularly at temperatures exceeding 80 °C.28 The peaks around 1040 cm−1, 962 cm−1, 603 cm−1, and 570 cm−1 are assigned to the PO43− group present in HA.22 These findings are congruent with the XRD patterns and collectively confirm that the synthesized HA samples are indeed carbonate-substituted HA.
 | ||
| Fig. 2 FE-SEM images of the products prepared under different hydrothermal times. (a–c) 0.5 h, (d–f) 1 h, (g–i) 3 h and (j–l) 8 h. (b), (e), (h), and (k) in the second column are high-magnification views of the areas within the red rectangular boxes in (a), (d), (g), and (j), respectively. | ||
3.2. Morphological characterization
The morphological evolution of the porous HA microspheres was investigated over time using FE-SEM, as illustrated in Fig. 2. At a hydrothermal duration of 0.5 h, the precipitated ACP phases are observed to be spherical with a smooth surface, ranging in size from 1 to 5 μm (Fig. 2a–c). The dimensions of the ACP particles are influenced by several factors, including the solution pH, the concentration of the mixed reagents, and the preparation temperature.29 Upon increasing the hydrothermal time to 1 h, the morphology of the microspheres undergoes a noticeable change, characterized by the emergence of a small number of particles on the surface and a slight reduction in the microsphere diameter (Fig. 2d–f). Notably, the surface of the microspheres becomes pitted and loses its smoothness. These morphological changes are in agreement with the XRD results. As the hydrothermal time extends to 3 h, the particles continue to grow and accumulate, forming a spherical shell with a discernible gap from the inner core, leading to the formation of HA core–shell microspheres with a diameter of approximately 2 μm (Fig. 2g–i). In some instances, the nuclei have begun to separate from the shell. The transformation from ACP to crystalline HA phase is suggested to occur through nucleation and/or the direct formation of crystalline phases within the ACP phases or on their surface, followed by outward growth via the Kirkendall process, with the internal ACP phases supplying the material flux necessary for phase conversion.30 Further extending the hydrothermal time to 8 h results in the HA maintaining a core–shell microsphere morphology (Fig. 2j and k). It is observed that the microspheres, post-core–shell separation, exhibit a porous structure (Fig. 2l). With prolonged hydrothermal treatment, the reaction becomes more complete, and the microspheres exhibit enhanced stability.Upon closer examination, it is observed that the synthesized HA core–shell microspheres are composed of an inner core and an outer shell (Fig. 3a). The shell is constructed from nano whiskers, which measure approximately 200–800 nm in length, and there are discernible gaps between the core and the shell, as shown in Fig. 3a–c. In certain regions, the core has become entirely detached from the shell, as depicted in Fig. 3d.
 | ||
| Fig. 3 FE-SEM images of the core–shell HA microspheres after broken. (a) Core–shell microspheres, (b) high magnification of the outer shells in (a), (c) hollow exterior shells and (d) isolated inner cores separated from the core–shell structure. | ||
To substantiate the transformation process from ACP to HA, element analysis of the samples prepared for 0.5 h and 2 h was conducted using EDS. As depicted in Fig. 4, S1 and 2 (ESI),† the presence of calcium (Ca), phosphorus (P), and oxygen (O) is observed in both the ACP and HA microspheres. The detection of aluminum (Al) and platinum (Pt) is attributed to the use of aluminum foil during sample preparation. A notable increase in the Ca/P atomic ratio is observed, rising from 1.034 to 1.642 as calculated from the EDS spectrum curves. This increase aligns with the expected chemical transformation from ACP to HA. It is noteworthy that ACP can be synthesized with a Ca/P molar ratio as low as approximately 1.0,31–33 whereas the stoichiometric ratio for HA is 1.67.
 | ||
| Fig. 4 Electronic images and corresponding EDS spectra of the products prepared under different hydrothermal times. (a and b) 0.5 h and (c and d) 2 h. | ||
3.3. Particle size distribution and specific surface area
The particle size distribution and physical gas adsorption properties are pivotal in determining the characteristics of synthesized microspheres. Granulometric analysis data for ACP and HA microspheres, prepared under varying hydrothermal conditions, are detailed in Fig. 5 and Table 2. The particle size distribution curves, as shown in Fig. 5, reveal that both types of microspheres have a concentrated size distribution, adhering to a Gaussian pattern, with two main size peaks suggesting uniformity in microsphere sizing. The observed shift from larger to smaller diameters marks the transition from solid ACP to core–shell HA microspheres, a finding corroborated by FE-SEM analysis, indicating that there is a sign of ACP shrinkage during HA growth. Notably, the particle size within the 3–10 μm range remains stable over hydrothermal treatments ranging from 3 to 8 h, indicating minimal alteration in HA microsphere size with extended treatment duration. Table 1 further illustrates that while the mean particle size decreases from 9.26 μm to approximately 5.72 μm as the hydrothermal treatment time increases from 0.5 to 3 h, there is a negligible change in mean particle size with additional treatment extending to 8 h.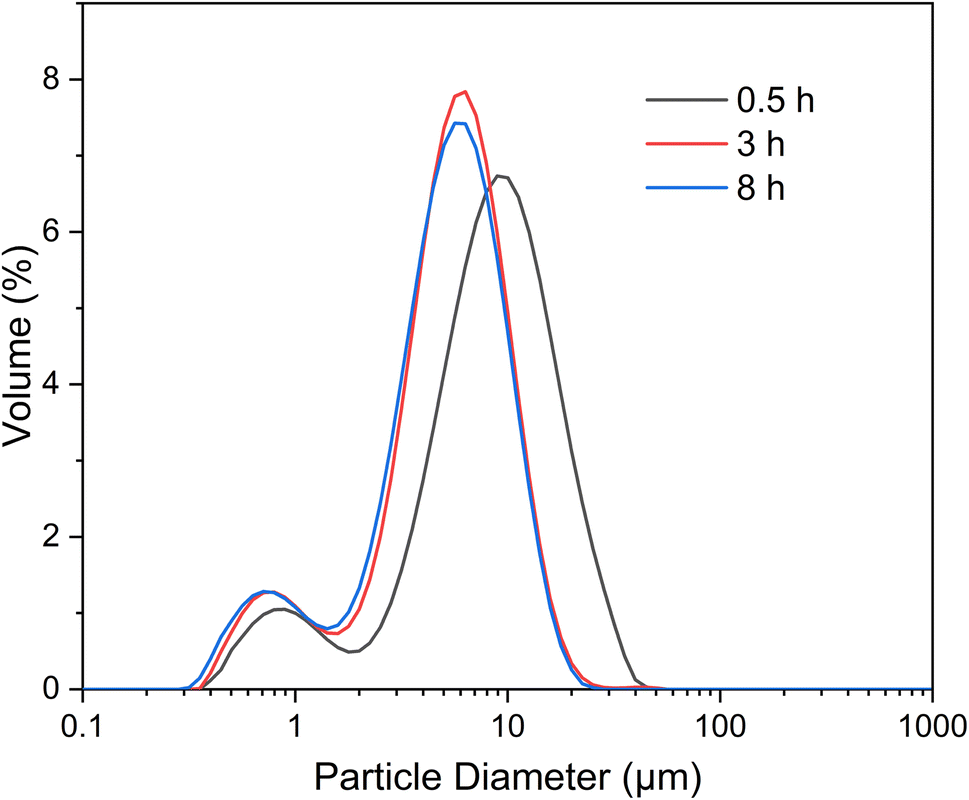 | ||
| Fig. 5 Particle size distribution curves of the microspheres prepared under different hydrothermal treatment times. | ||
| Hydrothermal treatment time (h) | d0.1 (μm) | d0.5 (μm) | d0.9 (μm) | Mean size (μm) |
|---|---|---|---|---|
| a d0.1, d0.5 and d0.9 means 10%, 50% and 90% of total particle size less than some value, respectively. | ||||
| 0.5 | 1.83 | 7.96 | 17.95 | 9.26 |
| 1 | 1.21 | 5.18 | 10.32 | 5.72 |
| 3 | 1.09 | 4.93 | 10.03 | 5.44 |
Fig. 6a illustrates the nitrogen adsorption–desorption isotherms for microspheres synthesized under varying hydrothermal durations. These isotherms, along with their type H3 hysteresis loops, are categorized as type IV according to the International Union of Pure and Applied Chemistry (IUPAC). This classification is indicative of a shell that self-assembles from clusters, creating slit-shaped pores. As depicted in Fig. 6b, the pore width distributions widen with increasing hydrothermal time, suggesting that more intricate pore channels are beneficial for drug loading and sustained release. Table 3 compiles the BET surface areas, total pore volumes, and average pore diameters for microspheres produced at different hydrothermal times of 0.5, 1, and 3 h. Initially, at a hydrothermal time of 0.5 h, the ACP microspheres exhibit a relatively low BET surface area of 3.00 m2 g−1. This specific surface area progressively increases to 5.17 m2 g−1 as the hydrothermal time is extended. Notably, the HA microspheres, once the core–shell structure is established, exhibit a significantly higher BET surface area of 35.24 m2 g−1. This elevated surface area can be ascribed to the porous structure and the self-assembly of nano whiskers that serve as the building blocks for the external shell, which is characteristic of a typical mesoporous structure.34 The average Barrett–Joyner–Halenda (BJH) pore size for the solid ACP microspheres is 7.24 nm, whereas for the HA core–shell microspheres, it expands to 14.91 nm. All the characteristic makes them an exemplary candidate for drug-delivery systems, presenting a strong potential in the realms of controlled drug release and targeted therapeutics.
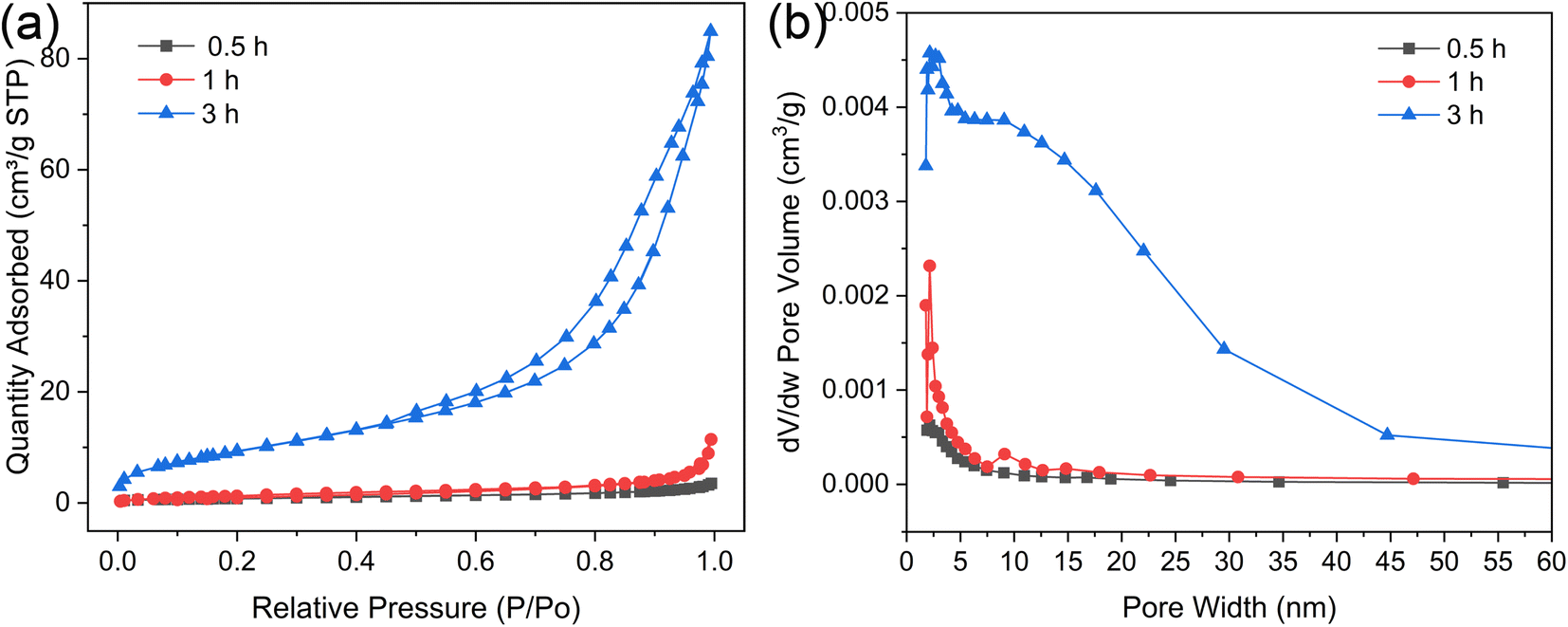 | ||
| Fig. 6 Physical gas adsorption performance analysis of the microspheres prepared under different hydrothermal times. (a) N2 adsorption–desorption isotherms and (b) BJH desorption pore size distribution curves. | ||
| Hydrothermal treatment time (h) | BET surface area (m2 g−1) | Total pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|
| 0.5 | 3.00 | 0.0054 | 7.24 |
| 1 | 5.17 | 0.0177 | 13.69 |
| 3 | 35.24 | 0.1314 | 14.91 |
3.4. From smooth ACP to core–shell HA: a proposed growth mechanism of the microspheres
The ACP-to-HA conversion in this experiment is a complex, multi-step process, with the structural evolutions depicted in Fig. 7.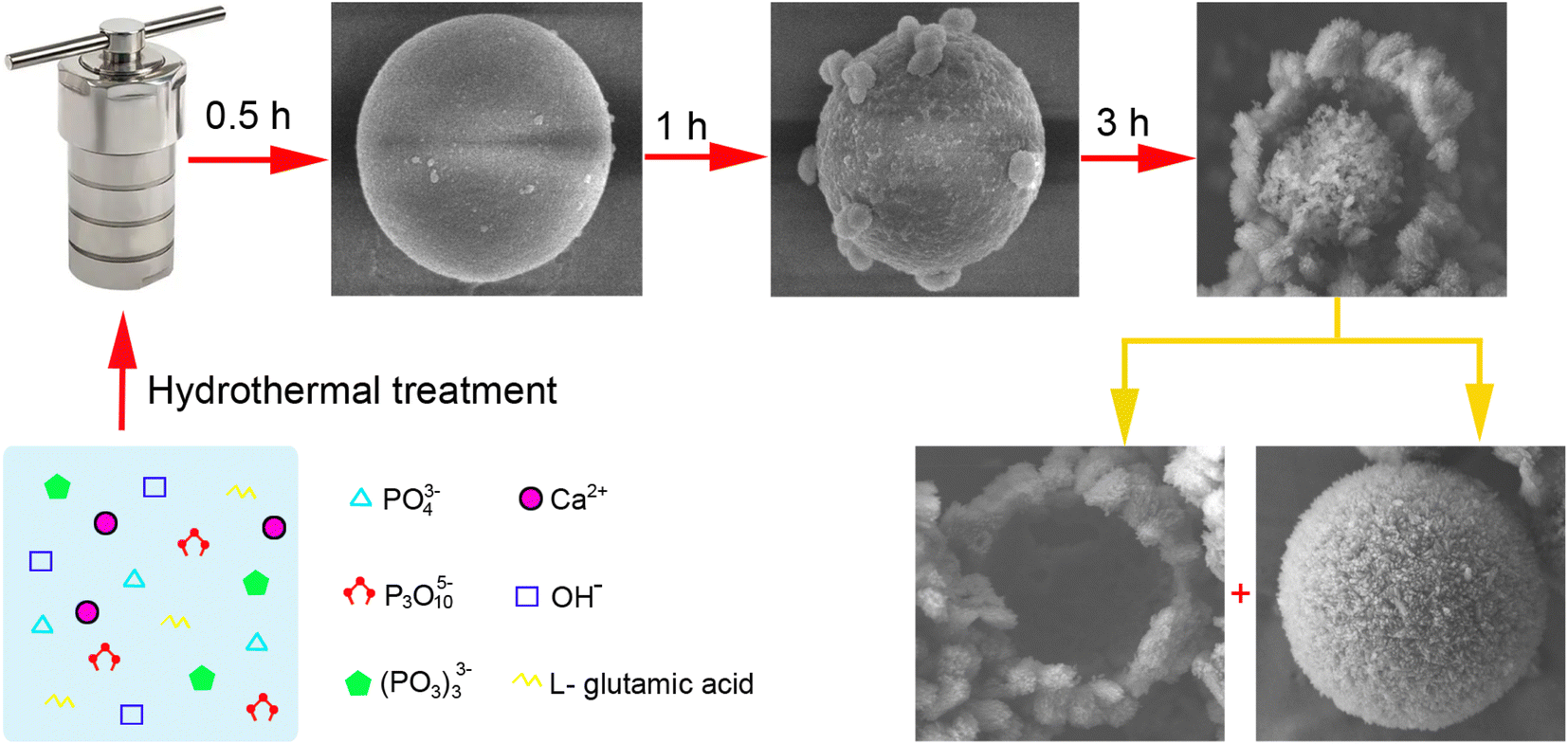 | ||
| Fig. 7 Formation mechanism of the HA microspheres transformed from ACP. | ||
This transformation process is significant for applications in biomaterials and drug delivery, as it results in microspheres with unique properties that can be tailored for specific uses, such as controlled drug release and bone growth promotion.
4. Conclusions
In summary, porous HA microspheres featuring a core–shell structure have been successfully synthesized from smooth ACP microspheres by using sodium trimetaphosphate and L-glutamic through a one-step hydrothermal method. A time-gradient experiment elucidated the progression from smooth to core–shell microspheres, along with the phase transition from ACP to HA. When subjected to hydrothermal conditions exceeding 3 h, pure core–shell HA microspheres with enhanced crystallinity, an average diameter of 5.44 μm, and a BET surface area of 35.24 m2 g−1 were obtained. A plausible mechanism for the crystallization of smooth ACP microspheres into core–shell HA microspheres has been proposed, involving a multi-step reaction with sodium trimetaphosphate and a dissolution–recrystallization process during nucleation and growth. These findings provide valuable insights into a pivotal yet previously obscure phase in the formation of HA crystals, particularly within the context of hydrothermal synthesis.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and the ESI.†Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
This study was supported by the Young Talent of Lifting Engineering for Science and Technology in Shandong, China (Award No. SDAST2021qt05), Shandong Provincial Natural Science Foundation (Award No. ZR2020QE070, ZR2020QA076 and ZR2021MB128) and the National Key R&D Program of China (2022YFB2601900).References
- S.-D. Jiang, Q.-Z. Yao, Y.-F. Ma, G.-T. Zhou and S.-Q. Fu, Phosphate-dependent morphological evolution of hydroxyapatite and implication for biomineralisation, Gondwana Res., 2015, 28(2), 858–868 CrossRef CAS.
- S. K. Avinashi, Shweta, B. Bohra, R. K. Mishra, S. Kumari, Z. Fatima, A. Hussain, B. Saxena, S. Kumar, M. Banerjee and C. R. Gautam, Fabrication of Novel 3-D Nanocomposites of HAp–TiC–h-BN–ZrO2: Enhanced Mechanical Performances and In Vivo Toxicity Study for Biomedical Applications, ACS Biomater. Sci. Eng., 2024, 10(4), 2116–2132 CrossRef CAS PubMed.
- M. L. Oyen, The materials science of bone: Lessons from nature for biomimetic materials synthesis, MRS Bull., 2008, 33(1), 49–55 CrossRef CAS.
- J. Aizenberg, D. A. Muller, J. L. Grazul and D. Hamann, Direct fabrication of large micropatterned single crystals, Science, 2003, 299(5610), 1205–1208 CrossRef CAS.
- D. C. Bassett, L. M. Grover, F. A. Müller, M. D. McKee and J. E. Barralet, Serum protein controlled nanoparticle synthesis, Adv. Funct. Mater., 2011, 21(15), 2968–2977 CrossRef CAS.
- S. Weiner, J. Mahamid, Y. Politi, Y. Ma and L. Addadi, Overview of the amorphous precursor phase strategy in biomineralization, Front. Mater. Sci. China, 2009, 3, 104–108 CrossRef.
- M. Sawada, K. Sridhar, Y. Kanda and S. Yamanaka, Pure hydroxyapatite synthesis originating from amorphous calcium carbonate, Sci. Rep., 2021, 11(1), 11546 CrossRef CAS.
- H. Ding, H. Pan, X. Xu and R. Tang, Toward a detailed understanding of magnesium ions on hydroxyapatite crystallization inhibition, Cryst. Growth Des., 2014, 14(2), 763–769 CrossRef CAS.
- S. Jiang, Y. Chen, H. Pan, Y.-J. Zhang and R. Tang, Faster nucleation at lower pH: amorphous phase mediated nucleation kinetics, Phys. Chem. Chem. Phys., 2013, 15(30), 12530–12533 RSC.
- S. Jiang, H. Pan, Y. Chen, X. Xu and R. Tang, Amorphous calcium phosphate phase-mediated crystal nucleation kinetics and pathway, Faraday Discuss., 2015, 179, 451–461 RSC.
- Z. Guo, Z. Liu and R. Tang, Applications of amorphous inorganics as novel functional materials, Mater. Chem. Front., 2024, 8(7), 1703–1730 RSC.
- Y. Xiao, J. Lin, Y. Zhao, X. Wang, Q. Lv, W. Li, R. Tang and G. Fu, Plaque-Specific Adhesive Balloons Coated with Calcium Phosphate Nanoparticles Loaded with Rapamycin for Atherosclerosis Therapy, Adv. Funct. Mater., 2024, 2315317 CrossRef CAS.
- B. Jin, Z. Liu, C. Shao, J. Chen, L. Liu, R. Tang and J. J. De Yoreo, Phase transformation mechanism of amorphous calcium phosphate to hydroxyapatite investigated by liquid-cell transmission electron microscopy, Cryst. Growth Des., 2021, 21(9), 5126–5134 CrossRef CAS.
- B. Xie, T. J. Halter, B. M. Borah and G. H. Nancollas, Tracking amorphous precursor formation and transformation during induction stages of nucleation, Cryst. Growth Des., 2014, 14(4), 1659–1665 CrossRef CAS.
- C.-G. Wang, J.-W. Liao, B.-D. Gou, J. Huang, R.-K. Tang, J.-H. Tao, T.-L. Zhang and K. Wang, Crystallization at multiple sites inside particles of amorphous calcium phosphate, Cryst. Growth Des., 2009, 9(6), 2620–2626 CrossRef CAS.
- M.-L. Qi, J. Qi, G.-Y. Xiao, K.-Y. Zhang, C.-Y. Lu and Y.-P. Lu, One-step hydrothermal synthesis of carbonated hydroxyapatite porous microspheres with a large and uniform size regulated by l-glutamic acid, CrystEngComm, 2016, 18(31), 5876–5884 RSC.
- M. Tavafoghi and M. Cerruti, The role of amino acids in hydroxyapatite mineralization, J. R. Soc., Interface, 2016, 13(123), 20160462 CrossRef.
- Z. Li, Q. Ren, J. Cui, D. Hu, T. Tian, T. He, K. Wang, W. Jiang and L. Zhang, Comparing the efficacy of hydroxyapatite nucleation regulated by amino acids, poly-amino acids and an amelogenin-derived peptide, CrystEngComm, 2020, 22(22), 3814–3823 RSC.
- Z. Wang, Z. Xu, W. Zhao and N. Sahai, A potential mechanism for amino acid-controlled crystal growth of hydroxyapatite, J. Mater. Chem. B, 2015, 3(47), 9157–9167 RSC.
- W.-L. Xu, B. Liu, Y.-C. Wang, G.-Y. Xiao, X. Chen, W.-H. Xu and Y.-P. Lu, A facile strategy for one-step hydrothermal preparation of porous hydroxyapatite microspheres with core–shell structure, J. Mater. Res. Technol., 2022, 17, 320–328 CrossRef CAS.
- M.-L. Qi, S. Qin, Y.-C. Wang, S. Yao, L. Qi, Y. Wu, Y.-P. Lu and F. Cui, Controlled synthesis of hydroxyapatite nanomaterials regulated by different phosphorus sources, Crystals, 2020, 10(8), 678 CrossRef CAS.
- Y.-C. Wang, Z.-C. Wang, G.-Y. Xiao, W.-L. Xu, K. Wang, Y. Jiao, M.-L. Qi and Y.-P. Lu, Investigation on [OH−]- responsive systems for construction of one-dimensional hydroxyapatite via a solvothermal method, New J. Chem., 2021, 45(1), 358–364 RSC.
- J. Jana, T. S. K. Sharma, J. S. Chung, W. M. Choi and S. H. Hur, Role of Physicochemical Features toward Bifunctional Redox Activity of Transition-Metal Molybdates, ACS Sustain. Chem. Eng., 2023, 11(35), 13013–13023 CrossRef CAS.
- S. V. Dorozhkin, Synthetic amorphous calcium phosphates (ACPs): Preparation, structure, properties, and biomedical applications, Biomater. Sci., 2021, 9(23), 7748–7798 RSC.
- T. S. K. Sharma, et al., Rational synthesis of alkaline earth metal vanadates: Structural origin of MgVO3 honeycomb lattice system and its electrochemical analysis for the detection of sulfadiazine, Chem. Eng. J., 2023, 464, 142673 CrossRef.
- S. Gadaleta, E. Paschalis, F. Betts, R. Mendelsohn and A. Boskey, Fourier transform infrared spectroscopy of the solution-mediated conversion of amorphous calcium phosphate to hydroxyapatite: new correlations between X-ray diffraction and infrared data, Calcif. Tissue Int., 1996, 58, 9–16 CrossRef CAS.
- M. Vignoles, G. Bonel, D. Holcomb and R. Young, Influence of preparation conditions on the composition of type B carbonated hydroxyapatite and on the localization of the carbonate ions, Calcif. Tissue Int., 1988, 43, 33–40 CrossRef CAS PubMed.
- S. P. Parthiban, I. Y. Kim, K. Kikuta and C. Ohtsuki, Effect of urea on formation of hydroxyapatite through double-step hydrothermal processing, Mater. Sci. Eng., C, 2011, 31(7), 1383–1388 CrossRef CAS.
- N. Blumenthal, A. Posner and J. Holmes, Effect of preparation conditions on the properties and transformation of amorphous calcium phosphate, Mater. Res. Bull., 1972, 7(11), 1181–1189 CrossRef CAS.
- J. Tao, H. Pan, J. Wang, J. Wu, B. Wang, X. Xu and R. Tang, Evolution of amorphous calcium phosphate to hydroxyapatite probed by gold nanoparticles, J. Phys. Chem. C, 2008, 112(38), 14929–14933 CrossRef CAS.
- Z. Zyman, A. Goncharenko and D. Rokhmistrov, Kinetics and mechanisms of the transformation of precipitated amorphous calcium phosphate with a Ca/P ratio of 1: 1 to calcium pyrophosphates, J. Cryst. Growth, 2017, 478, 117–122 CrossRef CAS.
- Z. Zyman, M. Epple, A. Goncharenko, D. Rokhmistrov, O. Prymak and K. Loza, Thermally induced crystallization and phase evolution in powders derived from amorphous calcium phosphate precipitates with a Ca/P ratio of 1: 1, J. Cryst. Growth, 2016, 450, 190–196 CrossRef CAS.
- Z. Zyman, M. Epple, A. Goncharenko, D. Rokhmistrov, O. Prymak and K. Loza, Peculiarities in thermal evolution of precipitated amorphous calcium phosphates with an initial Ca/P ratio of 1: 1, J. Mater. Sci.: Mater. Med., 2017, 28, 1–7 CrossRef CAS PubMed.
- R. Pierotti and J. Rouquerol, Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity, Pure Appl. Chem., 1985, 57(4), 603–619 CrossRef.
- Z. Xue, M. Yang and D. Xu, Nucleation of biomimetic hydroxyapatite nanoparticles on the surface of type I collagen: molecular dynamics investigations, J. Phys. Chem. C, 2019, 123(4), 2533–2543 CrossRef CAS.
- M. W. Swift, C. G. Van de Walle and M. P. Fisher, Posner molecules: from atomic structure to nuclear spins, Phys. Chem. Chem. Phys., 2018, 20(18), 12373–12380 RSC.
- A. S. Posner and F. Betts, Synthetic amorphous calcium phosphate and its relation to bone mineral structure, Acc. Chem. Res., 1975, 8(8), 273–281 CrossRef CAS.
- S. V. Dorozhkin, Calcium Orthophosphate-Based Bioceramics and Biocomposites, John Wiley & Sons, 2016 Search PubMed.
- S. V. Dorozhkin, Amorphous calcium orthophosphates: nature, chemistry and biomedical applications, Int. J. Mater. Chem., 2012, 2(1), 19–46 CrossRef CAS.
- E. D. Eanes, Amorphous calcium phosphate, Octacalcium phosphate, 2001, vol. 18, pp. 130–147 Search PubMed.
- F. Abbona and A. Baronnet, A XRD and TEM study on the transformation of amorphous calcium phosphate in the presence of magnesium, J. Cryst. Growth, 1996, 165(1–2), 98–105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04078c |
| This journal is © The Royal Society of Chemistry 2024 |