
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, molecular docking, and dynamics studies of novel thiazole-Schiff base derivatives containing a fluorene moiety and the assessment of their antimicrobial and antioxidant activity†
Sumita Saznin Marufaa,
Tasnim Rahmana,
Mohammad Mostafizur Rahman *a,
Md. Mizanur Rahmana,
Samira Jarin Khana,
Rownok Jahana,
Hiroshi Nishinob,
Mohammad Sayed Alam*a and
Md. Aminul Haque*a
*a,
Md. Mizanur Rahmana,
Samira Jarin Khana,
Rownok Jahana,
Hiroshi Nishinob,
Mohammad Sayed Alam*a and
Md. Aminul Haque*a
aDepartment of Chemistry, Jagannath University, Dhaka 1100, Bangladesh. E-mail: mostafiz@chem.jnu.ac.bd; msalam@chem.jnu.ac.bd; amin2k12@chem.jnu.ac.bd; Fax: +880-027113752; Tel: +880-029583794 Tel: +88-01711287600
bDepartment of Chemistry, Graduate School of Science and Technology, Kumamoto University, Kumamoto, Japan
First published on 4th November 2024
Abstract
In this study, a series of eighteen fluorene-containing substituted thiazole derivatives were synthesized and characterized via spectral analyses. The proposed compounds were screened for their in vitro antimicrobial activity, and it was found that compound 2a displayed a significant zone of inhibition (20.3 ± 0.6 mm) against B. subtilis and compound 2b exhibited inhibitory activity (30.3 ± 0.6 mm) against a C. albicans fungal strain. Furthermore, antioxidant activity was evaluated for all analogues, where 2f exhibited a four-fold higher antioxidant capability (11.73 ± 1.22 μg mL−1) than the standard ascorbic acid. Oral bioavailability and toxicological parameters were considered, and most of the compounds satisfied Lipinski's rule of five and Veber's rule, except for one violation by a few derivatives. Molecular docking and molecular dynamics simulation were performed, providing more explicit ideas on the binding interaction and stability of compounds that exhibited wet lab activity. Average RMSD and RMSF values ranged between 0.5 Å and 2.5 Å, which indicated the stability of ligands inside the complex, yielding some engrossing insights.
1. Introduction
The intensity of the antimicrobial drug resistance and rising inactivity of antibiotics can turn the world back to the pre-antibiotic era, where a simple infection would cause a pernicious situation.1,2 Additionally, inappropriate use and overuse of antibiotics are further deteriorating this condition, and the case fatality rate is escalating all over the world.3,4 To ameliorate these circumstances, we need an acute plan, systematic synthetic process, and detailed action regarding the accuracy and selectivity in drug design. Molecular hybridization is crucial in such situations for intermixing distinct active biological groups.5,6 From the history of different generations of antibiotics, Schiff bases are marked as a prominent figure in various drug designs.7 They have a wide range of uses as antimicrobial,8,9 anticancer,10 antioxidant,11 antiviral,12 and anti-Alzheimer's13 agents, indicating the significance of these bioactive scaffolds in the synthesis of new drugs to deal with this detrimental state of antimicrobial resistance. Many commercially available drugs, such as methisazone, thioacetazone, and triapine, contain Schiff bases and are used as a medication for cancer and tubercular (Fig. 1). Thiazole, another key moiety, can stimulate the activity of drugs, as established by numerous studies, generating a continuous research interest in the medicinal sector.14–16 Penicillin, the first invented antibiotic, carries the thiazole group and lactam ring, which brought huge optimism to the pharmaceutical sector. Other popular drugs, such as meloxicam (anti-inflammatory), epothilone D (anticancer), and abafungin (antifungal), bear the thiazole moiety, thus significantly protecting against infectious diseases. Its presence in many biologically active compounds makes it one of the extensively studied heterocycles.17–19 The strong S–C–N fragment of thiazole has a wide range of pharmacological properties, such as antitumor,20 anti-inflammatory,21 antimicrobial,22 antioxidant,23 neuroprotective,24 anti-diabetic25 and anticancer26 effects. In contrast, fluorene, a polycyclic aromatic hydrocarbon, is widely used in industries, including in the manufacturing of plastics, pesticides, and dyes, as well as for its promising implementation as a pharmacophore in the medicinal sector. Fluorene-containing derivatives carry an encouraging pharmacological profile, such as antimicrobial,27,28 antiviral,29 antimalarial,30 anticancer31 antioxidant,32 and anti-Alzheimer's33 effects, which position fluorene as a predominant synthetic moiety for future drug regimes. Fluorene-containing commercial drugs such as indecainide (anti-arrhythmic), tilorone (antiviral), and hexafluronium (muscle relaxant) behold its therapeutic potential for future drug design. By considering previous findings, we predicted the incorporation of Schiff base and thiazole scaffolds, along with fluorene, would be a credible step in anticipation of this ongoing crisis of antimicrobial drug resistance. A series of Schiff base-thiazole-fluorene derivatives were synthesized in this study and were evaluated for their antimicrobial and antioxidant activities. Free radicals and reactive oxidative species (ROS) expand many chronic diseases, including cancer, aging, anemia, inflammation and many cardiovascular diseases.34,35 To combat against this deleterious effect of oxidative stress, the antioxidant capability of the synthesized analogues was depicted in this study. The disc diffusion method was used for the antimicrobial activity of the novel analogues, and antioxidant activity was determined using the DPPH free radical scavenging assay. In silico study is an important approach for the investigation and development of drug design, which analyzes computer-based structures and considers thermodynamics through binding derivatives within protein receptors.36,37 As a part of the advancement of these novel organic molecules, an in silico study was performed on these new chemical entities, including optimization, molecular docking, ADMET and molecular dynamics (MD) simulation.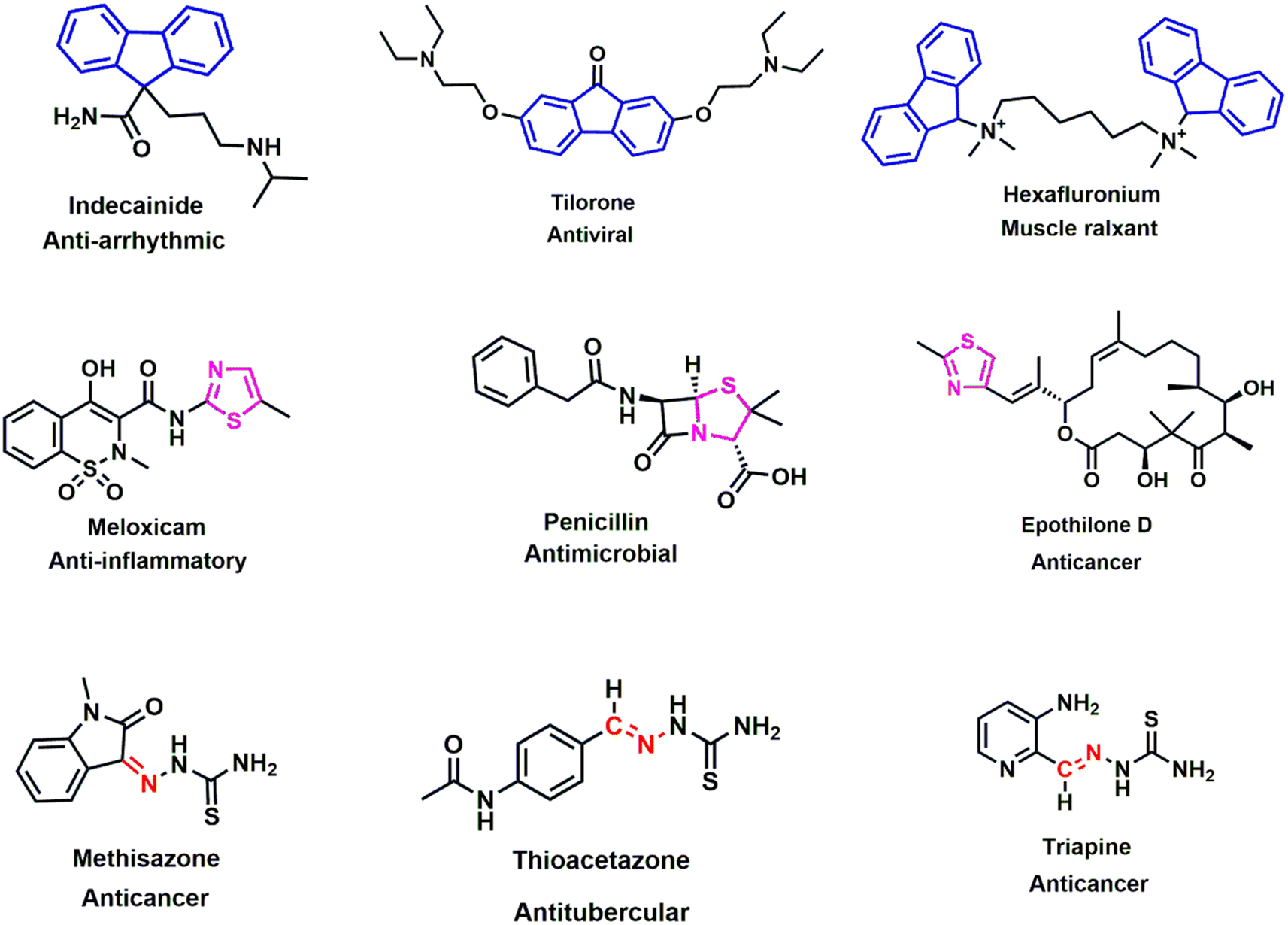 | ||
| Fig. 1 Chemical structures of several fluorene (blue), thiazole (pink) and Schiff base (red) bearing biologically active compounds. | ||
2. Experimental
2.1 General method
All the reagents and solvents were purchased from Sigma-Aldrich and TCI Chemical Industries, Ltd and used without further purification. Melting points of the derivatives were determined via an SMP10 melting point apparatus and were uncorrected. IR spectra (KBr pellets) were recorded by a SHIMADZU IRTracer-100 infrared spectrophotometer. 1H NMR (400 MHz) spectra of the samples were documented using a BRUKER NMR spectrometer with DMSO-d6 as a solvent. HRMS data using a JEOL JMS-700 MStation were obtained from the Instrumental Analysis Center, Kumamoto University, Kumamoto, Japan. The progress of the reactions was observed using aluminum-coated silica gel containing TLC plates (Merck, Germany), which was visualized by exposure to UV light (Scheme 1).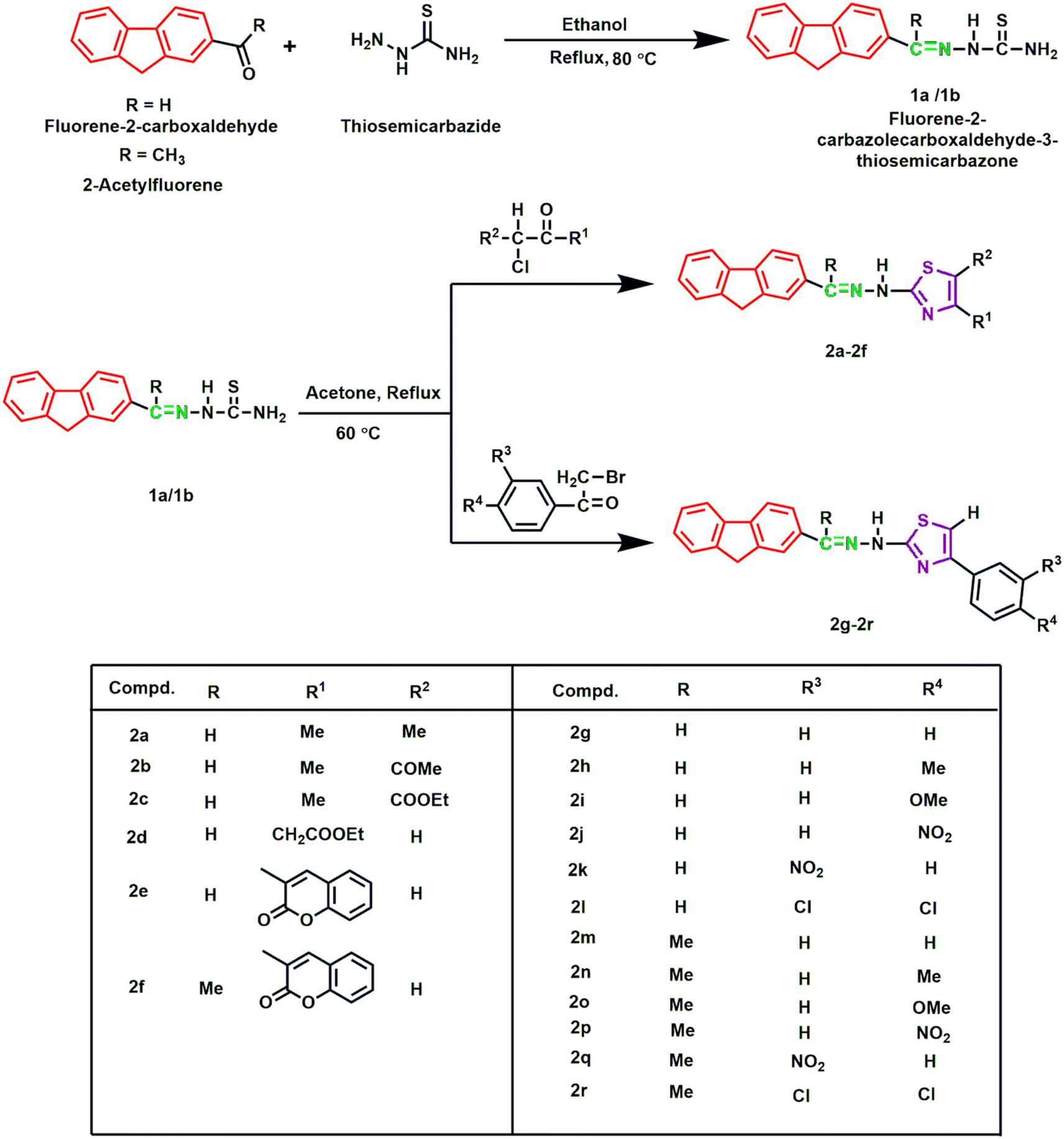 | ||
| Scheme 1 Synthetic routes to the target derivatives 2a–2r. | ||
2.2 Synthesis
2.2.1.1 2-((9H-Fluoren-2-yl)methylidene)hydrazine-1-carbothioamide (1a). Colorless solid, Rf value: 0.85 (EtOAc
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane = 1:2), m.p. = 257–258 °C. IR (KBr, νmax, cm−1): 3144 (NH), 1590 (C
:hexane = 1:2), m.p. = 257–258 °C. IR (KBr, νmax, cm−1): 3144 (NH), 1590 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1533 (CC, Ar), 1296 (CS). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 3.95 (s, 2H, CH2), 7.34–7.40 (m, 2H, Ar–H), 7.61 (d, J = 7.6 Hz, 1H, Ar–H), 7.77 (d, J = 8.0 Hz, 1H, Ar–H), 7.92–7.95 (m, 2H, Ar–H), 8.05–8.08 (m, 2H, NH2), 8.13 (s, 1H, Ar–H), 8.20 (s, 1H, HCN), 11.44 (s, 1H, NH). FAB HRMS (acetone/NBA) calcd. for C15H14N3S 268.0908 [M + H]+. Found 268.0933.
N), 1533 (CC, Ar), 1296 (CS). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 3.95 (s, 2H, CH2), 7.34–7.40 (m, 2H, Ar–H), 7.61 (d, J = 7.6 Hz, 1H, Ar–H), 7.77 (d, J = 8.0 Hz, 1H, Ar–H), 7.92–7.95 (m, 2H, Ar–H), 8.05–8.08 (m, 2H, NH2), 8.13 (s, 1H, Ar–H), 8.20 (s, 1H, HCN), 11.44 (s, 1H, NH). FAB HRMS (acetone/NBA) calcd. for C15H14N3S 268.0908 [M + H]+. Found 268.0933.
2.2.1.2 2-(1-(9H-Fluoren-2-yl)ethylidene)hydrazine-1-carbothioamide (1b). Pale yellow solid, Rf value: 0.76 (EtOAc
:hexane = 1:1), m.p. = 267–268 °C. IR (KBr, νmax, cm−1): 3166 (NH), 1591 (CN), 1503 (CC, Ar), 1272 (CS). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 3.35 (s, 3H, NC–CH3), 3.95 (s, 2H, CH2), 7.34–7.40 (m, 2H, Ar–H), 7.61 (d, J = 7.60 Hz, 1H, Ar–H), 7.88–7.98 (m, 4H, Ar–H), 8.19 (s, 2H, NH2), 10.23 (s, 1H, NH). FAB HRMS (acetone/NBA) calcd. for C16H16N3S 282.1065 [M + H]+. Found 282.1073.
2.2.2.1 2-{2-[(9H-Fluoren-2-yl)methylidene]hydrazine-1-yl}-4,5-dimethyl-1,3-thiazole (2a). Orange solid, Rf value: 0.85 (EtOAc
:hexane = 1:2), m.p. = 224–225 °C. IR (KBr, νmax, cm−1): 3018 (NH), 1588 (CN), 1534 (CC, Ar), 741 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 2.17 (s, 3H, CH3), 2.20 (s, 3H, CH3), 3.99 (s, 2H, CH2), 7.34–7.43 (m, 2H, Ar–H), 7.62 (d, J = 7.20 Hz, 1H, Ar–H), 7.77 (d, J = 8.00 Hz, 1H, Ar–H), 7.94–7.99 (m, 3H, Ar–H), 8.41 (s, 1H, NCH). FAB HRMS (acetone/NBA) calcd. for C19H18N3S 320.1221 [M + H]+. Found 320.1212.
2.2.2.2 1-{2-[2-[(9H-Fluoren-2-yl)methylidene]hydrazine-1-yl]-4-methyl-1,3-thiazol-5-yl}ethan-1-one (2b). Yellow solid, Rf value: 0.40 (EtOAc
:hexane = 3:4), m.p. = 273–274 °C. IR (KBr, νmax, cm−1): 3168 (NH), 1738 (CO), 1625 (CN), 1577 (CC, Ar), 738 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 2.43 (s, 3H, CH3), 2.49 (s, 3H, COCH3), 3.99 (s, 2H, CH2), 7.33–7.43 (m, 2H, Ar–H), 7.61 (d, J = 7.20 Hz, 1H, Ar–H), 7.69 (d, J = 7.60 Hz, 1H, Ar–H), 7.93–7.97 (m, 3H, Ar–H), 8.20 (s, 1H, NCH). FAB HRMS (acetone/NBA) calcd. for C20H17N3NaOS 370.0990 [M + Na]+. Found 370.1021.
2.2.2.3 Ethyl-{2-[2-[(9H-fluoren-2-yl)methylidene]hydrazine-1-yl]-4-methyl-1,3-thiazol-5-yl}carboxylate (2c). Colorless solid, Rf value: 0.76 (EtOAc
:hexane = 1:1), m.p. = 250–251 °C. IR (KBr, νmax, cm−1): 3032 (NH), 1714 (CO), 1618 (CN), 1540 (CC, Ar), 734 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 1.28 (t, J = 7.20 Hz, 3H, OCH2CH3), 2.48 (s, 3H, CH3), 3.99 (s, 2H, CH2), 4.21 (q, J = 7.20 Hz, 2H, OCH2CH3), 7.33–7.42 (m, 2H, Ar–H), 7.61 (d, J = 7.2 Hz, 1H, Ar–H), 7.68 (d, J = 8.00 Hz, 1H, Ar–H), 7.92–7.96 (m, 3H, Ar–H), 8.19 (s, 1H, NCH). FAB HRMS (acetone/NBA) calcd. for C21H19N3NaO2S 400.1096 [M + Na]+. Found 400.1101.
2.2.2.4 Ethyl-2-{2-[2-[(9H-fluoren-2-yl)methylidene]hydrazine-1-yl]-1,3-thiazol-4-yl}acetate (2d). Colorless solid, Rf value: 0.80 (EtOAc
:hexane = 1:1), m.p. = 215–216 °C. IR (KBr, νmax, cm−1): 3127 (NH), 1723 (CO), 1621 (CN), 1508 (CC, Ar), 732 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 1.21 (t, J = 7.20 Hz, 3H, OCH2CH3), 3.68 (s, 2H, CH2COO), 3.99 (s, 2H, CH2), 4.12 (q, J = 7.20 Hz, 2H, OCH2CH3), 6.76 (s, 1H, thiazole-H), 7.33–7.42 (m, 2H, Ar–H), 7.61 (d, J = 7.20 Hz, 1H, Ar–H), 7.69 (d, J = 8.00 Hz, 1H, Ar–H), 7.93–7.97 (m, 3H, Ar–H), 8.20 (s, 1H, NCH). FAB HRMS (acetone/NBA) calcd. for C21H20N3O2S 378.1276 [M + H]+. Found 378.1281.
2.2.2.5 3-{2-[2-[(9H-Fluoren-2-yl)methylidene]hydrazine-1-yl]-1,3-thiazol-4-yl}-2H-chromen-2-one (2e). Yellow solid, Rf value: 0.87 (EtOAc
:hexane = 1:2), m.p. = 275–276 °C. IR (KBr, νmax, cm−1): 3014 (NH), 1717 (CO), 1627 (CN), 1507 (CC, Ar), 767 (C–S–C). 1H NMR (DMSO- d6): 3.99 (s, 2H, CH2), 7.35–7.47 (m, 4H, Ar–H), 7.61–7.68 (m, 3H, Ar–H), 7.79 (s, 1H, thiazole-H), 7.85 (d, J = 7.20 Hz, 1H, Ar–H), 7.90–7.97 (m, 3H, Ar–H), 8.15 (s, 1H, Ar–H), 8.56 (s, 1H, CH). FAB HRMS (acetone/NBA) calcd. for C26H18N3O2S 436.1120 [M + H]+. Found 436.1117.
2.2.2.6 3-{2-[2-[(9H-Fluoren-2-yl)ethylidene]hydrazine-1-yl]-1,3-thiazol-4-yl}-2H-chromen-2-one (2f). Yellow solid, Rf value: 0.57 (EtOAc
:hexane = 1:1), m.p = 283–284 °C. IR (KBr, νmax, cm−1): 3028 (NH), 1717 (CO), 1627 (CN), 1546 (CC, Ar), 726 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 2.39 (s, 3H, NC–CH3), 3.98 (s, 2H, CH2), 7.33–7.46 (m, 4H, Ar–H), 7.59–7.61 (m, 2H, Ar–H), 7.79 (s, 1H, thiazole-H), 7.81–7.85 (m, 2H, Ar–H), 7.91–7.97 (m, 2H, Ar–H), 8.01 (s, 1H, Ar–H), 8.59 (s, 1H, Ar–H). FAB HRMS (acetone/NBA) calcd. for C27H20N3O2S 450.1276 [M + H]+. Found 450.1266.
2.2.2.7 2-{2-[(9H-Fluoren-2-yl)methylidene]hydrazine-1-yl]1,3-thiazol-4-yl} phenyl (2g). Cream-colored solid, Rf value: 0.93 (EtOAc
:hexane = 1:2), m.p. = 237–238 °C. IR (KBr, νmax, cm−1): 3055 (NH), 1624 (CN), 1507 (CC, Ar), 732 (C–S–C). 1H NMR (DMSO- d6): 3.98 (s, 2H, CH2), 7.31 (s, 1H, thiazole-H), 7.32–7.43 (m, 5H, Ar–H), 7.61 (d, J = 7.20 Hz, 1H, Ar–H), 7.66 (d, J = 8.00 Hz, 1H, Ar–H), 7.84–7.95 (m, 5H, Ar–H), 8.13 (s, 1H, NCH). FAB HRMS (acetone/NBA) calcd. for C23H18N3S 368.1221 [M + H]+. Found 368.1220.
2.2.2.8 2-{2-[(9H-Fluoren-2-yl)methylidene]hydrazine-1-yl}-1,3-thiazol-4-yl-4-methylphenyl (2h). Cream-colored solid, Rf value: 0.95 (EtOAc
:hexane = 1:2), m.p. = 255–256 °C. IR (KBr, νmax, cm−1): 3060 (NH), 1622 (CN), 1508 (CC, Ar), 732 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 2.32 (s, 3H, CH3), 3.99 (s, 2H, CH2), 7.22 (d, J = 8.00 Hz, 2H, Ar–H), 7.25 (s, 1H, thiazole-H), 7.33–7.41 (m, 2H, Ar–H), 7.59–7.67 (m, 2H, Ar–H), 7.74 (d, J = 8.00 Hz, 2H, Ar–H), 7.89–7.96 (m, 3H, Ar–H), 8.13 (s, 1H, NCH). FAB HRMS (acetone/NBA) calcd. for C24H20N3S 382.1378 [M + H]+. Found 382.1382.
2.2.2.9 2-{2-[(9H-Fluoren-2-yl)methylidene]hydrazine-1-yl}-1,3-thiazol-4-yl-4-methoxyphenyl (2i). Brown solid, Rf value: 0.88 (EtOAc
:hexane = 1:2), m.p. = 239–240 °C. IR (KBr, νmax, cm−1): 3069 (NH), 1626 (CN), 1573 (CC, Ar), 731 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 3.79 (s, 3H, OCH3), 3.99 (s, 2H, CH2), 6.99 (dd, J = 2.00 Hz, 6.80 Hz, 2H, Ar–H), 7.16 (s, 1H, thiazole-H), 7.33–7.43 (m, 2H, Ar–H), 7.62 (d, J = 0.80 Hz, 1H, Ar–H), 7.68 (d, J = 0.80 Hz, 1H, Ar–H), 7.79 (dd, J = 6.80 Hz, 2.00 Hz, 2H, Ar–H), 7.96–7.89 (m, 3H, Ar–H), 8.13 (s, 1H, NCH). FAB HRMS (acetone/NBA) calcd. for C24H20N3OS 398.1327 [M + H]+. Found 398.1311.
2.2.2.10 2-{2-[(9H-Fluoren-2-yl)methylidene]hydrazine-1-yl}-1,3-thiazol-4-yl-4-nitrophenyl (2j). Orange solid, Rf value: 0.75 (EtOAc
:hexane = 1:2), m.p. = 266–267 °C. IR (KBr, νmax, cm−1): 3068 (NH), 1632 (CN), 1574 (CC, Ar), 1526, 733 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 3.99 (s, 2H, CH2), 7.33–7.43 (m, 2H, Ar–H), 7.61 (d, J = 7.20 Hz, 1H, Ar–H), 7.67 (d, J = 8.00 Hz, 1H, Ar–H), 7.73 (s, 1H, thiazole-H), 7.90–7.97 (m, 3H, Ar–H), 8.10–8.16 (m, 2H, Ar–H), 8.14 (s, 1H, NCH), 8.27–8.30 (m, 2H, Ar–H). FAB HRMS (acetone/NBA) calcd. for C23H17N4O2S 413.1072 [M + H]+. Found 413.1068.
2.2.2.11 2-{2-[(9H-Fluoren-2-yl)methylidene]hydrazine-1-yl}-1,3-thiazol-4-yl-3-nitrophenyl (2k). Colorless solid, Rf value: 0.90 (EtOAc
:hexane = 1:2), m.p. = 256–257 °C. IR (KBr, νmax, cm−1): 3051 (NH), 1632 (CN), 1559 (CC Ar), 728 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 3.99 (s, 2H, CH2), 7.35–7.41 (m, 2H, Ar–H), 7.61 (d, J = 7.20 Hz, 1H, Ar–H), 7.67 (s, 1H, thiazole-H), 7.68–7.74 (m, 2H, Ar–H), 7.90–7.96 (m, 3H, Ar–H), 8.14–8.17 (m, 2H, Ar–H, CH), 8.32 (d, J = 8.40 Hz, 1H, Ar–H), 8.69 (s, 1H, Ar–H). FAB HRMS (acetone/NBA) calcd. for C23H17N4O2S 413.1072 [M + H]+. Found 413.1080.
2.2.2.12 2-{2-[(9H-Fluoren-2-yl)methylidene]hydrazine-1-yl}-1,3-thiazol-4-yl-3,4-dichlorophenyl (2l). Brown solid, Rf value: 0.41 (EtOAc
:hexane = 1:2), m.p. = 249–250 °C. IR (KBr, νmax, cm−1): 3075 (NH), 1681 (CN), 1507 (CC, Ar), 730 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 3.99 (s, 2H, CH2), 7.33–7.41 (m, 2H, Ar–H), 7.55 (s, 1H, thiazole-H), 7.59–7.68 (m, 3H, Ar–H), 7.83–7.96 (m, 4H, Ar–H), 8.02 (d, 1H, J = 0.2 Hz, Ar–H), 8.13 (s, 1H, NCH). FAB HRMS (acetone/NBA) calcd. for C23H16Cl2N3S 436.0442 [M + H]+. Found 436.0420.
2.2.2.13 2-{2-[1-(9H-Fluoren-2-yl)ethylidene]hydrazine-1-yl}-4-phenyl-1,3-thiazol (2m). Cream-colored solid, Rf value: 0.79 (EtOAc
:hexane = 1:1), m.p. = 270–271 °C. IR (KBr, νmax, cm−1): 3062 (NH), 1621 (CN), 1506 (CC, Ar), 731 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 2.39 (s, 3H, NC–CH3), 3.99 (s, 2H, CH2), 7.33 (t, J = 1.20 Hz, 1H, Ar–H), 7.35 (s, 1H, thiazole-H), 7.36–7.44 (m, 4H, Ar–H), 7.61 (d, J = 7.20 Hz, 1H, Ar–H), 8.01 (s, 1H, Ar–H), 7.84–7.95 (m, 5H, Ar–H). FAB HRMS (acetone/NBA) calcd. for C24H19N3S 381.1300 [M + H]+. Found 381.1304.
2.2.2.14 2-{2-[1-(9H-Fluoren-2-yl)ethylidene]hydrazine-1-yl}-4-(4-methylphenyl)-1,3-thiazole (2n). Cream-colored solid, Rf value: 0.76 (EtOAc
:hexane = 1:1), m.p. = 268–269 °C, IR (KBr, νmax, cm−1): 3046 (NH), 1623 (CN), 1507 (CC, Ar), 733 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 2.09 (s, 3H, NC–CH3), 2.51 (t, J = 2.00 Hz, 3H, CH3), 3.99 (s, 2H, CH2), 7.24 (d, J = 8.00 Hz, 2H, Ar–H), 7.26 (s, 1H, thiazole-H), 7.32–7.42 (m, 2H, Ar–H), 7.61 (d, J = 7.60 Hz, 1H, Ar–H), 7.76–7.86 (m, 3H, Ar–H), 7.92–7.95 (m, 2H, Ar–H), 8.01 (s, 1H, Ar–H). FAB HRMS (acetone/NBA) calcd. for C25H22N3S 396.1534 [M + H]+. Found 396.1518.
2.2.2.15 2-{2-[1-(9H-Fluoren-2-yl)ethylidene]hydrazine-1-yl}-4-(4-methoxyphenyl)-1,3-thiazol (2o). Brown solid, Rf value: 0.60 (EtOAc
:hexane = 1:1), m.p. = 258–259 °C. IR (KBr, νmax, cm−1): 3078 (NH), 1627 (CN), 1574 (CC, Ar), 736 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 2.39 (s, 3H, NC–CH3), 3.79 (s, 3H, –OCH3), 3.99 (s, 2H, CH2), 7.32–7.42 (m, 2H, Ar–H), 6.98–7.00 (m, 2H, Ar–H), 7.61 (d, J = 7.20 Hz, 1H, Ar–H), 7.82–7.86 (m, 3H, Ar–H), 7.92–7.95 (m, 2H, Ar–H), 8.01 (s, 1H, Ar–H). FAB HRMS (acetone/NBA) calcd. for C25H22N3OS 412.1484 [M + H]+. Found 412.1470.
2.2.2.16 2-{2-[1-(9H-Fluoren-2-yl)ethylidene]hydrazine-1-yl-}-4-(4-nitrophenyl)-1,3-thiazole (2p). Orange solid, Rf value: 0.73 (EtOAc
:hexane = 1:1), m.p. = 278–279 °C, IR (KBr, νmax, cm−1): 3341 (NH), 3113 (CC), 1625 (CN), 1598 (CC, Ar), 732 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 2.40 (s, 3H, NC–CH3), 3.99 (s, 2H, CH2), 7.17 (s, 1H, thiazole-H), 7.32–7.41 (m, 2H, Ar–H), 7.61 (d, J = 7.20 Hz, 1H, Ar–H), 7.76 (s, 1H, thiazole-H), 7.84–7.86 (m, 1H, Ar–H), 7.93–7.96 (m, 2H, Ar–H), 8.01 (d, J = 0.80 Hz,1H, Ar–H), 8.14–8.17 (m, 2H, Ar–H), 8.29–8.31 (m, 2H, Ar–H). FAB HRMS (acetone/NBA) calcd. for C24H19N4O2S 427.1229 [M + H]+. Found 427.1233.
2.2.2.17 2-{2-[1-(9H-Fluoren-2-yl)ethylidene]hydrazine-1-yl}-4-(3-nitrophenyl)-1,3-thiazole (2q). Cream-colored solid, Rf value: 0.69 (EtOAc
:hexane = 1:1), m.p. = 283–284 °C. IR (KBr, νmax, cm−1): 3055 (NH), 1628 (CN), 1592 (CC Ar), 729 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 2.39 (s, 3H, NC–CH3), 7.34–7.40 (m, 2H, Ar–H), 3.99 (s, 2H, CH2), 7.61 (d, J = 7.20 Hz, 1H, Ar–H), 7.67 (s, 1H, thiazole-H), 7.72 (t, J = 8.00 Hz, 1H, Ar–H), 7.85 (dd, J = 1.20, 8 Hz, 1H, Ar–H), 7.91–7.95 (m, 2H, Ar–H), 8.01 (s, 1H, Ar–H), 8.14–8.17 (m, 1H, Ar–H), 8.34 (d, J = 1.20 Hz, 1H, Ar–H), 8.73 (s, 1H, Ar–H). FAB HRMS (acetone/NBA) calcd. for C24H19N4O2S 427.1229 [M + H]+. Found 427.1223.
2.2.2.18 2-{2-[1-(9H-Fluoren-2-yl)ethylidene]hydrazine-1-yl}-4-(3,4-dichlorophenyl)-1,3-thiazole (2r). Brown solid, Rf value: 0.80 (EtOAc
:hexane = 1:1), m.p. = 273–274 °C, IR (KBr, νmax, cm−1): 3084 (NH), 1617 (CN), 1507 (CC, Ar), 730 (C–S–C). 1H NMR (400 MHz, δ, ppm, DMSO-d6): 2.38 (s, 3H, NC–CH3), 3.98 (s, 2H, CH2), 7.32–7.42 (m, 2H, Ar–H), 7.56 (s, 1H, thiazole-H), 7.62 (d, J = 7.60 Hz, 1H, Ar–H), 7.68 (d, J = 8.40 Hz, 1H, Ar–H), 7.83–7.88 (m, 2H, Ar–H), 7.91–7.94 (m, 2H, Ar–H), 8.00 (s, 1H, Ar–H), 8.12 (s, 1H, Ar–H). FAB HRMS (acetone/NBA) calcd. for C24H18Cl2N3S 450.0598 [M + H]+. Found 450.0599.
2.3 Antimicrobial activity assay
The agar disc diffusion method was used to evaluate the antimicrobial activity of the synthesized compounds.38 For the broth culture of the tested organism, Mueller Hinton Agar (for bacteria) and Potato Dextrose Agar (for fungi) were used as the media. Tested organisms were inoculated on the media in an even fashion using a sterilized cotton bar after 24 h of incubation. Synthesized compounds with a concentration of 50 μL (dissolved in DMSO) were injected into the pre-seeded sample disc. Discs were incubated for 24 h at 37 °C for antibacterial and for 48 h at 26 °C for antifungal assays after injecting tested compounds. This procedure was followed for the standard ceftriaxone and amphotericin B (10 μL) to determine the antibacterial and antifungal activity.The efficacy of the tested compounds was evaluated against different strains in this study, namely two-gram positive such as Staphylococcus aureus and Bacillus subtilis, two gram-negative Escherichia coli and Shigella flexneri bacteria, and two fungal strains Candida albicans and Aspergillus niger.
2.4 Antioxidant activity
The antioxidant capability of the synthesized compounds was determined by using the DPPH (2,2-diphenyl-1-picrylhydrazyl) radical scavenging method. A free radical stock solution was prepared in methanol (500 mL) with 3 mg of DPPH powder in a volumetric flask (covered with aluminum foil) and stirred for 2 h. This solution was kept in the refrigerator for 24 h under dark conditions. Then, the stock solution was taken in test tubes (4 mL each) against five different concentrations for a single tested compound. 100 μL of the prepared sample solution (in methanol) was added to each test tube and was kept in the dark for 20 minutes afterwards. This mixture was whirled in a vortex machine for a few seconds before recording the absorbance (at 517 nm) using a UV-visible spectrometer. The methanol solution was considered as the blank, and the percentage of inhibition was calculated using the following equation.here, Acontrol = absorbance of the DPPH radical and Asample = absorbance of DPPH with the sample. The IC50 values for ascorbic acid and synthesized compounds were calculated from the concentration–inhibition curves.
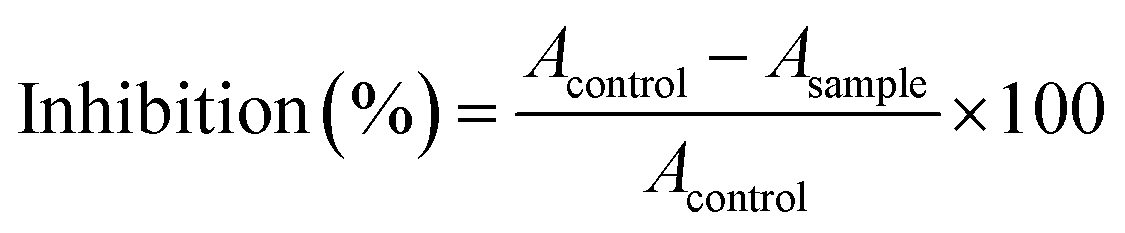
2.5 In silico analysis
3. Result and discussion
3.1 Chemistry
In this study, eighteen fluorene-based compounds were synthesized by substituting a thiazole ring in a two-step reaction methodology. Fluorene-2-carbaldehyde and 2-acetylfluorene reacted with thiosemicarbazide in an equimolar ratio to form two initial products 1a and 1b. Subsequently, α-haloketones and phenacyl bromide were cyclized with 1a and 1b to obtain the final products (2a–2r). All compounds were obtained in good yields, ranging from 70 to 96%, except compound 2a (50%). The structures of the synthesized analogues were established using different spectroscopic methods (HRMS, IR and NMR). Spectroscopic data of 2a, for instance, revealed IR absorption peaks at 3018, 1588, 1534, and 741 cm−1, confirming the presence of NH, CN, CC, and C–S–C functional groups.23 The 1H NMR spectrum of 2a displayed singlets at 2.17 ppm and 2.20 ppm for two thiazole-substituted methyl groups. Fluorene-ring containing CH2 group had a singlet at 3.99 ppm. Other substituent protons of the fluorene ring had peaks ranging from 7.34 to 7.99 ppm, where two doublets appeared at 7.62 ppm and 7.77 ppm, respectively. The predominant presence of the azomethene group (HCN) was confirmed at 8.41 ppm with a singlet peak. Respective structures of the synthesized derivatives agreed well with the calculated HRMS data.
3.2 Antimicrobial activity using the disc diffusion method
In vitro antimicrobial activities of the synthesized compounds and standards were evaluated using the agar disc diffusion method. The activity was shown as the zone of inhibition (mm) of the standard (ceftriaxone and amphotericin B) and tested compounds, which are represented in Table 1. Two-gram positive (Staphylococcus aureus and Bacillus subtilis), two-gram negative (Escherichia coli and Shigella flexneri) bacteria and two fungal (Aspergillus niger and Candida albicans) strains were used in this experiment. Compounds 2a and 2b displayed good activity against most of the strains. Specifically, 2a exhibited the highest activity against S. flexneri with an inhibition zone of 20.3 ± 0.6 mm and its activity against B. subtilis (20.3 ± 0.6 mm), which was bordering on the activity of the standard (20.7 ± 0.6). This extended antimicrobial activity of compound 2a might be influenced by the presence of thiazole-substituted methyl groups reported in previous studies.45 Like 2a, compound 2b, containing a methyl substituent along with an acetyl group, showed the highest activity against bacterial strain S. flexneri (17.0 ± 1.0) and B. subtilis (17.7 ± 0.6). Both compounds showed similar activity against S. aureus (16.3 ± 0.6 and 16.3 ± 0.6). In the case of compound 2c with an ester substituent, the activity declined noticeably, which could be related to its electron-withdrawing characteristics. Among others, compound 2q exhibited a zone of inhibition of 18.7 ± 0.6 against S. aureus, which was the maximum compared to other compounds. The nitro group presence in the aromatic ring of 2q might be the reason for its increased activity against S. aureus and S. flexneri. It was found that the presence of a nitro/chloro/fluoro group, while attached to the heterocyclic rings, leads to better antimicrobial properties and improved lipophilicity values.46| Compd. | Gram (+) bacteria | Gram (−) bacteria | Fungi | |||
|---|---|---|---|---|---|---|
| S. aureus | B. subtilis | E. coli | S. flexneri | C. albicans | A. niger | |
| a Data were expressed as mean ± SD of three experiments. – Represents no activity. | ||||||
| 2a | 16.3 ± 0.6 | 20.3 ± 0.6 | 12.0 ± 1.0 | 20.3 ± 0.6 | 22.3 ± 0.6 | 11.3 ± 0.6 |
| 2b | 16.3 ± 0.6 | 17.7 ± 0.6 | 15.7 ± 0.6 | 17.0 ± 1.0 | 30.3 ± 0.6 | 10.3 ± 0.6 |
| 2c | 11.7 ± 0.6 | 12.3 ± 0.6 | 9.7 ± 0.6 | 9.3 ± 0.6 | 11.3 ± 0.6 | — |
| 2d | 10.0 ± 1.0 | 13.3 ± 0.6 | 11.7 ± 0.6 | 16.3 ± 0.6 | 27.3 ± 0.6 | 12.3 ± 0.6 |
| 2e | — | 10.7 ± 0.6 | 8.3 ± 0.6 | 13.3 ± 0.6 | 11.3 ± 0.6 | 9.7 ± 0.6 |
| 2f | 11.7 ± 0.6 | 10.0 ± 1.0 | 11.0 ± 1.0 | 8.0 ± 1.0 | 12.3 ± 0.6 | 11.7 ± 0.6 |
| 2g | 12.3 ± 0.6 | 10.3 ± 0.6 | 12.3 ± 0.6 | 10.7 ± 0.6 | 12.7 ± 0.6 | 8.0 ± 1.0 |
| 2h | 9.0 ± 1.0 | 11.3 ± 0.6 | 13.0 ± 1.0 | 12.7 ± 0.6 | 14.3 ± 0.6 | 8.3 ± 0.6 |
| 2i | 13.0 ± 1.0 | 11.0 ± 1.0 | 9.3 ± 0.6 | 10.3 ± 0.6 | 9.7 ± 0.6 | 8.7 ± 0.6 |
| 2j | 14.3 ± 0.6 | 10.3 ± 0.6 | 12.3 ± 0.6 | 14.3 ± 0.6 | 12.0 ± 1.0 | 8.3 ± 0.6 |
| 2k | 13.3 ± 0.6 | 9.3 ± 0.6 | 10.0 ± 1.0 | 8.3 ± 0.6 | 13.3 ± 0.6 | 8.0 ± 1.0 |
| 2l | 14.0 ± 1.0 | 10.7 ± 0.6 | 9.7 ± 0.6 | 8.3 ± 0.6 | 10.7 ± 0.6 | 9.7 ± 0.6 |
| 2m | 9.3 ± 0.6 | — | 9.7 ± 0.6 | 9.0 ± 1.0 | 15.3 ± 0.6 | 18.0 ± 1.0 |
| 2n | 9.7 ± 0.6 | 9.0 ± 0.0 | 10.3 ± 0.6 | 9.3 ± 0.6 | 12.7 ± 0.6 | 20.3 ± 0.6 |
| 2o | — | — | 8.3 ± 0.6 | 10.0 ± 1.0 | 11.7 ± 0.6 | 18.3 ± 0.6 |
| 2p | — | — | 8.7 ± 0.6 | — | 12.0 ± 1.0 | 17.3 ± 0.6 |
| 2q | 18.7 ± 0.6 | 11.0 ± 1.0 | 13.3 ± 0.6 | 14.3 ± 0.6 | — | 10.3 ± 0.6 |
| 2r | 15.3 ± 0.6 | 15.7 ± 0.6 | 10.3 ± 0.6 | — | 12.0 ± 0.0 | 8.0 ± 1.0 |
| Cef. | 40.3 ± 0.6 | 20.7 ± 0.6 | 30.7 ± 0.6 | 45.3 ± 0.6 | ||
| Am | 9.3 ± 0.6 | 16.7 ± 0.6 | ||||
| DMSO | — | — | — | — | — | — |
In the case of fungal strains, all the compounds showed higher activity against C. albicans compared to standard amphotericin B, except 2q. The compounds 2a, 2b, and 2d showed significant influence on this fungal strain, where 2b displayed the maximum activity with an inhibition zone of 30.3 ± 0.6. In the case of A. niger, the compounds 2m, 2n, 2o, and 2p had higher activity comparing the zone of inhibition of amphotericin B (16.7 ± 0.6). The remaining compounds showed moderate to low activity in this study.
3.3 Antioxidant assay
DPPH free radical scavenging method was used to determine the antioxidant activity of the tested compounds in five different concentrations, which varied from 15 μL to 250 μL. The absorbance fluctuated and reduced significantly with the increased concentration, as represented in Fig. 2. These variations of the antioxidant data are the consequence of the mechanism of hydrogen atom transfer and lipophilicity of the compounds. Amidst the tested entities, 80% of DPPH free radical was inhibited by all the compounds except 2a and 2e at 225 μg mL−1. The compound 2m showed the highest inhibition at a concentration of 125 μg mL−1, which was around 83%. This DPPH free radical scavenging method was assessed by a UV-vis spectrophotometer at approximately 517 nm to collect data and calculate the IC50 values of the selected analogues and standard ascorbic acid, which are displayed in Table 2. The compound 2f revealed its predominant IC50 value (11.73 ± 1.22 μg mL−1), owing to the presence of the coumarin group as a thiazole ring substituent with maximum inhibition (98%) at the highest concentration of 250 μL. The compound 2n showed the second highest IC50 value of 38.35 ± 2.44, reasoning the presence of an electron-donating methyl group, which was assumed to stabilize the radicals with its inductive effect.47 However, compounds 2b (48.62 ± 2.41) and 2m (48.99 ± 2.11) showed moderate activity similar to ascorbic acid with a value of 49.68 ± 3.68 despite their different substituent groups. The presence of an alkyl group in 2b and a phenyl ring in 2m had significant attributes on antioxidant properties.48 The IC50 value of 2c (57.39 ± 2.47) and 2d (99.86 ± 0.38) increased consecutively compared to 2b (48.62 ± 2.41) due to the presence of the electron-withdrawing ester group. Compound 2j exhibited the lowest IC50 value of 140.87 ± 1.76; a thiazole-substituted phenyl ring with an electron-withdrawing nitro group at the meta position reduced the electron density in the compounds, which may lead to poor activity. Despite the presence of a phenyl ring substituted nitro group in 2q, it showed moderate activity with an IC50 value of 68.95 ± 0.56.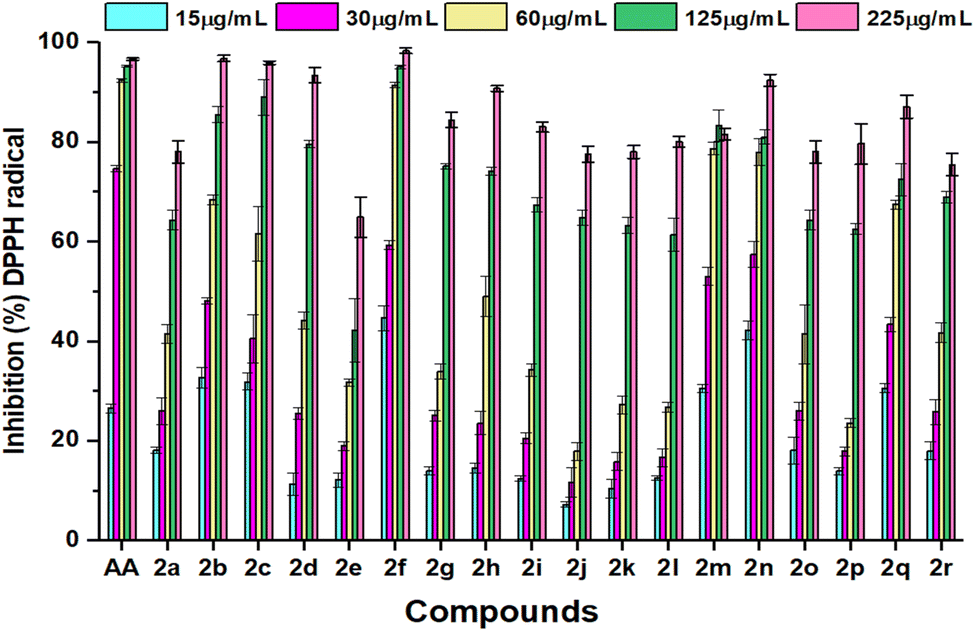 | ||
| Fig. 2 Comparison of inhibition (%) of DPPH radicals at various concentrations of ascorbic acid (AA) and synthesized compounds 2a–2r. Data are expressed as mean ± SD of three experiments. | ||
| Compound | IC50 (μg mL−1) | Compound | IC50 (μg mL−1) |
|---|---|---|---|
| 2a | 117.08 ± 1.43 | 2j | 171.73 ± 2.80 |
| 2b | 48.62 ± 2.41 | 2k | 134.8 ± 1.42 |
| 2c | 57.39 ± 2.47 | 2l | 133.62 ± 0.72 |
| 2d | 99.86 ± 0.38 | 2m | 48.99 ± 2.11 |
| 2e | 140.87 ± 1.76 | 2n | 38.35 ± 1.69 |
| 2f | 11.73 ± 1.22 | 2o | 118.15 ± 1.43 |
| 2g | 111.57 ± 1.22 | 2p | 132.48 ± 3.11 |
| 2h | 101.72 ± 0.69 | 2q | 68.95 ± 0.56 |
| 2i | 120.68 ± 0.80 | 2r | 118.44 ± 1.19 |
| Ascorbic acid | 49.68 ± 3.68 |
3.4 In silico analysis
 | ||
| Fig. 3 Optimized molecular structures of compounds 2a, 2b, and 2f on the basis of DFT/B3LYP/6-31+G(d,p). | ||
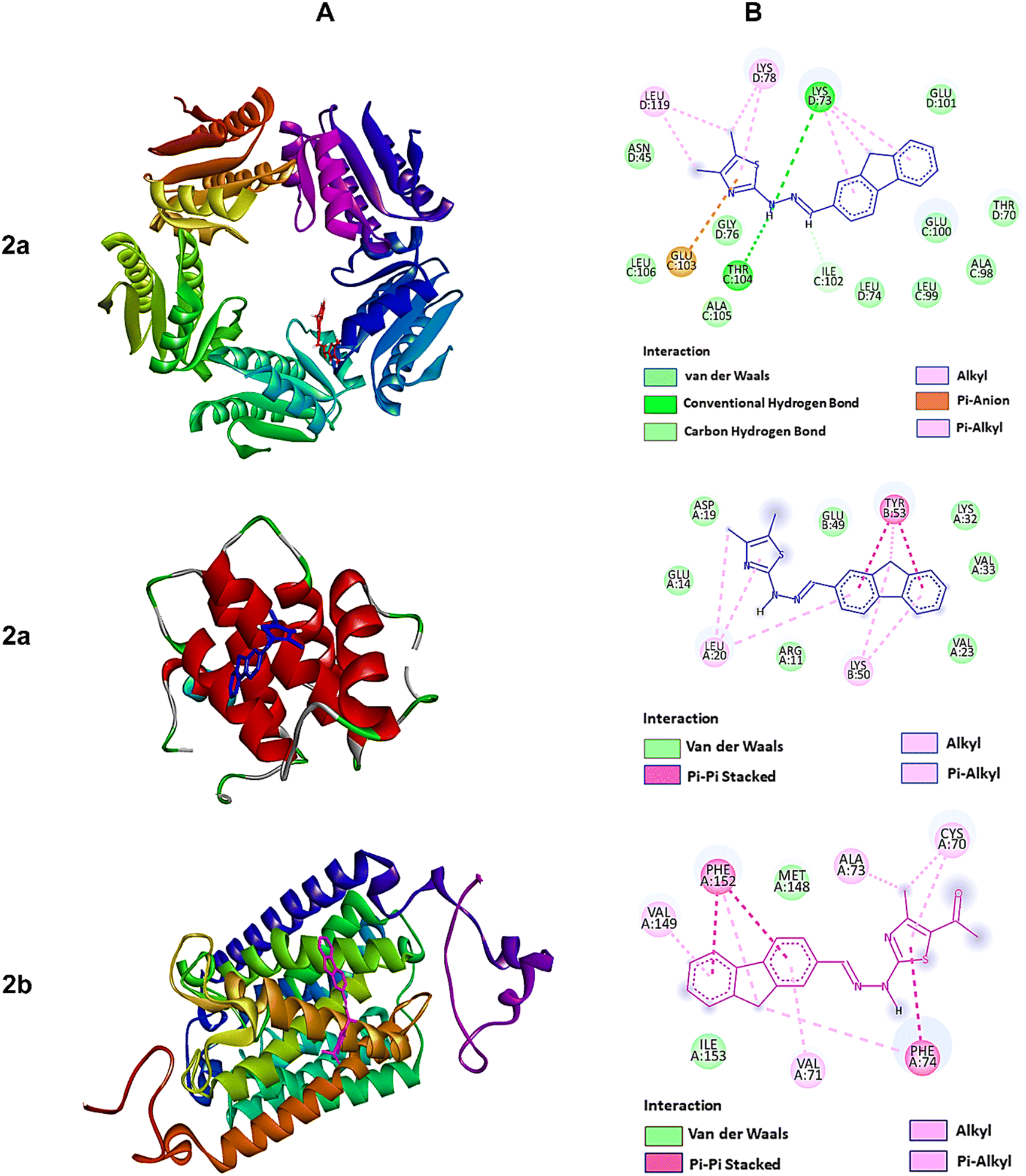 | ||
| Fig. 4 The molecular docking studies of 2a against 6JHK, 5H1N, and 2b against 5AEZ protein receptors. (A) 3D interaction profiles. (B) 2D docking simulations. | ||
On the other hand, compound 2f, with higher antioxidant ability, was docked against human antioxidant enzyme receptor 3MNG with a binding affinity of −8.9 kcal mol−1 (Fig. 5). Thiazole substituted coumarin displayed the maximum interaction with different amino acid residues. The coumarin ring and its ring containing oxygen atom displayed a pi–sigma and a conventional hydrogen bond with Val23 at a distance of 2.88 Å and 4.16 Å. Glu16 was involved in two pi–anion interactions with coumarin (4.99 Å) and fluorene ring (3.48 Å). Coumarin also had an electrostatic interaction with Leu28 at a distance of 5.37 Å. Leu96, at a distance of 5.29 Å, displayed a pi–alkyl interaction with the fluorene ring. This ring had more interaction with Arg86 via pi–cation (4.55 Å) and pi–alkyl (5.33 Å) interactions and with Ala90 at a distance of 5.00 Å through electrostatic bond interaction.
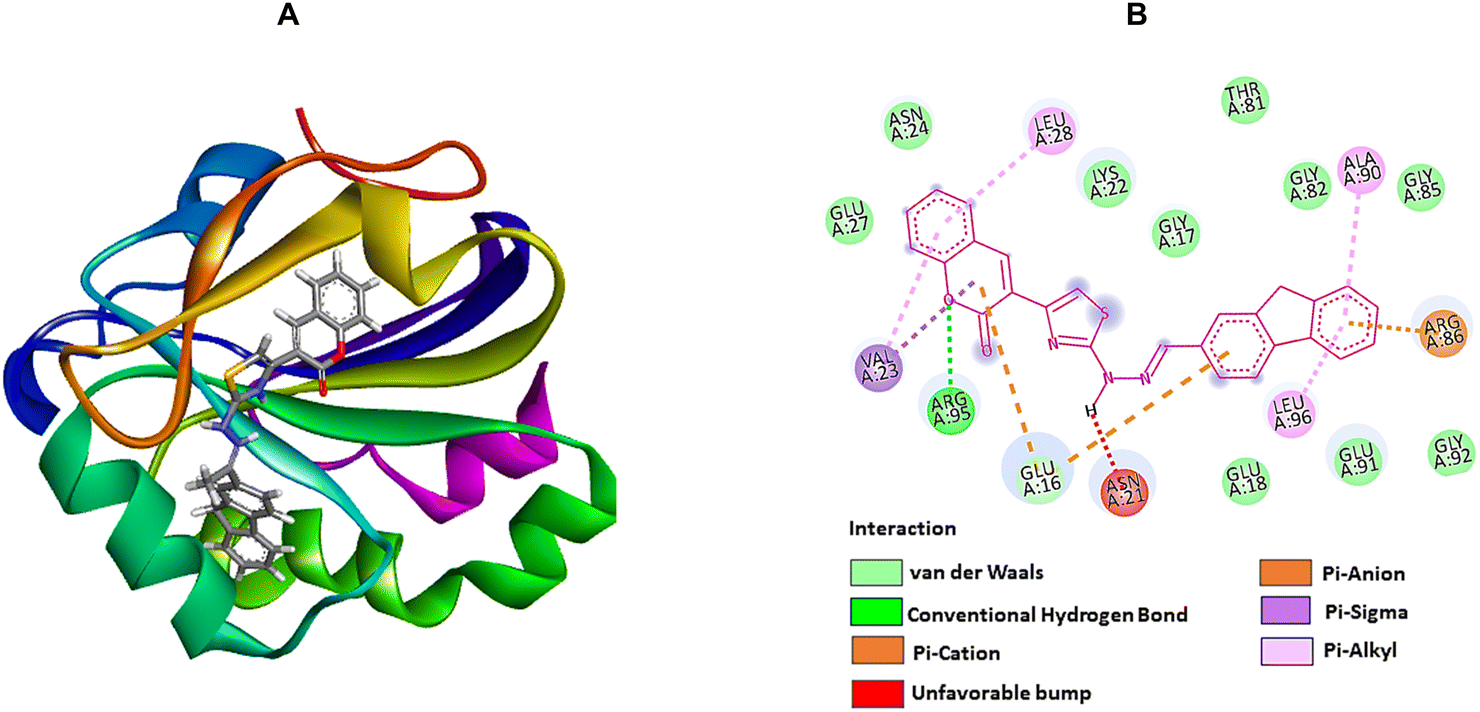 | ||
| Fig. 5 The molecular docking studies of 2f against the human antioxidant enzyme receptor 3MNG. (A) 3D interaction profile. (B) 2D docking simulation. | ||
logP), topological polar surface area (TPSA), solubility parameter (logS), and percentage of absorption (%ABS). Therefore, the suggested approach during the pharmaceutical development process is to increase the solubility of the synthesized compounds. Among the titled compounds, most followed the standard range of the mentioned criteria, while a few showed one violation, which is tabulated in Table 3. All the compounds showed a single hydrogen bond donor and showed a lipophilicity range from 2.87 to 4.12. The topological polar surface area (TPSA) value for all the compounds was below 140, which satisfied the limit range, inferring good intestinal absorption. Good gastrointestinal absorption was also forecasted from the low water solubility (−5.16 to −7.77).52 The toxicological effects of the compounds were represented as mutagenicity, tumorigenicity, irritancy, and reproductive effects, which are displayed in Table 4. Except for 2e and 2f, the remaining compounds had low irritancy and low reproductive effects. The presence of different substituent groups changed the toxicity of different compounds, which was compared with standards, ceftriaxone and amphotericin B, and drug-likeness of the compound was also compared with reference data. Compound 2e showed the highest drug-likeness (5.47), and 2b had the maximum drug score (0.11) compared to other compounds (Fig. 6).
| Compd. | Lipinski's violations | Lipinski's rule | Veber's rule | logSg |
%ABSh | ||||
|---|---|---|---|---|---|---|---|---|---|
| MWa (≤500) | HBAb (≤10) | HBDc (≤5) | clogPd (≤5) |
NROTBe (≤10) | TPSAf (140 Å2) | ||||
| a Molecular weight.b Number of hydrogen bond acceptors.c Number of hydrogen bond donors.d Lipophilicity.e Number of rotatable bonds.f Topological polar surface area.g Solubility parameter.h Percentage of absorption. | |||||||||
| 2a | 0 | 319 | 2 | 1 | 2.99 | 3 | 65.52 | −5.53 | 86.34 |
| 2b | 0 | 347 | 3 | 1 | 2.87 | 4 | 82.59 | −5.35 | 80.43 |
| 2c | 0 | 377 | 4 | 1 | 3.47 | 6 | 91.82 | −7.70 | 77.24 |
| 2d | 0 | 377 | 4 | 1 | 3.16 | 7 | 91.82 | −5.16 | 77.24 |
| 2e | 0 | 435 | 4 | 1 | 3.29 | 4 | 95.73 | −6.80 | 75.89 |
| 2f | 1 | 449 | 4 | 1 | 3.50 | 4 | 95.73 | −7.01 | 75.89 |
| 2g | 1 | 367 | 2 | 1 | 3.33 | 4 | 65.52 | −6.38 | 86.34 |
| 2h | 0 | 381 | 2 | 1 | 3.59 | 4 | 65.52 | −6.68 | 86.34 |
| 2i | 0 | 397 | 3 | 1 | 3.64 | 5 | 74.75 | −6.44 | 83.14 |
| 2j | 0 | 412 | 4 | 1 | 3.07 | 5 | 111.34 | −6.43 | 70.49 |
| 2k | 0 | 412 | 4 | 1 | 3.09 | 5 | 111.34 | −6.43 | 70.49 |
| 2l | 1 | 436 | 2 | 1 | 3.79 | 4 | 65.52 | −7.56 | 86.34 |
| 2m | 1 | 381 | 2 | 1 | 3.62 | 4 | 65.52 | −6.59 | 86.34 |
| 2n | 1 | 395 | 2 | 1 | 3.84 | 4 | 65.52 | −6.89 | 86.34 |
| 2o | 0 | 411 | 3 | 1 | 3.88 | 4 | 74.75 | −6.65 | 83.14 |
| 2p | 0 | 426 | 4 | 1 | 2.89 | 5 | 111.34 | −6.63 | 70.49 |
| 2q | 0 | 426 | 4 | 1 | 2.98 | 5 | 111.34 | −6.63 | 70.49 |
| 2r | 0 | 450 | 2 | 1 | 4.12 | 4 | 65.52 | −7.77 | 86.34 |
| Cf | 1 | 331 | 5 | 2 | −1.53 | 3 | 72.88 | −3.32 | 83.85 |
| Am | 1 | 416 | 3 | 0 | 4.85 | 6 | 27.05 | −5.08 | 99.67 |
| AA | 0 | 176 | 6 | 4 | −2.46 | 2 | 107.20 | −0.35 | 71.91 |
| Compd. | Toxicity effects | Drug-likeness | |||
|---|---|---|---|---|---|
| M | T | I | R | ||
| 2a | High | High | Low | Low | 1.43 |
| 2b | High | High | Low | Low | 3.96 |
| 2c | High | High | Low | Low | 0.44 |
| 2d | High | High | Low | Low | −6.18 |
| 2e | High | High | Low | Medium | 5.47 |
| 2f | High | High | Low | Medium | 5.01 |
| 2g | High | High | Low | Low | 4.08 |
| 2h | High | High | Low | Low | 2.81 |
| 2i | High | High | Low | Low | 4.13 |
| 2j | High | High | Low | Low | −6.29 |
| 2k | High | High | Low | Low | −1.26 |
| 2l | High | High | Low | Low | 4.09 |
| 2m | High | High | Low | Low | 3.44 |
| 2n | High | High | Low | Low | 2.22 |
| 2o | High | High | Low | Low | 3.56 |
| 2p | High | High | Low | Low | −6.9 |
| 2q | High | High | Low | Low | −1.88 |
| 2r | High | High | Low | Low | 3.46 |
| Ceftriaxone | Low | Low | Low | Low | 2.07 |
| Amphotericin B | Low | Low | Low | Low | 7.64 |
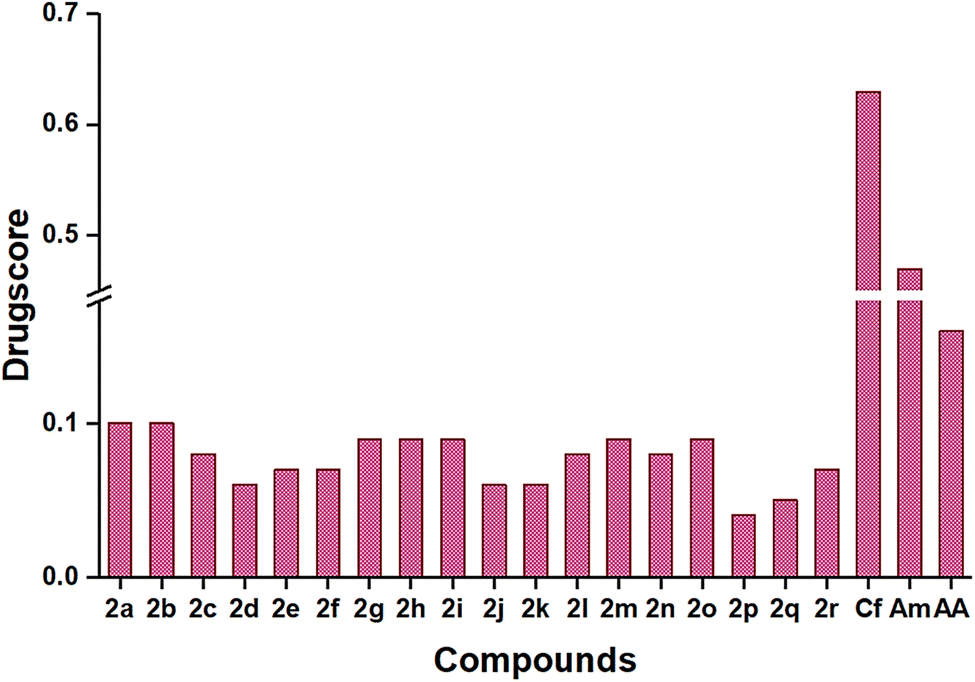 | ||
| Fig. 6 Predicted drug-score of synthesized compounds 2a–2m, ceftriaxone (Cf), amphotericin B (Am), and ascorbic acid (AA). | ||
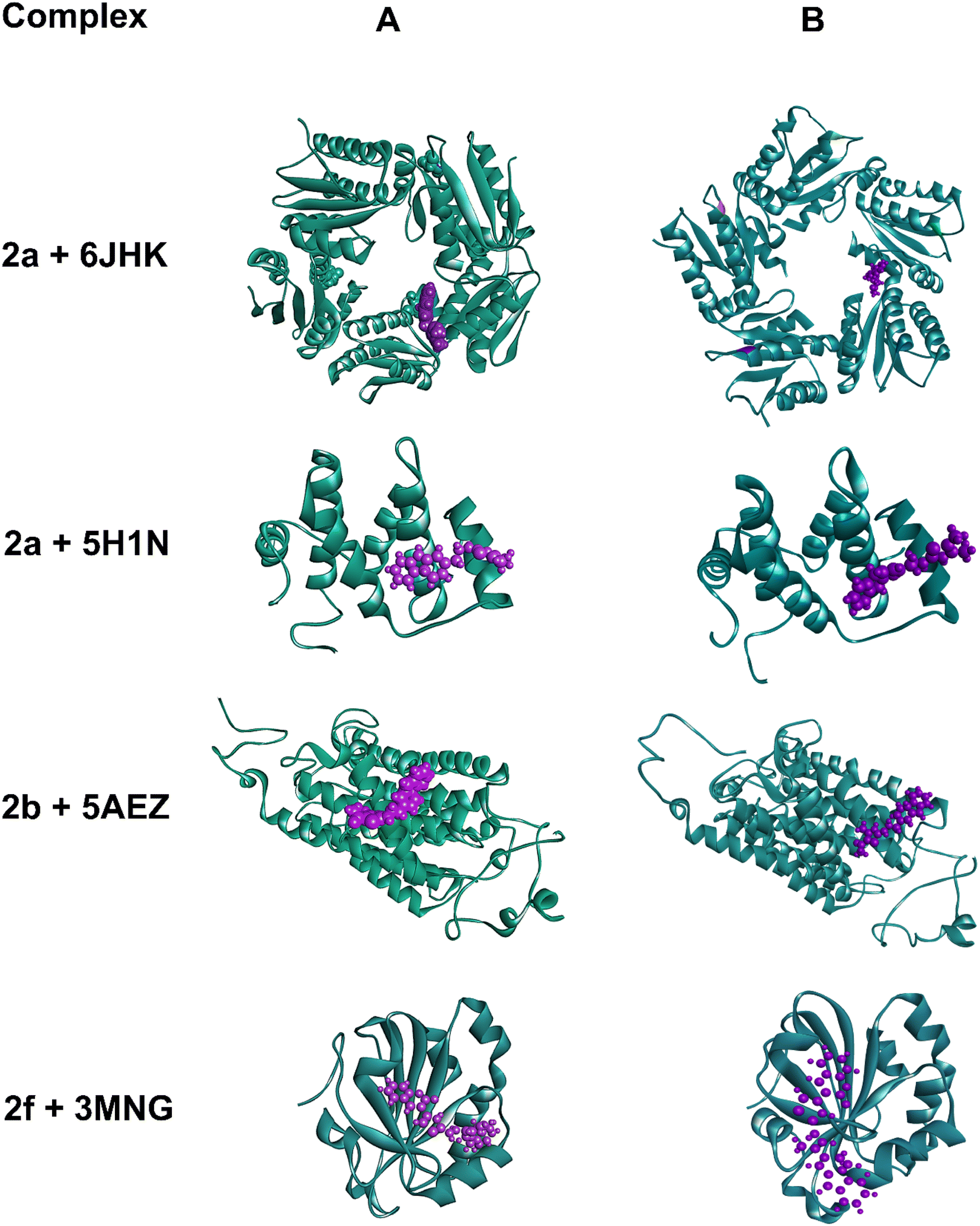 | ||
| Fig. 7 (A) Initial and (B) final snapshots of four different complexes during the 100 ns MD trajectory for 2a+6JHK, 2a+5H1N, and 2b+5AEZ complexes for antimicrobial studies and the 2f+3MNG complex for antioxidant studies. | ||
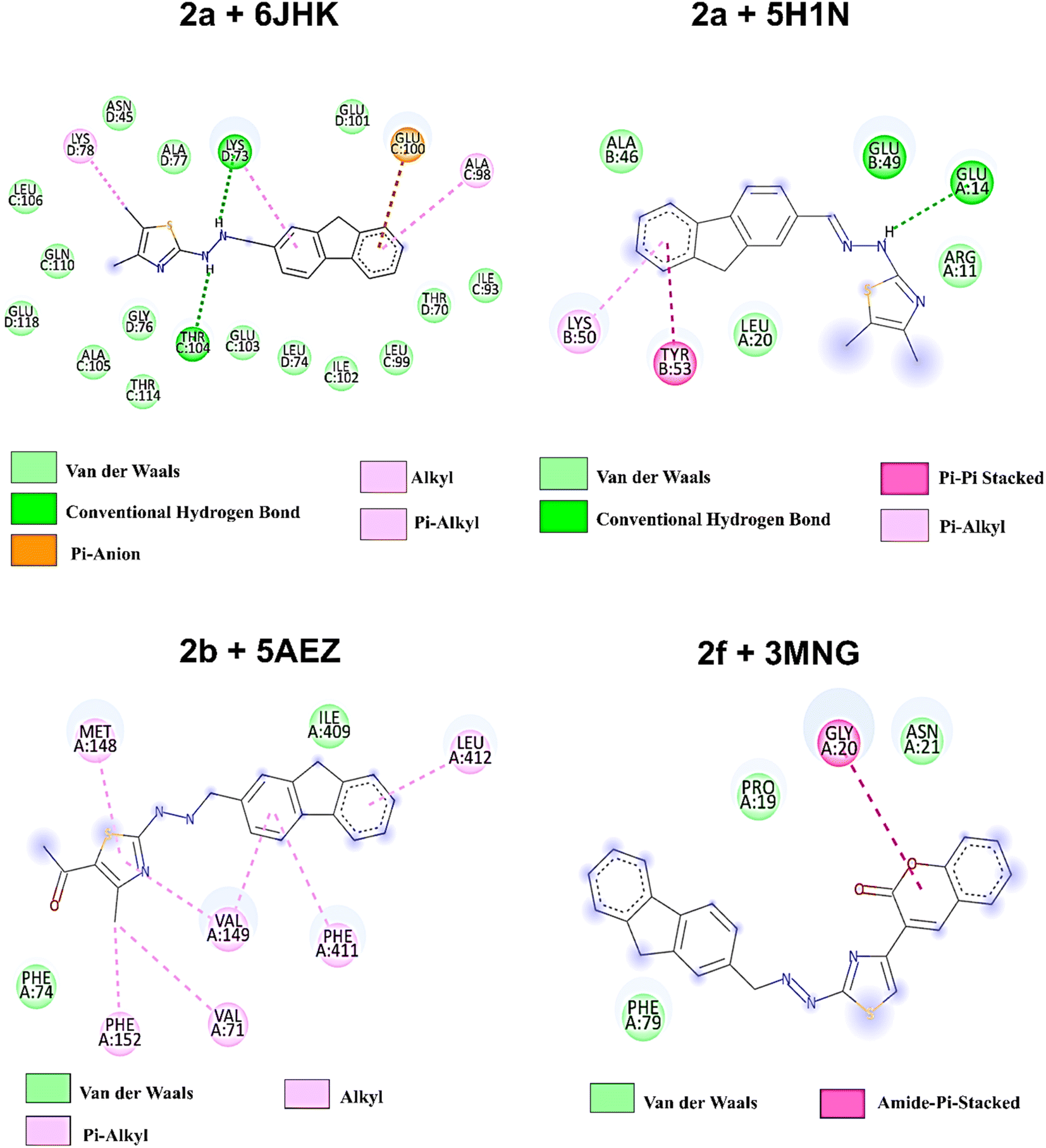 | ||
| Fig. 8 2D amino acid interaction profile of four protein–ligands complexes after 100 ns of simulation. | ||
To predict the stability of the protein during the simulation time, RMSD (root mean square displacement), radius of gyration (Rg), and RMSF (root mean square fluctuation) values were analyzed and represented graphically in Fig. 9–11. In complex, 2a + 6JHK, RMSD of the receptor protein went up from 0.47 Å to 2.35 Å to reach the equilibrium phase at 1.3 ns and carried an average deviation range between 1.5 Å to 2.4 Å. The value increased to 3.015 at 99 ns from the average range. Similarly, when compound 2a interacted with the 5H1N protein receptor, it attained equilibrium at 1 ns and remained in the range of 0.9 Å to 1.2 Å, which inferred minimal deviation of the compound from the reference structure. At 12 ns, this complex jumped suddenly and reached an RMSD value of 1.67 Å at 17 Å and remained in the range till 2 Å in the remaining simulation period. The RMSD value was maximum at 78 ns (1.95 Å) and 96 ns (2.07 Å), except that complex 2a + 51HN seemed stable throughout the 100 ns of trajectory (Fig. 9A). The RMSD value for the complex 2b + 5AEZ increased gradually till 21 ns and displayed no major changes afterward, with a deviation area of 0.8 Å to 2.22 Å till 32 ns. After 32 ns, the value crossed 2.5 Å, and the RMSD range was below 2.5 Å mostly till 55 ns. After 70 ns, the range increased and continued between 2.5 Å to 3.0 Å, representing considerable stability of the complex from the reference structure. In the case of compound 2f within the human antioxidant enzyme receptor 3MNG (Fig. 9B), the RMSD value of the post-equilibrium phase was retained in a constant range (1.0 Å to 1.3 Å) in the entire simulation time with an initial rise at 2.3 ns from 0.3 to 1.2 Å and 1.53 Å at 98 ns. Among all complexes, 2a + 3MNG displayed the maximum stability with minimal deviation.
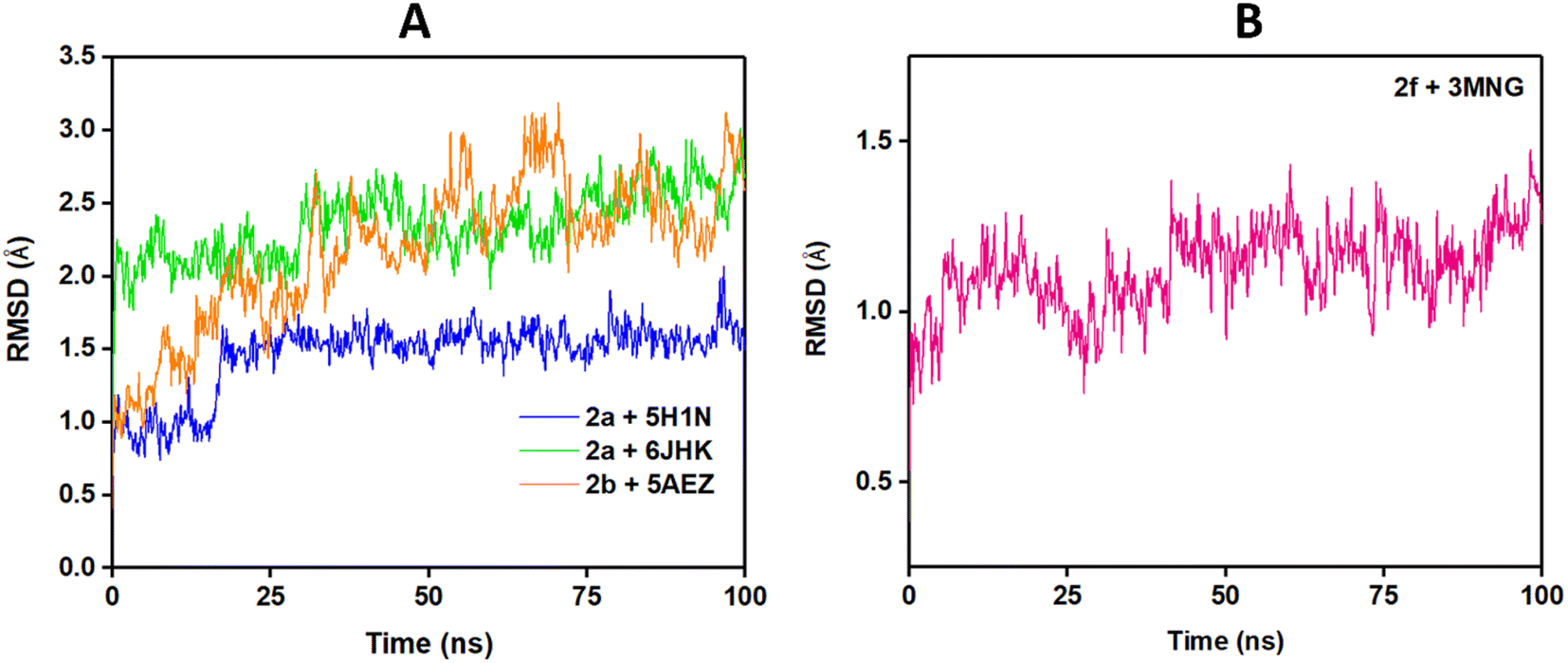 | ||
| Fig. 9 RMSD plot of protein–ligands complexes: (A) bacterial and fungal protein receptors and (B) antioxidant enzyme receptor. | ||
We also considered the Rg (radius of gyration) value of the four complexes to demonstrate their stability more explicitly during the MD simulation. Rg measures the compactness of the protein, which helps to assure the firmness of the protein and ligand interaction. Even though complex 2a + 5H1N (1.49–1.53 nm) and 2f + 3MNG (1.48–1.50 nm) displayed minimal flexibility, which meant its maximum stability, the average Rg value of 2b + 5AEZ and 2a + 6JHK indicated the firmness of the complexes as well. Rg value of 2b + 5AEZ was in the range of 2.28–2.32 nm, and the major clustering did not exceed the value of 2.26 nm. In the case of 2a+ 6JHK, the Rg value (2.72–2.76 nm) was maximum, expressing higher flexibility. Except for 2a+ 6JHK, the Rg value did not outdo the value of 2.5 nm, indicating more compactness of protein receptors even after binding with the respective ligands and made them more stable complexes than 2a+ 6JHK (Fig. 10).53
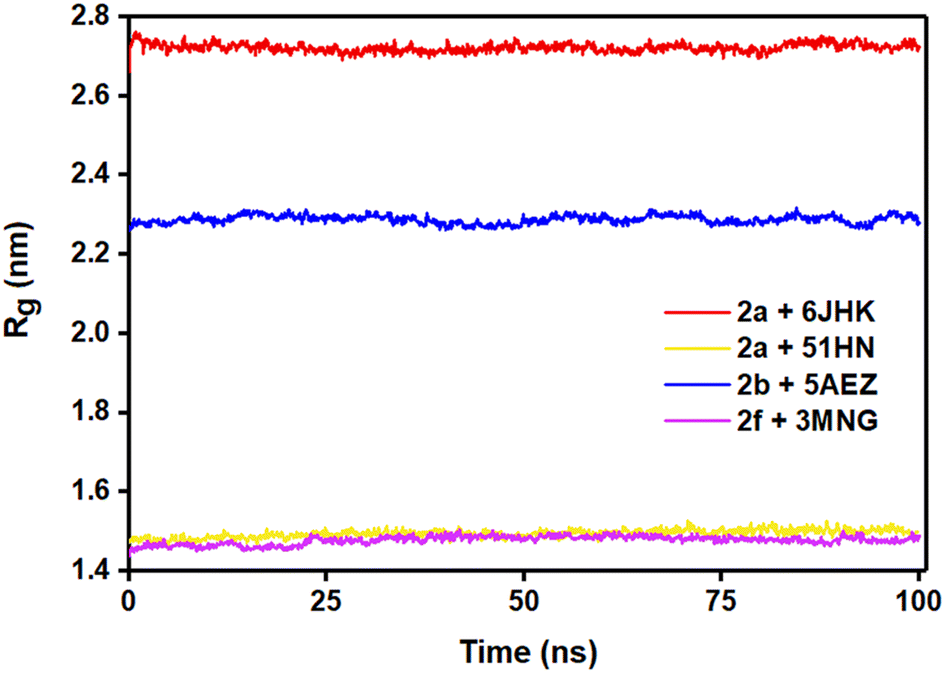 | ||
| Fig. 10 Radius of gyration (Rg) plot for the four complexes. | ||
RMSF (root mean square fluctuation) is crucial for MD simulation studies to understand the conformational changes of a protein from its mean structure. Four protein–ligand complexes were analyzed and plotted in this study in Fig. 11. Lys5 residue showed the maximum number of fluctuation and Leu119 had the highest RMSF value of 4.39 Å in the 2a + 6JHK complex. In addition, Try12, Leu99, and Glu100 displayed some major fluctuations in the protein while binding with ligand 2a. More than 96% of the residues exhibited variations below 2.5 Å in this complex. The complex 2a + 5H1N attained a 0.5 Å to 2.0 Å fluctuation range with the residues apart from Lys3, His 69, and His70. His69 and His70 climbed the maximum in the graph, which was around 4 Å, having the foremost deviation for this complex. Complex 2b + 5AEZ experienced a disordered zone at the beginning with a sharp rise to 11.48 Å by Met1, and the initial sixteen residues were in the most unstable position, ranging from 11 Å to 5 Å. Subsequently, the rest of the residues remained in the area of 0.5 Å to 2.4 Å without any major fluctuation, inferring conformational constancy during the simulation period. The protein 3MNG seemed to have a very sporadic movement of the residues with 2f during the 100 ns of simulation time. However, this complex had the most consistent range of RMSF values from 0.5 Å to 2.0 Å, considering the ref. 54. Glu16, Pro19, Pro100, and Val117 revealed maximum fluctuation above 2.0 Å during the simulation period. Most of the residues of the four complexes maintained the range of 0.5 Å to 2.5 Å throughout the whole trajectory, while complex 2f + 3MNG represented a minimal fluctuation range, indicating its maximum stability.
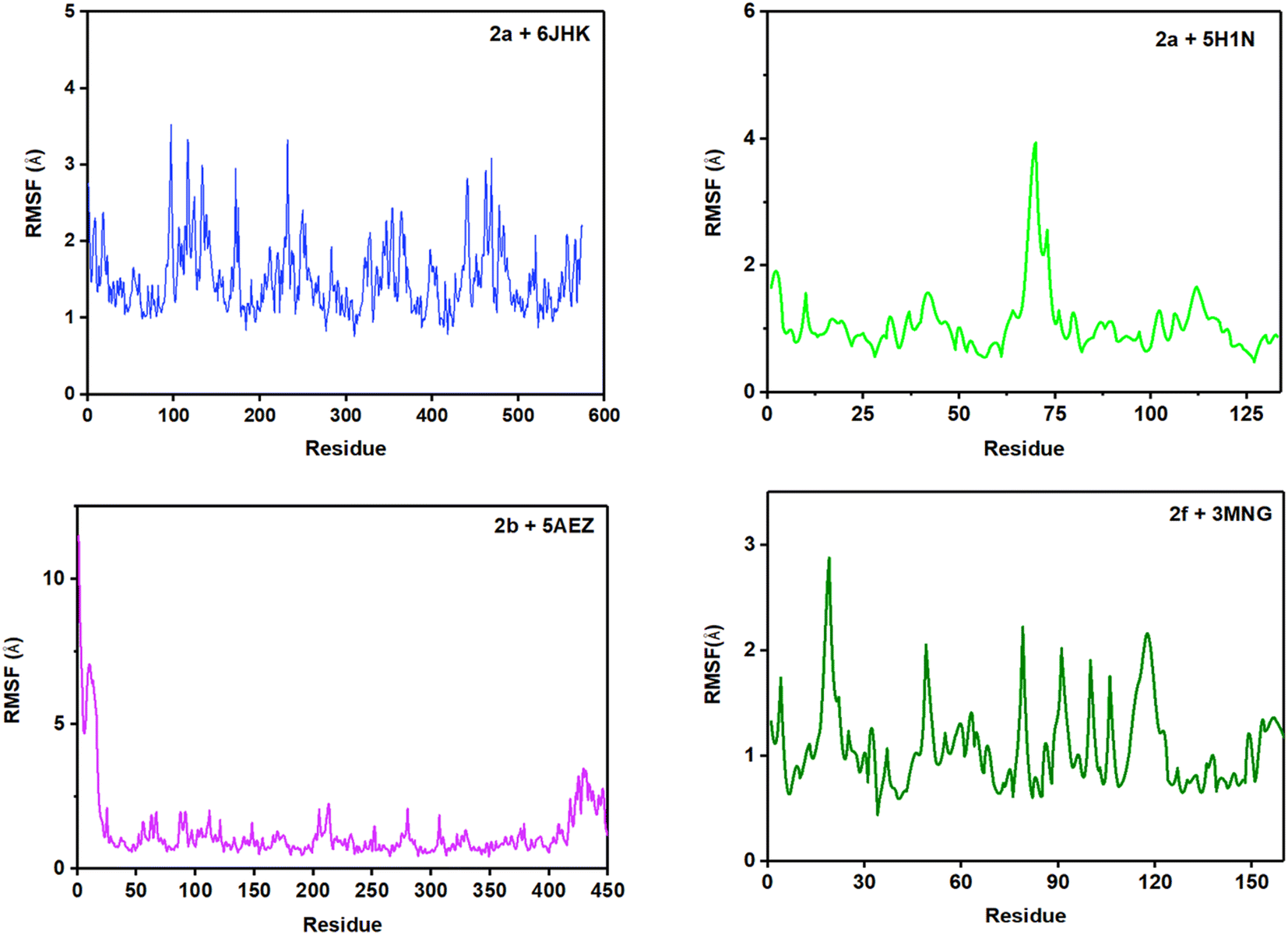 | ||
| Fig. 11 Root mean square fluctuation (RMSF) analysis for the four protein–ligand systems. | ||
The energy analysis was performed to demonstrate the favorable binding of the ligand molecules with the target proteins. Among the four complexes, 2a + 6JHK displayed maximum stability considering the energy plot during the simulation time with an average binding energy of −7.82 kcal mol−1. With the average free binding energy of −7.14 kcal mol−1, 2b + 5AEZ was the second most stable complex. Complex 2f + 3MNG had the minimal binding free energy with an average value of −6.40 kcal mol−1, with the most sporadic motion of energy during the simulation, even though all four complexes preserved the close average energy range. The initial 10 ns seemed similar for all four complexes, with rising binding free energy and progressing with a consistent energy range, indicating their stability during the simulation period. The stability of the complex on the basis of their average binding free energy is as follows 2a + 6JHK >2b + 5AEZ>2a + 5H1N > 2f + 3MNG (Fig. 12).
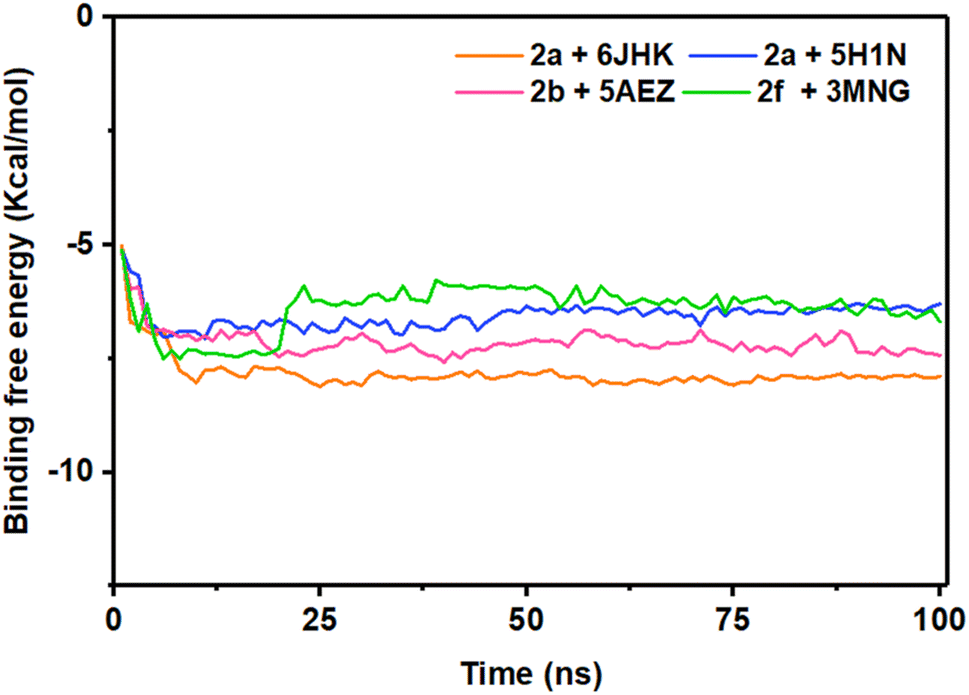 | ||
| Fig. 12 Binding free energy of four protein–ligand complexes over 100 ns of simulation. | ||
4. Conclusion
In summary, eighteen thiazole-Schiff base derivatives containing fluorene moiety via a two-step reaction procedure were synthesized and clarified the structures by different spectral analyses. These new chemical entities were screened for antimicrobial activity, and the results revealed the notable activity of compounds 2a and 2b against most of the strains. As antioxidant potency, the compound 2f (11.73 ± 1.22 μg mL−1) exhibited greater activity than standard ascorbic acid and the compounds 2b, 2m, and 2n also showed more activity compared to the standard. The pharmacokinetic and physicochemical properties were accounted for in all the analogues with their deleterious effects, where most of the compounds fulfilled the oral bioavailability parameters. In the molecular docking study, compound 2b showed the highest binding affinity (−9.0 kcal mol−1) and drug score, among others. Moreover, MD simulation was performed additionally, having average RMSD and RMSF values of 0.5 Å to 2.5 Å for most of the residues that suggested strong interactions of the compounds inside the protein receptor. Rg value indicated the strong binding stability of the complex 2a + 5H1N and 2f + 3MNG. The binding free energy of the complexes found compatible over 100 ns of simulation time referring the stability of the complexes. Considering both wet and dry lab outcomes, we proposed 2b as the lead compound since it showed moderate to high activity against all bacterial and fungal strains and displayed the second-highest antioxidant potency with binding affinity, drug-likeness, and drug-score values. All the data demonstrated here could play a pivotal role in further investigation for more accuracy and the development of novel drugs.Data availability
The research data associated with this article are included within the article. The research data associated with this article are included in the ESI† of this article.Author contributions
Sumita Saznin Marufa: data curation, formal analysis, visualization, software, writing – original draft, writing – review & editing. Tasnim Rahman: data curation, formal analysis, investigation. Mohammad Mostafizur Rahman: conceptualization, methodology, supervision, writing – review & editing. Md. Mizanur Rahman: data curation, formal analysis. Samira Jarin Khan: data curation, visualization. Rownok Jahan: data curation. Hiroshi Nishino: formal analysis, writing – review & editing. Mohammad Sayed Alam: conceptualization, methodology. Md. Aminul Haque: conceptualization, fund acquisition, methodology, project administration, supervision, writing – review & editing.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
The authors are thankful to the Grant of Advanced Research in Education (GARE), Ministry of Education, Bangladesh, for funding this project (Project ID PS20201511, 2020–2021). We are also indebted to the Instrumental Analysis Center, Kumamoto University, Kumamoto, Japan, for HRMS analyses.References
- M. Kaboré, I. Konaté, Y. Cissoko, I. Guindo, B. Coulibaly, M. Hermine, A. A. Oumar, M. Soumaré, A. Fofana, A. Zaré, M. A. Cissé, D. Sogoba, O. Magassouba, H. H. Issa and F. S. Dao, Microbiological Assessment and Antimicrobials' Use in an Infectious Diseases Department in Mali, J. Adv. Microbiol., 2021, 11, 8, DOI:10.4236/aim.2021.118029.
- J. Tanwar, S. Das, Z. Fatima and S. Hameed, Multidrug resistance: an emerging crisis, Int. J. Infect. Dis., 2014, 2014, 1–7, DOI:10.1155/2014/541340.
- S. Rachakonda and L. Cartee, Challenges in antimicrobial drug discovery and the potential of nucleoside antibiotics, Curr. Med. Chem., 2004, 11(6), 775–793, DOI:10.2174/0929867043455774.
- C. L. Ventola, The Antibiotic Resistance Crisis Part 1: Causes and Threats, Pharm. Ther., 2015, 40(4), 277–283 Search PubMed.
- L. R. Singh, S. R. Avula, S. Raj, A. Srivastava, G. R. Palnati, C. K. M. Tripathi, M. Pasupuleti and K. V. Sashidhara, Coumarin–benzimidazole hybrids as a potent antimicrobial agent: synthesis and biological elevation, J. Antibiot., 2017, 70, 954–961, DOI:10.1038/ja.2017.70.
- M. L. Sbaraglini and A. Talevi, Hybrid Compounds as Anti-infective Agents, Curr. Top. Med. Chem., 2017, 17(9), 108, DOI:10.2174/1568026616666160927160912.
- M. Durgun, C. Türkeş, M. Işık, Y. Demir, A. Saklı, A. Kuru, A. Güzel, Ş. Beydemir, S. Akocak, S. M. Osman, Z. AlOthman and C. T. Supuran, Synthesis, characterisation, biological evaluation and in silico studies of sulphonamide Schiff bases, J. Enzyme Inhib. Med. Chem., 2020, 35(1), 950–962, DOI:10.1080/14756366.2020.1746784.
- A. Kajal, S. Bala, S. Kamboj, N. Sharma and V. Saini, Schiff Bases: A Versatile Pharmacophore, J. Catal., 2013, 2013, 1–14, DOI:10.1155/2013/893512.
- T. Y. Fonkui, M. I. Ikhile, D. T. Ndinteh and P. B. Njobeh, Microbial activity of some heterocyclic Schiff bases and metal complexes: A review, Trop. J. Pharm. Res., 2018, 17(12), 2507–2518, DOI:10.4314/tjpr.v17i12.29.
- H. B. Shi, S. J. Zhang, Q. F. Ge, D. W. Guo, C. M. Cai and W. X. Hu, Synthesis and anticancer evaluation of thiazolyl-chalcones, Bioorg. Med. Chem. Lett., 2010, 20(22), 6555–6559, DOI:10.1016/j.bmcl.2010.09.041.
- M. H. Shih, Y. S. Su and C. L. Wu, Syntheses of aromatic substituted hydrazino-thiazole derivatives to clarify structural characterization and antioxidant activity between 3- arylsydnonyl and aryl substituted hydrazino-thiazoles, Chem. Pharm. Bull., 2007, 55(8), 1126–1135, DOI:10.1248/cpb.55.1126.
- H. Bulut, M. Karatepe, H. Temel, M. Sekerc and M. Koparir, Studies on the Antiviral and cytotoxic Activity of Schiff Bases Derived from 1,2-Bis-(o- and p-aminophenoxy)ethane ans Salicyladehyde, Asian J. Chem., 2005, 17(4), 2793–2796 Search PubMed.
- M. Taheri, S. Aslani, H. Ghafouri, A. Mohammadi, V. A. Moghaddam, N. Moradi and H. Naeimi, Synthesis, in vitro biological evaluation and molecular modelling of new 2-chloro- 3-hydrazinopyrazine derivatives as potent acetylcholinesterase inhibitors on PC12 cells, BMC Chem., 2022, 16(7), 1–12, DOI:10.1186/s13065-022-00799-w.
- R. G. Franzen, Recent advances in the preparation of heterocycles on solid support: a review of the literature, J. Comb. Chem., 2000, 2(3), 195–214, DOI:10.1021/cc000002f.
- S. Eryılmaz, E. T. Çelikoğlu, O. İdil, E. İnkaya, Z. Kozak, E. Mısır and M. Gül, Derivatives of pyridine and thiazole hybrid: synthesis, DFT, biological evaluation via antimicrobial and DNA cleavage activity, Bioorg. Chem., 2020, 95, 103476, DOI:10.1016/j.bioorg.2019.103476.
- Y. Demir, P. Taslimi, Ü. M. Koçyiğit, M. Akkuş, M. S. Özaslan, H. E. Duran, Y. Budak, B. Tüzün, M. B. Gürdere, M. Ceylan, S. Taysi, İ. Gülçin and Ş. Beydemir, Determination of the inhibition profiles of pyrazolyl–thiazole derivatives against aldose reductase and α-glycosidase and molecular docking studies, Arch. Pharm., 2020, 353(12), 2000118, DOI:10.1002/ardp.202000118.
- T. H. Namitha, S. N. Saranya, A. Kumar, B. Vinod and P. A. Daisy, A Review on Synthesis and Biological Activity of Thiazole and its Derivatives, Int. J. Pharm. Sci. Rev. Res., 2021, 70(1), 189–193, DOI:10.47583/ijpsrr.2021.v70i01.029.
- A. M. Alotaibi, A. A. Alotaibi, S. Kumar, S. M. B. Asdaq, M. Imran, P. K. Deb, K. N. Venugopala and S. Jomah, Thiazole: A Versatile Standalone Moiety Contributing to the Development of Various Drugs and Biologically Active Agents, Molecules, 2022, 27, 3994, DOI:10.3390/molecules27133994.
- B. Sever, M. D. Altıntop, Y. Demir, G. A. Çiftçi, S. Beydemir and A. Özdemir, Design, synthesis, in vitro and in silico investigation of aldose reductase inhibitory effects of new thiazole-based compounds, Bioorg. Chem., 2020, 102, 104110, DOI:10.1016/j.bioorg.2020.104110.
- E. Gursoy and N. U. Guzeldemirci, Synthesis and primary cytotoxicity evaluation of new imidazo[2,1-b]thiazole derivatives, Eur. J. Med. Chem., 2007, 42(3), 320–326, DOI:10.1016/j.ejmech.2006.10.012.
- R. N. Sharma, F. P. Xavier, K. K. Vasu, S. C. Chaturvedi and S. S. Pancholi, Synthesis of 4- Benzyl-1,3-Thiazole Derivatives as Potential Anti-Inflammatory Agents: An Analogue- Based Drug Design Approach, J. Enzyme Inhib. Med. Chem., 2009, 24(3), 890–897, DOI:10.1080/14756360802519558.
- I. Althagafi, N. El-Metwaly and T. A. Farghaly, New Series of Thiazole Derivatives: Synthesis, structural elucidation, antimicrobial activity, molecular modeling and MOE docking, Molecules, 2019, 24(9), 1741, DOI:10.3390/molecules24091741.
- M. S. Shah, M. M. Rahman, M. D. Islam, A. A. Macktuf, J. U. Ahmed, H. Nishino and M. A. Haque, Synthesis, antimicrobial and antioxidant evaluation with in silico studies of new thiazole Schiff base derivatives, J. Mol. Struct., 2022, 1248, 131465, DOI:10.1016/j.molstruc.2021.131465.
- B. N. Saglık, D. Osmaniye, U. A. Çevik, S. Levent, B. K. Çavusoglu, Y. Özkay and Z. A. Kaplancıklı, Design, Synthesis, and Structure–Activity Relationships of Thiazole Analogs as Anticholinesterase Agents for Alzheimer's Disease, Molecules, 2020, 25, 4312, DOI:10.3390/molecules25184312.
- T. V. Sravanthi, S. S. Lulu, S. Vino, M. A. Jayasri, A. Mohanapriya and S. L. Manju, Synthesis, docking, and evaluation of novel thiazoles for potent antidiabetic activity, Med. Chem. Res., 2017, 26(6), 1306–1315, DOI:10.1007/s00044-017-1851-8.
- M. A. T. Nguyen, A. K. Mungara, J. A. Kim, K. D. Lee and S. Park, Synthesis, Anticancer and Antioxidant Activity of Novel Carbazole-based Thiazole Derivatives, Phosphorus, Sulfur, Silicon Relat. Elem., 2015, 190(2), 191–199, DOI:10.1080/10426507.2014.914933.
- T. Ahmad, F. Kandil and C. Moustapha, Synthesis, Characterization, Biological Evaluation and Antibacterial Activity of some Heterocyclic Fluorene Compounds Derived from Schiff Base, Int. J. ChemTech Res., 2015, 7(6), 2752–2762 Search PubMed.
- R. I. Alsantali, E. M. Hussein, R. J. Obaid, M. Morad, H. M. Altass, A. Alharbi, A. M. Hameed, R. S. Jassas, M. A. S. Abourehab, B. H. Asghar, Z. Moussa and S. A. Ahmed, Bioactive Fluorenes. Part II. Unprecedented biologically active thiazole derivatives based- 2,7-dichlorofluorene as competent DHFR inhibitors: Design, synthesis, and molecular docking approaches, Arabian J. Chem., 2020, 13(5), 5251–5462, DOI:10.1016/j.arabjc.2020.03.024.
- H. S. Kim, Y. Youc, J. Muna, C. G. Gadhe, H. Moon, J. S. Lee, A. N. Pae, M. Kohara, G. Keum, B. M. Kim and S. K. Jang, Structure-activity relationships of fluorene compounds inhibiting HCV variants, Antiviral Res., 2020, 174, 104678, DOI:10.1016/j.antiviral.2019.104678.
- S. Savir, Z. J. Wei, J. W. K. Liew, I. Vythilingam, Y. A. L. Lim, H. M. Saad, K. S. Sim and K. W. Tan, Synthesis, cytotoxicity and antimalarial activities of thiosemicarbazones and their nickel (II) complexes, J. Mol. Struct., 2020, 1211, 128090, DOI:10.1016/j.molstruc.2020.128090.
- E. M. Hussein, R. I. Alsantali, S. M. Abd El-Galil, R. J. Obaid, A. Alharbi, M. A. S. Abourehab and S. A. Ahmed, Bioactive fluorenes. part I. Synthesis, pharmacological study and molecular docking of novel dihydrofolate reductase inhibitors based-2,7- dichlorofluorene, Heliyon, 2019, 5, e01982, DOI:10.1016/j.heliyon.2019e01982.
- I. M. Vlad, D. C. Nut, R. V. Ancuceanu, T. Costea, M. Coanda, M. Popa, L. G. Marutescu, I. Zarafu, P. Ionita, C. E. D. Pirvu, C. Bleotu, M. C. Chifiriuc and C. Limban, Insights into the Microbicidal, Antibiofilm, Antioxidant and Toxicity Profile of New O-Aryl-Carbamoyl- Oxymino-Fluorene Derivatives, Int. J. Mol. Sci., 2023, 24, 7020, DOI:10.3390/ijms24087020.
- A. Pasieka, D. Panek, P. Zaręba, E. Sługocka, N. Gucwa, A. Espargaro, G. Latacz, N. Khan, A. Bucki, R. Sabate, A. Więckowska and B. Malawska, Novel drug-like fluorenyl derivatives as selective butyrylcholinesterase and β-amyloid inhibitors for the treatment of Alzheimer's disease, Bioorg. Med. Chem., 2023, 88–89, DOI:10.1016/j.bmc.2023.117333.
- C. Zehiroglu, S. Beyza and O. Sarikaya, The importance of antioxidants and place in today's scientific and technological studies, J. Food Sci. Technol., 2019, 56(11), 4757–4774, DOI:10.1007/s13197-019-03952-x.
- Y. Demir, Naphthoquinones, benzoquinones, and anthraquinones: Molecular docking, ADME and inhibition studies on human serum paraoxonase-1 associated with cardiovascular diseases, Drug Dev. Res., 2020, 81(5), 628–636, DOI:10.1002/ddr.21667.
- X. Y. Meng, H. X. Zhang, M. Mezei and M. Cui, Molecular Docking: A powerful approach for structure-based drug discovery, Curr. Comput.-Aided Drug Des., 2011, 7(2), 146–157 CrossRef CAS PubMed.
- B. Sever, M. D. Altıntop, Y. Demir, N. Yılmaz, G. A. Çiftçi, S. Beydemir and A. Ozdemir, Identification of a new class of potent aldose reductase inhibitors: Design, microwave-assisted synthesis, in vitro and in silico evaluation of 2-pyrazolines, Chem.-Biol. Interact., 2021, 345, 109576, DOI:10.1016/j.cbi.2021.109576.
- M. Balouiri, M. Sadiki and S. K. Ibnsouda, Methods for in vitro evaluating antimicrobial activity: A review, J. Pharm. Anal., 2016, 6, 71–79, DOI:10.1016/j.jpha.2015.11.005.
- E. Kwon, D. Pathak, H. Kim, P. Dahal, S. C. Ha, S. S. Lee, H. Jeong, D. Jeoung, H. W. Chang, H. S. Jung and D. Y. Kim, Structural insights into stressosome assembly, IUCrJ, 2019, 6, 938–947, DOI:10.1107/S205225251900945X.
- C. TT Su, C. Schönbach and C. K. Kwoh, Molecular docking analysis of 2009-H1N1 and 2004-H5N1 influenza virus HLA-B*4405-restricted HA epitope candidates: implications for TCR cross-recognition and vaccine development, BMC Bioinf., 2013, 14(2), S21, DOI:10.1186/1471-2105-14-S2-S21.
- B. V. D. Berg, A. Chembath, D. Jefferies, A. Basl, S. Khalid and J. C. Rutherford, Structural basis for Mep2 ammonium transceptor activation by phosphorylation, Nat. Commun., 2016, 7, 11337, DOI:10.1038/ncomms11337.
- A. Hall, D. Parsonage, L. B. Poole and P. A. Karplus, Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization, J. Mol. Biol., 2010, 402, 194–209, DOI:10.1016/j.jmb.2010.07.022.
- G. Yadav, S. Ganguly, S. Murugesan and A. Dev, Synthesis, Anti-HIV, Antimicrobial Evaluation and Structure Activity Relationship Studies of Some Novel Benzimidazole Derivatives, Anti-Infect. Agents, 2015, 13, 65–77 CrossRef CAS.
- L. C. Xue, J. P. Rodrigues, P. L. Kastritis, A. M. Bonvin and A. Vangone, PRODIGY: A Web Server for Predicting the Binding Affinity of Protein−Protein Complexes, Bioinformatics, 2016, 32, 3676–3678, DOI:10.1093/bioinformatics/btw514.
- D. Ungureanu, B. Tiperciuc, C. Nastasă, I. Ionut, G. Marc, I. Oniga and O. Oniga, An Overview of the Structure–Activity Relationship in Novel Antimicrobial Thiazoles Clubbed with Various Heterocycles (2017–2023), Pharmaceutics, 2024, 16, 89, DOI:10.3390/pharmaceutics16010089.
- N. C. Desai, N. Bhatt, H. Somani and A. Trivedi, Synthesis, antimicrobial and cytotoxic activities of some novel thiazole clubbed 1,3,4-oxadiazoles, Eur. J. Med. Chem., 2013, 67, 54–59, DOI:10.1016/j.ejmech.2013.06.029.
- N. C. Charlton, M. Mastyugin, B. Török and M. Török, Structural Features of Small Molecule Antioxidants and Strategic Modifications to Improve Potential Bioactivity, Molecules, 2023, 28(3), 1057, DOI:10.3390/molecules28031057.
- E. Bendary, R. R. Francis, H. M. G. Ali, M. I. Sarwat and S. El Hady, Antioxidant and structure–activity relationships (SARs) of some phenolic and anilines compounds, Ann. Agric. Sci., 2013, 58(2), 173–181, DOI:10.1016/j.aoas.2013.07.002.
- A. Rauf, H. Khan, M. Khan, A. Abusharha, G. Serdarouglu and M. Daglia, In Silico, SwissADME, and DFT Studies of Newly Synthesized Oxindole Derivatives Followed by Antioxidant Studies, J. Chem., 2023, 2023, 16, DOI:10.1155/2023/5553913.
- H. Muglu, H. Yakan, M. Erdogan, F. Topal, M. Topal, C. Turkes and S. Beydemir, Novel asymmetric biscarbothioamides as Alzheimer's disease associated cholinesterase inhibitors: synthesis, biological activity, and molecular docking studies, New J. Chem., 2024, 48, 10979, 10.1039/d4nj01462f.
- Ö. Güleç, C. Türkeş, M. Arslan, Y. Demir, B. Dincer, A. Ece and Ş. Beydemir, Novel beta-lactam substituted benzenesulfonamides: in vitro enzyme inhibition, cytotoxic activity and in silico interactions, J. Biomol. Struct. Dyn., 2024, 42(12), 6359–6377, DOI:10.1080/07391102.2023.2240889.
- F. Lemilemu, M. Bitew, T. B. Demissie, R. Eswaramoorthy and M. Endale, Synthesis, antibacterial and antioxidant activities of Thiazole-based Schiff base derivatives: a combined experimental and computational study, BMC Chem., 2021, 15(67), 1–18, DOI:10.1186/s13065-021-00791-w.
- A. Kumar, C. Prasun, E. Rathi, M. S. Nair and S. G. Kini, Identification of potential DNA gyrase inhibitors: virtual screening, extra-precision docking and molecular dynamics simulation study, Chem. Pap., 2023, 77, 6717–6727, DOI:10.1007/s11696-023-02971-5.
- A. Castro-Alvarez, A. M. Costa and J. Vilarrasa, The Performance of Several Docking Programs at Reproducing Protein–Macrolide-Like Crystal Structures, Molecules, 2017, 22, 136, DOI:10.3390/molecules22010136.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04197f |
| This journal is © The Royal Society of Chemistry 2024 |