
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrgano NHC catalyzed aqueous synthesis of 4β-isoxazole-podophyllotoxins: in vitro anticancer, caspase activation, tubulin polymerization inhibition and molecular docking studies†
Rajkumar Nagavatha,
Murali Krishna Thupuranib,
Vinitha Badithapurama,
Ravinder Manchala,
Chandra Sekhar Vasam*c and
Narasimha Swamy Thirukovela *a
*a
aDepartment of Chemistry, Chaitanya (Deemed to be University), Himayathnagar (V), Moinabad (M), Ranga Reddy (D), Hyderabad, India. E-mail: thirukovelann@chaitanya.edu.in
bDepartment of Biotechnology, Chaitanya (Deemed to be University), Himayathnagar (V), Moinabad (M), Ranga Reddy (D), Hyderabad, India
cDepartment of Pharmaceutical Chemistry, Telangana University, Nizamabad, TS, India. E-mail: csvasam@telanganauniversity.ac.in
First published on 26th July 2024
Abstract
We present, for the first time, the organo-N-heterocyclic carbene (NHC) catalyzed 1,3-dipolar cycloaddition of 4β-O-propargyl podophyllotoxin (1) with in situ aromatic nitrile oxides to afford regioselective 4β-isoxazolepodophyllotoxin hybrids (6a–n) in benign aqueous-organic media. Preliminary anticancer activity results showed that compound 6e displayed superior activity against MCF-7, HeLa and MIA PaCa2 human cell lines compared with podophyllotoxin. Compounds 6j and 6n showed greater activity against the MCF-7 cell line than the positive control. Caspase activation studies revealed that compound 6e at 20 μg ml−1 concentration had greater caspase 3/7 activation in MCF-7 and MIAPaCa2 cells than podophyllotoxin. Furthermore, in vitro tubulin polymerization inhibition studies revealed that compound 6e showed comparable activity with podophyllotoxin. Finally, in silico molecular docking studies of compounds 6e, 6j, 6n and podophyllotoxin on α,β-tubulin (pdb id 1SA0) revealed that compound 6n showed excellent binding energies and inhibition constants compared with podophyllotoxin.
Introduction
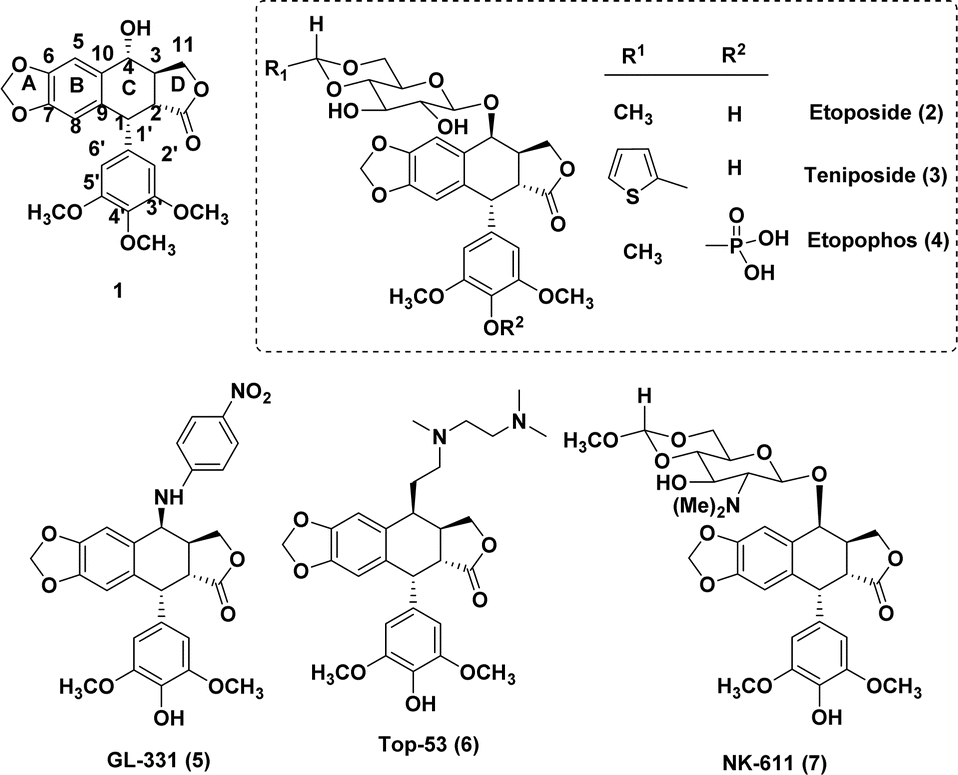
Nature has consistently offered us a wide variety of bioactive products to treat serious diseases such as cancer, immune system diseases, neurological conditions, and infections.1 Among these products is podophyllotoxin (1), one of the most prevalent naturally occurring cyclolignans isolated from Podophyllum peltatum L. and Podophyllum hexandrum,2 which has been broadly used in clinical studies on diverse malignancies. The mechanisms of action of semi-synthetic derivatives etoposide (2) and teniposide (3) are significantly different from those of the parent podophyllotoxin.3,4 These two semi-synthetic derivatives inhibit DNA topoisomerase II, whereas the parent podophyllotoxin inhibits the assembly in the microtubulin.5,6 Even yet, there have been reports on etoposide's toxicity and limitations, including its moderate efficacy, low solubility in water, potential for drug resistance, metabolic inactivation, and other adverse consequences.7,8These findings have prompted numerous investigations into the structural modification of etoposide, which includes etopophos (4), that addresses the bioavailability aspect. The substitution at the 4β-position resulting in strong inhibition of topoisomerase II was the most significant change. After a study9,10 on the substitution of heterocycles for etoposide's C-4 sugar unit, MacDonald and colleagues11 produced a composite pharmacophore model that identified the C-4 molecular area of podophyllotoxin (1) as a potentially variable position for further research. Bulky substituents at C-4 may be advantageous for DNA topo-II inhibition, as further evidenced by comparative molecular field analysis (CoMFA) models revealed by Lee and the group.11,12 These hypotheses align with the outstanding activity profiles of GL-331 (5), TOP-53 (6), and NK 611 (7).13 Interestingly, drug-resistance profiles of GL-331 and TOP-53 differed significantly from that of the parent compound 1, and both compounds demonstrated increased DNA topo-II inhibition and antitumor potential. The activity profiles of these classes of compounds suggest that substitution at position C-4 plays a crucial role and rational modifications at the C-4 position are feasible.14
One of the main challenges in medicinal chemistry is the development of anticancer agents, as evidenced by severe side effects of current chemotherapeutics, which include non-specific targeting, low solubility and incapability to enter tumour cells.15 These factors suggest the need for ongoing efforts to develop desired anti-cancer drug-like candidates with minimal side effects.16,17 As a result, current research is concentrated on the development of novel, safer therapeutic agents that are crucial for clinical use.18,19 Remarkably, because of their numerous biological uses, N-heterocycles with an oxygen atom are regarded as an important class of compounds in medicinal chemistry.20 In view of this, the selection of an isoxazole ring in the design and synthesis of biologically active compounds is considered to be a better choice, since in isoxazole both O and N atoms are present in adjacent positions and have low bond dissociation energy.21 Also, isoxazole consists weak basic character and due to a weak N–O bond, this ring breaks simply by photolysis and thermolysis. Predominantly, deprotonation of isoxazole leads to ring-opening and additional substitutions would lead to well therapeutic activity.22 Many research groups have been working on the development of new isoxazole-based anticancer active compounds (Fig. 1).23
 | ||
| Fig. 1 Structures of a few anticancer-active podophyllotoxin derivatives. | ||
Synthesis of isoxazole derivatives is extensively carried out through24 cyclomerization, cycloaddition, condensation and functionalization etc. In particular, 1,3-dipolar cycloaddition of nitrile oxides with alkynes has good synthetic value, since it produces biologically useful isoxazoles.22,23 Thus, Cu(I) and Ru(II) catalyzed 1,3-dipolar cycloaddition of nitrile oxides with terminal alkynes to access regioselective 3,5-di- and 3,4-disubstituted isoxazoles correspondingly have been well-developed.24,25 However, considering the probable disproportionation in metal catalysis and advantages observed with organo-catalysis in homogeneous catalysis,26 in 2011, our group developed an organo-N-heterocycliccarbenes (NHCs) catalyzed 1,3-dipolar cycloaddition of nitrile oxides with alkynes to obtain regioselective 3,5-di- and 3,4,5-trisubstituted isoxazoles in DCM solvent,27 since, NHCs are distinct Lewis base (nucleophilic) organocatalysts, they have both σ-basicity and π-acidity properties.28 Starting from initial studies on thiamine-derived NHCs in benzoin,28a and Stetter reactions,28b the mechanistic variety of NHCs contingent on their properties has led to the progress of numerous extraordinary C–C and C–X (X = heteroatom) bond formations. Certainly, the isolation of the first stable NHC by Arduengo in 1991 (ref. 29) from imidazolium salts revealed their tunable steric and electronic properties by changing N-substituents on the imidazole ring. In addition, the imidazolium salts, precursors to NHCs, remain stable and easy to handle.
Conversely, organocatalytic reactions have appeared as a substitute synthetic strategy that may eventually lead to large-scale pharmaceutical synthesis applications.30 The development of organocatalytic reactions in aqueous media is especially promising because, generally speaking, organocatalysts are stable in the presence of aqueous media. The use of water as the solvent in N-heterocyclic carbene (NHC)-catalyzed polarity inversion reactions is restricted when compared to enamine catalysis.28c,31 In 2004, Bode revealed the use of aq. organic (THF–H2O, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in organo-NHC-catalyzed addition of enals and aldehydes.28p,32 The similar group then used a stoichiometric amount of water as a reagent or co-solvent in the organo-NHC catalyzed reaction of formylcyclopropanes and α-chloroaldehyde bisulfite salts.33 In 2010, Rovis and group developed an organo-NHC catalyzed reaction of α-chloro aldehydes in toluene–water solvent media comprising around 1.0 equivalent of water.34
:1) in organo-NHC-catalyzed addition of enals and aldehydes.28p,32 The similar group then used a stoichiometric amount of water as a reagent or co-solvent in the organo-NHC catalyzed reaction of formylcyclopropanes and α-chloroaldehyde bisulfite salts.33 In 2010, Rovis and group developed an organo-NHC catalyzed reaction of α-chloro aldehydes in toluene–water solvent media comprising around 1.0 equivalent of water.34
In another study, Hoveyda and group developed an enantioselective addition between dimethylphenylsilyls and to α,β-unsaturated carbonyls in the aqueous medium using organo-NHC-catalysis and results displayed that water is comfortable to organo-NHC catalysis.35 Certainly, traces of water are usually presumed to exist even when anhydrous organic solvents are utilized in organo-NHC-catalyzed reactions. Also, results of the experimental and theoretical studies of Amyes, Diver, Gudat, and Nyulaszi groups show that NHCs are reasonably stable in aqueous environment.36
Considering that thiamine, an NHC precursor, is indispensable for several biological processes that arise mainly in an aqueous atmosphere.37 In 2013, Y. R. Chi and co-workers first time utilized catalytic efficacy of organo-NHC to promote the reaction of enals with enones in pure water.38 Following nature's lead, the use of aqueous solvent media would be an appropriate choice in NHC-catalyzed reactions.
Based on all the above findings and in our ongoing effort to develop aqueous organic synthesis39a–d and anticancer agents,39e–k we report an organo-NHC catalyzed 1,3-dipolar cycloaddition between in situ nitrile oxides and 4β-O-propargyl podophyllotoxin to construct new C4-modified isoxazole linked podophyllotoxins in aq. MecCN media. As well, we also investigated the in vitro anticancer activity, caspases activation and in vitro tubulin polymerization of newly synthesized isoxazole-linked podophyllotoxins. Finally, molecular docking studies were carried out for the most potent compounds found in in vitro activity studies. To the best of our knowledge, this is the first report concerning the organo-NHC catalyzed 1,3-dipolar alkyne-nitrile oxide cycloaddition in aqueous organic media to obtain new biologically active isoxazole compounds.
Results and discussion
At first, the synthesis of the key starting materials of the current work such as 4β-O-propargyl podophyllotoxin (1)39f,40 and chlorooximes (hydroximoyl chlorides) (6a–n)41 was achieved according to literature procedures.Later, we concentrated on the development of the optimization reaction conditions from 1,3-dipolar cycloaddition of terminal alkyne 1 with N-hydroxybenzimidoyl chloride (6a) as a model reaction using K2CO3 and diverse 5 mol% NHC pre-catalysts (A–G) at RT in (7:3) H2O/MeCN solvent media.
The results revealed that the NHC pre-catalysts (imidazolium salts) (A–G) (Fig. 2) used in this reaction gave desired cycloaddition product (5R,8aR,9S)-9-((3-phenylisoxazol-5-yl)methoxy)-5-(3,4,5-trimethoxyphenyl)-5,8,8a,9-tetrahydrofuro[3′,4′:6,7]naphtha [2,3-d][1,3]dioxol-6(5aH)-one (6a) in 22–76% yields (Table 1, entries 1–7). Out of all, NHC pre-catalyst B gave the best yield (76%) of 6a (Table 1, entry 2). The molecular structure of NHC pre-catalysts had a robust effect on the catalytic efficacy. The NHC pre-catalysts comprising bulky aromatic groups provided good yields of 6a (Table 1, entries 1–2), while, NHC pre-catalysts containing aliphatic groups gave low to moderate yields of 6a (Table 1, entries 3–7). Moreover, lowering the NHC pre-catalyst B concentration from 5 mol% led to a lower yield of 6a (Table 1, entry 8), whereas, increasing NHC pre-catalyst B loading from 5 mol% product did not show any effect on the yield of the product 6a (Table 1, entry 9). Using the optimal NHC pre-catalyst B (Table 1, entry 2), the effect of the bases on product 6a yield was also examined. The results revealed that KHCO3, DIEA and Et3N provided low yields (38–52%) of 6a (Table 1, entries 10–12), while, DBU gave a moderate (68%) yield of 6a (Table 1, entry 13). Interestingly, K2CO3 was found to be suitable, as it provided a good yield (76%) of 6a (Table 1, entry 2).
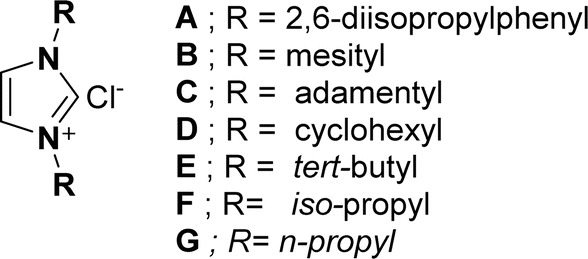 | ||
| Fig. 2 Structures of NHC pre-catalysts used in the present study. | ||
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst precursor (n mmol) | Base | Solvent | Time (h) | Yieldb (%) |
| a Reaction conditions: (i) intermediate 1 (1 mmol), NHC pre-catalysts A–G (0.05 mmol), base (0.15 mmol), (7:3) aqueous/organic solvent media RT and 20 min. (ii) 5a (1 mmol), base (1.5 mmol) and remaining time according to Table 1.b Isolated yields after column chromatography. |
|||||
| 1 | A (5) | K2CO3 | H2O/MeCN | 2 | 71 |
| 2 | B (5) | K2CO3 | H2O/MeCN | 2 | 76 |
| 3 | C (5) | K2CO3 | H2O/MeCN | 3 | 46 |
| 4 | D (5) | K2CO3 | H2O/MeCN | 3 | 40 |
| 5 | E (5) | K2CO3 | H2O/MeCN | 5 | 31 |
| 6 | F (5) | K2CO3 | H2O/MeCN | 5 | 22 |
| 7 | G (5) | K2CO3 | H2O/MeCN | 5 | 27 |
| 8 | B (3) | K2CO3 | H2O/MeCN | 5 | 35 |
| 9 | B (7) | K2CO3 | H2O/MeCN | 2 | 76 |
| 10 | B (5) | KHCO3 | H2O/MeCN | 3 | 38 |
| 11 | B (5) | DIEA | H2O/MeCN | 2 | 56 |
| 12 | B (5) | Et3N | H2O/MeCN | 3 | 52 |
| 13 | B (5) | DBU | H2O/MeCN | 2 | 68 |
| 14 | B (5) | K2CO3 | H2O/DCM | 6 | Trace |
| 15 | B (5) | K2CO3 | H2O/toluene | 6 | Trace |
| 16 | B (5) | K2CO3 | H2O/MeOH | 2 | 62 |
| 17 | B (5) | K2CO3 | H2O/EtOH | 2 | 65 |
| 18 | B (5) | K2CO3 | H2O | 3 | 31 |
Finally, we investigated the effect of the solvent system on the yields of 6a, under optimized reaction conditions. The trace amount of 6a was isolated using bi-phasic systems such as aq. DCM (Table 1, entry 14) and aq. toluene (Table 1, entry 15). The yield of 6a was slightly good in the case of aq. MeOH (Table 1, entry 16) and aq. EtOH (Table 1, entry 17). However, the use of pure water gave a low yield (31%) of the product 6a (Table 1, entry 18).
With the above optimal conditions (5 mol% NHC pre-catalyst B, K2CO3 and 7:3 water/MeCN) in our hand, the current approach was extended to a variety of aromatic aldehydes. In general, aromatic aldehydes containing electron-withdrawing groups provided better yields of the corresponding isoxazoles (Scheme 1, entries 6f–n) than the remaining aldehydes (Scheme 1, entries, 6a–g).
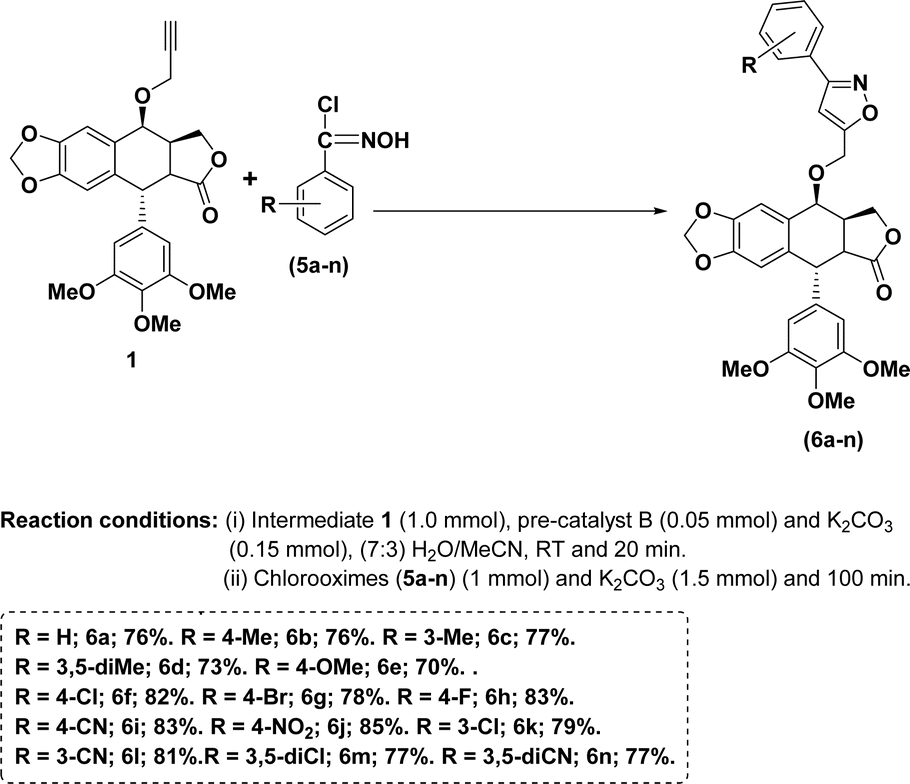 | ||
| Scheme 1 Synthesis of 4β-isoxazolepodophyllotoxin hybrids (6a–n). | ||
Based on the literature survey,27,42 we propose a plausible mechanism as shown in Scheme 2. At first, an in situ organo-NHC catalyst (obtained from pre-catalyst B and K2CO3) would react with alkyne (1) to form the zwitterion intermediate (H). Later this reactive intermediate interacts with in situ nitrile oxides (obtained from the reaction between chlorooximes (5a–n) and K2CO3) through a nucleophilic attack to give additional zwitterion intermediate (I), which, finally undergo C–O hetero-cyclization to give regioselective 3,5-di-substituted 4β-isoxazolepodophyllotoxin hybrids (6a–n).
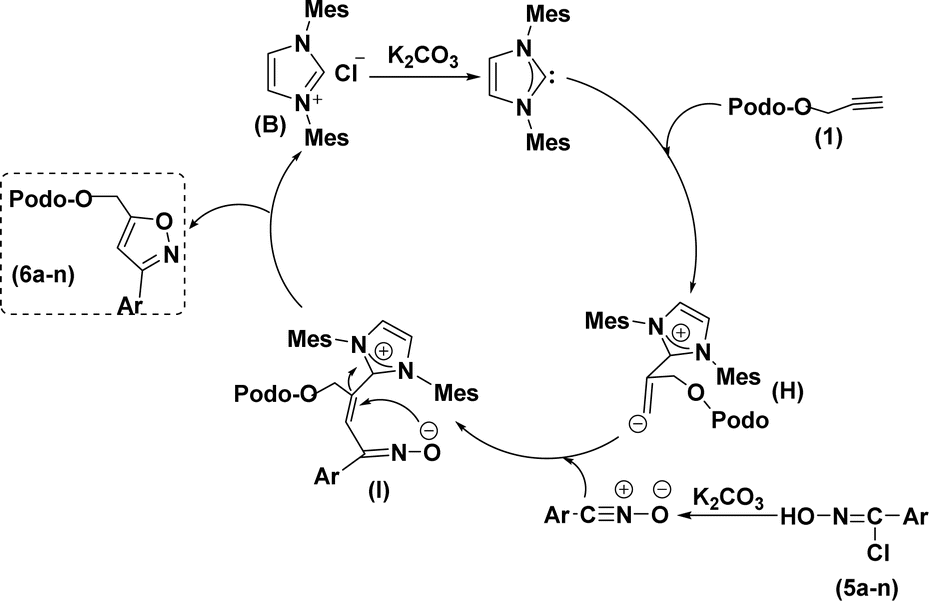 | ||
| Scheme 2 A possible mechanism. | ||
In vitro anticancer activity
All the above 4β-isoxazolepodophyllotoxin hybrids (6a–n) were examined for their in vitro anticancer activity against three human cancer cell lines (MCF-7, HeLa and MIAPACA) using podophyllotoxin as a positive control. The results (Table 2) revealed that compound 6e (GI50 = 0.18–0.32 μM) showed greater activity than the podophyllotoxin (GI50 = 0.31–0.51 μM) against three cell lines. As well, compounds 6j and 6n exhibited greater activity (GI50 values 0.23 and 0.12 μM, respectively) than the positive control (GI50 = 0.31 μM) against MCF-7, while, the same compounds (GI50 = 1.03–1.54 μM) had promising activity in comparison to podophyllotoxin (GI50 = 0.51–0.74 μM) against HeLa and MIAPACA. Furthermore, compounds 6i (GI50 = 0.42 μM) and 6m (GI50 = 0.36 μM) had almost similar activity with the podophyllotoxin (GI50 = 0.31 μM) against MCF-7.| Compound | R | MCF-7b | HeLac | MIAPACAd |
|---|---|---|---|---|
| a 50% growth inhibition and values are the mean of three independent experiments.b Breast cancer.c Cervical cancer.d Pancreatic cancer. | ||||
| 6a | H | 2.19 ± 0.33 | 4.45 ± 1.17 | 3.98 ± 1.26 |
| 6b | 4-CH3 | 3.08 ± 0.54 | 3.92 ± 1.18 | 3.22 ± 1.02 |
| 6c | 3-CH3 | 3.92 ± 0.82 | 3.83 ± 1.54 | 4.67 ± 1.93 |
| 6d | 3,5-diCH3 | 1.34 ± 0.23 | 2.74 ± 0.52 | 2.28 ± 0.29 |
| 6e | 4-OCH3 | 0.18 ± 0.07 | 0.32 ± 0.11 | 0.17 ± 0.05 |
| 6f | 4-Cl | 2.13 ± 0.37 | 3.05 ± 1.02 | 3.14 ± 0.32 |
| 6g | 4-Br | 4.32 ± 0.83 | 7.48 ± 0.69 | 6.84 ± 2.01 |
| 6h | 4-F | 4.87 ± 0.79 | 6.12 ± 1.08 | 13.13 ± 3.27 |
| 6i | 4-CN | 0.42 ± 0.15 | 3.64 ± 0.93 | 4.02 ± 0.79 |
| 6j | 4-NO2 | 0.23 ± 0.12 | 1.11 ± 0.29 | 1.54 ± 0.18 |
| 6k | 3-Cl | 3.15 ± 0.43 | 9.68 ± 3.12 | 18.32 ± 2.87 |
| 6l | 3-CN | 2.52 ± 0.91 | 12.25 ± 2.24 | 10.22 ± 3.06 |
| 6m | 3,5-diCl | 0.36 ± 0.17 | 2.19 ± 0.84 | 2.33 ± 0.18 |
| 6n | 3,5-diCN | 0.12 ± 0.08 | 1.28 ± 0.25 | 1.03 ± 0.19 |
| Podophyllotoxin | 0.31 ± 0.19 | 0.74 ± 0.23 | 0.51 ± 0.12 | |
The nature of the phenyl ring that is attached to isoxazole moiety, affecting in vitro anticancer activities was revealed using structure–activity relationship (SAR) studies. Introducing a strong electron-releasing group (OCH3) at the 4th position resulted in compound 6e displayed enhanced activity. The compound 6d containing a weak electron-donating group at 3rd and 5th positions (3,5-diCH3) was ranked second in this category. However, simple phenyl ring compound 6a or compounds 6b and 6c bearing mono-CH3 groups irrespective of their positions had reduced potency as compared to compounds 6d and 6e (Fig. 3).
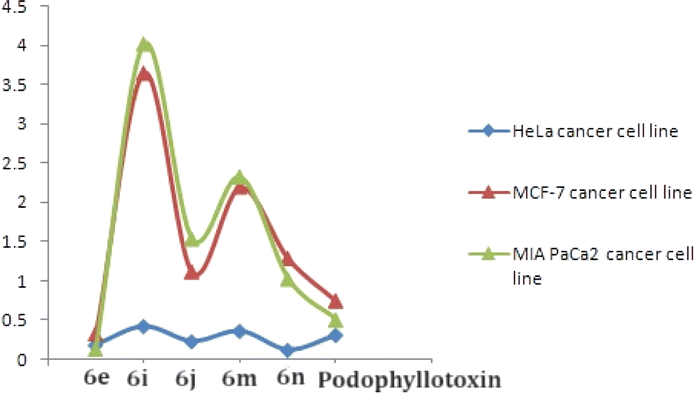 | ||
| Fig. 3 GI50 curves of compounds 6e, 6i, 6j, 6m and 6n and podophyllotoxin. | ||
With respect to the electron-withdrawing group series, compounds 6j and 6n bearing 4-NO2 and 3,5-diCN groups, respectively, displayed improved activity. The next better activity was shown by compound 6m having the 3,5-diCl group. Compounds 6f and 6i with 4-Cl and 4-CN groups, respectively, had slightly weaker activity than the compound 6m. However, compounds 6g, 6h and 6k containing mono-halogen groups such as 4-Br, 4-F and 3-Cl, respectively, or compound 6l with 3-CN group displayed poorer activity than the remaining compounds in this series (Fig. 4).
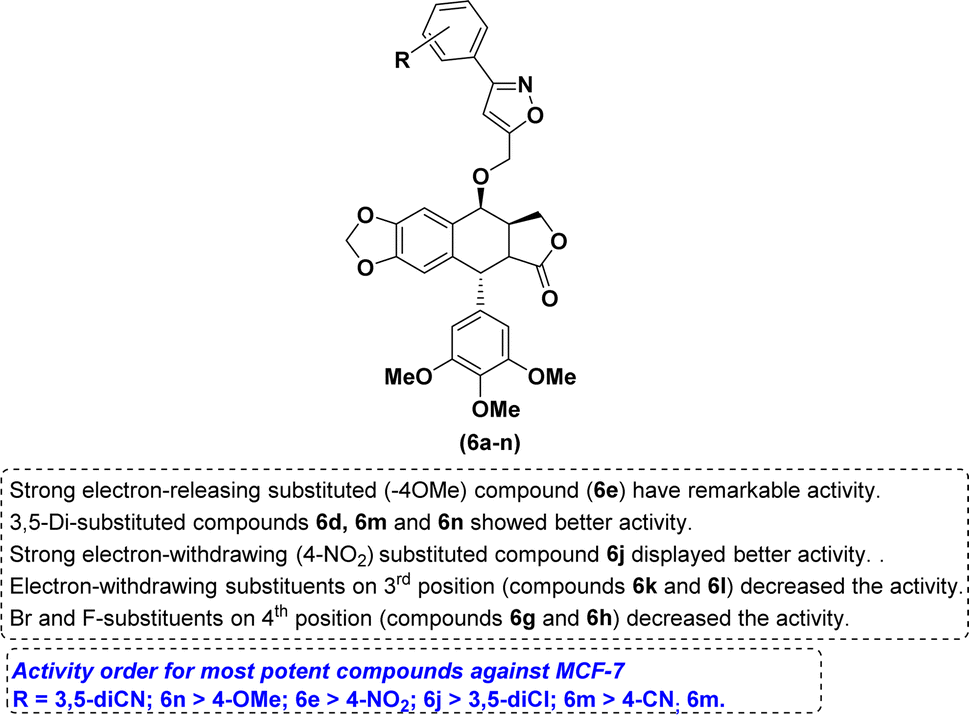 | ||
| Fig. 4 SAR summary. | ||
Caspases activation study
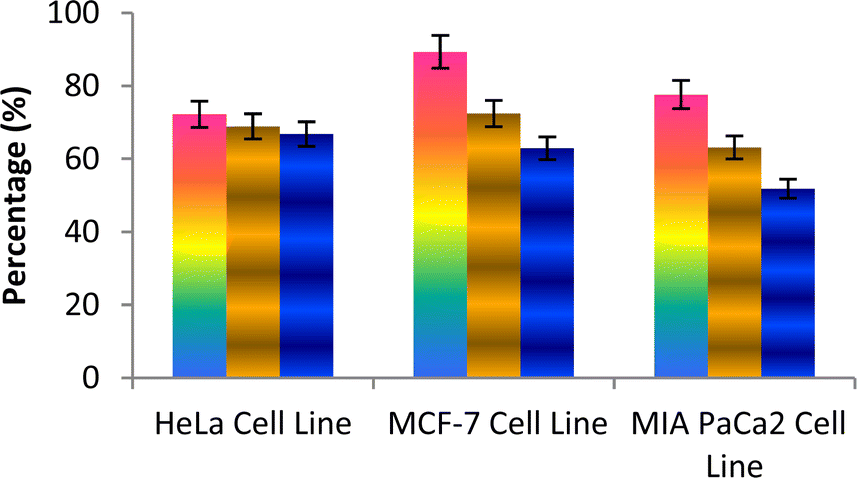
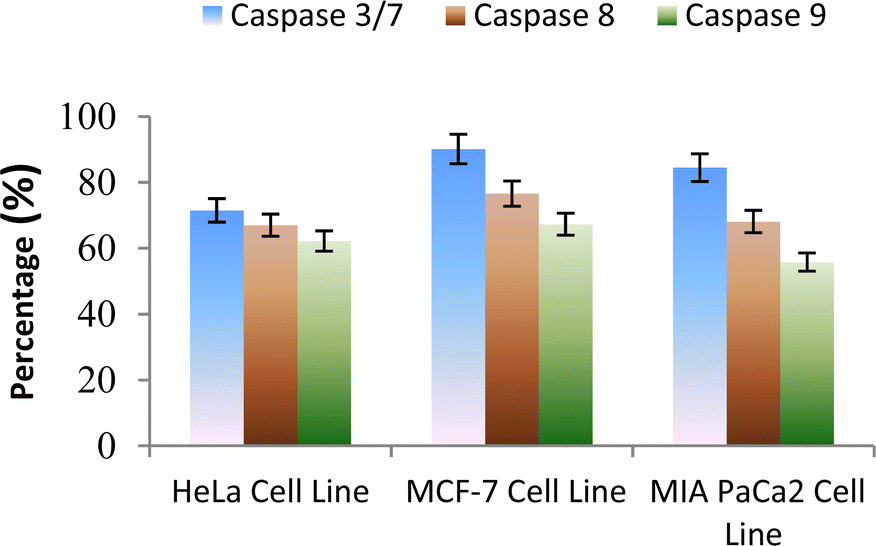
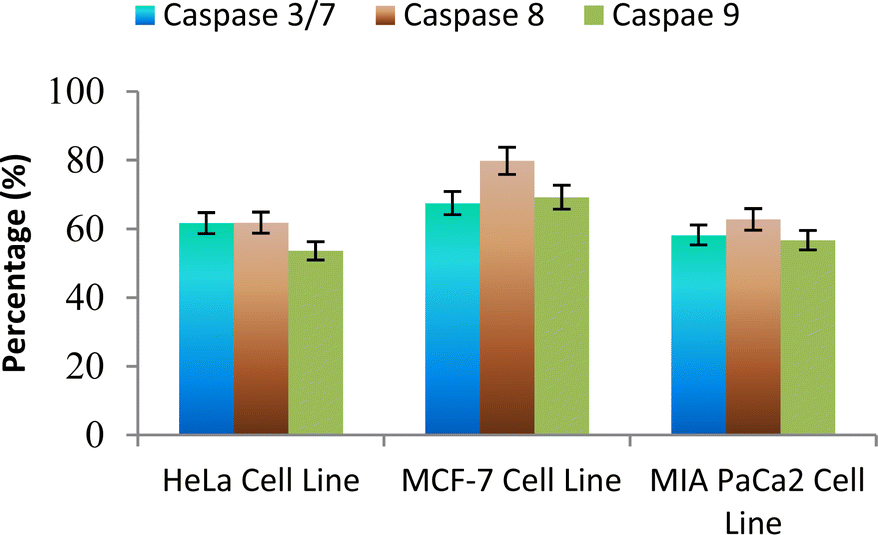
The caspase protease family of enzymes is essential for both the start and completion of apoptosis. When they become active, they cleave many regulatory and structural proteins, which causes the cell to break down internally.43 The traditional indicators of apoptosis, including nuclear condensation, DNA fragmentation, and plasma membrane blebbing, are caused by these proteolytic processes.44 The elimination of damaged cells and preservation of cellular homeostasis are two processes in which apoptosis is essential.45 The terms “intrinsic pathway” and “extrinsic pathway” refer to the two primary caspase activation mechanisms that lead to apoptosis; they are so named because pressures from the outside or inside of the cell, respectively, often activate them. Cellular stressors such as end plasmicreticulum stress, metabolic stress, and DNA damage are the main causes of the intrinsic pathway's activation. This route is activated by several chemotherapy medications that are used to treat different types of cancer. When these stimuli come together, the mitochondria experience outer membrane permeabilization (MOMP), which releases cytochrome c from the intermembrane space of the mitochondria into the cytosol.Hence the podophyllotoxin and most potent compounds 6e, 6i, 6j, 6m and 6n observed in the above in vitro anticancer activities were further tested for their caspase activation and results are shown in Table S1† (Fig. 5–9). Out of all, compound 6e showed paramount caspase activation. In accordance with the results, we noted that the activation of caspases by the compounds was found to be concentration-dependent. Among all, compound 6e highly activated caspase 3/7 in MCF-7 cells with 94.5%, which is even higher than the percentage of the podophyllotoxin 91.9% recorded at 20 μg ml−1. Subsequently, the 3/7 caspase activation by 6e was also found significant in MIA PaCa2 cells with 92.3%, which is higher than the percentage value of podophyllotoxin 88.6% recorded at 20 μg ml−1. On the other hand, the caspase 3/7 activation by 6e in HeLa cells was found slightly lower at 81.3% compared to podophyllotoxin at 86.9% recorded at 20 μg ml−1. These results suggest that compound 6e is a good selection for the activation of 3/7 caspase. Next to 6e, compounds 6j and 6i also highly induced caspase 3/7 activation in MCF-7 cells with 90.1% and 89.3%, respectively, recorded at 20 μg ml−1. The results are compared with 91.9% recorded with podophyllotoxin at 20 μg ml−1. While the different caspase action by the remaining compounds was found average or least. Compound 6m exhibited the least caspase 3/7, 8, and 9 activations with 44.8%, 53.4, 65.0%, 63.0%, 41.9% and 51.7%, respectively, in HeLa, MCF-7 and MIA PaCa2 cancer cells.
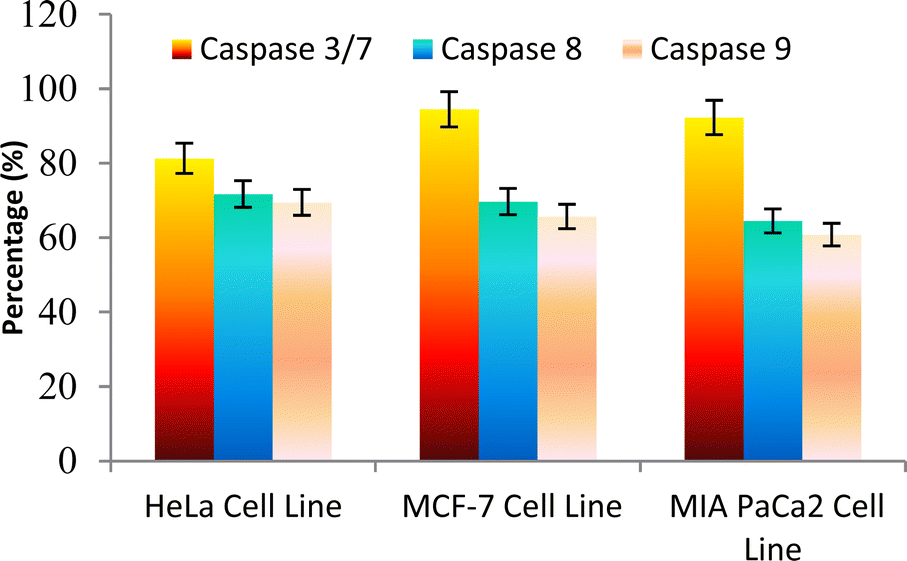 | ||
| Fig. 5 Effect of compound 6e against the activation of different caspases in selected cancer cell lines. | ||
 | ||
| Fig. 6 Effect of compound 6i against the activation of different caspases in the selected cancer cell lines. | ||
 | ||
| Fig. 7 Effect of compound 6j against the activation of different caspases in selected cancer cell lines. | ||
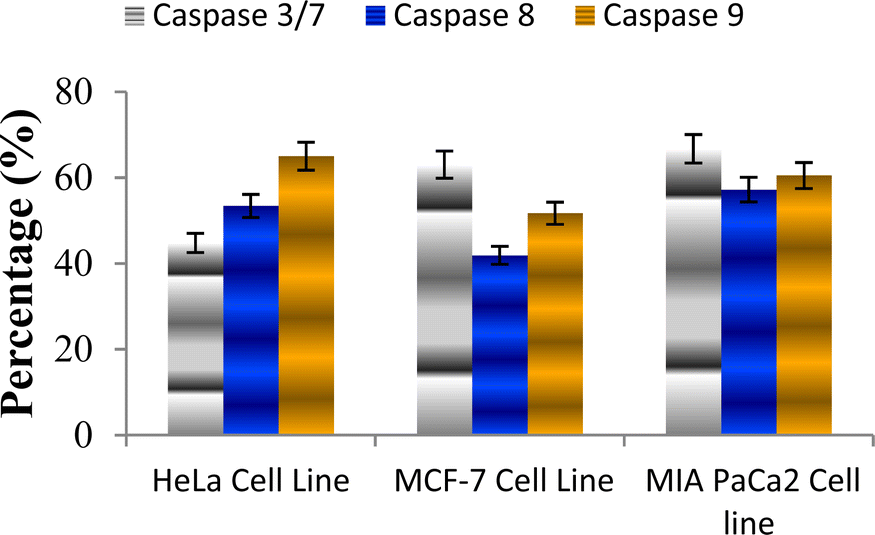 | ||
| Fig. 8 Effect of compound 6m against the activation of different caspases in selected cancer cell lines. | ||
 | ||
| Fig. 9 Effect of compound 6n against the activation of different caspases in selected cancer cell lines. | ||
Tubulin polymerization inhibition
Targeting the microtubules in the development of anticancer drugs was considered to be an important goal in current medicinal chemistry,46 as they are involved in a few cellular functions, such as cell division, organelle transport, motility and keeping signal transduction.47 A few antimitotic agents such as Vinblastine, Colchicine, and Vincristine inhibit tubulin polymerization via their binding to the colchicine or vinca-binding sites of tubulins.48 Curiously, for the past few years, few anti-mitotic agents such as taxanes and vinca alkaloids were used for clinical trials of varied cancer patients.46,49 However, little solubility and oral bioavailability and slightly high toxicity make these anti-mitotic agents less in clinical trials of cancer,46–50 which were made a crucial need to develop novel anti-mitotic agents. Interestingly, a few reports of C4-ring-modified podophyllotoxin51 and a few isoxazole52 derivatives have been recognised to inhibit tubulin polymerization.Hence, we studied the in vitro tubulin polymerization inhibition studies of compounds 6e, 6i, 6j, 6m and 6n using a tubulin assembly assay. The results revealed that compound 6e (IC50 = 0.88 μM) showed comparable activity with the podophyllotoxin (IC50 = 0.84 μM). As well, compounds 6j (IC50 = 1.32 μM) and 6n (IC50 = 1.43 μM) have shown promising potency in comparison to positive control. However, compounds 6i (IC50 = 4.85 μM) and 6m (IC50 = 3.79 μM) had moderate activity as compared to the positive control (Table 3).
| Compound | IC50 (μM) |
|---|---|
| 6e | 0.88 |
| 6i | 4.85 |
| 6j | 1.32 |
| 6m | 3.79 |
| 6n | 1.43 |
| Podophyllotoxin | 0.84 |
Molecular docking studies
The molecular docking studies of most potent compounds 6e, 6j and 6n found in the above in vitro studies and positive control podophyllotoxin were carried out by taking α,β-tubulin (pdb id 1SA0) as target53 and results are shown in Table 4. It was found that compound 6n displayed greater binding energy (−9.39 kcal mol−1) and showed 130.79 nM inhibition constant. Compound 6e was ranked second in this study, with its binding energy as −7.99 kcal mol−1 and 1.38 μM as the inhibition constant. Compound 6j displayed binding energy (−7.84 kcal mol−1) and showed a 1.84 μM as the inhibition constant. On the other aspect, positive control podophyllotoxin showed binding energy = −9.22 kcal mol−1 and 173.47 nM as the inhibition constant.| Entry | Binding energy (kcal mol−1) | No. of H-bonds | Inhibition constant |
|---|---|---|---|
| 6e | −7.99 | — | 1.38 μM |
| 6j | −7.84 | — | 1.80 μM |
| 6n | −9.39 | — | 130.79 nM |
| Podophyllotoxin | −9.22 | — | 173.47 nM |
With respect to binding interaction, podophyllotoxin showed π–π stacking with TYR224 residue and compound 6j formed salt bridge with ARG2 residue.
Overall results revealed that the compound 6n showed an encouraging binding energy and inhibition constant than the podophyllotoxin (Fig. 10 and 11).
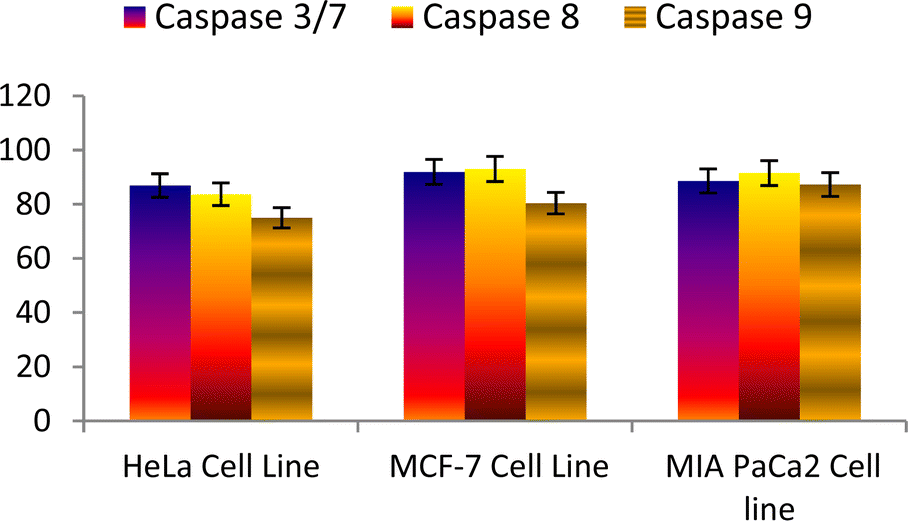 | ||
| Fig. 10 Effect of podophyllotoxin against the activation of different caspases in the selected cancer cell lines. | ||
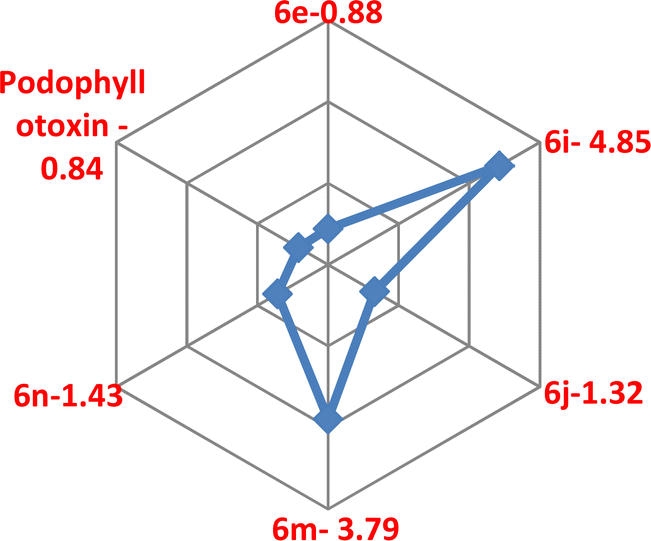 | ||
| Fig. 11 Tubulin polymerization inhibition studies. | ||
Conclusions
Herein, we, first time developed an aqueous organo-NHC catalyzed 1,3-dipolar cycloaddition between 4β-O-propargyl podophyllotoxin (1) and in situ nitrile oxides to afford regioselective 3,5-di-substituted 4β-isoxazolepodophyllotoxin hybrids (6a–n). Compound 6e was the most potent analogue showing greater anti-cancer activity against MCF-7, HeLa and MIAPACA cell lines with GI50 values of 0.18, 0.32 and 0.17 μM, respectively, compared to the standard drug podophyllotoxin (GI50; 0.31, 0.74 and 0.51 μM, respectively). Compounds 6j and 6n showed greater activity (GI50 = 0.23 and 0.12 μM, respectively) than the positive control (GI50 = 0.31 μM) against the MCF-7 cell line. Furthermore, compounds 6i (GI50 = 0.42 μM) and 6m (GI50 = 0.36 μM) had closer activity to the podophyllotoxin (GI50 = 0.31 μM) against the MCF-7 cell line. The results of in vitro tubulin polymerization inhibition revealed that compound 6e (IC50 = 0.88 μM) displayed almost similar activity with podophyllotoxin (IC50 = 0.84 μM). The molecular docking studies of potent compounds 6e, 6j, 6n and podophyllotoxin as tubulin polymerization inhibitors were found to be supportive of the corresponding in vitro activity data.Data availability
Data will be made available on the reader's request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to Chaitanya (Deemed to be University), Himayathnagar (V), Moinabad (M), Ranga Reddy (D), Hyderabad, India, for providing research facilities.Notes and references
- L. M. L. Slevin, Cancer, 1991, 67, 319–329 CrossRef PubMed.
- K. R. Hande, Eur. J. Cancer, 1998, 34, 1514–1521 CrossRef CAS PubMed.
- B. A. Caner and D. L. Longo, Cancer therapy and biotherapy, Lippincott-Raven Publishers, New York, 1996 Search PubMed.
- L. Huang, R. L. Wu and A. M. Xu, Am. J. Transl. Res., 2015, 7, 2141–2158 CAS.
- S. Desben and S. G. Renault, Curr. Med. Chem., 2002, 2, 71–90 Search PubMed.
- D. Leroy, A. V. Kajava, C. Frei and S. M. Gasser, Biochemistry, 2001, 40, 1624–1634 CrossRef CAS PubMed.
- K. Kobayashi and M. J. Ratain, Cancer Chemother. Pharmacol., 1994, 34, S64–S68 CrossRef.
- J. Y. Chang, F. S. Han, S. Y. Liu, Z. Q. Wang, K. H. Lee and Y. C. Cheng, Cancer Res., 1991, 51, 195–198 Search PubMed.
- R. M. Moraes, F. E. Dayan and C. Canel, Stud. Nat. Prod. Chem., 2002, 26, 149–182 CAS.
- T. L. MacDonald, E. K. Lehnert, J. T. Loper, K. C. Chow and W. E. Ross, Oxford University Press, New York, 1991, p. 119.
- S. J. Cho, A. Tropsha, M. Suffness, Y. C. Cheng and K. H. J. Lee, Med. Chem., 1996, 39, 1383–1395 CrossRef CAS PubMed.
- Z. Xiao, Y. D. Xiao, J. Feng, A. Golbraikh, A. Tropsha and K. H. J. Lee, Med. Chem., 2002, 45, 2294–2309 CrossRef CAS PubMed.
- T. Terada, K. Fujimoto, M. Nomura, J. Yamashita, K. Wierzba, R. Yamazaki, J. Shibata, Y. Sugimoto, Y. Yamada, T. Kobunai, S. Takeda, Y. Minami, K. Yoshida and H. J. Yamaguchi, Med. Chem., 1993, 36, 1689–1699 CrossRef CAS PubMed.
- (a) A. Kamal, B. A. Kumar, M. Arifuddin and S. G. Dastidar, Bioorg. Med. Chem., 2003, 11, 5135–5142 CrossRef CAS PubMed; (b) A. Kamal, N. L. Gayatri, D. R. Reddy, P. S. M. M. Reddy, M. Arifuddin, S. G. Dastidar, A. K. Kondapi and M. Rajkumar, Bioorg. Med. Chem., 2005, 13, 6218–6225 CrossRef CAS PubMed; (c) A. Kamal, E. Laxman, G. B. R. Khanna, P. S. M. M. Reddy, T. Rehana, M. Arifuddin, K. Neelima, A. K. Kondapi and S. G. Dastidar, Bioorg. Med. Chem., 2004, 12, 4197–4201 CrossRef CAS PubMed; (d) A. Kamal, B. A. Kumar, M. Arifuddin and S. G. Dastidar, Lett. Drug Des. Discovery, 2006, 3, 205–209 CrossRef CAS; (e) A. Kamal and B. A. Kumar, WO136018, 2008; (f) A. Kamal, B. A. Kumar and M. Arifuddin, WO07339, 2004.
- P. Dong, K. Rakesh, H. Manukumar, Y. H. E. Mohammed, C. Karthik, S. Sumathi, P. Mallu and H. L. Qin, Bioorg. Chem., 2019, 85, 325–336 CrossRef CAS PubMed.
- X. Zhang, K. Rakesh, C. Shantharam, H. Manukumar, A. Asiri, H. Marwani and H. L. Qin, Bioorg. Med. Chem., 2018, 26, 340–355 CrossRef CAS PubMed.
- W. Y. Fang, L. Ravindar, K. Rakesh, H. Manukumar, C. Shantharam, N. S. Alharbi and H. L. Qin, Eur. J. Med. Chem., 2019, 173, 117–153 CrossRef CAS PubMed.
- H. Liu, S. Long, K. Rakesh and G. F. Zha, Eur. J. Med. Chem., 2020, 185, 111804 CrossRef CAS PubMed.
- M. Xu, Y. Peng, L. Zhu, S. Wang, J. Ji and K. P. Rakesh, Eur. J. Med. Chem., 2019, 180, 656–672 CrossRef CAS PubMed.
- (a) M. R. Sivala, V. Chintha, K. M. Potla, S. Chinnam and N. R. Chamarthi, Signal Transduction Res., 2020, 40, 486–492 CrossRef CAS PubMed; (b) Md. Elagawany, L. Maram and B. Elgendy, J. Org. Chem., 2023, 88, 17062–17068 CrossRef CAS PubMed.
- F. Hu and M. Szostak, Adv. Synth. Catal., 2015, 357, 2583–2614 CrossRef CAS.
- G. Boyd, E. Fangheanel, D. Gudat, D. Kikelj and C. T. Pedersen, Hetarenes and Related Ring Systems, in Science of Synthesis: Houben-Weyl Methods of Molecular Transformations, vol. 11, ed. E. Schaumann, Thieme Chemistry, 2014 Search PubMed.
- (a) J. Zhua, J. Moa, H. Z. Lina, Y. Chenb and H. Suna, Bioorg. Med. Chem., 2018, 26, 3065–3075 CrossRef; (b) G. Ch. Arya, K. Kaur and V. Jaitak, Eur. J. Med. Chem., 2021, 221, 113511 CrossRef CAS PubMed; (c) A. Sysak and B. O. Mrukowicz, Eur. J. Med. Chem., 2017, 137, 292–309 CrossRef CAS PubMed.
- F. Hua and M. Szostak, Adv. Synth. Catal., 2015, 357, 2583–2614 CrossRef.
- (a) G. Scott and V. V. Fokin, Angew. Chem., 2008, 120, 8409–8411 CrossRef; (b) F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed; (c) T. V. Hansen, P. Wu and V. V. Fokin, J. Org. Chem., 2005, 70, 7761–7764 CrossRef CAS PubMed.
- (a) B. List, Chem. Rev., 2007, 107, 5413–5415 CrossRef CAS; (b) S. Bertelsen and K. A. Jorgensen, Chem. Soc. Rev., 2009, 38, 2178–2189 RSC; (c) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS PubMed; (d) W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef CAS PubMed; (e) L. S. Hegedus, J. Am. Chem. Soc., 2009, 131, 17995–17997 CrossRef CAS PubMed.
- S. Kankala, R. Vadde and C. S. Vasam, Org. Biomol. Chem., 2011, 9, 7869–7876 RSC.
- (a) R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS; (b) H. Stetter, Angew. Chem., Int. Ed. Engl., 1976, 15, 639–647 CrossRef; (c) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (d) N. Marion, S. Diez-Gonzalez and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS PubMed; (e) W. D. Jones, J. Am. Chem. Soc., 2009, 131, 15075–15077 CrossRef CAS PubMed; (f) P. Chiang, M. Rommel and J. W. Bode, J. Am. Chem. Soc., 2009, 131, 8714–8718 CrossRef CAS PubMed; (g) S. D. Sarkar and A. Studer, Org. Lett., 2010, 12, 1992–1995 CrossRef PubMed; (h) J. Pinaud, K. Vijayakrishna, D. Taton and Y. Gnanou, Macromolecules, 2009, 42, 4932–4936 CrossRef CAS; (i) N. E. Kamber, W. Jeong, S. Gonzalez, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2009, 42, 1634–1639 CrossRef CAS; (j) E. M. Phillips, A. Chan and K. A. Scheid, Aldrichim. Acta, 2009, 42, 55–66 CAS; (k) X. Bugaut, F. Liu and F. Glorius, J. Am. Chem. Soc., 2011, 133, 8130–8133 CrossRef CAS PubMed; (l) A. T. Biju, N. E. Wurz and F. Glorius, J. Am. Chem. Soc., 2010, 132, 5970–5971 CrossRef CAS PubMed; (m) J. Mahatthananchai, P. Zheng and J. W. Bode, Angew. Chem., Int. Ed., 2011, 50, 1673–1677 CrossRef CAS PubMed; (n) V. Nair, S. Bindu and V. Sreekumar, Angew. Chem., Int. Ed., 2004, 43, 5130–5135 CrossRef CAS PubMed; (o) A. B. Powell, Y. Suzuki, M. Ueda, C. W. Bielawski and A. H. Cowley, J. Am. Chem. Soc., 2011, 133, 5218–5220 CrossRef CAS PubMed; (p) Y. Wang, Y. Xie, M. Y. Abraham, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2010, 132, 14370–14372 CrossRef CAS PubMed; (q) S. Kankala, R. Edulla, S. Modem, R. Vadde and C. S. Vasam, Tetrahedron Lett., 2011, 52, 3828–3831 CrossRef CAS; (r) D. Du, L. Li and Z. Wang, J. Org. Chem., 2009, 74, 4379–4382 CrossRef CAS PubMed.
- (a) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS; (b) A. J. Arduengo III, Acc. Chem. Res., 1999, 32, 913–921 CrossRef; (c) D. Bourissou, O. Guerret, F. P. Gabba and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS; (d) L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. Cesar, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed; (e) D. Tapu, D. A. Dixon and C. Roe, Chem. Rev., 2009, 109, 3385–3407 CrossRef CAS PubMed; (f) A. J. Arduengo III and L. I. Iconaru, Dalton Trans., 2009, 6903–6914 RSC.
- (a) R. M. de Figueiredo and M. Christmann, Eur. J. Org Chem., 2007, 2575–2600 CrossRef CAS; (b) E. Marques-Lopez, R. P. Herrera and M. Christmann, Nat. Prod. Rep., 2010, 27, 1138–1167 RSC; (c) C. Vaxelaire and P. W. M. Christmann, Angew. Chem., Int. Ed., 2011, 50, 3605–3607 CrossRef CAS PubMed.
- (a) N. Marion, S. Diez-Gonzalez and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS PubMed; (b) A. J. Arduengo III and L. I. Iconaru, Dalton Trans., 2009, 6903–6914 RSC; (c) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011, 44, 1182–1195 CrossRef CAS PubMed; (d) V. Nair, R. S. Menon, A. T. Biju, C. R. Sinu, R. R. Paul, A. Jose and V. Sreekumar, Chem. Soc. Rev., 2011, 40, 5336–5346 RSC; (e) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314–325 CrossRef CAS PubMed; (f) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC.
- C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205–6208 CrossRef CAS PubMed.
- (a) S. S. Sohn and J. W. Bode, Angew. Chem., Int. Ed., 2006, 45, 6021–6024 CrossRef CAS PubMed; (b) M. He, B. J. Beahm and J. W. Bode, Org. Lett., 2008, 10, 3817–3820 CrossRef CAS PubMed.
- H. U. Vora and T. Rovis, J. Am. Chem. Soc., 2010, 132, 2860–2861 CrossRef CAS PubMed.
- J. M. Obrien and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 7712–7715 CrossRef CAS PubMed.
- (a) T. L. Amyes, S. L. Diver, J. P. Richard, F. M. Rivas and K. J. Toth, J. Am. Chem. Soc., 2004, 126, 4366–4374 CrossRef CAS PubMed; (b) O. Holloczki, P. Terleczky, D. Szieberth, G. Mourgas, D. Gudat and L. Nyulaszi, J. Am. Chem. Soc., 2011, 133, 780–789 CrossRef CAS PubMed.
- R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 CrossRef CAS.
- W. Wen, Y. Leong, X. Chen and Y. R. Chi, Green Chem., 2013, 15, 1505–1508 RSC.
- (a) N. S. Thirukovela, R. Balaboina, V. Botla, R. Vadde, S. B. Jonnalagadda and C. S. Vasam, Catal. Sci. Technol., 2019, 9, 6471–6481 RSC; (b) N. S. Thirukovela, R. Balaboina, S. Kankala, R. Vadde and C. S. Vasam, Tetrahedron, 2019, 75, 2637–2641 CrossRef CAS; (c) N. S. Thirukovela, R. Balaboina, R. Vadde and C. S. Vasam, Tetrahedron Lett., 2018, 59, 3749–3752 CrossRef CAS; (d) M. Muqeed, R. Manchal, V. Botla, Md. Azam, C. S. Vasam and N. S. Thirukovela, Asian J. Org. Chem., 2023, 12, e202300356 CrossRef CAS; (e) R. R. Sagam, S. K. Nukala, R. Nagavath, N. Sirassu, M. Mohammod, R. Manchal and N. S. Thirukovela, J. Mol. Struct., 2022, 1268, 133692 CrossRef CAS; (f) R. Nagavath, S. K. Nukala, N. Sirassu, R. R. Sagam, R. Manchal, S. Paidakula and N. S. Thirukovela, J. Mol. Struct., 2022, 1250, 131724 CrossRef CAS; (g) P. K. Kannekanti, S. K. Nukala, M. Bangaru, N. Sirassu, R. Manchal and N. S. Thirukovela, ChemistrySelect, 2023, 8, e202204010 CrossRef CAS; (h) M. Bangaru, S. K. Nukala, P. K. Kannekanti, N. Sirassu, R. Manchal and N. S. Thirukovela, ChemistrySelect, 2023, 8, e202204414 CrossRef CAS; (i) N. S. Thirukovela, S. K. Nukala, N. Sirassu, R. Manchal, P. Gundepaka and S. Paidakula, ChemistrySelect, 2020, 5, 12317–12319 CrossRef CAS; (j) R. R. Sagam, S. K. Nukala, R. Nagavath, N. Sirassu, P. Gundepaka, R. Manchal and N. S. Thirukovela, ChemistrySelect, 2021, 6, 7670–7673 CrossRef CAS; (k) R. Nagavath, S. K. Nukala, R. R. Sagam, N. Sirassu, V. Guguloth, P. Kamarajugadda, S. Paidakula and N. S. Thirukovela, ChemistrySelect, 2022, 7, e202202200 CrossRef CAS.
- N. Srinivas, K. Shravan and G. Brahmeshwari, Nat. Prod. Res., 2019, 35, 9–16 Search PubMed.
- (a) G. C. Senadi, M. R. Mutra, T. Y. Lu and J. J. Wang, Green Chem., 2017, 19, 4272–4277 RSC; (b) Y. H. T. Sai, C. M. B. Etichetti, S. Cicetti, J. E. Girardini, R. A. Spanevello, A. G. Suarez and A. M. Sarotti, Bioorg. Med. Chem. Lett., 2020, 30, 127247 CrossRef PubMed.
- (a) V. Nair, A. N. Pillai, P. B. Beneesh and E. Suresh, Org. Lett., 2005, 7, 4625–4628 CrossRef CAS PubMed; (b) V. Nair, A. N. Pillai, R. S. Menon and E. Suresh, Org. Lett., 2005, 7, 1189–1191 CrossRef CAS PubMed; (c) V. Nair, S. Bindu, V. Sreekumar and N. P. Rath, Org. Lett., 2003, 5, 665–667 CrossRef CAS PubMed; (d) Y. L. Wu, P. D. Jarowski, W. B. Schweizer and F. Diederich, Chem.–Eur. J., 2010, 16, 202–211 CrossRef CAS PubMed; (e) V. Nair, R. S. Menon, P. B. Beneesh, V. Sreekumar and S. Bindu, Org. Lett., 2004, 6, 767–769 CrossRef CAS PubMed; (f) X. F. Zhu, C. E. Henry and O. Kwon, J. Am. Chem. Soc., 2007, 129, 6722–6723 CrossRef CAS PubMed; (g) M. R. Siebert, A. K. Yudin and D. J. Tantillo, Org. Lett., 2008, 10, 57–60 CrossRef CAS PubMed.
- S. J. Martin and D. R. Green, Cell, 1995, 82, 349–352 CrossRef CAS PubMed.
- J. F. Kerr, A. H. Wyllie and A. R. Currie, Br. J. Cancer, 1972, 26, 239–257 CrossRef CAS PubMed.
- S. Arandjelovic and K. S. Ravichandran, Nat. Immunol., 2015, 16, 907–917 CrossRef CAS PubMed.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed.
- (a) J. Ceramella, A. Caruso, M. A. Occhiuzzi, D. Iacopetta, A. Barbarossa, B. Rizzuti, P. Dallemagne, S. Rault, H. El. Kashef, C. Saturnino and M. S. Sinicropi, Eur. J. Med. Chem., 2019, 1, 111583 CrossRef PubMed; (b) K. H. Downing, Annu. Rev. Cell Dev. Biol., 2000, 16, 89–111 CrossRef CAS PubMed; (c) H. S. Mohamed, N. H. Amin, M. T. El-Saadi and H. M. A. Rahman, Bioorg. Chem., 2022, 16, 105687 CrossRef PubMed.
- H. Chen, Z. Lin, K. E. Arnst, D. D. Miller and W. Li, Molecules, 2017, 22, 1281 CrossRef PubMed.
- G. Wang, C. Li, L. He, K. Lei, F. Wang, Y. Pu, Z. Yang, D. Cao, L. Ma, J. Chen, Y. Sang, X. Liang, M. Xiang, A. Peng, Y. Wei and L. Chen, Bioorg. Med. Chem., 2014, 22, 2060–2079 CrossRef CAS PubMed.
- S. Messaoudi, B. Treguier, A. Hamze, O. Provot, J. F. Peyrat, J. R. De Losada, J. M. Liu, J. Bignon, J. W. Bakala, S. Thoret, J. Dubois, J. D. Brion and M. Alami, J. Med. Chem., 2009, 52, 4538–4542 CrossRef CAS PubMed.
- (a) I. Hyder, Y. Deepthi, K. Shasi, J. Khazir, N. Naresh, M. Sreekanth, M. Halmuthur and S. Kumar, Bioorg. Med. Chem. Lett., 2015, 14, 2860–2863 CrossRef PubMed; (b) Y. Liu, D. Wei, Y. Zhao, W. Cheng, Y. Lu, Y. Ma, L. Xin, C. Han, Y. Wei, H. Cao and Z. Chunyan, Bioorg. Med. Chem. Lett., 2012, 20, 6285–6295 CrossRef CAS PubMed; (c) L. Zhang, X. Zeng, X. Ren, N. Tao, C. Yang, Y. Xu, Y. Chen and J. Wang, Med. Chem. Res., 2019, 28, 81–94 CrossRef CAS; (d) Z. Zhen Wang, W. Xue Sun, X. Wang, Z. Y. Han, Q. H. Yue, Q. J. Liang, P. Y. Jun, L. G. Hua, X. W. Ming, Y. F. Gen and Y. Y. H. Yang, Chem. Biol. Drug Des., 2017, 2, 236–243 CrossRef PubMed; (e) M. V. P. S Vishnuvardhan, V. S. Reddy, K. Chandrasekhar, V. Lkshma Nayak, S. D. I. Bin, A. Abdullah and A. Kamal, MedChemComm, 2017, 8, 1817–1823 RSC; (f) Y. Chengli, X. Qiongli, X. Zeng, T. Nengyin, Y. Xu, C. Yongzhen, W. Jing and L. Zhang, Bioorg. Chem., 2019, 85, 445–454 CrossRef PubMed.
- (a) D. J. Fu, S. M. Liu, F. H. Li, J. J. Yang and J. Li, J. Enzyme Inhib. Med. Chem., 2020, 35, 1050–1059 CrossRef CAS PubMed; (b) G. Wang, W. Liu, Y. Huang, Y. Li and Z. Peng, Arabian J. Chem., 2020, 13, 5765–5775 CrossRef CAS; (c) K. D. Shin, Y. J. Yoon and Y. R. Kang, Biochem. Pharmacol., 2008, 75, 383–394 CrossRef CAS PubMed.
- R. Ravelli, B. Gigant, P. Curmi, I. Jourdain, S. Lachkar, A. Sobel and M. Knossow, Nature, 2004, 428, 198–202 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04297b |
| This journal is © The Royal Society of Chemistry 2024 |