DOI:
10.1039/D4RA04453C
(Paper)
RSC Adv., 2024,
14, 28659-28668
Synthesis of quinoxalines and assessment of their inhibitory effects against human non-small-cell lung cancer cells†
Received
18th June 2024
, Accepted 3rd September 2024
First published on 9th September 2024
Abstract
Twenty-six quinoxalin derivatives were synthesized to assess their biological activities against human non-small-cell lung cancer cells (A549 cells). Compound 4b (IC50 = 11.98 ± 2.59 μM) and compound 4m (IC50 = 9.32 ± 1.56 μM) possess anticancer activity comparable to 5-fluorouracil (clinical anticancer drug) (IC50 = 4.89 ± 0.20 μM). Western blot tests further confirmed that compound 4m effectively induced apoptosis of A549 cells through mitochondrial- and caspase-3-dependent pathways. The introduction of bromo groups instead of nitro groups into the quinoxaline skeleton has been shown to provide better inhibition against lung cancer cells in this article. This modification in the molecular structure could enhance the biological activity and effectiveness of quinoxaline derivatives in the design and synthesis of anticancer drugs, making bromo-substituted quinoxalines a promising avenue for further research and development in anticancer therapeutics.
1 Introduction
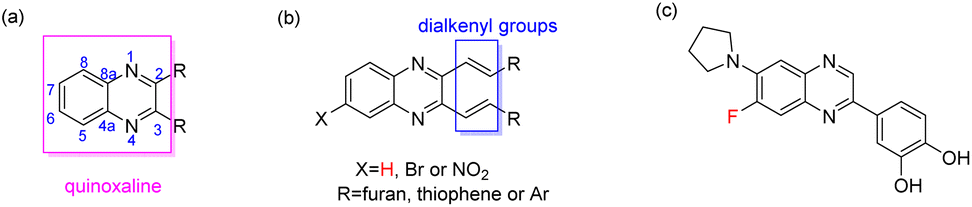
Quinoxaline, known as benzopyrazine, is naphthalene with some carbon atoms replaced with nitrogen atoms1 (Fig. 1a). The nitrogen atoms of quinoxalines contribute to the stabilization of radical ion species. Naturally occurring quinoxalines are scarce,2,3 and their derivatives are mostly derived from synthetic products. Quinoxaline's skeleton is in frame with some natural antibiotics, such as echinomycin,4,5 quinornycin A,6 triostins7 and actinoleutin which show activities against Gram-positive bacteria8 and tumors.9 Furthermore, the synthetic quinoxaline derivatives possess antimicrobial,1,10,11 anti-inflammatory,12,13 antiviral,1,14 antioxidant,13,15 antitumor,1,9 receptor antagonist,16 antitubercular,1,17,18 antifungal,1,11,15,19–22 antiparasitic23 and antidiabetic1,24–28 activities. Besides the pharmacological interest in quinoxalines, they are also employed in material sciences, such as in OLEDs29 and dye-sensitized solar cells.30
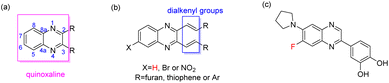 |
| | Fig. 1 The skeletons of quinoxaline (a) and C6-substituted alkenyl quinoxalines (b) are reported in this article, and a quinoxaline derivative (c) is reported in the literature with potent biological activity. | |
Quinoxaline urea derivatives were evaluated in the treatment of pancreatic therapy.31 Therefore, we have reported the synthesis of cinnamils and 2,3-dialkenyl-substituted quinoxalines to evaluate their activities against pancreatic cancer cells.32 Among them, one of cinnamils exhibited a more potent anticancer effect than that of quinoxalines. However, that series of quinoxalines presented a moderate inhibitory effect. Compared with other quinoxalines, synthesizing 2,3-dialkenyl-substituted quinoxalines is relatively rare.33,34 They exhibited photophysical properties34 or as a detector,35 however, their biological activity is not well studied. A C6-fluoro substituted quinoxaline was reported as a potent inhibitor of JNK stimulatory phosphatase-1 (JSP-1) in an in vitro biological assay (Fig. 1c).36 Due to quinoxalines for their pharmacological interests, it prompted us to introduce an electron-withdrawing group to the C6-position of 2,3-dialkenyl-substituted quinoxalines. Therefore, the bromo or nitro groups were selected to assess their activities against non-small-cell lung cancer cells (Fig. 1b, X = Br, or NO2) and pursue hit compounds. According to reports, the NO2 functional group may be genotoxic, so the genotoxicity of 5a–5m needs further discussion. However, this article aims to investigate the apoptotic mechanisms of the most effective compound 4m.
2 Results and discussion
2.1 Chemistry
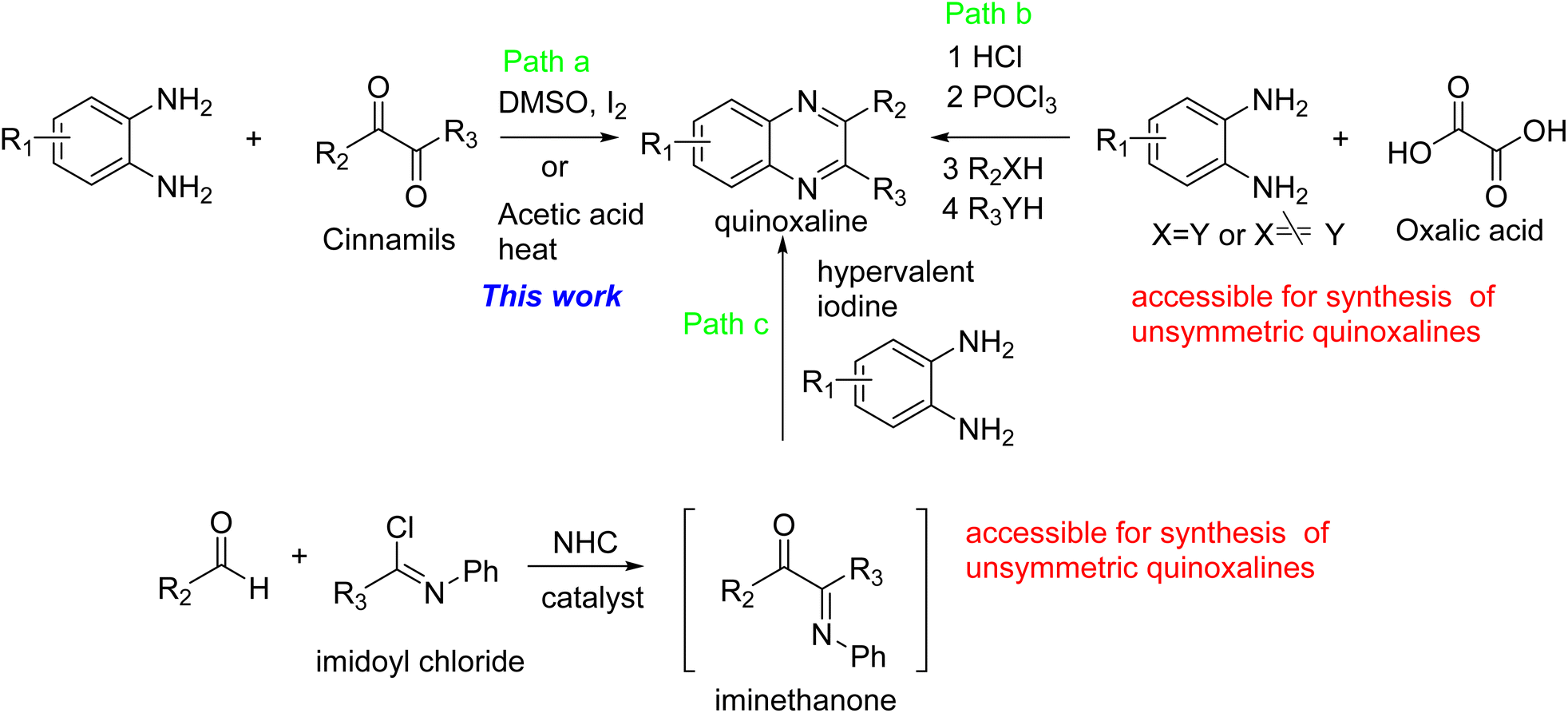
There are at least three methods available for the synthesis of quinoxalines. First, the condensation of cinnamils with 1,2-phenylenediamine derivatives under either the catalytic amount of I2 in DMSO37 or reflux acetic acid,38 which condition is used for the synthesis of target molecules in this article (Path a). Secondly, oxalic acid is condensed with 1,2-phenylenediamine in an acidic condition and then treated with POCl3 followed by reacting with nucleophiles (Path b).10 Finally, the key intermediate, iminoethanone, was prepared from aldehyde and imidoyl chloride under the NHC catalyst (path c). The intermediate, iminoethanone, was condensed with 1,2-phenylenediamine catalyzed by hypervalent iodine39 to obtain quinoxaline. It is noticed that Path b and Path c are more readily available for the synthesis of unsymmetric quinoxalines (Scheme 1).
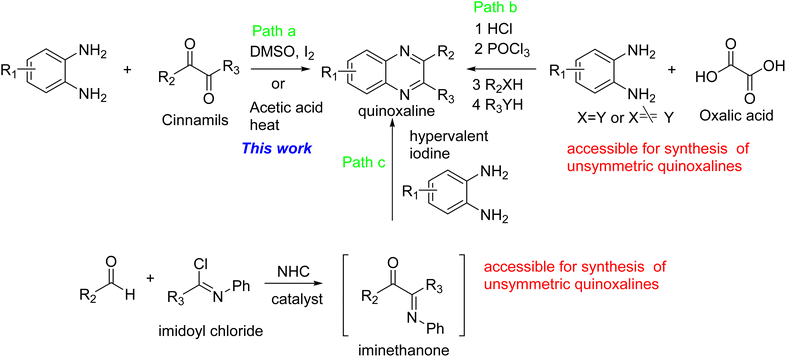 |
| | Scheme 1 The reported strategies for the synthesis of quinoxalines. | |
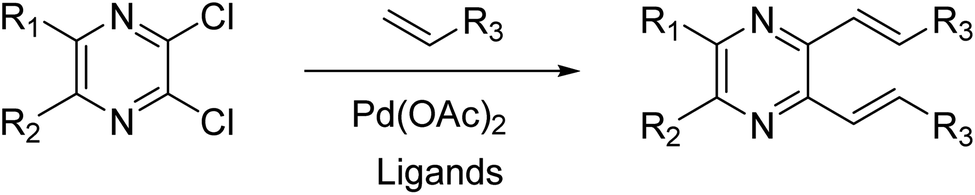
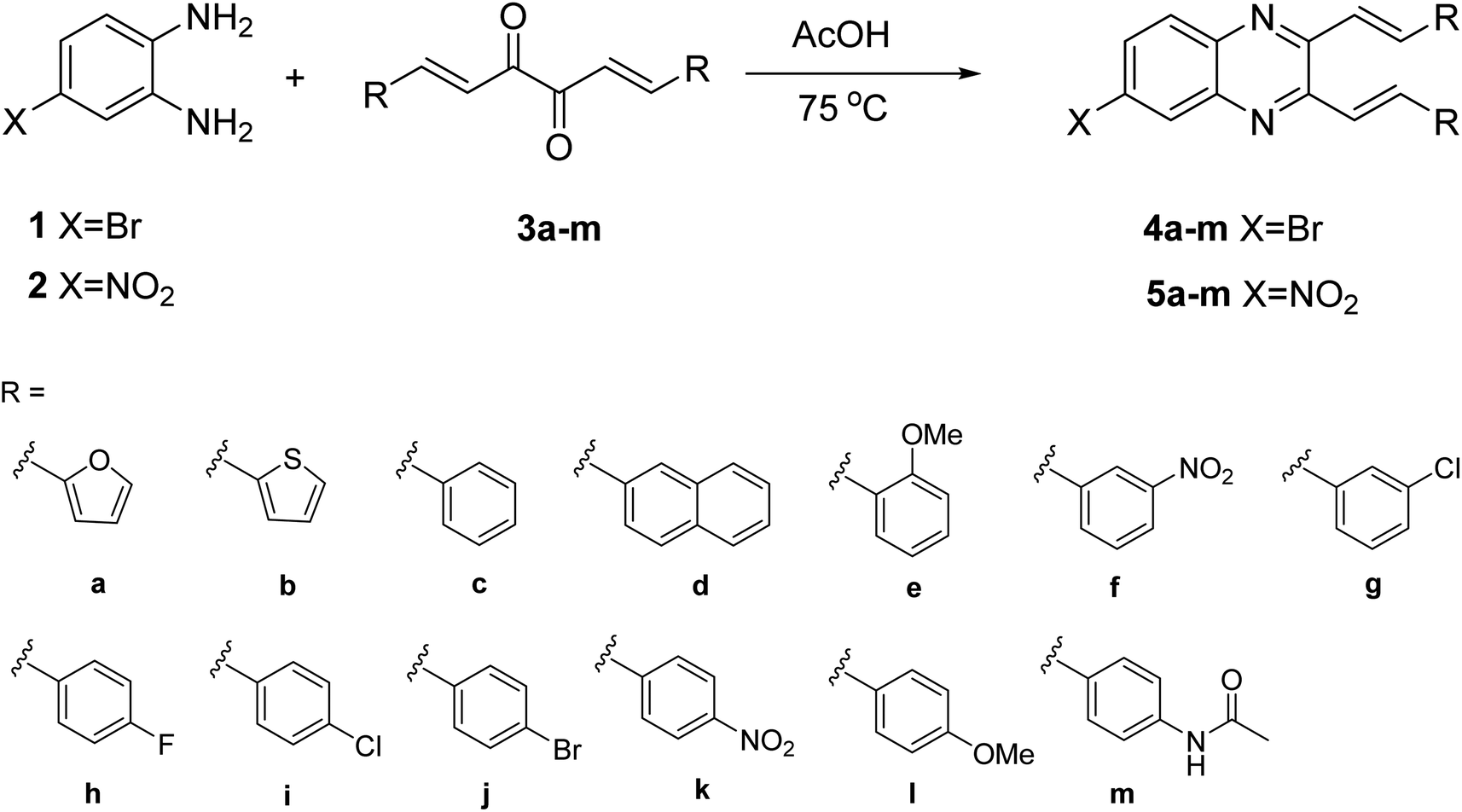
The previous synthesis of 2,3-dialkenyl-substituted quinoxalines employed a Heck method (Fig. 2).33 We applied the condensation of 1 or 2 with the corresponding 3a–m in acetic acid under reflux conditions.32 The resulting solid was filtered by suction and washed with distilled water to obtain 4a–m and 5a–m in moderate to high yields without column chromatography (Scheme 2, see experimental section).
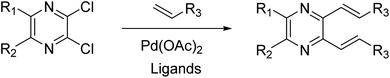 |
| | Fig. 2 The synthesis of 2,3-dialkenyl-substituted quinoxalines via the Heck method. | |
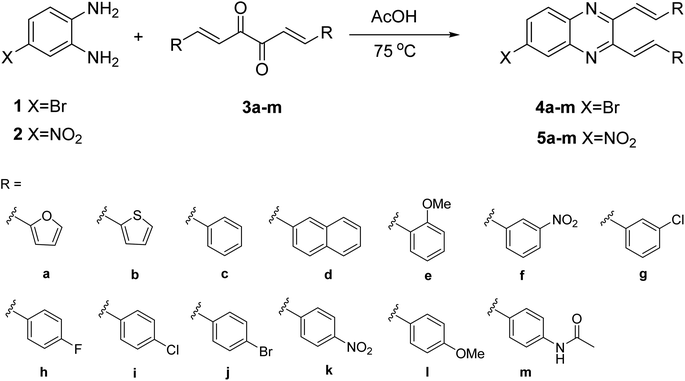 |
| | Scheme 2 Synthesis of quinoxalines 4a–m and 5a–m. | |
2.2 Biological evaluation
The cytotoxic effects of all twenty-six synthesized compounds were evaluated by their activity to suppress human non-small cell lung cancer cells (A549 cells). As shown in Table 1, compound 4m (IC50 = 9.32 ± 1.56 μM) exhibited the most potent inhibitory activity against A549 cells. Although compound 4m showed slightly weaker anticancer activity than 5-fluorouracil (5-FU) against A549 cells. We further compared the cytotoxicity of 4m and the clinical anticancer drug 5-FU on normal human cells. The cytotoxicity of 4m (IC50 = 9.29 ± 1.25 μM) to human foreskin fibroblast (Hs68) cells is approximately 3 times lower than that of 5-FU (IC50 = 3.08 ± 1.40 μM) (Table 2). Therefore, it shows that 4m possesses research and development value. In addition, compounds 4a, 4b, 4e, 4g, 4l, and 5g also exhibited cytotoxic activities against A549 cells.
Table 1 Inhibitory effects of compounds against human non-small-cell lung cancer cells (A549)a
| Compounds |
IC50b (μM) |
| Results are presented as averages ±SD (n = 3). Concentration necessary for 50% inhibition (IC50). 5-Fluorouracil (5-FU) was used as a positive control. |
| 4a |
54.94 ± 2.13** |
| 4b |
11.98 ± 2.59*** |
| 4c |
>100 |
| 4d |
>100 |
| 4e |
35.23 ± 9.29* |
| 4f |
>100 |
| 4g |
42.39 ± 1.40** |
| 4h |
>100 |
| 4i |
>100 |
| 4j |
>100 |
| 4k |
>100 |
| 4l |
38.17 ± 0.48* |
| 4m |
9.32 ± 1.56** |
| 5a |
>100 |
| 5b |
>100 |
| 5c |
>100 |
| 5d |
>100 |
| 5c |
>100 |
| 5d |
>100 |
| 5e |
>100 |
| 5f |
>100 |
| 5g |
75.02 ± 4.50** |
| 5h |
>100 |
| 5i |
>100 |
| 5j |
>100 |
| 5k |
>100 |
| 5l |
>100 |
| 5m |
>100 |
| 5-FUc |
4.89 ± 0.20** |
Table 2 In vitro cytotoxic effects of compound 4m and 5-fluorouracil against Hs68 (human foreskin fibroblast) cellsa
| Compounds |
IC50b (μM) |
| Results are presented as averages ±SD (n = 3). Concentration necessary for 50% inhibition (IC50). 5-Fluorouracil (5-FU) was used as a positive control. |
| 4m |
9.29 ± 1.25 |
| 5-FUc |
3.08 ± 0.40 |
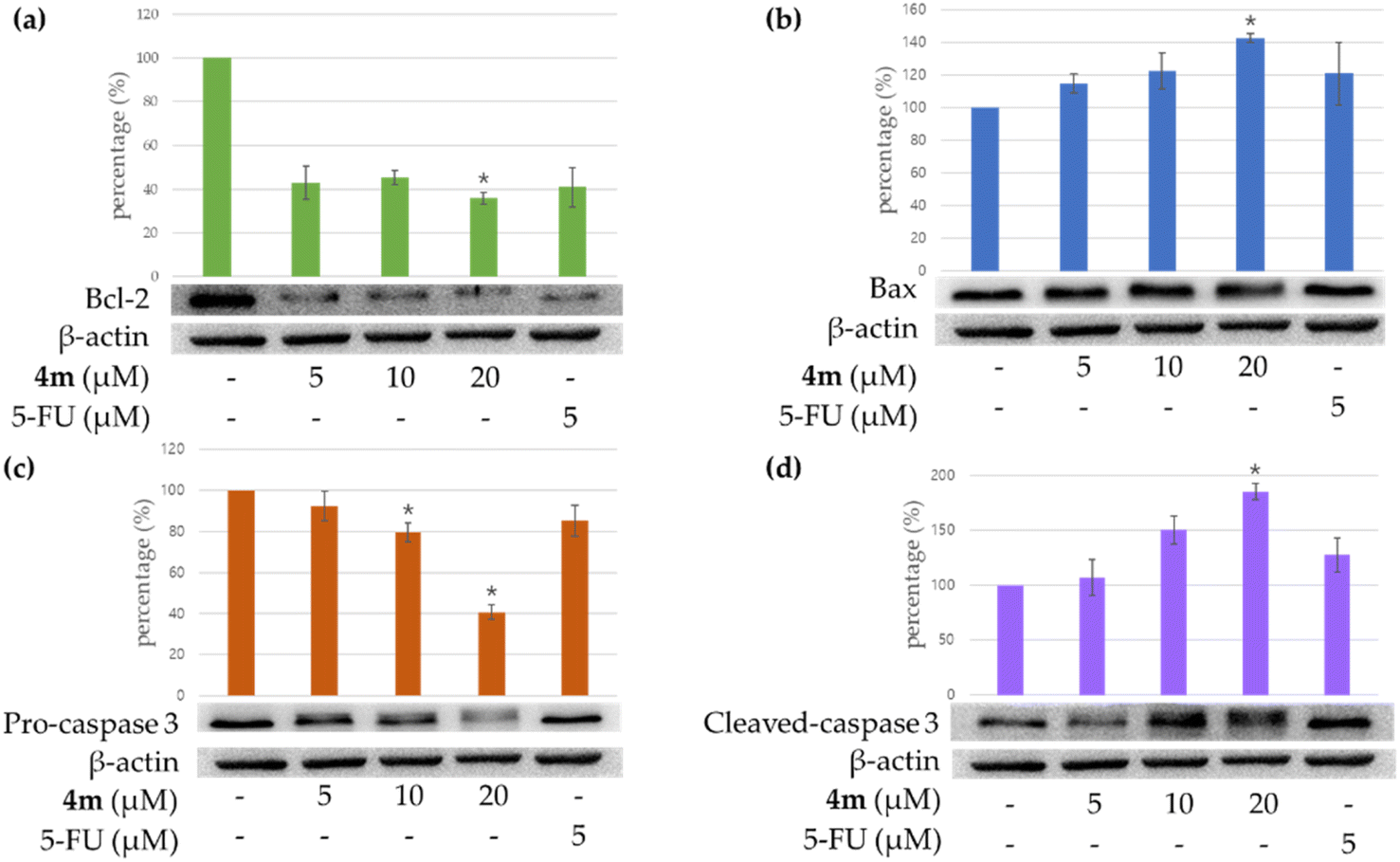
The effect of treating A549 cells with compound 4m on the expression of apoptosis-related proteins was investigated. Fig. 3a shows that the expression of the pro-apoptotic protein Bax treated with 20 μM of compound 4m was higher than that treated with 5 or 10 μM of 4m. In contrast, cells treated with 20 μM of compound 4m showed lower Bcl-2 (anti-apoptotic protein) expression than those treated with 5 or 10 μM (Fig. 3b). The results show that compound 4m suppressed the expression of Bcl-2 and increased the expression of Bax.
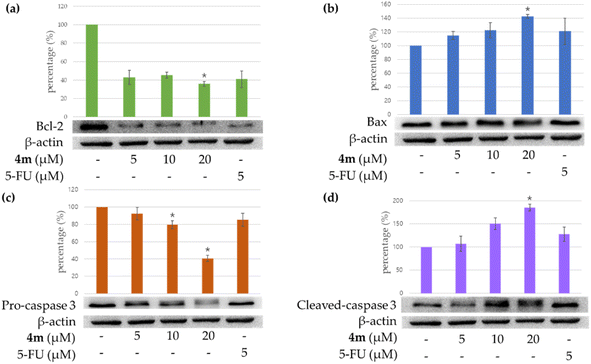 |
| | Fig. 3 Western blot analysis for Bcl-2 (a), Bax (b), pro-caspase 3 (c), and cleaved caspase 3 (d) in each group on A549 cells. 4m means compound 4m. 5-Fluorouracil (5-FU) was used as a positive control. Asterisks indicate significant differences (*p < 0.05) compared with the control group. | |
Besides, caspase 3 activation is a hallmark of apoptosis. We further investigated whether compound 4m could influence the enzymatic activities of caspase-3. The results show that compound 4m suppressed pro-caspase 3 and increased the cleaved caspase-3 (Fig. 3c and d). Furthermore, compound 4m markedly induced apoptosis of A549 cells through caspase-3-dependent pathways.
3 Conclusions
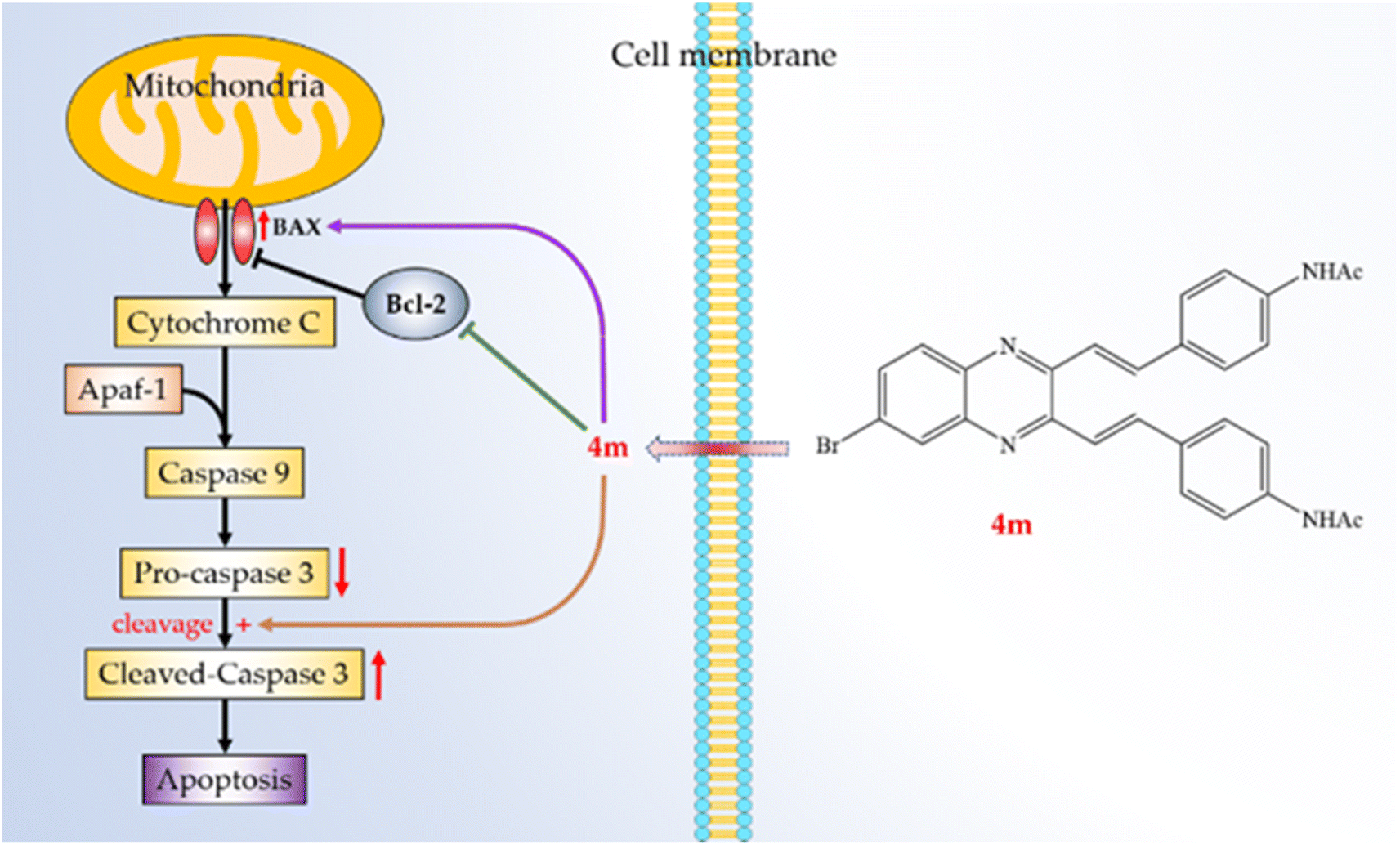
The development of quinoxalines as anticancer agents has gained attention in recent years.40,41 Synthesis of quinoxalines 4a–m and 5a–m was facile. Their purifications were simple filtration, then washed with water to obtain the pure target molecules. This series of compounds showed less potency on A549 cells than 5-FU. However, it provides important information that the C6-bromo rather than C6-nitro group is a suitable quinoxaline substitute for designing potential drug candidates. Furthermore, we found compounds 4f, 4k, 5f, and 5k, which are with two nitro groups, reduced the solubility. Compound 4m was found to have the highest cytotoxic activity against A549 cells out of all synthesized molecules. The activity of compound 4m was comparable to that of 5-FU (clinical anticancer drug). Furthermore, compound 4m markedly induced apoptosis of A549 cells through the mitochondrial- and caspase-3-dependent pathways (Fig. 4). This suggests that the halo substituted at C6-position of 2,3-dialkenyl quinoxalines may be necessary. Thus, compound 4m is worth further investigation and might be developed as a candidate for treating or preventing human non-small-cell lung cancer.
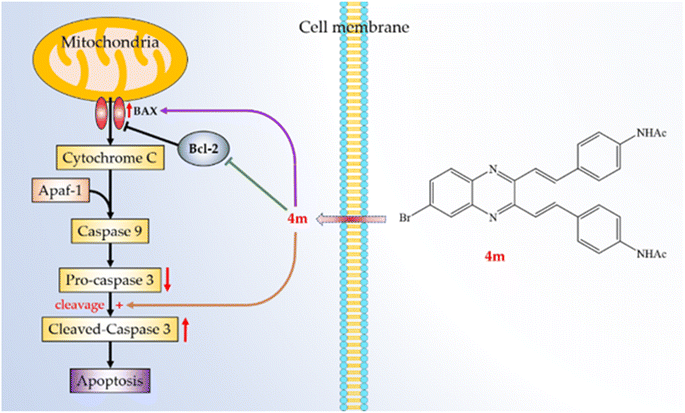 |
| | Fig. 4 Schematic diagram for cancer cell apoptosis mechanism of compound 4m in A549 cells. | |
4 Experimental
All chemicals were purchased from either Acros or Alfa from Uniworld in Taiwan and used from received without further purification except otherwise mentioned. The structure's determinations were based on 1H and 13C-NMR data recorded on a 600 MHz Bruker Ultrashield instrument. The chemical shifts were reported in part per million (ppm) for the residual solvent (CDCl3: 7.26 ppm for 1H; 77.0 ppm for 13C). The splitting constant (J) was represented in hertz (Hz), and the splitting patterns were designated as s (singlet), br s (broad singlet), d (doublet), dd (doublet of doublets), t (triplet), and m (multiplet). The purity of compounds was performed by high-resolution mass spectrometry (HRMS) using a Bruker UltraFlex II instrument (from Bruker Taiwan Co. Ltd) for electrospray ionization (ESI). The reaction processes were monitored by thin layer chromatography (Analtech Silica gel HLF UV254) and visualized by UV (256 or 365 nm) or stained with KMnO4 or p-anisaldehyde solution. The melting points were determined using an MP-2D apparatus and were uncorrected.
4.1 General procedure for the synthesis of compounds 4a–m and 5a–m
Compound 1 (4-bromo-1,2-diaminobenzene, 1.0 equivalent) and the corresponding cinnamil 3a–m (1.2 equivalent) were dissolved in acetic acid (0.2 M). This resulting mixture was heated at 75 °C for 6 h and then cooled in an ice bath until a precipitate formed. The precipitate was filtrated by suction and washed with H2O to obtain the target molecules 4a–m, respectively. Synthesis of compounds 5a–m followed the method mentioned above.
4.2 Spectra data
4.2.1 6-Bromo-2,3-bis[(E)-2-(furan-2-yl)vinyl]quinoxaline (4a). Use compound 1 (0.093 g, 0.495 mmol) and compound 3a (0.10 g, 0.413 mmol) to afford compound 4a (0.144 g, 0.366 mmol). Yield: 89%. Mp 213–214 °C. A brown solid. 1H NMR (600 MHz, CDCl3) δ 8.15 (d, J = 1.7 Hz, 1H), 7.83 (d, J = 13.4 Hz, 2H), 7.82 (d, J = 9.6 Hz, 1H), 7.70 (dd, J = 8.9, 1.6 Hz, 1H), 7.55 (d, J = 3.2 Hz, 1H), 7.54 (d, J = 15.2 Hz, 2H), 7.53 (s, 1H), 6.62 (t, J = 4.8 Hz, 2H), 6.51 (d, J = 1.4 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 152.9, 152.8, 149.4, 148.9, 143.8, 143.7, 142.1, 140.3, 132.7, 131.0, 130.0, 125.1, 124.8, 123.1, 119.8, 119.7, 113.1, 112.9, 112.3. HRMS (ESI) calculated for C20H14BrN2O2 [M + H]+ 393.0239. Found: 393.0225.
4.2.2 6-Bromo-2,3-bis[(E)-2-(thiophen-2-yl)vinyl]quinoxaline (4b). Use compound 1 (0.082 g, 0.437 mmol) and compound 3b (0.10 g, 0.364 mmol) to afford compound 4b (0.118 g, 0.276 mmol). Yield: 76%. Mp 210–211 °C. A yellow solid. 1H NMR (600 MHz, CDCl3) δ 8.17 (d, J = 2.1 Hz, 1H), 8.15 (d, J = 3.5 Hz, 1H), 8.12 (d, J = 3.5 Hz, 1H), 7.84 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 8.8, 2.1 Hz, 1H), 7.38 (d, J = 15.3 Hz, 2H), 7.37 (d, J = 15.3 Hz, 2H), 7.32 (t, J = 3.4 Hz, 2H), 7.11–7.08 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 149.2, 148.7, 142.1, 141.9, 141.8, 140.3, 132.8, 131.2, 131.0, 130.9, 129.4, 129.3, 128.1, 126.9, 126.8, 123.2, 121.1, 121.0. HRMS (ESI) calculated for C20H14BrN2S2 [M + H]+ 424.9782. Found: 424.9798.
4.2.3 6-Bromo-2,3-di[(E)-styryl]quinoxaline (4c). Use compound 1 (0.257 g, 1.373 mmol) and compound 3c (0.30 g, 1.144 mmol) to afford compound 4c (0.448 g, 1.084 mmol). Yield: 95%. Mp 190–191 °C. A yellow solid. 1H NMR (600 MHz, CDCl3) δ 8.20 (d, J = 2.0 Hz, 1H), 8.00 (d, J = 15.5 Hz, 1H), 7.99 (d, J = 15.5 Hz, 1H), 7.87 (d, J = 8.9 Hz, 1H), 7.72 (d, J = 8.9, 2.0 Hz, 1H), 7.67 (d, J = 7.7 Hz, 4H), 7.60 (d, J = 15.5 Hz, 1H), 7.59 (d, J = 15.5 Hz, 1H), 7.44 (d, J = 7.4 Hz, 1H), 7.43 (d, J = 7.4 Hz, 2H), 7.38 (t, J = 7.3 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 149.7, 149.2, 142.0, 140.3, 138.6, 138.3, 136.3, 136.2, 132.9, 131.1, 130.1, 129.3, 129.2, 128.9, 127.7, 127.6, 123.3, 122.2, 122.1. HRMS (ESI) calculated for C24H18BrN2 [M + H]+ 413.0653. Found: 413.0640.
4.2.4 6-Bromo-2,3-bis[(E)-2-(naphthalen-2-yl)vinyl]quinoxaline (4d). Use Compound 1 (0.031 g, 0.165 mmol) and compound 3d (0.050 g, 0.138 mmol) to afford compound 4d (0.065 g, 0.126 mmol). Yield: 91%. Mp 234–235 °C. A yellow solid. 1H NMR (600 MHz, CDCl3) δ 8.25 (d, J = 1.5 Hz, 1H), 8.20 (d, J = 15.5 Hz, 2H), 8.07 (s, 2H), 7.93–7.85 (m, 9H), 7.78–7.75 (m, 3H), 7.55–7.51 (br m, 4H). 13C NMR (150 MHz, CDCl3) δ 149.8, 149.4, 142.2, 140.4, 138.8, 138.5, 133.9, 133.8, 133.6, 132.9, 131.2, 130.2, 129.1, 129.0, 128.6, 128.5, 128.4, 127.8, 126.8, 126.7, 126.6, 123.7, 123.3, 122.4, 122.3. HRMS (ESI) calculated for C32H22BrN2 [M + H]+ 513.0966. Found: 513.0971.
4.2.5 6-Bromo-2,3-bis[(E)-2-methoxystyryl]quinoxaline (4e). Use compound 1 (0.139 g, 0.744 mmol) and compound 3e (0.20 g, 0.620 mmol) to afford compound 4e (0.284 g, 0.601 mmol). Yield: 97%. Mp 182–183 °C. A yellow solid. 1H NMR (600 MHz, CDCl3) δ 8.28 (d, J = 15.7 Hz, 1H), 8.27 (d, J = 15.7 Hz, 1H), 8.24 (d, J = 2.1 Hz, 1H), 7.91 (d, J = 8.9 Hz, 1H), 7.75 (d, J = 15.7 Hz, 1H), 7.73 (d, J = 15.7 Hz, 1H), 7.72 (dd, J = 8.7, 2.2 Hz, 1H), 7.70 (d, J = 7.6 Hz, 2H), 7.34 (t, J = 7.6 Hz, 2H), 7.02 (t, J = 7.5 Hz, 2H), 6.97 (d, J = 8.3 Hz, 2H). 13C NMR (150 MHz, CDCl3) d 158.0, 150.5, 150.1, 142.2, 140.3, 133.9, 133.6, 132.5, 131.2, 130.3, 130.2, 130.1, 128.2, 125.6, 125.5, 123.5, 123.3, 122.9, 120.8, 111.1, 55.5. HRMS (ESI) calculated for C26H22BrN2O2 [M + H]+ 473.0865. Found: 473.0858.
4.2.6 6-Bromo-2,3-bis[(E)-3-nitrostyryl]quinoxaline (4f). Use compound 1 (0.032 g, 0.170 mmol) and compound 3f (0.050 g, 0.14 mmol) to afford compound 4f (0.061 g, 0.121 mmol). Yield: 85%. Mp 290–291 °C. A yellow solid. 1H and 13C-NMR spectra were not obtained in perfect forms because of insolubility in CDCl3, DMSO-d6, CD3OD, and acetone-d6. HRMS (ESI) calculated for C24H16BrN4O4 [M + H]+ 503.0355. Found: 503.0335.
4.2.7 6-Bromo-2,3-bis[(E)-3-chlorostyryl]quinoxaline (4g). Use compound 1 (0.068 g, 0.362 mmol) and compound 3g (0.10 g, 0.302 mmol) to afford compound 4g (0.123 g, 0.254 mmol). Yield: 85%. Mp 94–95 °C. A yellow-brown solid.1H NMR (600 MHz, CDCl3) δ 8.21 (d, J = 1.9 Hz, 1H), 7.94 (d, J = 15.5 Hz, 2H), 7.88 (d, J = 8.9 Hz, 1H), 7.76 (dd, J = 8.9, 2.0 Hz, 1H), 7.66 (s, 2H), 7.57 (d, J = 3.6 Hz, 1H), 7.55 (d, J = 9.2 Hz, 1H), 7.54 (s, 1H), 7.39–7.34 (m, 4H). 13C NMR (150 MHz, CDCl3) δ 149.1, 148.7, 142.2, 140.4, 138.1, 138.0, 137.3, 137.0, 134.9, 133.3, 131.2, 130.2, 130.1, 129.2, 129.1, 127.4, 125.9, 125.8, 123.7, 123.2, 123.1. HRMS (ESI) calculated for C24H16BrCl2N2 [M + H]+ 480.9874. Found: 480.9870.
4.2.8 6-Bromo-2,3-bis[(E)-4-fluorostyryl]quinoxaline (4h). Use compound 1 (0.113 g, 0.603 mmol) and compound 3h (0.150 g, 0.503 mmol) to afford compound 4h (0.179 g, 0.399 mmol). Yield: 80%. Mp 212–213 °C. A yellow solid. 1H NMR (600 MHz, CDCl3) δ 8.21 (s, 1H), 7.97 (d, J = 15.5 Hz, 1H), 7.96 (d, J = 15.5 Hz, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.75 (dd, J = 8.9, 1.2 Hz, 1H), 7.66 (d, J = 7.9 Hz, 2H), 7.65 (d, J = 7.9 Hz, 2H), 7.52 (d, J = 15.5 Hz, 1H), 7.50 (d, J = 15.5 Hz, 1H), 7.13 (t, J = 8.3 Hz, 4H). 13C NMR (150 MHz, CDCl3) δ 164.1, 163.3 (1JC–F = 249.0 Hz), 149.5, 149.0, 142.1, 140.3, 137.4, 137.1, 133.0, 132.6, 132.5, 131.1, 130.1, 129.4 (3JC–F = 7.5 Hz), 129.3, 123.4, 121.8, 121.7, 116.0 (2JC–F = 22.5 Hz). HRMS (ESI) calculated for C24H16BrF2N2 [M + H]+ 449.0465. Found: 449.0455.
4.2.9 6-Bromo-2,3-bis[(E)-4-chlorostyryl]quinoxaline (4i). Use compound 1 (0.068 g, 0.362 mmol) and compound 3i (0.10 g, 0.302 mmol) to afford compound 4i (0.081 g, 0.168 mmol). Yield: 56%. Mp 219–220 °C. A brown solid. 1H NMR (600 MHz, CDCl3) δ 8.22 (s, 1H), 7.96 (d, J = 15.5 Hz, 2H), 7.89 (d, J = 8.8 Hz, 1H), 7.76 (dd, J = 8.8, 1.7 Hz, 1H), 7.61 (d, J = 8.3 Hz, 4H), 7.57 (d, J = 15.5 Hz, 1H), 7.56 (d, J = 15.5 Hz, 1H), 7.41 (d, J = 8.1 Hz, 4H). 13C NMR (150 MHz, CDCl3) δ 149.3, 148.9, 142.2, 140.4, 137.4, 137.1, 135.1, 134.8, 134.7, 133.2, 131.1, 130.1, 129.2, 128.8, 128.7, 123.6, 122.5, 122.4. HRMS (ESI) calculated for C24H16BrCl2N2 [M + H]+ 480.9880. Found: 480.9874.
4.2.10 6-Bromo-2,3-bis[(E)-4-bromostyryl]quinoxaline (4j). Use compound 1 (0.053 g, 0.286 mmol) and compound 3j (0.10 g, 0.238 mmol) to afford compound 4j (0.116 g, 0.203 mmol). Yield: 85%. Mp 241–242 °C. A yellow solid. 1H NMR (600 MHz, CDCl3) δ 8.22 (d, J = 2.1 Hz, 1H), 7.95 (d, J = 15.5 Hz, 1H), 7.94 (d, J = 15.5 Hz, 1H), 7.89 (d, J = 8.9 Hz, 1H), 7.76 (dd, J = 8.9, 2.1 Hz, 1H), 7.59–7.52 (m, 10H). 13C NMR (150 MHz, CDCl3) δ 149.3, 148.9, 142.2, 140.4, 137.5, 137.2, 135.2, 135.1, 133.2, 132.1, 131.9, 131.5, 131.2, 130.4, 130.2, 129.1, 129.0, 123.6, 123.4, 123.3, 122.6, 122.5. HRMS (ESI) calculated for C24H16Br3N2 [M + H]+ 568.8864. Found: 568.8877.
4.2.11 6-Bromo-2,3-bis[(E)-4-nitrostyryl]quinoxaline (4k). Use compound 1 (0.032 g, 0.170 mmol) and compound 3k (0.050 g, 0.142 mmol) to afford compound 4k (0.061 g, 0.121 mmol). Yield: 85%. Mp 307–308 °C. A yellow solid. 1H NMR (600 MHz, CDCl3) δ 8.32–8.28 (m, 5H), 8.11 (d, J = 15.2 Hz, 2H), 7.95 (d, J = 8.6 Hz, 1H), 7.84 (d, J = 8.0 Hz, 5H), 7.75 (d, J = 15.2 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 148.0, 147.9, 147.8, 142.3, 142.2, 140.5, 136.2, 135.9, 133.9, 131.2, 130.2, 128.1, 128.0, 125.7, 125.6, 124.4, 124.2. HRMS (ESI) calculated for C24H16BrN4O2 [M + H]+ 503.0355. Found: 503.0356.
4.2.12 6-Bromo-2,3-bis[(E)-4-methoxystyryl]quinoxaline (4l). Use compound 1 (0.069 g, 0.372 mmol) and compound 3l (0.10 g, 0.310 mmol) to afford compound 4l (0.092 g, 0.195 mmol). Yield: 63%. Mp 190–191 °C. A yellow solid. 1H NMR (600 MHz, CDCl3) δ 8.20 (d, J = 2.0 Hz, 1H), 7.97 (d, J = 15.5 Hz, 1H), 7.96 (d, J = 15.5 Hz, 1H), 7.87 (d, J = 8.9 Hz, 1H), 7.71 (dd, J = 8.9, 2.0 Hz, 1H), 7.64 (d, J = 8.3 Hz, 4H), 7.50 (d, J = 15.5 Hz, 1H), 7.49 (d, J = 15.5 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 3.87 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 160.6, 160.5, 150.0, 149.6, 142.0, 140.2, 138.1, 137.8, 132.5, 131.0, 130.0, 129.2, 129.1, 122.8, 120.0, 119.9, 114.3, 55.4. HRMS (ESI) calculated for C26H22BrN2O2 [M + H]+ 473.0865. Found: 473.0864.
4.2.13 N,N′-[{(1E,1′E)-(6-bromoquinoxaline-2,3-diyl)bis(ethene-2,1-diyl)}bis(4,1-phenylene)] diacetamide (4m). Use compound 1 (0.029 g, 0.159 mmol) and compound 3m (0.050 g, 0.133 mmol) to afford compound 4m (0.057 g, 0.108 mmol). Yield: 81%. Mp 239 °C (decomposed). A yellow solid. 1H NMR (600 MHz, CDCl3) δ 10.12 (s, 1H), 10.1 (s, 1H), 8.18 (d, J = 1.6 Hz, 1H), 7.95–7.82 (m, 10H), 7.68 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 8.0 Hz, 2H), 2.07 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 168.6, 149.7, 149.3, 141.5, 140.4, 140.3, 139.7, 137.6, 132.6, 130.8, 130.7, 130.4, 130.3, 128.9, 128.7, 122.3, 120.3, 120.3, 119.0. HRMS (ESI) calculated for C28H24BrN4O2 [M + H]+ 527.1083. Found: 527.1087.
4.2.14 2,3-Bis[(E)-2-(furan-2-yl)vinyl]-6-nitroquinoxaline (5a). Use compound 2 (0.076 g, 0.495 mmol) and compound 3a (0.10 g, 0.413 mmol) to afford compound 5a (0.124 g, 0.345 mmol). Yield: 84%. Mp 218–219 °C. 1H NMR (600 MHz, CDCl3) δ 8.83 (d, J = 2.2 Hz, 1H), 8.37 (dd, J = 9.1, 2.2 Hz, 1H), 8.03 (d, J = 9.1 Hz, 1H), 7.92 (d, J = 15.2 Hz, 1H), 7.89 (d, J = 15.2 Hz, 1H), 7.56 (s, 1H), 7.55 (s, 1H), 7.52 (d, J = 15.2 Hz, 1H), 7.51 (d, J = 15.2 Hz, 1H), 6.68 (d, J = 3.1 Hz, 1H), 6.67 (d, J = 3.1 Hz, 1H), 6.53 (d, J = 1.6 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 152.6, 151.4, 150.8, 147.3, 144.3, 144.1, 144.0, 140.2, 130.0, 126.6, 126.0, 125.1, 122.5, 119.0, 118.8, 114.2, 113.9, 112.5, 112.4. HRMS (ESI) calculated for C20H14N3O4 [M + H]+ 360.0984. Found: 360.0995.
4.2.15 6-Nitro-2,3-bis[(E)-2-(thiophen-2-yl)vinyl]quinoxaline (5b). Use compound 2 (0.067 g, 0.437 mmol) and compound 3b (0.10 g, 0.364 mmol) to afford compound 5b (0.109 g, 0.279 mmol). Yield: 91%. Mp 223–224 °C. 1H NMR (600 MHz, CDCl3) δ 8.86 (d, J = 2.4 Hz, 1H), 8.26 (d, J = 15.2 Hz, 1H), 8.23 (d, J = 15.2 Hz, 1H), 8.06 (d, J = 9.1 Hz, 1H), 7.41 (d, J = 5.1 Hz, 1H), 7.40 (d, J = 5.1 Hz, 1H), 7.38–7.34 (m, 4H), 7.13–7.10 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 151.1, 150.6, 147.3, 144.0, 141.6, 140.2, 133.0, 132.4, 130.3, 130.1, 130.0, 128.3, 128.2, 127.8, 127.5, 125.1, 122.6, 120.3, 120.1. HRMS (ESI) calculated for C20H14N3O2S2 [M + H]+ 392.0527. Found: 392.0540.
4.2.16 6-Nitro-2,3-di[(E)-styryl]quinoxaline (5c). Use compound 2 (0.070 g, 0.457 mmol) and compound 3c (0.10 g, 0.381 mmol) to afford compound 5c (0.109 g, 0.286 mmol). Yield: 75%. Mp 195–196 °C. A brown solid. 1H NMR (600 MHz, CDCl3) δ 8.91 (d, J = 2.5 Hz, 1H), 8.41 (dd, J = 9.1, 2.5 Hz, 1H), 8.13 (d, J = 15.5 Hz, 1H), 8.11 (d, J = 9.1 Hz, 1H), 8.09 (d, J = 15.5 Hz, 1H), 7.70 (d, J = 7.4 Hz, 1H), 7.62 (d, J = 15.5 Hz, 1H), 7.61 (d, J = 15.5 Hz, 1H), 7.46 (t, J = 7.4 Hz, 4H), 7.42–7.39 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 151.6, 151.1, 147.4, 144.0, 140.5, 140.3, 140.0, 136.0, 135.9, 130.2, 129.9, 129.7, 129.0, 127.9, 127.8, 125.2, 122.7, 121.4, 121.1. HRMS (ESI) calculated for C24H18N3O2 [M + H]+ 380.1399. Found: 380.1405.
4.2.17 2,3-Bis[(E)-2-(naphthalen-2-yl)vinyl]-6-nitroquinoxaline (5d). Use compound 2 (0.051 g, 0.331 mmol) and compound 3d (0.10 g, 0.276 mmol) to afford compound 5d (0.094 g, 0.196 mmol). Yield: 70%. Mp 228–229 °C. An orange solid. 1H NMR (600 MHz, CDCl3) δ 8.93 (d, J = 2.3 Hz, 1H), 8.42 (dd, J = 9.1, 2.8 Hz, 1H), 8.31 (d, J = 15.4 Hz, 1H), 8.28 (d, J = 15.4 Hz, 1H), 8.14 (d, J = 9.1 Hz, 1H), 8.08 (s, 2H), 7.92–7.86 (m, 9H), 7.78 (d, J = 15.4 Hz, 1H), 7.77 (d, J = 15.4 Hz, 1H), 7.55–7.53 (m, 5H). 13C NMR (150 MHz, CDCl3) δ 151.7, 151.2, 147.4, 144.1, 140.7, 140.3, 140.0, 134.1, 134.0, 133.5, 133.4, 130.2, 130.0, 129.5, 128.8, 128.7, 128.5, 127.8, 127.1, 127.0, 126.8, 126.7, 125.2, 123.6, 122.7, 121.6, 121.4. HRMS (ESI) calculated for C32H22N3O2 [M + H]+ 480.1712. Found: 480.1719.
4.2.18 2,3-Bis[(E)-2-methoxystyryl]-6-nitroquinoxaline (5e). Use compound 2 (0.057 g, 0.372 mmol) and compound 3e (0.10 g, 0.310 mmol) to afford compound 5e (0.122 g, 0.278 mmol). Yield: 90%. Mp 219–220 °C. An orange solid. 1H NMR (600 MHz, CDCl3) δ 8.95 (s, 1H), 8.40 (dd, J = 9.4, 2.5 Hz, 1H), 8.39 (d, J = 15.7 Hz, 1H), 8.38 (d, J = 15.7 Hz, 1H), 8.14 (d, J = 9.1 Hz, 1H), 7.78 (d, J = 15.7 Hz, 1H), 7.77 (d, J = 15.7 Hz, 1H), 7.71 (d, J = 7.5 Hz, 2H), 7.40–7.35 (m, 2H), 7.04 (t, J = 7.4 Hz, 2H), 6.99 (d, J = 8.3 Hz, 2H), 3.97 (s, 3H), 3.96 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 158.3, 158.2, 152.5, 152.0, 147.2, 135.8, 135.1, 130.9, 130.7, 130.2, 128.6, 128.4, 125.3, 125.2, 125.1, 122.7, 122.5, 122.4, 120.9, 120.8, 119.2, 55.6. HRMS (ESI) calculated for C26H22N3O4 [M + H]+ 440.1610. Found: 440.1594.
4.2.19 6-Nitro-2,3-bis[(E)-3-nitrostyryl]quinoxaline (5f). Use compound 2 (0.052 g, 0.341 mmol) and compound 3f (0.10 g, 0.284 mmol) to afford compound 5f (0.069 g, 0.146 mmol). Yield: 86%. Mp 197–198 °C. A yellow solid. 1HNMR (600 MHz, CDCl3) δ 8.99 (d, J = 2.4 Hz, 1H), 8.57 (d, J = 2.1 Hz, 2H), 8.51 (dd, J = 9.3, 2.7 Hz, 1H), 8.28–8.24 (m, 3H), 8.21 (dd, J = 9.3, 1.8 Hz, 2H), 8.06–8.01 (m, 2H), 7.76 (d, J = 15.6, 3.0 Hz, 2H), 7.67 (t, J = 7.8 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 150.6, 150.1, 148.9, 148.0, 144.1, 140.6, 138.2, 137.6, 137.5, 133.7, 133.5, 130.6, 130.1, 125.4, 124.2, 124.1, 124.0, 123.8, 123.5, 122.4, 122.2. HRMS (ESI) calculated for C24H16N5O6 [M + H]+ 470.1101. Found: 470.1094.
4.2.20 2,3-Bis[(E)-3-chlorostyryl]-6-nitroquinoxaline (5g). Use compound 2 (0.044 g, 0.289 mmol) and compound 3g (0.080 g, 0.242 mmol) to afford compound 5g (0.092 g, 0.205 mmol). Yield: 85%. Mp 200–201 °C. A brown solid. 1H NMR (600 MHz, CDCl3) δ 8.93 (s, 1H), 8.45 (d, J = 8.9 Hz, 1H), 8.15 (d, J = 8.9 Hz, 1H), 8.08 (d, J = 15.6 Hz, 1H), 8.05 (d, J = 15.6 Hz, 1H), 7.69 (s, 2H), 7.62–7.57 (br m, 4H), 7.45–7.38 (br m, 4H). 13C NMR (150 MHz, CDCl3) δ 151.1, 150.6, 147.7, 144.1, 140.4, 139.2, 138.6, 137.7, 135.0, 130.4, 129.7, 129.6, 127.7, 127.5, 126.2, 125.3, 123.0, 122.6, 122.4. HRMS (ESI) calculated for C24H16Cl2N3O2 [M + H]+ 448.0620. Found: 448.0521.
4.2.21 2,3-Bis[(E)-4-fluorostyryl]-6-nitroquinoxaline (5h). Use compound 2 (0.062 g, 0.402 mmol) and compound 3h (0.10 g, 0.335 mmol) to afford compound 5h (0.080 g, 0.195 mmol). Yield: 83%. Mp 258–259 °C. An orange-yellow solid. 1H NMR (600 MHz, CDCl3) δ 8.95 (s, 1H), 8.45 (d, J = 9.5 Hz, 1H), 8.15 (d, J = 9.5 Hz, 1H), 8.13 (d, J = 15.5 Hz, 1H), 8.09 (d, J = 15.5 Hz, 1H), 7.70 (br s, 4H), 7.56 (d, J = 15.5 Hz, 1H), 7.55 (d, J = 15.5 Hz, 1H), 7.16 (d, J = 8.3 Hz, 2H), 7.15 (d, J = 8.3 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 167.0, 163.7 (1JC–F = 249.0 Hz), 163.6 (1JC–F = 249.0 Hz), 151.5, 151.0, 147.5, 144.0, 140.3, 139.4, 138.7, 132.2, 130.2, 130.0, 129.7 (2JC–F = 24.0 Hz), 129.6 (3JC–F = 7.5 Hz), 125.2, 122.8, 121.1 (2JC–F = 28.5 Hz), 116.2 (4JC–F = 4.5 Hz), 116.0 (4JC–F = 4.5 Hz). HRMS (ESI) calculated for C24H16F2N3O2 [M + H]+ 416.1222. Found: 416.1215.
4.2.22 2,3-Bis[(E)-4-chlorostyryl]-6-nitroquinoxaline (5i). Use compound 2 (0.031 g, 0.199 mmol) and compound 3h (0.055 g, 0.166 mmol) to afford compound 5h (0.062 g, 0.138 mmol). Yield: 89%. Mp 221–222 °C. A yellow-brown solid. 1H NMR (600 MHz, CDCl3) δ 8.93 (s, 1H), 8.44 (d, J = 7.4 Hz, 1H), 8.14 (d, J = 8.8 Hz, 1H), 8.11 (d, J = 15.8 Hz, 1H), 8.06 (d, J = 15.8 Hz, 1H), 7.64 (d, J = 6.8 Hz, 1H), 7.59 (d, J = 15.8 Hz, 1H), 7.58 (d, J = 15.8 Hz, 1H), 7.43 (d, J = 7.4 Hz, 4H). 13C NMR (150 MHz, CDCl3) δ 151.3, 150.8, 147.6, 144.1, 140.4, 139.3, 138.6, 135.7, 135.6, 134.4, 130.3, 129.3, 129.1, 129.0, 125.3, 122.9, 121.9, 121.7. HRMS (ESI) calculated for C24H16Cl2N3O2 [M + H]+ 448.0620. Found: 448.0617.
4.2.23 2,3-Bis[(E)-4-bromostyryl]-6-nitroquinoxaline (5j). Use compound 2 (0.022 g, 0.143 mmol) and compound 3j (0.050 g, 0.119 mmol) to afford compound 5h (0.059 g, 0.111 mmol). Yield: 93%. Mp 247–248 °C. A yellow-brown solid. 1H NMR (600 MHz, CDCl3) δ 8.93 (d, J = 2.3 Hz, 1H), 8.44 (dt, J = 9.2, 1.2 Hz), 8.14 (d, J = 9.2 Hz, 1H), 8.08 (d, J = 15.4 Hz, 1H), 8.04 (d, J = 15.4 Hz, 1H), 7.62–7.55 (m, 11H). 13C NMR (150 MHz, CDCl3) δ 150.9, 150.4, 147.2, 143.7, 140.0, 138.9, 138.3, 134.5, 134.4, 131.9, 131.8, 129.9, 128.9, 128.8, 124.9, 123.7, 123.5, 122.6, 121.5, 121.3. HRMS (ESI) calculated for C24H16Br2N3O2 [M + H]+ 535.9609. Found: 535.9607.
4.2.24 6-Nitro-2,3-bis[(E)-4-nitrostyryl]quinoxaline (5k). Use compound 2 (0.052 g, 0.341 mmol) and compound 3k (0.10 g, 0.284 mmol) to afford compound 5k (0.108 g, 0.231 mmol). Yield: 81%. Mp 297–298 °C. A yellow solid. 1H NMR (600 MHz, CDCl3) δ 9.00 (s, 1H), 8.53 (d, J = 9.1 Hz, 1H), 8.34 (d, J = 8.5 Hz, 4H), 8.25–8.20 (m, 3H), 7.87 (d, J = 8.2 Hz, 4H), 7.78 (d, J = 16.2 Hz, 1H), 7.76 (d, J = 16.2 Hz, 1H). No satisfaction of the 13C NMR spectrum was obtained due to solubility. HRMS (FAB) calculated for C24H16N5O6 [M + H]+ 470.1101. Found: 470.1101.
4.2.25 2,3-Bis[(E)-4-methoxystyryl]-6-nitroquinoxaline (5l). Use compound 2 (0.057 g, 0.372 mmol) and compound 3l (0.10 g, 0.335 mmol) to afford compound 5l (0.069 g, 0.184 mmol). Yield: 89%. Mp 144–145 °C. A dark-orange solid. 1H NMR (600 MHz, CDCl3) δ 8.85 (d, J = 2.4 Hz, 1H), 8.36 (dd, J = 9.1, 2.4 Hz, 1H), 8.05 (d, J = 15.2 Hz, 1H), 8.04 (d, J = 9.1 Hz, 1H), 8.02 (d, J = 15.2 Hz, 1H), 7.63 (d, J = 8.5 Hz, 4H), 7.45 (d, J = 15.4 Hz, 1H), 7.44 (d, J = 15.4 Hz, 1H), 6.97 (d, J = 8.5 Hz, 4H). 13C NMR (150 MHz, CDCl3) δ 161.0, 160.9, 151.9, 151.4, 147.1, 144.0, 140.1, 140.0, 139.3, 129.9, 129.5, 129.4, 128.9, 128.8, 125.0, 122.3, 119.1, 118.9, 114.4, 114.3, 55.4. HRMS (ESI) calculated for C26H22N3O4 [M + H]+ 440.1610. Found: 440.1613.
4.2.26 N,N′-[(1E,1′E)-(6-nitroquinoxaline-2,3-diyl)bis(ethene-2,1-diyl)bis(4,1-phenylene)]diacetamide (5m). Use compound 2 (0.024 g, 0.159 mmol) and compound 3m (0.050 g, 0.133 mmol) to afford compound 5m (0.060 g, 0.121 mmol). Yield: 91%. Mp 325–326 °C. An orange solid. 1H NMR (600 MHz, CDCl3) δ 10.15 (s, 1H), 10.14 (s, 1H), 8.66 (d, J = 2.1 Hz, 1H), 8.34 (d, J = 9.1 Hz, 1H), 8.09 (d, J = 9.1 Hz, 1H), 7.99 (d, J = 15.6 Hz, 1H), 7.95 (d, J = 15.6 Hz, 1H), 7.85–7.81 (m, 6H), 7.67 (d, J = 8.0 Hz, 4H). 13C NMR (150 MHz, CDCl3) δ 168.7, 151.7, 151.1, 146.8, 143.6, 140.9, 140.7, 139.6, 139.5, 138.9, 130.6, 130.5, 130.1, 129.3, 129.2, 124.4, 122.6, 119.9, 119.7, 119.0, 24.2. HRMS (FAB) calculated for C28H24N5O4 [M + H]+ 494.1828. Found: 494.1824.
4.3 Cell culture
Prof. Y. Su of National Yang-Ming Chiao Tung University, Taipei, Taiwan kindly provided human non-small lung cancer cells (A549 cells). The cells were stored in liquid nitrogen at −196 °C. After the cells were thawed, the cells were incubated at 37 °C in 5% CO2 and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U per mL penicillin, 100 μg per mL streptomycin, 2 μM L-glutamine, and 1 mM sodium pyruvate.42,43
4.4 In vitro cytotoxicity assay
The cell viability was evaluated with MTT assay, as previously reported to further assess the cytotoxicity.32,43 In brief, the cells were plated in 96-well culture plates at a concentration of 3 × 103 cells in 200 μl per well. After 24 hours, cells were treated with different concentrations (6.25, 12.5, 25, 50, and 100 μM) of all twenty-six synthesized compounds, and 5-fluorouracil (5-FU) was used as a positive control. After 72 hours, the attached cells were added with MTT reagent at 0.5 mg mL−1 in 100 μL per well and incubated at 37 °C for 3 h. Then, the MTT reagent was removed, 100 μL of DMSO was added per well to dissolve the formazan metabolite, and the amount of formazan was measured by the absorbance at 570 nm, using an ELISA plate reader, TECAN Spark (Tecan Group Ltd) (μ Quant). The optical density recorded previously in the control group (untreated cells) was considered to be 100% cell viability.
4.5 Western blot
Western blot analysis was performed according to the method previously described.32,43 Briefly, the cells were seeded to 6-well culture plates. After reaching 85–90% confluence, cells were treated with compound 4m (5, 10, and 20 μM) and 5-FU (5 μM). The cells were incubated for 48 h after treatment. Afterward, the cells were collected and lysed using radioimmunoprecipitation assay (RIPA) buffer. Lysates of total protein were separated by 12.5% sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes. Then, the membranes were blocked with 2% bovine serum albumin (BSA) solution and incubated with anti-Bax, anti-Bcl-2 (Cell Signaling Inc., Danvers, MA, USA), anti-caspase-3, and anti-β-actin (GeneTex Inc., Irvine, CA, USA) primary antibodies at 4 °C overnight. Each membrane was washed with tris-buffered saline containing 0.1% Tween 20 (TBST) three times and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 2 h. Finally, the membranes were developed with an enhanced chemiluminescence (ECL) detection kit and visualized by ImageQuant LAS 4000 Mini bio-molecular imager (GE Healthcare, MA, USA). ImageJ software quantified the band densities (BioTechniques, NY, USA).
Data availability
The data supporting this article have been included as part of the ESI.† No code was produced as part of this review.
Author contributions
Jia-Hua Liang performed the bioassay and analyzed the data and manuscript writing. Shu-Tse Cho conducted the isolation and structure elucidation of the constituents. Jih-Jung Chen and Tzenge-Lien Shih planned, designed, and organized this study's research and the manuscript's preparation. All authors read and approved the final version of the manuscript.
Conflicts of interest
The authors declare no conflicts of financial interest.
Acknowledgements
This research was supported by grants from the National Science and Technology Council (NSTC) and the Ministry of Science and Technology (MOST) of Taiwan (No. NSTC 112-2320-B-A49-028-MY3 and MOST 111-2113-M-032-004), awarded to J.-J. C. and T.-L. S., respectively.
Notes and references
- J. A. Pereira, A. M. Pessoa, M. N. D. S. Cordeiro, R. Fernandes, C. Prudêncio, J. P. Noronha and M. Vieira, ‘Quinoxaline, its derivatives and applications: A state of the art review’, Eur. J. Med. Chem., 2015, 97, 664–672 CrossRef CAS PubMed.
- K. Watanabe, H. Oguri and H. Oikawa, ‘Diversification of echinomycin molecular structure by way of chemoenzymatic synthesis and heterologous expression of the engineered echinomycin biosynthetic pathway’, Curr. Opin. Chem. Biol., 2009, 13, 189–196 CrossRef CAS.
- B. Dietrich and U. Diederichsen, ‘Synthesis of cyclopeptidic analogues of triostin a with quinoxalines or nucleobases as chromophores’, Eur. J. Org Chem., 2005, 13, 147–153 CrossRef.
- D. Kong, E. J. Park, A. G. Stephen, M. Calvani, J. H. Cardellina, A. Monks, R. J. Fisher and R. H. Shoemaker, ‘Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity’, Cancer Res., 2005, 65, 9047–9055 CrossRef CAS PubMed.
- Y. B. Kim, Y. H. Kim, J. Y. Park and S. K. Kim, ‘Synthesis and biological activity of new quinoxaline antibiotics of echinomycin analogues’, Bioorg. Med. Chem. Lett., 2004, 14, 541–544 CrossRef CAS.
- N. Steinerová, H. Lipavská, K. Stajner, J. Čáslavská, M. Blumauerová, J. Cudlín and Z. Vank, ‘Production of quinomycin a in Streptomyces lasaliensis, Folia Microbiol., 1987, 32, 1–5 CrossRef PubMed.
- A. Cornish, M. J. Waring and R. D. Nolan, ‘Conversion of triostins to quinomycins by protoplasts of Streptomyces echinatus’, J. Antibiot., 1983, 36, 1664–1670 CrossRef CAS PubMed.
- H. Ishikawa, T. Sugiyama, K. Kurita and A. Yokoyama, ‘Synthesis and antimicrobial activity of 2,3-bis(bromomethyl)quino-xaline derivatives’, Bioorg. Chem., 2012, 41–42, 1–5 CrossRef CAS.
- M. Montana, F. Mathias, T. Terme and P. Vanelle, ‘Antitumoral activity of quinoxaline derivatives: a systematic review’, Eur. J. Med. Chem., 2019, 163, 136–147 CrossRef CAS PubMed.
- M. A. El-Atawy, E. A. Hamed, M. Alhadi and A. Z. Omar, ‘Synthesis and antimicrobial activity of some new substituted quinoxalines’, Molecules, 2019, 24, 4198 CrossRef CAS PubMed.
- H. Ishikawa, T. Sugiyama and A. Yokoyama, ‘Synthesis and antimicrobial activity of some new substituted quinoxalines’, Chem. Pharm. Bull., 2013, 61, 438–444 CrossRef CAS.
- E. A. Ahmed, M. F. A. Mohamed and O. A. Omrana, ‘Novel quinoxaline derivatives as dual EGFR and COX-2 inhibitors: synthesis, molecular docking and biological evaluation as potential anticancer and anti-inflammatory agents’, RSC Adv., 2022, 12, 25204–25216 RSC.
- A. Burguete, E. Pontiki, D. Hadjipavlou-Litina, S. Ancizu, R. Villar, B. Solano, E. Moreno, E. Torres, S. Pérez, I. Aldana and A. Monge, ‘Synthesis and biological evaluation of new quinoxaline derivatives as antioxidant and anti-inflammatory agents’, Chem. Biol. Drug Des., 2011, 77, 255–267 CrossRef CAS.
- M. Montana, V. Montero, O. Khoumeri and P. Vanelle, ‘Quinoxaline derivatives as antiviral agents: a systematic review’, Molecules, 2020, 25, 2784 CrossRef CAS PubMed.
- M. Zhang, Z.-C. Dai, S.-S. Qian, J.-Y. Liu, Y. Xiao, A.-M. Lu, H.-L. Zhu, J.-X. Wang and Y.-H. Ye, ‘Design, synthesis, antifungal, and antioxidant activities of (E)-6-((2-phenylhydrazono) methyl) quinoxaline derivatives’, J. Agric. Food Chem., 2014, 62, 9637–9643 CrossRef CAS.
- M. Kessler, M. Baudry and G. Lynch, ‘Quinoxaline derivatives are high-affinity antagonists of the NMDA receptor-associated glycine sites’, Brain Res., 1989, 489, 377–382 CrossRef CAS.
- S. Ancizu, E. Moreno, B. Solano, R. Villar, B. Asunción, E. Torres, S. Pérez-Silanes, I. Aldana and A. Monge, ‘New 3-methylquinoxaline-2-carboxamide 1, 4-di-N-oxide derivatives as anti-Mycobacterium tuberculosis agents’, Bioorg. Med. Chem., 2010, 18, 2713–2719 CrossRef CAS.
- M. Montana, V. Montero, O. Khoumeri and P. Vanelle, ‘Quinoxaline moiety: a potential scaffold against Mycobacterium tuberculosis’, Molecules, 2021, 26, 4742 CrossRef CAS.
- X. Tang, Q. Zhou, W. Zhan, D. Hu, R. Zhou, N. Sun, S. Chen, W. Wu and W. Xue, ‘Synthesis of novel antibacterial and antifungal quinoxaline derivatives’, RSC Adv., 2022, 12, 2399–2407 RSC.
- E. A. Fayed, M. A. Ebrahim, U. Fathy, H. S. El Saeed and W. S. Khalaf, ‘Evaluation of quinoxaline derivatives as potential ergosterol biosynthesis inhibitors: design, synthesis, ADMET, molecular docking studies, and antifungal activities’, J. Mol. Struct., 2022, 1267, 133578 CrossRef CAS.
- M. F. El Shehry, S. Y. Abbas, A. M. Farrag, S. I. Eissa, S. A. Fouad and Y. A. Ammar, ‘Design, synthesis and biological evaluation of quinoxaline N-propionic and O-propionic hydrazide derivatives as antibacterial and antifungal agents’, Med. Chem. Res., 2018, 27, 2287–2296 CrossRef CAS.
- M. J. Waring, B. H. Taibi, A. T. Kotchevar, A. Ramdani, R. Touzani, S. Elkadiri, A. Hakkou, M. Bouakka and T. Ellis, ‘2,3-Bifunctionalized quinoxalines: synthesis, DNA interactions and evaluation of anticancer, anti-tuberculosis and antifungal activity’, Molecules, 2002, 7, 641–656 CrossRef CAS.
- J. Soto-Sánchez and J. D. Ospina-Villa, ‘Current status of quinoxaline and quinoxaline 1, 4-di-N-oxides derivatives as potential antiparasitic agents’, Chem. Biol. Drug Des., 2021, 98, 683–699 CrossRef.
- S. A. Shintre, D. Ramjugernath, M. S. Islam, R. Mopuri, C. Mocktar and N. A. Koorbanally, ‘Synthesis, in vitro antimicrobial, antioxidant, and antidiabetic activities of thiazolidine–quinoxaline derivatives with amino acid side chains’, Med. Chem. Res., 2017, 26, 2141–2151 CrossRef CAS.
- Y. Yang, S. Zhang, B. Wu, M. Ma, X. Chen, X. Qin, M. He, S. Hussain, C. Jing, B. Ma and C. Zhu, ‘An efficient synthesis of quinoxalinone derivatives as potent inhibitors of aldose reductase’, ChemMedChem, 2012, 7, 823–835 CrossRef CAS.
- N. V. Kulkarni, V. K. Revankar, B. N. Kirasur and M. H. Hugar, ‘Transition metal complexes of thiosemicarbazones with quinoxaline hub: an emphasis on antidiabetic property’, Med. Chem. Res., 2012, 21, 663–671 CrossRef CAS.
- R. H. Bahekar, M. R. Jain, A. A. Gupta, A. Goel, P. A. Jadav, D. N. Patel, V. M. Prajapati and P. R. Patel, ‘Synthesis and antidiabetic activity of 3,6,7-trisubstituted-2-(1H-imidazol-2-ylsulfanyl) quinoxalines and quinoxalin-2-yl isothioureas’, Arch. Pharm., 2007, 340, 359–366 CrossRef CAS.
- D. Gupta, N. N. Ghosh and R. Chandra, ‘Synthesis and pharmacological evaluation of substituted [5-[4-[2-(6, 7-dimethyl-1, 2, 3, 4-tetrahydro-2-oxo-4-quinoxalinyl) ethoxy] phenyl] methylene] thiazolidine-2,4-dione derivatives as potent euglycemic and hypolipidemic agents’, Bioorg. Med. Chem. Lett., 2005, 15, 1019–1022 CrossRef CAS PubMed.
- P. Ledwon, R. Motyka, K. Ivaniuk, A. Pidluzhna, N. Martyniuk, P. Stakhira, G. Baryshnikov, B. F. Minaev and H. Ågren, ‘The effect of molecular structure on the properties of quinoxaline-based molecules for OLED applications’, Dyes Pigm., 2020, 173, 108008 CrossRef CAS.
- D. Kitazawa, N. Watanabe, S. Yamamoto and J. Tsukamoto, ‘Quinoxaline-based donor polymers for organic solar cells’, J. Photopolym. Sci. Technol., 2010, 23, 293–296 CrossRef CAS.
- S. Sagar, S. Singh, J. R. Mallareddy, Y. A. Sonawane, J. V. Napoleon, S. Rana, J. I. Contreras, C. R. E. L. Ezell, S. Kizhake, J. C. Garrison, P. Radhakrishnan and A. Natarajan, ‘Structure activity relationship (SAR) study identifies a quinoxaline urea analog that modulates IKKβ phosphorylation for pancreatic cancer therapy’, Eur. J. Med. Chem., 2021, 222, 113579 CrossRef CAS.
- R.-Y. Wang, C.-W. Li, S.-T. Cho, C.-H. Chang, J.-J. Chen and T.-L. Shih, ‘Synthesis of cinnamils and quinoxalines and their biological evaluation as anticancer agents’, Arch. Pharm., 2022, 355, 2100448 CrossRef CAS.
- I. Malik, M. Hussain, A. Ali, S.-M. T. Togue, F. Z. Bash, C. Fischer and P. Langer, ‘Synthesis of 2,3-disubstituted pyrazines and quinoxalines by Heck cross-coupling reactions of 2,3-dichloropyrazine and 2,3-dichloroquinoxaline. Influence of thetemperature on the product distribution’, Tetrahedron, 2010, 66, 1637–1642 CrossRef CAS.
- P. Thirumurugan, D. Muralidharan and P. T. Perumal, ‘The synthesis and photophysical studies of quinoxaline and pyridopyrazine derivatives’, Dyes Pigm., 2009, 81, 245–253 CrossRef CAS.
- X. Luo and L.-T. Lim, ‘Cinnamil- and Quinoxaline-Derivative Indicator Dyes for Detecting Volatile Amines in Fish Spoilage’, Molecules, 2019, 24, 3673 CrossRef CAS.
- L. Zhan, B. Qi, X. L, X. Wan, J. L, Y. Zhang, J. Li, J. Li and J. Shen, ‘Preparation of 6-substituted quinoxaline JSP-1 inhibitors by microwave accelerated nucleophilic substitution’, Molecules, 2006, 11, 988–999 CrossRef PubMed.
- R. S. Bhosale, S. R. Sarda, S. S. Ardhapure, W. N. Jadhav, S. R. Bhusare and R. P. Pawar, ‘An efficient protocol for the synthesis of quinoxaline derivatives at room temperature using molecular iodine as the catalyst’, Tetrahedron Lett., 2005, 46, 7183–7186 CrossRef CAS.
- Z. Zhao, D. D. Wisnoski, S. E. Wolkenberg, W. H. Leister, Y. Wang and C. W. Lindsley, ‘General microwave-assisted protocols for the expedient synthesis of quinoxalines and heterocyclic pyrazines’, Tetrahedron Lett., 2004, 45, 4873–4876 CrossRef CAS.
- Y. Suzuki, R. Takehara, K. Miura, R. Ito and N. Suzuki, ‘Regioselective synthesis of trisubstituted quinoxalines mediated by hypervalent iodine reagents’, J. Org. Chem., 2021, 86, 16892–16900 CrossRef CAS.
- S. K. Suthar, N. S. Chundawat, G. P. Singh, J. M. Padrón and Y. K. Jhala, ‘Quinoxaline: A comprehension of current pharmacological advancement in medicinal chemistry’, Eur. J. Med. Chem. Rep., 2022, 5, 100040 CAS.
- M. S. Nafie, S. H. Kahwash, M. M. Youssef and K. M. Dawood, ‘Recent advances on quinoxalines as target-oriente chemotherapeutic anticancer agents through apoptosis’, Arch. Pharm., 2024, 357, e2400225 CrossRef PubMed.
- D. K. Magozwi, M. Dinala, N. Mokwana, X. Siwe-Noundou, R. W. M. Krause, M. Sonopo, L. J. McGaw, W. A. Augustyn and V. J. Tembu, ‘Flavonoids from the genus euphorbia: Isolation, structure, pharmacological activities and structure–activity relationships’, Pharmaceuticals, 2021, 14, 428 CrossRef CAS.
- T. Mosmann, ‘Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays’, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *acde
*acde