
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of surface modification on silica supported Ti catalysts for cyclohexene oxidation with vapor-phase hydrogen peroxide†
Sol Ahn *a,
Sarah K. Friedmanb and
Justin M. Notestein*bc
*a,
Sarah K. Friedmanb and
Justin M. Notestein*bc
aDepartment of Chemical Engineering, Chung-Ang University, 84 Heukseok-ro, Dongjak-gu, Seoul, 06974, Republic of Korea. E-mail: solahn@cau.ac.kr
bDepartment of Chemical and Biological Engineering, USA. E-mail: j-notestein@northwestern.edu
cCenter for Catalysis and Surface Science, 2145 Sheridan Rd, Evanston, Illinois 60208, USA
First published on 13th August 2024
Abstract
Surface modification via grafting of organic moieties on a Lewis acid catalyst (silica supported Ti catalyst, Ti-SiO2) alters the activation of H2O2 in vapor-phase cyclohexene epoxidation. Grafting a fluorous group (1H,1H-perfluoro-octyl) suppresses activity of Ti-SiO2. Conversely, grafting either a nonpolar group (octyl) or a polar aprotic group (triethylene glycol monomethyl ether) enhances rates and shifts selectivity toward trans-1,2-cyclohexanediol.
Post-synthetic modification provides an opportunity to tune the surface properties (e.g. hydrophilicity/hydrophobicity) of previously-synthesized, supported catalysts. For example, surface modification to remove surface hydroxyls and to increase hydrophobicity of a surface is an effective way to reduce the negative effects of water in liquid-phase selective oxidation chemistry.1,2 This increase in hydrophobicity is particularly useful when water adsorbs onto the active metal site competingly with a reactant. Beyond altering surface hydrophilicity/hydrophobicity, other post-synthetic modifications include overcoating of metal oxide layers,3,4 grafting of functional organic groups,5,6 and depositing additional active metal oxide sites.7,8 For liquid phase reactions, any surface modifications must compete against the solvent for any changes to the local environment around the active site. Here, we report grafting of organic molecules to change surface properties for vapor phase cyclohexene epoxidation with vaporized H2O2, where we hypothesized that surface modification might have a more direct impact on the elementary steps of catalysis. By condensing the corresponding terminal alcohols with surface silanols, we grafted (Scheme 1) three different types of functional groups on a pre-synthesized Ti-SiO2 Lewis acid catalyst: octyl groups (Ti-SiO2-o, nonpolar), triethylene glycol monomethyl ether (Ti-SiO2-tg, polar aprotic), and 1H,1H-perfluoro-1-octyl (Ti-SiO2-F, fluorous).
 | ||
| Scheme 1 Schematic diagram of grafting of 1-octanol (nonpolar), triethylene glycol monomethyl ether (polar aprotic), and 1H,1H-perfluoro-1-octanol (polar protic) on Ti-SiO2. | ||
We prepared a highly dispersed silica supported Ti catalyst (Ti-SiO2) via liquid-phase grafting of trichloro(pentamethylcyclopentadienyl)titanium(IV) onto a mesoporous silica support at 0.2 Ti atoms per nm2, followed by calcination, which is known to give high specific activity in H2O2 activation.9 We modified the parent Ti-SiO2 by grafting the corresponding terminal alcohol in refluxing toluene for 24 h, Soxhlet extraction for 24 h in toluene to remove any ungrafted species,2 and drying at 100 °C under vacuum.
Successful grafting is indicated by slight decreases in BET surface area (Fig. 1(a) and Table 1) without changes to the shape of physisorption isotherm and by mass losses in thermogravimetric analysis (Fig. 1(b)). Mass losses beyond the low-temperature desorption of water are due to combustion or decomposition of the grafted species. The water desorption temperatures of modified catalysts are similar, which agree with the previous study.2 Mass losses beyond the shaded regime in Fig. 1(b) correspond to loadings of 0.75, 0.42, and 0.30 groups per nm2, for Ti-SiO2-o, Ti-SiO2-tg, and Ti-SiO2-F, respectively. Grafting 1-octanol on a Ta-SiO2 catalyst was previously reported to give 0.39–0.64 groups per nm2.2 Most importantly, all these values are higher than the surface Ti loading of parent supported catalyst, which is 0.2 Ti atoms per nm2, so that they should be sufficient to affect catalytic behavior of Ti-SiO2.
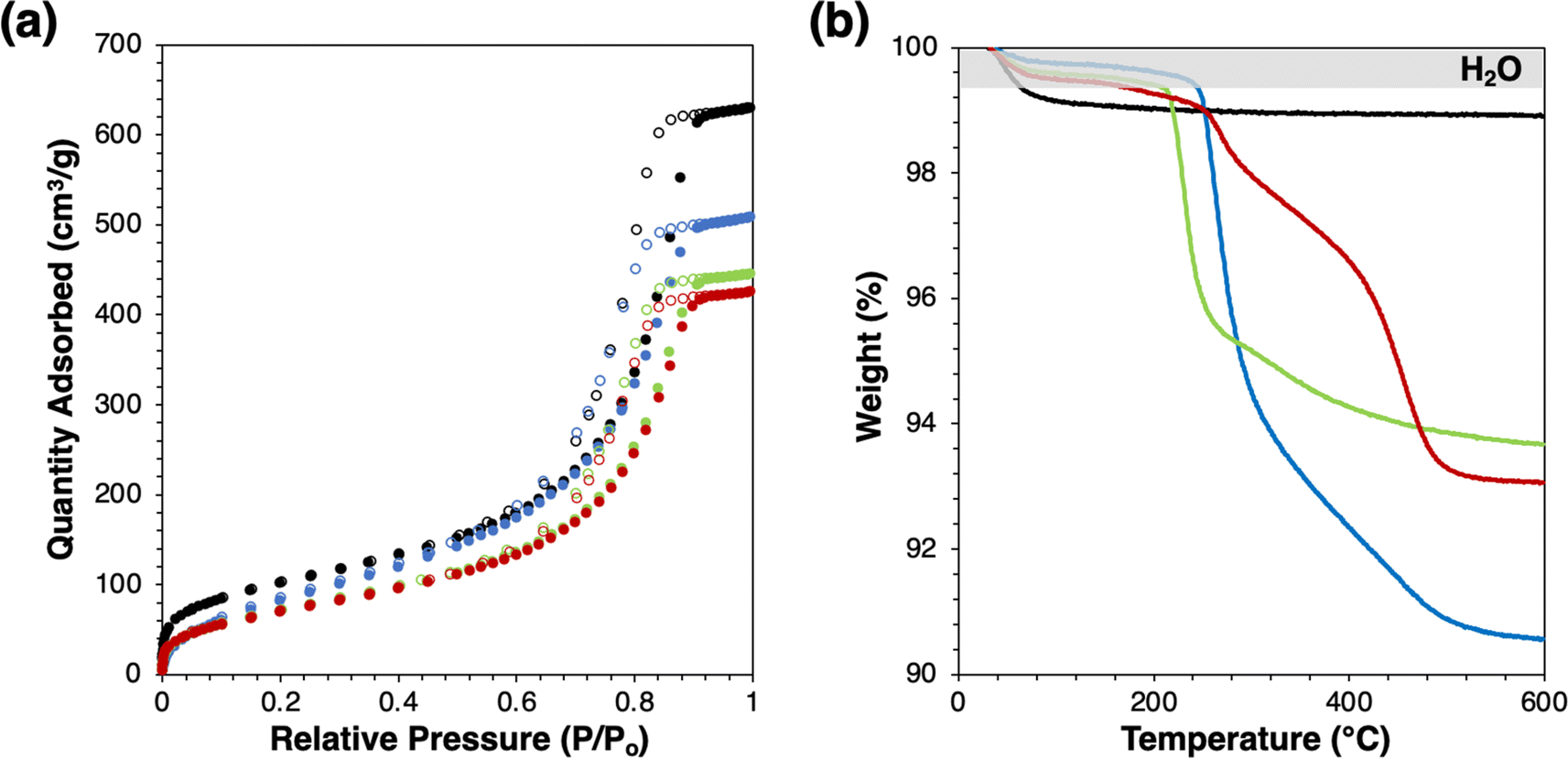 | ||
| Fig. 1 (a) N2 physisorption isotherm and (b) mass loss profile by thermogravimetric analysis of catalysts (black: Ti-SiO2, blue: Ti-SiO2-o, green: Ti-SiO2-tg, red: Ti-SiO2-F). | ||
| Catalyst | BET surface area [m2 g−1] | Organic surface density [# nm−2] | Steady state TOFa [molC6 molTi−1 h−1] | Steady state selectivitya [%] (epoxide/diol/COx) |
|---|---|---|---|---|
| a TOF values of production of epoxide and diol values at 600 min. Steady state operation is reached after 200–400 minutes at these conditions. | ||||
| Ti-SiO2 | 370 | — | 6.5 | 39/42/19 |
| Ti-SiO2-o | 340 | 0.75 | 15.5 | 6/86/8 |
| Ti-SiO2-tg | 270 | 0.42 | 11.0 | 4/85/11 |
| Ti-SiO2-F | 290 | 0.30 | 1.6 | 18/32/50 |
We performed vapor-phase cyclohexene epoxidation at 120 °C, 3 kPa of cyclohexene, and 3 kPa of vaporized H2O2 employing our custom built reactor.10 Here, we used H2O2 in acetonitrile, dried over MgSO4, to minimize initial water content.11 In this study, products were detected with online GC-FID and an in-jet methanizer. We do not observe any C6 derived products other than cyclohexene epoxide (epoxide) and trans-1,2-cyclohexanediol (diol), consistent with our previous work with Ti-SiO2 at similar conditions.10 In these systems, cyclohexene first converts to epoxide, and then hydrolyzes to the trans-diol (Scheme 2). This stepwise conversion of cyclohexene is consistent with our previous studies,7,10 as we do not observe any cis-diol that is the product of direct cis-dihydroxylation of cyclohexene. Radical oxidation to cyclohexenone or cyclohexenol is not observed. Background over-oxidation to CO and CO2 occurs at a rate of approximately 1.2 to 4.1 molcyclohexene molTi−1 h−1 or 0.2 to 0.8% conversion at these conditions, regardless of catalyst.
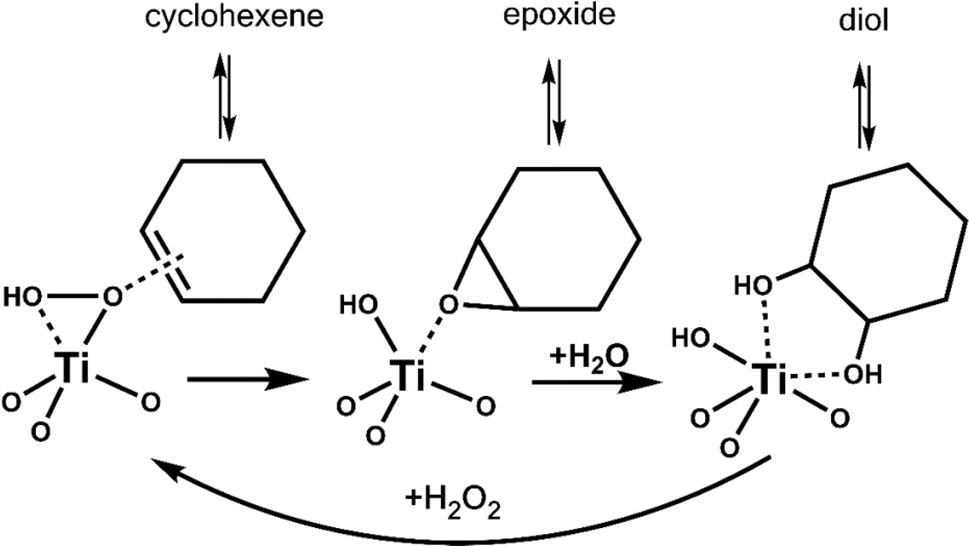 | ||
| Scheme 2 Reaction network of cyclohexene epoxidation to cyclohexene oxide (epoxide), and its hydrolysis to yield trans-1,2-cyclohexane diol (diol). Surface modifications near the active site can alter the strength of adsorption of reactants and intermediates, altering product selectivity. | ||
The parent Ti-SiO2 shows an initial turnover frequency (TOF = mol(epoxide+diol) molTi−1 hr−1) of 19.6 h−1 at 50 min time-on-stream (TOS), which decays to a steady-state rate of 6.5 h−1 at 600 min TOS. As seen mostly clearly in the selectivity plot, steady-state is reached after ∼200 minutes, with only slow catalyst deactivation thereafter (Fig. 2). The steady state selectivity is 39%/42% to epoxide and diol respectively, with the remainder going to background overoxidation to COx. Grafting of a fluorous group (Ti-SiO2-F) almost totally suppresses C6 product formation. The small amount of remaining C6 formation has a selectivity of 18%/32% to epoxide and diol, relatively similar to the parent catalyst and suggesting the existence of small patches of unfunctionalized surface. Otherwise, the conclusion is that the fluorous surface makes binding and activation of cyclohexene unfavorable by inhibiting the adsorption of cyclohexene on the surface. Conversely, grafting of either nonpolar octyl or polar aprotic tri(ethylene glycol) groups on Ti-SiO2 increases rates by at least 1.7-fold at steady-state, relative to the parent catalyst. These enhanced catalytic rates have two effects. First, the loss of C6 to background overoxidation drops dramatically, from 19% in the parent catalyst to 8–11% in the modified catalysts. Moreover, the C6 selectivity shifts to substantially favor hydrolysis of the epoxide to the diol, giving approximately 5%/85% for epoxide and diol, respectively, at steady state.
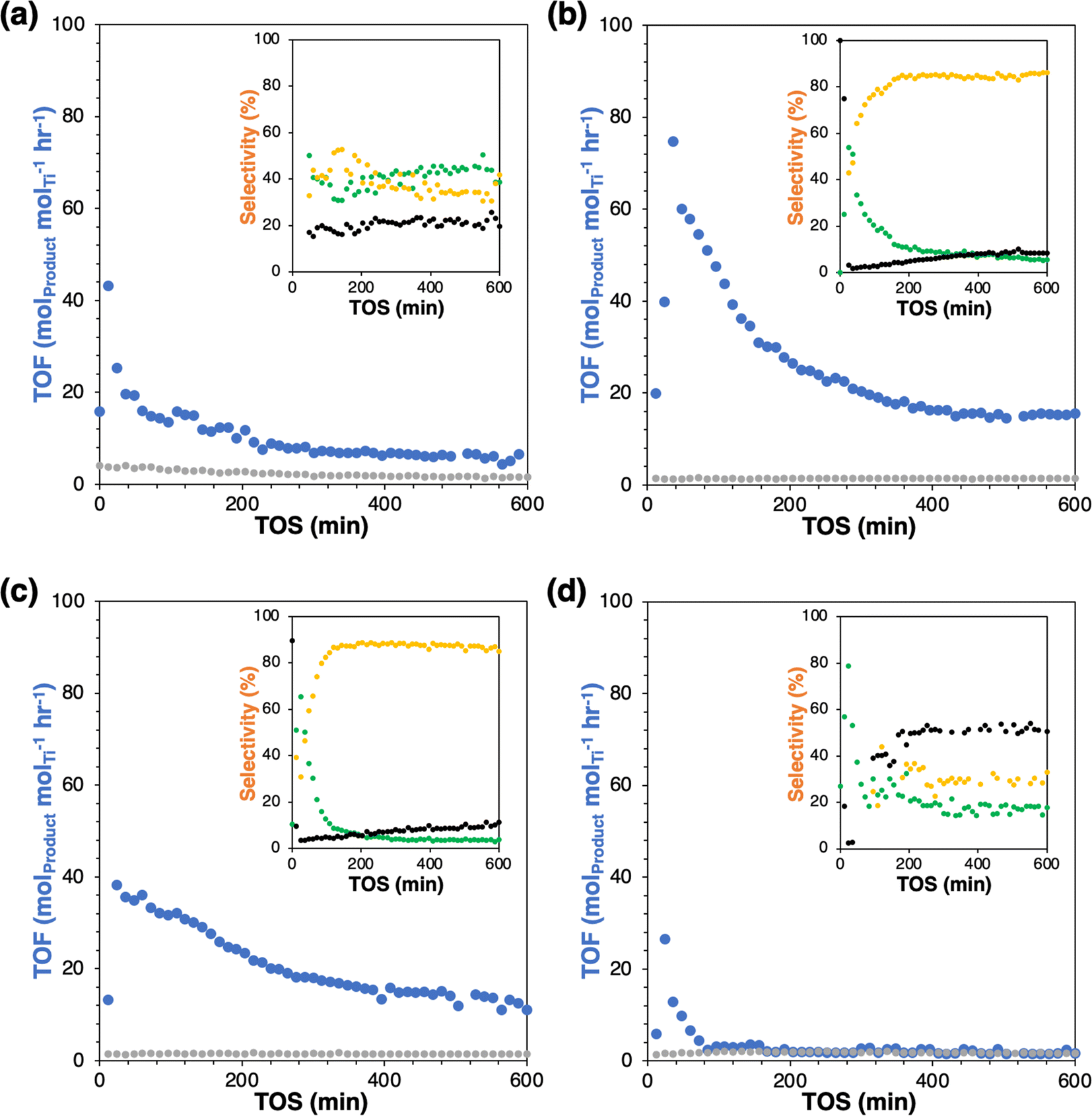 | ||
| Fig. 2 Time-on-stream turnover frequency (TOF) and selectivity (inset) of (a) Ti-SiO2, (b) Ti-SiO2-o, (c) Ti-SiO2-tg and (d) Ti-SiO2-F (blue: TOF of C6 products, grey: TOF of COx/6, green: epoxide selectivity, yellow: diol selectivity, black: COx/6 selectivity). | ||
This behavior is quite different from that observed in the condensed phase, where grafting groups to remove surface silanols tends to decrease yields slightly and increase epoxide selectivity relative to diol by suppressing water sorption at the active site.2 In the vapor phase and for these wide-pore materials, the lack of a liquid solvent phase means that surface modification can more directly influence the stability of reaction intermediates. As suggested in Scheme 2, the surface modifications appear to be strengthening the adsorption of cyclohexene and the intermediate epoxide, leading to corresponding increases in rate and selectivity to the hydrolysis product. In addition, a recent study by Leonhardt et al. proposed computationally that an epoxide molecule can remain adsorbed to one facet of the Ti-OH site while still leaving another coordination site available for oxidation of an incoming cyclohexene.12 In that mechanism, enhancing epoxide adsorption at the active site increases hydrolysis to the diol without inhibiting – or even enhancing – overall product formation rates, such as we have observed with the octyl- and tg-modified surfaces. Overall, these observations show that surface grafting can play a significant role in modifying the reactivity of catalysts in the nascent field of selective oxidation with vaporized H2O2. Also, the results presented here contribute/expand to the current design strategy of post-modification of heterogeneous catalysts with simple method. Additional studies will be carried out to understand the precise mechanistic origins of these changes in rate and selectivity and develop further strategies to tune catalyst surface properties.
Data availability
The data supporting this article have been included solely in the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge support from the Institute for Catalysis in Energy Processes, funded by DOE, Office of Science, Office of Basic Energy Sciences (DE-FG02-03ER15457). This research was supported by the Chung-Ang University Research Grants in 2023.References
- D. A. Ruddy and T. D. Tilley, Highly selective olefin epoxidation with aqueous H2O2 over surface-modified TaSBA15 prepared via the TMP method, Chem. Commun., 2007,(32), 3350–3352 RSC
.
- N. Morlanés and J. M. Notestein, Grafted Ta–calixarenes: Tunable, selective catalysts for direct olefin epoxidation with aqueous hydrogen peroxide, J. Catal., 2010, 275(2), 191–201 CrossRef
- A. T. Y. Wolek, M. A. Ardagh, H. N. Pham, S. Alayoglu, A. K. Datye and J. M. Notestein, Creating Brønsted acidity at the SiO2-Nb2O5 interface, J. Catal., 2021, 394, 387–396 CrossRef CAS
- A. T. Y. Wolek, K. E. Hicks and J. M. Notestein, Tuning acidity in silica-overcoated oxides for hydroalkoxylation, J. Catal., 2023, 426, 113–125 CrossRef CAS
- C. Khoury, S. Holton, D. Shpasser, E. Hallo, A. Kulkarni, F. C. Jentoft and O. M. Gazit, Elucidating Cooperative Interactions between Grafted Amines and Tin or Titanium Sites on Silica, ACS Catal., 2022, 12(16), 9846–9856 CrossRef CAS
- C. A. Contreras, R. A. Hackler, J. M. Notestein and P. C. Stair, Orientation of 1,1′-Bi-2-naphthol Grafted onto TiO2, J. Phys. Chem. C, 2022, 126(18), 7980–7990 CrossRef CAS
- S. Ahn, N. E. Thornburg, Z. Li, T. C. Wang, L. C. Gallington, K. W. Chapman, J. M. Notestein, J. T. Hupp and O. K. Farha, Stable Metal–Organic Framework-Supported Niobium Catalysts, Inorg. Chem., 2016, 55(22), 11954–11961 CrossRef CAS
- I. S. Kim, S. Ahn, N. A. Vermeulen, T. E. Webber, L. C. Gallington, K. W. Chapman, R. L. Penn, J. T. Hupp, O. K. Farha, J. M. Notestein and A. B. F. Martinson, The Synthesis Science of Targeted Vapor-Phase Metal–Organic Framework Postmodification, J. Am. Chem. Soc., 2020, 142(1), 242–250 CrossRef CAS
- M. A. Ardagh, D. T. Bregante, D. W. Flaherty and J. M. Notestein, Controlled Deposition of Silica on Titania-Silica to Alter the Active Site Surroundings on Epoxidation Catalysts, ACS Catal., 2020, 10(21), 13008–13018 CrossRef CAS
- S. Ahn, S. L. Nauert, K. E. Hicks, M. A. Ardagh, N. M. Schweitzer, O. K. Farha and J. M. Notestein, Demonstrating the Critical Role of Solvation in Supported Ti and Nb Epoxidation Catalysts via Vapor-Phase Kinetics, ACS Catal., 2020, 10(4), 2817–2825 CrossRef CAS
- N. E. Thornburg, A. B. Thompson and J. M. Notestein, Periodic Trends in Highly Dispersed Groups IV and V Supported Metal Oxide Catalysts for Alkene Epoxidation with H2O2, ACS Catal., 2015, 5(9), 5077–5088 CrossRef CAS
- B. E. Leonhardt, M. Head-Gordon and A. T. Bell, The Effects of ≡Ti–OH Site Distortion and Product Adsorption on the Mechanism and Kinetics of Cyclohexene Epoxidation over Ti/SiO2, ACS Catal., 2024, 14(5), 3049–3064 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04552a |
| This journal is © The Royal Society of Chemistry 2024 |