
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen synthesis and antitumor activity of (E)-diethyl 2-styrylquinoline-3,4-dicarboxylates†
Hong Zhanga,
Jianyuan Wangb,
Cheng Lic,
Di Zhaoa,
Tianyu Lianga and
Yang Li *a
*a
aCollege of Chemistry and Materials Engineering, Bohai University, No. 19, Keji Street, Jinzhou, P. R. China. E-mail: bhuzh@163.com
bDepartment of Chemical Engineering, Fushun Teacher's College, No. 103, Wenhua Street, Fushun, P. R. China
cLiaoyang Ecological Environment Monitoring Center, Liaoyang, P. R. China
First published on 23rd August 2024
Abstract
In this work, a green, efficient and catalyst-free synthesis of a series of structurally novel (E)-diethyl 2-styrylquinoline-3,4-dicarboxylates via a direct olefination reaction between diethyl 2-methylquinoline-3,4-dicarboxylate and various aromatic aldehydes was successfully accomplished by employing eco-friendly 1,3-dimethylurea/L-(+)-tartaric acid (DMU/LTA) as an inexpensive, non-toxic and reusable reaction medium. This methodology has the attractive advantages of mild reaction conditions, simple experimental operation, and the absence of any dangerous catalysts or unsafe volatile organic solvents, with satisfactory to good yields. Subsequently, a primary in vitro evaluation for their anti-proliferative activity against human cancer cell lines A549, HT29 and T24 revealed that the compound with the 3,4,5-trimethoxystyryl moiety exhibited potent anti-tumor activity with IC50 values of 2.38, 4.52 and 9.86 μmol L−1, respectively, thereby being equipotent or even better than the reference cisplatin.
Introduction
Among the quinoline derivatives, the 2-styrylquinoline (SQ) structure forms an important type of structural motif and represents an elite scaffold and a wonderful pharmacophore in drug discovery.1 Many members of this family, such as FZ-41 (I), VUF5017 (II), WK14 (III), and L-660,771 (IV), have been widely applied as antiproliferative,2 antiviral,3 anti-HIV,4 and antimicrobial5 agents and antagonists,6 as shown in Fig. 1. Recent anti-proliferative activity evaluation of 2-styrylquinoline derivatives against tumor cell lines has validated the importance of this class of compounds as a new hope in developing anti-cancer drugs.7,8 For example, 8-hydroxyl-2-styrylquinoline (V, Fig. 1) was reported to demonstrate a marked anti-cancer activity against the human neuroblastoma cell line SK-N-MC,9 and the newly-synthesized 5,6,7-trimethoxy-N-aryl-2-styrylquinolin-4-amines (VI, Fig. 1) were reported to exhibit potent anti-cancer activity against human cancer cell lines A-2780 and MCF-7.10 Owing to their striking biological activities and in order to have structurally diversified molecules for bio-screening, considerable synthetic efforts have been directed toward developing synthetic approaches for the construction of 2-styrylquinoline derivatives. Considering the fact that 2-methylquinoline derivatives could be readily converted into an enamine tautomeric form via C(sp3)–H activation by the nitrogen atom under various catalytic conditions,11 the direct olefination of 2-methylquinolines via the Knoevenagel condensation reaction with aromatic aldehydes appears to be the most simple and straightforward approach in comparison with other possible methods such as Wittig reactions using expensive 2-quinolinecarboxaldehydes12 and reductive olefinations of quinoline N-oxides.13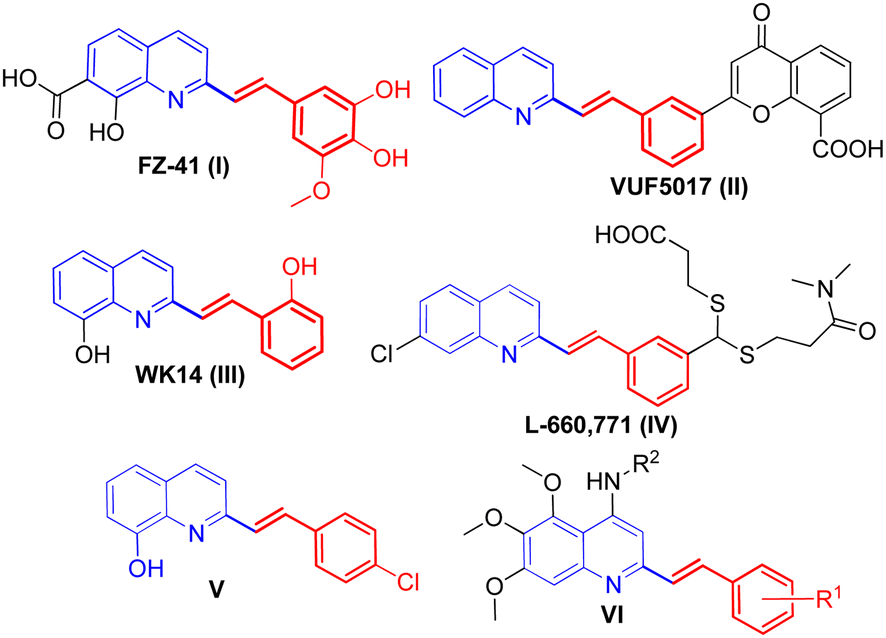 | ||
| Fig. 1 Structures of some bioactive 2-styrylquinolines I–VI. | ||
The conventional approach for the direct olefination of 2-methylquinolines with aldehydes involves the use of acetic anhydride as the reaction medium,14 but the protocol is plagued by constraints such as high reaction temperature, the use of a large excess of aldehydes and low product yields. Owing to the great significance of direct olefination in organic synthesis, great efforts have been devoted during the past decades to develop more convenient and effective synthetic methods, including (1) the utilization of NaOAc as a base in water-acetic acid binary solvents;5 (2) the use of metal catalysts such as La(Pfb)3,15 Fe3O(BPDC)3,16 or Fe(OAc)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 in toluene; InCl3 in THF;18 CoCl2 in H2O;19 Ca(OTf)2/Bu4NPF6 under solvent-free conditions;20 and ZnCl2 under microwave irradiation;21 (3) solvent-free microwave irradiation under inert gas atmosphere (N2) at high temperatures (170–175 °C);22 (4) employment of 1,4-dioxane as the reaction medium under catalyst-free conditions at 140 °C;23 and (5) the combination of 1,3-dimethylbarbituric acid and HOAc as synergistic catalysts,24 as summarized in Scheme 1a. Although these elegant methods for the direct olefination of 2-methylquinolines have emerged, they were invariably associated with certain limitations such as harsh reaction conditions; toxic chemical reagents; expensive or unavailable catalysts; and volatile, flammable and harmful organic solvents. In addition, 2-styrylquinolines could also be facilely accessed using 1-methylimidazolium trifluoroacetate ([Hmim]TFA)25 or Brønsted acidic imidazolium ionic liquids as the reaction medium,26 which involved in situ generated 2-methylquinoine from o-aminoketone and β-ketoester, followed by its olefination reaction with aromatic aldehydes. However, the high cost and environmental toxicity have limited their practical application.
17 in toluene; InCl3 in THF;18 CoCl2 in H2O;19 Ca(OTf)2/Bu4NPF6 under solvent-free conditions;20 and ZnCl2 under microwave irradiation;21 (3) solvent-free microwave irradiation under inert gas atmosphere (N2) at high temperatures (170–175 °C);22 (4) employment of 1,4-dioxane as the reaction medium under catalyst-free conditions at 140 °C;23 and (5) the combination of 1,3-dimethylbarbituric acid and HOAc as synergistic catalysts,24 as summarized in Scheme 1a. Although these elegant methods for the direct olefination of 2-methylquinolines have emerged, they were invariably associated with certain limitations such as harsh reaction conditions; toxic chemical reagents; expensive or unavailable catalysts; and volatile, flammable and harmful organic solvents. In addition, 2-styrylquinolines could also be facilely accessed using 1-methylimidazolium trifluoroacetate ([Hmim]TFA)25 or Brønsted acidic imidazolium ionic liquids as the reaction medium,26 which involved in situ generated 2-methylquinoine from o-aminoketone and β-ketoester, followed by its olefination reaction with aromatic aldehydes. However, the high cost and environmental toxicity have limited their practical application.
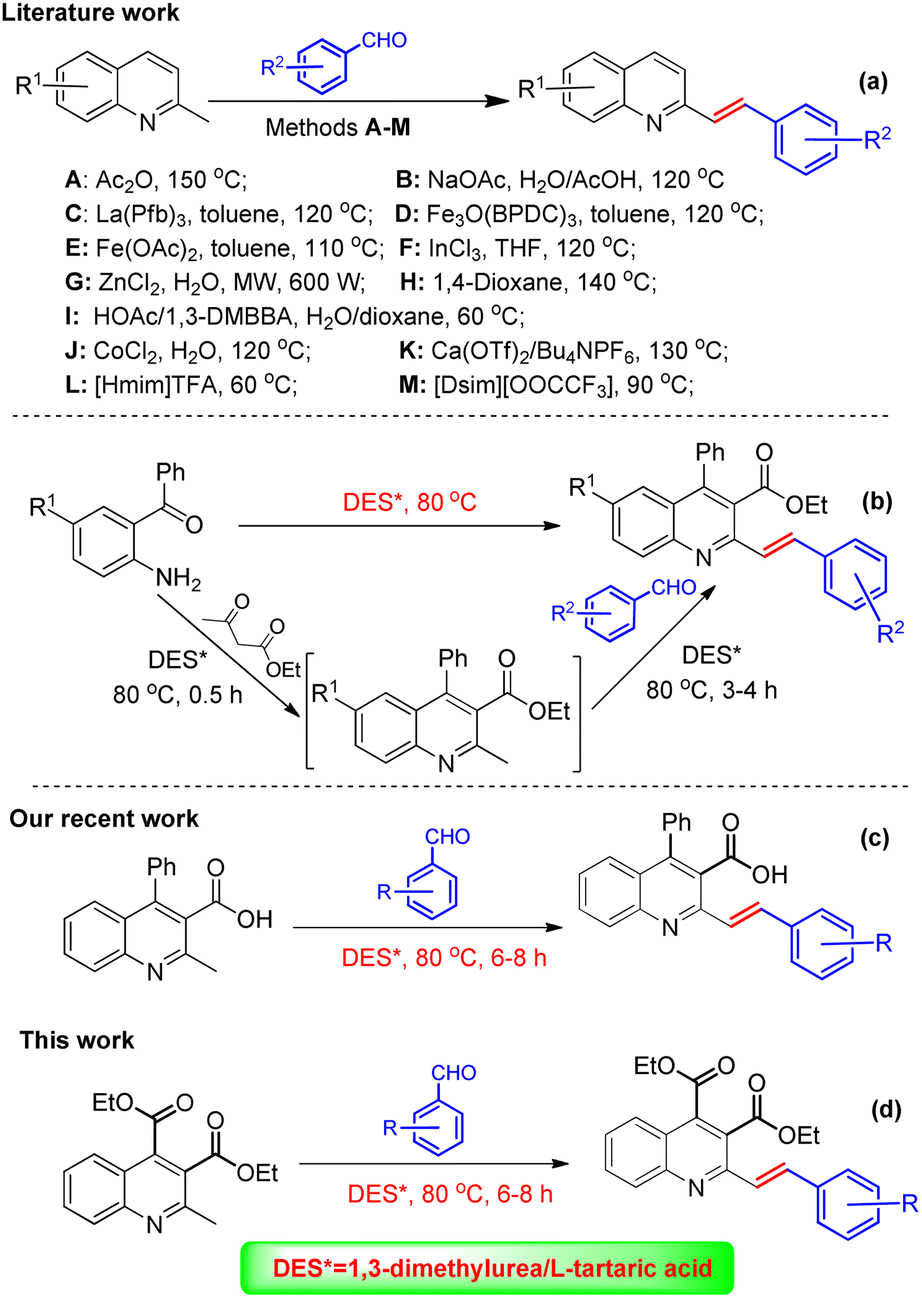 | ||
| Scheme 1 Synthesis of 2-styrylquinoline derivatives by using distinct methods (a)–(d). | ||
Nowadays, in light of the stringent environmental requirements and safety considerations in chemical production, increasing research efforts have been focused on the development of sustainable and environmentally benign reaction procedures to replace those efficient but somewhat outdated methods, particularly, to replace volatile, flammable and harmful organic solvents.27 Deep eutectic solvents (DESs), as an emerging class of unconventional solvents derived from the combination of two or three safe and cheap components of Lewis or Brønsted acids and bases through hydrogen bond formation, have attracted enormous attention due to their unique properties, including a wide liquid range, negligible vapor pressure, low toxicity, non-flammability, high solvation capacity, high biodegradability, low cost of components and convenient preparation,28 thereby rendering them acknowledged widely as an excellent alternative to volatile organic solvents in the development of environmentally friendly organic reactions.29 For example, Zhang's group has developed the application of choline chloride (ChCl)/urea,30 choline chloride/lactic acid (ChCl/LAC),31 choline chloride/glycerol (ChCl/Gly),32 choline chloride (ChCl)/malonic acid,33 and ChCl/L-(+)-tartaric acid34 as a biodegradable, recycled and reusable media in the green synthesis of heterocyclic compounds. Therefore, the synthesis of this class of 2-styrylquinoline derivative using deep eutectic solvents would be highly sought after. There was an impressive report from Kashinath group,35 who described a green, metal-free, one-pot synthesis of 2-styrylquinolines using a combination of 1,3-dimethyl urea (1,3-DMU) and L-tartaric acid (LTA) (in a 3:1 ratio) as a deep eutectic solvent (DES) via Friedlander annulation, followed by Knoevenagel condensation, as shown in Scheme 1b. Inspired by the report, our group in this regard has recently achieved the green synthesis of (E)-2-styrylquinoline-3-carboxylic acid by using the non-toxic deep eutectic solvent (DES) of 1,3-dimethylurea (DMU)/L-tartaric acid (LTA) as medium (Scheme 1c).36 In view of the structural diversity playing a prominent role in new drug discovery37 and in the context of our ongoing studies concerning the green synthesis of quinoline derivatives, we envisioned that 2-methylquinoline-3,4-dicarboxylate as a very intriguing scaffold might be amenable to the green strategy to access a new class of 2-styrylquinoline compounds, which might exhibit interesting biological activities. Thus, we would like to report, herein, a new and environmentally benign synthesis and preliminary in vitro anti-tumor activity evaluation of a series of structurally novel diethyl 2-styrylquinoline-3,4-dicarboxylate derivatives by using the safe, eco-friendly and unconventional 1,3-DMU/LTA as a solvent (Scheme 1d). To the best of our knowledge, the green synthesis and biological activity evaluation of such quinoline derivatives has not been achieved so far and might be employed as potential candidates for future drug discovery.
Results and discussion
In the past decades, the synthesis of quinoline carboxylate derivatives has been a very attractive synthetic target due to their potent biological activities.38–40 For example, Lu et al. recently reported a TMSCl-mediated Pfitzinger reaction of isatin with ethyl acetoacetate for the synthesis of diethyl 2-methylquinoline-3,4-dicarboxylate.41 On the basis of the report, we devised that if its 2-methyl group could be further transformed into styrene moiety, the resulting diethyl 2-styrylquinoline-3,4-dicarboxylate derivatives might lead to a new dimension of structural diversity as potential candidates for biological evaluations or provide more opportunities for further synthetic manipulations. On the other hand, our group has recently developed a green, efficient and catalyst-free strategy for the synthesis of (E)-2-styrylquinoline-3-carboxylic acid derivatives via direct olefination of 2-methylquinoline-3-carboxylic acid with aromatic aldehydes by using an environmentally benign and non-toxic deep eutectic solvent (DES) of 1,3-dimethylurea (DMU)/L-(+)-tartaric acid (LTA) as both catalyst and reaction medium.36 Building on the evolving expertise, we were very keen to explore whether 2-methylquinoline-3,4-dicarboxylate could be converted into the desired diethyl 2-styrylquinoline-3,4-dicarboxylate derivatives under the same reaction conditions as well as the subsequent evaluation of their anti-tumor activity. Accordingly, following the line of green chemistry, we investigated the model reaction of diethyl 2-methylquinoline-3,4-dicarboxylate (1) with benzaldehyde (2a) by using the DES, namely DMU/LTA (7/3, mol mol−1) as the reaction medium at 80 °C, as shown in Scheme 2. To our great delight, the application of DES was found to be very suitable to prompt the reaction proceeding efficiently, giving the corresponding product 3a in a remarkably high yield of 83% within 6 h.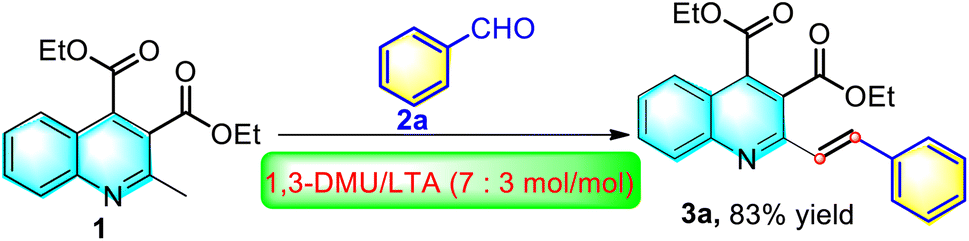 | ||
| Scheme 2 Various methods for the synthesis of 2-styrylquinoline derivatives. | ||
Due to the satisfactory yield obtained and in order to retain the simplicity of the procedure, no further optimization in reaction conditions was necessary. In order to highlight the advantages of the DES solvent, a control experiment was carried out, in which the model reaction was conducted employing the widely used Ac2O as the medium based on the literature method.2 However, from the experiment, product 3a was obtained only in 38% yield. Thus, the use of DMU/LTA as the green reaction medium not only avoids the disadvantages of conventional organic solvents but also results in greatly enhanced reactivity. In addition, it is worth noting that we also carried out the model reaction in the presence of either L-(+)-tartaric acid or 1,3-dimethylurea in some common organic solvents at refluxing temperatures. However, we found that the product formed from these tests was only in a trace amount, as observed by TLC after 24 hours, which suggested that the reaction gave a good yield due to the interaction of DMU/L-(+)-tartaric acid system and not due to its individual component. The reason for the effectiveness of DMU/LTA might be its good solubility, high stability and positive synergic effect on the reaction through extensive hydrogen bonding. Finally, the recyclability of DMU/LTA for the model reaction was investigated by subjecting the fresh substrates 1 and 2a to the recovered DMU/LTA obtained by evaporating the aqueous layer under vacuum after product removal to repeat the model reaction. We found that the recovered DMU/LTA could be re-used up to three consecutive runs, with the 3a yields being 82%, 81% and 77%, respectively, demonstrating negligible changes in the synthetic efficiency, though a slight darkening of the eutectic mixture was observed after recycling. Thus, the DES could be successfully re-used over three cycles with a decrease of 6% in the final 3a yield, and the corresponding E-factor value (total mass of waste/mass of product) from the three re-used cycles was calculated to be 93.68. However, starting from the fourth cycle, an obvious decrease in the product yield of 65% was noticed.
To demonstrate the synthetic potential by applying DMU/LTA as the privileged reaction medium in the Knoevenagel condensation reaction, we extended the reaction to other substituted aromatic aldehydes in a similar fashion. Satisfactorily, these aldehydes were equally amenable to the reaction process without any experimental difficulties, successfully delivering the corresponding 3b–o in satisfactory yields of 61–86%, as listed in Table 1. It is worth noting that in all cases, the acid-sensitive ester and the newly formed olefin groups remain unaffected under the reaction conditions.
|
||||
|---|---|---|---|---|
| Entry | Compd | Ar | Yielda/% | Mp/°C |
| a Isolated yield. | ||||
| 1 | 3a | Ph | 83 | 92–94 |
| 2 | 3b | 3-MeC6H4 | 82 | 78–79 |
| 3 | 3c | 4-MeC6H4 | 85 | 89–90 |
| 4 | 3d | 2,5-(Me)2C6H3 | 72 | Oily liquid |
| 5 | 3e | 3,4-(Me)2C6H3 | 81 | 108–109 |
| 6 | 3f | 2-MeOC6H4 | 70 | 98–100 |
| 7 | 3g | 3-MeOC6H4 | 78 | Oily liquid |
| 8 | 3h | 2,3-(MeO)2C6H3 | 65 | 72–73 |
| 9 | 3i | 2,5-(MeO)2C6H3 | 67 | Oily liquid |
| 10 | 3j | 3,4-(MeO)2C6H3 | 80 | Oily liquid |
| 11 | 3k | 3,4,5-(MeO)3C6H3 | 83 | Oily liquid |
| 12 | 3l | 2-ClC6H4 | 69 | Oily liquid |
| 13 | 3m | 3-BrC6H4 | 81 | 69–71 |
| 14 | 3n | 2-NO2C6H4 | 61 | 119–120 |
| 15 | 3o | 4-NO2C6H4 | 86 | 161–162 |
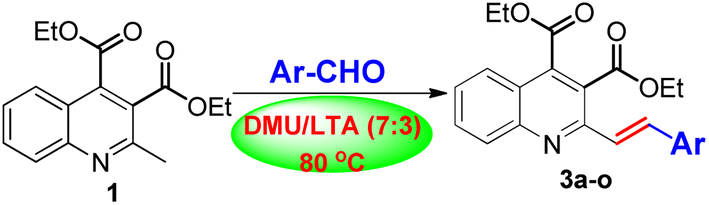
As shown in Table 1, the electronic nature of the substituent present in the aromatic aldehydes appeared not to affect the transformation, neither in product yield nor in reaction rate. For example, compound 3c with an electron-donating methyl group and 3o bearing an electron-withdrawing nitro group were obtained in comparable yields of 85% and 86%, respectively, showing little distinction (entries 3 vs. 15, Table 1). Conversely, the site of the substituent present in the aromatic aldehydes had a significant steric hindrance effect on the product yields. The reaction with ortho-substituted aromatic aldehydes generally gave the corresponding products 3d, 3f, 3h, 3i, 3l and 3n in relatively lower yields with longer reaction times compared with those of meta- and para-substituted ones (entries 4, 6, 8, 9, 12 and 14, Table 1). Particularly, we found that the reaction with di-ortho substituted aromatic aldehydes such as 2,6-dimethyl-, 2,6-dimethoxy and 2,6-dihalobenzaldehydes scarcely proceeded, from which the desired products were detected only in negligible amounts that did not warrant isolation. In addition, the reaction with aliphatic aldehydes such as butyraldehyde, iso-butyraldehyde and cyclohexanecarboxaldehyde was also tested. However, the reaction was found to be fraught with difficulties associated with the combination of starting materials and numerous products, from which we could not separate the desired alkenylation products in any appreciable yield.
With the aim of further diversifying our synthetic work, we became interested in seeing whether quinoline aldehydes would exhibit a similar reactivity. To our delight, the two chosen 2-chloroquinoline-3-carbaldehydes were viable substrates for this transformation as well, invariably furnishing the corresponding vinyl-linked bisquinolines 3p and 3q though in low yields of 53% and 51%, respectively, with a longer reaction time of 8 hours, as shown in Scheme 3. Their structures would be very attractive as many bisquinoline derivatives usually exhibit potent biological activities.42 Work is currently ongoing in this regard, and more studies on extending the reaction scope will be part of our future efforts.
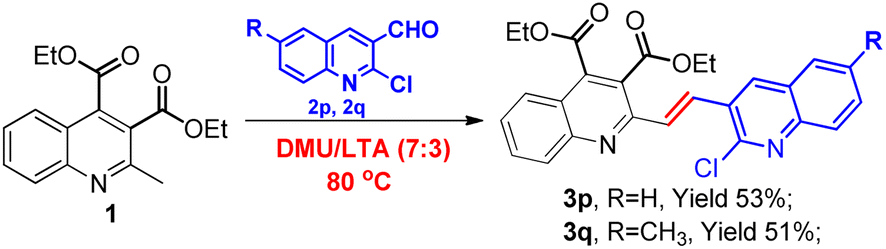 | ||
| Scheme 3 Synthesis of vinyl-linked bisquinolines 3p and 3q. | ||
To the best of our knowledge, all these newly synthesized products 3a–q have never been reported, and their structures have been explicitly characterized based on their spectral and analytical data. Theoretically, these structures should exist as (E)- and/or (Z)-geometry due to the presence of the exocyclic vinyl double bond. The most diagnostic evidence for the geometry of vinyl moiety was the characteristic resonances of the arising two vinylic proton CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH doublets at δ 7.42–7.78 ppm and 8.01–8.38 ppm with large spin–spin coupling constants Jab ∼16.0 Hz in their 1H NMR spectra, which clearly established the stereochemistry of the product as an E-stereoisomer.
CH doublets at δ 7.42–7.78 ppm and 8.01–8.38 ppm with large spin–spin coupling constants Jab ∼16.0 Hz in their 1H NMR spectra, which clearly established the stereochemistry of the product as an E-stereoisomer.
Mechanistically, on the basis of the reports from Alvi et al.,43 who recently described the resonance equation of the DES acting as a proton source, and Krishnakumar et al.,44 who determined the structure, thermal stability and pH value of DMU/LTA (7:3), a proposed reaction mechanism for the synthesis of the title compounds is outlined in Scheme 4. First, the ability of the acidic DMU/LTA (7:3) (pH = 3.7) to N-protonation might play an important role in the activation of 2-methylquinoline 1 to generate the enamine intermediate A. The formed intermediate A could behave as a nucleophile to attack the aromatic aldehydes and give rise to the corresponding adduct B. In this nucleophilic addition reaction, there was a consensus that the DES might also assist in improving the electrophilic reactivity of the aldehydes by hydrogen bonding with its carbonyl group.45 Subsequently, the generated hydroxyl group readily underwent the elimination reaction with the ortho-position hydrogen proton with the loss of one molecule of water to produce the corresponding 2-styrylquinoline derivatives 3. In the sequences of steps, DES played dual roles of solvent and catalyst.
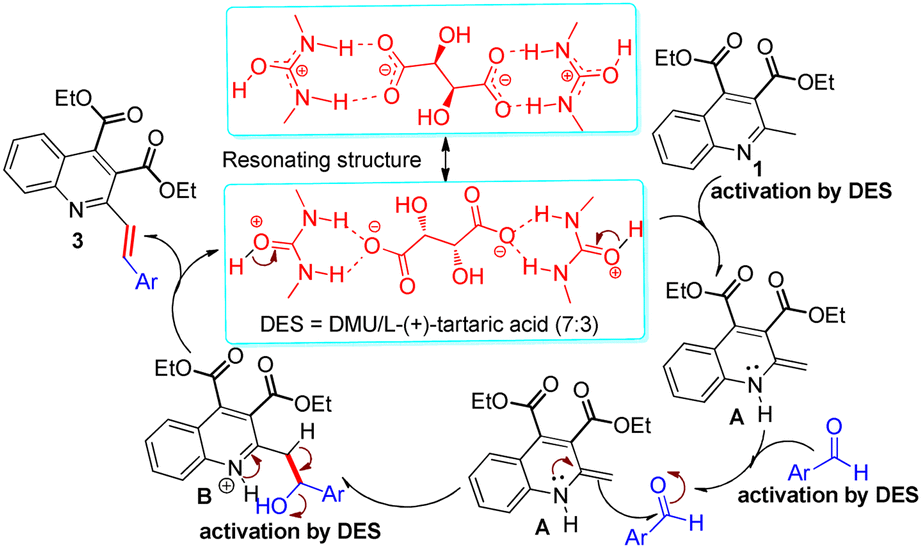 | ||
| Scheme 4 Proposed mechanistic pathway for the synthesis of 3. | ||
With the series of newly synthesized (E)-diethyl 2-styrylquinoline-3,4-dicarboxylate derivatives in hand, we became interested in evaluating their anti-tumor activity. Thus, a preliminary screening for their in vitro anti-tumor activities against human cancer cell lines A549, HT29 and T24 was conducted by the methylthiazolyldiphenyltetrazolium bromide (MTT) conversion assay using the known anti-cancer cisplatin as a reference drug. As listed in Table 2, the unsubstituted 2-styrylquinoline-3,4-dicarboxyalte (3a) exhibited moderate anti-proliferative activity against A549 and HT29 and poor inhibitory effect towards T24 (entry 1, Table 2). The activity was not further potentiated by the introduction of methyl, chloro, bromo and nitro substituents as in 3b–e and 3l–o (entries 2–5 and 12–15, Table 2), which exhibited moderate inhibitory effects. Interestingly, it was observed that the introduction of methoxy substituent appeared to be beneficial in terms of the anti-tumor activity as in compounds 3f–k, which exhibited superior activity (entries 6–11 Table 2). Moreover, the poly-methoxy substituted ones have higher inhibitory activity than the mono-substituted counterparts, and especially, the compound 3k with the 3,4,5-trimethoxystyryl fragment had the best anti-proliferative activity against A549, HT29 and T24 cell lines with the IC50 values of 2.38, 4.52 and 9.86 μmol L−1, respectively (entry 11, Table 2), being equipotent or even better than the reference cisplatin. Additionally, the vinylene-linked bisquinolines 3p and 3q were also found to exhibit satisfactory inhibition properties against the growth of three tested cancer cell lines (entries 16 and 17, Table 2), having the potential to further exploit new drug discovery.
| Entry | Compd | IC50a/μmol L−1 | ||
|---|---|---|---|---|
| A549b | HT29b | T24b | ||
| a IC50 is defined as the drug concentration causing a 50% decrease in cell population using MTT assay as described in the Experimental section.b Cell lines: A549: human lung tumor; HT29: human colon tumor cells, and T24: human urinary bladder tumor cells. | ||||
| 1 | 3a | 18.47 | 24.86 | >40 |
| 2 | 3b | 20.58 | 30.94 | >40 |
| 3 | 3c | 18.61 | 28.15 | >40 |
| 4 | 3d | 18.15 | 25.92 | 30.88 |
| 5 | 3e | 23.49 | >40 | 35.89 |
| 6 | 3f | 16.85 | 14.12 | >40 |
| 7 | 3g | 8.27 | 13.51 | 18.78 |
| 8 | 3h | 9.27 | 15.76 | 26.43 |
| 9 | 3i | 12.84 | 19.56 | 17.69 |
| 10 | 3j | 5.24 | 10.32 | 15.16 |
| 11 | 3k | 2.38 | 4.52 | 9.86 |
| 12 | 3l | 19.45 | 23.08 | >40 |
| 13 | 3m | 25.40 | 21.34 | 30.61 |
| 14 | 3n | >40 | >40 | >40 |
| 15 | 3o | >40 | >40 | >40 |
| 16 | 3p | 6.79 | 8.04 | 16.55 |
| 17 | 3q | 7.41 | 4.33 | 11.18 |
| 18 | Cisplatin | 2.73 | 6.82 | 7.69 |
It is interesting to mention that some significant anti-tumor agents, such as (E)-5,6,7-trimethoxy-N-phenyl-2-styrylquinolines,10 combretastatin A4,46 and its indolin-2-one-based analogue,47 as shown in Fig. 2, also contain the 3,4,5-trimethoxyphenyl fragment as the key pharmacophore. Thus, these insights from the in vitro anti-tumor activity might provide valuable information for further optimization of the series of derivatives, and hopefully, contribute to the development of new and effective anti-tumor candidates.
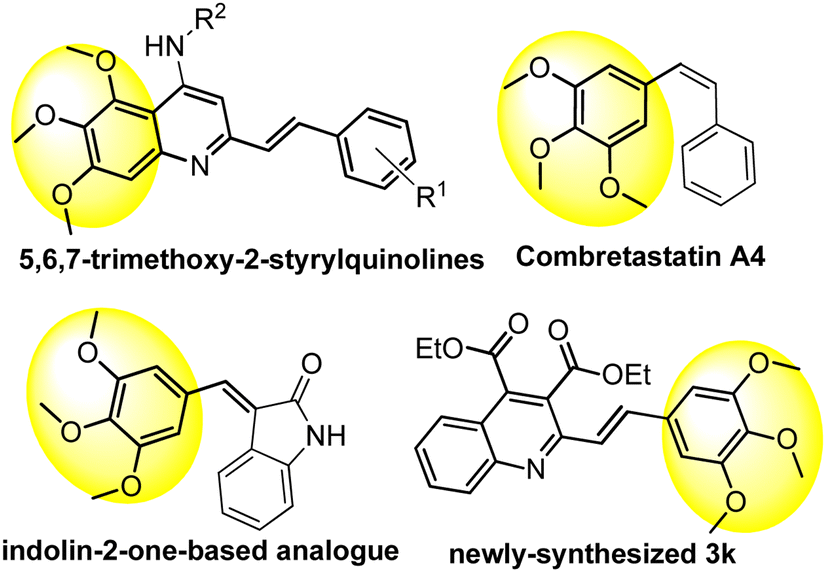 | ||
| Fig. 2 Structures of some 3,4,5-trimethoxyphenyl-containing antitumor compounds. | ||
Conclusion
In summary, we have employed the environmentally benign and non-toxic DES DMU/LTA as a mildly acidic reaction medium for the green synthesis of a new series of structurally intriguing (E)-diethyl 2-arylvinylquinoline-3,4-dicarboxylates via the direct olefination of 2-methylquinolines with aromatic aldehydes. The recyclability and biodegradability of DMU/LTA make this methodology highly sustainable and reliable. The merits of the synthetic protocol described here include experimental simplicity, the use of inexpensive reagents, easy work-up procedure and satisfactory yields, which would contribute to the usefulness of this method. A preliminary evaluation for their in vitro anti-tumor activity bioassay revealed that the 3,4,5-trimethoxy substituted compound exhibited potent anti-proliferative activity against the human cancer cell lines A549, HT29 and T24, being equipotent or even better than the reference drug. These results might give an important insight into the future optimization of the series of 2-styrylquinolines. Currently, work is ongoing, mainly focusing on the further elaboration and application of these compounds, which represent an intriguing goal that we are contemplating, and these results will be a part of future reports.Experimental section
General information
The chemicals used in this report were obtained from Energy Chemical and were used without further purification. Melting points were determined using a WRS-1B melting point apparatus. The 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded on an Agilent 400-MR spectrometer using CDCl3 as the solvent. The reported chemical shifts (δ values) are given in parts per million downfield from tetramethylsilane (TMS) as the internal standard (NMR abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, m = multiplet, J = coupling constant). Elemental analyses were carried out on an EA 2400II elemental analyzer (PerkinElmer, Waltham, MA). The progress of reactions was monitored by TLC on silica gel GF254 using ethyl acetate/petroleum ether (1:10) as eluent.
Preparation of deep eutectic solvent
The deep eutectic solvent 1,3-dimethylurea/L-(+)-tartaric acid was prepared as follows:44 a mixture of 1,3-dimethylurea and L-(+)-tartaric acid with a molar ratio of 7:3 was heated at 100 °C in the air with stirring until a clear colourless liquid was obtained. After cooling to room temperature and vacuum drying for 5 h, the resulting 1,3-dimethylurea/L-(+)-tartaric acid deep eutectic solvent was sealed for later use.
General procedure for the synthesis of (E)-diethyl 2-arylvinylquinoline-3,4-dicarboxylates (3a–q)
Diethyl 2-methylquinoline-3,4-dicarboxylate (1) (0.5 mmol, 0.144 g) and respective aromatic aldehydes (2a–o) or 2-chloroquinoline-3-carbaldehydes (2p and 2q) (0.55 mmol) was added into the medium of 1,3-dimethylurea/L-(+)-tartaric acid (7:3 mol mol−1) (1.5 g). The resulting reaction mixture was stirred at 80 °C for 6–8 hours (as monitored by TLC). After the reaction was completed, the mixture was diluted with an equal volume of water and extracted using EtOAc (3 × 5 mL). The deep eutectic solvent could be easily isolated after removing H2O from the aqueous layer under a vacuum and could be further used for the next reaction run. The combined organic layer was dried over Na2SO4, followed by evaporation of the solvent under reduced vacuum and washing with EtOH or column chromatography over silica gel using petroleum ether/EtOAc (12:1) as eluent to afford the desired 3a–q.
(E)-Diethyl 2-styrylquinoline-3,4-dicarboxylate (3a)
White solid, yield 83.6%, m.p. 92.4–93.8 °C. IR (KBr, cm−1) v 2981, 1740, 1704, 1246, 1194, 1027, 745; 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 8.4 Hz, 1H, ArH), 8.28 (d, J = 15.6 Hz, 1H, CHCH), 8.23 (d, J = 8.8 Hz, 1H, ArH), 8.02 (t, J = 7.6 Hz, 1H, ArH), 7.85 (d, J = 8.0 Hz, 2H, ArH), 7.80 (t, J = 7.2 Hz, 1H, ArH), 7.77 (d, J = 15.6 Hz, 1H, CHCH), 7.62 (t, J = 7.6 Hz, 2H, ArH), 7.55 (t, J = 7.2 Hz, 1H, ArH), 4.75 (q, J = 7.2 Hz, 2H, CH2), 4.71 (q, J = 7.2 Hz, 2H, CH2), 1.67 (t, J = 7.2 Hz, 3H, CH3), 1.66 (t, J = 7.2 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.64, 165.99, 151.73, 148.26, 138.94, 136.61, 136.25, 131.08, 129.46, 128.56, 128.44, 127.41, 127.34, 125.13, 123.86, 123.48, 122.08, 62.08, 61.95, 13.88, 13.85. Anal. calcd for C23H21NO4: C, 73.58; H, 5.64; N, 3.73%. Found: C, 73.74; H, 5.61; N, 3.91%.
(E)-Diethyl 2-(3-methylstyryl)quinoline-3,4-dicarboxylate (3b)
Yellow solid, yield 82.4%, m.p. 78.2–79.5 °C. IR (KBr, cm−1) v 2972, 1731, 1546, 1379, 1255, 1203, 1080, 1027, 965, 763; 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 8.4 Hz, 1H, ArH), 8.24 (d, J = 16.0 Hz, 1H, ArH), 8.18 (d, J = 8.4 Hz, 1H, ArH), 7.98 (t, J = 8.0 Hz, 1H, ArH), 7.78 (d, J = 15.6 Hz, 1H, CHCH), 7.75 (t, J = 7.6 Hz, 1H, ArH), 7.63 (s, 1H, ArH), 7.61 (d, J = 7.2 Hz, 1H, ArH), 7.48 (t, J = 7.6 Hz, 1H, ArH), 7.34 (d, J = 7.2 Hz, 1H, ArH), 4.73–4.64 (m, 4H, CH2), 2.57 (s, 3H, CH3), 1.65 (t, J = 7.2 Hz, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.85, 166.15, 151.97, 148.42, 139.07, 138.15, 136.98, 136.36, 131.23, 129.60, 128.50, 128.18, 127.53, 125.28, 124.72, 123.79, 123.66, 122.22, 62.24, 62.11, 21.31, 14.03. Anal. calcd for C24H23NO4: C, 74.02; H, 5.95; N, 3.60%. Found: C, 73.81; H, 6.06; N, 3.83%.
(E)-Diethyl 2-(4-methylstyryl)quinoline-3,4-dicarboxylate (3c)
Yellow solid, yield 84.7%, m.p. 89.3–90.8 °C. IR (KBr, cm−1) v 2981, 1722, 1546, 1379, 1246, 1194, 1027, 974, 763; 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 8.4 Hz, 1H, ArH), 8.26 (d, J = 15.6 Hz, 1H, CHCH), 8.23 (d, J = 7.6 Hz, 1H, ArH), 8.01 (t, J = 7.6 Hz, 1H, ArH), 7.79 (t, J = 7.6 Hz, 1H, ArH), 7.77 (d, J = 15.6 Hz, 1H, CHCH), 7.75 (d, J = 8.0 Hz, 2H, ArH), 7.42 (d, J = 7.6 Hz, 2H, ArH), 4.75–4.67 (m, 4H, CH2), 2.59 (s, 3H, CH3), 1.68–1.64 (m, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.94, 166.24, 152.14, 148.50, 139.07, 138.95, 136.85, 133.73, 131.26, 129.63, 129.40, 127.53, 127.51, 125.34, 123.71, 123.06, 122.23, 62.29, 62.15, 21.34, 14.11, 14.08. Anal. calcd for C24H23NO4: C, 74.02; H, 5.95; N, 3.60%. Found: C, 74.19; H, 5.78; N, 3.67%.
(E)-Diethyl 2-(2,5-dimethylstyryl)quinoline-3,4-dicarboxylate (3d)
Yellow oily liquid, yield 71.6%; 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 15.2 Hz, 1H, CHCH), 8.13 (d, J = 8.4 Hz, 1H, ArH), 7.99 (d, J = 8.4 Hz, 1H, ArH), 7.77 (t, J = 8.4 Hz, 1H, ArH), 7.55 (t, J = 7.6 Hz, 1H, ArH), 7.45 (d, J = 15.2 Hz, 1H, CHCH), 7.43 (s, 1H, ArH), 7.08 (d, J = 7.6 Hz, 1H, ArH), 7.02 (d, J = 7.6 Hz, 1H, ArH), 4.51–4.41 (m, 4H, CH2), 2.45 (s, 3H, CH3), 2.32 (s, 3H, CH3), 1.43–1.36 (m, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 167.00, 166.26, 152.20, 148.49, 139.02, 135.45, 135.30, 134.97, 134.13, 131.24, 130.47, 129.79, 129.51, 127.59, 126.63, 125.34, 124.97, 123.84, 122.29, 62.32, 62.19, 21.02, 19.53, 14.09, 14.08. Anal. calcd for C25H25NO4: C, 74.42; H, 6.25; N, 3.47%. Found: C, 74.24; H, 6.29; N, 3.31%.
(E)-Diethyl 2-(3,4-dimethylstyryl)quinoline-3,4-dicarboxylate (3e)
Yellow solid, yield 80.7%, m.p. 108.1–110.0 °C. IR (KBr, cm−1) v 2981, 1713, 1546, 1246, 1185, 1027, 754; 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 8.4 Hz, 1H, ArH), 8.13 (d, J = 16.0 Hz, 1H, CHCH), 8.09 (d, J = 7.6 Hz, 1H, ArH), 7.87 (t, J = 8.0 Hz, 1H, ArH), 7.64–7.60 (m, 2H, ArH), 7.49–7.45 (m, 2H, CHCH, ArH), 7.23 (s, 1H, ArH), 4.62–4.55 (m, 4H, CH2), 2.37 (s, 6H, CH3), 1.54–1.52 (m, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.99, 166.27, 152.20, 148.52, 139.04, 137.74, 137.08, 136.79, 134.16, 131.25, 129.99, 129.64, 128.82, 127.48, 125.35, 125.19, 123.75, 122.87, 122.22, 62.29, 62.15, 19.77, 19.68, 14.12, 14.10. Anal. calcd for C25H25NO4: C, 74.42; H, 6.25; N, 3.47%. Found: C, 74.61; H, 6.13; N, 3.18%.
(E)-Diethyl 2-(2-methoxystyryl)quinoline-3,4-dicarboxylate (3f)
Yellow solid, yield 70.4%, m.p. 98.0–100.4 °C. IR (KBr, cm−1) v 2972, 1722, 1573, 1493, 1246, 1027, 745; 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 16.0 Hz, 1H, CHCH), 8.13 (d, J = 8.4 Hz, 1H, ArH), 7.98 (d, J = 8.4 Hz, 1H, ArH), 7.74 (t, J = 7.6 Hz, 1H, ArH), 7.64 (d, J = 16.0 Hz, 1H, CHCH), 7.61 (d, J = 7.6 Hz, 1H, ArH), 7.53 (t, J = 7.6 Hz, 1H, ArH), 7.27 (t, J = 7.6 Hz, 1H, ArH), 6.95 (t, J = 7.2 Hz, 1H, ArH), 6.89 (d, J = 8.4 Hz, 1H, ArH), 4.49–4.40 (m, 4H, CH2), 3.87 (s, 3H, CH3), 1.42–1.36 (m, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 167.07, 166.27, 157.92, 152.51, 148.53, 138.81, 132.21, 131.13, 129.94, 129.80, 128.07, 127.47, 125.56, 125.30, 124.82, 123.98, 122.20, 120.63, 111.00, 62.27, 62.10, 55.45, 14.08. Anal. calcd for C24H23NO5: C, 71.10; H, 5.72; N, 3.45%. Found: C, 71.31; H, 5.60; N, 3.62%.
(E)-Diethyl 2-(3-methoxystyryl)quinoline-3,4-dicarboxylate (3g)
Yellow oily liquid, yield 77.6%. IR (KBr, cm−1) v 2981, 1731, 1546, 1379, 1246, 1045, 780; 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.4 Hz, 1H, ArH), 8.01 (d, J = 15.6 Hz, 1H, CHCH), 7.99 (d, 1H, J = 8.0 Hz, ArH), 7.78 (t, J = 7.6 Hz, 1H, ArH), 7.56 (d, J = 15.6 Hz, 1H, CHCH), 7.54 (t, 1H, J = 8.0 Hz, ArH), 7.30 (t, J = 8.0 Hz, 1H, ArH), 7.22 (d, J = 7.6 Hz, 1H, ArH), 7.12 (s, 1H, ArH), 6.87 (d, J = 8.0 Hz, 1H, ArH), 4.47 (q, J = 7.2 Hz, 2H, CH2), 4.45 (q, J = 7.2 Hz, 2H, CH2), 3.82 (s, 3H, CH3), 1.41 (t, J = 7.2 Hz, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.79, 166.14, 159.73, 151.82, 148.42, 139.12, 137.85, 136.70, 131.26, 129.63, 129.56, 127.61, 125.30, 124.37, 123.67, 122.28, 120.10, 114.47, 112.77, 62.25, 62.12, 55.17, 14.05, 14.03. Anal. calcd for C24H23NO5: C, 71.10; H, 5.72; N, 3.45%. Found: C, 71.21; H, 5.87; N, 3.57%.
(E)-Diethyl 2-(2,3-dimethoxystyryl)quinoline-3,4-dicarboxylate (3h)
Yellow solid, yield 65.3%, m.p. 71.7–72.6 °C. IR (KBr, cm−1) v 2981, 1731, 1475, 1238, 1062, 780; 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 15.6 Hz, 1H, CHCH), 7.90 (d, J = 8.4 Hz, 1H, ArH), 7.75 (d, J = 8.8 Hz, 1H, ArH), 7.52 (t, J = 7.6 Hz, 1H, ArH), 7.42 (d, J = 16.0 Hz, 1H, CHCH), 7.31 (t, J = 7.6 Hz, 1H, ArH), 7.00 (d, J = 7.6 Hz, 1H, ArH), 6.81 (t, J = 8.0 Hz, 1H, ArH), 6.63 (d, J = 8.0 Hz, 1H, ArH), 4.26–4.16 (m, 4H, CH2), 3.64 (s, 3H, CH3), 3.61 (s, 3H, CH3), 1.18–1.13 (m, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.76, 165.98, 152.85, 152.01, 148.26, 147.82, 138.63, 131.57, 130.94, 130.45, 129.63, 127.38, 125.62, 125.06, 123.78, 123.71, 122.03, 119.27, 112.34, 62.05, 61.93, 60.86, 55.58, 13.84. Anal. calcd for C25H25NO6: C, 68.95; H, 5.79; N, 3.22%. Found: C, 69.17; H, 5.83; N, 3.07%.
(E)-Diethyl 2-(2,5-dimethoxystyryl)quinoline-3,4-dicarboxylate (3i)
Yellow oily liquid, yield 66.7%; 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 16.0 Hz, 1H, CHCH), 8.01 (d, J = 8.4 Hz, 1H, ArH), 7.86 (d, J = 8.4 Hz, 1H, ArH), 7.63 (t, J = 7.6 Hz, 1H, ArH), 7.49 (d, J = 15.6 Hz, 1H, CHCH), 7.42 (t, J = 7.6 Hz, 1H, ArH), 7.03 (s, 1H, ArH), 6.72 (d, J = 7.2 Hz, 1H, ArH), 6.70 (d, J = 7.2 Hz, 1H, ArH), 4.37–4.28 (m, 4H, CH2), 3.72 (s, 3H, CH3), 3.65 (s, 3H, CH3), 1.30–1.25 (m, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.93, 166.15, 153.42, 152.42, 152.29, 148.42, 138.73, 131.96, 131.04, 129.72, 127.42, 126.19, 125.20, 125.08, 123.88, 122.13, 115.06, 113.05, 112.18, 62.18, 62.00, 56.02, 55.65, 13.99, 13.98. Anal. calcd for C25H25NO6: C, 68.95; H, 5.79; N, 3.22%. Found: C, 68.73; H, 5.64; N, 3.26%.
(E)-Diethyl 2-(3,4-dimethoxystyryl)quinoline-3,4-dicarboxylate (3j)
Brown oily liquid, yield 79.9%; 1H NMR (400 MHz, CDCl3) δ 8.13 (d, J = 8.4 Hz, 1H, ArH), 8.03 (d, J = 15.2 Hz, 1H, CHCH), 8.01 (d, J = 8.8 Hz, 1H, ArH), 7.80 (t, J = 7.6 Hz, 1H, ArH), 7.58 (t, J = 7.6 Hz, 1H, ArH), 7.47 (d, J = 15.2 Hz, 1H, CHCH), 7.23 (d, J = 8.4 Hz, 1H, ArH), 7.16 (s, 1H, ArH), 6.90 (d, J = 8.4 Hz, 1H, ArH), 4.54–4.45 (m, 4H, CH2), 3.95 (s, 3H, CH3), 3.92 (s, 3H, CH3), 1.47 (t, J = 7.2 Hz, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.99, 166.25, 152.14, 149.93, 149.02, 148.52, 139.08, 136.77, 131.28, 129.60, 129.56, 127.45, 125.36, 123.62, 122.18, 122.10, 121.29, 111.12, 109.88, 62.30, 62.11, 55.89, 55.83, 14.13, 14.08. Anal. calcd for C25H25NO6: C, 68.95; H, 5.79; N, 3.22%. Found: C, 68.69; H, 5.86; N, 3.14%.
(E)-Diethyl 2-(3,4,5-trimethoxystyryl)quinoline-3,4-dicarboxylate (3k)
Yellow oily liquid, yield 83.0%; 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.4 Hz, 1H, ArH), 8.01 (d, J = 8.0 Hz, 1H, ArH), 7.96 (d, J = 15.6 Hz, 1H, CHCH), 7.79 (t, J = 7.6 Hz, 1H, ArH), 7.58 (t, J = 7.6 Hz, 1H, ArH), 7.49 (d, J = 16.0 Hz, 1H, CHCH), 6.84 (s, 2H, ArH), 4.53–4.44 (m, 4H, CH2), 3.91 (s, 6H, CH3), 3.88 (s, 3H, CH3), 1.45 (t, J = 7.2 Hz, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.96, 166.25, 153.32, 151.85, 148.51, 139.26, 138.92, 136.90, 132.18, 131.43, 129.62, 127.69, 125.42, 123.62, 123.49, 122.31, 104.67, 62.40, 62.20, 60.98, 56.10, 14.18, 14.12. Anal. calcd for C26H27NO7: C, 67.09; H, 5.85; N, 3.01%. Found: C, 66.91; H, 6.04; N, 3.17%.
(E)-Diethyl 2-(2-chlorostyryl)quinoline-3,4-dicarboxylate (3l)
Yellow oily liquid, yield 68.5%; 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 15.6 Hz, 1H, CHCH), 8.14 (d, J = 8.4 Hz, 1H, ArH), 7.98 (d, J = 8.4 Hz, 1H, ArH), 7.77 (t, J = 8.4 Hz, 1H, ArH), 7.70 (d, J = 7.2 Hz, 1H, ArH), 7.58 (d, J = 15.6 Hz, 1H, CHCH), 7.54 (t, J = 8.4 Hz, 1H, ArH), 7.38 (d, J = 7.6 Hz, 1H, ArH), 7.24 (t, J = 7.6 Hz, 1H, ArH), 7.20 (t, J = 7.6 Hz, 1H, ArH), 4.50 (q, J = 7.6 Hz, 2H, CH2), 4.43 (q, J = 7.6 Hz, 2H, CH2), 1.42 (t, J = 7.6 Hz, 3H, CH3), 1.38 (t, J = 7.6 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.78, 166.19, 151.67, 148.50, 139.31, 134.76, 134.47, 132.88, 131.36, 129.99, 129.95, 129.58, 127.88, 127.29, 126.87, 126.84, 125.31, 123.59, 122.43, 62.33, 62.23, 14.09, 14.06. Anal. calcd for C23H20ClNO4: C, 67.40; H, 4.92; N, 3.42%. Found: C, 67.23; H, 4.79; N, 3.33%.
(E)-Diethyl 2-(3-bromostyryl)quinoline-3,4-dicarboxylate (3m)
Yellow solid, yield 80.7%, m.p. 68.9–70.8 °C. IR (KBr, cm−1) v 2981, 1722, 1255, 1185, 1027, 772; 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 8.4 Hz, 1H, ArH), 7.98 (d, J = 8.4 Hz, 1H, ArH), 7.96 (d, J = 15.6 Hz, 1H, CHCH), 7.79 (t, J = 8.0 Hz, 1H, ArH), 7.74 (s, 1H, ArH), 7.58 (d, J = 15.6 Hz, 1H, CHCH), 7.54 (d, J = 7.6 Hz, 1H, ArH), 7.51 (d, J = 8.0 Hz, 1H, ArH), 7.43 (d, J = 8.0 Hz, 1H, ArH), 7.24 (t, J = 7.2 Hz, 1H, ArH), 4.52–4.43 (m, 4H, CH2), 1.44 (t, J = 7.2 Hz, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.43, 165.92, 151.24, 148.21, 139.20, 138.40, 134.92, 131.29, 131.20, 129.97, 129.92, 129.49, 127.62, 125.96, 125.24, 125.13, 123.28, 122.59, 122.20, 62.10, 62.03, 13.91, 13.84. Anal. calcd for C23H20BrNO4: C, 60.81; H, 4.44; N, 3.08%. Found: C, 60.69; H, 4.25; N, 3.16%.
(E)-Diethyl 2-(2-nitrostyryl)quinoline-3,4-dicarboxylate (3n)
Yellow solid, yield 61.3%, m.p. 118.7–119.6 °C. IR (KBr, cm−1) v 2981, 1713, 1519, 1353, 1220, 772; 1H NMR (400 MHz, CDCl3) δ 8.43 (d, J = 16.0 Hz, 1H, CHCH), 8.14 (d, J = 8.4 Hz, 1H, ArH), 7.97 (d, J = 8.4 Hz, 1H, ArH), 7.94 (d, J = 8.0 Hz, 1H, ArH), 7.79 (d, J = 15.6 Hz, 1H, CHCH), 7.77 (t, J = 7.6 Hz, 1H, ArH), 7.61–7.55 (m, 3H, CHCH, ArH), 7.45 (t, J = 7.6 Hz, 1H, ArH), 4.50 (q, J = 7.2 Hz, 2H, CH2), 4.44 (q, J = 7.2 Hz, 2H, CH2), 1.41 (t, J = 7.6 Hz, 3H, CH3), 1.37 (t, J = 7.6 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.30, 165.87, 150.78, 148.25, 148.20, 139.35, 132.80, 132.10, 131.44, 131.25, 129.83, 128.81, 128.63, 128.51, 127.88, 125.02, 124.41, 123.02, 122.29, 62.06, 62.00, 13.81, 13.73. Anal. calcd for C23H20N2O6: C, 65.71; H, 4.79; N, 6.66%. Found: C, 65.48; H, 4.84; N, 6.52%.
(E)-Diethyl 2-(4-nitrostyryl)quinoline-3,4-dicarboxylate (3o)
Yellow solid, yield 85.8%, m.p. 161.5–162.3 °C. IR (KBr, cm−1) v 2990, 1731, 1519, 1335; 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 8.4 Hz, 2H, ArH), 8.12 (d, J = 8.4 Hz, 1H, ArH), 8.06 (d, J = 15.6 Hz, 1H, CHCH), 7.98 (d, J = 8.4 Hz, 1H, ArH), 7.80 (t, J = 8.4 Hz, 1H, ArH), 7.78 (d, J = 15.6 Hz, 1H, CHCH), 7.70 (d, J = 8.4 Hz, 2H, ArH), 7.60 (t, J = 8.0 Hz, 1H, ArH), 4.52–4.42 (m, 4H, CH2), 1.44–1.38 (m, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.48, 166.07, 151.00, 148.47, 147.48, 142.85, 139.84, 134.05, 131.69, 129.83, 128.42, 128.28, 127.98, 125.46, 124.06, 123.43, 122.68, 62.43, 62.39, 14.09. Anal. calcd for C23H20N2O6: C, 65.71; H, 4.79; N, 6.66%. Found: C, 65.64; H, 4.82; N, 6.81%.
(E)-Diethyl 2-(2-(2-chloroquinolin-3-yl)vinyl)quinoline-3,4-dicarboxylate (3p)
Yellow solid, yield 53.4%, m.p. 143.8–144.2 °C. IR (KBr, cm−1) v 2981, 1731, 1246, 1194, 1045, 737; 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1H, ArH), 8.43 (d, J = 15.6 Hz, 1H, CHCH), 8.22 (d, J = 8.8 Hz, 1H, ArH), 8.03 (d, J = 8.4 Hz, 2H, ArH), 7.89 (d, J = 8.0 Hz, 1H, ArH), 7.85 (t, J = 8.0 Hz, 1H, ArH), 7.78 (d, J = 16.0 Hz, 1H, CHCH), 7.75 (t, J = 8.0 Hz, 1H, ArH), 7.64 (t, J = 8.0 Hz, 1H, ArH), 7.59 (t, J = 8.0 Hz, 1H, ArH), 4.56 (q, J = 7.2 Hz, 2H, CH2), 4.51 (q, J = 7.2 Hz, 2H, CH2), 1.48 (t, J = 7.2 Hz, 3H, CH3), 1.45 (t, J = 7.2 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 166.64, 166.14, 151.16, 150.46, 148.51, 147.23, 139.66, 135.04, 131.68, 131.58, 130.69, 130.04, 129.65, 128.61, 128.29, 128.16, 127.77, 127.30, 127.26, 125.36, 123.29, 122.58, 62.37, 62.34, 14.10, 14.03. Anal. calcd for C26H21ClN2O4: C, 67.75; H, 4.59; N, 6.08%. Found: C, 67.86; H, 4.62; N, 5.88%.
(E)-Diethyl 2-(2-(2-chloro-6-methylquinolin-3-yl)vinyl)quinoline-3,4-dicarboxylate (3q)
Yellow solid, yield 51.1%, m.p. 158.6–161.4 °C. IR (KBr, cm−1) v 2981, 1731, 1379, 1246; 1H NMR (400 MHz, CDCl3) δ 8.46 (d, J = 15.6 Hz, 1H, CHCH), 8.38 (s, 1H, ArH), 8.23 (d, J = 8.4 Hz, 1H, ArH), 8.04 (d, J = 8.0 Hz, 1H, ArH), 7.92 (d, J = 8.4 Hz, 1H, ArH), 7.86 (t, J = 8.0 Hz, 1H, ArH), 7.76 (d, J = 15.6 Hz, 1H, CHCH), 7.64 (s, 1H, ArH), 7.64 (t, J = 7.2 Hz, 1H, ArH), 7.58 (d, J = 8.4 Hz, 1H, ArH), 4.57–4.46 (m, 4H, CH2), 2.55 (s, 3H, CH3), 1.49–1.42 (m, 6H, CH3). 13C NMR (100 MHz, CDCl3) δ 161.51, 161.02, 146.10, 144.44, 143.32, 140.74, 134.53, 132.22, 129.34, 127.91, 126.81, 126.48, 124.86, 124.35, 123.19, 123.03, 122.81, 122.17, 121.47, 120.23, 118.18, 117.43, 57.27, 57.22, 16.44, 8.98, 8.93. Anal. calcd for C27H23ClN2O4: C, 68.28; H, 4.88; N, 5.90%. Found: C, 68.47; H, 4.63; N, 5.93%.
Experimental procedure for cancer cell growth inhibition assay (MTT assay)
The anti-proliferative activity of the target compounds on the human non-small cell lung cancer cell (A549), human colon cancer cell (HT29) and human bladder carcinoma cells (T24) was tested using the MTT assay, in comparison to cisplatin. A549, HT29 and T24 cell lines were obtained from the Cell Resource Center (Shanghai Institutes for Biological Sciences, China Academy of Sciences). Briefly, the three cell types were seeded in a 96-well culture plate at the cell density of 5 × 104 cells per well in 100 μL of culture medium at 37 °C in a 5% CO2 incubator for 24 h seeding. The stock solutions of test compounds 3a–q were prepared in DMSO. After incubation, the target compounds were added to the culture medium at five times the concentration gradient. The final concentration of DMSO in the medium was less than 0.5%. Triplicates of each concentration were used. After 48 h incubation, the supernatant was removed and 5 mg mL−1 of a freshly prepared solution of MTT was added to each well. The plates were then incubated with the cells at 37 °C for another 4 h. The medium was then removed and 100 μL DMSO was added to each well to dissolve the formazan. The OD values were measured using SPECTRA max 190 Cell microplate reader under 490 nm (for absorbance of MTT formazan) and 630 nm (for the reference wavelength). The cell growth inhibition rate formula is (AC − AT)/AC × 100%. AC is the absorbance value of the blank control group; AT is the absorbance value of the experimental group. The average 50% inhibitory concentration (IC50) was calculated using GraphPadPrism version 6.00 software from the non-linear curve.Consent to participate
All authors participated directly in the current research work.Consent to publish
The authors agree to publish the article under the Creative Commons Attribution License.Data availability
All relevant data are within the manuscript and available from the corresponding author upon request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to thank the Scientific Research Foundation of the Education Department of Liaoning Province (Grant no. JTTMS1618) and Open Fund of Key Laboratory of Biotechnology and Bioresources Utilization (Dalian Minzu University), Ministry of Education (no. KF2024009) for financial support.References
- M. Dhanawat, D. K. Mehta and R. Das, Mini-Rev. Med. Chem., 2021, 21, 1849–1864 CrossRef CAS PubMed.
- F. S. Chang, W. Chen, C. Wang, C. C. Tzeng and Y. L. Chen, Bioorg. Med. Chem., 2010, 18, 124–133 CrossRef CAS PubMed.
- J. Szczepaniak, W. Cieslik, A. Romanowicz, R. Musioł and A. Krasowska, Int. J. Antimicrob. Agents, 2017, 50, 171–176 CrossRef CAS PubMed.
- R. Musiol, Curr. Pharm. Des., 2013, 19, 1835–1849 CrossRef CAS PubMed.
- A. Kamal, A. Rahim, S. Riyaz, Y. Poornachandra, M. Balakrishna, C. G. Kumar, S. M. A. Hussaini, B. Sridhar and P. K. Machiraju, Org. Biomol. Chem., 2015, 13, 1347–1357 RSC.
- M. E. Zwaagstra, S. H. Schoenmakers, P. H. Nederkoorn, E. Gelens, H. Timmerman and M. Q. Zhang, J. Med. Chem., 1998, 41, 1439–1445 CrossRef CAS PubMed.
- A. Mrozek-Wilczkiewicz, M. Kuczak, K. Malarz, W. Cieślik, E. Spaczynska and R. Musiol, Eur. J. Med. Chem., 2019, 177, 338–349 CrossRef CAS PubMed.
- R. K. Mittal and P. urohit, Anti-Cancer Agents Med. Chem., 2020, 20, 1981–1991 CrossRef CAS PubMed.
- R. Musiol, J. Jampilek, K. Kralova, D. R. Richardson, D. Kalinowski, B. Podeszwa, J. Finster, H. Niedbala, A. Palka and J. Polanski, Bioorg. Med. Chem., 2007, 15, 1280–1288 CrossRef CAS PubMed.
- S. Mirzaei, F. Eisvand, F. Hadizadeh, F. Mosaffa, A. Ghasemi and R. Ghodsi, Bioorg. Chem., 2020, 98, 103711 CrossRef CAS PubMed.
- M. A. Fakhfakh, X. Franck, A. Fournet, R. Hocquemiller and B. Figadere, Synth. Commun., 2002, 32, 2863–2875 CrossRef CAS.
- H. Xia, Y. Liu, P. Zhao, S. Gou and J. Wang, Organ. Lett., 2016, 18, 1796–1799 CrossRef CAS PubMed.
- D. S. Latha and S. Yaragorla, Eur. J. Org Chem., 2020, 2155–2179 CrossRef CAS.
- M. V. Rubtsov, G. N. Pershin, N. A. Yanbuktin, L. A. Pelenitsina, T. J. Gurevich, N. A. Novitskaya, S. N. Milovanova and S. A. Vichkanova, J. Med. Chem., 1959, 2, 113–131 CrossRef PubMed.
- D. Mao, G. Hong, S. Wu, X. Liu, J. Yu and L. Wang, Eur. J. Org Chem., 2014, 3009–3019 CrossRef CAS.
- H. T. Dang, T. N. Lieu, T. Truong and N. T. Phan, J. Mol. Catal. A: Chem., 2016, 420, 237–245 CrossRef CAS.
- D. Pi, K. Jiang, H. Zhou, Y. Sui, Y. Uozumi and K. Zou, RSC Adv., 2014, 4, 57875–57884 RSC.
- Z. Jamal, Y. C. Teo and G. S. Lim, Tetrahedron, 2016, 72, 2132–2138 CrossRef CAS.
- Z. Jamal and Y. C. Teo, Synlett, 2014, 25, 2049–2053 CrossRef CAS.
- S. Yaragorla, G. Singh and R. Dada, Tetrahedron Lett., 2015, 56, 5924–5929 CrossRef CAS.
- V. M. Li, T. N. Gavrishova and M. F. Budyka, Russ. J. Org. Chem., 2012, 48, 823–828 CrossRef CAS.
- R. Musiol, B. Podeszwa, J. Finster, H. Niedbala and J. Polanski, Monatsh. Chem., 2006, 137, 1211–1217 CrossRef CAS.
- L. Xu, Z. Shao, L. Wang, H. Zhao and J. Xiao, Tetrahedron Lett., 2014, 55, 6856–6860 CrossRef CAS.
- E. Liang, J. Wang, Y. Wu, L. Huang, X. Yao and X. Tang, Adv. Synth. Catal., 2019, 361, 3619–3623 CrossRef CAS.
- M. Dabiri, P. Salehi, M. Baghbanzadeh and M. S. Nikcheh, Tetrahedron Lett., 2008, 49, 5366–5368 CrossRef CAS.
- P. Sarma, S. Saikia and R. Borah, Synth. Commun., 2016, 46, 1187–1196 CrossRef CAS.
- S. Fu, L. Wang, H. Dong, J. Yu, L. Xu and J. Xiao, Tetrahedron Lett., 2016, 57, 4533–4536 CrossRef CAS.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- D. A. Alonso, A. Baeza, R. Chinchilla, G. Guillena, I. M. Pastor and D. J. Ramón, Eur. J. Org Chem., 2016, 612–632 CrossRef CAS.
- M. Zhang, Y. H. Liu, Z. R. Shang, H. C. Hu and Z. H. Zhang, Catal. Commun., 2017, 88, 39–44 CrossRef CAS.
- W. H. Zhang, M. N. Chen, Y. Hao, X. Jiang, X. L. Zhou and Z. H. Zhang, J. Mol. Liq., 2019, 278, 124–129 CrossRef CAS.
- L. Xu, W. H. Zhang, Z. S. Cui and Z. H. Zhang, Curr. Organocatal., 2021, 8, 249–257 CrossRef CAS.
- H. C. Hu, Y. H. Liu, B. L. Li, Z. S. Cui and Z. H. Zhang, RSC Adv., 2015, 5, 7720–7728 RSC.
- P. Wang, F. P. Ma and Z. H. Zhang, J. Mol. Liq., 2014, 198, 259–262 CrossRef CAS.
- N. Satyanarayana, K. Sathish, S. Nagaraju, R. Pawar, M. Faizan, M. Arumugavel, T. Shirisha and D. Kashinath, New J. Chem., 2022, 46, 1637–1642 RSC.
- Y. Li, J. D. Yuan and D. Zhao, Chin. J. Org. Chem., 2023, 43, 3268–3276 CrossRef CAS.
- S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed.
- S. K. Gadakh, S. Dey and A. Sudalai, Org. Biomol. Chem., 2016, 14, 2969–2977 RSC.
- J. J. Kalbfleisch, C. W. Reed, C. Park, P. K. Spearing, M. C. Quitalig, M. T. Jenkins, A. L. Rodriguez, A. L. Blobaum, P. J. Conn, C. M. Niswender and C. W. Lindsley, Bioorg. Med. Chem. Lett., 2020, 30, 127529 CrossRef CAS PubMed.
- R. K. Mittal and P. Purohit, Anti-Cancer Agents Med. Chem., 2021, 21, 1708–1716 CrossRef CAS PubMed.
- L. Lu, P. Zhou, B. Hu, X. Li, R. Huang and F. Yu, Tetrahedron Lett., 2017, 58, 3658–3661 CrossRef CAS.
- F. R. B. Bokosi, R. M. Beteck, M. Mbaba, T. E. Mtshare, D. Laming, H. C. Hoppe and S. D. Khanye, Bioorg. Med. Chem. Lett., 2021, 38, 127855 CrossRef CAS PubMed.
- S. Alvi and R. Ali, Org. Biomol. Chem., 2021, 19, 9732–9745 RSC.
- V. Krishnakumar, N. G. Vindhya, B. K. Mandal and F. R. Nawaz Khan, Ind. Eng. Chem. Res., 2014, 53, 10814–10819 CrossRef CAS.
- S. Gore, S. Baskaran and B. Koenig, Green Chem., 2011, 13, 1009–1013 RSC.
- C. Borrel, S. Thoret, X. Cachet, D. Guénard, F. Tillequin, M. Kocha and S. Michel, Bioorg. Med. Chem., 2005, 13, 3853–3864 CrossRef CAS PubMed.
- A. Andreani, S. Burnelli, M. Granaiola, A. Leoni, A. Locatelli, R. Morigi, M. Rambaldi, L. Varoli and M. W. Kunkel, J. Med. Chem., 2006, 49, 6922–6924 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04588b |
| This journal is © The Royal Society of Chemistry 2024 |