
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of an eco-friendly capillary electrophoresis method for the simultaneous determination of piperacillin, tazobactam and ibuprofen in plasma samples: application to a pharmacokinetic study in rats
Weam M. Othman *a,
Nourah Z. Al-Zomanb,
Ibrahim A. Darwishb,
Aliyah Almomenb,
Samah S. Saada,
Fatma F. Abdallahc and
Nehal F. Faridc
*a,
Nourah Z. Al-Zomanb,
Ibrahim A. Darwishb,
Aliyah Almomenb,
Samah S. Saada,
Fatma F. Abdallahc and
Nehal F. Faridc
aPharmaceutical Analytical Chemistry Department, Faculty of Pharmacy, Misr University for Science and Technology, 6th October City, Egypt. E-mail: weam.mahmoud@must.edu.eg; Dr.weamelfergan@gmail.com
bDepartment of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia
cPharmaceutical Analytical Chemistry Department, Faculty of Pharmacy, Beni-Suef University, Egypt
First published on 24th July 2024
Abstract
Piperacillin (PIP) and tazobactam (TAZ) are broad-spectrum beta-lactam antimicrobial agents, which are frequently co-prescribed in intensive care units (ICUs) worldwide. Ibuprofen (IBU) is a potent pain killer which is commonly co-prescribed with PIP and TAZ postoperatively. The combination therapy of PIP, TAZ, and IBU has been administered commonly after surgical procedures to combat aerobic and anaerobic microbes and exert anti-inflammatory and analgesic effects. This study describes, for the first time, the development of a new capillary electrophoresis (CE) method with a photodiode array detector for the simultaneous determination of PIP, TAZ, and IBU in plasma samples. The experimental factors affecting the elution of analytes were carefully optimized. The final analysis was achieved using a fused silica capillary (58 cm effective length and 75 μm ID) and a background electrolyte solution containing a methanol/borax buffer solution (15 mM and pH 9.3) in a ratio of (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 v/v) with a driving voltage of 30 kV and detection at 210 nm. The relationship between the peak area and concentration was linear from 1 to 200 μg mL−1 for both PIP and TAZ and from 3 to 200 μg mL−1 for TAZ. The method used was thoroughly validated in accordance with the validation requirements set out by the Food and Drug Administration (FDA) for bio-analytical processes. The proposed CE method was employed to conduct pharmacokinetic and bioavailability studies of the drugs in rat models. The pharmacokinetic results revealed that there is a significant impact upon prescribing this combination concurrently when compared to their single administration. To illustrate, the time required to reach their maximum concentrations (Tmax) was increased by 0.25 h for both PIP and TAZ, whereas it was increased by 0.5 for IBU. When it comes to their maximum concentration (Cmax), it was increased by 13.7%, 55.5%, and 44% for PIP, TAZ, and IBU, respectively. Furthermore, the bioavailabilities of PIP, TAZ, and IBU were significantly increased by 55.4%, 19.7%, and 35.6%, respectively. These findings require caution when these drugs are co-prescribed as there is a noticeable augmentation in their therapeutic impacts. Additionally, the greenness of the proposed method was assessed by three metric tools. In conclusion, the method is a valuable tool for further studies on drug–drug interaction in humans.
:90 v/v) with a driving voltage of 30 kV and detection at 210 nm. The relationship between the peak area and concentration was linear from 1 to 200 μg mL−1 for both PIP and TAZ and from 3 to 200 μg mL−1 for TAZ. The method used was thoroughly validated in accordance with the validation requirements set out by the Food and Drug Administration (FDA) for bio-analytical processes. The proposed CE method was employed to conduct pharmacokinetic and bioavailability studies of the drugs in rat models. The pharmacokinetic results revealed that there is a significant impact upon prescribing this combination concurrently when compared to their single administration. To illustrate, the time required to reach their maximum concentrations (Tmax) was increased by 0.25 h for both PIP and TAZ, whereas it was increased by 0.5 for IBU. When it comes to their maximum concentration (Cmax), it was increased by 13.7%, 55.5%, and 44% for PIP, TAZ, and IBU, respectively. Furthermore, the bioavailabilities of PIP, TAZ, and IBU were significantly increased by 55.4%, 19.7%, and 35.6%, respectively. These findings require caution when these drugs are co-prescribed as there is a noticeable augmentation in their therapeutic impacts. Additionally, the greenness of the proposed method was assessed by three metric tools. In conclusion, the method is a valuable tool for further studies on drug–drug interaction in humans.
1. Introduction
Piperacillin (PIP) is a fourth-generation broad-spectrum β-lactam antimicrobial agent, which is frequently prescribed in intensive care units (ICUs) worldwide to manage sepsis, especially for patients with related organ dysfunction.1–3 The combination of a β-lactamase inhibitor, tazobactam (TAZ), with PIP greatly enhances PIP's activity against β-lactamase-producing bacteria [https://www.mdpi.com/2079-6382/12/2/321]. The combination therapy of PIP and TAZ is marketed as an 8:1 fixed-dose combination.4 Inflammation and pain are the core symptoms in infected patients;5,6 therefore, analgesics and anti-inflammatory agents are commonly prescribed concomitantly with PIP/TAZ therapy.7 Ibuprofen (IBU) is a nonsteroidal anti-inflammatory drug (NSAID) commonly prescribed for the management of pain and inflammation. The FDA proposed IBU as a better NSAID to manage infected outpatients and prevent complications.8 It is a nonselective blocker of cyclooxygenase enzyme, and it is frequently prescribed for children with diverse therapeutic indications, including fever, postoperative pain, tumors, and inflammatory disorders, such as juvenile idiopathic arthritis.9 The chemical structures of PIP, TAZ and IBU are given in Fig. 1.
 | ||
| Fig. 1 Chemical structures of piperacillin (PIP), tazobactam (TAZ) and ibuprofen (IBU). | ||
PIP and TAZ were determined by several chromatographic methods either individually or in combination with other drugs. These methods include HPLC-UV,1,10–12 UPLC-MS/MS,13,14 and capillary electrophoresis (CE).15 Additionally, IBU was determined by numerous analytical techniques either individually or in combination with other drugs, such as HPLC-UV,16 HPLC-MS/MS,17,18 GC-MS,19 GC,20 LC-MS/MS,21,22 TLC,23 spectrofluorimetry,24 and spectrophotometry.25 All these techniques were developed for the individual determination of PIP, TAZ and IBU; however, there is no published method for their simultaneous determination. Due to the popular use of the combination of PIP-TAZ-IBU for in-patients26 and post operatively,27,28 it is of utmost importance to study the influence of each drug when concurrently prescribed with the other one through a full pharmacokinetic study. Given that the proposed CE method will be the first innovative approach to be followed by a comprehensive pharmacokinetic study to demonstrate the therapeutic impact of the three medications on one another. By gathering information for the determination of the three drugs in biological fluids, PIP and TAZ were determined in human plasma by HPLC-UV,10,12 human plasma and urine by UPLC-MS/MS,13 human plasma and pleural fluid by LC-MS/MS,29 and rat plasma by LC-MS/MS.30 IBU was detected in human plasma by HPLC-MS/MS,17,18 human plasma by LC-MS/MS,21,22 human plasma by TLC,23 human plasma and urine by GC-MS,19 rabbit plasma by GC,20 mouse plasma and tissue by HPLC-FLU,31 and rat plasma by HPLC.32 All the above-mentioned methods were applied for the determination of each drug individually.
CE is now utilized for the investigation of different aspects of pharmacokinetics because of its unique features including high resolving power, versatility, short analysis time, low consumption of chemicals and samples, small sample volume for cell-based investigations, and fast and simplified sample processing, especially when compared with methods that require tedious sample preparation and sample stacking for increasing the detection sensitivity.33 This research demonstrates, for the first time, the detailed description for the development and validation of a new ecologically friendly CE method equipped with a diode array detector for the simultaneous quantification of PIP, TAZ and IBU in rat plasma samples followed by a full pharmacokinetic profile. The proposed method combined numerous privileges including a simple and straightforward one-step protein precipitation method for preparing plasma samples, the reduction of sample size, the use of a minimal number of solvents, and a short analysis time (<12 min). The method's straightforward extraction procedure and quick run time contribute to its high throughput, enabling the efficient processing of diverse samples in several clinical laboratories.
Studying the pharmacokinetics of drugs provides critical data on the behavior of molecules within organisms and aids in providing more effective and safer therapies. In the proposed CE approach, the pharmacokinetic findings of the three drugs were compared when administered singly and when administered concurrently. The latter demonstrated the accumulative impact of co-administering the drugs in terms of rapid onset of action with a maximized concentration. This necessitates caution when prescribing the compounds together, as there is a significant change in their bioavailabilities. The entire pharmacokinetic study's data can be employed for therapeutic drug monitoring and as a foundation for future researchers who are interested in conducting additional studies on drug–drug interactions in humans.
In addition, the proposed analytical procedure was evaluated using three green metric tools: Analytical Eco-scale (ESA), Green Analytical Procedure Index (GAPI), and Analytical GREEnness Calculator (AGREE). By the ESA tool, the proposed method scored 89 out of 100. The GAPI pictogram demonstrated 7 green parameters, 7 yellow parameters, and only 1 red parameter. By the AGREE tool, the method scored 0.77 out of 1. These results indicated that the CE procedure had a minimal environmental impact while maintaining the analytical parameters.
2. Experimental
2.1. Instrumentation and electrophoretic conditions
This investigation employed a capillary electrophoresis instrument manufactured by Agilent in Germany, which has a PDA detector adjusted at 210 nm. Data processing was performed using the Agilent ChemStation software. A deactivated fused silica capillary (Agilent Technologies, Santa Clara, CA) was employed for electrophoretic analysis. The capillary measured 66 cm in total length, 58 cm in effective length, and 75 μm in internal diameter. Both the capillary and the sample were maintained at a temperature of 24 °C. A background electrolyte (BGE) solution containing a methanol/borax buffer solution (15 mM, pH 9.3) in a ratio of 10/90 v/v was used to perform separations. Samples have been injected into the capillary through the anodic side under a pressure of 40 mbar for 30 seconds. In order to conduct electrophoresis, a 30 kV potential was introduced into the capillary, with the cathode positioned at the detector end. To assure the analysis's reproducibility, the capillary was rinsed for two minutes among runs with deionized water and equilibrated for an additional four minutes with a running buffer. The capillary was conditioned with 0.1 M sodium hydroxide for 5 minutes, deionized water for 5 minutes, and a running buffer for 10 minutes prior to sample injection. Prior to analysis, each solution was filtered through 0.2 μm Millipore membrane filters (Millipore, Nihon, Japan).2.2. Materials used and reagents
Reference standard materials of TAZ and PIP were purchased from Fluka, Sigma Aldrich (GmbH, Germany), with a purity >99%. IBU was obtained from Amgen Inc. (Thousand Oaks, CA, USA). The internal standard (IS) Paracetamol (PAR) was obtained from Jigs Chemical Limited (Ellisbridge, Ahmedabad) with a purity of 99%. Acetonitrile of analytical grade (Sigma-Aldrich, St Louis, MO, USA) was used for protein precipitation. Analytical grade borax was purchased from Winlab (Leicestershire, UK) and sodium hydroxide was obtained from Merck (Darmstadt, Germany). All solvents and other materials used throughout this study were of analytical grade. The investigations were conducted using deionized water and membrane filters (0.2 μm) obtained from Nihon, Millipore (Yonnezawa, Japan).2.3. Standard solutions
Stock standard solutions containing each of TAZ, PIP, IBU and PAR (IS) (1 mg mL−1) were prepared separately in four separate 10 mL calibrated flasks in methanol. These stock solutions were further diluted with the BGE to obtain 10 mL of working standards of 0.5 mg mL−1 of each compound. When stored in a refrigerator, the methanolic stock solutions remained stable for a minimum of three weeks.2.4. Construction of calibration curves and method validation
Analytes of different concentrations in the range of 1–200 μg mL−1 were prepared from the working standard solutions (0.5 mg mL−1) of PIP, TAZ and IBU in three series of test tubes. Each sample solution was mixed with 200 μL of PAR (IS) from 0.5 mg mL−1 standard working solution, and the volume was then adjusted to 1 mL with the BGE and injected into the CE in triplicate under the above-mentioned optimized conditions. Subsequently, calibration curves were generated for the pure samples by using these prepared samples. In the in vitro experiments, calibration standards were created by properly transferring different volumes of the medications being tested from their working standard solutions (0.5 mg mL−1) into two separate sets of test tubes. Spiking of each sample was performed with 200 μL blank plasma and 100 μL of working IS solution (0.5 mg mL−1), and then vortexed for 1 min. To each sample, 1 mL acetonitrile was added for plasma protein precipitation by centrifugation at 5000 rpm for 10 min.A similar procedure was used for obtaining the quality control samples, which are low-, medium- and high-level quality control samples (50, 100, and 150 μg mL−1 for PIP, TAZ and IBU). Until the analysis time, all samples and prepared solutions were kept at −20 °C in the refrigerator for storage.
For each drug, two calibration curves were generated: one for spiked plasma samples and the other for pure standards. After preparing multiple samples, they were analyzed in triplicate in accordance with the prescribed methodology and the operating conditions of the electrophoresis instruments. The corrected peak area was calculated using the following formula: peak divided of analyte/analyte migration time. To generate the calibration curves, the corrected peak area ratios, calculated as the corrected peak areas of analyte/corrected peak areas of IS, were plotted as a function of analyte concentrations. The data set was analyzed by regression analysis, and the linear fitting equations were derived, along with their parameters (intercepts, slopes, and determination coefficients). For calculations of sample solutions, the automatic integration system (data acquisition and processing software) was used. The percentage of a sample analyte was calculated by determining the peak area as a percentage of the total corrected areas of all peaks, excluding those due to BGE. The quality control samples that had been previously generated were analyzed by the proposed method. To determine the concentrations, regression equations were applied to the injected plasma calibration curves. The results were subsequently utilized to validate the method according to the US-FDA criteria for Industry and Bioanalytical Method Validation.34
2.5. Pharmacokinetic study
For the experiment, 28 Wistar albino rats (250 ± 30 g) were divided into four groups (7 rats per group), and all rats were housed under the conditions of 25 °C and 50% relative humidity. The rats received the following treatment through intraperitoneal injection: group I received 160/40 mg kg−1 of PIP/TAZ dissolved in dimethyl sulfoxide-saline (5%, v/v),35 group II received 30 mg kg−1 of IBU dissolved in dimethyl sulfoxide–saline (5%, v/v),36 group III received mixed doses of 160/40 mg kg−1 PIP/TAZ and 30 mg per kg IBU, while group IV served as the control. Blood samples (∼400 μL) were collected from each rat from the tail vein and transferred into heparinized polythene tubes at predetermined time points of 0.25, 0.5, 0.45, 1, 1.5, 2, 3, 5 and 8 h after drug administration. Plasma was extracted from blood samples by centrifuging them at 5000 rpm for 30 minutes. Plasma samples were obtained and stored in a refrigerated state at −20 °C until the analysis was performed. Protein precipitation using acetonitrile was used to extract the proposed drugs from the plasma samples. Then, 200 μL of each sample was prepared in a clean centrifuge tube and spiking was performed with 100 μL of the IS's working solution (0.5 mg mL−1). Following a 1 min vortex mixing step, a plasma protein precipitant (1 mL of acetonitrile) was added, followed by precipitation at 5000 rpm for separating the plasma protein. After conducting three distinct analyses of the samples according to the methodology specified, plasma concentration–time curves were constructed, and subsequently, all pharmacokinetic parameters were calculated.3. Results and discussion
3.1. Optimization of the electrophoretic parameters
An investigation was conducted to examine the parameters that influence the electrophoretic separation of the analytes. The aim of the study was to obtain sharp and symmetrical peaks of PIP, TAZ, and IBU, with a higher signal-to-noise ratio. To assess the purity of the peaks, the absorption spectra of the analyte peaks were compared to those of standard solutions of each analyte using the diode array detector. The analysis was performed using a detection wavelength of 210 nm as it resulted in the highest response for all components.For CE analysis, a variety of circulating buffers at various molarities and pH levels, including phosphate, acetate, and borate, were evaluated. The overlapping of PIP and TAZ could not be resolved by employing phosphate and acetate buffers (pH ranges from 7 to 9.3). The investigation involved using a borate buffer, which exhibited a pH range of 7 to 10. It was observed that complete separation between the medications did not occur for values below 9.3; nevertheless, peaks that were not completely separated at values of pH 10, and the TAZ peak was eluted after a duration of 20 minutes, as demonstrated in Fig. 2(A). It was noticed that a borate buffer with a pH value of 9.3 exhibited sharp and symmetric peaks, indicating optimal separation, and consequently, it was selected as the final background electrolyte considering the additional parameters to be optimized (resolution, migration time, peak shape and height, baseline noise, and electric current generated).
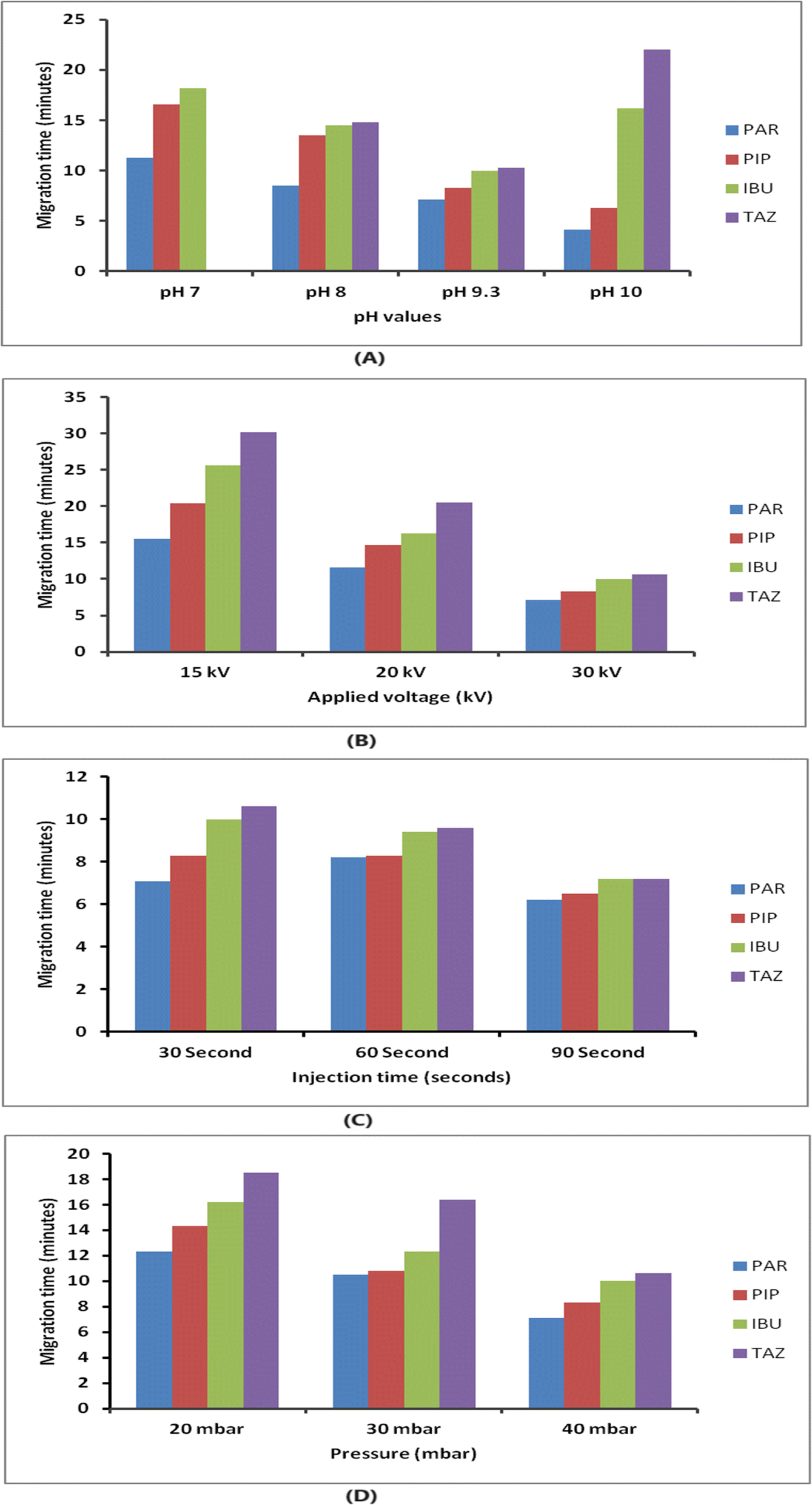 | ||
| Fig. 2 (A) Effect of borate buffer at different pH values on the compounds' migration time. (B) Effect of applied voltage (kV) on the compounds' migration time. (C) Effect of injection time (seconds) on the compounds' migration time. (D) Effect of pressure (mbar) on the compounds' migration time. | ||
The impact of borate concentration on the separation was examined by varying the amounts from 5 to 50 mM at a pH of 9.3. Buffer concentrations below 15 mM led to a notable overlap between PIP and IBU. Borate buffers at a concentration of 15 mM or more exhibited enhanced separation efficiency. The analysis was conducted with a 15 mM borate buffer with a pH of 9.3 due to its ability to generate analyte peaks that are both well-resolved and symmetrical within an acceptable time of analysis.
The applied pressure was systematically adjusted within the range of 20 to 40 mbar in order to assess its impact on the separation process, as depicted in Fig. 2(D). Raising the pressure led to improved efficiency and reduced migration time. For subsequent analysis, a pressure of 40 mbar and an injection period of 30 seconds were chosen. These parameters were picked because they offered a balance between achieving the best separation, obtaining well-defined peak shapes, and keeping the analysis time within ten minutes.
In summary, the separation of the analytes was successfully accomplished by employing a fused silica capillary with an effective length of 58 cm and an ID of 75 mm. The background electrolyte employed was a mixture of borax buffer (15 mM, pH 9.3) and methanol (90:10, v/v) at a temperature of 25 °C. This information is presented in Table 1. The ideal separation was achieved by employing a voltage of 30 kV, a pressure of 40 mbar, and an injection duration of 30 seconds.
| Parameter investigated | Range | Optimum value |
|---|---|---|
| Buffer pH | 7 to 10 | 9.3 |
| Buffer concentration (mM) | 5 to 50 mM | 15 mM |
| Organic modifier | 5 to 20% | 10% |
| Applied voltage (kV) | 15 to 30 kV | 30 kV |
| Injection time (seconds) | 30 to 90 seconds | 30 seconds |
| Pressure (mbar) | 20 to 40 mbar | 40 mbar |
| Temperature (°C) | 15 to 50 °C | 25 °C |
Fig. 3 displays the standard electropherogram of a mixture of PIP, TAZ, and IBU, which was obtained using the optimized approach. The resulting peaks exhibited optimum sharpness and demonstrated complete baseline separation. The employed methodology achieved sufficient resolution of the components within an acceptable time for analysis. The PIP, IBU, and TAZ elution times were 8.3 ± 0.551, 10 ± 0.451, and 10.6 ± 0.843 min, respectively. Table 2 displays the system parameters. The use of the CE approach enabled sufficient resolution within appropriate capacity factors. Furthermore, the presence of a significant number of theoretical plates indicates the great efficiency of CE. The degree of peak asymmetry was assessed by calculating the tailing factor, and it was not exceeding the crucial value (1.12), which verified the permitted level of peak asymmetry.
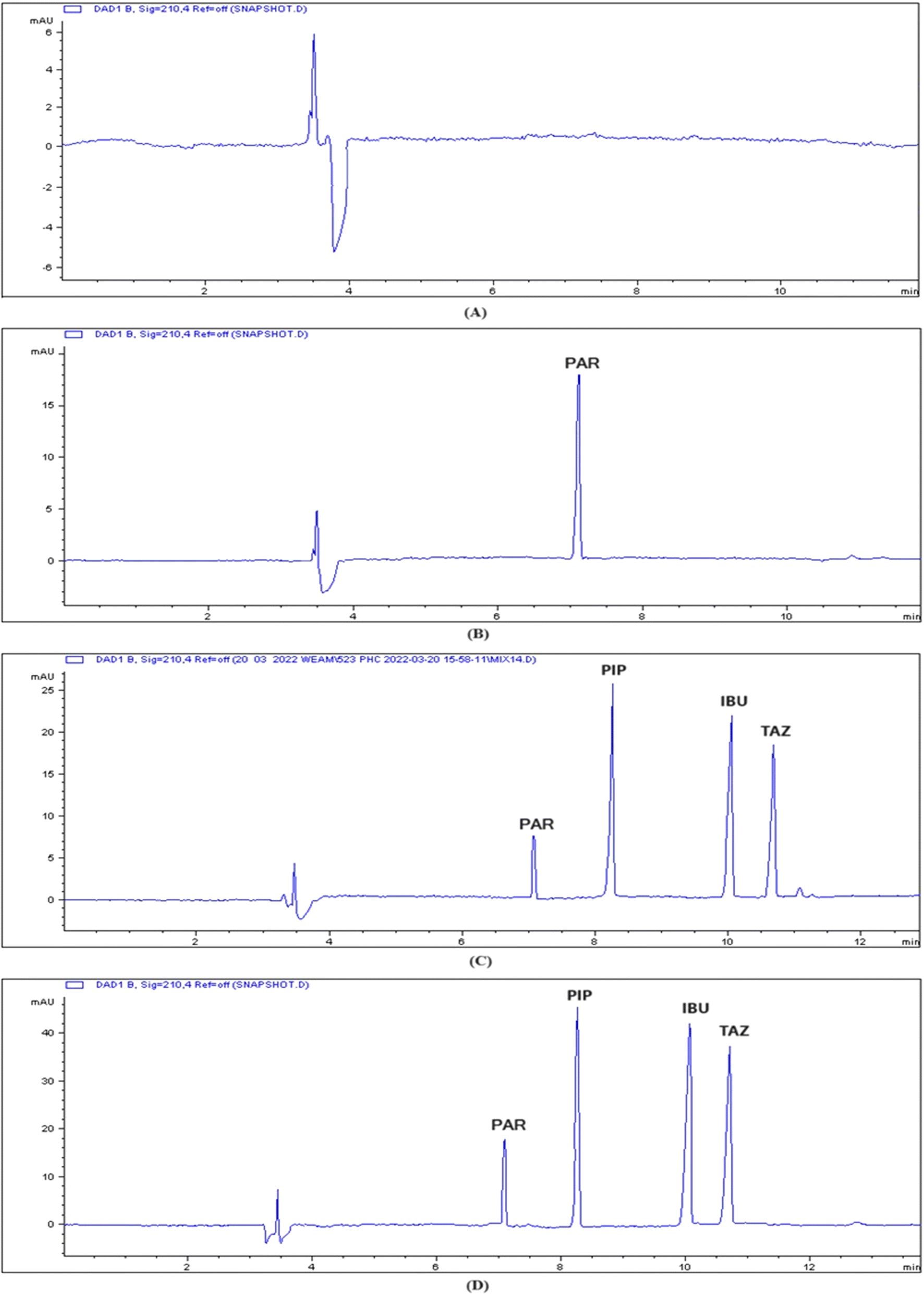 | ||
| Fig. 3 Capillary electropherogram of (A) blank plasma; (B) plasma sample spiked with 50 μg per mL IS, and scanning at 210 nm; (C) plasma sample spiked with 50 μg per mL IS, 60 μg per mL PIP, 50 μg per mL IBU, 20 μg per mL TAZ, and scanning at 210 nm; (D) real rat plasma sample spiked with 50 μg per mL IS and elution of the studied drugs PIP, IBU, and TAZ, and scanning at 210 nm. | ||
| Analyte | Migration time (min) | Number of theoretical plates | Capacity factor (1–10) | Selectivity (>1) | Resolution (>1.5) | Tailing factor (0.9–1.2) |
|---|---|---|---|---|---|---|
| PAR | 7.1 ± 0.756 | 17203 |
1.03 | 1.10 | ||
| 1.33 | 6 | |||||
| PIP | 8.3 ± 0.551 | 15876 |
1.37 | 1.12 | ||
| 1.35 | 11.3 | |||||
| IBU | 10 ± 0.451 | 16796 |
1.85 | 1.02 | ||
| 1.09 | 4 | |||||
| TAZ | 10.6 ± 0.843 | 14231 |
2.03 | 1.11 |
Under these conditions, the system suitability parameters are summarized and presented in Table 2.
3.2. Method validation
The CE technique underwent validation in adherence to the US-FDA guidelines for Industry-Bioanalytical technique Validation.34 Based on the FDA requirements, the method validation was performed on three quality control solutions (QCs) covering the method's range: low QC (LOQ, 50 μg mL−1), middle QC (MOQ, 100 μg mL−1), and high QC (HOQ, 150 μg mL−1) for the three drugs, using these suggested parameters in terms of linearity, selectivity, accuracy, precision, stability, extraction recovery, and LOD and LOQ.| For PIP: P = 0.0290C − 0.0512 (r2 = 0.9998) |
| For TAZ: P = 0.0314C + 0.1198 (r2 = 0.9994) |
| For IBU: P = 0.0270C − 0.02465 (r2 = 0.9990) |
The results are given in Table 3. The obtained small intercepts indicated the minimum background signals, and the high determination coefficients indicated the excellent linearity of the calibration lines, and their reliability for the accurate analysis of the three analytes. Every concentration that was computed for the in vitro calibration standards remained within 15% of its nominal value.
| Parameters | PIP | TAZ | IBU |
|---|---|---|---|
| Calibration range (μg mL−1) | 1–200 | 3–200 | 1–200 |
| Slope | 0.0290 | 0.0314 | 0.0270 |
| Intercept | 0.0512 | −0.1198 | 0.02465 |
| Determination coefficient (r2) | 0.9998 | 0.9994 | 0.9990 |
| LOD (μg mL−1) | 0.3 | 0.7 | 0.3 |
| LOQ (μg mL−1) | 1.0 | 2.3 | 0.9 |
| Drug concentration (μg mL−1) | Intra–day | Inter–day | ||||
|---|---|---|---|---|---|---|
| Recoverya (%) | RSDa (%) | Biasb (%) | Recoverya (%) | RSDa (%) | Biasb (%) | |
| a Average of 3 determinations.b Bias = [(measured concentration - nominal concentration)/nominal concentration] × 100. The values are an average of 3 determinations. | ||||||
| PIP | ||||||
| 50 | 95.81 | 3.50 | −4.19 | 103.02 | 6.22 | 3.02 |
| 100 | 96.90 | 2.50 | −3.13 | 98.30 | 2.04 | −1.71 |
| 150 | 97.78 | 3.14 | −2.32 | 99.09 | 3.81 | −0.91 |
|
||||||
| TAZ | ||||||
| 50 | 93.76 | 3.31 | −3.25 | 93.75 | 6.31 | −6.25 |
| 100 | 95.54 | 1.10 | −4.46 | 96.55 | 2.65 | −3.45 |
| 150 | 99.47 | 1.20 | −0.53 | 99.24 | 2.01 | −0.76 |
|
||||||
| IBU | ||||||
| 50 | 96.01 | 5.28 | −3.99 | 99.16 | 3.25 | −0.84 |
| 100 | 94.40 | 2.17 | −5.60 | 94.40 | 2.17 | −5.60 |
| 150 | 101.45 | 0.68 | 1.45 | 100.58 | 1.06 | 0.58 |
| Drug concentration (μg mL−1) | Recoverya (%) | |
|---|---|---|
| Three freeze thaw cyclesb | Bench top stability | |
| a Average of 3 determinations.b Freezing was done at −20 °C. | ||
| PIP | ||
| 50 | 110.57 | 106.10 |
| 100 | 87.23 | 116.20 |
| 150 | 107.85 | 100.37 |
| Mean ± RSD (%) | 101.88 ± 1.24 | 107.55 ± 8.01 |
|
||
| TAZ | ||
| 50 | 95.94 | 98.09 |
| 100 | 103.81 | 107.05 |
| 150 | 111.70 | 99.43 |
| Mean ± RSD (%) | 103.82 ± 7.15 | 101.53 ± 4.83 |
|
||
| IBU | ||
| 50 | 94.82 | 97.83 |
| 100 | 109.03 | 100.07 |
| 150 | 112.12 | 99.76 |
| Mean ± RSD (%) | 105.32 ± 5.27 | 99.22 ± 1.21 |
The extraction efficiency was taken into account, and the extraction recoveries for TAZ, PIP, and IBU were computed to be 104.96 ± 3.35, 99.68 ± 7.63, and 107.85 ± 3.81, respectively, as shown in Table 6. The results validated the efficacy of the used extraction procedure.
| PIP | TAZ | IBU | ||||
|---|---|---|---|---|---|---|
| Concentration (μg mL−1) | Recoverya (%) | Concentration (μg mL−1) | Recoverya (%) | Concentration (μg mL−1) | Recoverya (%) | |
| a Average of 3 determinations. | ||||||
| 50 | 108.65 | 50 | 90.95 | 50 | 109.91 | |
| 100 | 102.10 | 100 | 105.05 | 100 | 110.19 | |
| 150 | 104.15 | 150 | 103.07 | 150 | 103.46 | |
| Mean ± RSD (%) | 104.97 ± 3.35 | 99.69 ± 7.63 | 107.85 ± 3.81 | |||
3.3. Results of pharmacokinetic study
The assessed pharmacokinetic characteristics of the medications administered singly were utilized as comparison points for evaluating the combined effects of the medications when administered concurrently. The pharmacokinetic characteristics of the administered drugs were calculated by plotting the plasma concentration of the drugs at various time intervals using regression equations. A graph showing the average plasma concentration over time was then plotted, as shown in Fig. 4. PK Solver42 was used to conduct a non-compartmental pharmacokinetic study (NCA), and the results of all groups were compared, as shown in Table 7. | ||
| Fig. 4 Mean plasma concentration–time curves of PIP after intraperitoneal administration of mixed doses of 160/40 PIP/TAZ mg kg−1 (A), PIP after intraperitoneal administration of mixed doses of 160/40 mg per kg PIP/TAZ and 30 mg per kg IBU (B), TAZ after intraperitoneal administration of mixed doses of 160/40 mg per kg PIP/TAZ (C), TAZ after intraperitoneal administration of mixed doses of 160/40 mg per kg PIP/TAZ and 30 mg per kg IBU (D), IBU after single intraperitoneal administration of a dose of 30 mg kg−1 (E), and IBU after intraperitoneal administration of mixed doses of 160/40 mg per kg PIP/TAZ and 30 mg per kg IBU (F). | ||
| PIP | TAZ | IBU | ||||
|---|---|---|---|---|---|---|
| a Vd = Volume of distribution.b Cl = Clearance. | ||||||
| Parameters | Group administered with PIP and TAZ | Group administered with PIP,TAZ and IBU | Group administered with TAZ and PIP | Group administered with PIP,TAZ and IBU | Group administered with IBU | Group administered with PIP,TAZ and IBU |
| Tmax (h) | 0.5 | 0.75 | 0.5 | 0.75 | 1.5 | 1 |
| T1/2 (h) | 1.08 | 0.67 | 2.28 | 1.66 | 1.30 | 1.41 |
| Cmax (μg mL−1) | 117.46 | 132.82 | 18.35 | 28.41 | 59.23 | 84.56 |
| Elimination constant (k) | 0.64 | 1.03 | 0.30 | 0.42 | 0.53 | 0.49 |
| Vd,a (mg) (μg−1 mL−1) | 1.55 | 0.61 | 2.17 | 1.32 | 0.45 | 0.36 |
| Cl,b (mg) (μg−1 mL−1) h−1 | 0.99 | 0.64 | 0.66 | 0.55 | 0.24 | 0.18 |
| AUC 0 → t (μg mL−1 h−1) | 199.72 | 313.33 | 55.62 | 70.42 | 202.94 | 277.78 |
| AUC 0 → ∞ (μg mL−1 h−1) | 201.59 | 313.87 | 60.75 | 72.84 | 208.59 | 282.13 |
From the pharmacokinetic profiles in Fig. 4, it is obvious that all drugs were quickly absorbed almost after 0.25 h in all groups; however, their onset concentrations in group (III) were more than those in groups (I) and (II) by 21.7%, 80% and 14.2% for PIP, TAZ and IBU, respectively. It is noticed that the time required for IBU to record its maximum concentration in group (III) was higher than that in group (II) by 0.5 h, albeit the time attained by PIP and TAZ to reach their upper concentrations in group (III) was raised by 0.25 h when compared to group (I). The half-lives (t1/2) of the drugs PIP and TAZ have reduced obviously (from 1.07 to 0.66 and from 2.06 to 1.66 for PIP and TAZ, respectively). The IBU half-life (t1/2) was declined somewhat (from 1.20 to 1.09). With regard to the maximum plasma concentration (Cmax) of all drugs, it was incredibly increased in group (III) compared to groups (I) and (II). PIP and TAZ in group (III) have higher Cmax than group (I) by about 13.7% and 55.5%, whereas IBU in group (III) have higher Cmax than group (II) by about 44%. When it comes to bioavailability which is represented by the area under the curve (AUC), the bioavailability of PIP and TAZ was increased incrementally when combined with IBU in group (III) compared to group (I) with 55.4% and 19.7% increase in their plasma levels. Furthermore, the IBU plasma concentration was increased by 35.6% in group (III) than in group (II). Additionally, the distribution volumes (Vd) and clearance rates (Cl) were considerably affected by the concomitant administration of the examined drugs. PIP and TAZ distribution volumes declined by 145.9% and 66.2% and their clearance values were reduced by 57.1% and 20%. For IBU, the distribution volume was minimized by 20%, while its clearance value was reduced by 33.3%. It is now clear that the concurrent administration of the drugs changed their pharmacokinetic parameters and prescribing them together for in-patients or postoperatively has an accumulative therapeutic impact. This requires dose regimen regulation since there was a noticeable change in their bioavailability. All the pharmacokinetic parameters are given in Table 7.
3.4. Greenness profile of the CE method
Green Analytical Chemistry (GAC) is the design and development of analytical procedures to determine certain compounds in a given sample that generate less hazardous substances without impairing the efficiency even at low concentrations of analytes in complex matrix compositions.43 Three metric assessment tools were used to assess the greenness of the proposed method: the eco-scale assessment, ESA,44 green analytical procedure index, GAPI,45 and analytical greenness, AGREE.46The ESA tool assesses the environmental influence of the analytical procedure/approach. The procedure/approach encompasses various factors such as the solvents and reagents employed, the energy consumption of the analytical instrument, the occupational dangers involved, and the generation of waste. The negative results of each procedure/approach are assigned as penalty points (PPs), which are then deducted from a perfect score of 100 points (representing an optimum eco-friendly procedure with no penalty points) in order to calculate the final result. The result of the calculation is ranked on a scale, where the score >75 represents excellent green analysis, >50 represents acceptable green analysis, and <50 represents inadequate green analysis.44 The CE approach, as proposed, achieved a cumulative ESA score of 89. To illustrate: PP (penalty points) of solvent = subtotal PP × number of pictograms × signal word. For methanol = 1 × 3 × 2 = 6, while for Acetonitrile = 1 × 2 × 2 = 4. As the energy used was <0.1 kW h per sample (PP = 0), there was no analytical process hermetization (PP = 0), and the wastes were less than 10 mL (PP = 1), the total penalty points were 11. After deducting 11 from 100, the net ESA score was 89, as indicated in Table 8. Based on the ESA scale, the score designates the suggested CE technique as an optimum environmentally friendly procedure.
| ESA tool (PPs) | GAPI tool | AGREE tool | ||
|---|---|---|---|---|
| Methanol | 6 |  |
 |
|
| Acetonitrile | 4 | Details of GAPI tool | 1- Sample treatment | |
| Distilled water | 0 | Sample preparation | 2- Sample amount | |
| Energy used (instrument) | 0 | 1-Offline (red) | 2-None (green) | 3- Device positioning |
| Waste | 1 | 3-None (green) | 4-Storage (yellow) | 4- Sample prep. Stages |
| Occupational hazards | Total | 5- Simple procedure (yellow) | 6-Micro-extraction (yellow) | 5- Method automation/miniaturization |
| PPs | 11 | 7- Solvents and reagents (yellow) | 8-None (green) | 6- Derivatization |
| ESA score | 89 | Reagent and solvents | 7- Waste | |
| 9- Less than 10 mL (green) | 8- Analysis throughput | |||
| 10- NFPA = 3, moderate toxicity (yellow) | 9- Energy consumption | |||
| 11- NFPA = 3; high flammability (yellow) | 10- Source of reagent | |||
| Instrumentation | 11- Toxicity | |||
| 12- ≤ 1.5 kW h per sample (yellow) | 12- Operator's safety | |||
| 13- Hermetic sealing of analytical procedure (green) | ||||
| 14- 1–10 (green) | ||||
| 15- Recycling possible (green) | ||||
The GAPI instrument is responsible for assessing the overall analytical approach and delivering a comprehensive evaluation of 15 parameters, pertaining to the procedures' greenness, as shown in Table 8. The results of GAPI pentagrams revealed that the proposed method had 7 parameters colored green, 7 parameters colored yellow, and 1 parameter shaded red. Because the samples were prepared offline and stored at −20 degrees Celsius prior to analysis, parameter 1 (relating to sample preparation) in the GAPI pictogram of the proposed CE method was colored red. Due to the absence of chemical, physical, and physico–chemical preservation, parameter 2 was assigned the color green. Similarly, since there is no transportation, parameter 3 is also colored green. Parameter 4 was tinted yellow due to storage being conducted under standard conditions. The reason parameter 5 is colored yellow because the CE method involves operations such as filtration and do not require extraction. Parameter 6 was marked as yellow due to the successful drug detection using the micro-scale in the CE method. Parameter 7 was also marked as yellow because the CE method relies heavily on the use of green solvents. Parameter (8) was assigned the color green since the process of preparing the sample does not necessitate any simple or advanced treatment. While the remaining parameters were colored yellow or green, the majority of the parameters were green. Hence, the suggested CE approach may be regarded as a relatively environmentally friendly method.
The AGREE metric, which is widely used, assesses analytical methodologies by considering a set of 12 parameters that are scaled from 0 to 12 on a common scale. The metric provides a visual result in the form of a number and a color. The final evaluation result is obtained by adding up the assessment findings for each concept. The AGREE pictograms indicate the degree to which specific principles are adhered to, as shown in Table 8. The final score, which is generated automatically in the center, is presented alongside the twelve evaluation parameters in the form of a colored circular pictogram representing the results. The AGREE pictogram represents the assumed CE technique. Table 8 presents the level of adherence to the principles, with a value of 0.77 out of 1, and a middle color of light green which ascertains that the performance of the principles is quite excellent, and the CE method is an eco-friendly method.
In summary, the proposed CE method satisfies the GAC's criteria for routine application in the analysis of TAZ, PIP and IBU in plasma samples. The comprehensive assessment of the method's environmental impact, as evidenced by the ESA, GAPI, and AGREE tools, substantiates its ecological friendliness and greenness.
4. Conclusion
For the first time, a sensitive, selective, and green capillary electrophoresis method equipped with diode array detection was employed to determine the co-administered drugs PIP, TAZ, and IBU in the plasma of rats. The proposed technique combined numerous privileges as plasma samples were prepared by simple straightforward one-step protein precipitation with miniaturizing of sample size, high efficiency, few solvents required and a short analysis time. The method's straightforward extraction procedure and short run time contribute to its high throughput, enabling the efficient processing of diverse samples across several clinical laboratories. The CE method complied with FDA recommendation, and it was used for the full pharmacokinetic study of the drugs on rats. The pharmacokinetics of the drugs when administered singly was compared with that of the drugs when administered concurrently, and it showed the accumulative impact from co-administering the drugs in terms of rapid onset of action with a maximized concentration, which requires caution when prescribing the compounds together as there is a massive change in their bioavailability. The data resulting from the full pharmacokinetic study can be used for therapeutic drug monitoring and as basics for future researchers who are interested in further studies on drug–drug interaction in humans. Furthermore, the proposed analytical method was assessed using three green metric tools, namely ESA, GAPI and AGREE. The resulted outcomes revealed that the method has minimum environmental impacts while maintaining analytical parameters.The proposed method is the first capillary electrophoretic method for simultaneous analysis of PIP, TAZ, and IBU and applied for pharmacokinetic studies. A comparison of the performance and features of the proposed method with the reported separation-based methods for simultaneous analysis of binary mixtures is summarized in Table 9.
| Method | Analyte | Linearity range | Remarks | Ref. |
|---|---|---|---|---|
| CE-PDA | PIP, TAZ, and IBU | 1–200 μg mL−1 for both PIP and TAZ, and 3–200 μg mL−1 for IBU | The first method for ternary mixture and applied to pharmacokinetic studies | Proposed study |
| UHPLC-MS/MS | PIP, TAZ, and IBU | 0.5–500 μg mL−1 for PIP and 0.625–62.5μgmL−1 for TAZ. |
The method was not applied to pharmacokinetic studies | 13 |
| HPLC-UV | PIP and TAZ | 0.5–400 μg mL−1 and 1–100 μgmL−1 for PIP and TAZ, respectively | The method was not applied to pharmacokinetic studies | 1 |
| RP-HPLC | PIP and TAZ | 1.0–200 μg mL−1 for PIP and 0.78–50 μg mL−1 for TAZ | The method was not applied to pharmacokinetic studies | 10 |
| RP-HPLC | PIP and TAZ | 50–100 ppm for both PIP and TAZ | The method was not applied to pharmacokinetic studies | 11 |
| Capillary zone electrophoresis | PIP and TAZ | 20–400 for PIP and 10–100 μg mL−1 for TAZ | The method was not applied to pharmacokinetic studies | 15 |
Ethical statement
The Committee of Beni-Suef University, Faculty of Pharmacy approved the study protocol from an ethical point of view. Approval no.: REC-H-PhBSU-022-234.Data availability
The data that support the findings of this study are included within the article, and any additional data will be available upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project number (RSP2024R215), King Saud University, Riyadh, Saudi Arabia, for funding this work.References
- K. Abdelkawy, T. Le and S. H. Mahmoud, Simple HPLC-UV method for piperacillin/tazobactam assay in human plasma, Antibiotics, 2023, 12, 321 CrossRef CAS PubMed.
- J. L. Vincent, Y. Sakr, M. Singer, I. Martin-Loeches, F. R. Machado, F. Marshall and J. C. Simon, Prevalence and outcomes of infection among patients in intensive care units in 2017, JAMA, 2020, 323, 1478–1487 CrossRef PubMed.
- S. Hagel, F. Bach, T. Brenner, H. Bracht, A. Brinkmann, T. Annecke and M. W. Pletz, Effect of therapeutic drug monitoring-based dose optimization of piperacillin/tazobactam on sepsis-related organ dysfunction in patients with sepsis: a randomized controlled trial, Intensive Care Med., 2022, 48, 311–321 CrossRef CAS PubMed.
- C. J. Clancy and M. H. Nguyen, Management of Highly Resistant Gram-Negative Infections in the Intensive Care Unit in the Era of Novel Antibiotics, Infect. Dis. Clin., 2022, 36, 791–823 Search PubMed.
- S. P. Cohen, E. J. Wang, T. L. Doshi, L. Vase, K. A. Cawcutt and N. Tontisirin, Chronic pain and infection: mechanisms, causes, conditions, treatments, and controversies, BMJ Medicine, 2022, 1, 1 CrossRef PubMed.
- Y. Li, L. Yin, Z. Fan and Z. Fang, Microglia, a potential therapeutic target for sepsis-associated encephalopathy and sepsis-associated chronic pain, Front. Pharmacol, 2020, 11, 600421 CrossRef CAS PubMed.
- K. C. Carpenter, J. M. Hakenjos, C. D. Fry and J. A. Nemzek, The influence of pain and analgesia in rodent models of sepsis, Comp. Med., 2019, 69, 546–554 CrossRef CAS PubMed.
- K. D. Rainsford, Ibuprofen: Pharmacology, Therapeutics and Side Effects, Springer Science & Business Media, 2013 Search PubMed.
- B. J. Anderson, The Nonsteroidal Anti-inflammatory Drugs and Acetaminophen, Oxford Textbook of Pediatric Pain, 2021, p. 449 Search PubMed.
- J. J. Veillette, S. A. Winans, S. C. Forland and V. K. Maskiewicz, A simple and rapid RP-HPLC method for the simultaneous determination of piperacillin and tazobactam in human plasma, J. Pharm. Biomed. Anal., 2016, 131, 80–86 CrossRef CAS PubMed.
- P. R. K. Veni, N. Sharmila, K. J. P. Narayana, B. H. Babu and P. V. V. Satyanarayana, Simultaneous determination of piperacillin and tazobactam in pharmaceutical formulations by RP-HPLC method, J. Pharm. Res., 2013, 7, 127–131 Search PubMed.
- N. Murao, H. Ohge, K. Ikawa, Y. Watadani, S. Uegami, N. Shigemoto, N. Shimada, R. Yano, T. Kajihara, K. Uemura and Y. Murakami, Pharmacokinetics of piperacillin-tazobactam in plasma, peritoneal fluid and peritoneum of surgery patients, and dosing considerations based on site-specific pharmacodynamic target attainment, Int. J. Antimicrob. Agents, 2017, 50, 393–398 CrossRef CAS PubMed.
- S. Naicker, Y. C. G. Valero, J. L. O. Meija, J. Lipman, J. A. Roberts, S. C. Wallis and S. L. Parker, A UHPLC–MS/MS method for the simultaneous determination of piperacillin and tazobactam in plasma (total and unbound), urine and renal replacement therapy effluent, J. Pharm. Biomed. Anal., 2018, 148, 324–333 CrossRef CAS PubMed.
- A. M. Nicasio, B. D. VanScoy, R. E. Mendes, M. Castanheira, C. C. Bulik, O. O. Okusanya, S. M. Bhavnani, A. Forrest, R. N. Jones, L. V. Friedrich and J. N. Steenbergen, Pharmacokinetics-pharmacodynamics of tazobactam in combination with piperacillin in an in vitro infection model, Antimicrob. Agents Chemother., 2016, 60, 2075–2080 CrossRef CAS PubMed.
- A. Al-Attas, J. J. Nasr, N. El-Enany and F. Belal, A green capillary zone electrophoresis method for the simultaneous determination of piperacillin, tazobactam and cefepime in pharmaceutical formulations and human plasma, Biomed. Chromatogr., 2015, 29, 1811–1818 CrossRef CAS PubMed.
- S. O. Eraga, M. I. Arhewoh, R. N. Chibuogwu and M. A. Iwuagwu, A comparative UV-HPLC analysis of ten brands of ibuprofen tablets, Asian Pac. J. Trop. Biomed., 2015, 5, 880–884 CrossRef CAS.
- T. Chen, Q. Li, J. Lu, C. Yu, C. Chen and Z. Li, Determination of ibuprofen enantiomers in human plasma by HPLC–MS/MS: Validation and application in neonates, Bioanalysis, 2016, 8, 1237–1250 CrossRef CAS PubMed.
- N. Nakov, R. Petkovska, L. Ugrinova, Z. Kavrakovski, A. Dimitrovska and D. Svinarov, Critical development by design of a rugged HPLC-MS/MS method for direct determination of ibuprofen enantiomers in human plasma, J. Chromatogr. B, 2015, 992, 67–75 CrossRef CAS PubMed.
- B. Yilmaz and A. F. Erdem, Determination of ibuprofen in human plasma and urine by gas chromatography/mass spectrometry, J. AOAC Int., 2014, 97, 415–420 CrossRef CAS PubMed.
- B. Yilmaz and V. Akba, Determination and Pharmacokinetics of Ibuprofen in Rabbit Plasma by GC-MS Method, Res. J. Pharm. Technol., 2011, 4, 52–56 CAS.
- C. Wang, Y. Wang, H. Xie, C. Zhan, X. He, R. Liu, R. Hu, J. Shen and Y. Jia, Establishment and validation of an SIL-IS LC–MS/MS method for the determination of ibuprofen in human plasma and its pharmacokinetic study, Biomed. Chromatogr., 2022, 36, e5287 CrossRef CAS PubMed.
- A. Puangpetch, A. Limrungsikul, S. Prommas, P. Rukthong and C. Sukasem, Development and validation of a liquid chromatography-tandem mass spectrometry method for determination of ibuprofen in human plasma, Clin. Mass Spectrom., 2020, 15, 6–12 CrossRef.
- N. F. Farid, M. O. Elgendy and N. S. Abdelwahab, Sustainable TLC-densitometric method for pharmacokinetic study of the concurrently used ibuprofen and metronidazole: Green metric assessment, Microchem. J., 2022, 179, 107582 CrossRef CAS.
- G. Magdy, F. Belal, A. M. Abdel-Megied and A. F. Abdel Hakiem, Two different synchronous spectrofluorimetric approaches for simultaneous determination of febuxostat and ibuprofen, R. Soc. Open Sci., 2021, 8, 210354 CrossRef CAS PubMed.
- M. Bahram, T. Madrakian and S. Alizadeh, Simultaneous colorimetric determination of morphine and ibuprofen based on the aggregation of gold nanoparticles using partial least square, J. Pharm. Anal., 2017, 7, 411–416 CrossRef PubMed.
- F. Abacıgil, S. G. Turan, F. Adana, P. Okyay and B. Demirci, Rational use of drugs among inpatients and its association with health literacy, Meandros Med. Dental J., 2019, 20, 64 CrossRef.
- H. Cho, A. J. Lynham and E. Hsu, Postoperative interventions to reduce inflammatory complications after third molar surgery: review of the current evidence, Aust. Dent. J., 2017, 62, 412–419 CrossRef CAS PubMed.
- N. Aldamluji, A. Burgess, E. Pogatzki-Zahn, J. Raeder, H. Beloeil, E. Albrecht, H. Beloeil, F. Bonnet, S. Freys, G. P. Joshi and M. Van de Velde, Prospect guideline for tonsillectomy: systematic review and procedure-specific postoperative pain management recommendations, Anaesthesia, 2021, 76, 947–961 CrossRef CAS PubMed.
- N. D. Popowicz, S. J. O'Halloran, D. Fitzgerald, Y. G. Lee and D. A. Joyce, A rapid LC-MS/MS assay for quantification of piperacillin and tazobactam in human plasma and pleural fluid; application to a clinical pharmacokinetic study, J. Chromatogr. B, 2018, 1081, 58–66 CrossRef PubMed.
- S. Yang, Z. Liu, C. Wang, S. Wen, Q. Meng, X. Huo and K. Liu, Piperacillin enhances the inhibitory effect of tazobactam on β-lactamase through inhibition of organic organic anion transporter 1/3 in rats, Asian J. Pharm. Sci., 2019, 14, 677–686 CrossRef PubMed.
- K. Przejczowska-Pomierny, M. Włodyka, A. Cios and E. Wyska, Enantioselective analysis of ibuprofen enantiomers in mice plasma and tissues by high-performance liquid chromatography with fluorescence detection: Application to a pharmacokinetic study, Chirality, 2017, 29, 500–511 CrossRef CAS PubMed.
- M. M. Abdel Moneim, Simultaneous determination of dantrolene with ibuprofen and diclofenac in plasma by HPLC-DAD: Application to comparative pharmacokinetic study, Acta Chromatogr., 2024, 36, 23–30 CAS.
- S. H. Chen and Y. H. Chen, Pharmacokinetic applications of capillary electrophoresis, Electrophoresis, 1999, 20, 3259–3268 CrossRef CAS PubMed.
- FDA, Bioanalytical Method Validation Guidance for Industry, Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry.
- T. Sato, E. A. Lochry, A. M. Hoberman and M. S. Christian, Reproductive and developmental toxicity studies of tazobactam/piperacillin or tazobactam (2): Teratological study in rats with intravenously administration, J. Toxicol. Sci., 1994, 19(SupplementII), 215–232 CrossRef CAS PubMed.
- M. K. Park, S. H. Kang, J. Y. Son, M. K. Lee, J. S. Ju, Y. C. Bae and D. K. Ahn, Co-administered low doses of ibuprofen and dexamethasone produce synergistic antinociceptive effects on neuropathic mechanical allodynia in rats, J. Pain Res., 2019, 2959–2968 CrossRef CAS PubMed.
- H. M. Maher, N. Z. Al-Zoman, M. M. Al-Shehri, H. Al-Showiman, A. M. Al-Taweel, G. A. Fawzy and S. Perveen, Determination of luteolin and apigenin in herbs by capillary electrophoresis with diode array detection, Instrum. Sci. Technol., 2015, 43, 611–625 CrossRef CAS.
- S. Oh, W. S. Shin and H. T. Kim, Effects of pH, dissolved organic matter, and salinity on ibuprofen sorption on sediment, Environ. Sci. Pollut. Res., 2016, 23, 22882–22889 CrossRef CAS PubMed.
- N. Z. Alzoman, M. M. Alshehri, M. A. Sultan, H. M. Maher, I. V. Olah and I. A. Darwish, Micellar electrokinetic capillary chromatographic determination of a polypill combination containing, lisinopril, hydrochlorothiazide, aspirin, and atorvastatin, Anal. Methods, 2013, 5, 1238–1244 RSC.
- N. Z. Alzoman, M. A. Sultan, H. M. Maher, M. M. AlShehri and I. V. Olah, Validated stability-indicating capillary electrophoresis method for the separation and determination of a fixed-dose combination of carvedilol and hydrochlorothiazide in tablets, J. AOAC Int., 2013, 96, 951–959 CrossRef CAS PubMed.
- N. Z. Alzoman, M. M. Alshehri, M. A. Sultan, H. M. Maher, I. V. Olah and I. A. Darwish, Micellar electrokinetic capillary chromatographic determination of a polypill combination containing, lisinopril, hydrochlorothiazide, aspirin, and atorvastatin, Anal. Methods, 2013, 5, 1238–1244 RSC.
- Y. Zhang, M. Huo, J. Zhou and S. Xie, PK Solver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel, Comput. Methods Programs Biomed., 2010, 99, 306–314 CrossRef PubMed.
- M. Sajid and J. Płotka-Wasylka, Green analytical chemistry metrics: A review, Talanta, 2022, 238, 123046 CrossRef CAS PubMed.
- A. Gałuszka, Z. M. Migaszewski, P. Konieczka and J. Namieśnik, Analytical Eco-Scale for assessing the greenness of analytical procedures, TrAC, Trends Anal. Chem., 2012, 37, 61–72 CrossRef.
- J. Płotka-Wasylka, A new tool for the evaluation of the analytical procedure: Green Analytical Procedure Index, Talanta, 2018, 181, 204–209 CrossRef PubMed.
- F. Pena-Pereira, W. Wojnowski and M. Tobiszewski, AGREE—Analytical GREEnness metric approach and software, Anal. Chem., 2020, 92, 10076–10082 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |