
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of mesoporous silica supported metallosalphen-azobenzene complexes: efficient photochromic heterogeneous catalysts for the oxidation of cyclohexane to produce KA oil
Salimah Alshehri and
Mohamed Abboud *
*
Catalysis Research Group (CRG), Department of Chemistry, College of Science, King Khalid University, Abha 61413, Saudi Arabia. E-mail: mabboud@kku.edu.sa; Tel: +966 53 48 46 782
First published on 27th August 2024
Abstract
The oxidation of cyclohexane to produce KA oil (cyclohexanone and cyclohexanol) is important industrially but faces challenges such as low cyclohexane conversion at high KA oil selectivity, and difficult catalyst recyclability. This work reports the synthesis and evaluation of new heterogeneous catalysts consisting of Co(II), Mn(II), Ni(II) and Cu(II) salphen-azobenzene complexes [ML1] immobilized on amino-functionalized mesoporous silica (SBA-15, MCM-41, MCM-48) through coordination bonding. In the first step, the salphen-azobenzene ligand was synthesized and complexed with Co, Mn, Ni and Cu metal ions. In the second step, aminopropyltriethoxysilane (APTES) was grafted onto the surface of different types of commercial mesoporous silica. The immobilization of [ML1] onto the mesoporous silica surface and the thermal stability of the obtained materials were confirmed using different characterization techniques such as FT-IR, powder XRD, SEM, TEM, BET, and TGA. The obtained results revealed high dispersion of [ML1] through the silica surface. The catalytic activity of the prepared materials Silica-N-ML1 was evaluated on the cyclohexane oxidation to produce KA oil using various oxidants. The cis–trans isomerization of the azobenzene upon UV irradiation was found to affect the catalytic performance of Silica-N-ML1. The cis isomer of SBA-15-N-CoL1 exhibited the highest cyclohexane conversion (93%) and KA selectivity (92%) under mild conditions (60 °C, 6 h) using m-CPBA as oxidant. Moreover, The SBA-15-N-CoL1 showed high stability during four successive cycles.
1. Introduction
The oxidation of cyclohexane is an important industrial chemical reaction. This transformation produces cyclohexanol (A) and cyclohexanone (K), commonly referred to together as ketone-alcohol (KA) oil.1 KA oil serves as a critical feedstock in the manufacture of nylon 6,6 fibers (Scheme 1).2 The production of nylon 6,6 involves further oxidation of KA oil with nitric acid to form adipic acid. Adipic acid then acts as an important building block monomer in the preparation of nylon 6,6.3 This nylon polymer finds extensive application in textiles due to their desirable mechanical properties.4,5 Additionally, adipic acid itself is an important intermediate chemical in various industrial processes. It is commonly used to manufacture other polymeric materials, resins, polyesters, and plasticizers.5–8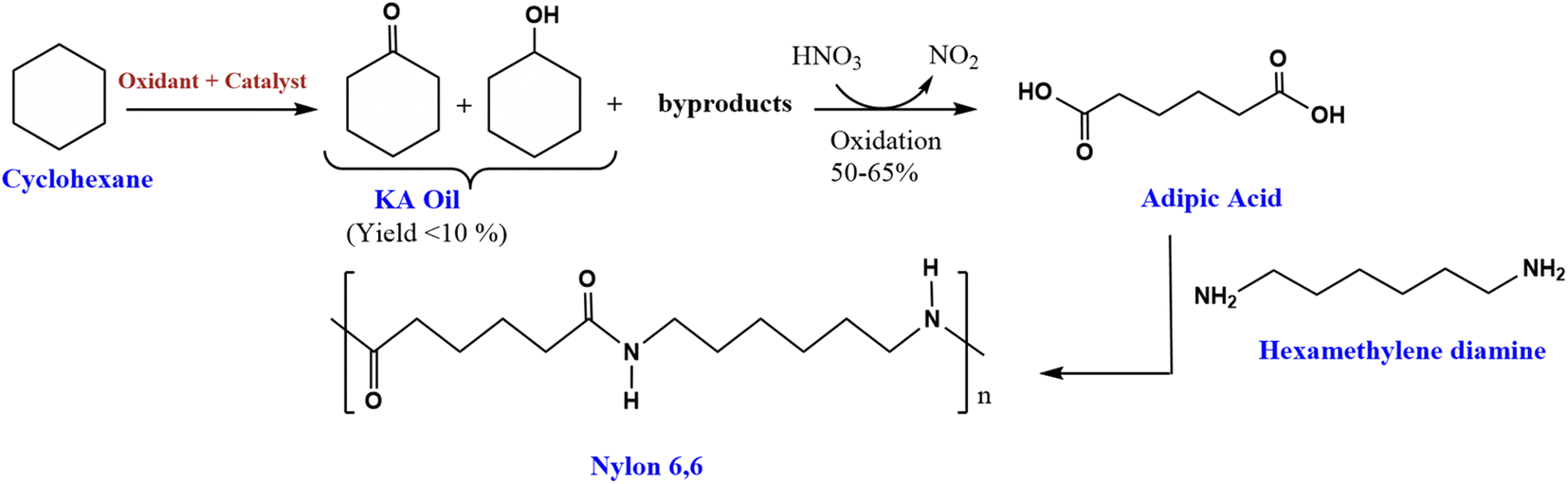 | ||
| Scheme 1 Synthesis route of nylon 6,6 from cyclohexane.2 | ||
The current industrial process for the oxidation of cyclohexane to produce KA oil involves cobalt or manganese salt as homogenous catalysts under high-temperature (150–160 °C) and high-pressure conditions (10–20 atm) of air or oxygen gas.9 However, cyclohexane is quite stable under these conditions while the desired products cyclohexanol and cyclohexanone are less stable, resulting in numerous undesirable by-products forming at the elevated temperatures and pressures.10 Another limitation of this industrial process is that the conversion of cyclohexane must be kept at less than 10% to ensure high selectivity toward KA oil (around 80%).11 Additionally, this process also faces challenges in regenerating and reusing the homogeneous catalysts.12 Therefore, many efforts have been made to develop more efficient catalysts to covert cyclohexane to KA with higher conversion and selectivity under mild conditions.13
Over the past decades, extensive research efforts have been focused on developing heterogeneous catalysts as alternatives to address issues such as low conversion, selectivity, and the high-cost.14,15 In addition, heterogeneous catalysis provides important advantages over homogeneous conditions, including improved catalyst activity, easy separation from reaction mixtures, and reuse over multiple cycles.16–20
Various transition metal-based systems have shown promise as catalysts, including transition metal ions, organotransition metal complexes (OTMCs), and transition metal-oxo and -peroxo complexes.21–23 Some OTMCs have shown more promise as effective catalysts for the oxidation of cyclohexane.24 Notably, the most extensively investigated OTMCs for cyclohexane oxidation are Schiff base and metallo-porphyrins metal complexes.22,25 Using common oxidizing agents such as hydrogen peroxide (H2O2), oxygen gas (O2), tert-butyl hydroperoxide (t-BuOOH), and meta-chloroperbenzoic acid (m-CPBA).26–29
Salphen, one of the Schiff base ligands, it has proven to be a promising ligand for OTMCs used as catalyst in the oxidation of hydrocarbons, because of its simple synthesis and structural tunability. Salphen are organic compounds with azomethine (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) groups.30 They are often made by condensing carbonyl compounds with o-phenylenediamine. The introduction of various metal centres, functional groups, and substituents is made possible by their facile synthesis and structural diversity. This enabled the performance of the obtained catalysts to be optimized for certain reactions.31 Transition metals such as cobalt, copper, manganese, and nickel are of more interest due to their efficiency in the oxidation of hydrocarbons, relative abundance, and lower cost, compared to precious metals such as silver and gold.32
N–) groups.30 They are often made by condensing carbonyl compounds with o-phenylenediamine. The introduction of various metal centres, functional groups, and substituents is made possible by their facile synthesis and structural diversity. This enabled the performance of the obtained catalysts to be optimized for certain reactions.31 Transition metals such as cobalt, copper, manganese, and nickel are of more interest due to their efficiency in the oxidation of hydrocarbons, relative abundance, and lower cost, compared to precious metals such as silver and gold.32
Modification of metal–ligand combinations aims to develop sustainable oxidation catalysts. One promising strategy is to insert photoactive azo group into metal–ligand complexes to impart light-responsive properties.33 Azobenzene derivatives have many useful applications. Specifically, the cis/trans photoisomerization of azobenzene moiety can find important utilizations in optics, photochemistry, and biomaterials.34 Inserting such photochromic moiety into salphen ligand scaffold could impart new light-responsive functionality to the resulting OTMC catalyst.35,36 However, the effect of cis/trans photoisomerization of the azobenzene on the catalytic activity remains unexplored. Moreover, only few studies have investigated salphen-azobenzene-based OTMCs as catalysts in the oxidation reaction of hydrocarbons.37 Salem and coworkers are the most reported the synthesized and characterization of salphen-azobenzene complexes.35 However, on the best of our knowledge, this type of OTMCs have never been successfully employed as heterogeneous catalysts.27
Various solid supports have been reported for the heterogenization of salphen-based OTMCs. This is including mesoporous silicas, porous carbons, zeolites, polymers, clays and resins.38 Mesoporous silica materials (silica) such as MCM-41, MCM-48 and SBA-15 are considered more efficient as support due to their high surface areas and large pore sizes, narrow pore size distribution, easy functionalization, highly ordered nanostructure, different pores-network dimension (1D, 2D, and 3D), which allow easy diffusion of reactants and products, without the need for swelling agents.39–43
Different techniques have been used to immobilize OTMCs in silica, either onto surface or into framework, such as physical adsorption, grafting, co-condensation, periodic mesoporous organosilica (PMOs).44,45 The physical adsorption of OMTCs into silica surface is the easiest technique. However, due to the very weak physical bonds between the catalyst and support, this technique suffers from a major problem, which is the rapid leaching of the catalyst from the support. Previous studies have investigated immobilizing some metal–salphen complexes on solid supports via physical adsorption. However, challenges remained, such as leaching, lower activity, selectivity and recyclability.44–47 While the immobilization of OTMCs in silica through chemical bonding, including grafting, co-condensation, PMOs methods, affords more stable OTMCs@silica heterogeneous catalyst.46,47 However, these methods are usually more complicated than the physical approach.
Therefore, developing an efficient, stable, and reusable heterogeneous catalyst via a simple immobilization method, under mild conditions, remains the main object of many researchers in the field of catalysis.48–50
Herein, we report the synthesis, characterization, and catalytic activity evaluation of silica supported Co(II), Mn(II), (Ni(II) and Cu(II) metallosalphen-azobenzene derivatives [ML1]. [ML1] were immobilized onto silica surface via coordination-assisted grafting method. In this approach, 3-aminopropyltriethoxysilane (APTES) was first grafted onto the surface of different type of silica (i.e., SBA-15, MCM-41, and MCM-48) to afford amino-functionalized silica (i.e., SBA-15-N, MCM-41-N, and MCM-48-N). Then [ML1] were added to Silica-NH2 to form stable Silica-N-ML1 materials via coordinate bond between NH2 group of APTES and the metal ions (e.g., SBA-15-NH2-Co(II) salphen-azobenzene). The obtained nanocatalysts Silica-N-ML1 were characterized by TEM, SEM, FT-IR, BET, TGA and XRD. The catalytic activity of these new nanocatalysts were evaluated in the oxidation of cyclohexane using different oxidants such as meta-chloroperoxybenzoic acid (m-CPBA), tert-butyl hydroperoxide (tBuOOH, TBHP), hydrogen peroxide (H2O2).
2. Experimental section
2.1 Materials
Hydrochloric acid (99.8%), sodium nitrite (NaNO2) (99.99%), sodium hydroxide (NaOH) (≥98%), salicylaldehyde (99%), aniline (99.5%), o-phenylenediamine (99.5%), and glacial acetic acid (99.9%) were used as the starting materials for the preparation of ligands H2L. The metal salts employed were cobalt(II) acetate tetrahydrate Co(CO2CH3)2·4H2O) (≥98%), manganese(II)acetate tetrahydrate Mn(CO2CH3)2·4H2O) (≥99%), nickel(II) acetate tetrahydrate Ni(CO2CH3)2·4H2O) (≥99%), copper(II) acetate tetrahydrate Cu(CO2CH3)2·4H2O) (≥98%), 3-aminopropyltriethoxysilane (APTES), commercial silica (SBA-15, MCM-41, MCM-48) cyclohexane (99%), cyclohexanone (99%), cyclohexanol (99%), chlorobenzene (99%), meta-chloroperoxybenzoic acid (m-CPBA), tert-butyl hydroperoxide (tBuOOH, TBHP), hydrogen peroxide 30% (H2O2), dichloromethane (99.8%), acetonitrile (99.9%), ethanol absolute (99.8%), chloroform (99.9%), diethyl ether (99.9%), ether (99.9%), all were purchased from Sigma Aldrich. All reagents were of analytical grade and used without further purification.2.2. Methods
Proton nuclear magnetic resonance spectrum (1H-NMR) and (13C NMR) of the salphen ligand were acquired in DMSO-d6 solution using a Brucker AMx 600 MHz spectrometer. Elemental analysis and ICP-mass were used to determine the composition of metallosalphen-azobenzene complexes and the metal content, respectively. The UV/vis spectra of the free salphen ligand and complexation were recorded on a Shimadzu UV-1600 UV/vis spectrophotometer in the wavelength range of 250–700 nm. The morphology of the immobilized catalysts was identified by scanning electron microscopy (SEM; JSM-7100F) (JEOL (Germany) GmbH). Transmission electron microscopy (TEM) micrographs were obtained using an FEI Tecnai G2 F30 TEM operating at 200 kV using a CCD camera. TEM samples were prepared by suspending the material in ethanol using bath sonication for a few minutes, then adding a drop of the resulting suspension solution on carbon coated copper grids with lacey carbon (Ted Pella Inc.) and then letting it dry at room temperature. Important functional groups of salphen ligand complexes and immobilized systems were determined using an FTIR spectrometer (Bruker Vector 22, Ettlingen, Germany) with a wavelength range of 4000 to 500 cm−1. A Shimadzu Lab-XRD-6000 with CuKα radiation and a secondary monochromator was applied to measure the X-ray diffraction pattern. The thermal stability of the immobilized material was demonstrated under air using a STARe System thermogravimetric analyzer (TGA) operating at a rate of 30 mL min−1 from 25 to 900 °C. The specific surface area, pore volume, pore size, and pore-size distribution of immobilized samples were identified via using a Micrometrics ASAP 2010 apparatus (Norcross, GA). The oxidation reaction was monitored using a Shimadzu GC-2014 gas chromatography (GC) instrument equipped with a flame ionization detector and an FFAP15%CW60/80 column that was 4.0 m long, 0.32 mm in diameter and had a 1 mm film thickness. Nitrogen was used as the carrier gas at a flow rate of 35 mL min−1. Samples were withdrawn from the reaction mixture. The injection volume was 1 μL and the total flow rate was 35 mL min−1. The oven temperature was initially held at 120 °C for 1 minute then increased to 150 °C at 20 °C min−1 and held for 10 minutes. It was then increased to 280 °C at 50 °C min−1 and held for 4 minutes. The injector temperature was 190 °C and the detector temperature was 300 °C.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of hydrochloric acid and nitric acid. The vessels were heated to completely dissolve the catalyst matrices. The resultant digests were then filtered before analysis by ICP-MASS. The ICP-MS was first calibrated using metal standard solutions to correlate elemental emission intensities to concentration.
:1 mixture of hydrochloric acid and nitric acid. The vessels were heated to completely dissolve the catalyst matrices. The resultant digests were then filtered before analysis by ICP-MASS. The ICP-MS was first calibrated using metal standard solutions to correlate elemental emission intensities to concentration.2.3 Synthesis
O) 1675, ν(NN) 1471, ν(CC) 1463, ν(O–H) 3447, ν(C–H, aromatic), 2963. 1H NMR (DMSO-d6 as solvent, δ ppm): δ 13 (s, 1H, O–H), δ 10.3 (s, 1H, CHO), 8.52–7.95 (m, 8H, aromatic C–H). 13C NMR (DMSO-d6, ppm): 193 (CHO), 163.98 (C–OH), 153 (N–C), 152 (C–N), 115–153 (aromatic C atoms), (kmax, nm): UV-Vis (CHCl3) (kmax, nm): 330, 441.
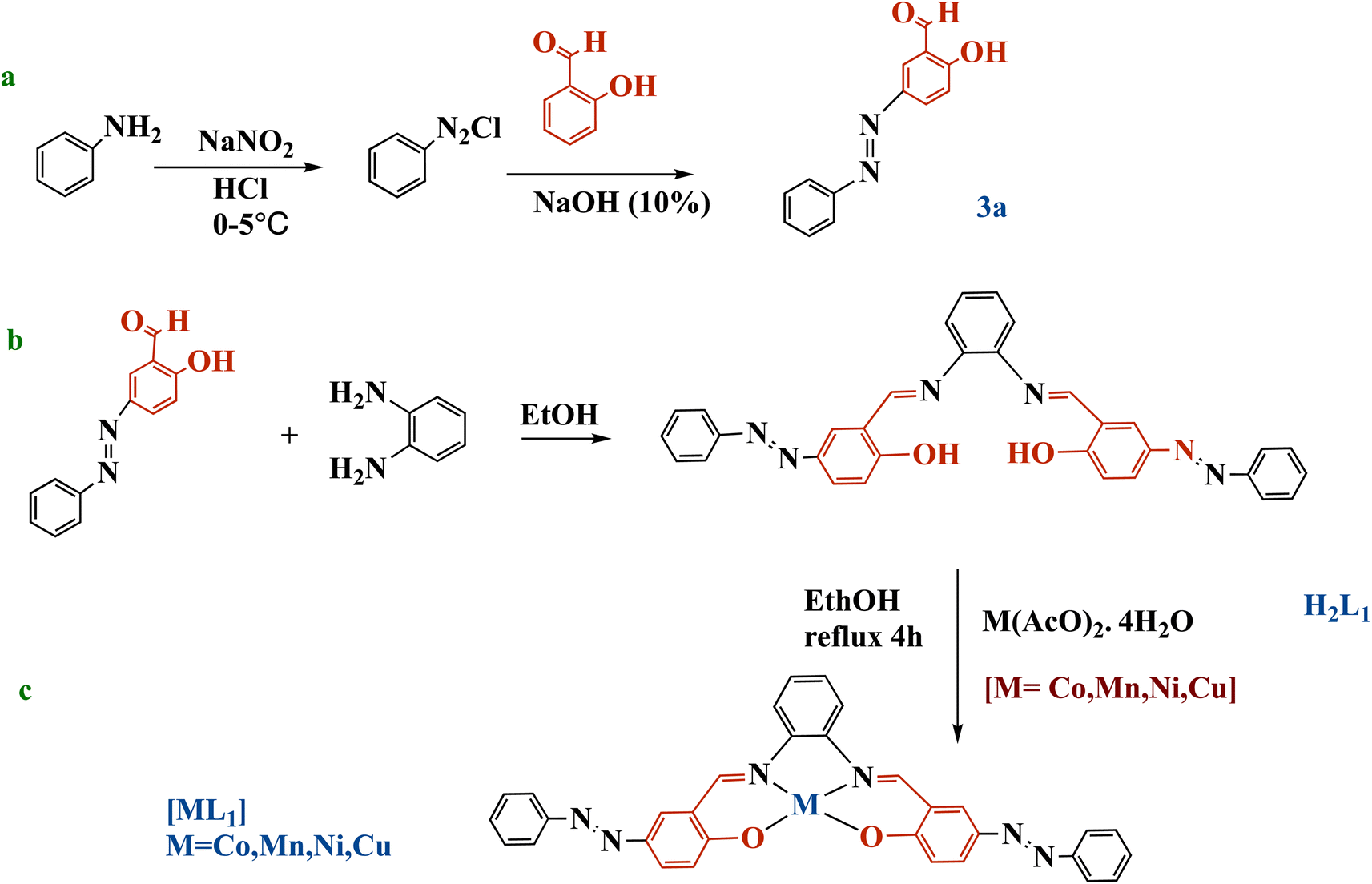 | ||
| Scheme 2 Synthesis of azobenzene precursor 3a (a), salphen-azobenzene ligand H2L1 (b), and metal complexes [ML1] (M: Co, Mn, Ni, or Cu) (c). | ||
Ligand H2L1 was synthesized through the condensation between (0.04 mol) of obtained salicylaldehyde azobenzene (3a) and (0.02 mmol) of o-phenylenediamine, using ethanol as solvent (Scheme 2b). The reaction was stirred and refluxed for 6 h. Then the dark yellowish brown precipitated product was cooled down at room temperature, then filtered, rinsed with ethanol, and recrystallized in ethanol. The obtained orange crystals were also filtered, and then dried in air. Yield: 82%; mp: 250 °C; FT-IR ν(CN) 1606, ν(NN) 1446, 1284, ν(CC) 1463, ν(OH) 3447. 1H NMR (DMSO-d6 as solvent, δ ppm): 13.71 (s, 2H, O–H); 8.99 (s, 2H, CHN), 7.95–7.06 (m, 20 H, aromatic C–H). 13C NMR (DMSO-d6), (δ ppm):163.98 (C–OH): 160 (CN), 163.98, 118.01–150.89 (aromatic C atoms). UV-Vis (CHCl3) (kmax, nm): 357, 456.
2.3.2.1 Synthesis of [CoL1]. (0.012 mol) of H2L1 was first dissolved in 50 mL of ethanol. Then (0.012 mol) of cobalt(II) acetate tetrahydrate solution was added dropwise to H2L1 solution, and the mixture was refluxed for 4 h. The complex was isolated from mixture as the dark brown by filtration, then washed with ethanol and ether. Drying in air yielded [CoL1] complex as a dark brown solid. Yield: 70%; mp: 273 °C; FT-IR: ν(C
N) 1592, ν(NN)1435, ν(C–O) 1287, ν(CC) 1520, ν(Co–O) 678, ν(Co–N) 499. UV-Vis (CHCl3) (kmax, nm): 381, 460, and 467. Elemental and ICP-MS analysis for compound (C32H22CoN6O2) MW (581.50 g mol−1): calculated: C, 66.10%; H, 3.81%; Co, 10.13%; N, 14.45%; O, 5.50%, found C, 67.10; H, 4.51; N, 15.05; O, 6.50 Co, 7.4%
2.3.2.2 Synthesis of [MnL1]. (0.012 mol) of H2L1 was first dissolved in 50 mL of ethanol. Then (0.012 mol) of manganese(II) acetate tetrahydrate solution was added dropwise to H2L1 solution, and the mixture was refluxed for 4 h. The complex [MnL1] was isolated from mixture as a deep brown solid by filtration, then washed with ethanol and ether. Yield: 69% C; FT-IR ν(C
N) 1597, ν(NN) 1430, ν(C–O) 1287, ν(CC) 1520, ν(Mn–O) 774, (Mn–N) 506; UV-Vis (CHCl3) (kmax, nm): 400, 462. Elemental and ICP-MS analysis for compound (C32H22Mn N6O2) MW (577.51 g mol−1) calculated: C, 66.55%; H, 3.84%; Mn, 9.51%; N, 14.55%; O, 5.54%, found C, 65.99%; H, 4.96%; N, 14.10%; O, 10.20%; Mn, 8.2%.
2.3.2.3 Synthesis of [NiL1]. (0.012 mol) of H2L1 was dissolved in 50 mL of ethanol. (0.012 mol) of nickel(II) acetate tetrahydrate solution was added dropwise to the ligand solution and refluxed for 4 hours. After refluxing, the solution had turned dark red. The complex was isolated by filtration and purified by washing with ethanol and ether. Drying yielded [NiL1] as a dark red solid. Yield: 72%; mp: 274 °C; FT-IR: ν(C
N) 1594, ν(NN) 1432, ν(C–O) 1284, ν(CC) 1517, ν(Ni–O) 667, ν(Ni–N) 495. UV-Vis (CHCl3) (kmax, nm): 381, 460, and 467. Elemental and ICP-MS analysis for compound (C32H22N6NiO2) MW (581.26 g mol−1): calculated: C, 66.12%; H, 3.82%; N, 14.46%; Ni, 10.10%; O, 5.50%, found C, 65.99%; H, 4.96%; N, 15.3%; O, 10.28%; Ni, 9.1%
2.3.2.4 Synthesis of [CuL1]. The ligand H2L1 (0.012 mol) was dissolved in ethanol. A solution of copper(II) acetate tetrahydrate (0.012 mol) was added dropwise to the ligand solution. The mixture was refluxed for 4 hours. During refluxing, the solution turned dark yellowish brown, indicating the formation of the complex. The complex was isolated from mixture as a dark yellowish brown solid by filtration, then washed with ethanol and ether. Yield: 70%; mp: 276 °C; FT-IR: ν(C
N)1590, ν(NN)1430, ν(C–O) 1285, ν(CC) 1518, ν(Cu–O) 663, ν(Cu–N) 492. UV-Vis (CHCl3) (kmax, nm): 380, 457, and 467. Elemental and ICP-MS analysis for compound (C32H22CuN6O2) MW (586.11 g mol−1): calculated: C, 65.58%; H, 3.78%; Cu, 10.84%; N, 14.34%; O, 5.46%, found C, 65.1%; H, 4.98%; N, 15.3%; O, 10.88%; Co, 8.1%
:1 v/v). Then 0.5 g of [ML1] was added to the mixture. The desired catalysts Silica-N-ML1 (Silica: SBA-15, MCM-41 or MCM-48, M: Co, Mn, Ni or Cu) were obtained after stirring the mixture at 50 °C for 15 h, followed by filtration and washing thoroughly with CH2Cl2 and EtOH to remove the excess of [ML1], and drying the obtained solid overnight at 80 °C for 15 h.
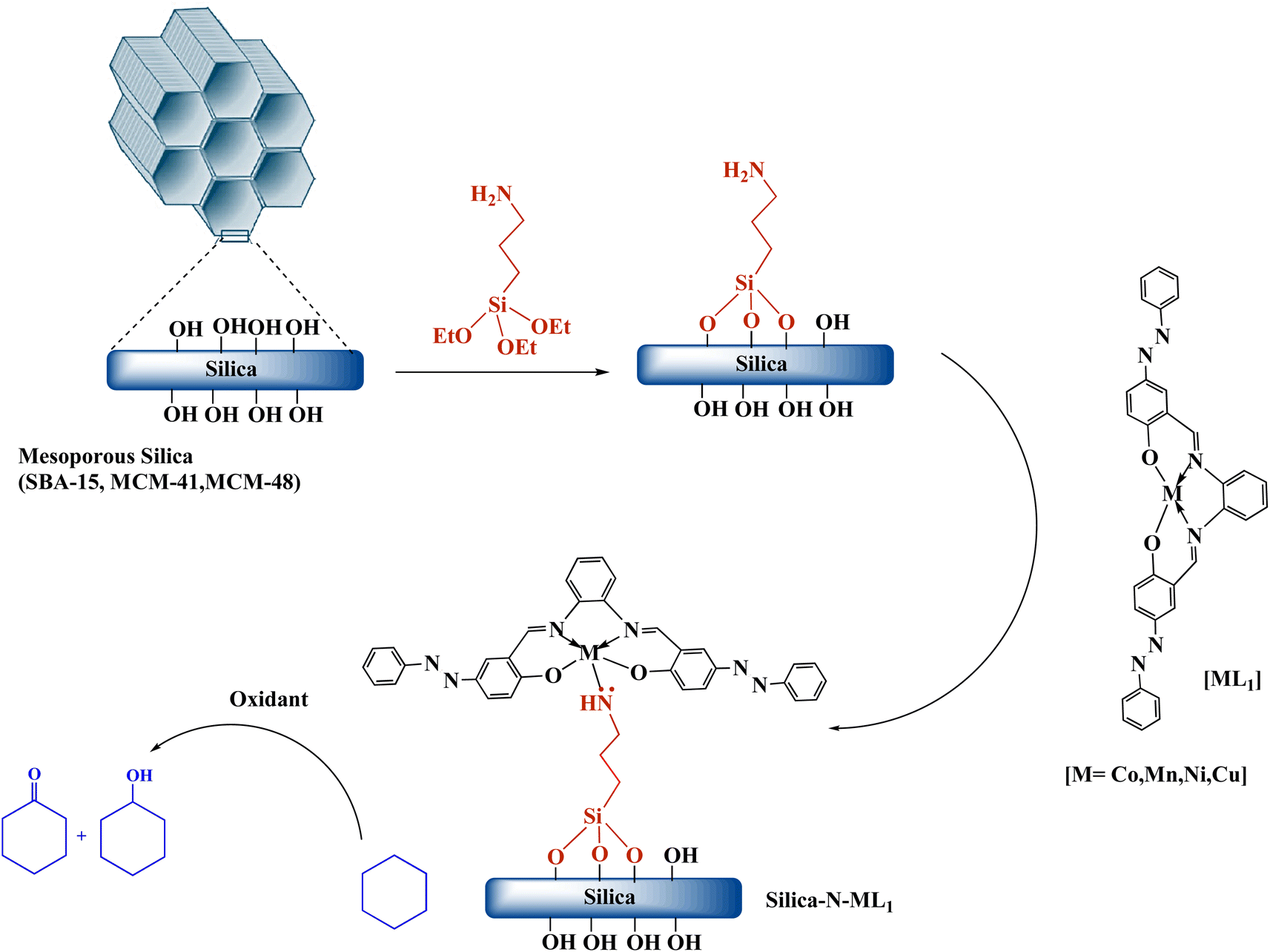 | ||
| Scheme 3 Synthesis of the nanocatalysts Silica-N-ML1 (Silica: SBA-15, MCM-41 or MCM-48; M: Co, Mn, Ni or Cu). | ||
The conversion of cyclohexane and selectivity of cyclohexanone and cyclohexanol were calculated according to the following equations:
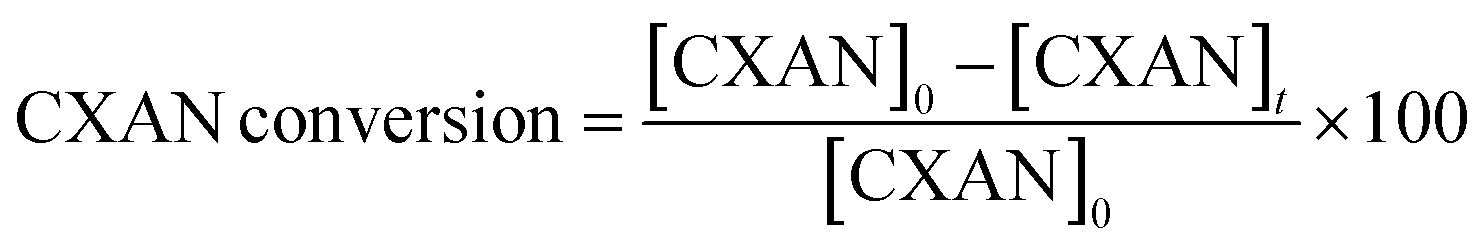
where [CXON]t is the cyclohexanone concentration after 6 hours
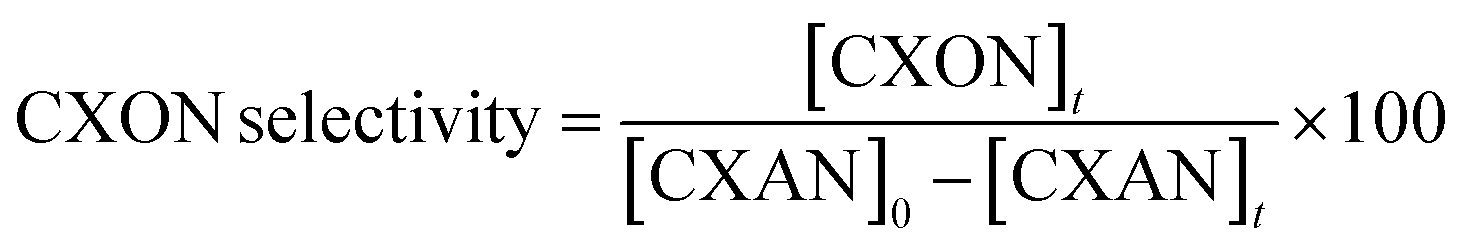
where [CXOL]t is the cyclohexanone concentration after 6 hours.
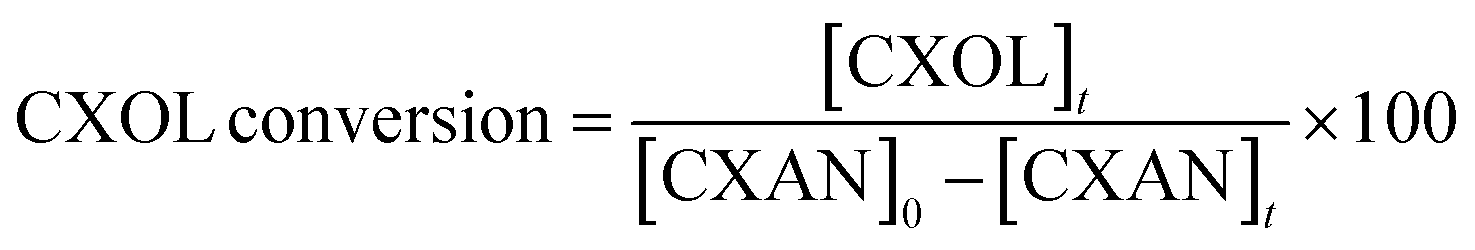
3. Results and discussion
3.1 Synthesis and characterization
FT-IR spectroscopy was used to characterize the structures of the synthesized salphen-azobenzene ligand H2L1 and its Co, Mn, Ni, and Cu complexes. The IR spectrum of H2L1 (Fig. 1a) confirmed the formation of the salphen backbone, due the disappearance of the aldehyde carbonyl peak at 1703 cm−1 (◄) of the azobenzene compound (3a), and appearance of a new imine stretching band at 1606 cm−1 (⊙). Additional characteristic peaks were observed at 1446 cm−1 (Δ) assigned to the NN stretching, and 3200 cm−1 (▲) corresponding to aromatic C–H stretching. Furthermore, the broad peak at 3447 cm−1 (□) was assigned to O–H stretching.
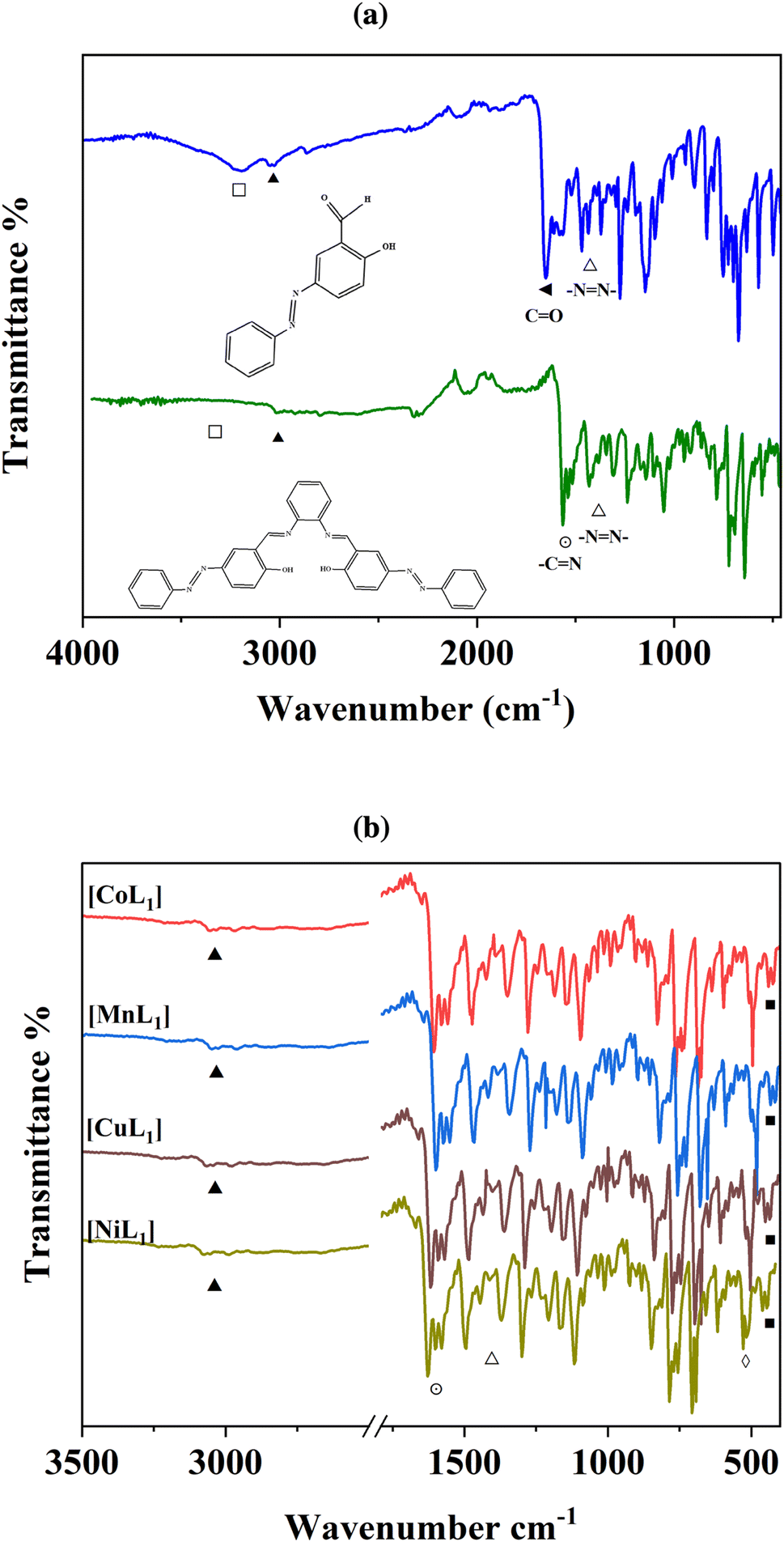 | ||
| Fig. 1 FTIR spectra of (3a) and H2L1 (a), and its corresponding Co, Mn, Ni, and Cu complexes (b). | ||
The coordination of Co(II), Mn(II), Ni(II), and Cu(II) to H2L1 was indicated by shifts in the CN stretching bands to lower wavenumbers of 13–25 cm−1 (⊙) in the spectra of [CoL1], [MnL1], [NiL1], and [CuL1] compared to H2L1 (Fig. 1b). The O–H stretching in H2L1 is absent in the complexes, confirming the deprotonation and coordination of O–H to the metal centers. Finally, new weak bands in the low wavenumber region of 678–674 cm−1 (◊) and 499–506 cm−1 (■) provide evidence of M–O and M–N bonding in the Co, Mn, Ni, and Cu complexes. Overall, the FT-IR analyses indicated the successful synthesis of the salphen-azobenzene ligand and its Co(II), Mn(II), Ni(II), and Cu(II) metal complexes.
The structure of the synthesized salphen-azobenzene ligand H2L1 was confirmed by the 1H and 13C NMR analysis. As shown in Fig. 2, the 1H NMR spectrum displays a singlet at δ 13.23 ppm assigned to the phenolic –OH protons, indicating an enolic contribute to the structure as reported previously.54 Additionally, a characteristic singlet signal observed at δ 8.98 ppm attributed to the azo-methine (–CHN–) protons. The aromatic proton signals are observed at δ 6.90–8 ppm.55 The 13C NMR spectrum presented in Fig. 3 shows the aromatic carbon signals at δ 160.20–118.19 ppm. The signals observed at δ 163.88 and δ165.90 ppm correspond to the phenolic C–OH and CHN carbons, respectively. These findings confirmed the proposed structure of the synthesized salphen-azobenzene ligand H2L1.
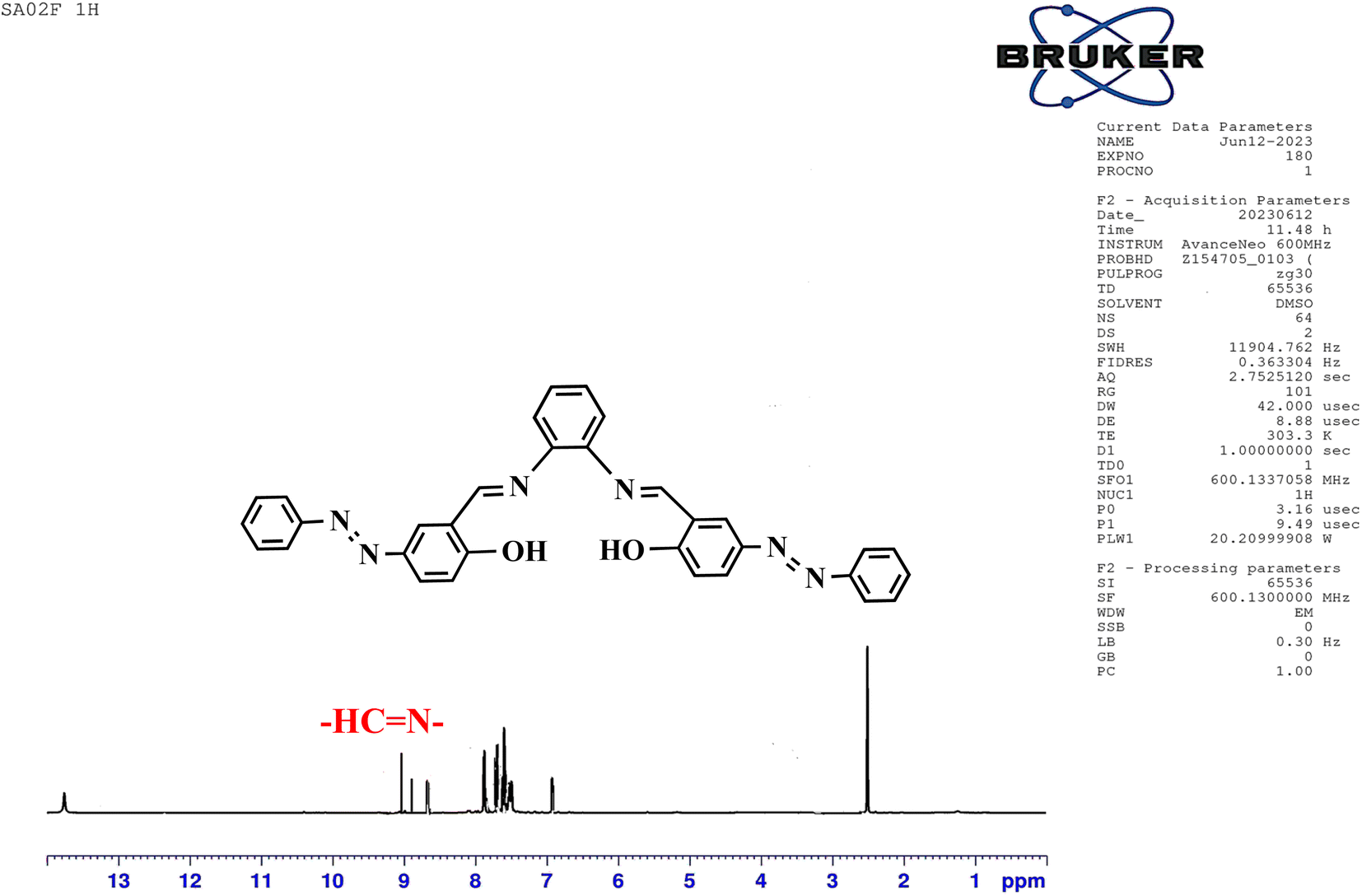 | ||
| Fig. 2 1H NMR spectra of H2L1. | ||
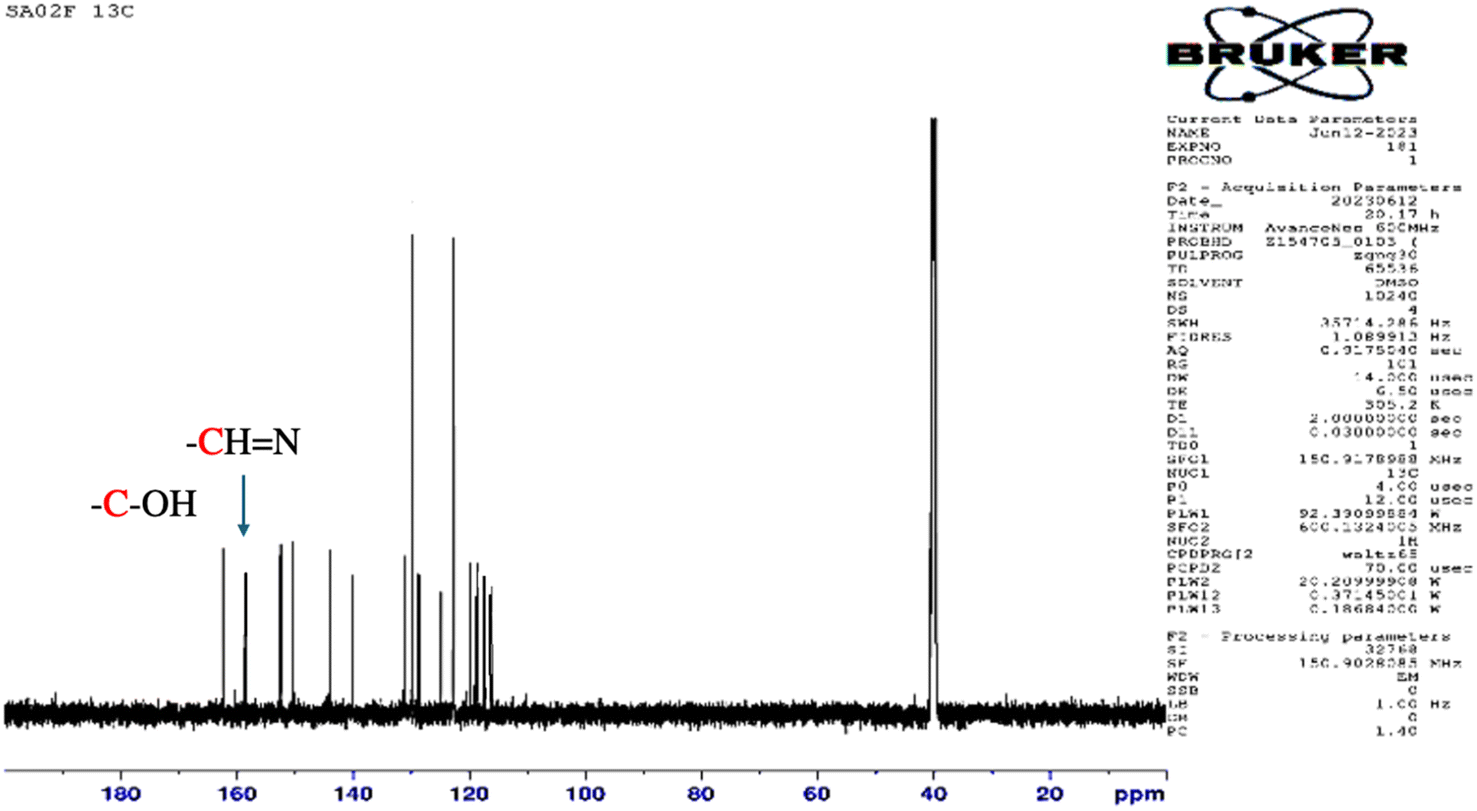 | ||
| Fig. 3 13C NMR spectra of H2L1. | ||
UV-Visible absorption spectra were collected for azobenzene (3a), salphen-azobenzene ligand H2L1, and its Co, Mn, Ni, and Cu complexes from 260–700 nm in chloroform solution at room temperature, and the obtained results are presented in Fig. 4. The azobenzene intermediate (3a) exhibited characteristic π–π* and n–π* electronic transitions, with the prominent trans π–π* absorption band appearing sharply at 350 nm (Fig. 4a). While the spectrum of ligand H2L1 displayed broader absorption bands that were red-shifted, which probably attributed the extension of the conjugated π system. The trans π–π* transition was observed at around 390 nm (Fig. 4a). Upon complexation of ligand H2L1 with Co(II), Mn(II), Ni(II) and Cu(II), the spectra of [CoL1], [MnL1], [NiL1] and [CuL1] showed a perturbation of the ligand-centered orbitals as evidenced by decreased intensities and shifting of the π–π* and n–π* bands to longer wavelengths. This suggests metal–ligand orbital mixing upon coordination. Additionally, d–d transition bands ascribed to the metal ions emerged in the higher wavelength region, observed at around 485 nm for [CoL1], 530 nm for [MnL1], 550 nm for [NiL1] and 525 nm for [CuL1] (Fig. 4b).
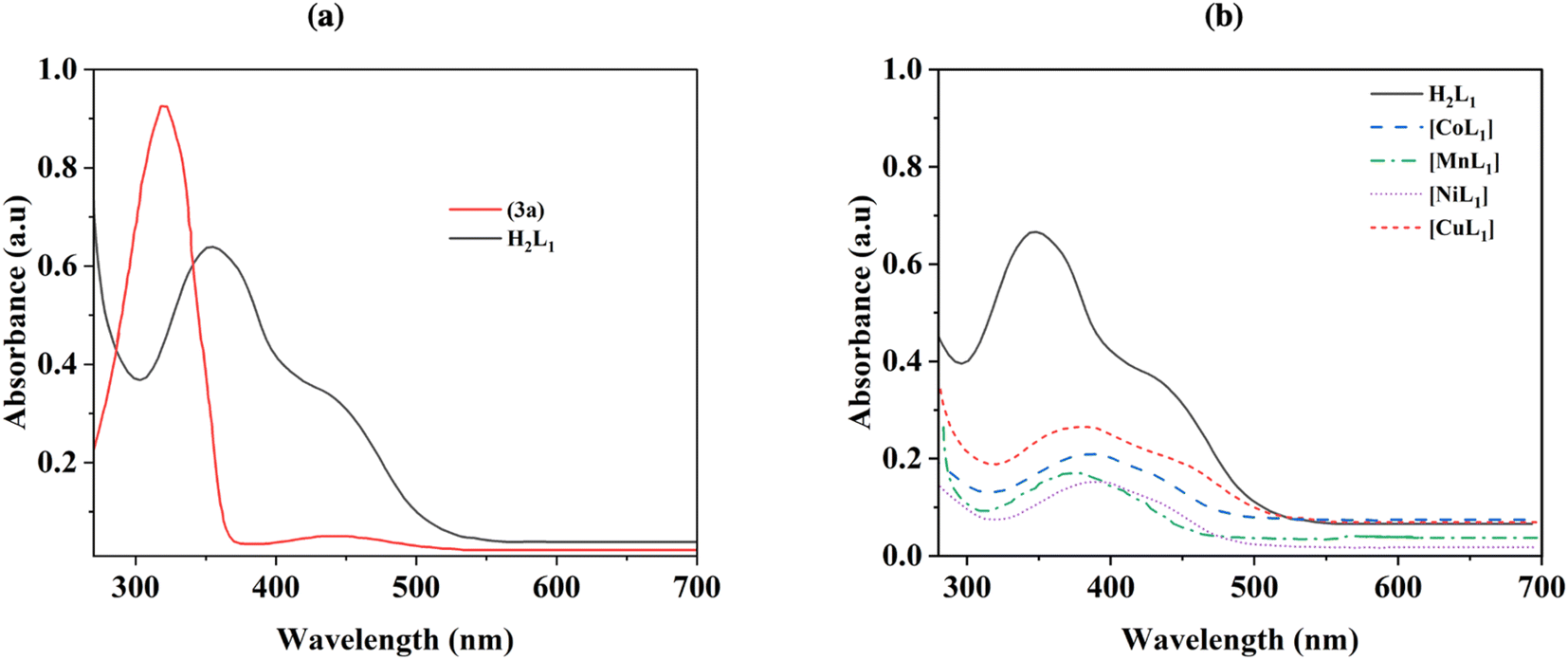 | ||
| Fig. 4 UV-Visible spectra of 3a and H2L1 (a), and its corresponding Co, Mn, Ni, and Cu complexes (b). | ||
The thermal behavior of the complexes [CoL1], [MnL1], [NiL1], and [CuL1] was analyzed using thermogravimetric analysis (TGA) over a temperature range of 30–800 °C. The obtained results are displayed in Fig. 5. The initial weight loss step observed below 200 °C, which can be attributed to the evaporation of physically adsorbed water and solvent. The second weight loss step observed after 200 °C attributed to the breakdown of the organic ligand. Complexes [CoL1] and [NiL1] showed slight thermal stability compared to [CuL1] and [MnL1]. The complete breakdown of the salphen-azobenzene ligand occurred in the temperature ranges of 200–330 °C for [NiL] (85.90 wt%), 200–320 °C for [CoL1] (86 wt%), 200–270 °C for [MnL1] (86.20 wt%), and 200–260 °C for [CuL1] (85.1 wt%).
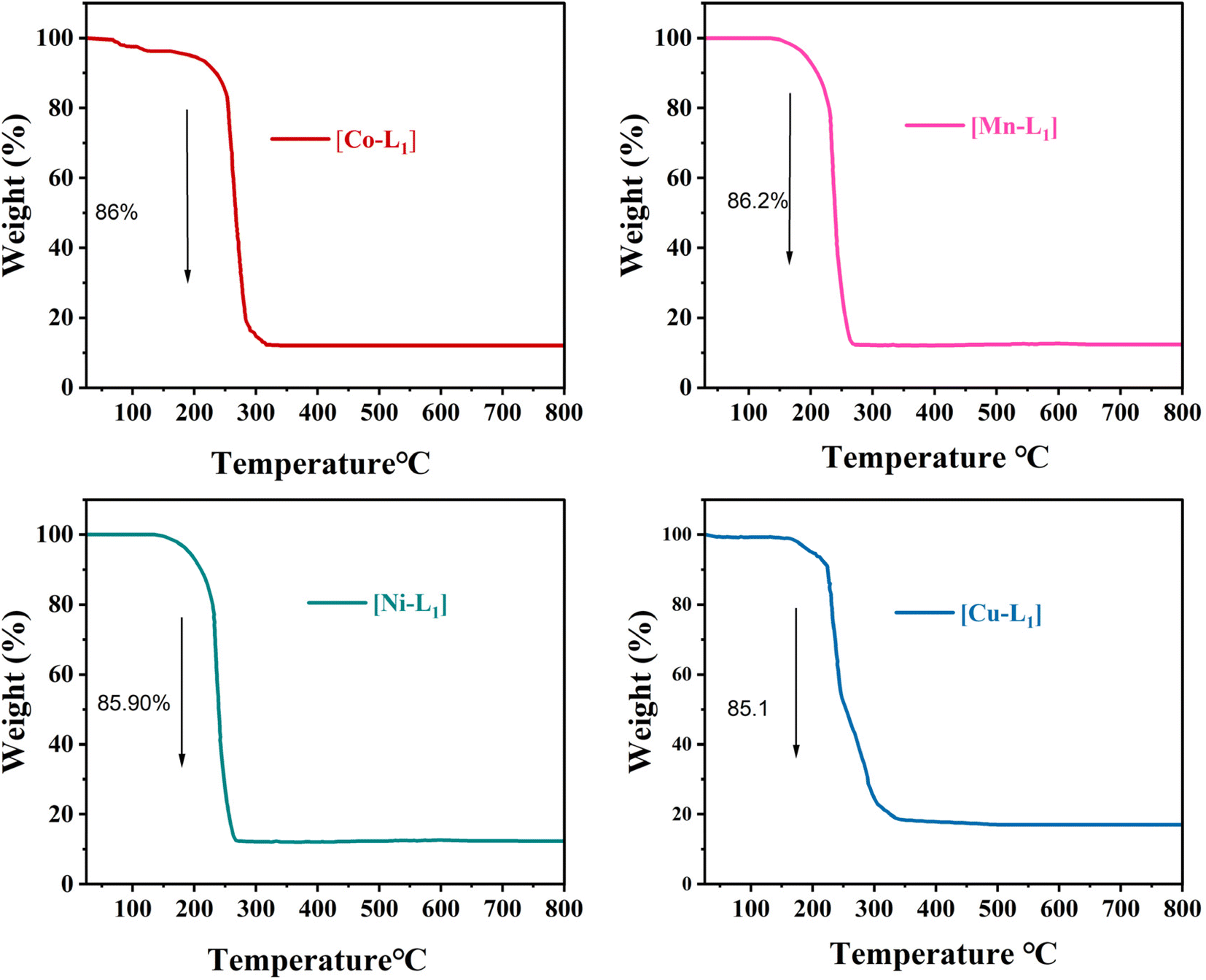 | ||
| Fig. 5 TGA curves of [CoL], [MnL], [NiL], and [CuL] complexes under air. | ||
Amino-functionalized materials were prepared by grafting aminopropyltriethoxysilane (APTES) onto the surface of SBA-15, MCM-41, and MCM-48, according to literature procedures.52 The FTIR spectra of the commercial silica materials (i.e., SBA-15, MCM-41, and MCM-48), and their corresponding modified silica materials (i.e., SBA-15-N, MCM-41-N, and MCM-48-N) are presented in Fig. 6. In the fingerprint region (700–1300 cm−1) of the mesoporous silica spectra, the symmetric (○) and asymmetric (●) stretching vibrations of the Si–O–Si linkages forming the silica frameworks showed two bands at ∼1045 cm−1 and 800 cm−1, respectively (Fig. 6). Upon grafting of APTES onto the silica surfaces, the intensities of both peaks corresponding to the OH stretching (■) and bending (□) vibrations noticeably decreased. This reduction occurred because the silica surface silanol groups (Si–OH) transformed to Si–O–Si–(CH2)3–NH2 after successful reaction with APTES. Additionally, a new peak at ∼2980 cm−1 (∇) appeared, which can be assigned to the CH2 groups of the grafted APTES. Further peaks (◊) at ∼700 cm−1 can be attributed to the stretching vibrations of the N–H bond of the APTES. All these findings demonstrate the successful covalent attachment of APTES onto the surfaces of the silica materials.
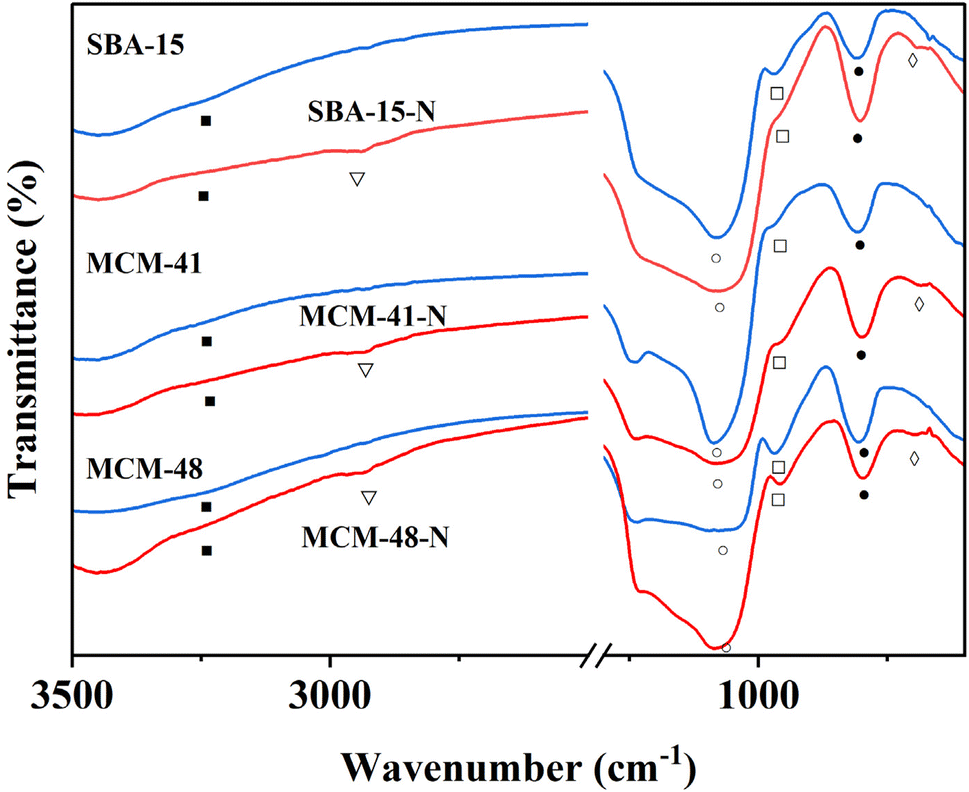 | ||
| Fig. 6 FTIR of SBA-15, MCM-41, and MCM-48 and modified silica SBA-15-N, MCM-41-N, and MCM-48-N. | ||
After the addition of [ML1] complexes to the silica materials, FTIR analysis were performed to the obtained materials Silica-N-ML (i.e., Silica: SBA-15, MCM-41, and MCM-48), and the obtained results are presented in Fig. 7a–c. The intensity of the peaks around 2980–3000 cm−1 (▲) corresponding to C–H increased compared to the same peaks in Silica-N. This can be due to the additional peaks of C–H groups in [ML1] complexes. Three peaks were observed at ∼1506 cm−1 (⨀), ∼1400 (●), and ∼1370 (Δ) which can be assigned to CN, CC, and NN bonds in the metal complexes, respectively. The N–H bending (□) of the NH2 group was shifted to a lower frequency of ∼675 cm−1 after the addition of [ML1] complexes, which can be due to the formation of a coordination bond between the electron lone pair of NH2 groups and vacant orbital of the metal's ions (i.e., Co, Mn, Ni, and Cu). Moreover, other tiny peaks appeared between 525 and 550 cm−1 (■), can be attributed to metal–O bonds. Co–O peak appeared at ∼542 cm−1, Mn–O at ∼547 cm−1, Ni–O at ∼556 cm−1, and Cu–O at ∼525 cm−1.
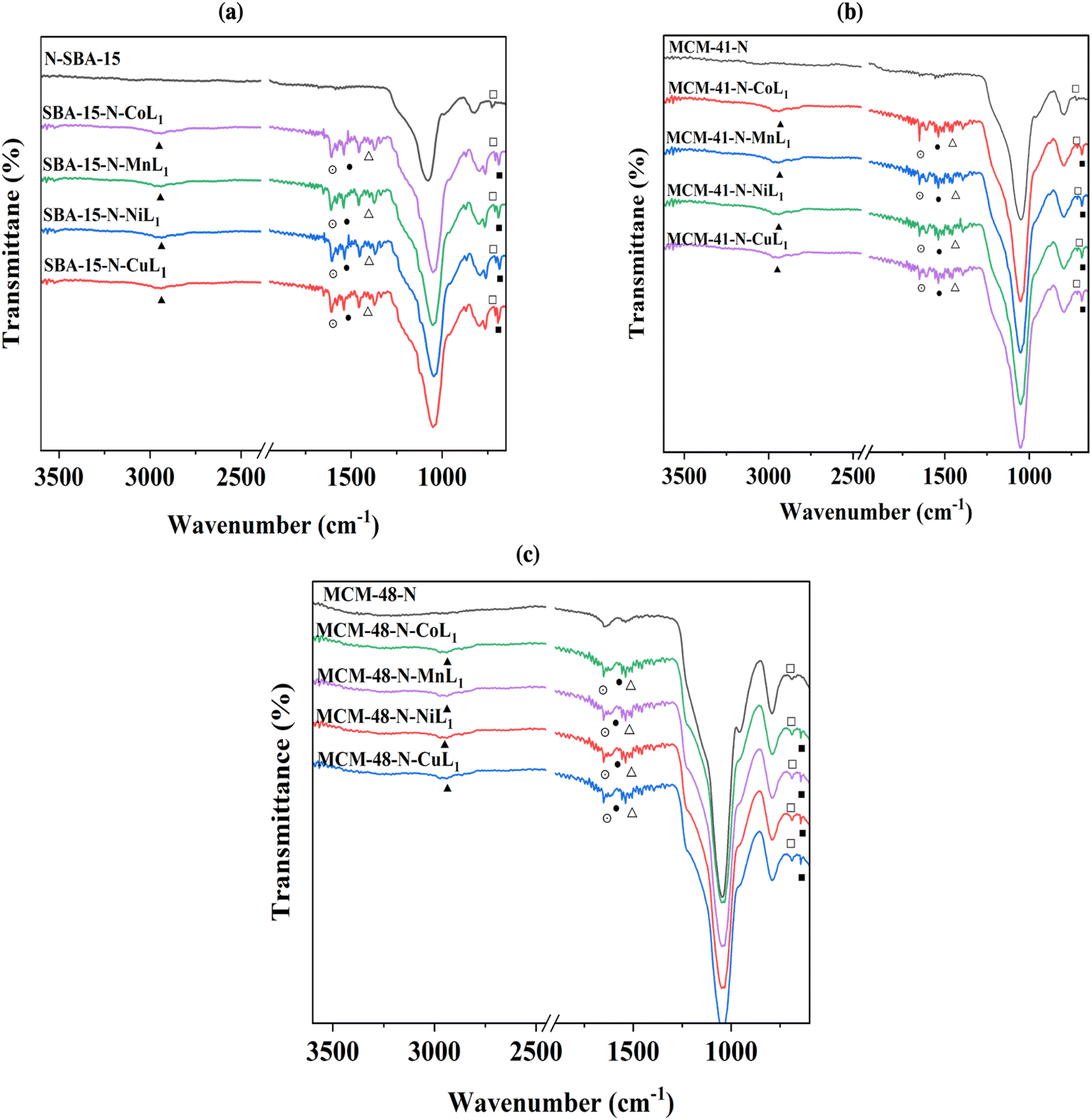 | ||
| Fig. 7 FTIR spectra of SBA-15-N-ML1 (a), MCM-41-N-ML1 (b), and MCM-48-N-ML1 (c), with M = Co, Mn, Cu or Ni. | ||
These observations confirmed the successful immobilization of [CoL1], [MnL1], [NiL1], and [CuL1] complexes onto the silica surfaces.
N2 adsorption/desorption analysis was carried out to determine the surface area, pore size, and pore volumes of the different mesoporous silica materials before and after functionalization and immobilization of the metal complexes [ML1]. The average pore diameter and pore volume were derived using the Barrett, Joyner, and Halenda (BJH) method. Fig. 8, 9 and 10 present the resulting Brunauer, Emmett, and Teller (BET) isotherms and the pore size distribution curves of commercial silica (i.e., SBA-15, MCM-41, MCM-48) as well as the amino-functionalized silica (i.e., SBA-15-N, MCM-41-N, MCM-48-N), and the silica supported metallosalphen-azobenzene complexes Silica-N-ML1 (M: Co, Mn, Ni, or Cu; Silica: SBA-15, MCM-41, or MCM-48). Moreover, Table 1 summarizes the textural properties of all samples.
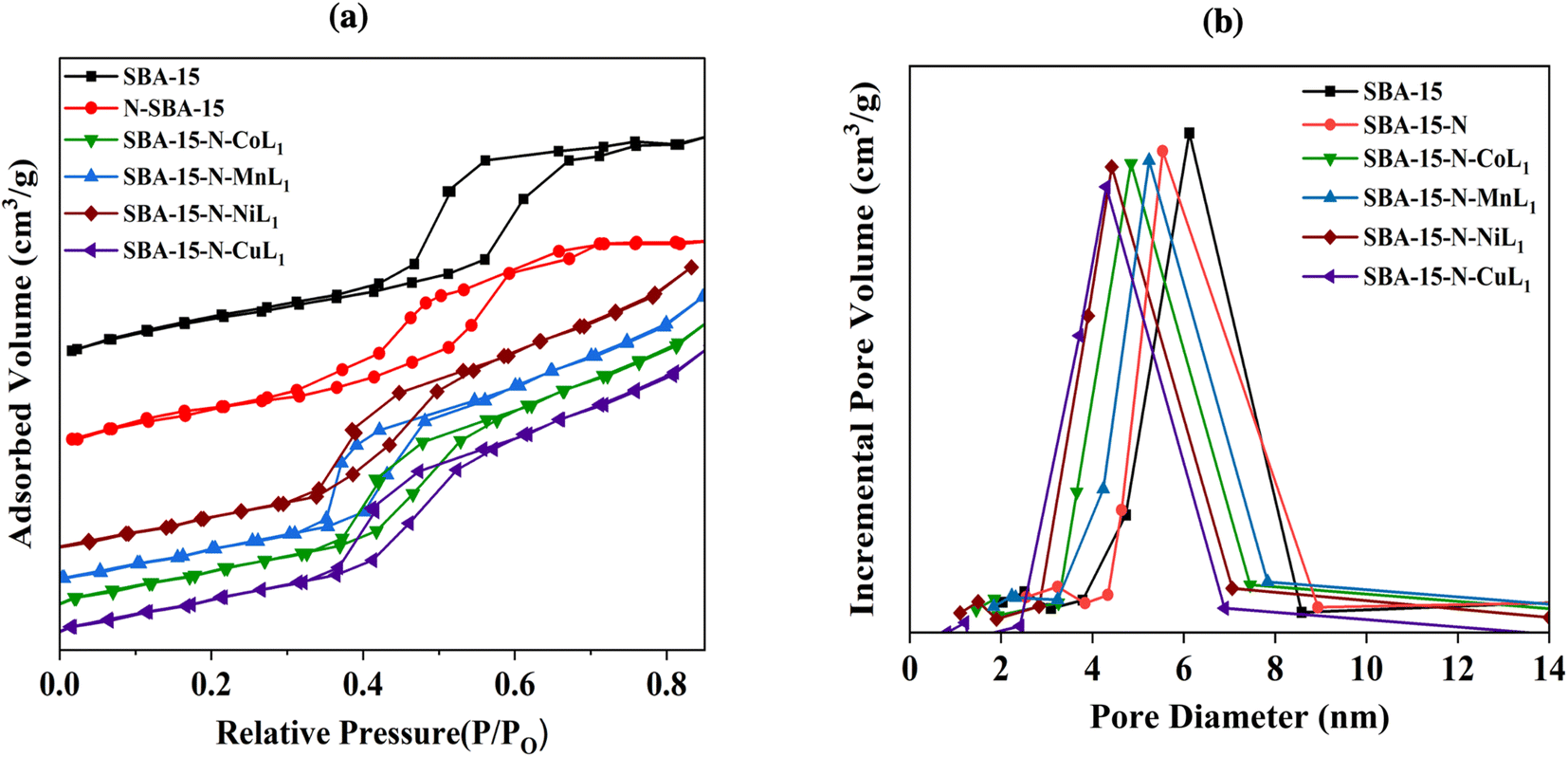 | ||
| Fig. 8 BET isotherms (a) and pore size distribution curves (b) of samples SBA-15, SBA-15-N, and SBA-15-N-ML1 (M: Mn, Co, Ni, or Cu). | ||
| Samples | SBETa BJHb (m2 g−1) | Pore diameter BJHb (nm) | Pore volume BJHb (cm3 g−1) |
|---|---|---|---|
| a Brunauer, Emmett, and Teller.b Barrett, Joyner, and Halenda. | |||
| SBA-15 | 663.19 | 5.60 | 0.61 |
| SBA-15-N | 320.90 | 4.80 | 0.46 |
| SBA-15-N-CoL1 | 248.30 | 3.88 | 0.37 |
| SBA-15-N-Mn L1 | 249.20 | 3.98 | 0.38 |
| SBA-15-N-NiL1 | 247.20 | 3.85 | 0.36 |
| SBA-15-N-CuL1 | 247.60 | 3.88 | 0.37 |
| MCM-41 | 1130.00 | 2.40 | 0.65 |
| MCM-41-N | 455.80 | 1.8 | 0.27 |
| MCM-41-N-CoL1 | 372.90 | 1.50 | 0.19 |
| MCM-41-N-MnL1 | 380.18 | 1.50 | 0.18 |
| MCM-41-N-NiL | 378.20 | 1.48 | 0.17 |
| MCM-41-N-CuL1 | 377.99 | 1.47 | 0.17 |
| MCM-48 | 1000.00 | 2.70 | 0.56 |
| MCM-48-N | 510.08 | 1.60 | 0.39 |
| MCM-48-N-CoL1 | 406.05 | 0.98 | 0.21 |
| MCM-48-N-MnL1 | 409.03 | 1.00 | 0.23 |
| MCM-48-N-NiL1 | 406.00 | 0.97 | 0.21 |
| MCM-48-N-CuL1 | 405.01 | 0.96 | 0.20 |
The obtained N2 adsorption/desorption isotherms of all samples demonstrated type IV isotherms with H1 hysteresis loops, indicating the maintenance of the mesoporous structures after the amino-functionalization and [ML] complexes immobilization (Fig. 8a). Commercial SBA-15 had a BET surface area of approximately 663.19 m2 g−1 and pore volume of 0.61 cm3 g−1 (Table 1). Its pore size distribution curve exhibited a peak at ∼5.6 nm (Fig. 8b). After APTES grafting, SBA-15-N's surface area and pore volumes decreased to 320.90 m2 g−1 and 0.46 cm3 g−1, respectively, with an average pore diameter of ∼4.8 nm (Fig. 8b), indicating a successful grafting of APTES onto SBA-15 surface.
After addition of [ML1], the surface area, pore size, and pore volume of SBA-15-N were all decreased (Table 1). Specifically, the surface areas of SBA-15-N-CoL1, SBA-15-N-MnL1, SBA-15-N-NiL1, and SBA-15-N-CuL1 were decreased to 248.30 m2 g−1, 249.20 m2 g−1, 247.20 m2 g−1 and 247.60 m2 g−1, respectively. Pore sizes of SBA-15-N-CoL1, SBA-15-N-MnL1, SBA-15-N-NiL1, and SBA-15-N-CuL1 were reduced to 3.88 nm, 3.98, 3.85 and 3.88 nm, respectively (Table 1). Additionally, pore volumes of SBA-15-N-CoL1, SBA-15-N-MnL1, SBA-15-N-NiL1, and SBA-15-N-CuL1 reduced to 0.37 cm3 g−1, 0.38 cm3 g−1, 0.36 cm3 g−1 and 0.37 cm3 g−1, respectively. This finding indicated a successful immobilization of the [ML1] complexes onto SBA-15-N surface.
Similar trends were observed for the MCM-41 samples. The commercial MCM-41 exhibited a BET surface area of approximately 1130.00 m2 g−1, with a pore volume of 0.65 cm3 g−1 (Table 1). The pore size distribution of MCM-41 showed a sharp peak centered around 2.40 nm (Fig. 9b), confirming a uniform pores size distribution. After the amino-functionalization of MCM-41 with APTES (N-MCM-41), the surface area was decreased to approximately 455.80 m2 g−1. The average pore diameter of N-MCM-41 was also decreased to 1.8 nm (Fig. 9b). Additionally, the average pore volume of MCM-41-N was reduced to 0.27 cm3 g−1. These findings indicated the successful grafting of APTES onto MCM-41 surface.
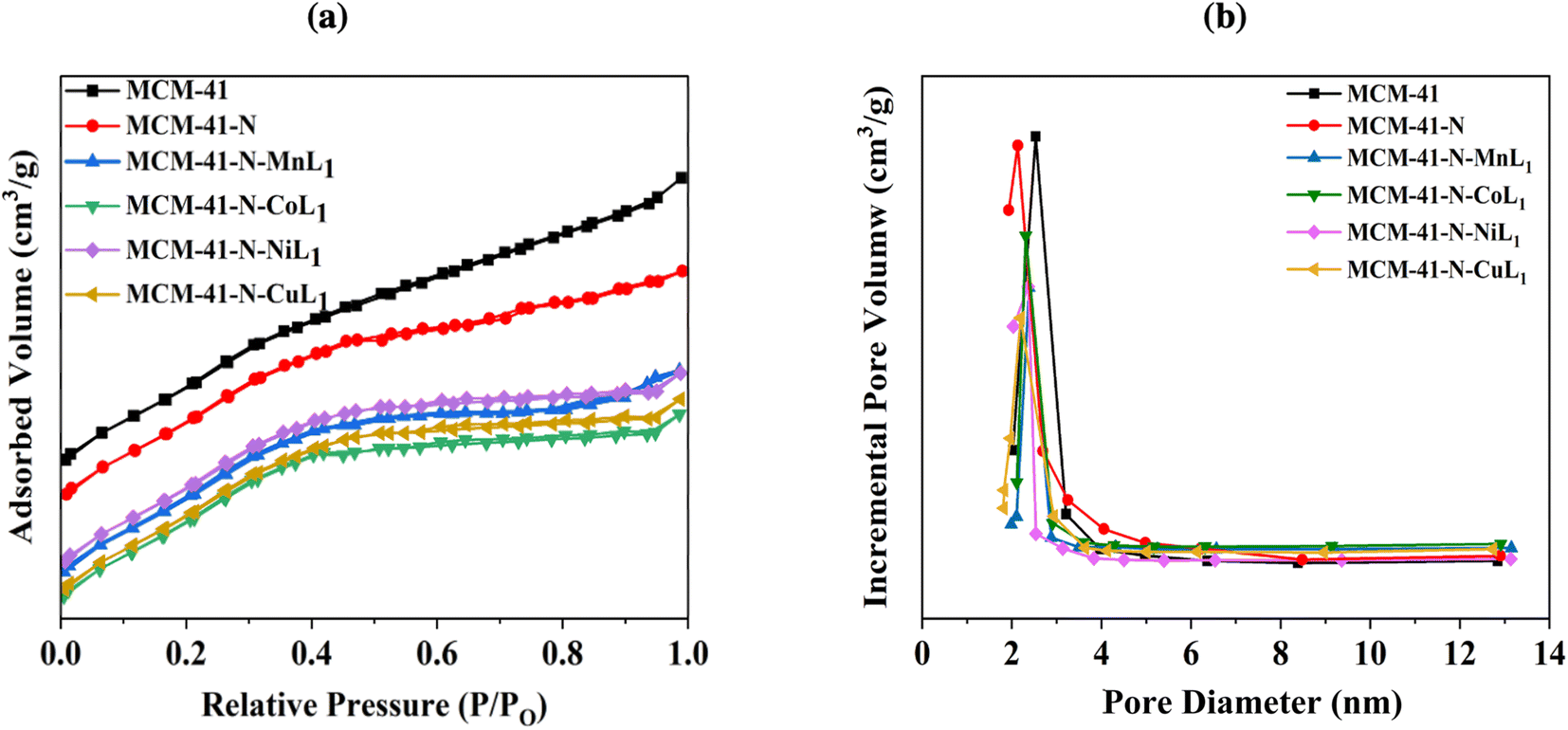 | ||
| Fig. 9 BET isotherms (a) and pore size distribution curves (b) of samples MCM-41, MCM-41-N, and MCM-41-N-ML1 (M: Mn, Co, Ni, or Cu). | ||
Furthermore, similar behavior was observed after the introduction of the metal complexes [ML1] into MCM-41-N. Specifically, the surface area of MCM-41-N was reduced to 380.18 m2 g−1, 372.90 m2 g−1, 378.20 m2 g−1, and 377.98 m2 g−1 for samples MCM-41-N-CoL1, MCM-41-N-MnL1, MCM-41-N-NiL1, and MCM-41-N-CuL1, respectively (Table 1). The pore sizes were also reduced to 1.50 1.50 nm, 1.48 nm, and 1.47 nm, respectively. Additionally, the pore volumes were reduced to 0.18 cm3 g−1, 0.19 cm3 g−1, 0.17 cm3 g−1, and 0.17 cm3 g−1, respectively (Fig. 9b).These results indicated the successful immobilization of the [ML1] complexes onto MCM-41-N surface.
Comparable results were found for the MCM-48 samples. The commercial MCM-48 exhibited a BET surface area of approximately 1000.00 m2 g−1, and an average pore volume of 0.56 cm3 g−1 (Table 1). The pore size distribution of MCM-48 showed a sharp peak at 2.7 nm (Fig. 10b), confirming uniform mesopores. After grafting APTES onto surface of MCM-48 its surface dramatically decreased to 510 m2 g−1. Similarly, the average pore diameter and average pore volume of N-MCM-48 was measured to be approximately 1.6 nm and 0.39 cm3 g−1, respectively (Fig. 10b, Table 1). This result indicated the successful grafting of APTES onto MCM-48 surface. As expected, after the incorporation of [ML1] complexes into MCM-48-N the surface area was decreased again to 409 m2 g−1, 406 m2 g−1, 406 m2 g−1 and 405 m2 g−1 for samples MCM-48-N-CoL1, MCM-48-N-MnL1, and MCM-48-N-NiL1, and MCM-48-N-CuL1, respectively (Table 1). In addition, the average pore diameter of the obtained samples were also reduced to 1 nm, 0.98 nm, 0.96 nm, and 0.97 nm respectively (Fig. 10b). Furthermore, similar behavior was observed for the average pore volumes of the prepared samples. The average pore volume of the final materials was also reduced to 0.23 cm3 g−1, 0.23 cm3 g−1, 0.20 cm3 g−1, and 0.21 cm3 g−1, respectively (Table 1). These findings indicate a successful immobilization of the metal complexes [ML1] onto the amino-functionalized MCM-48-N surface.
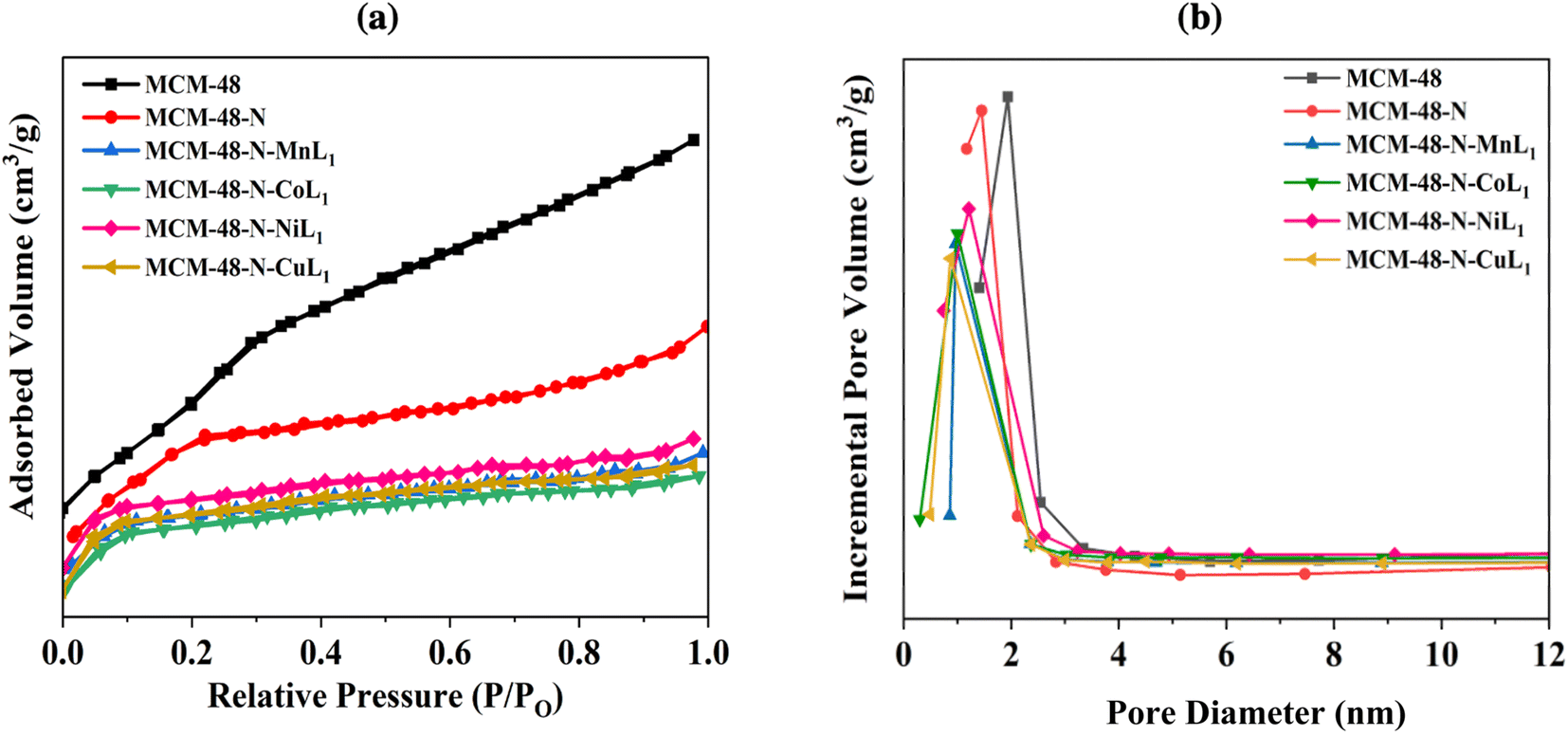 | ||
| Fig. 10 BET isotherms (a) and pore size distribution curves (b) of samples MCM-48, MCM-48-N, and MCM-48-N-ML1 (M: Mn, Co, Ni, or Cu). | ||
TGA analysis was conducted on all silica samples, before and after the incorporation of the metallosalphen-azobenzene complexes to investigate their thermal behavior and determine the [ML1] content in silica. The obtained results are presented in Fig. 11a–c. The thermograms of all samples showed two main steps of weight loss. The initial step below 220 °C presents a weight loss of 2–5%, which is attributed to absorbed solvents and water residues. The second step observed above 220 °C related to the decomposition of the salphen-azobenzene ligand and APTES linker. Notably, the decomposition step of all silica supported [ML1] complexes was started at around 200 °C but extended to around 750 °C, compared to unsupported [ML1] which exhibited a rapid decomposition between 200 °C and 300 °C. This suggests that the silica wall acted as a thermal insulator for the [ML1] complexes.
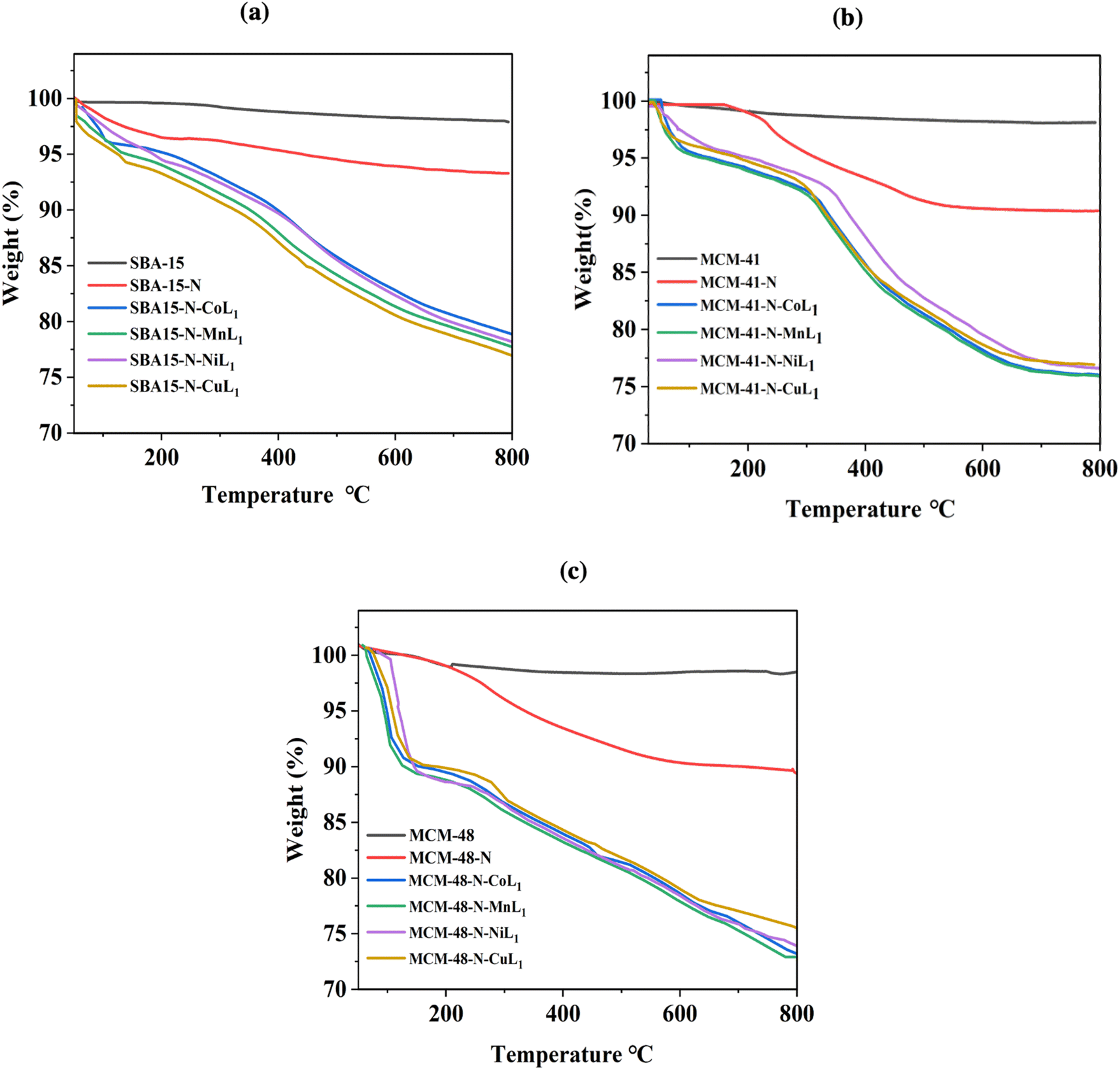 | ||
| Fig. 11 TGA thermographs of SBA-15 samples (a), MCM-41 samples (b), and MCM-48 samples (c). | ||
The thermogravimetric analysis of SBA-15-N, MCM-41-N and MCM-48-N was performed to serve references. For all three materials, the APTES-propylamine groups decomposed between 330–450 °C, with a weight loss of around 6–7%. Specifically, SBA-15-N exhibited a weight loss of 4.5% in this temperature range. MCM-41-N showed a similar weight loss of 4.3%. Similar weight loss of 4.7% was also observed for MCM-48-N. The remaining insignificant weight loss occurring up to 850 °C for all materials could be assigned to condensation reactions between silanol groups on the silica surface.56,57
The weight loss observed between 220–750 °C corresponds to the decomposition of the salphen-azobenzene ligand and APTES linker, with organic weight losses (APTES + L) of 13.10% for SBA-15-N-CoL, 13.6% for SBA-15-N-MnL, 12.75% for MCM-41-N-CoL1, 13.5% for MCM-41-N-MnL1, 12.94% for MCM-48-N-CoL1 and 12.01% for MCM-48-N-MnL1. This is consistent with the ML1 content in silica determined to be in the range of 0.97–1.25 wt% as presented in Table 2, which is determined by ICP-MASS.
The morphology of the amino-functionalized silica SBA-15-N, MCM-41-N, and MCM-48-N, along with their corresponding Silica-N-ML samples were investigated using scanning electron microscopy (SEM). The obtained results are presented in Fig. 12. The SEM images SBA-15-N, MCM-41-N, and MCM-48-N (a, f and k), depict particles with length ranging 0.5–5.0 μm, 0.10–0.27 μm, and 0.12–0.24 μm. Their corresponding SBA-15-N-ML1 (b–e), MCM-41-N-ML1 (g–j), and MCM-48-N-ML1 (l–o) depict particles with length ranging (0.5–0.22), (0.29–0.21), and (0.19–0.22) μm respectively. Samples SBA-15-N, MCM-41-N, and MCM-48-N exhibited some degree of aggregation and formation of tubular agglomerates due to formation of hydrogen bonding between the amine groups and the surface silanols. However, after the addition [ML1] complexes (Fig. 12b–e, g–j and l–o) the aggregation was reduced, and isolated and smaller particles were observed. This decrease provides direct evidence that coordination bonding stabilizes the nanoparticles. Without crosslinking, Silica-N particles could aggregate and grow larger over time. However, the metal–amine coordination bonds restrict this by rigidly connecting particles at a smaller set size. Therefore, the size reductions upon addition of the ML1 complexes directly support our hypothesis that coordination bonding counteracts aggregative processes and enhances nanoparticle stability.
 | ||
| Fig. 12 SEM micrographs of SBA-15-N (a), SBA-15-N-CoL1 (b), SBA-15-N-MnL1 (c), SBA-15-N-NiL1 (d), SBA-15-N-CuL1 (e), MCM-41-N (f), MCM-41-N-CoL1 (g), MCM-41-N-MnL1 (h), MCM-41-N-NiL1 (i), MCM-41-N-CuL1 (j), MCM-48-N (k), MCM-48-N-CoL1 (l), MCM-48-N-MnL1 (m), MCM-48-N-NiL1 (n), and MCM-48-N-CuL1 (o). | ||
Transmission electron microscopy (TEM) was used to visualize the nanostructures and dispersion of [ML1] complexes trough silica surface. However, TEM images were obtained only for Silica-N-CoL1 and Silica-N-MnL1 and their corresponding silica materials. Because these two catalysts exhibited higher catalytic activity in the oxidation reaction of cyclohexane compared to their Ni and Cu analogues. The obtained TEM images are presented in Fig. 13. Images (a) and (d) display the parallel mesoporous channels, characteristic of the highly nano-ordered SBA-15 and MCM-41 materials, respectively. The TEM images obtained for these two materials after the incorporation of [CoL1] (Fig. 13b and e) and [MnL1] (Fig. 13c and f) revealed the preservation of the silica nanostructure. More importantly, the absence of any dark spots in the TEM images of Silica-N-CoL1 and Silica-N-MnL1 (Silica: SBA-15 or MCM-41) indicates the high dispersion of [CoL1] and [MnL1] through SBA-15 and MCM-41 surface.
 | ||
| Fig. 13 TEM images of commercial silica SBA-15 (a), MCM-41 (d), and MCM-48 (g), and silica supported [CoL] (b, e and h) and [MnL] (c, f and i). | ||
The obtained TEM images showed a spherical morphology of MCM-48 particles (Fig. 13g), with the average particle's diameters of 245 nm. However, images at more than 200000× were not taken to visualize clearly the very tiny nanochannels of MCM-41 material.58 After the addition of [CoL1] and [MnL1] to prepared MCM-4-N-CoL1 and MCM-4-N-MnL1, TEM images (Fig. 13h and i) revealed the preservation of the spherical morphology of MCM-48 with average particles diameter of 220 nm and 222 nm, respectively. Moreover, the absence of any dark spots in the obtained TEM images indicates high dispersion of [CoL1] and [MnL1] through MCM-48 surface.
Powder XRD was performed for the salphen-azobenzene ligand H2L1 and its metal complexes [ML1] where M = Mn, Co, Ni, Cu (Fig. 14). All samples presented distinct peaks corresponding to their crystalline nature. This confirms the formation of metal complexes [ML1]. The complexes display less intense peak reflections compared to the free ligand which indicates that the crystallinity of the H2L1 decreases upon complexation with M(II) ion = Mn, Co, Ni, and Cu. This observation agrees with previous reports on the impact of metal coordination on ligand crystallinity.59,60
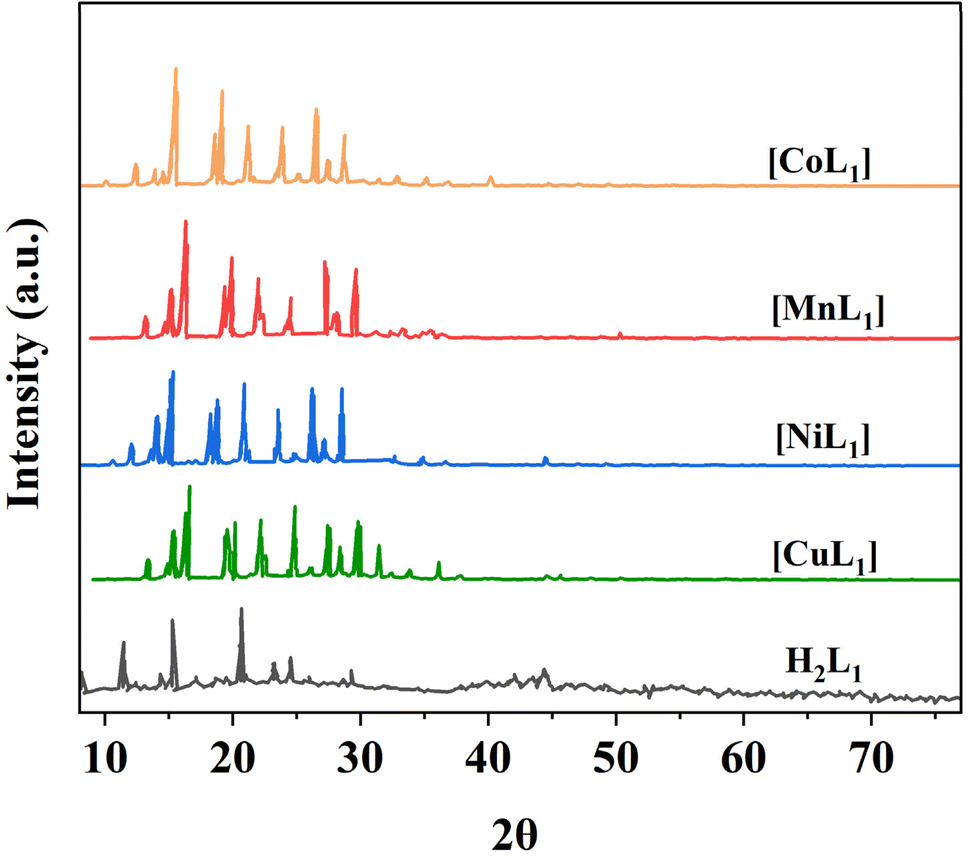 | ||
| Fig. 14 XRD patterns of salphen-azobenzene ligand H2L1 and its metal complexes [ML1] (M: Mn, Co, Ni, and Cu). | ||
After incorporation of these metal complexes [ML1] into silica Silica-N-ML1 (Silica: SBA-15, MCM-41 or MCM-48; M: Mn, Co, Ni or Cu), as shown in Fig. 15, all samples displayed a single broad peak around 2θ = 22.9°, characteristic of amorphous silica. Notably, no distinctive peaks for crystalline complexes [ML1] were observed. This indicates the metal complexes were highly dispersed on the mesoporous silica surfaces.61 This is in agreement with the TEM results described above. This confirms our approach to prepare Silica-N-ML1 materials with only isolated [CoL1], [MnL1], [NiL1] and [CuL1] molecules coordinated to surface NH2 groups, without aggregation phase, and to remove the free molecules by filtration and frequent washing process.
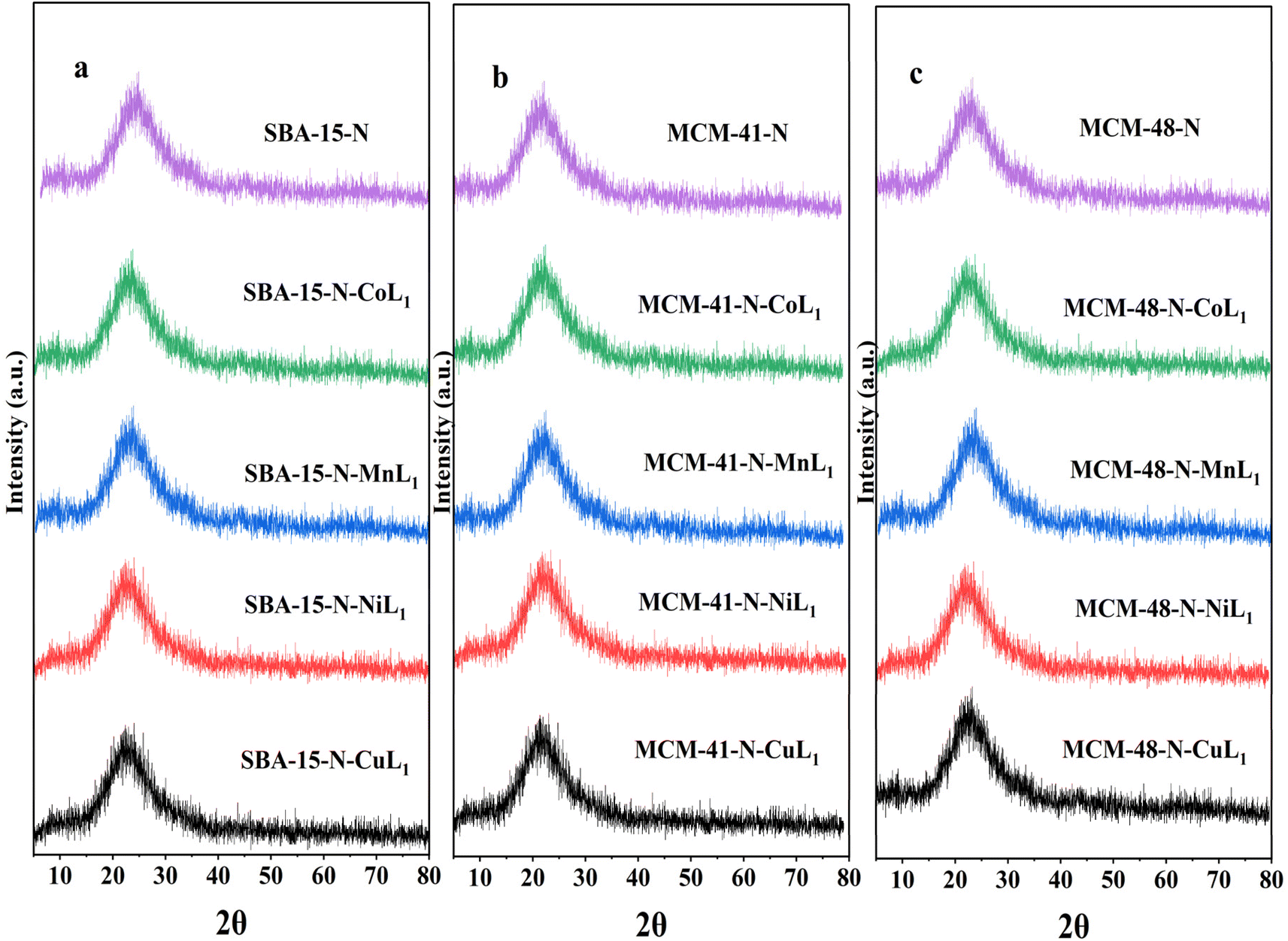 | ||
| Fig. 15 XRD patterns of SBA-15-N and SBA-15-N-ML1 (a), MCM-41-N and MCM-41-N-ML1 (b), MCM-48-N and MCM-48-N-ML1 (c), with M = Mn, Co, Ni, or Cu. | ||
3.2 Oxidation of cyclohexane
The catalytic activity of the prepared Silica-N-ML1 materials was evaluated in the oxidation reaction of cyclohexane to produce KA oil. The reaction was performed in a sealed autoclave. In this study different parameters such as type of oxidant, temperature, reaction time, catalyst dose, solvent. The reaction was monitored by gas chromatography (GC) using chlorobenzene as an internal standard.3.2.1.1 m-CPBA. Table 3 summarize the obtained results with different Silica-N-ML1 catalysts compared to unsupported [ML1], using 1.5 eq. of m-CPBA (2.5 g, 15 mmol) as an oxidant. Without a catalyst (entry 1), no conversion was observed. The unsupported catalysts [CoL1] (entry 2), [MnL1] (entry 3), [NiL1] (entry 4), and [CuL1] (entry 5) showed a cyclohexane conversion ranging from 40% to 55%, and KA oil selectivity in the range of 41–54%. The heterogeneous catalysts SBA-15-N-CoL1 (entry 6), SBA-15-N-MnL1 (entry 7), SBA-15-N-NiL1 (entry 8), and SBA-15-N-CuL1 (entry 9), exhibited higher conversion of 69–79%, and improved selectivity of 68–78%. When SAB-15 was replaced by MCM-41 the conversion and selectivity were slightly improved. 89%/90% and 85%/87% (conversion/selectivity) were obtained with MCM-41-N-CoL1 (entry 10) and MCM-41-N-MnL1 (entry 11) respectively. However, no significant improvement was observed with MCM-41-N-NiL1 (entry 12) and MCM-41-N-CuL1 (entry 13). Furthermore, when SBA-15 was replaced by MCM-48 the conversion and selectivity were slightly decreased. We can conclude that the best results were obtained with [CoL1] and [MnL1], using SBA-15 as a support. Compared to the literature (entries 18–21), despite performing the reaction at higher temperature (70–80 °C) and for more time (12–24 h) the obtained results are less important than those obtained in this work.
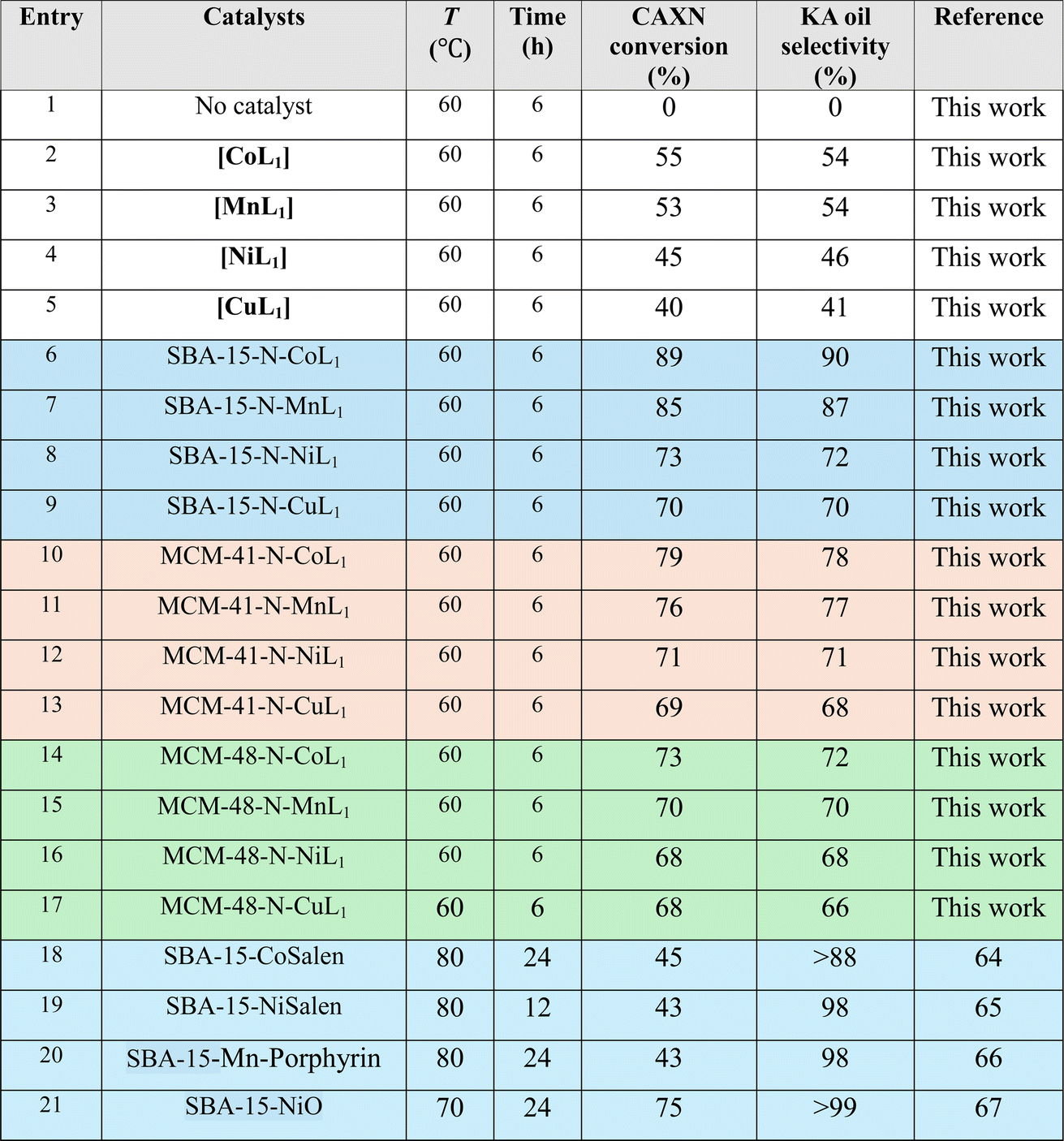 |
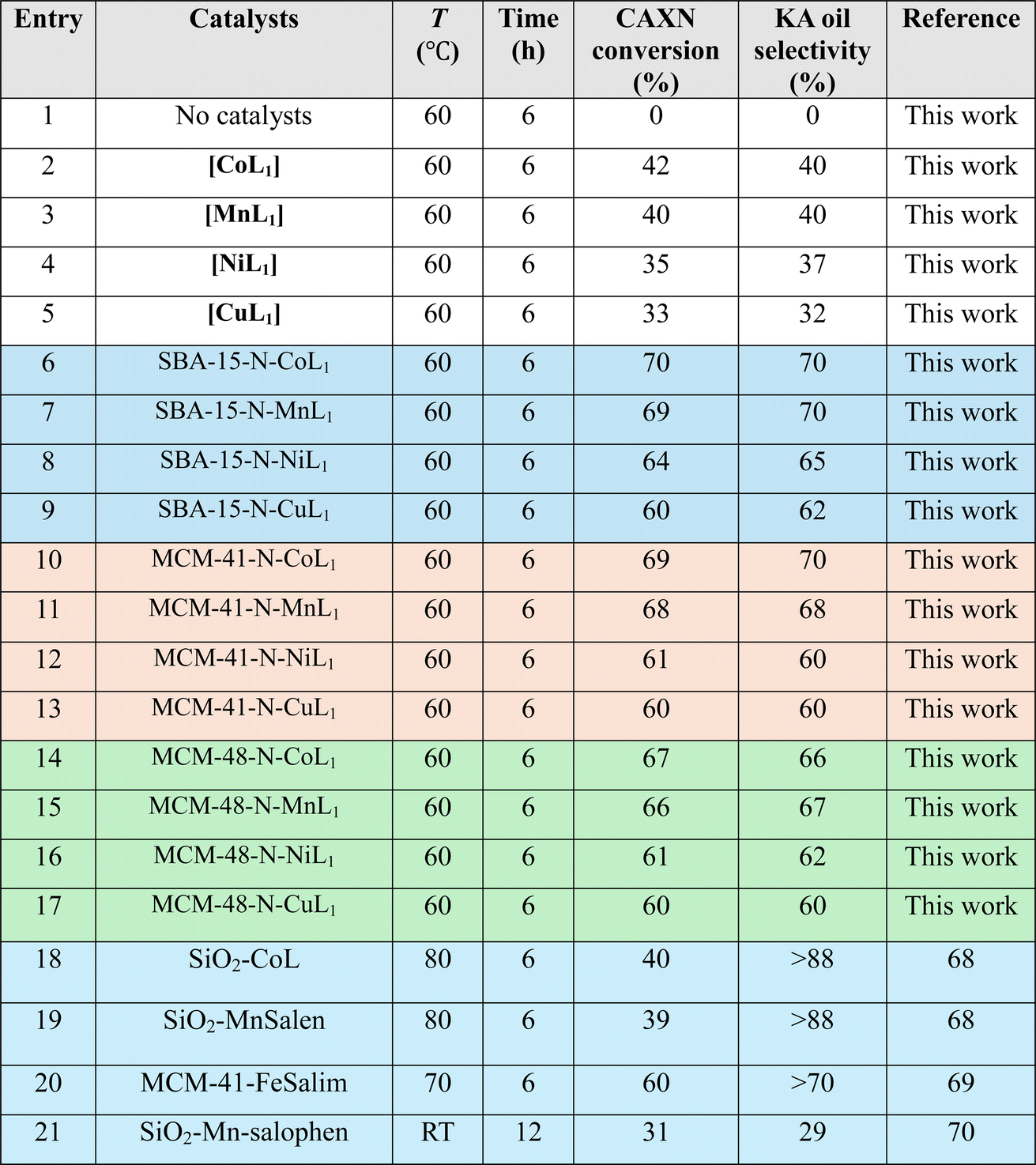
3.2.1.2 TBHP. Table 4 summarize the obtained results with different Silica-N-ML1 catalysts compared to unsupported [ML1], using 2 eq. of TBHP (1.80 mL, 20 mmol) as oxidant. As expected, the conversion and selectivity results obtained with TBHP are generally lower than those obtained with m-CPBA. The unsupported complexes [ML1] (entries 2–5) showed low conversions ranging from 42 to 33%, and selectivity toward KA oil in the range of 40–32%. The silica supported complexes exhibited higher conversion and selectivity (entries 6–17), with cyclohexane conversion in the range of 60–70%, and KA oil selectivity between 70–62%. The best results were obtained with [CoL1] and [MnL1] supported on SBA-15 and MCM-41 (entries 6, 7, 10, and 11). Lower conversions and selectivity were obtained with the other catalytic systems. Literature catalysts (entries 18–21) such as SiO2-CoL and SiO2-Mn-salophen tested at different conditions showed conversions ranging from 40–31% and selectivity over 88–29%. Moreover, compared to the literature, low conversion was obtained even at higher temperatures 70–80 °C during 6 h (entries 18–20). Very low conversion and selectivity obtained at room temperature for 12 h using SiO2-Mn-salophen (entry 21).
 |
3.2.1.3 H2O2. H2O2 was tested as an eco-friendly oxidant, using 2.0 mL (20 mmol) of 30% H2O2. However, SBA-15-CoL was chosen among the heterogeneous catalysts exhibited the best catalytic activity with m-CPBA and TBHP. The obtained results were compared with some selected results from the literature and presented in Table 5. 21% cyclohexane conversion and 50% KA oil selectivity were obtained with SBA-15-N-CoL1 as catalyst (entry 1). This result was similar to that obtained with Co-(complex) SiO2 at 70 °C (entry 2). Better results were obtained by increasing the time or/and temperature (entries 3 and 4). However, even using a cobalt-based catalyst, and increasing the reaction temperature to 100 °C, only 12% of cyclohexane was converted to product, with 80% selectivity (entry 5).
To better understand factors influencing the catalytic performance of SBA-15-N-CoL1, and to optimize the reaction conditions, other parameters were also investigated, such as the reaction temperature, reaction time, catalysts dose, amount of m-CPBA, and cis/trans isomerization of the azobenzene moiety of the ligand H2L1.
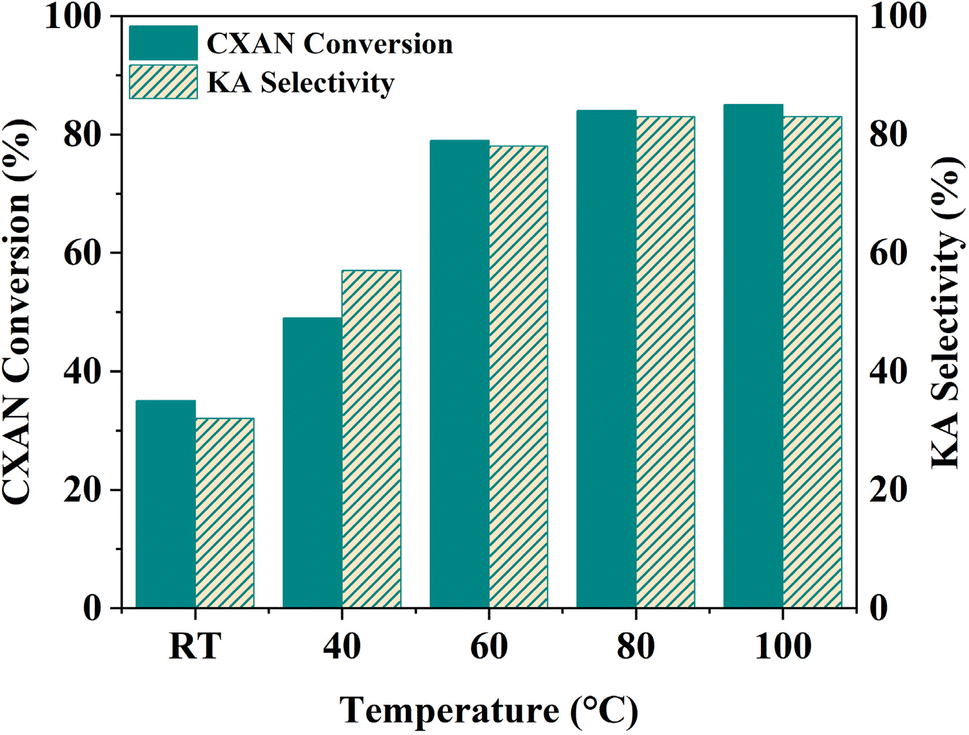 | ||
| Fig. 16 Effect of temperature on the cyclohexane oxidation over SBA-15-N-CoL1. Cyclohexane: 1.0 mL, catalyst: 100 mg, oxidant: 2.50 g of m-CPBA, chlorobenzene: 1.0 mL, time: 6 h, solvent: 5 mL of acetonitrile. | ||
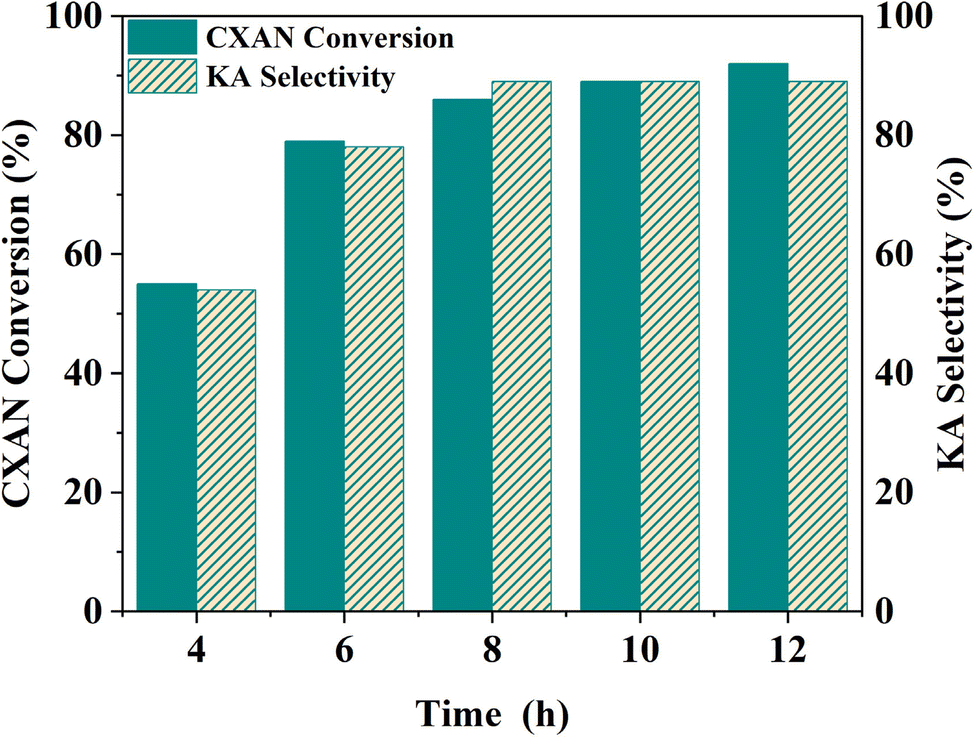 | ||
| Fig. 17 Effect of reaction time on the oxidation of cyclohexane. Cyclohexane: 1.0 mL, chlorobenzene: 1.0 mL, catalyst: 100 mg of SBA-15-N-CoL, oxidant: 2.50 g of m-CPBA, solvent: 5 mL of acetonitrile, T: 60 °C. | ||
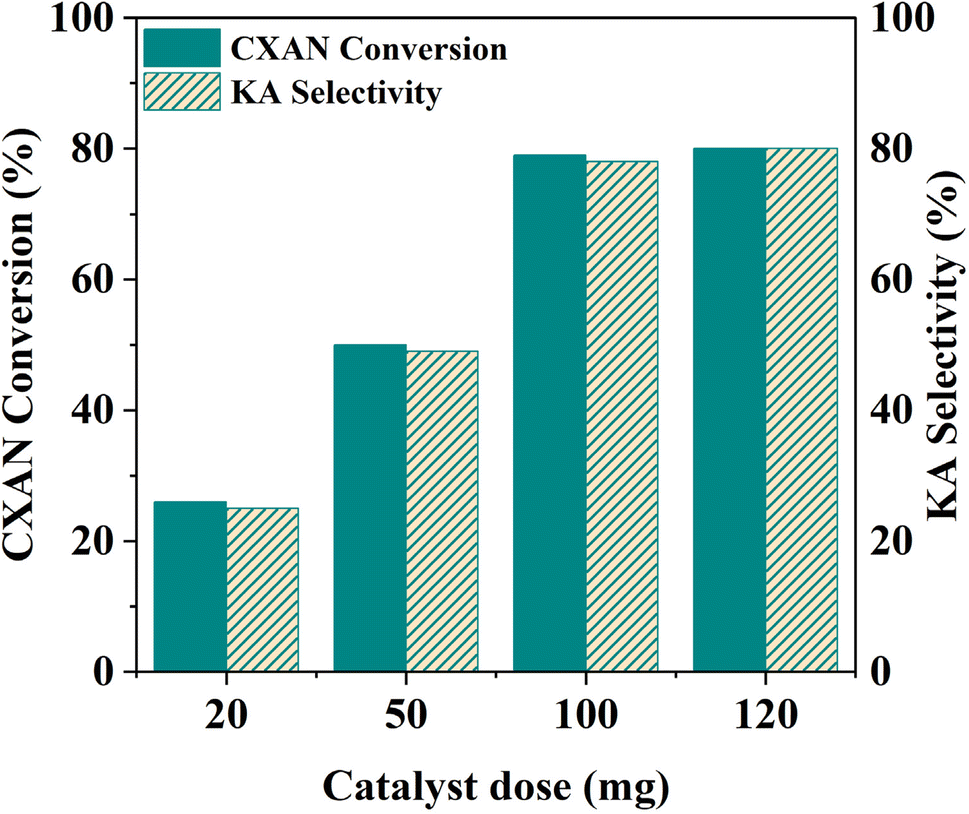 | ||
| Fig. 18 Effect of catalysts dose on the oxidation of cyclohexane. Cyclohexane: 1.0 mL, chlorobenzene: 1.0 mL, catalyst: 20–120 mg of SBA-15-N-CoL1, oxidant: 2.50 g of m-CPBA, solvent: 5 mL of acetonitrile, T: 60 °C, time: 6 h. | ||
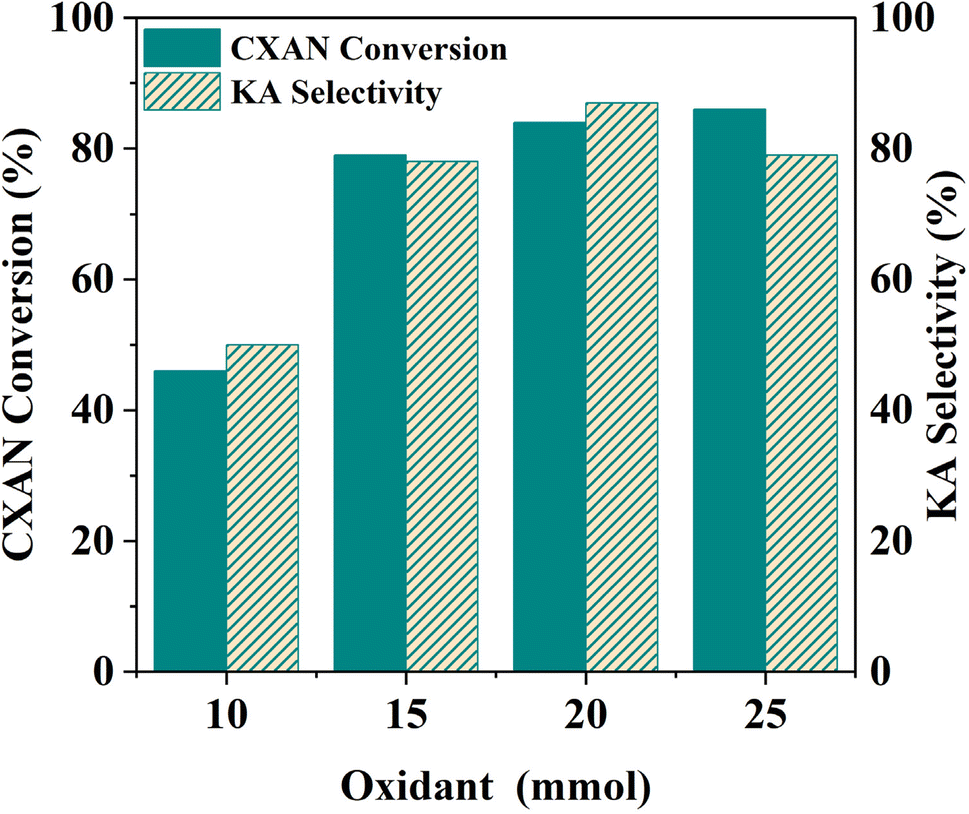 | ||
| Fig. 19 Effect of oxidant amount on the oxidation of cyclohexane. Cyclohexane: 1.0 mL, chlorobenzene: 1.0 mL, catalyst: 100 mg of SBA-15-N-CoL1, oxidant: 1–2.5 eq. of m-CPBA, solvent: 5 mL of acetonitrile, T: 60 °C, time: 6 h. | ||
Furthermore, by-products of oxidation of cyclohexane include over-oxidized products resulting from oxidation of the cyclohexanone or cyclohexanol. Common by-products include cyclohexanone oxime, cyclohexenone, and dicarboxylic acids such as glutaric and adipic acid.78,79 m-CPBA is commonly used as the oxidant for cyclohexane oxidation due to its ability to perform the reaction cleanly with few by-products such as dicarboxylic acid.3,5 m-CPBA is commonly used as the oxidant for cyclohexane oxidation due to its ability to perform the reaction cleanly with few by-products such as dicarboxylic acid.3,5 m-CPBA oxidizes cyclohexane to cyclohexanone and cyclohexanol in a stereospecific catalyst without cleaving the ring. This allows the reaction to obtain high yields of the desired mono-oxidation products with minimal over-oxidation.80,81 The ratio of cyclohexanone to cyclohexanol products (K/A ratio) is affected by several reaction conditions. Previous work has shown that increasing the amount of catalyst, leads to higher conversions but also increases over-oxidation reactions, lowering the K/A ratio.82 Extending the reaction time beyond 7 hours has a similar effect, as longer reactions enhance further oxidation of cyclohexanone and cyclohexanol into by-products.81,83,84 Other studies have also demonstrated the influence of reaction parameters on cyclohexane oxidation for example, Lesbani and coworker, found that temperatures below 80 °C using m-CPBA resulted in higher cyclohexanone selectivity while also suppressing by-product formation.76 Additionally, Maciuk et al. 2023, showed that the use of an m-CPBA to cyclohexane ratio of 1.5:1 improved conversion without significantly impacting selectivity or yield of by-products.80 Optimal conditions such as a catalyst dose of 100 mg, reaction time between 6–8 hours, and temperature below 80 °C, and a 1.5:1 m-CPBA:cyclohexane ratio can minimize by-product formation.
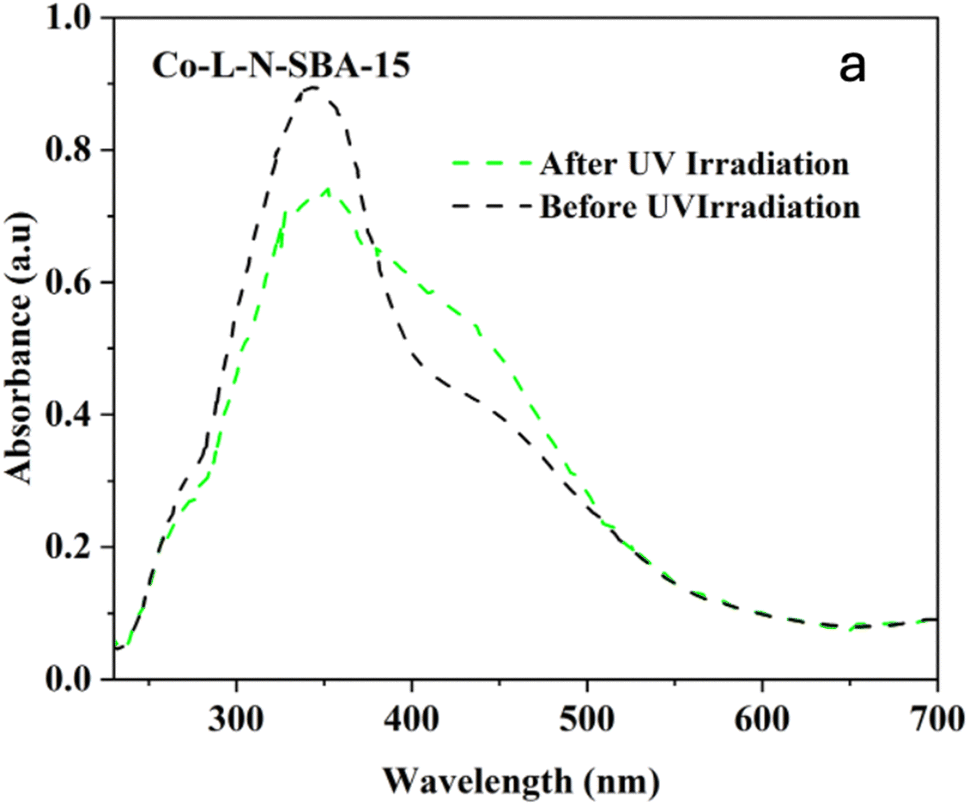 | ||
| Fig. 20 UV-Visible diffuse reflectance (DR) spectra of SBA-15-N-CoL1 before UV irradiation (black line) and after UV irradiation at 365 nm for 45 min (green line). | ||
The effect of azobenzene cis/trans isomerization on the catalytic activity of SBA-15-N-CoL1 was investigated by performing the oxidation reaction of cyclohexane with fresh catalyst and UV irradiated catalyst under the optimized conditions. Actually, in the first step the catalyst (100 mg) was first dispersed in acetonitrile (5 mL), then the mixture was exposed to UV light 365 nm for 45 minutes to induce the trans-to-cis isomerization of the azobenzene groups. In the second step, cyclohexane (1.0 mL, 10 mmol), chlorobenzene (1.0 mL, 10 mmol) as an internal standard, and m-CPBA (2.50 mg, 15 mmol), were added to the catalyst mixture. In the last step, the mixture obtained was poured into a sealed autoclave and heated to 60 °C for 2–8 h. The obtained results (Table 6) revealed that UV irradiation was clearly improved the catalytic activity of SBA-15-N-CoL1. For example, the cyclohexane conversion and KA oil selectivity were increased from 86%/85% before the UV irradiation to 93%/92% after UV irradiation. The high cyclohexane conversion and KA oil selectivity achieved simultaneously over the SBA-15-N-CoL catalysts can be attributed to key features of the photoactive cis-azobenzene complex. Compared to the trans isomer, simulations using density functional theory (DFT) showed the cis conformation offers more accessible active sites for substrate oxidation.85,86 Molecular dynamics simulations further explain that the flexible azobenzene ligands in the cis form allow for ideal substrate orientation within the porous framework.87 Additionally, two DFT investigations indicate the photoinduced cis isomer has a narrower HOMO–LUMO gap than trans-azobenzene, consistent with its higher activity in oxidizing cyclohexane.88 These effects are complemented by the flexible cis structure enabling dynamic accommodation and orientation of reactant/product molecules, as revealed through experimental kinetic isotope effect measurements.89 The synergistic impact of factors such as the accessible active sites, dynamic substrate positioning, and electronic structure modulation provided by the light-responsive cis complex, helps account for the high conversion and selectivity achieved under mild conditions. This new class of photoactive heterogeneous catalysts extends opportunities for remote performance optimization through photoisomerization.90
| Time (h) | Before UV irradiation | After UV irradiation | ||
|---|---|---|---|---|
| CAXN conversion (%) | KA oil selectivity (%) | CAXN conversion (%) | KA oil selectivity (%) | |
| 2 | 25 | 23 | 40 | 43 |
| 4 | 55 | 54 | 67 | 68 |
| 6 | 79 | 78 | 84 | 85 |
| 8 | 86 | 85 | 93 | 92 |
The kinetics of the reaction were also studied to determine the catalyst performance. Under the optimized conditions (cyclohexane 1.0 mL (10 mmol), chlorobenzene 1.0 mL, SBA-15-N-CoL1 100 mg, m-CPBA 2.50 g as an oxidant, acetonitrile 5 mL as a solvent, 60 °C, 6 h), the molar consumption rate of cyclohexane and generation rate of KA oil over SBA-15-N-CoL1 were determined. The molar consumption of cyclohexane was calculated using the equation:
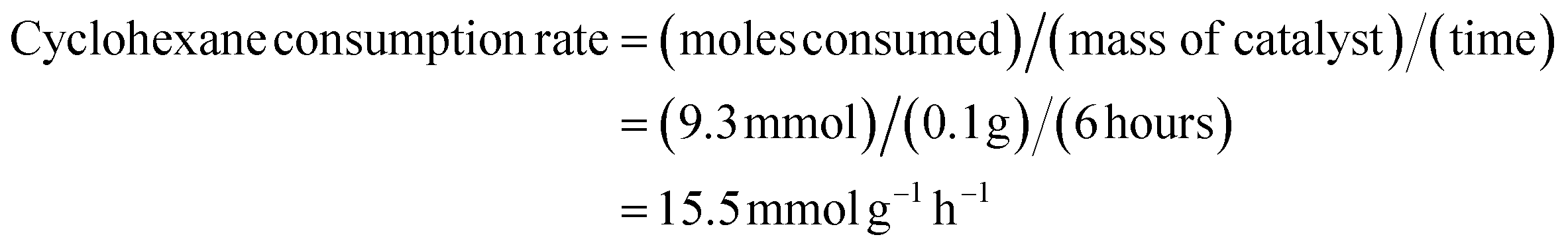
The generation rate of KA oil was calculated using the equation below:
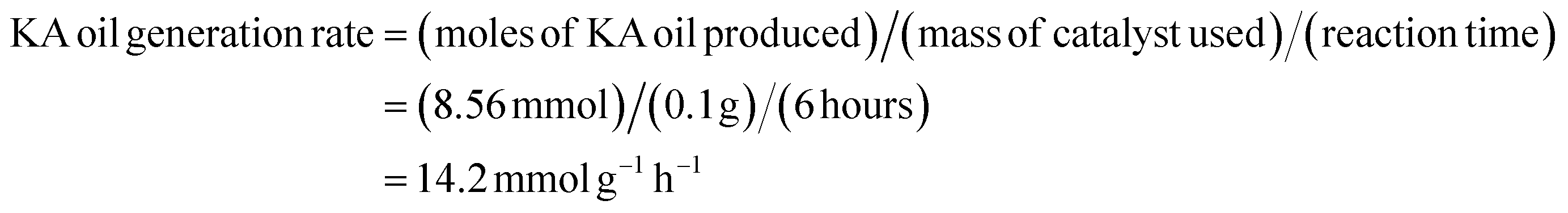
These kinetic parameters of the SBA-15-N-CoL1 catalyst confirm its high performance for cyclohexane oxidation.
3.3. Catalyst reuse and stability
The recyclability of SBA-15-N-CoL1 was evaluated through consecutive reaction cycles using the optimized condition. After each run, the catalyst was isolated via filtration, washed with chloroform, and dried overnight at 70 °C. The obtained are presented in Fig. 21a. Notably, after 4 consecutive runs, the catalyst maintained high catalytic activity, exhibiting only minor decreases in the conversion and selectivity. Moreover, the nanostructure of the recycled catalyst after the 4 runs was analyzed by TEM and ICP-MS. The characterization outcomes indicated that the structure of the recovered catalyst remained relatively unchanged after 4 cycles, as evidenced by TEM images (Fig. 21b) and ICP-MS results (1.08 wt%).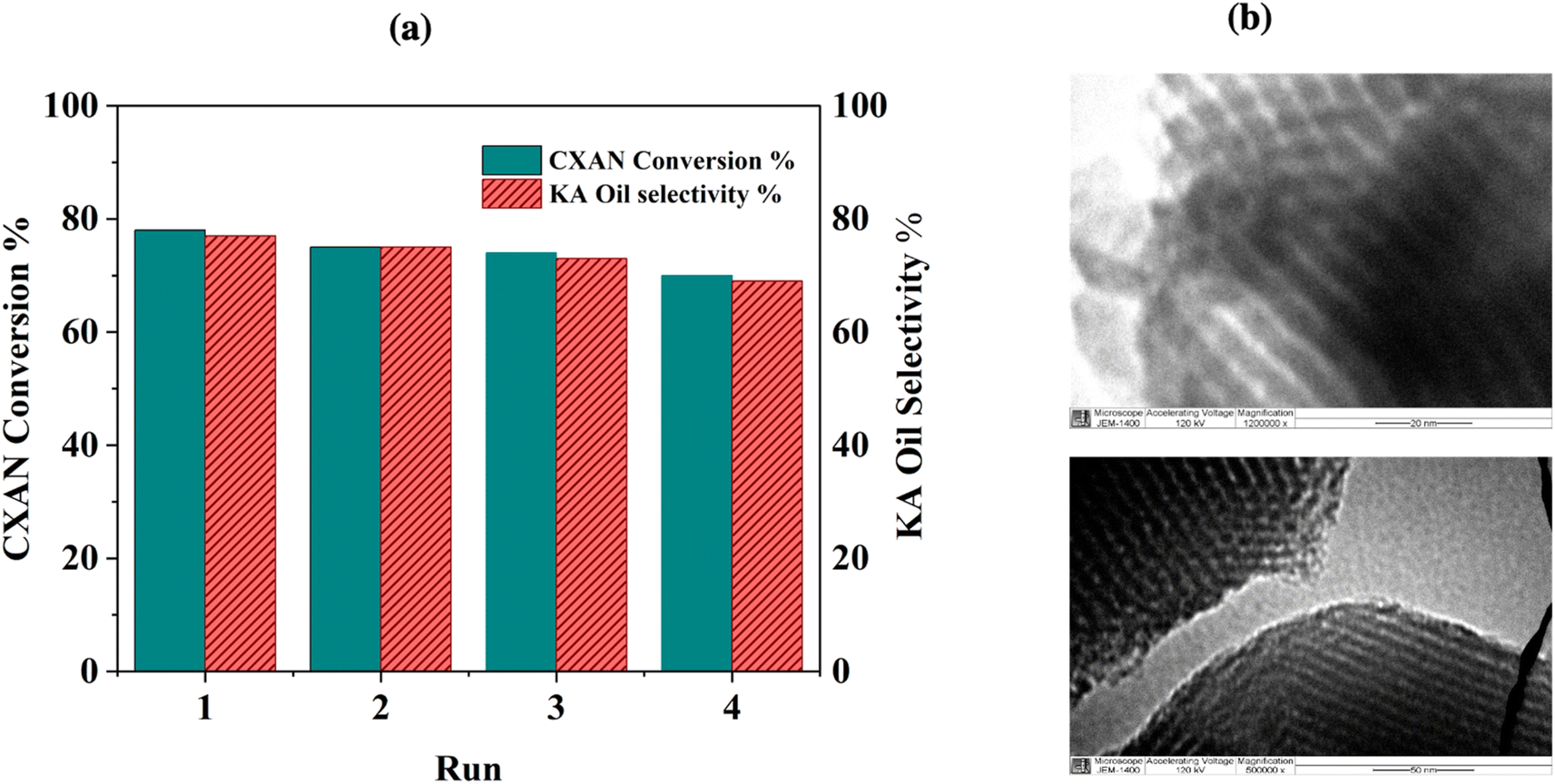 | ||
| Fig. 21 SBA-15-N-CoL1 recycling results (a), and TEM images of the spent catalyst (b). | ||
3.4 Proposed mechanism
Meta-chloroperoxybenzoic acid (m-CPBA) is a widely used and effective organic peroxide that serves as a strong oxidizing agent for many different organic reactions. m-CPBA has been effectively used to activate the typically unreactive C–H bonds found in hydrocarbons.91 The oxidation of C–H bonds is one of the most compelling challenges in organic chemistry. Compared to other common oxidants such as H2O2 and TBHP, m-CPBA exhibits greater stability and selectivity.92 These properties are highly advantageous for organic syntheses. m-CPBA can also form specialized intermediates when used in conjunction with auxiliary reagents, and these intermediates display enhanced reactivity. Moreover, m-CPBA is straightforward to handle as a terminal oxidizing agent.93,94 However, in some cases m-CPBA may non-selectively generate a variety of radical species. Therefore, a catalyst is needed to activate the O–O bond in a targeted manner and control the reaction pathway.95 The catalyst ensures oxidation occurs suitably while suppressing unwanted side and secondary reactions.96 Based on prior theoretical studies of transition metal-catalyzed hydrocarbon oxidation and our experimental results, the following mechanism is proposed over Silica-N-ML catalyst (Scheme 4).97–99 Through O–O bond homolysis of m-CPBA, facilitated by Silica-N-ML active sites, M-oxo (MO) species and m-CBOO˙ radicals are generated (Scheme 4, step 1). The m-CBOO˙ acts as cyclohexane (CXAN) C–H bond abstracting agents, producing CXA˙ radicals and m-CBA (Scheme 4, step 2). CXA˙ then reacts with additional m-CPBA, regenerating m-CBOO˙ while forming CXAOL (Scheme 4, step 3). Some CXAOL further undergoes oxidation at Silica-N-ML, yielding CXON (Scheme 4, step 4).
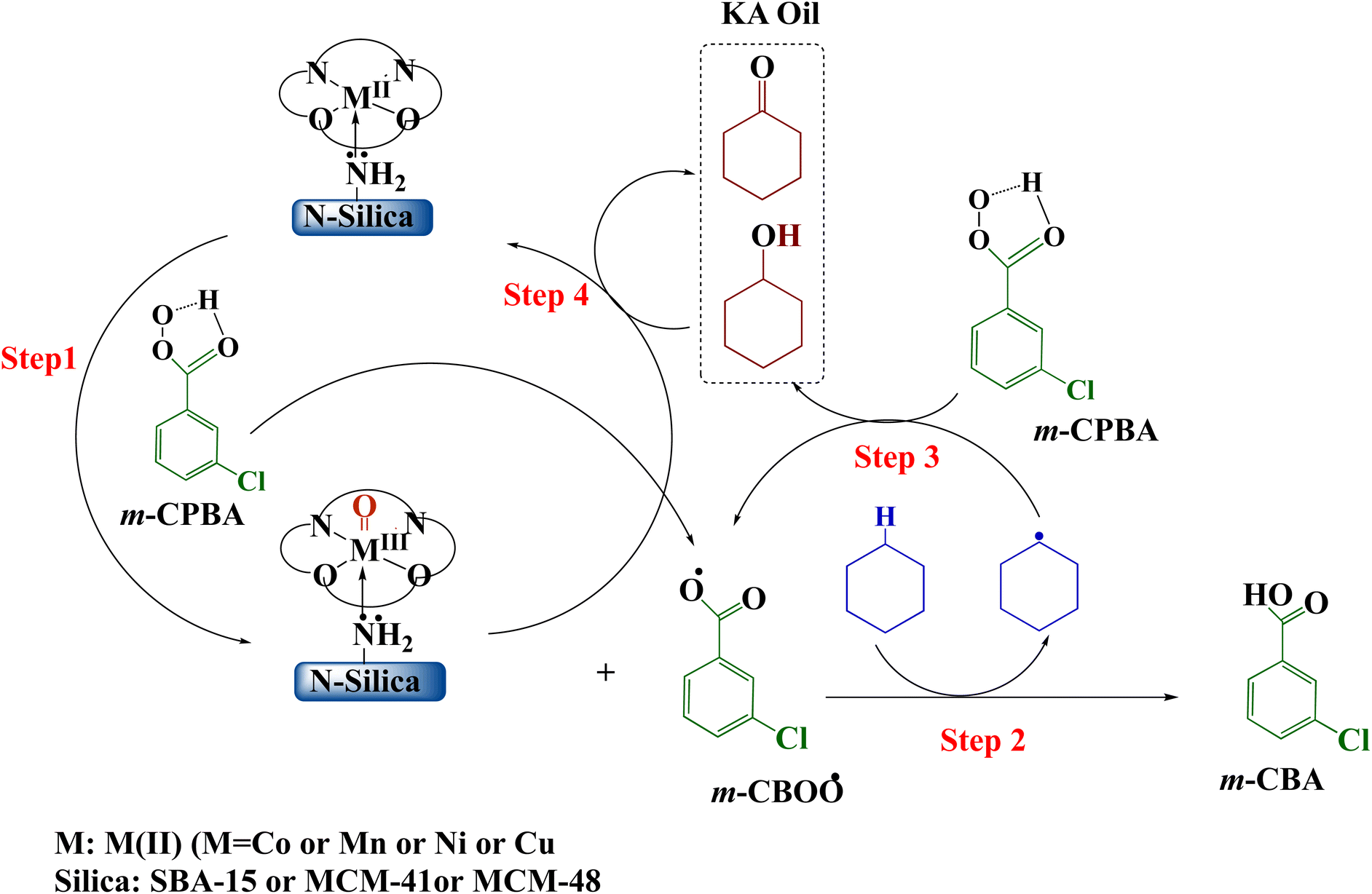 | ||
| Scheme 4 Mechanism of oxidation of cyclohexane over Silica-N-ML1 (Silica: SBA-15, MCM-41, MCM-48; M: Co, Mn, Ni, and Cu; L: salphen-azobenzene). | ||
4. Conclusion
New and efficient photochromic heterogeneous nanocatalysts were successfully synthesized by immobilizing metallosalphen-azobenzene complexes onto different mesoporous silica surface. A salphen-azobenzene H2L1 derivative was firs synthetized and complexed with four different transition metals (M: Mn, Co, Ni and Cu). The structure of the obtained complexes [ML1] was confirmed by NMR, IR and elemental analysis and PXRD. Then [ML1] complexes were incorporated into three different pre-prepared amino-functionalized mesoporous silica (N-Silica: SBA-15-N, MCM-41-N, and MCM-48-N) via coordination bonds. The twelve prepared catalysts Silica-N-ML1 were fully characterized by different techniques such as FT-IR, SEM, TEM, XRD, ICP-MS, DR UV-Vis and N2 physisorption. The obtained results confirmed the successful grafting of APTES and immobilization of [ML1] complexes onto silica surface, with the preservation of the silica mesoporosity and nanostructure order. Results revealed also the presence of trans configuration of the azobenzene group as the major isomer in Silica-N-ML1 materials, which was easily transformed to cis isomer upon UV irradiation. The catalytic activity of the prepared nanocatalyst (Silica-N-ML1) was evaluated in the oxidation reaction of cyclohexane to produce KA oil. Different parameters were investigated to determine the optimized conditions, such as type of oxidant, type of silica, type of metal, catalyst dose, reaction time, temperature, and UV light. The best results were obtained with 100 mg of SBA-15-N-CoL1, using m-CPBA as oxidant, at 60 °C, for 6 h, and under UV light. A superior catalytic activity was observed for the cis conformation under UV light, achieving 93% conversion and 92% selectivity toward KA oil. Moreover, the SBA-15-N-CoL1 nanocatalyst exhibited a good catalytic activity performance and high stability in four consecutive cycles. Leaching measurement using ICP-MS and TEM images of the spent catalyst confirmed an excellent stability of this photochromic nanocatalyst.Data availability
Data available upon request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through a large group Research Project under grant number RGP2/280/45.References
- K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646–1647 CrossRef.
- R. S. Alnefaie, M. Abboud, A. Alhanash and M. S. Hamdy, Molecules, 2022, 27, 3145 CrossRef PubMed.
- S. Liu, M. Pasha, M. Shang, Y. Wang, G. Qian, Z.-H. Luo and Y. Su, Chem. Eng. Sci., 2023, 266, 118273 CrossRef.
- M. Najafi, L. Nasri and R. Kotek, in Structure and Properties of High-Performance Fibers, Elsevier, 2017, pp. 199–244 Search PubMed.
- M. Shakiba, E. Rezvani Ghomi, F. Khosravi, S. Jouybar, A. Bigham, M. Zare, M. Abdouss, R. Moaref and S. Ramakrishna, Polym. Adv. Technol., 2021, 32, 3368–3383 CrossRef.
- I. N. Vikhareva, G. K. Aminova and A. K. Mazitova, Molecules, 2021, 26, 4833 CrossRef CAS.
- M. Bratychak, O. Astakhova, O. Mykhailiv, A. Stryzhachuk and O. Shyshchak, Chem. Chem. Technol., 2012, 6, 51–57 CrossRef CAS.
- J. Rios, J. Lebeau, T. Yang, S. Li and M. D. Lynch, Green Chem., 2021, 23, 3172–3190 RSC.
- K. Ono and A. Erhard, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Wiley, 1st edn, 2011 Search PubMed.
- U. Schuchardt, D. Cardoso, R. Sercheli, R. Pereira, R. S. Da Cruz, M. C. Guerreiro, D. Mandelli, E. V. Spinacé and E. L. Pires, Appl. Catal., A, 2001, 211, 1–17 CrossRef CAS.
- A. P. C. Ribeiro, E. Spada, R. Bertani and L. M. D. R. S. Martins, Catalysts, 2020, 10, 1443 CrossRef CAS.
- I. L. Librando, A. Paul, A. G. Mahmoud, A. V. Gurbanov, S. A. C. Carabineiro, M. F. C. Guedes Da Silva, C. F. G. C. Geraldes and A. J. L. Pombeiro, RSC Sustainability, 2023, 1, 147–158 RSC.
- H. Yu, F. Peng, J. Tan, X. Hu, H. Wang, J. Yang and W. Zheng, Angew. Chem., Int. Ed., 2011, 50, 3978–3982 CrossRef PubMed.
- W. Deng, L. Yan, B. Wang, Q. Zhang, H. Song, S. Wang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 4712–4719 CrossRef PubMed.
- H. Li, Y. She and T. Wang, Front. Chem. Sci. Eng., 2012, 6, 356–368 CrossRef.
- A. Bhattacharjee, T. Das, H. Uyama, P. Roy and M. Nandi, ChemistrySelect, 2017, 2, 10157–10166 CrossRef.
- A. Gualandi, F. Calogero, S. Potenti and P. G. Cozzi, Molecules, 2019, 24, 1716 CrossRef CAS.
- J. Goscianska, A. Olejnik and I. Nowak, Colloids Surf., A, 2017, 533, 187–196 CrossRef CAS.
- C. Li, Catal. Rev., 2004, 46, 419–492 CrossRef CAS.
- V. Ganesan and S. Yoon, Inorg. Chem., 2020, 59, 2881–2889 CrossRef CAS PubMed.
- A. R. Silva, T. Mourão and J. Rocha, Catal. Today, 2013, 203, 81–86 CrossRef.
- A. A. Alshaheri, M. I. M. Tahir, M. B. A. Rahman, T. B. S. A. Ravoof and T. A. Saleh, Chem. Eng. J., 2017, 327, 423–430 CrossRef.
- Y. Hong, J. Peng, Z. Sun, Z. Yu, A. Wang, Y. Wang, Y.-Y. Liu, F. Xu and L.-X. Sun, Materials, 2020, 13, 829 CrossRef PubMed.
- W. Al Zoubi and Y. G. Ko, J. Organomet. Chem., 2016, 822, 173–188 CrossRef.
- R. Iwanejko, T. Mulodnicka and J. Potowicz, in Studies in Surface Science and Catalysis, Elsevier, 1990, vol. 55, pp. 195–203 Search PubMed.
- J. Li, Y. Shi, L. Xu and G. Lu, Ind. Eng. Chem. Res., 2010, 49, 5392–5399 CrossRef CAS.
- X. Zhang, Z. Chen, J. Chen and J. Xu, Chem. Eng. Sci., 2024, 288, 119777 CrossRef CAS.
- A. Vomeri, M. Stucchi, A. Villa, C. Evangelisti, A. Beck and L. Prati, J. Energy Chem., 2022, 70, 45–51 CrossRef CAS.
- D. G. Montjoy, E. A. K. Wilson, H. Hou, J. D. Graves and N. A. Kotov, Nat. Commun., 2023, 14, 857 CrossRef CAS.
- A. K. Asatkar, M. Tripathi and D. Asatkar, in Stability and Applications of Coordination Compounds, ed. A. Nanda Srivastva, IntechOpen, 2020 Search PubMed.
- S. M. Elbert and M. Mastalerz, Org. Mater., 2020, 02, 182–203 CrossRef.
- H. Wang, Y. Pei, K. Wang, Y. Zuo, M. Wei, J. Xiong, P. Zhang, Z. Chen, N. Shang, D. Zhong and P. Pei, Small, 2023, 19, 2304863 CrossRef.
- P. J. Coelho, L. M. Carvalho, J. C. V. P. Moura and M. M. M. Raposo, Dyes Pigm., 2009, 82, 130–133 CrossRef.
- T. Akitsu, B. Miroslaw and S. Sudarsan, Int. J. Mol. Sci., 2022, 23, 10005 CrossRef PubMed.
- Z. Salem, Synthesis and Study of an Azo-azomethine Dyes with N,O Donor Set of Atoms and Their Cu (II), Co (II) and Ni(II) Complexes, Chem. Mater. Res., 2017, 9, 10–16 Search PubMed.
- G. Markiewicz, A. Walczak, F. Perlitius, M. Piasecka, J. M. Harrowfield and A. R. Stefankiewicz, Dalton Trans., 2018, 47, 14254–14262 RSC.
- W. Zhang, L. Hu, H. Zhang, C. Pan and J. Tang, Polymers, 2020, 12, 1076 CrossRef CAS PubMed.
- S. M. Elbert and M. Mastalerz, Org. Mater., 2020, 02, 182–203 CrossRef CAS.
- A. Ramirez, J.-M. Clacens, C. Lorentz and Y. Pouilloux, COC, 2012, 16, 2774–2781 CrossRef CAS.
- P. Verma, Y. Kuwahara, K. Mori, R. Raja and H. Yamashita, Nanoscale, 2020, 12, 11333–11363 RSC.
- J. Andas, S. H. Ekhbal and T. H. Ali, Environ. Technol. Innovation, 2021, 21, 101308 CrossRef CAS.
- T. Prasomsri, W. Jiao, Z. Weng and J. Garcia Martinez, Chem. Commun., 2015, 51, 8900–8911 RSC.
- K. An and G. A. Somorjai, Catal. Lett., 2015, 145, 233–248 CrossRef.
- M. F. Al-Samarraie and W. Steedman, Liq. Fuels Technol., 1985, 3, 55–71 CrossRef.
- S. Rayati, E. Khodaei, P. Nafarieh, M. Jafarian, B. Elmi and A. Wojtczak, RSC Adv., 2020, 10, 17026–17036 RSC.
- A. P. C. Ribeiro, E. Spada, R. Bertani and L. M. D. R. S. Martins, Catalysts, 2020, 10, 1443 CrossRef.
- R. Kumar, S. Sithambaram and S. L. Suib, J. Catal., 2009, 262, 304–313 CrossRef.
- R. Wang, J. Wang, H. Zi, Y. Xia, H. Wang and X. Liu, Mol. Catal., 2017, 441, 168–178 CrossRef.
- P. A. Carvalho, J. W. Comerford, K. J. Lamb, M. North and P. S. Reiss, Adv. Synth. Catal., 2019, 361, 345–354 CrossRef.
- J. Ortiz-Bustos, A. Martín, V. Morales, R. Sanz and R. A. García-Muñoz, Microporous Mesoporous Mater., 2017, 240, 236–245 CrossRef.
- A. Sheykhi-Estalkhjani, N. O. Mahmoodi, A. Yahyazadeh and M. P. Nadamani, Tetrahedron, 2018, 74, 4868–4874 CrossRef CAS.
- M. Abboud, N. Al-Zaqri, T. Sahlabji, M. Eissa, A. T. Mubarak, R. Bel-Hadj-Tahar, A. Alsalme, F. A. Alharthi, A. Alsyahi and M. S. Hamdy, RSC Adv., 2020, 10, 35407–35418 RSC.
- S. Arumugam, V. Singh, A. P. Tathod, S. Daniel and N. Viswanadham, Ind. Eng. Chem. Res., 2022, 61, 18372–18381 CrossRef CAS.
- T. Eren, M. Kose, K. Sayin, V. McKee and M. Kurtoglu, J. Mol. Struct., 2014, 1065–1066, 191–198 CrossRef CAS.
- K. Rezaeian and H. Khanmohammadi, Spectrochim. Acta, Part A, 2014, 133, 31–37 CrossRef CAS PubMed.
- M. Espinosa, S. Pacheco and R. Rodriguez, J. Non-Cryst. Solids, 2007, 353, 2573–2581 CrossRef CAS.
- M. Moritz and M. Łaniecki, Appl. Surf. Sci., 2012, 258, 7523–7529 CrossRef CAS.
- W. Qian, H. Wang, J. Chen and Y. Kong, Materials, 2015, 8, 1752–1765 CrossRef CAS.
- J. J. Rani, A. M. I. Jayaseeli, S. Rajagopal, S. Seenithurai, J.-D. Chai, J. D. Raja and R. Rajasekaran, J. Mol. Liq., 2021, 328, 115457 CrossRef CAS.
- A. K. Babaheydari, M. Salavati-Niasari and A. Khansari, Particuology, 2012, 10, 759–764 CrossRef CAS.
- M. Abboud, N. Al-Zaqri, T. Sahlabji, M. Eissa, A. T. Mubarak, R. Bel-Hadj-Tahar, A. Alsalme, F. A. Alharthi, A. Alsyahi and M. S. Hamdy, RSC Adv., 2020, 10, 35407–35418 RSC.
- R. S. Alnefaie, M. Abboud, A. Alhanash and M. S. Hamdy, Molecules, 2022, 27, 3145 CrossRef.
- M. Abboud, R. S. Alnefaie, A. A. AL-Zahrani, N. Al-Zaqri, M. A. Haija, A. Al-Ghamdi, M. Alsaiari, M. Jalalah, O. Albormani and M. S. Hamdy, Sustainability, 2023, 15, 5817 CrossRef.
- J. Nakazawa, A. Yata, T. Hori, T. D. P. Stack, Y. Naruta and S. Hikichi, Chem. Lett., 2013, 42, 1197–1199 CrossRef.
- J. Nakazawa, T. Hori, T. D. P. Stack and S. Hikichi, Chem.–Asian J., 2013, 8, 1191–1199 CrossRef PubMed.
- G. M. Ucoski, V. H. A. Pinto, G. DeFreitas-Silva, J. S. Rebouças, R. Marcos Da Silva, I. Mazzaro, F. S. Nunes and S. Nakagaki, Microporous Mesoporous Mater., 2018, 265, 84–97 CrossRef CAS.
- M. Abboud, R. S. Alnefaie, A. A. AL-Zahrani, N. Al-Zaqri, M. A. Haija, A. Al-Ghamdi, M. Alsaiari, M. Jalalah, O. Albormani and M. S. Hamdy, Sustainability, 2023, 15, 5817 CrossRef CAS.
- S. Khare and P. Shrivastava, J. Mol. Catal. A: Chem., 2016, 411, 279–289 CrossRef CAS.
- S. Singha, K. M. Parida and A. C. Dash, J. Porous Mater., 2011, 18, 707–714 CrossRef CAS.
- V. Mirkhani, M. Moghadam, S. Tangestaninejad and B. Bahramian, Appl. Catal., A, 2006, 313, 122–129 CrossRef CAS.
- R. Antony, S. T. David Manickam, P. Kollu, P. V. Chandrasekar, K. Karuppasamy and S. Balakumar, RSC Adv., 2014, 4, 24820–24830 RSC.
- J. Zhao, W. Wang and Y. Zhang, J. Inorg. Organomet. Polym., 2008, 18, 441–447 CrossRef.
- W. Trakarnpruk, Int. J. Chem. Eng. Appl., 2015, 6, 120–124 Search PubMed.
- M. A. Andrade and L. M. D. R. S. Martins, Catalysts, 2019, 10, 2 CrossRef.
- R. Antony, S. T. David Manickam, P. Kollu, P. V. Chandrasekar, K. Karuppasamy and S. Balakumar, RSC Adv., 2014, 4, 24820–24830 RSC.
- A. Lesbani, F. Fatmawati, R. Mohadi, N. A. Fithri and D. Rohendi, Indones. J. Chem., 2018, 16, 175 CrossRef.
- J. J. Zhang, H. L. Song, J. Wang and H. Song, Adv. Mater. Res., 2012, 549, 411–414 CAS.
- A. Vomeri, M. Stucchi, A. Villa, C. Evangelisti, A. Beck and L. Prati, J. Energy Chem., 2022, 70, 45–51 CrossRef CAS.
- I. Hermans, J. Peeters and P. A. Jacobs, J. Phys. Chem. A, 2008, 112, 1747–1753 CrossRef CAS PubMed.
- S. Maciuk, S. H. Wood, V. K. Patel, P. D. P. Shapland and N. C. O. Tomkinson, Chem.–Eur. J., 2023, 29, e202204007 CrossRef CAS PubMed.
- G. Shul’pin, D. Loginov, L. Shul’pina, N. Ikonnikov, V. Idrisov, M. Vinogradov, S. Osipov, Y. Nelyubina and P. Tyubaeva, Molecules, 2016, 21, 1593 CrossRef.
- X. Fang, Z. Yin, H. Wang, J. Li, X. Liang, J. Kang and B. He, J. Catal., 2015, 329, 187–194 CrossRef.
- G. Shul’pin, Catalysts, 2016, 6, 50 CrossRef.
- A. Lesbani, M. Setyowati, R. Mohadi and D. Rohendi, Molekul, 2016, 11, 53 CrossRef.
- R. Mogale, J. Conradie and E. H. G. Langner, Molecules, 2022, 27, 1370 CrossRef PubMed.
- D. Das, M. K. Yadav, L. Singla, A. Kumar, M. Karanam, S. Dev and A. R. Choudhury, ChemistrySelect, 2020, 5, 13957–13962 CrossRef CAS.
- J. Tian, L. Fu, Z. Liu, H. Geng, Y. Sun, G. Lin, X. Zhang, G. Zhang and D. Zhang, Adv. Funct. Mater., 2019, 29, 1807176 CrossRef.
- Y.-P. Wang, Z.-X. Zhang, M. Xie, F.-Q. Bai, P.-X. Wang and H.-X. Zhang, Dyes Pigm., 2016, 129, 100–108 CrossRef CAS.
- H. M. D. Bandara, T. R. Friss, M. M. Enriquez, W. Isley, C. Incarvito, H. A. Frank, J. Gascon and S. C. Burdette, J. Org. Chem., 2010, 75, 4817–4827 CrossRef CAS PubMed.
- C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475–2532 CrossRef CAS.
- M. A. Behnajady and S. Bimeghdar, Chem. Eng. J., 2014, 239, 105–113 CrossRef CAS.
- O. Y. Lyakin, K. P. Bryliakov and E. P. Talsi, Coord. Chem. Rev., 2019, 384, 126–139 CrossRef CAS.
- F. V. Singh and T. Wirth, Chem.–Asian J., 2014, 9, 950–971 CrossRef CAS PubMed.
- P. Ghosh, B. Ganguly and S. Das, Org. Biomol. Chem., 2021, 19, 2146–2167 RSC.
- X.-F. Wu, J.-L. Gong and X. Qi, Org. Biomol. Chem., 2014, 12, 5807–5817 RSC.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef PubMed.
- H. Hussain, A. Al-Harrasi, I. R. Green, I. Ahmed, G. Abbas and N. U. Rehman, RSC Adv., 2014, 4, 12882–12917 RSC.
- M. S. Hamdy, N. Al-Zaqri, T. Sahlabji, M. Eissa, M. A. Haija, A. M. Alhanash, A. Alsalme, F. A. Alharthi and M. Abboud, Catal. Lett., 2021, 151, 1612–1622 CrossRef.
- D. S. Nesterov and O. V. Nesterova, Catalysts, 2021, 11, 1148 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |