
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew N-phenylpyrrolamide inhibitors of DNA gyrase with improved antibacterial activity†
Andrej Emanuel Cotman a,
Federica Fulgheri‡
a,
Martina Pigaa,
Peter Peršoljaa,
Davide Benedetto Tiza,
Žiga Skoka,
Martina Durcika,
Maša Sterlea,
Jaka Dernovšeka,
Cristina D. Cruzb,
Päivi Tammelab,
Petra Éva Szilic,
Lejla Darukac,
Csaba Pálc,
Anamarija Zegaa,
Lucija Peterlin Mašiča,
Janez Ilaša,
Tihomir Tomašiča,
Danijel Kikelja and
Nace Zidar*a
a,
Federica Fulgheri‡
a,
Martina Pigaa,
Peter Peršoljaa,
Davide Benedetto Tiza,
Žiga Skoka,
Martina Durcika,
Maša Sterlea,
Jaka Dernovšeka,
Cristina D. Cruzb,
Päivi Tammelab,
Petra Éva Szilic,
Lejla Darukac,
Csaba Pálc,
Anamarija Zegaa,
Lucija Peterlin Mašiča,
Janez Ilaša,
Tihomir Tomašiča,
Danijel Kikelja and
Nace Zidar*a
aUniversity of Ljubljana, Faculty of Pharmacy, Aškerčeva cesta 7, 1000 Ljubljana, Slovenia. E-mail: nace.zidar@ffa.uni-lj.si
bDrug Research Program, Division of Pharmaceutical Biosciences, Faculty of Pharmacy, University of Helsinki, P.O. Box 56, Viikinkaari 5E, Helsinki 00014, Finland
cSynthetic and Systems Biology Unit, Institute of Biochemistry, Biological Research Centre of the Hungarian Academy of Sciences, Szeged, H-6726, Hungary
First published on 6th September 2024
Abstract
This study presents the discovery of a new series of N-phenylpyrrolamide inhibitors of bacterial DNA gyrase with improved antibacterial activity. The most potent inhibitors had low nanomolar IC50 values against Escherichia coli DNA gyrase (IC50; 2–20 nM) and E. coli topoisomerase IV (22i, IC50 = 143 nM). Importantly, none of the compounds showed activity against human DNA topoisomerase IIα, indicating selectivity for bacterial targets. Among the tested compounds, 22e emerged as the most effective against Gram-positive bacteria with minimum inhibitory concentration (MIC) values of 0.25 μg mL−1 against Staphylococcus aureus ATCC 29213 and MRSA, and 0.125 μg mL−1 against Enterococcus faecalis ATCC 29212. For Gram-negative bacteria, compounds 23b and 23c showed the greatest efficacy with MIC values ranging from 4 to 32 μg mL−1 against E. coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853, Acinetobacter baumannii ATCC 17978 and A. baumannii ATCC 19606. Notably, compound 23b showed promising activity against the clinically relevant Gram-negative pathogen Klebsiella pneumoniae ATCC 10031, with an MIC of 0.0625 μg mL−1. Furthermore, compounds 23a and 23c exhibited significantly lower susceptibility to resistance development compared to novobiocin in S. aureus ATCC 29213 and K. pneumoniae ATCC 10031. Overall, the most promising compounds of this series showed excellent on-target potency, marking a significant improvement over previous N-phenylpyrrolamide inhibitors.
1. Introduction
Antimicrobial resistance (AMR) is among the top ten global health threats of the 21st century.1 Antibiotic-resistant bacteria are projected to cause at least 10 million deaths annually by 2050.2 The World Health Organization (WHO) identifies six bacterial species as critical or high priority: the Gram-negative Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii and Pseudomonas aeruginosa, and the Gram-positive Staphylococcus aureus and Streptococcus pneumoniae.3 Among these, methicillin-resistant Staphylococcus aureus (MRSA) accounted for the most AMR-related deaths in 2019,3 and the incidence of community-acquired MRSA infections is continuing to rise.4,5DNA gyrase is an essential enzyme for bacterial survival as it plays an important role in DNA replication, transcription, and recombination by facilitating the cleavage and rejoining of DNA strands.6 Topoisomerase IV, structurally and functional similar enzyme to DNA gyrase, is vital for bacterial cell division. Both enzymes form tetrameric complexes necessary for DNA binding, cleavage and transport: DNA gyrase consists of two GyrA and two GyrB subunits, while topoisomerase IV comprises two ParC and two ParE subunits.6,7 The cleavage and rejoining of DNA require the hydrolysis of an ATP molecule. The ATP-binding site is located on the GyrB subunit of DNA gyrase and the ParE subunit of topoisomerase IV. Due to their structural similarities, these enzymes offer exceptional opportunities for dual targeting with new antibacterial compounds, potentially reducing the likelihood of bacteria developing resistance.8
The most well-known inhibitors of DNA gyrase and topoisomerase IV are fluoroquinolones, which act by binding to the GyrA and/or ParC subunit and stabilizing the enzyme-DNA complex.9,10 Fluoroquinolones are among the most common and effective antibacterial agents, but side effects and increasing bacterial resistance limit their use.11,12 Currently, no GyrB inhibitors that work through an ATP-competitive mechanism are available in clinical practice.13 Although several classes of ATP-competitive inhibitors targeting DNA gyrase and/or topoisomerase IV have been identified in recent decades, including pyridylureas,14 pyrimidinoindoles,15 benzimidazoleureas,16 benzothiazoles,17–22 pyrazolopyridones,23 and pyrrolamides,24 none have been approved for clinical use. Two ATP-competitive DNA gyrase inhibitors are currently in clinical trials: fobrepodacin or SPR720 for non-tuberculous mycobacterial infections25 and DS-2969b for Clostridium difficile infections.26 Further research is needed to develop GyrB/ParE inhibitors with enhanced antibacterial activity, reduced toxicity and favourable ADME (absorption, distribution, metabolism, and excretion) properties.
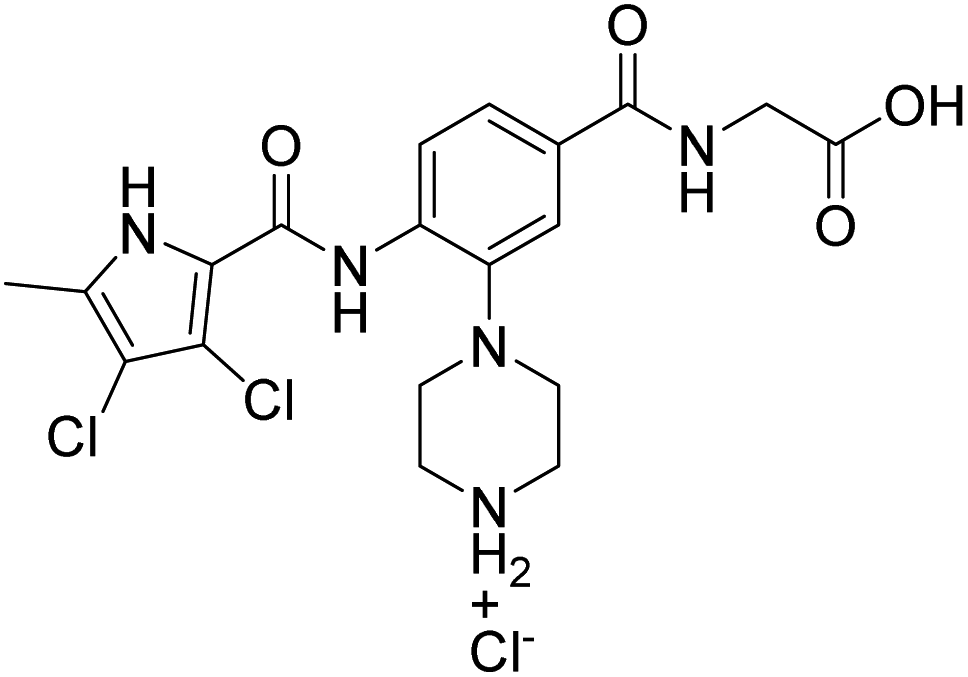
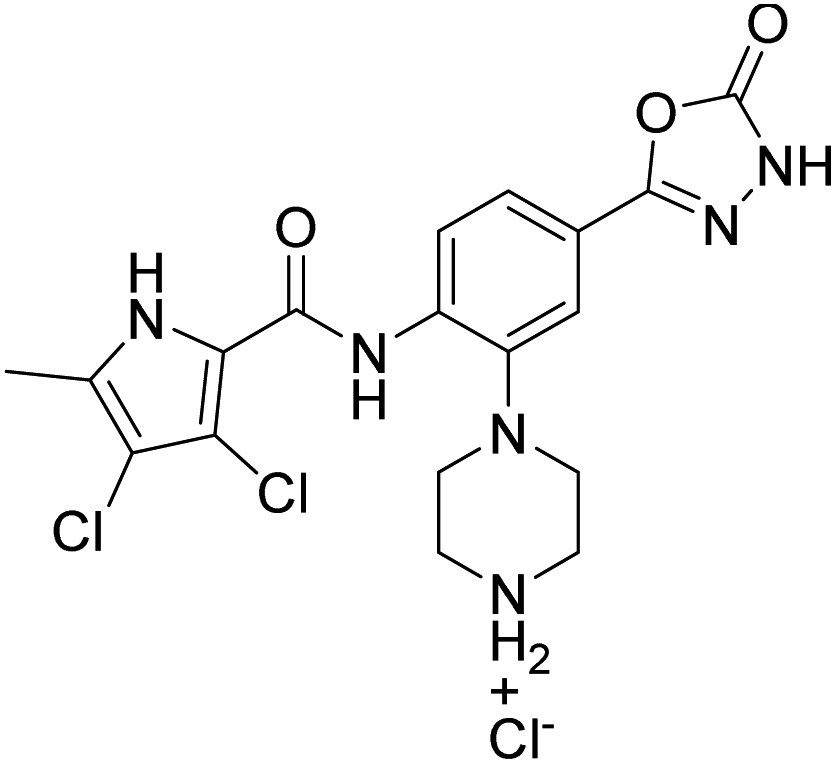
We have recently discovered a new class of DNA gyrase inhibitors, the N-phenylpyrrolamides (Fig. 1).27–32 Building on the first X-ray crystal structure of an N-phenylpyrrolamide inhibitor A bound to the active site of GyrB (Fig. 1),27 we developed several classes of optimised inhibitors. By substituting the 4,5-dibromopyrrole ring in compound A with a 3,4-dichloro-5-methyl-substituted pyrrole (Fig. 1, compound B), we achieved a twofold increase in activity against E. coli DNA gyrase, improving an IC50 value from 450 nM to 280 nM.28 Further enhancement was achieved by introducing an isopropoxy group on the central phenyl ring (Fig. 1, compound C), resulting in an IC50 of 47 nM against E. coli DNA gyrase, along with weak activity against Gram-positive E. faecalis.28 The antibacterial activity was further improved by replacing the carboxyl group with an isosteric group on the right-hand side of the molecule (Fig. 1, compound D).29 Structural optimisation at the three key positions marked in Fig. 1 (compound D) proved critical for the potent on-target and antibacterial activity of this class of DNA gyrase inhibitors. In this study, we present the synthesis and evaluation of a new series of N-phenylpyrrolamide DNA gyrase inhibitors, featuring a basic substituent at the central benzene core, which exhibit improved antibacterial activity. For selected promising inhibitors, we performed an in-depth evaluation, including determination of selectivity, frequency of resistance and thermodynamic solubility.
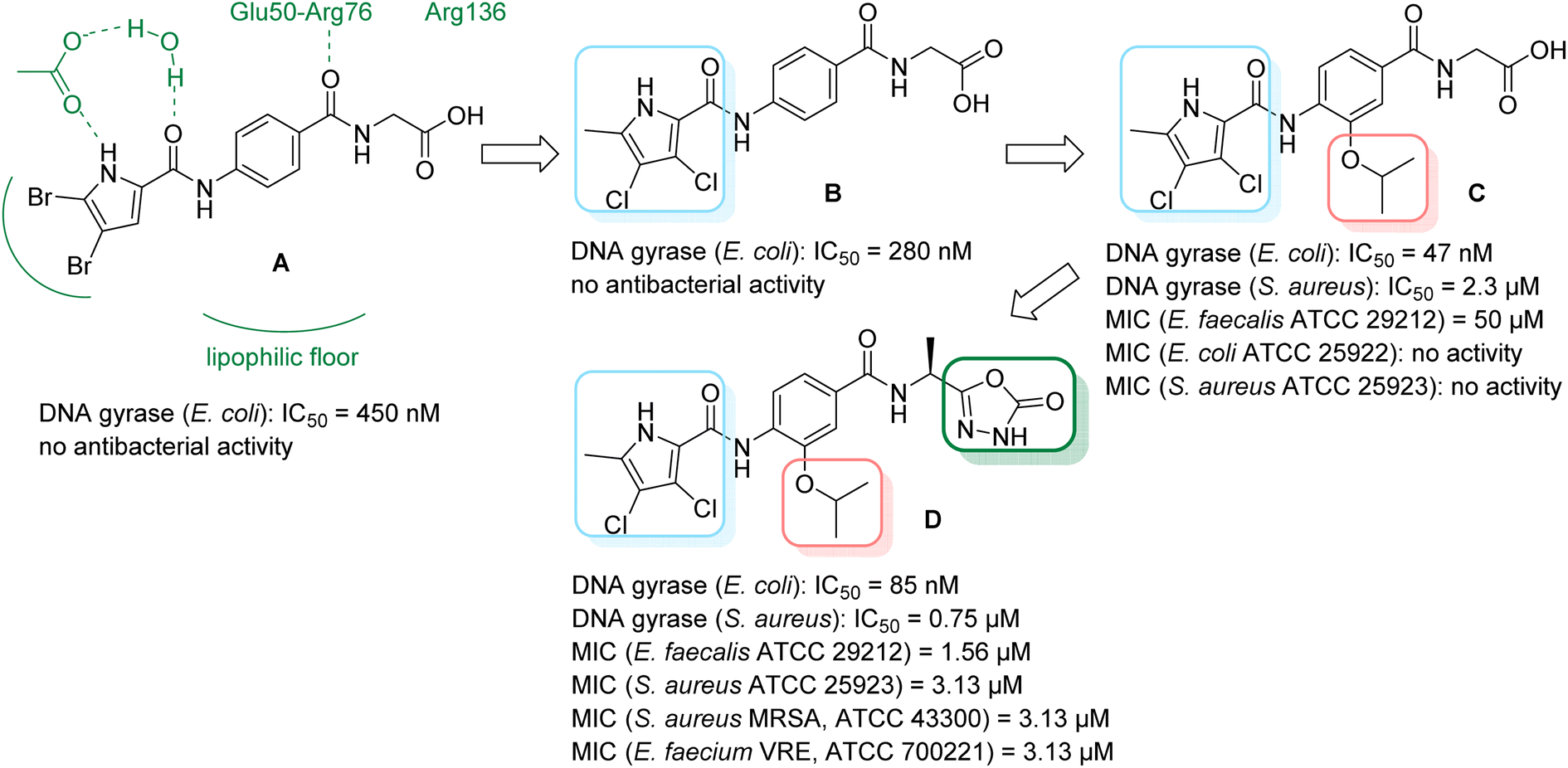 | ||
| Fig. 1 A brief summary of the optimisation pathway for N-phenylpyrrolamide inhibitors of DNA gyrase. Starting from compound A,27 the on-target and antibacterial activities increased through compounds B and C,28 to compound D.29 | ||
2. Results and discussion
2.1. Structure-based design
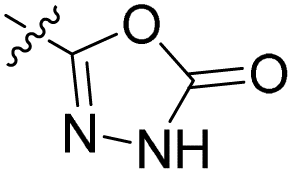
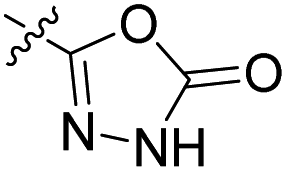
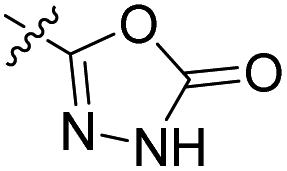
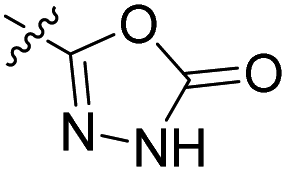
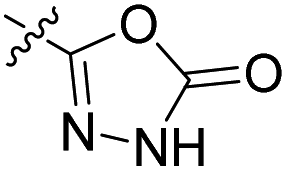
We have designed a series of new N-phenylpyrrolamide DNA gyrase inhibitors by systematically varying the substituents R1, R2, R3 and R4 (Fig. 2). In most of these new compounds, we used the 3,4-dichloro-5-methyl-substituted pyrrole as R1, as this has been shown to be optimal in our previous inhibitor series.28,29,32 To investigate the effects of different substituents on the pyrrole ring, we also synthesised a small set of 4,5-dibromopyrrole analogues. The NH group of the amide bond was left unsubstituted in most compounds (R2 = H, Fig. 2), although we also prepared two compounds with a methyl or benzyl substituent at this position. In this way, we aimed to disrupt the planarity of the central core and thus improve the physicochemical properties of the compounds. As R3 (Fig. 2), we introduced various nitrogen-containing heterocycles, including piperazine, 3-aminopyrrolidine, 3-aminopiperidine, piperidin-4-amine, morpholine, and 3-aminomethylpiperidine. These groups were chosen to (i) promote additional interactions with the lipophilic floor of the enzyme, (ii) enable additional hydrogen bonding through the NH group of the ring, and/or (iii) increase antibacterial activity, as the presence of a positive charge has been associated with enhanced accumulation in E. coli.33–35 At the right end of the molecules (R4, Fig. 2), we introduced either a carboxyl group or an isosteric 5-oxo-1,3,4-oxadiazole group, both capable of interacting with the positively charged Arg136 residue or forming cation-π interactions with the Glu50–Arg76 salt bridge. In some compounds, these groups were directly attached to the central phenyl ring, while in others they were connected via an additional three-atom linker to modulate the spatial arrangement and binding interactions.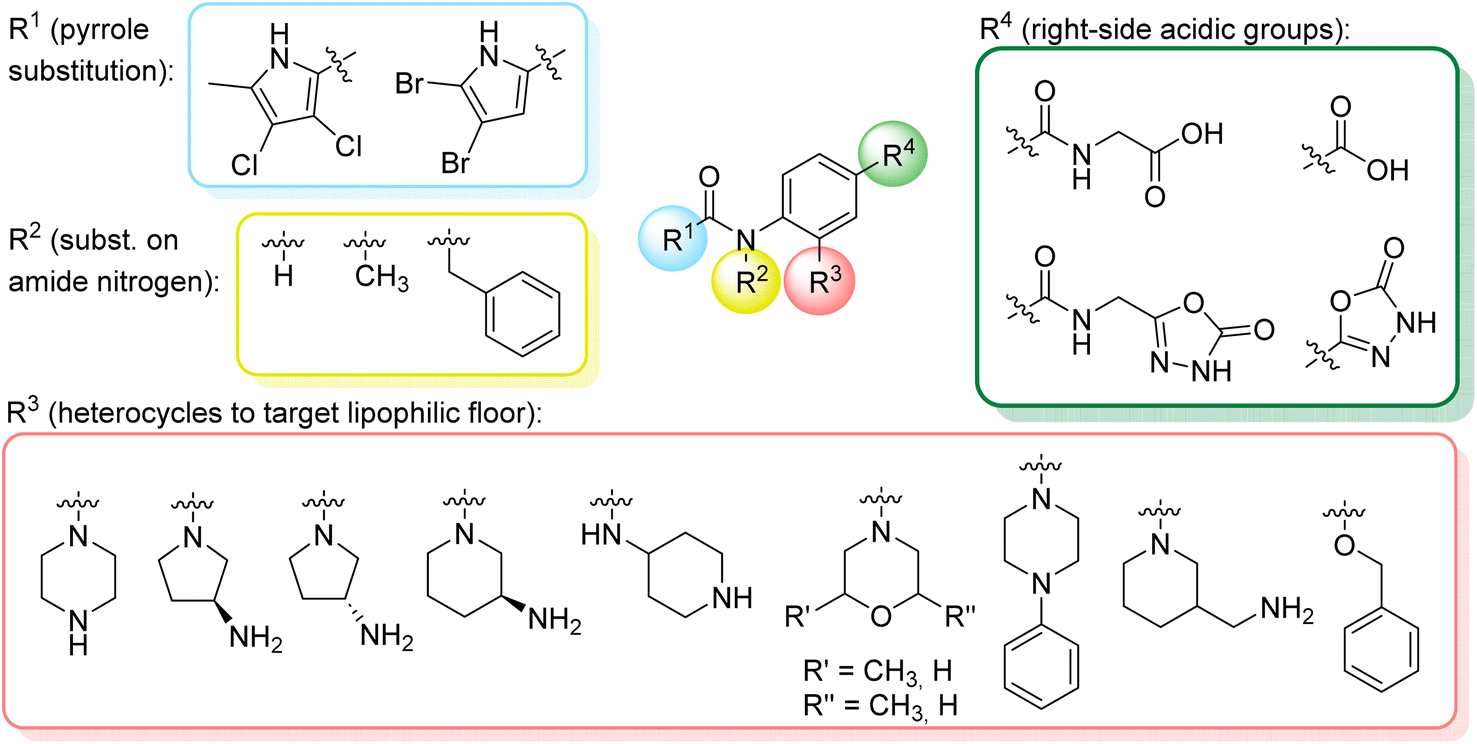 | ||
| Fig. 2 Optimisation plan of N-phenylpyrrolamide inhibitors of DNA gyrase. | ||
2.2. Chemistry
The synthesis of compounds 8–11 is shown in Scheme 1. In the first step, the fluorine in the methyl 3-fluoro-4-nitrobenzoate (1) was substituted with either Boc-piperazine or Boc-3-aminopyrrolidine as nucleophile. Next, the methyl ester groups of 2a–c were hydrolysed with sodium hydroxide solution, and the glycine methyl ester was attached to the free carboxyl groups of 3a–c via TBTU-promoted coupling. The nitro groups of 4a–c were then reduced to the amino groups by hydrogenation with Pd/C as catalyst. Either 3,4-dichloro-5-methyl-1H-pyrrole-2-carboxylic acid (for the synthesis of 6a–c) or 4,5-dibromo-1H-pyrrole-2-carboxylic acid (for the synthesis of 7a–c) were attached to the amino groups of 5a–c via the amide bond by first activating the carboxyl group with oxalyl chloride to form the acid chloride. The final compounds were obtained by first hydrolysing the methyl ester groups of 6a–c and 7a–c to the carboxyl groups (8a–c and 9a–c), followed by the removal of the Boc protecting groups with hydrochloric acid in 1,4-dioxane, resulting in compounds 10a–c and 11a–c. For compound 10a, an analogue (14) was also synthesised, in which the terminal carboxyl group on the right-hand side of the molecule was replaced with a 5-oxo-1,3,4-oxadiazole group. The synthesis of this compound is shown in Scheme 2. In the first step, the methyl ester group of 6c was converted to the hydrazide group by reaction with hydrazine monohydrate in a mixture of MeOH and THF under reflux to give 12. The hydrazide group was then cyclised to the 5-oxo-1,3,4-oxadiazole ring using 1,1′-carbonyldiimidazole (CDI) as a reagent. To obtain the final compound 14, the Boc protecting group was removed by acidolysis with hydrochloric acid in 1,4-dioxane.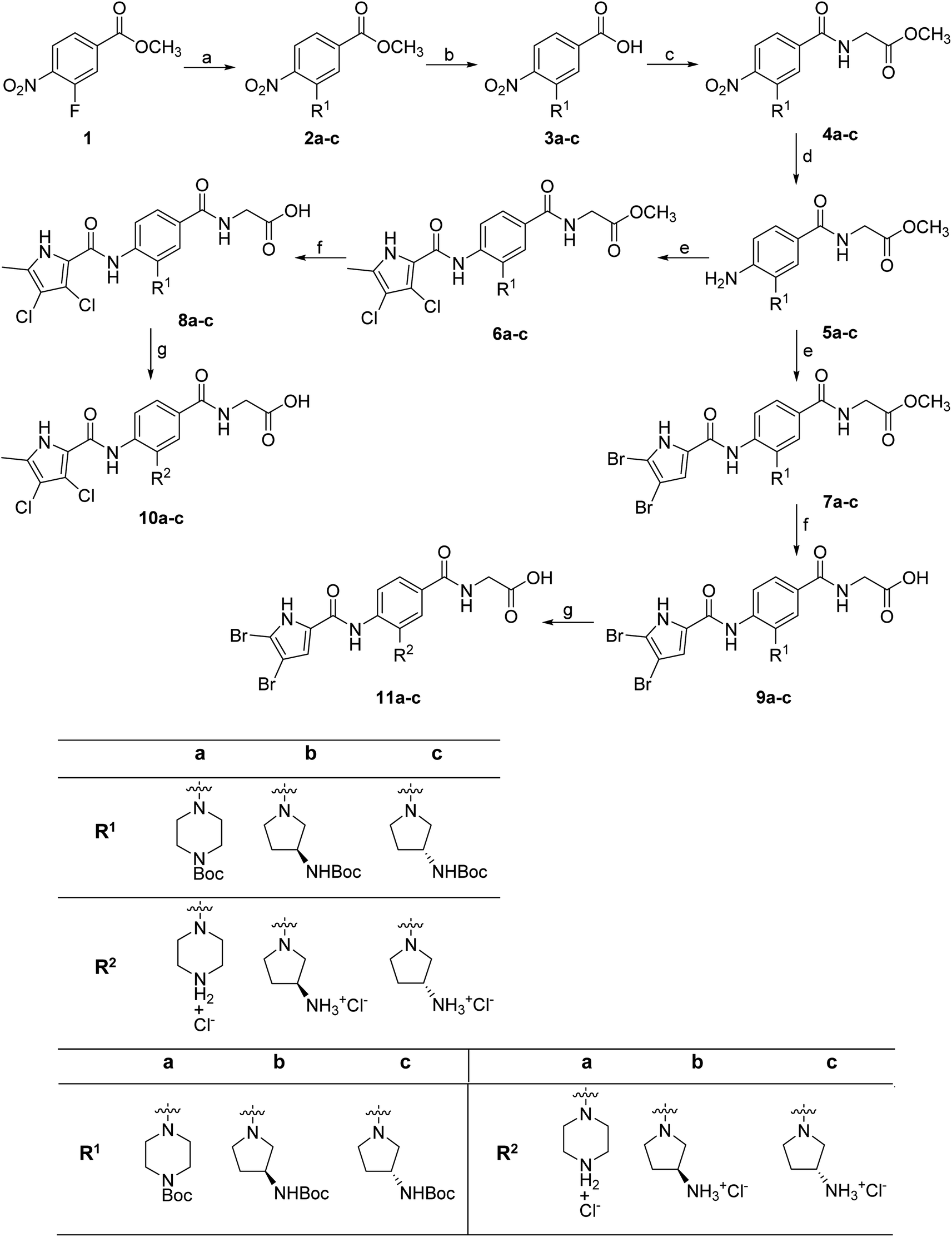 | ||
| Scheme 1 Reagents and conditions: (a) corresponding amine, K2CO3, DMF, 70 °C, 15 h; (b) 1 M NaOH, MeOH or MeOH and THF, rt, 15 h; (c) glycine methyl ester hydrochloride, TBTU, NMM, DMF, rt, 20 h; (d) H2, Pd/C, MeOH, rt, 4–5 h; (e) i: 3,4-dichloro-5-methylpyrrole-2-carboxylic acid or 4,5-dibromo-1H-pyrrole-2-carboxylic acid, oxalyl chloride, anhydrous CH2Cl2, rt, 15 h, then ii: 5a–c, anhydrous pyridine, anhydrous CH2Cl2, rt, 15 h; (f) 1 M NaOH, MeOH or MeOH and THF, rt, 15 h; (g) 4 M HCl in 1,4-dioxane, THF (for some compounds), rt, 1–2 h. | ||
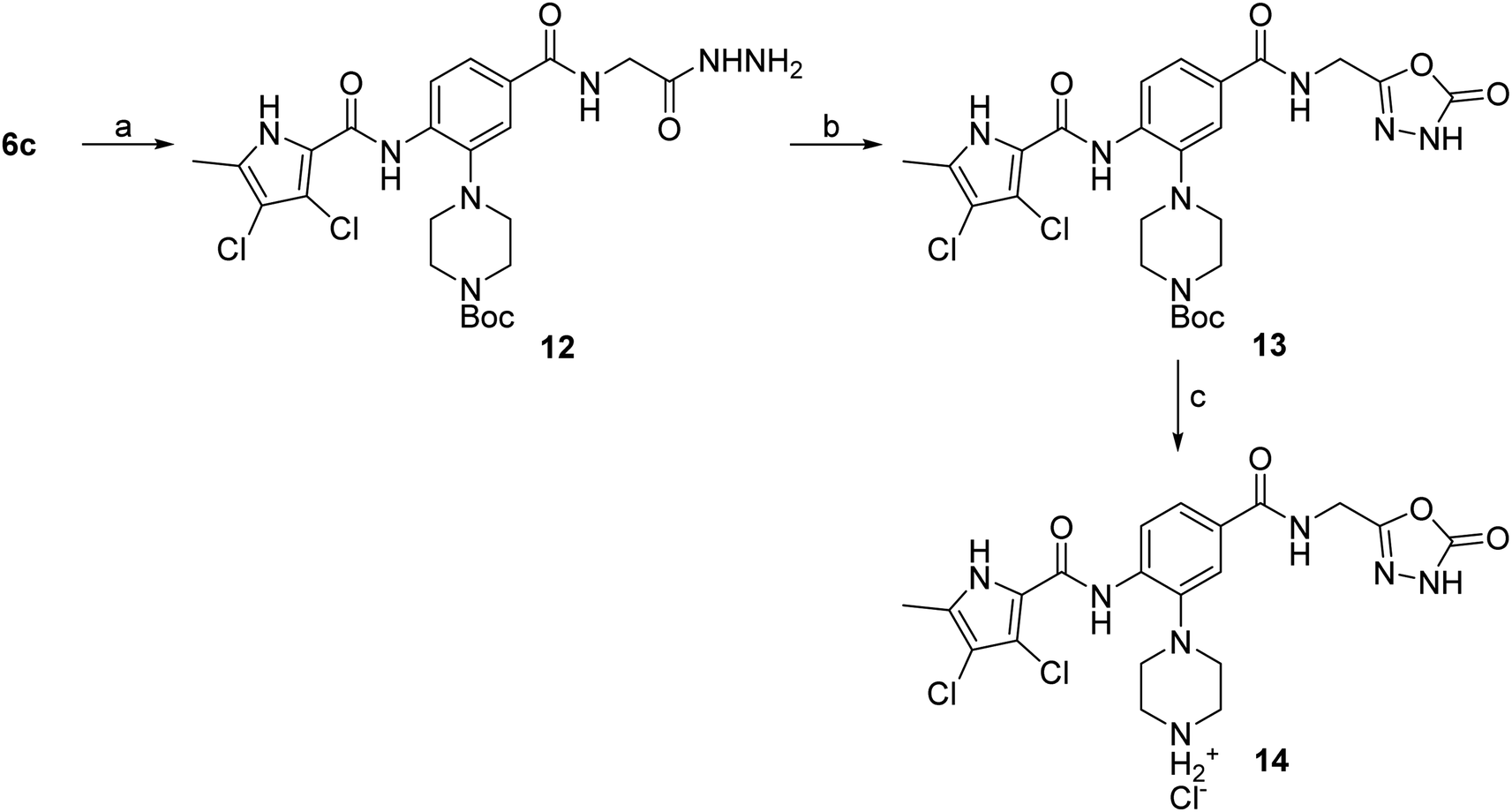 | ||
Scheme 2 Reagents and conditions: (a) hydrazine monohydrate, MeOH/THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), reflux, 20 h; (b) CDI, 1,4-dioxane/DMF (3:1), 101 °C, 20 h; (c) 4 M HCl in 1,4-dioxane, rt, 2 h. :1), reflux, 20 h; (b) CDI, 1,4-dioxane/DMF (3:1), 101 °C, 20 h; (c) 4 M HCl in 1,4-dioxane, rt, 2 h. | ||
To prepare compounds 17–20, we used the procedure shown in Scheme 3. Starting from 3-fluoro-4-nitrobenzoic acid methyl ester (1), we prepared a series of analogues (2a–b, 15c–i) by introducing an amine-containing heterocycle through nucleophilic substitution of fluorine at the central benzene core. After reduction of the nitro groups to the amino groups to form 16a–i, the 3,4-dichloro-5-methyl-1H-pyrrole-2-carboxylic acid was attached via the amide bond by first activating the carboxyl group with oxalyl chloride or thionyl chloride to give 17a–i. Compounds 18, 19 and 20 were prepared from 17a–i by hydrolysing the methyl ester groups and removing the Boc protecting groups by acidolysis with hydrochloric acid. Compounds 17a–g were further converted into analogues in which the 5-oxo-1,3,4-oxadiazole group was introduced in place of the carboxyl group on the right-hand side (Scheme 4). First, the methyl ester group of 17a–g was converted to a hydrazide group using hydrazine monohydrate as reagent (21a–g), which was followed by a reaction with CDI to give compounds 22a–g. The final step involved the acidolytic cleavage of the Boc protecting group in 22a–d, resulting in the desired compounds 23a–d.
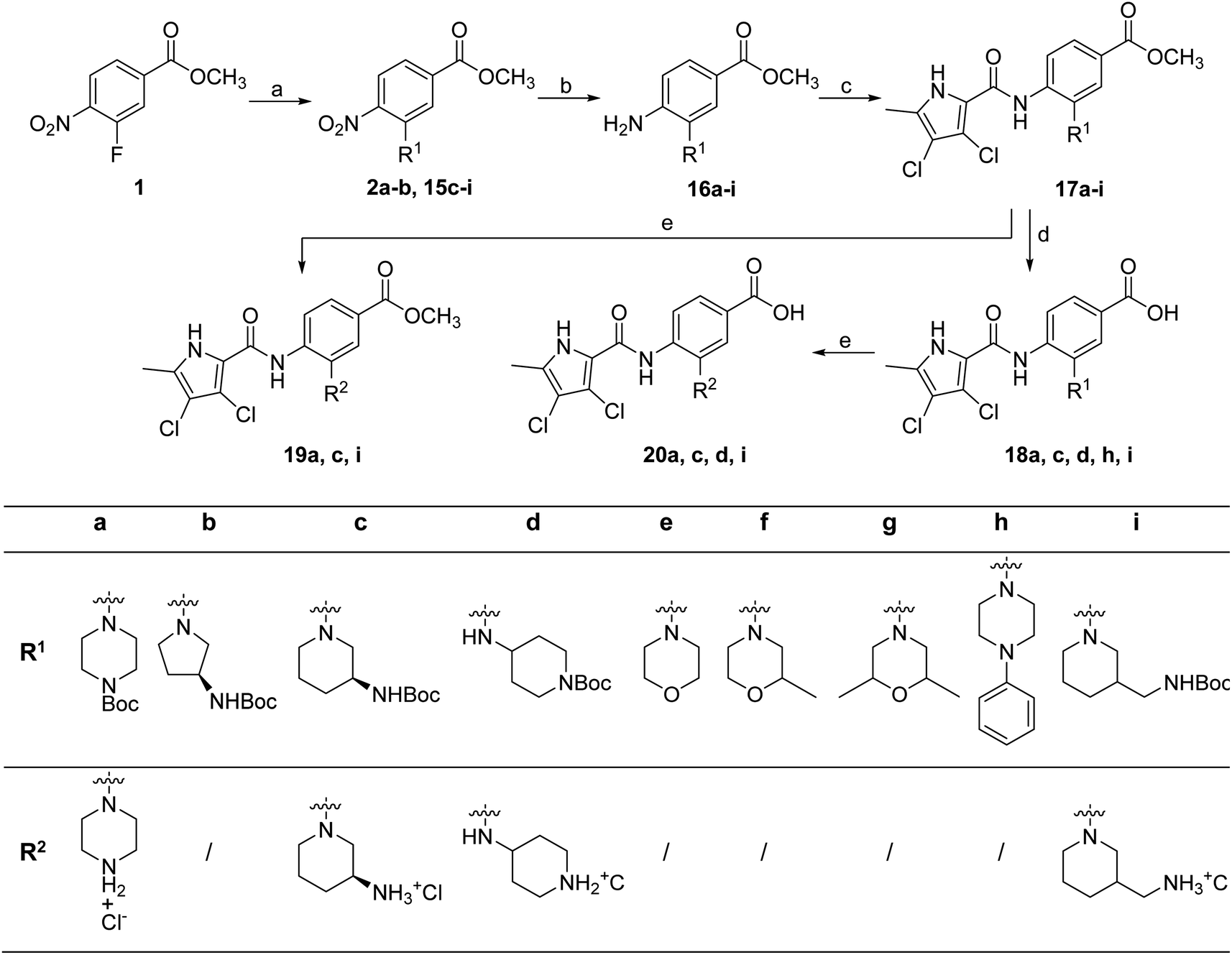 | ||
| Scheme 3 Reagents and conditions: (a) corresponding amine, K2CO3, DMF, 70 °C, 15 h; (b) H2, Pd/C, MeOH or MeOH and THF, rt, 1–5 h; (c) i: 3,4-dichloro-5-methylpyrrole-2-carboxylic acid, oxalyl chloride, anhydrous CH2Cl2, rt, 15 h (or thionyl chloride, 75 °C, 1 h, for the synthesis of 17e), then ii: 16a–i, anhydrous pyridine, anhydrous CH2Cl2, rt, 15 h (or toluene, 130 °C, 15 h, for the synthesis of 17e); (d) 1 M NaOH, MeOH or MeOH and THF, rt (or 60 °C for 18d and 18h), 15 h; (e) 1 M HCl in acetic acid or 4 M HCl in 1,4-dioxane, THF or CH2Cl2 (for some compounds), rt, 5–15 h. | ||
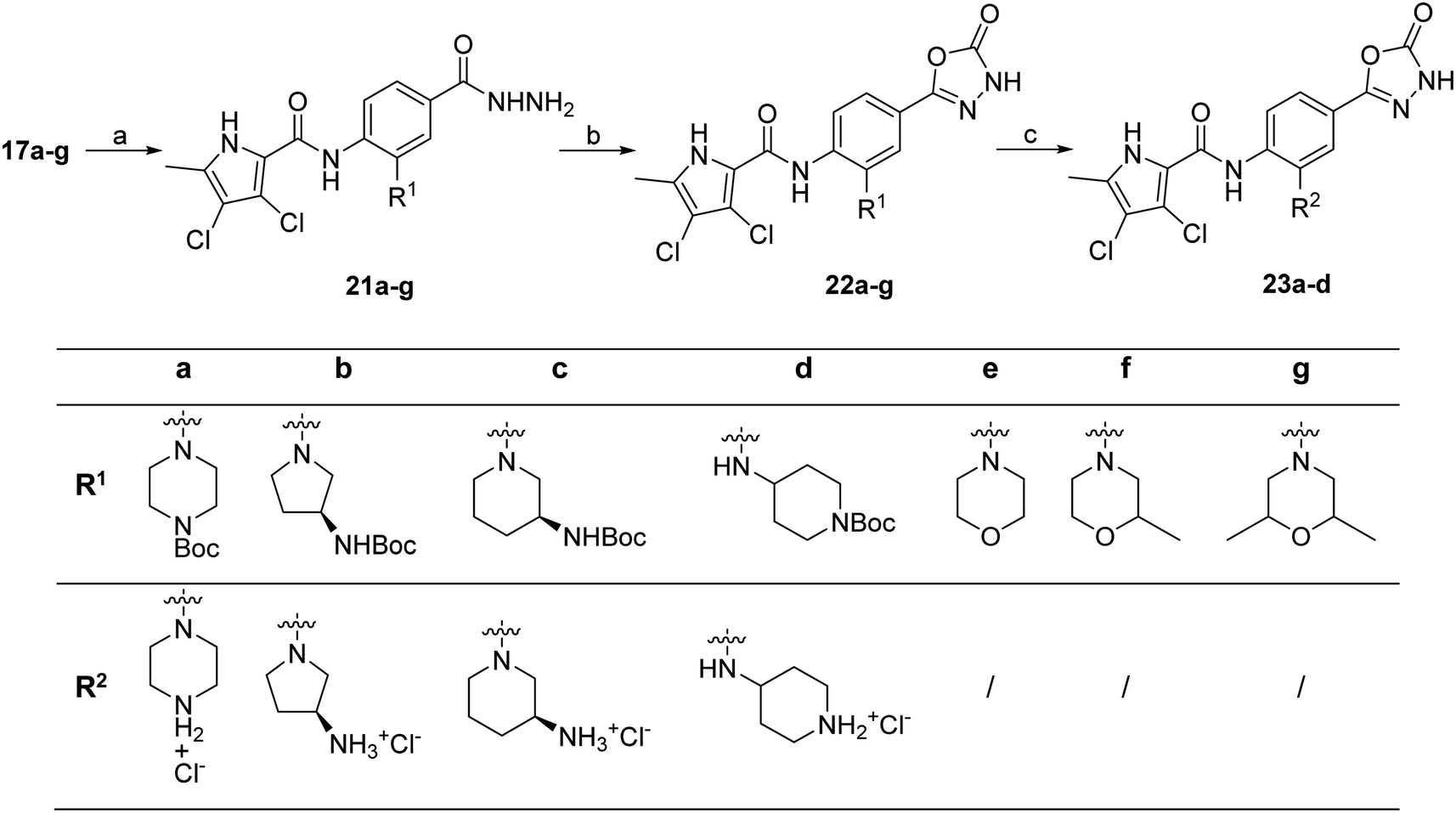 | ||
| Scheme 4 Reagents and conditions: (a) hydrazine monohydrate, MeOH, THF, high-pressure tube, 120 °C, 15–48 h; (b) CDI, 1,4-dioxane, DMF, 101 °C, 15–24 h; (c) 4 M HCl in 1,4-dioxane, DMF (for some compounds), rt, 3–15 h. | ||
The synthesis of the morpholine derivative 27a with the 4,5-dibromopyrrole group on the left-hand side, and the synthesis of the 3,4-dichloro-5-methylpyrrole analogue 27b with the methyl group on the amide nitrogen, are shown in Scheme 5. To introduce the methyl group on the amino group of 16e, a reductive amination reaction was employed. First, the methylenimine intermediate was formed using paraformaldehyde as a reagent, which was then reduced with sodium borohydride to give the methylamino compound 24. In the next step, 3,4-dichloro-5-methyl-1H-pyrrole-2-carboxylic acid or 4,5-dibromo-1H-pyrrole-2-carboxylic acid was coupled with the amino groups of 16e and 24 to obtain the amides 25a–b. After hydrazinolysis of the methyl ester groups of 26a–b and subsequent cyclization to 5-oxo-1,3,4-oxadiazole using CDI as reagent, the final compounds 27a–b were obtained.
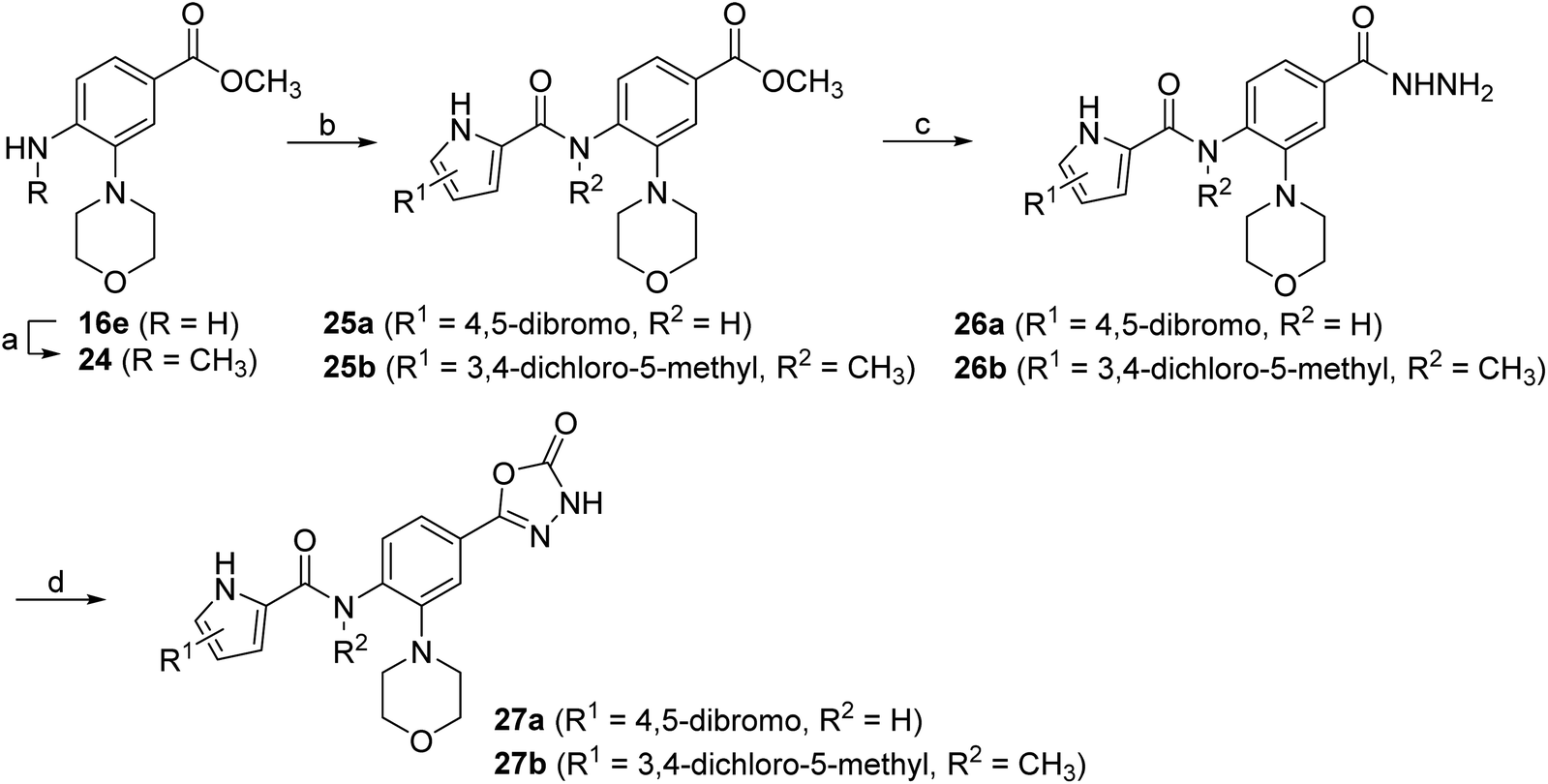 | ||
| Scheme 5 Reagents and conditions: (a) i: paraformaldehyde, NaOMe, MeOH, 40 °C, 15 h, then ii: NaBH4, 40 °C, 20 h; (b) i: 3,4-dichloro-5-methylpyrrole-2-carboxylic acid or 4,5-dibromo-1H-pyrrole-2-carboxylic acid, oxalyl chloride, anhydrous CH2Cl2, rt, 15 h, then ii: 16e or 24, anhydrous pyridine, anhydrous CH2Cl2, rt, 15 h; (c) hydrazine monohydrate, MeOH, THF, high-pressure tube, 120 °C, 15 h; (d) CDI, 1,4-dioxane, DMF, 101 °C, 15 h. | ||
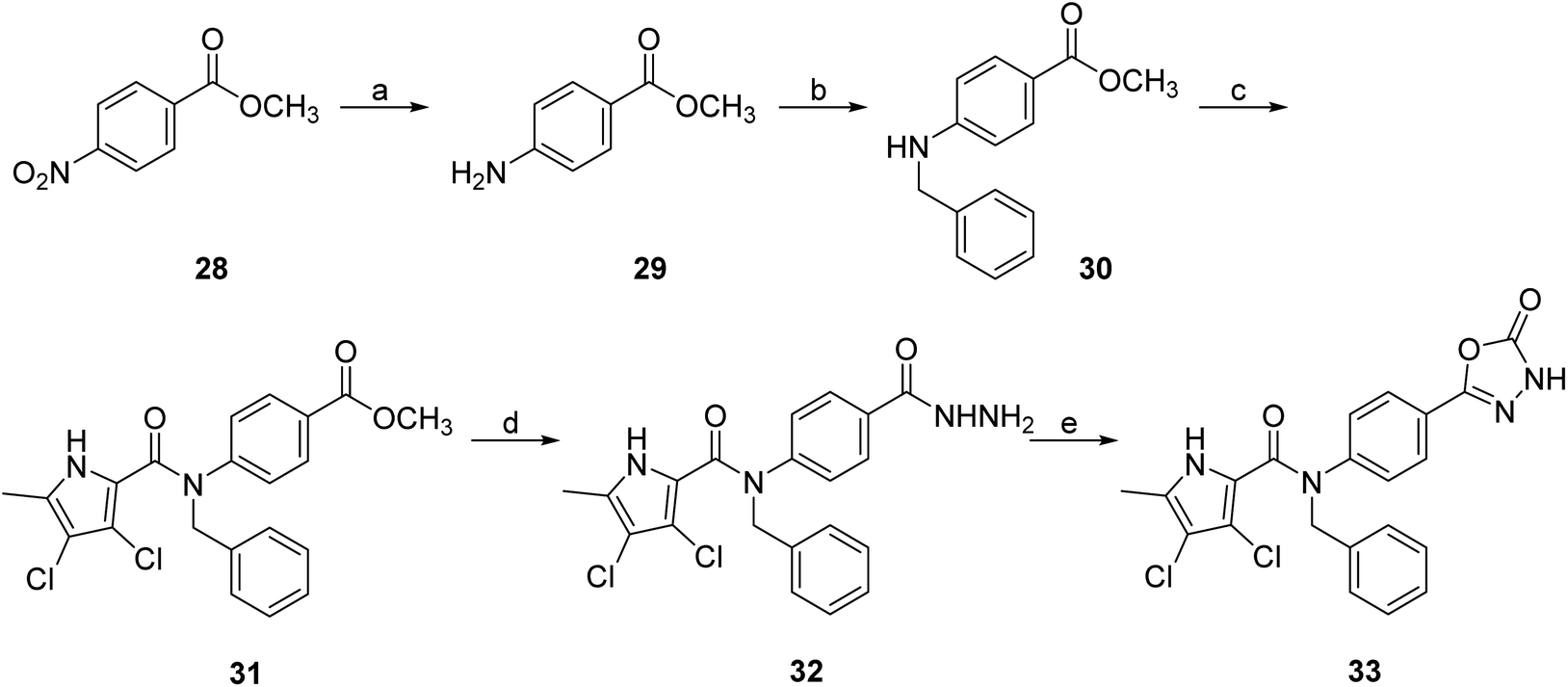
To prepare compound 33, which features a benzyl group on the amide nitrogen, the procedure shown in Scheme 6 was followed. After reduction of the nitro group of 4-nitrobenzoic acid methyl ester (28) by catalytic hydrogenation, the benzyl substituent was added in two steps. First, the benzylimine was formed in the reaction between 29 and benzaldehyde, and then the imine group was reduced with sodium borohydride to give 30. Next, the 3,4-dichloro-5-methyl-1H-pyrrole-2-carboxylic acid was coupled with the amino group of 30 via an amide bond to give 31. To obtain the final compound 33, the methyl ester group of 31 was first converted to the hydrazide group to give 32 in the reaction with hydrazine monohydrate. This was followed by cyclisation with CDI to form the 5-oxo-1,3,4-oxadiazole group. An analogue of 33 with the benzyloxy substituent attached to the benzene core (39) was prepared according to the procedure shown in Scheme 7. In the first step, the benzyl group was attached to the phenolic hydroxyl group of 34 using benzyl bromide as reagent and potassium carbonate as base. Subsequently, compound 35 was reacted with hydrazine monohydrate to form hydrazide 36, which was then converted to the 5-oxo-1,3,4-oxadiazole group with CDI. The nitro group of 37 was then reduced to the amino group, and finally the 4-dichloro-5-methyl-1H-pyrrole-2-carboxylic acid was attached to 38 via an amide bond. This was achieved by first activating the carboxyl group with oxalyl chloride to give the final compound 39.
 | ||
| Scheme 6 Reagents and conditions: (a) H2, Pd/C, MeOH, THF, rt, 3 h; (b) i: benzaldehyde, Na2SO4, CH2Cl2, reflux, 15 h, ii: NaBH4, MeOH, rt, 15 h; (c) i: 3,4-dichloro-5-methylpyrrole-2-carboxylic acid, oxalyl chloride, anhydrous CH2Cl2, rt, 15 h, then ii: 30, anhydrous pyridine, anhydrous CH2Cl2, rt, 15 h; (d) hydrazine monohydrate, MeOH, THF, high-pressure tube, 100 °C, 2 d; (e) CDI, 1,4-dioxane, 101 °C, 15 h. | ||
 | ||
| Scheme 7 Reagents and conditions: (a) K2CO3, benzyl bromide, CH3CN, rt, 60 °C, 3 h; (b) hydrazine monohydrate, MeOH, THF, 65 °C, 15 h; (c) CDI, 1,4-dioxane, 101 °C, 15 h; (d) Fe, glacial acetic acid, rt, 1.5 h; (e) i: 3,4-dichloro-5-methylpyrrole-2-carboxylic acid, oxalyl chloride, anhydrous CH2Cl2, rt, 15 h, then ii: 38, anhydrous pyridine, anhydrous CH2Cl2, rt, 15 h. | ||
2.3. Biological evaluation
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. | R1 | R2 | R3 | IC50a (nM) or RAb (%) | ||||
| E. coli gyrase | S. aureus gyrase | E. coli topo IV | S. aureus topo IV | Human topo IIα | ||||
| a Concentration of compound that inhibits the enzyme activity by 50%.b Residual activity of the enzyme at 1 μM or 10 μM concentration of the compound.c Not determined. Compounds 10a–c, 11a–c and 14 were prepared as hydrochloride salts. | ||||||||
| 6a | 3,4-Dichloro-5-methyl |  |
COOCH3 | 4080 nM | n.d.c | n.d. | n.d. | n.d. |
| 8a | 3,4-Dichloro-5-methyl |  |
COOH | 84.1 nM | 37% (1 μM) | 93% (1 μM) | 22% (1 μM) | n.d. |
| 8b | 3,4-Dichloro-5-methyl |  |
COOH | 196 nM | n.d. | n.d. | n.d. | n.d. |
| 8c | 3,4-Dichloro-5-methyl |  |
COOH | 162 nM | n.d. | n.d. | n.d. | n.d. |
| 9a | 4,5-Dibromo |  |
COOH | 362 nM | n.d. | n.d. | n.d. | n.d. |
| 9b | 4,5-Dibromo |  |
COOH | 4110 nM | n.d. | n.d. | n.d. | n.d. |
| 9c | 4,5-Dibromo |  |
COOH | 5390 nM | n.d. | n.d. | n.d. | n.d. |
| 10a | 3,4-Dichloro-5-methyl | 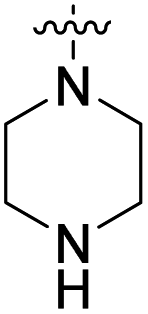 |
COOH | 40.3 nM | 621 nM | 93% (1 μM) | 4190 nM | 78% (10 μM) |
| 10b | 3,4-Dichloro-5-methyl | 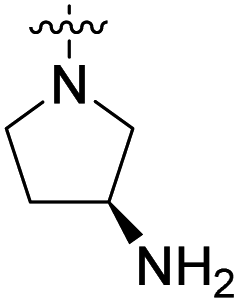 |
COOH | 19.3 nM | 112 nM | 958 nM | 2560 nM | 82% (10 μM) |
| 10c | 3,4-Dichloro-5-methyl | 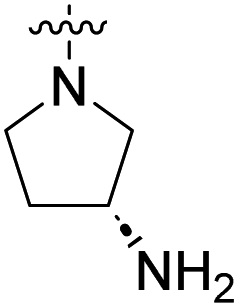 |
COOH | 21.3 nM | 1240 nM | 2370 nM | 2030 nM | 112% (40 μM) |
| 11a | 4,5-Dibromo | 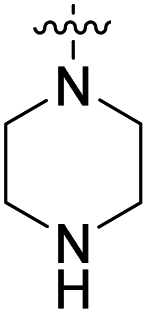 |
COOH | 841 nM | n.d. | 97% (10 μM) | 73% (10 μM) | 100% (40 μM) |
| 11b | 4,5-Dibromo | 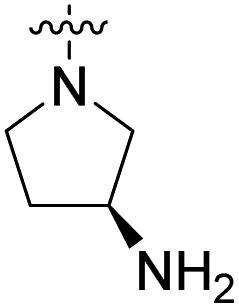 |
COOH | 235 nM | 46% (10 μM) | 84% (10 μM) | 91% (10 μM) | n.d. |
| 11c | 4,5-Dibromo | 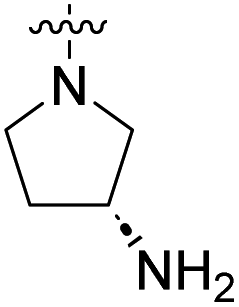 |
COOH | 996 nM | 78% (10 μM) | 75% (10 μM) | n.d. | n.d. |
| 13 | 3,4-Dichloro-5-methyl |  |
 |
483 nM | n.d. | n.d. | n.d. | n.d. |
| 14 | 3,4-Dichloro-5-methyl |  |
 |
22.4 nM | 37% (1 μM) | 2970 nM | 2700 nM | 111% (10 μM) |

|
||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. | R1 | R2 | R3 | IC50a (μM) or RAb (%) | ||||
| E. coli gyrase | S. aureus gyrase | E. coli topo IV | S. aureus topo IV | Human topo IIα | ||||
| a Concentration of compound that inhibits the enzyme activity by 50%.b Residual activity of the enzyme at 1 μM or 10 μM concentration of the compound.c Not determined. Compounds 19a, 19c, 19i, 20a, 20c–d, 20i and 23a–d were prepared as hydrochloride salts. | ||||||||
| 18a | H |  |
COOH | 159 nM | 54% (1 μM) | 99% (1 μM) | 21% (1 μM) | n.d.c |
| 18c | H |  |
COOH | 67% (1 μM) | n.d. | n.d. | n.d. | n.d. |
| 18h | H | 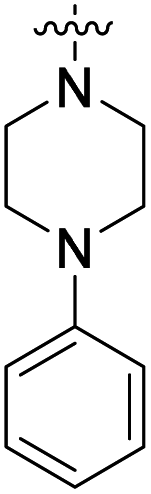 |
COOH | 199 nM | n.d. | 85% (10 μM) | n.d. | n.d. |
| 18i | H | 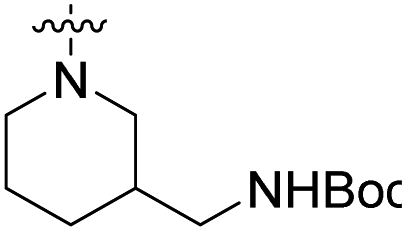 |
COOH | 89% (0.1 μM) | n.d. | n.d. | n.d. | 128% (100 μM) |
| 19a | H | 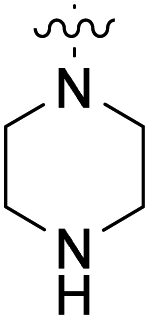 |
COOCH3 | 100 nM | 45% (1 μM) | 102% (1 μM) | 95% (1 μM) | n.d. |
| 19c | H | 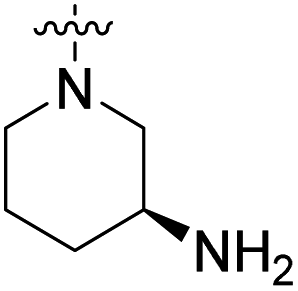 |
COOCH3 | 120 nM | 27% (1 μM) | 79% (1 μM) | n.d. | n.d. |
| 19i | H | 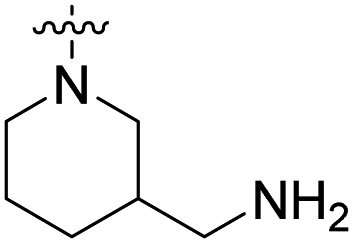 |
COOCH3 | 14.2 nM | n.d. | 533 nM | n.d. | n.d. |
| 20a | H | 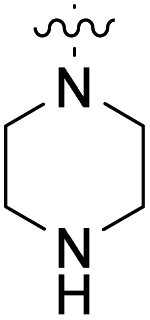 |
COOH | 67% (1 μM) | n.d. | 106% (1 μM) | 92% (1 μM) | 116% (40 μM) |
| 20c | H | 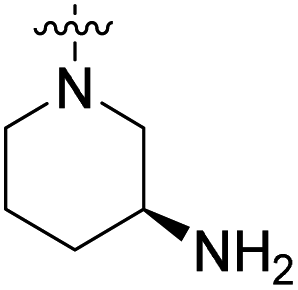 |
COOH | 134 nM | n.d. | 78% (1 μM) | n.d. | n.d. |
| 20d | H | 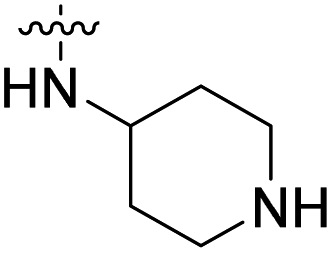 |
COOH | 532 nM | n.d. | 89% (10 μM) | n.d. | n.d. |
| 20i | H | 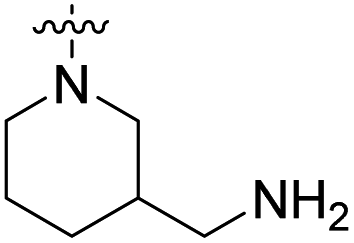 |
COOH | 6.34 nM | 28% (0.1 μM) | 143 nM | 0% (1 μM) | 28% (100 μM) |
| 22a | H | 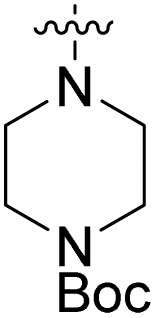 |
 |
59.0 nM | n.d. | 104% (1 μM) | n.d. | 100% (100 μM) |
| 22c | H | 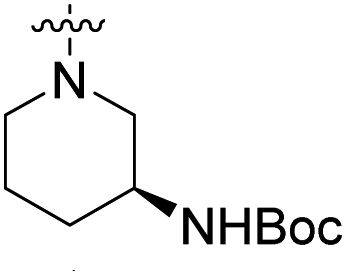 |
 |
90% (0.1 μM) | n.d. | n.d. | n.d. | n.d. |
| 22e | H | 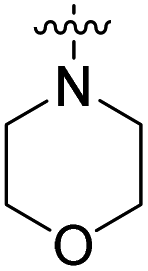 |
 |
82.2 nM | n.d. | 1230 nM | n.d. | 93% (100 μM) |
| 22f | H |  |
 |
53.5 nM | n.d. | 103% (1 μM) | n.d. | 98% (100 μM) |
| 22g | H | 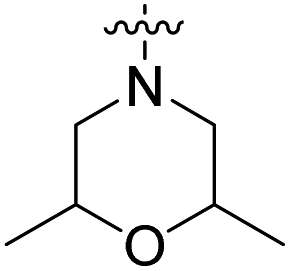 |
 |
109 nM | n.d. | 105% (1 μM) | n.d. | 97% (100 μM) |
| 23a | H | 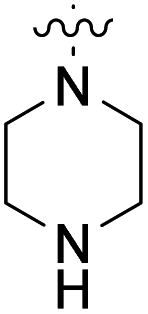 |
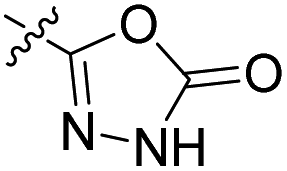 |
17.9 nM | n.d. | 71% (1 μM) | n.d. | 86% (100 μM) |
| 23b | H | 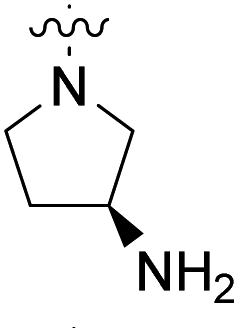 |
 |
2.80 nM | 39% (0.1 μM) | 105% (1 μM) | 99% (0.1 μM) | 86300 nM |
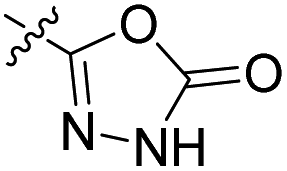
| 23c | H | 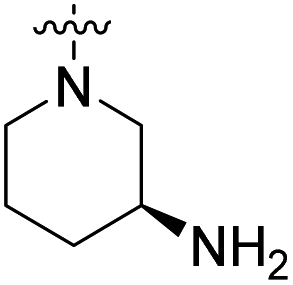 |
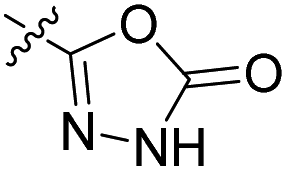 |
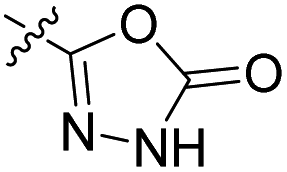
27.2 nM | 41% (0.1 μM) | 448 nM | 406 nM | 90% (50 μM) |
| 23d | H | 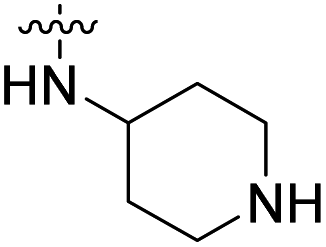 |
 |
4.40 nM | n.d. | 1257 nM | n.d. | 99% (10 μM) |
| 27a | CH3 | 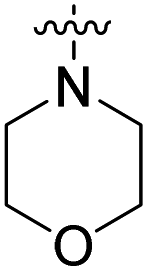 |
 |
98% (10 μM) | n.d. | 103% (1 μM) | n.d. | n.d. |
| 27b | — | — | — | 433 nM | n.d. | 103% (1 μM) | n.d. | 98% (100 μM) |
| 33 | Bzl | H |  |
81% (1 μM) | n.d. | n.d. | n.d. | 92% (100 μM) |
| 39 | — | — | — | 886 nM | n.d. | 101% (1 μM) | n.d. | 104% (100 μM) |
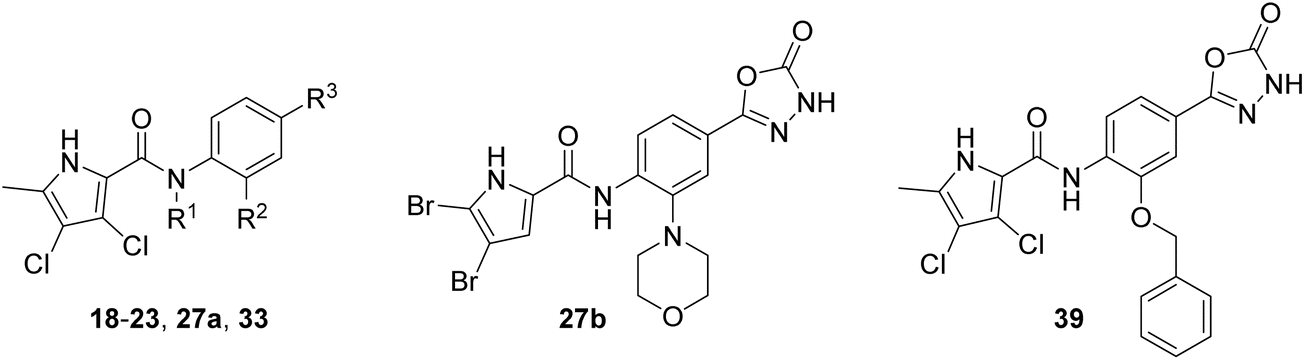
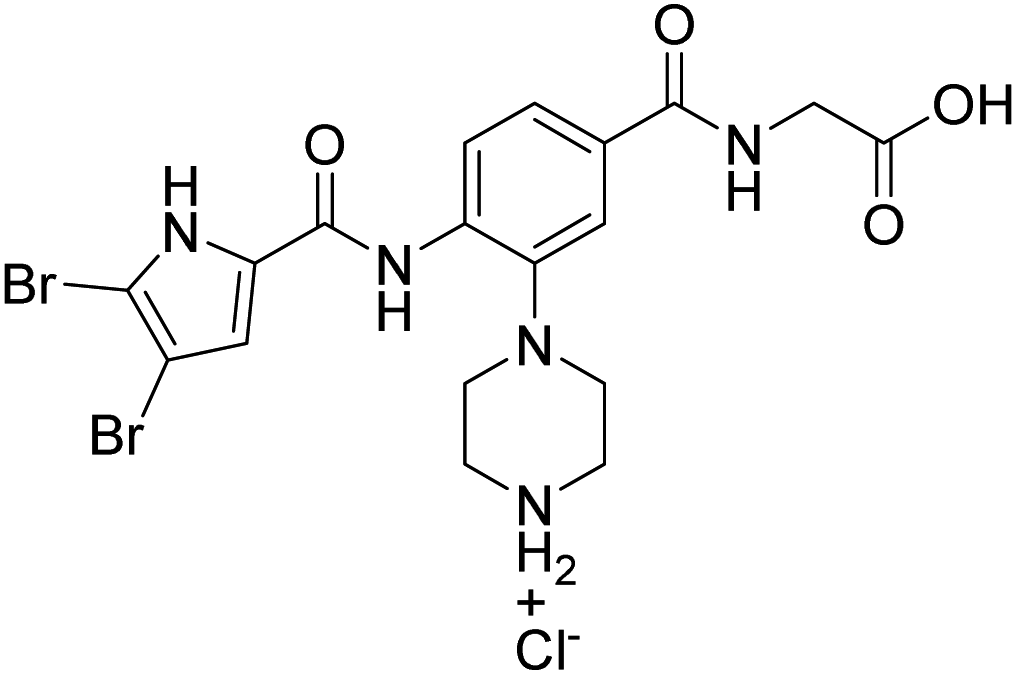
Table 1 shows the enzyme inhibitory activities of compounds 6–14, which feature carboxyl or 5-oxo-1,3,4-oxadiazole groups connected to the central phenyl ring through a three-atom linker. The comparison of compound 8a (IC50 = 84.1 nM) with its methyl ester analogue 6a (IC50 = 4080 nM) highlights the critical role of the terminal acidic group for the potent activity on E. coli gyrase. Furthermore, Table 1 shows that compounds containing the 3,4-dichloro-5-methyl-substituted pyrrole ring on the left-hand side of the molecules (8a–c, 10a–c) exhibit, on average, 10- to 50-fold greater potency on E. coli gyrase than those with the 4,5-dibromopyrrole ring (9a–c, 11a–c). A similar difference in activity is observed when the compounds with the Boc protecting group at the nitrogen-containing heterocycle (8a–c, 9a–c, 13) are compared with the Boc-deprotected analogues (10a–c, 11a–c, 14). This difference may be attributed to the free NH group in the Boc-deprotected compounds, which potentially forms additional interactions with the enzyme, or to steric hindrance caused by the Boc group. An important observation from Table 1 is that compound 10a (IC50 = 40.3 nM) with the terminal carboxyl group has about two times lower potency than its analogue 14 (IC50 = 22.4 nM) with the 5-oxo-1,3,4-oxadiazole ring as the carboxyl isostere. This difference likely arises from stronger interactions between the 5-oxo-1,3,4-oxadiazole ring and the positively charged Arg136 residue in the enzyme's binding site. Among the compounds shown in Table 1, 10b, 10c and 14 exhibit the highest potency against E. coli DNA gyrase with IC50 values around 20 nM. Compound 10b also proves to be the most potent inhibitor of S. aureus DNA gyrase, with an IC50 value of 112 nM. However, the inhibitory effects of these compounds on topoisomerase IV in both E. coli and S. aureus are generally less potent, with IC50 values in the low micromolar range. Importantly, all compounds tested exhibit exceptional selectivity for human DNA topoisomerase IIα.
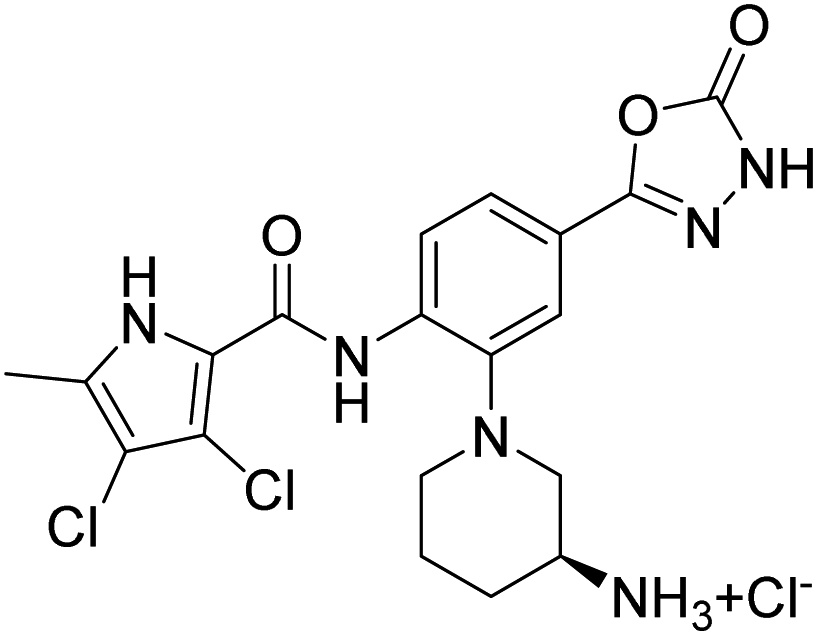
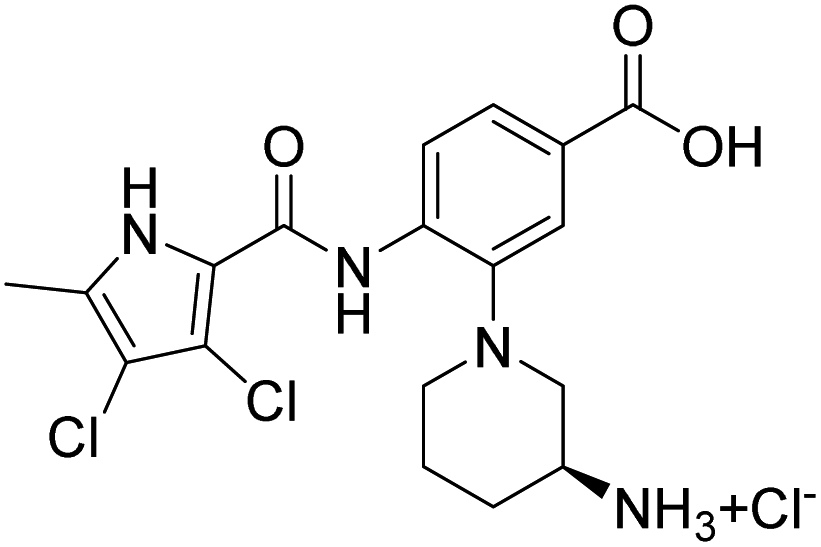
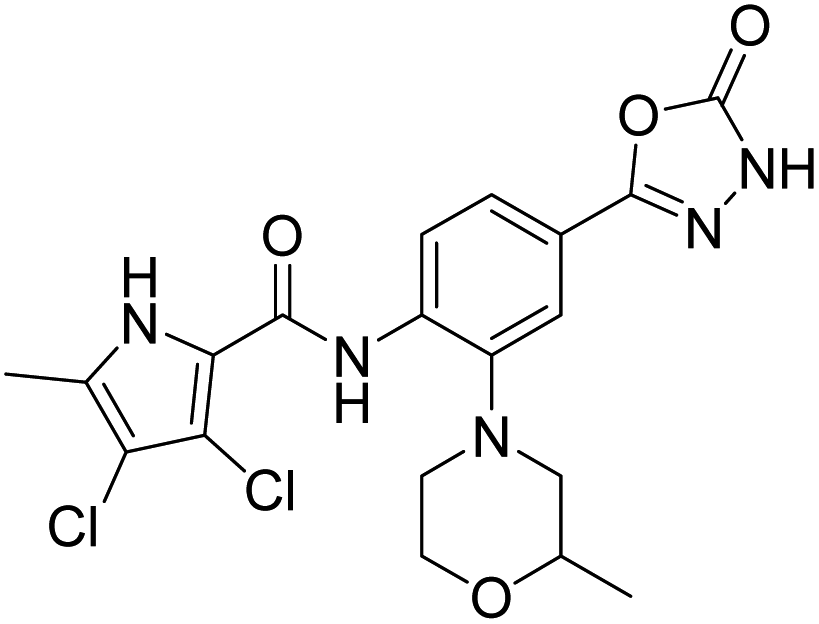
Table 2 shows the enzyme inhibitory activities of compounds 18–39, which differ from 6–14 by having the carboxyl or 5-oxo-1,3,4-oxadiazole groups directly attached to the central benzene ring. Consistent with the results in Table 1, the compound with a 3,4-dichloro-5-methyl-substituted pyrrole ring (22e, IC50 = 82.2 nM) exhibits approximately five times greater potency than its counterpart with the 4,5-dibromopyrrole ring (27b, IC50 = 433 nM). On the other hand, although compound 20i (IC50 = 6.34 nM), which contains a free carboxyl group, is about twice as potent as its methyl ester analogue 19i (IC50 = 14.2 nM), the activities of the methyl esters are slightly weaker in similar pairs, 20c (IC50 = 134 nM) and 19c (IC50 = 120 nM), and 20a (RA = 67% at 1 μM) and 19a (IC50 = 100 nM). As observed in Table 1, compounds with the Boc-protecting group on the nitrogen-containing heterocycle (18c, 18i, 22a, 22c) generally exhibit lower efficacy against E. coli DNA gyrase compared to their Boc-deprotected analogues (20c, 20i, 23a, 23c). Moreover, compounds with an additional heterocyclic basic centre, such as the piperazine derivative 23a (IC50 = 17.9 nM), the 3-aminopyrrolidine derivative 23b (IC50 = 2.80 nM), the 3-aminopiperidine derivative 23c (IC50 = 27.2 nM), and the piperidin-4-amine derivative 23d (IC50 = 4.40 nM), demonstrate slightly greater potency than those with a morpholine ring (22e–g, IC50 = 53.5–109 nM). This increased potency may be attributed to additional interactions formed by the NH group in the heterocyclic rings. Docking studies of compounds 20i and 23d with the active site of GyrB (Fig. 3) support this hypothesis, revealing potential ionic interactions between the amino groups of these compounds and the Asp49 residue.
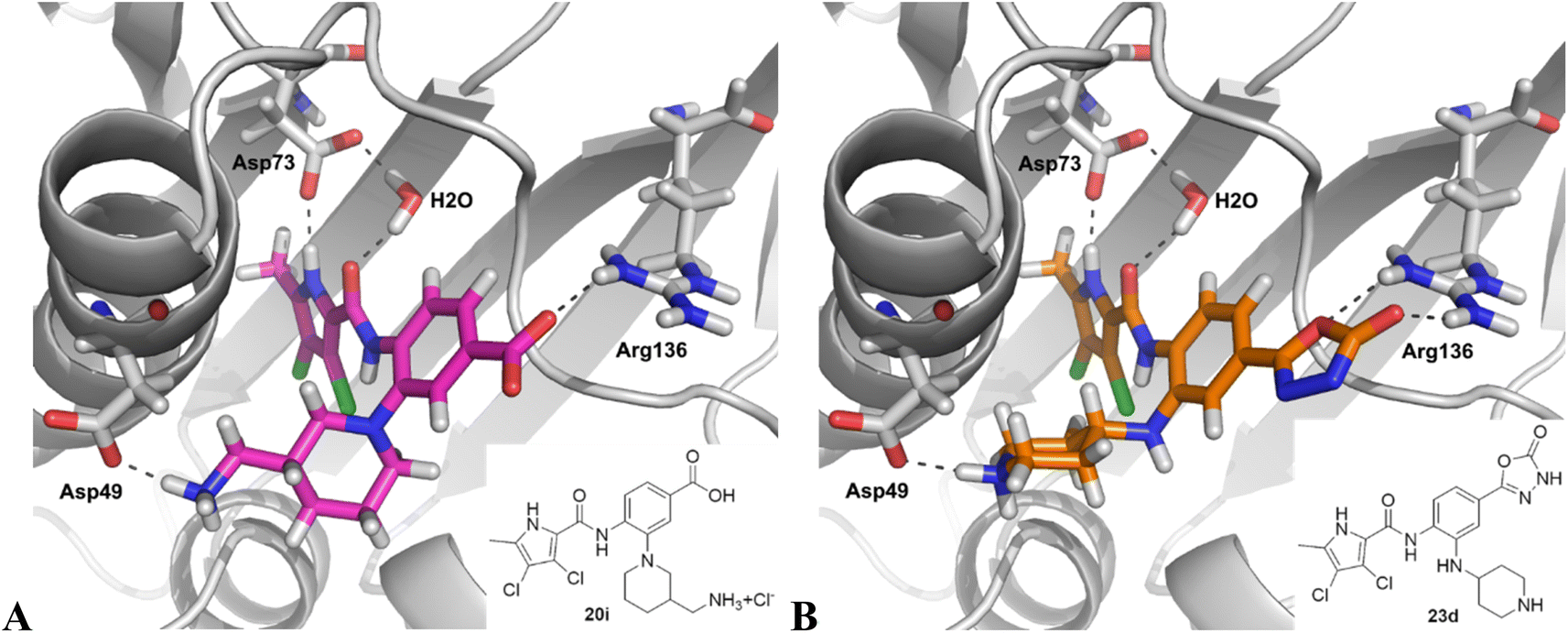 | ||
| Fig. 3 Binding modes of compounds 20i (A) and 23d (B) in the E. coli GyrB (grey) (PDB code: 4ZVI),27 as predicted by docking with Glide. The ligand and the key amino acid side chains are shown as stick models coloured according to the chemical atom type. The predicted H-bonds are shown as grey dotted lines. The figure was created with PyMOL.36 | ||
Compound 39 (IC50 = 886 nM), featuring a benzyloxy substituent, exhibits lower activity compared to other compounds in the series. This reduced activity may be due to the bulkiness of the benzyloxy group or its inability to form additional polar interactions with the enzyme. Similarly, compounds 27a and 33 with a methyl or benzyl substituent on the amide nitrogen (R2, Fig. 2) show weak enzyme inhibitory activities, suggesting that the free amide NH group is crucial for effective binding. Overall, the compounds listed in Table 2 (18–39), in which the carboxyl or 5-oxo-1,3,4-oxadiazole groups are directly attached to the central phenyl ring, exhibit slightly greater potency than those listed in Table 1 (6–14), which include an additional three-atom linker. The most effective DNA gyrase inhibitors in this series are compound 20i with a carboxyl group and the 5-oxo-1,3,4-oxadiazoles 23a–d. Compound 20i is also the most effective against topoisomerase IV, with a promising IC50 value of 143 nM against E. coli topoisomerase IV. Additionally, compound 23c shows significant activity against topoisomerase IV with IC50 values of 448 nM and 406 nM for E. coli and S. aureus topoisomerase IV, respectively. Importantly, none of the compounds are active on human DNA topoisomerase IIα. Compound 23b was further evaluated in a supercoiling assay against DNA gyrase from A. baumannii and P. aeruginosa. The IC50 values were 15.3 nM for A. baumannii DNA gyrase and 3.63 nM for P. aeruginosa DNA gyrase (Table 2S, ESI†).
2.3.2.1. Activity against selected Gram-positive and Gram-negative bacteria. The most potent inhibitors were tested against a range of Gram-positive (S. aureus ATCC 29213, S. aureus ATCC 43300 – MRSA, S. aureus ATCC 700699 – VISA, E. faecalis ATCC 29212 and E. faecium ATCC 35667) and Gram-negative (E. coli ATCC 25922, P. aeruginosa ATCC 27853, A. baumannii ATCC 17978, A. baumannii ATCC 19606, K. pneumoniae ATCC 10031, K. pneumoniae ATCC 700603, E. spp. cloacae ATCC 13047 and K. aerogenes ATCC 13408) bacterial strains using the broth microdilution method according to the Clinical and Laboratory Standards Institute guidelines.37 The antibacterial activities, expressed as minimum inhibitory concentrations (MICs), are shown in Tables 1S (ESI),† 3, 4 and Fig. 1S (ESI).† The dose–response curves for the selected most promising compounds are shown in Fig. 4.
| Species | MICa (μg mL−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | 18a | 18c | 18h | 18i | 19a | 19c | 19i | 20a | 20c | 20d |
| a Minimum inhibitory concentration.b Not determined. | ||||||||||
| Gram-positive bacteria | ||||||||||
| S. aureus ATCC 29213 | 4 | 16 | >64 | 32 | >32 | 8 | 8 | >32 | >32 | >64 |
| S. aureus ATCC 43300 (MRSA) | n.d.b | n.d. | >64 | n.d. | n.d. | n.d. | 2 | >64 | >64 | >64 |
| S. aureus ATCC 700699 (VISA) | n.d. | n.d. | >64 | n.d. | n.d. | n.d. | 1 | >64 | >64 | >64 |
| E. faecalis ATCC 29212 | n.d. | n.d. | >64 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | >64 |
| E. faecium ATCC 35667 | 16 | >32 | n.d. | >32 | >32 | 8 | 1 | >32 | >32 | n.d. |
|
||||||||||
| Gram-negative bacteria | ||||||||||
| E. coli ATCC 25922 | >32 | >32 | >64 | >32 | >32 | >32 | 4 | >32 | >32 | >64 |
| P. aeruginosa ATCC 27853 | >32 | >32 | >64 | >32 | >32 | >32 | >32 | >32 | >32 | >64 |
| A. baumannii ATCC 17978 | n.d. | n.d. | >64 | n.d. | n.d. | n.d. | 8 | >64 | >64 | >64 |
| A. baumannii ATCC 19606 | >32 | >32 | n.d. | >32 | 16 | 16 | 8 | >32 | >32 | n.d. |
| K. pneumoniae ATCC 10031 | n.d. | n.d. | >64 | n.d. | n.d. | 4 | 8 | >64 | >64 | >64 |
| K. pneumoniae ATCC 700603 | >32 | >32 | n.d. | >32 | >32 | >32 | >32 | >32 | >32 | n.d. |
| E. spp. cloacae ATCC 13047 | n.d. | n.d. | >64 | n.d. | n.d. | n.d. | >64 | >64 | >64 | >64 |
| K. aerogenes ATCC 13408 | >32 | >32 | n.d. | >32 | >32 | >32 | >32 | >32 | >32 | n.d. |
| Species | MICa (μg mL−1) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | 20i | 22c | 22e | 22f | 23a | 23b | 23c | 23d | 39 | |||||||
| a Minimum inhibitory concentration.b Not determined. | ||||||||||||||||
| Gram-positive bacteria | ||||||||||||||||
| S. aureus ATCC 29213 | 32 | 32 | 0.25 | >64 | 2 | 1 | 2 | >64 | >32 | |||||||
| S. aureus ATCC 43300 (MRSA) | n.d.b | n.d. | 0.25 | >64 | 4 | 2 | 2 | >64 | >32 | |||||||
| S. aureus ATCC 700699 (VISA) | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | >64 | >32 | |||||||
| E. faecalis ATCC 29212 | n.d. | n.d. | 0.125 | 1 | 0.5 | 1 | 2 | >64 | >32 | |||||||
| E. faecium ATCC 35667 | 32 | 16 | n.d. | n.d. | n.d. | n.d. | 2 | n.d. | n.d. | |||||||
|
||||||||||||||||
| Gram-negative bacteria | ||||||||||||||||
| E. coli ATCC 25922 | >32 | >32 | >64 | >64 | >64 | 8 | 16 | >64 | >32 | |||||||
| P. aeruginosa ATCC 27853 | >32 | >32 | >64 | >64 | >64 | 32 | 16 | >64 | >32 | |||||||
| A. baumannii ATCC 17978 | n.d. | n.d. | >64 | >64 | >64 | 8 | 16 | >64 | >32 | |||||||
| A. baumannii ATCC 19606 | >32 | >32 | n.d. | n.d. | n.d. | n.d. | 4 | n.d. | n.d. | |||||||
| K. pneumoniae ATCC 10031 | n.d. | n.d. | >64 | >64 | 0.25 | 0.0625 | 0.25 | 2 | >32 | |||||||
| K. pneumoniae ATCC 700603 | >32 | >32 | n.d. | n.d. | n.d. | >32 | >32 | n.d. | n.d. | |||||||
| E. spp. cloacae ATCC 13047 | n.d. | n.d. | >64 | >64 | >64 | 32 | >64 | >64 | >32 | |||||||
| K. aerogenes ATCC 13408 | >32 | >32 | n.d. | n.d. | n.d. | >32 | >32 | n.d. | n.d. | |||||||
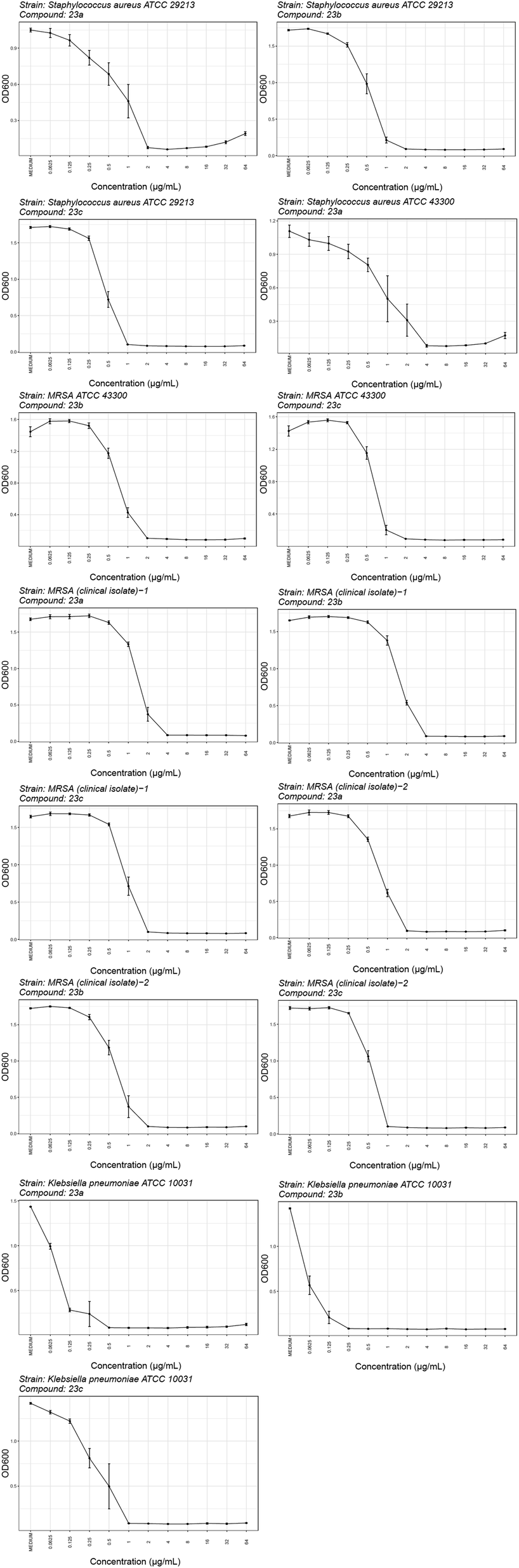 | ||
| Fig. 4 Dose–response curves for compounds 23a–c against the selected Gram-positive (S. aureus ATCC 29213, S. aureus ATCC 43300 – MRSA, MRSA clinical isolate-1, MRSA clinical isolate-2) and Gram-negative strains (K. pneumoniae ATCC 10031). | ||
Unfortunately, none of the compounds shown in Table 1S† exhibit significant antibacterial activity at concentrations below 32 μg mL−1, with the exception of compound 13. This compound demonstrates promising activity, showing a potency of 1 μg mL−1 against S. aureus ATCC 29213 and 32 μg mL−1 against E. faecium (ATCC 35667). However, the antibacterial activity of compounds shown in Tables 3 and 4 is more promising. In general, compounds with 5-oxo-1,3,4-oxadiazole group (22c, 22e, 22f, 23a–d and 39) exhibit stronger antibacterial activity than compounds with free carboxyl group (18a, 18c, 18h, 18i, 20a, 20c, 20d and 20i). The 5-oxo-1,3,4-oxadiazole group can interact with Arg136 similarly as the carboxyl group, but due to its higher pKa value, the compounds are likely to have better permeability through bacterial membranes. Among the carboxyl derivatives, 18h, 20a, 20c and 20d show no activity, while 18a, 18c, 18i and 20i show weak inhibition of growth of Gram-positive S. aureus ATCC 29213 (MIC ∼ 4–32 μg mL−1), and 18a and 20i also of E. faecium ATCC 35667 (MIC ∼16–32 μg mL−1). None of these compounds are active against Gram-negative strains up to 32 μg mL−1. Interestingly, three active carboxyl derivatives (18a, 18c, 18i) possess the Boc protecting group at the heterocycle and are more potent than their Boc-deprotected analogues 20a, 20c, and 20i, despite having weaker on-target activity. This suggests that while the Boc group may hinder binding to the enzyme, it potentially enhances accumulation in Gram-positive bacteria by facilitating cellular entry or reducing efflux. Furthermore, methyl esters 19a, 19c and 19i show higher antibacterial activity than their carboxyl analogues 20a, 20c and 20i, although their on-target activity is lower. This is likely due to their increased lipophilicity, which improves penetration through the bacterial cell wall and membrane. Among the entire series, the 5-oxo-1,3,4-oxadiazole derivatives 22e, 22f and 23a–d show the most promising antibacterial activity (Table 4), particularly against Gram-positive bacteria. The MIC values against S. aureus ATCC 29213, S. aureus ATCC 43300 (MRSA), E. faecalis ATCC 29212, and E. faecium ATCC 35667 fall within the low micromolar range. Notably, the morpholine derivative 22e exhibits the highest potency, with MIC values of 0.25 μg mL−1 against S. aureus ATCC 29213 and MRSA, and 0.125 μg mL−1 against E. faecalis ATCC 29212. Interestingly, compounds 23a–d with an additional basic centre in the heterocycle bound to the R3 position (Fig. 1), are the only ones showing activity against Gram-negative bacteria. This aligns with recent findings that the presence of a positive charge can enhance accumulation in Gram-negative bacteria.33–35 Compounds 23b and 23c demonstrate activity against E. coli ATCC 25922, P. aeruginosa ATCC 27853, A. baumannii ATCC 17978, and A. baumannii ATCC 19606 with MIC values from 4 to 32 μg mL−1. The most notable result is the very high potency of compounds 23b–d against the clinically relevant Gram-negative strain K. pneumoniae ATCC 10031, with MIC values below 1 μg mL−1. The most potent compound against K. pneumoniae ATCC 10031 is 23b with an MIC of 0.0625 μg mL−1. However, none of the tested compounds exhibit activity against the extended-spectrum β-lactamase (ESBL)-producing strain K. pneumoniae ATCC 700603.
2.3.2.2. Activity against fluoroquinolone-resistant strains. Four compounds with the most promising antibacterial activities (22e and 23a–c) were further evaluated against fluoroquinolone-resistant clinical isolates of S. aureus and K. pneumonia (Table 5). Two different S. aureus and four different K. pneumonia isolates were used, originating from patient samples from multiple hospitals in Hungary. The fluoroquinolone-resistant phenotype of the clinical isolates was previously confirmed in-house. The results presented in Table 5 show that while the compounds were inactive against fluoroquinolone-resistant strains of K. pneumonia, they exhibited similar activity against both fluoroquinolone-sensitive and fluoroquinolone-resistant strains of S. aureus. This underscores their potential for the treatment of high-priority MRSA infections.
| Species | MICc (μg mL−1) | ||||||
|---|---|---|---|---|---|---|---|
| Comp. | 22e | 23a | 23b | 23c | Novo.a | Zoli.b | |
| a Novobiocin.b Zoliflodacin.c Minimum inhibitory concentration. Fluoroquinolone-sensitive control strains are shown in bold. Each measurement was performed in at least four replicates. | |||||||
| S. aureus ATCC 29213 | 0.25 | 2 | 1 | 2 | 0.25 | 1 | |
| S. aureus ATCC 43300 (MRSA) | 0.25 | 4 | 2 | 2 | 0.5 | 0.125 | |
| MRSA clinical isolate-1 | 0.5 | 4 | 4 | 4 | 0.25 | 0.125 | |
| MRSA clinical isolate-2 | 0.5 | 2 | 2 | 2 | 0.25 | <0.0625 | |
| K. pneumoniae ATCC 10031 | >64 | 0.25 | 0.0625 | 0.25 | 0.5 | 0.25 | |
| K. pneumoniae NCTC 13440 | >64 | >64 | >64 | >64 | >64 | 64 | |
| K. pneumoniae SE K13 | >64 | >64 | 8 | 32 | >64 | 32 | |
| K. pneumoniae SE K14 | >64 | >64 | >64 | >64 | >64 | >64 | |
| K. pneumoniae K9–K25 | >64 | >64 | >64 | >64 | >64 | >64 | |
2.3.2.3. Frequency of resistance. The spontaneous frequency of resistance was determined for compounds 23a and 23c in the strains of S. aureus ATCC 29213 and K. pneumoniae ATCC 10031 (Fig. 5 and Table 3S†). The results show that both compounds are significantly less resistance-prone compared to novobiocin in both bacterial strains. The frequency of resistance was generally lower in S. aureus than in K. pneumoniae. Notably, no resistant colonies of S. aureus were detected for compound 23c when selected up to 4-fold MIC. In addition, we determined the MIC values of colonies isolated from the frequency of resistance plates after selection to assess the resistance level of single mutational events (Table 4S†). The change in MIC values compared to the ancestor is only 4-fold for 23c in S. aureus.
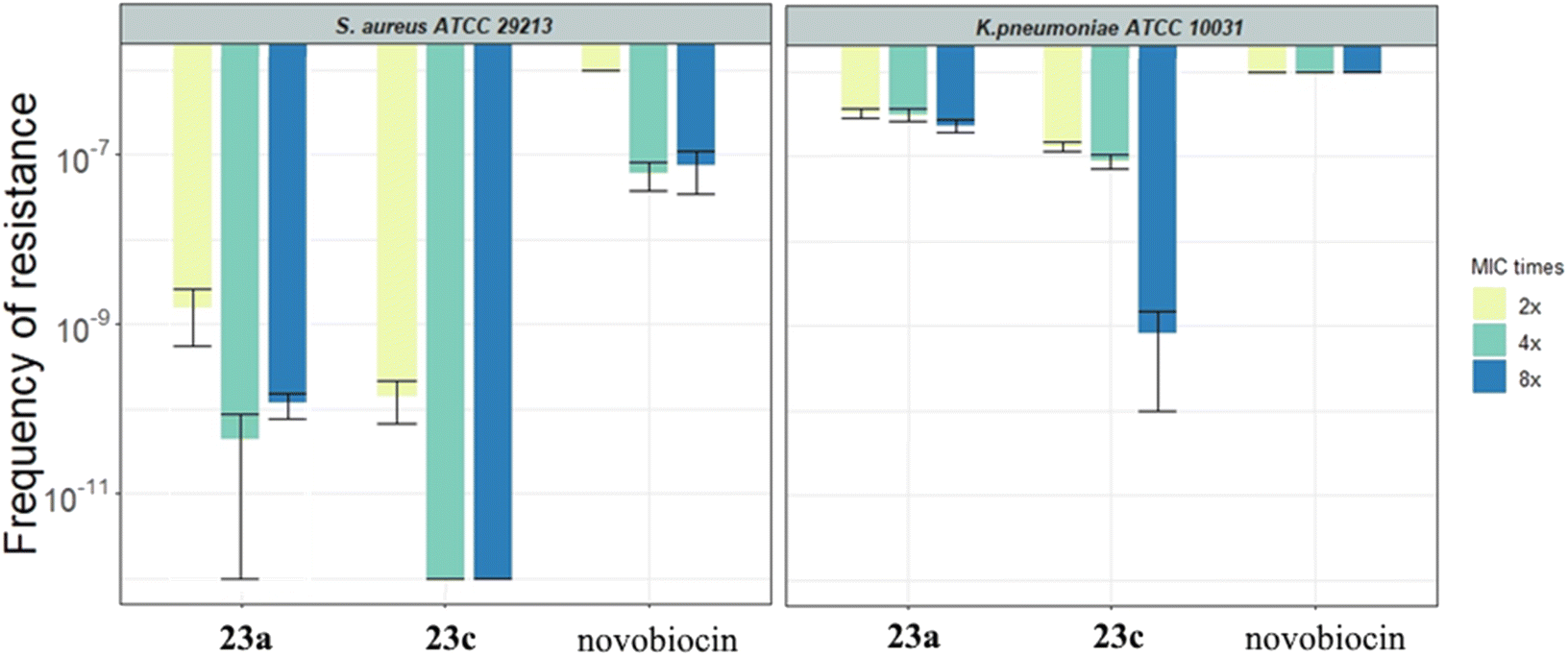 | ||
| Fig. 5 Spontaneous frequency of resistance to 23a, 23c and novobiocin in S. aureus ATCC 29213 (left panel) and K. pneumoniae ATCC 10031 (right panel). The bars represent the average of 3 independent biological replicates. The error bars indicate the 95% confidence interval. Values at 10−6 indicate that the plates contained an uncountable abundance of colonies (therefore the frequency of resistance is above the limit of detection), while 10−12 indicate that no colonies were observed under the experimental conditions (the frequency of resistance is below the limit of detection). | ||
3. Conclusions
We have designed and synthesised a new series of N-phenylpyrrolamide inhibitors of DNA gyrase with improved antibacterial activity compared to earlier compounds of this structural class. Among these, the most potent inhibitors of E. coli DNA gyrase were compounds 10b, 10c, and 20i with a carboxyl group on the right-hand side (IC50; 6.3–21 nM) and 14 and 23a–d with a 5-oxo-1,3,4-oxadiazole group at this position (IC50; 2.8–27 nM). Compound 21i also showed a promising IC50 value of 143 nM against a structurally similar enzyme, E. coli topoisomerase IV. Importantly, none of the compounds showed activity against human DNA topoisomerase IIα, indicating selectivity for bacterial targets. The most effective compound against Gram-positive bacteria was the morpholine derivative 22e with MIC values of 0.25 μg mL−1 against S. aureus ATCC 29213 and MRSA, and 0.125 μg mL−1 against E. faecalis ATCC 29212. Compounds 23b and 23c were most effective against Gram-negative E. coli ATCC 25922, P. aeruginosa ATCC 27853, A. baumannii ATCC 17978, and A. baumannii ATCC 19606 with MIC values from 4 to 32 μg mL−1. The most effective compound against the clinically relevant Gram-negative K. pneumoniae ATCC 10031 was 23b with a MIC of 0.0625 μg mL−1. Importantly, compounds 22e and 23a–c retained similar levels of activity against both fluoroquinolone-sensitive and fluoroquinolone-resistant clinical isolates of MRSA, highlighting their potential for the treatment of high-priority MRSA infections. Moreover, compounds 23a and 23c demonstrated markedly lower susceptibility to resistance development in S. aureus ATCC 29213 and K. pneumoniae ATCC 10031 compared to novobiocin. Overall, the most promising compounds in the series showed excellent on-target activity and improved antibacterial activity compared to previous series of N-phenylpyrrolamides.4. Experimental
4.1. Chemistry – general information
Chemicals were purchased from Sigma-Aldrich (St. Louis, USA), Acros Organics (Geel, Belgium), TCI (Tokyo, Japan) and Apollo Scientific (Stockport, UK). Thin layer chromatography (TLC) was performed using Merck 60 F254 silica gel plates (0.25 mm) and visualized with UV light and spray reagents. Silica gel 60 (particle size 240–400 mesh) was used for column chromatography. Infrared (IR) spectra were recorded using a Thermo Nicolet Nexus 470 ESP FT-IR spectrometer (Thermo Fisher Scientific, Waltham, USA). HPLC analyses were performed using a Dionex Ultimate 3000 binary rapid separation LC system (Thermo Scientific, Thermo Fisher Scientific, Waltham, MA, USA). Method A (for compounds 18h, 20d, and 23d): Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm) or Agilent Extend-C18 column (3.5 μm, 4.6 × 150 mm), flow rate of 1.0 mL min−1 and sample injection volume of 10 μL. The mobile phase consisted of acetonitrile (solvent A) and 0.1% trifluoroacetic acid (TFA) in ultrapure water (solvent B). The gradient (defined for solvent A) was: 0–16 min, 30–90%; 16–20 min, 90%. The compounds were detected at 254 and 280 nm. Method B (for compounds 22a, 22e, 22f, 22g, 23a, 27a, and 27b): Waters Acquity UPLC HSS C18 column (1.8 μm, 2.1 × 50 mm), flow rate of 0.4 mL min−1 and sample injection volume of 1.75 μL. The mobile phase consisted of acetonitrile (solvent A) and 0.1% trifluoroacetic acid (TFA) in ultrapure water (solvent B). The gradient (defined for solvent A) was: 0–10 min, 10–90%; 10–11 min, 90%. The compounds were detected at 254 and 280 nm. Melting points were determined using a Reichert hot stage microscope. Proton (1H), carbon-13 (13C) and DEPT nuclear magnetic resonance (NMR) spectra were recorded at frequencies of 400 and 100 MHz, respectively. The recordings were performed with a Bruker AVANCE III 400 spectrometer (Bruker Corporation, Billerica, USA) in solutions of CDCl3 or DMSO-d6, with TMS as internal standard. Mass spectra were obtained using a VG Analytical Autospec Q mass spectrometer (Fisons, VG Analytical, Manchester, UK). The purity of the analysed key compounds was determined to be ≥95%.4.2. Synthetic procedures and analytical data
4.2.1.1 Synthesis of tert-butyl 4-(5-(methoxycarbonyl)-2-nitrophenyl)piperazine-1-carboxylate (2a). To a suspension of methyl 3-fluoro-4-nitrobenzoate (1, 5.00 g, 25.0 mmol) and potassium carbonate (4.16 g, 30.0 mmol) in DMF (100 mL), piperazine N1-Boc protected (4.68 g, 25.0 mmol) was added. The mixture was heated at 70 °C for 15 h. The solvent was evaporated under reduced pressure. EtOAc (150 mL) and H2O (75 mL) were added and the compound was extracted into the organic layer. The organic phase was washed with water (50 mL) and brine (2 × 50 mL), dried over Na2SO4, filtered and the solvent was removed under reduced pressure to give 2a as a yellow solid. Yield 7.56 g (83%); yellow solid; mp 88–90 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.96 (d, 1H, J = 8.4 Hz, ArH), 7.80 (d, 1H, J = 1.6 Hz, ArH), 7.68 (dd, 1H, J = 8.4, 1.7 Hz, ArH), 3.90 (s, 3H, CH3), 3.40–3.50 (m, 4H, 2 × CH2), 2.98–3.08 (m, 4H, 2 × CH2), 1.43 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 164.8, 153.8, 145.4, 144.9, 133.8, 125.9, 122.6, 122.3, 79.1, 52.8, 50.8, 42.8, 28.0 ppm.
4.2.1.2 3-(4-(tert-Butoxycarbonyl)piperazin-1-yl)-4-nitrobenzoic acid (3a). To the solution of 2a (200 mg, 0.55 mmol) in methanol (10 mL), 1 M NaOH (0.81 mL, 0.82 mmol) was added and the reaction mixture was stirred for 15 h at rt. 1 M HCl was added dropwise to pH 7 and the solvent was removed under reduced pressure. To the crude residue were added 1 M HCl to pH 4, water (20 mL) and ethyl acetate (25 mL). The phases were separated and organic phase was washed with water (20 mL) and brine (20 mL), dried over Na2SO4, filtered and evaporated to give 171 mg of the product as orange crystals. Yield 171 mg (89%); orange crystals; mp 170–175 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.75–7.73 (m, 2H, ArH-2, ArH-5), 7.58 (dd, 1H, J = 6.2, 1.4 Hz, ArH-6), 3.41–3.47 (m, 4H, 2 × CH2), 2.93–2.95 (m, 4H, 2 × CH2), 1.42 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 166.6, 153.8, 153.6, 144.7, 144.4, 125.2, 122.9, 122.1, 79.1, 51.1, 42.8, 28.0 ppm. IR (ATR): ν 2976, 2931, 1686, 1570, 1419, 1365, 1231, 1160, 1126, 966, 749 cm−1. MS (ESI) m/z = 350.13 ([M − H]−).
4.2.1.3 tert-Butyl 4-(5-((2-methoxy-2-oxoethyl)carbamoyl)-2-nitrophenyl)piperazine-1-carboxylate (4a). To the solution of 3a (0.900 g, 2.4 mmol) and TBTU (1.01 g, 3.1 mmol) in DMF (20 mL) NMM (0.53 mL, 4.8 mmol) was added and the solution was stirred for 30 min at rt. Glycine methyl ester hydrochloride (333 mg, 2.7 mmol) was added and reaction mixture was stirred at rt for 20 h. The solvent was removed under reduced pressure, the residue was dissolved in ethyl acetate (20 mL) and washed with water (2 × 20 mL), saturated NaHCO3 solution (2 × 20 mL) and brine (20 mL), then organic phase was dried over Na2SO4, filtered and evaporated. The crude product was purified with flash column chromatography using ethyl acetate/petroleum ether 1
:1 as mobile phase to give 4a as orange oil (0.852 g). Yield 0.852 g (84%); orange oil. 1H NMR (400 MHz, DMSO-d6): δ 9.22 (t, 1H, J = 5.8 Hz, NH), 7.94 (d, 1H, J = 8.5 Hz, ArH-5), 7.76 (d, 1H, J = 1.7 Hz, ArH-2), 7.59 (dd, 1H, J = 8.5, 1.7 Hz, ArH-6), 4.05 (d, 2H, J = 5.8 Hz, CH2), 3.67 (s, 3H, CH3), 3.43–3.48 (m, 4H, 2 × CH2, overlapped with the signal of water), 3.00–3.03 (m, 4H, 2 × CH2), 1.42 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 170.1, 165.0, 153.8, 144.9, 144.4, 137.8, 125.7, 121.1, 120.8, 79.1, 51.8, 51.0, 43.7, 41.3, 28.0 ppm. IR (ATR): ν 3324, 2978, 1750, 1691, 1668, 1521, 1418, 1365, 1229, 1209, 1661, 1000, 834, 744 cm−1. MS (ESI) m/z = 445.01 ([M + Na]+).
4.2.1.4 tert-Butyl 4-(2-amino-5-((2-methoxy-2-oxoethyl)carbamoyl)phenyl)piperazine-1-carboxylate (5a). The solution of 4a (2.51 g, 5.9 mmol) in methanol (90 mL) was stirred under argon for 15 min. Pd/C (0.500 g) was added, the solution was saturated with hydrogen and the reaction mixture was stirred for 4 h under hydrogen atmosphere. The catalyst was filtered off and the solvent was evaporated. The crude product was purified with flash column chromatography using ethyl acetate/petroleum ether 2
:1 as mobile phase to obtain 2.08 g of 5a as white crystals. Yield 2.08 g (90%); white crystals; mp 80–84 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.59 (t, 1H, J = 5.8 Hz, NH), 7.53 (d, 1H, J = 1.9 Hz, ArH-6), 7.48 (dd, 1H, J = 8.4, 1.9 Hz, ArH-4), 6.74 (d, 1H, J = 8.4 Hz, ArH-3), 4.58–6.07 (br s, 2H, NH2), 3.99 (d, 2H, J = 5.8 Hz, CH2), 3.69 (s, 3H, CH3), 3.51–3.63 (m, 4H, 2 × CH2, overlapped with the signal of water), 2.75–2.85 (m, 4H, 2 × CH2), 1.49 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 170.8, 166.5, 153.9, 145.8, 136.7, 124.5, 121.0, 119.1, 113.1, 78.8, 51.6, 50.6, 43.2, 41.1, 28.1 ppm. IR (ATR): ν 3479, 3367, 3308, 2935, 1743, 1693, 1610, 1504, 1415, 1365, 1248, 1205, 1163, 1119, 768 cm−1. MS (ESI) m/z = 415.07 ([M + Na]+).
4.2.2.1 4-(2-(3,4-Dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-((2-methoxy-2-oxoethyl)carbamoyl)phenyl)piperazine-1-carboxylate (6a). To a solution of 3,4-dichloro-5-methyl-1H-pyrrole-2-carboxylic acid (0.178 g, 0.92 mmol) in anhydrous dichloromethane (10 mL), oxalyl chloride (0.31 mL, 3.7 mmol) was added dropwise and the solution was stirred at rt for 15 h under argon atmosphere. The solvent was evaporated under reduced pressure. Fresh anhydrous dichloromethane (5 mL), 5a (0.300 g, 0.76 mmol) and pyridine (2 mL) were added and the reaction mixture was stirred under argon at rt for 15 h. Solvent was removed under reduced pressure, the residue was dissolved in ethyl acetate (15 mL) and washed with H2O (10 mL), HCl 1 M solution (15 mL) and brine (2 × 15 mL). During the extraction the product precipitated and was filtered off. The crude product was triturated with water and the undissolved solid was filtered off. The product was then triturated with diethyl ether and the undissolved solid was filtered off to give 6a as a grey solid. Yield 246 mg (57%); grey solid; mp 194–197 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.43 (s, 1H, NH), 9.79 (s, 1H, NH), 8.93 (t, 1H, J = 5.7 Hz, NH), 8.48 (d, 1H, J = 8.4 Hz, ArH-3), 7.91 (d, 1H, J = 1.9 Hz, ArH-6), 7.75 (dd, 1H, J = 8.6, 1.9 Hz, ArH-4), 4.02 (d, 2H, J = 5.7 Hz, CH2), 3.67 (s, 3H, CH3), 3.50–3.57 (m, 4H, 2 × CH2), 2.82–2.85 (m, 4H, 2 × CH2), 2.24 (s, 3H, CH3), 1.44 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 170.5, 165.6, 156.5, 153.7, 141.0, 136.7, 129.8, 128.4, 125.3, 121.0, 118.8, 118.6, 109.7, 108.6, 79.1, 52.1, 51.7, 41.2, 28.0, 10.8 ppm. One peak not seen. IR (ATR): ν 3299, 2978, 1757, 1672, 1647, 1621, 1510, 1410, 1367, 1250, 1201, 1170, 1122, 947, 761 cm−1. MS (ESI) m/z = 566 ([M − H]−). HRMS for C25H30Cl2N5O6: calculated 566.1573, found 566.1572. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 30–90% of acetonitrile in TFA (0.1%) in 16 min, 90% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 14.083 min (96.1% at 280 nm).
4.2.2.2 tert-Butyl 4-(2-(4,5-dibromo-1H-pyrrole-2-carboxamido)-5-((2-methoxy-2-oxoethyl)carbamoyl)phenyl)piperazine-1-carboxylate (7a). Synthesised according to General procedure B from 5a (0.600 g, 1.5 mmol), 4,5-dibromo-1H-pyrrole-2-carboxylic acid (493 mg, 1.8 mmol) and oxalyl chloride (0.629 mL, 7.3 mmol). The precipitate that was formed during the extraction was filtered off to obtain crude product 1. The two phases of the mother liquor were separated and organic phase was washed with water (20 mL), saturated NaHCO3 solution (2 × 20 mL) and brine (2 × 20 mL), dried over Na2SO4, filtered and the solvent evaporated to obtain crude product 2. The combined crude products were triturated with diethyl ether and the undissolved solid was filtered off and dried to give 7a (801 mg) as a grey solid. Yield 801 mg (83%); grey solid; mp 140–143 °C. 1H NMR (400 MHz, DMSO-d6): δ 13.08 (d, 1H, J = 2.7 Hz, NH), 9.19 (s, 1H, NH), 8.93 (t, 1H, J = 5.7 Hz, NH), 8.10 (d, 1H, J = 8.4 Hz, ArH-3), 7.75 (d, 1H, J = 1.9 Hz, ArH-6), 7.68 (dd, 1H, J = 8.4, 1.9 Hz, ArH-4), 7.17 (d, 1H, J = 2.7 Hz, ArH), 4.01 (d, 2H, J = 5.7 Hz, CH2), 3.67 (s, 3H, CH3), 3.52–3.57 (m, 4H, 2 × CH2), 2.81–2.83 (m, 4H, 2 × CH2), 1.43 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 170.5, 165.9, 156.9, 153.8, 142.7, 135.1, 129.2, 127.5, 123.9, 121.1, 119.8, 113.5, 106.8, 98.7, 79.0, 51.7, 51.4, 41.2, 28.0 ppm. One peak not seen. IR (ATR): ν 3201, 2950, 1755, 1736, 1645, 1549, 1516, 1430, 1250, 1204, 1168, 937, 759, 680 cm−1. MS (ESI) m/z = 640.0 ([M − H]−). HRMS for C24H28Br2N5O6: calculated 640.0406, found 640.0403.
4.2.2.3 (3-(4-(tert-Butoxycarbonyl)piperazin-1-yl)-4-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzoyl)glycine (8a). To the solution of 6a (0.160 g, 0.28 mmol) in methanol (10 mL) 1 M NaOH (0.84 mL, 0.84 mmol) was added and the mixture was stirred at rt for 15 h. The mixture was neutralized with 1 M HCl and methanol was removed under reduced pressure. The pH was adjusted to 4 with 1 M HCl, ethyl acetate was added and the resulting precipitate was filtered off and dried to obtain 8a as a grey solid. Yield 138 mg (89%); grey solid; mp >300 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.63 (br s, 1H, COOH), 12.43 (s, 1H, NH), 9.79 (s, 1H, NH), 8.81 (t, 1H, J = 5.7 Hz, NH), 8.48 (d, 1H, J = 8.6 Hz, ArH-5), 7.91 (d, 1H, J = 1.9 Hz, ArH-2), 7.75 (dd, 1H, J = 8.6, 1.9 Hz, ArH-6), 3.93 (d, 2H, J = 5.7 Hz, CH2), 3.50–3.58 (m, 4H, 2 × CH2), 2.82–2.85 (m, 4H, 2 × CH2), 2.24 (s, 3H, CH3), 1.44 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 171.4, 165.5, 156.5, 153.7, 140.9, 136.6, 129.8, 128.6, 125.3, 121.0, 118.8, 118.5, 109.7, 108.6, 79.1, 52.1, 41.1, 28.0, 10.8 ppm. One peak not seen. DEPT 45 NMR (100 MHz, DMSO-d6) δ 125.8, 121.5, 119.1, 52.6, 41.7, 28.5, 11.2 ppm. One peak not seen. DEPT 135 NMR (100 MHz, DMSO-d6) δ 125.8, 121.5, 119.0, 52.6 (negative), 41.7 (negative), 28.5, 11.2 ppm. One peak not seen. IR (ATR): ν 3369, 3287, 3108, 2973, 1748, 1632, 1505, 1481, 1409, 1366, 1253, 1170, 1138, 1083, 759, 621 cm−1. MS (ESI) m/z = 552.0 ([M − H]−). HRMS for C24H28Cl2N5O6: calculated 552.1417, found 552.1411. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 30–90% of acetonitrile in TFA (0.1%) in 16 min, 90% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 12.420 min (98.1% at 280 nm).
4.2.2.4 (S)-(3-(3-((tert-Butoxycarbonyl)amino)pyrrolidin-1-yl)-4-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzoyl)glycine (8b). To the solution of 6b (155 mg, 0.27 mmol) in a mixture of methanol (10 mL) and tetrahydrofuran (2 mL) 1 M NaOH (1.09 mL, 1.09 mmol) was added and the mixture was stirred at rt for 15 h. To the mixture 1 M HCl was added to reach pH 7 and methanol was removed under reduced pressure. 1 M HCl was added to the aqueous residue to reach pH 4, after which ethyl acetate (15 mL) was added. The undissolved precipitate was filtered off and dried to give 8b (81 mg). The mother liquor was poured into a separating funnel and the two phases were separated. The organic phase was washed with brine (2 × 10 mL), dried with Na2SO4, filtered and evaporated. Diethyl ether was added to the residue, the resulting suspension was sonicated and the precipitate filtered off and dried (26 mg). The pure products were combined to give 8b (107 mg) as brown crystals. Yield 107 mg (71%); brown crystals; mp 210–215 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.57 (br s, 1H, COOH), 12.38 (s, 1H, NH), 9.50 (s, 1H, NH), 8.82 (t, 1H, J = 5.8 Hz, NH), 8.27 (d, 1H, J = 8.5 Hz, Ar–H-5), 7.76 (d, 1H, J = 1.8 Hz, Ar–H-2), 7.63 (dd, 1H, J = 8.5, 1.8 Hz, Ar–H-6), 7.19 (d, 1H, J = 6.5 Hz, NH), 4.07–4.14 (m, 1H, CH), 3.92 (d, 2H, J = 5.8 Hz, CH2), 3.27–3.33 (m, 1H, CH, overlapping with the signal for water), 3.14–3.19 (m, 1H, CH), 3.00–3.06 (m, 1H, CH), 2.89–2.94 (m, 1H, CH), 2.15–2.24 (m, 4H, CH, CH3), 1.81–1.89 (m, 1H, CH), 1.39 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 171.4, 165.9, 156.6, 155.2, 139.8, 135.3, 129.5, 129.2, 123.1, 119.7, 119.2, 118.8, 110.1, 108.6, 77.8, 57.5, 50.9, 49.8, 41.2, 31.3, 28.2, 10.8 ppm. IR (ATR): ν 3311, 3177, 2976, 1710, 1694, 1634, 1585, 1506, 1331, 1169, 860, 765 cm−1. [α]25D −12.0 (c 0.125, MeOH). MS (ESI) m/z = 552.0 ([M − H]−). HRMS for C24H28Cl2N5O6: calculated 552.1417, found 552.1412. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 30–90% of acetonitrile in TFA (0.1%) in 16 min, 90% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 11.366 min (97.0% at 280 nm).
4.2.2.5 (R)-(3-(3-((tert-Butoxycarbonyl)amino)pyrrolidin-1-yl)-4-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzoyl)glycine (8c). To the solution of 6c (200 mg, 0.35 mmol) in a mixture of methanol (8 mL) and tetrahydrofuran (3 mL) 1 M NaOH (1.41 mL, 1.4 mmol) was added and the mixture was stirred at rt for 15 h. Water (10 mL) was added to the mixture and the pH was adjusted to 4 with 1 M HCl, after which the obtained precipitate was filtered off and dried to give 8c (91 mg) as a grey solid. Yield 91 mg (47%); grey solid; mp 210–214 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.57 (br s, 1H, COOH), 12.38 (s, 1H, NH), 9.50 (s, 1H, NH), 8.81 (t, 1H, J = 5.8 Hz, NH), 8.27 (d, 1H, J = 8.5 Hz, ArH-5), 7.76 (d, 1H, J = 1.8 Hz, ArH-2), 7.63 (dd, 1H, J = 8.5, 1.8 Hz, ArH-6), 7.19 (d, 1H, J = 6.5 Hz, NH), 4.07–4.14 (m, 1H, CH), 3.92 (d, 2H, J = 5.8 Hz, CH2), 3.27–3.33 (m, 1H, CH, overlapping with the signal for water), 3.14–3.19 (m, 1H, CH), 3.00–3.06 (m, 1H, CH), 2.89–2.94 (m, 1H, CH), 2.15–2.24 (m, 4H, CH, CH3), 1.81–1.89 (m, 1H, CH), 1.39 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 171.4, 166.1, 156.6, 155.4, 139.9, 135.1, 129.6, 129.3, 122.9, 120.0, 119.0, 118.7, 110.2, 108.7, 78.0, 57.4, 50.7, 49.7, 41.5, 31.2, 28.1, 10.7 ppm. IR (ATR): ν 3440, 3292, 2977, 2931, 2840, 1733, 1687, 1636, 1506, 1403, 1244, 1063, 1041, 764, 605 cm−1. [α]25D +11.8 (c 0.187, MeOH). MS (ESI) m/z = 552.1 ([M − H]−). HRMS for C24H28Cl2N5O6: calculated 552.1417, found 552.1414. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 30–90% of acetonitrile in TFA (0.1%) in 16 min, 90% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 11.366 min (96.9% at 280 nm).
4.2.2.6 (3-(4-(tert-Butoxycarbonyl)piperazin-1-yl)-4-(4,5-dibromo-1H-pyrrole-2-carboxamido)benzoyl)glycine (9a). To the solution of 7a (200 mg, 0.32 mmol) in methanol (10 mL) 1 M NaOH (1.24 mL, 1.2 mmol) was added and the mixture was stirred at rt for 15 h. The mixture was neutralized with 1 M HCl and the methanol was removed under reduced pressure. Water (10 mL) was added, the pH was adjusted to 4 with 1 M HCl, ethyl acetate (20 mL) was added, and the organic phase was washed with water (10 mL) and brine (2 × 10 mL), dried over Na2SO4, filtered and the solvent removed. To the residue diethyl ether was added, the resulting suspension was sonicated and the precipitate was filtered off to give 9a (115 mg) as a grey solid. Yield 115 mg (59%); grey solid; mp >300 °C. 1H NMR (400 MHz, DMSO-d6): δ 13.08 (s, 1H, NH), 12.59 (br s, 1H, COOH), 9.20 (s, 1H, NH), 8.81 (t, 1H, J = 5.8 Hz, NH), 8.10 (d, 1H, J = 8.5 Hz, ArH-5), 7.76 (d, 1H, J = 1.9 Hz, ArH-2), 7.68 (dd, 1H, J = 8.5, 1.9 Hz, ArH-6), 7.17 (d, 1H, J = 2.8 Hz, ArH), 3.93 (d, 2H, J = 5.8 Hz, CH2), 3.51–3.57 (m, 4H, 2 × CH2), 2.81–2.83 (m, 4H, 2 × CH2), 1.43 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 171.4, 165.7, 156.9, 153.8, 142.6, 135.0, 129.4, 127.6, 123.9, 121.0, 119.8, 113.5, 106.8, 98.7, 79.0, 51.4, 41.2, 28.0 ppm. One peak not seen. IR (ATR): ν 3101, 3053, 2972, 1745, 1630, 1505, 1406, 1368, 1172, 1140, 830, 756, 631 cm−1. MS (ESI) m/z = 626.0 ([M − H]−). HRMS for C23H26Br2N5O6: calculated 626.0250, found 626.0255. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 30–90% of acetonitrile in TFA (0.1%) in 16 min, 90% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 11.169 min (95.8% at 280 nm).
4.2.2.7 (S)-(3-(3-((tert-Butoxycarbonyl)amino)pyrrolidin-1-yl)-4-(4,5-dibromo-1H-pyrrole-2-carboxamido)benzoyl)glycine (9b). To the solution of 7b (250 mg, 0.38 mmol) in methanol (10 mL) 1 M NaOH (1.52 mL, 1.52 mmol) was added and the mixture was stirred at rt for 15 h. The mixture was neutralized with 1 M HCl and methanol was removed under reduced pressure. The pH was adjusted to 4 with 1 M HCl, ethyl acetate was added, and the organic phase was washed with water (10 mL) and brine (2 × 10 mL), dried over Na2SO4, filtered and the solvent removed. To the residue diethyl ether was added, the resulting suspension was sonicated and the precipitate was filtered off to give 9b (188 mg) as a grey solid. Yield 188 mg (77%); grey solid; mp 219–223 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.89 (s, 1H, NH), 12.47 (br s, 1H, COOH), 9.52 (s, 1H, NH), 8.80 (t, 1H, J = 5.8 Hz, NH), 7.29–7.35 (m, 3H, 3 × ArH), 7.13–7.20 (m, 1H, ArH), 4.01–4.06 (m, 1H, CH), 3.91 (d, 2H, J = 5.8 Hz, CH2), 3.31–3.43 (m, 2H, 2 × CH, overlapping with the signal for water), 3.21–3.27 (m, 1H, CH), 3.04–3.08 (m, 1H, CH), 2.03–2.11 (m, 1H, CH), 1.75–1.84 (m, 1H, CH), 1.37 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 171.4, 166.3, 157.6, 155.2, 144.4, 131.7, 128.3, 128.0, 127.8, 117.5, 114.5, 113.5, 105.7, 98.2, 77.8, 55.9, 49.8, 48.3, 41.2, 30.5, 28.2 ppm. IR (ATR): ν 3376, 3276, 3104, 1735, 1692, 1666, 1628, 1509, 1399, 1383, 1236, 1153, 976, 767 cm−1. [α]25D −37.3 (c 0.126, MeOH). MS (ESI) m/z = 626.0 ([M − H]−). HRMS for C23H26Br2N5O6: calculated 626.0250, found 626.0239. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 30–90% of acetonitrile in TFA (0.1%) in 16 min, 90% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 9.325 min (98.6% at 280 nm).
4.2.2.8 (R)-(3-(3-((tert-Butoxycarbonyl)amino)pyrrolidin-1-yl)-4-(4,5-dibromo-1H-pyrrole-2-carboxamido)benzoyl)glycine (9c). To the solution of 7c (300 mg, 0.47 mmol) in a mixture of methanol (10 mL) and tetrahydrofuran (1 mL) 1 M NaOH (1.86 mL, 1.8 mmol) was added and the mixture was stirred at rt for 15 h. The mixture was neutralized with 1 M HCl and methanol was removed under reduced pressure. Water (10 mL) was added and the pH was adjusted to 4 with 1 M HCl. The precipitate was filtered off and dried to obtain 9c (234 mg) as a grey solid. Yield 234 mg (80%); grey solid; mp 218–223 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.89 (s, 1H, NH), 12.47 (br s, 1H, COOH), 9.52 (s, 1H, NH), 8.80 (t, 1H, J = 5.8 Hz, NH), 7.29–7.35 (m, 3H, 3 × ArH), 7.13–7.20 (m, 2H, NH, ArH), 4.01–4.06 (m, 1H, CH), 3.91 (d, 2H, J = 5.8 Hz, CH2), 3.39–3.44 (m, 2H, 2 × CH, overlapping with the signal for water), 3.20–3.26 (m, 1H, CH), 3.04–3.07 (m, 1H, CH), 2.03–2.10 (m, 1H, CH), 1.75–1.84 (m, 1H, CH), 1.37 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 171.4, 166.3, 157.6, 155.2, 144.3, 135.1, 129.6, 128.0, 127.8, 117.5, 114.5, 113.5, 105.8, 98.2, 77.8, 55.9, 49.8, 48.3, 41.2, 30.5, 28.2 ppm. Signal for one aromatic carbon not seen. IR (ATR): ν 3376, 3276, 3104, 1735, 1692, 1666, 1628, 1509, 1399, 1383, 1236, 1153, 976, 767 cm−1. [α]25D +35.3 (c 0.133, MeOH). MS (ESI) m/z = 626.0 ([M − H]−). HRMS for C23H26Br2N5O6: calculated 626.0250, found 626.0252. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 30–90% of acetonitrile in TFA (0.1%) in 16 min, 90% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 9.325 min (95.1% at 280 nm).
4.2.2.9 4-(5-((Carboxymethyl)carbamoyl)-2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)phenyl)piperazin-1-ium chloride (10a). Compound 8a (50 mg, 0.09 mmol) was suspended in 4 M HCl in 1,4-dioxane (5 mL) and THF (2 mL), and the mixture was stirred at rt for 1 h. The solvents were evaporated, to the solid residue diethyl ether was added, the resulting suspension was sonicated and the solid was filtered off to give 10a (39 mg) as a grey solid. Yield 39 mg (89%); grey solid; mp 262–265 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.59 (br s, 1H, COOH), 12.49 (s, 1H, NH), 9.59 (s, 1H, NH), 9.23 (br s, 2H, NH2+), 8.96 (t, 1H, J = 5.7 Hz, NH), 8.45 (d, 1H, J = 8.6 Hz, ArH-3), 7.83 (d, 1H, J = 1.8 Hz, ArH-6), 7.79 (dd, 1H, J = 8.6, 1.8 Hz, ArH-4), 3.93 (d, 2H, J = 5.7 Hz, CH2), 3.24–3.31 (m, 4H, 2 × CH2), 3.09–3.12 (m, 4H, 2 × CH2), 2.24 (s, 3H, CH3) ppm. 13C NMR (100 MHz, DMSO-d6): δ 171.4, 165.5, 156.6, 140.3, 136.4, 129.7, 128.9, 125.3, 120.7, 118.9, 118.8, 110.0, 108.6, 48.9, 43.4, 41.2, 10.8 ppm. DEPT 45 NMR (100 MHz, DMSO-d6) δ 125.7, 121.2, 119.4, 49.5, 44.0, 41.7, 11.2 ppm. DEPT 135 NMR (100 MHz, DMSO-d6) δ 125.7, 121.2, 119.4, 49.5 (negative), 44.0 (negative), 41.7 (negative), 11.2 ppm. IR (ATR): ν 3402, 3307, 3240, 2834, 1734, 1645, 1588, 1504, 1412, 1211, 1040, 920, 763 cm−1. MS (ESI) m/z = 452.0 ([M − H]−). HRMS for C19H20Cl2N5O4: calculated 452.0892, found 452.0891. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 20–40% of acetonitrile in phosphate buffer (pH = 6.8) in 16 min, 40% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 9.087 min (99.6% at 280 nm).
4.2.2.10 (S)-1-(5-((Carboxymethyl)carbamoyl)-2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)phenyl)pyrrolidin-3-aminium chloride (10b). The solution of 8b (40 mg, 0.072 mmol) in a mixture of 4 M HCl in 1,4-dioxane (1 mL) and THF (1 mL) was stirred at rt for 2 h. The solvent was removed, to the residue diethyl ether was added, the resulting suspension was sonicated and the undissolved solid was filtered off and dried to give 10b (34 mg) as a beige solid. Yield 24 mg (97%); beige solid; mp 216–220 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.6 (s, 1H, NH), 9.47 (s, 1H, NH), 8.92 (t, 1H, J = 5.8 Hz, NH), 8.43 (s, 3H, NH3+), 8.17 (d, 1H, J = 8.5 Hz, ArH-3), 7.79 (d, 1H, J = 1.8 Hz, ArH-6), 7.65 (dd, 1H, J = 8.5, 1.8 Hz, ArH-4), 3.92 (d, 2H, J = 5.8 Hz, CH2), 3.66–3.71 (m, 1H, CH, overlapping with the signal for water), 3.47–3.52 (m, 1H, CH, overlapping with the signal for water), 3.36–3.42 (m, 1H, CH), 3.08–3.12 (m, 1H, CH), 2.95–3.00 (m, 1H, CH), 2.24–2.34 (m, 4H, CH, CH3), 1.96–2.06 (m, 1H, CH) ppm. Signal for COOH not seen. 13C NMR (100 MHz, DMSO-d6): δ 171.4, 165.8, 156.7, 139.3, 134.8, 129.5, 129.1, 122.9, 121.0, 119.2, 118.7, 111.2, 108.7, 55.1, 50.3, 49.0, 41.2, 29.6, 10.8 ppm. DEPT 45 NMR (100 MHz, DMSO-d6) δ 175.7, 173.2, 123.4, 121.4, 119.8, 55.6, 50.8, 49.5, 41.7, 30.1, 11.2 ppm. DEPT 135 NMR (100 MHz, DMSO-d6) δ 123.4, 121.4, 119.8, 55.6 (negative), 50.8, 49.5 (negative), 41.7, 30.1 (negative), 11.2 (negative) ppm. IR (ATR): ν 3365, 3263, 2937, 1729, 1640, 1509, 1507, 1410, 1317, 1259, 1222, 1041, 762 cm−1. [α]25D −11.9 (c 0.117, MeOH). MS (ESI) m/z = 452.0 ([M − H]−). HRMS for C19H20Cl2N5O4: calculated 452.0892, found 452.0881. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 20–40% of acetonitrile in phosphate buffer (pH = 6.8) in 16 min, 40% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 9.252 min (99.3% at 280 nm).
4.2.2.11 (R)-1-(5-((Carboxymethyl)carbamoyl)-2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)phenyl)pyrrolidin-3-aminium chloride (10c). The solution of 8c (35 mg, 0.063 mmol) in 4 M HCl in 1,4-dioxane (5 mL) was stirred at rt for 2 h. The solvent was removed, to the residue diethyl ether was added, the resulting suspension was sonicated and the undissolved solid was filtered off and dried to give 10c (22 mg) as a grey solid. Yield 22 mg (71%); grey solid; mp 217–221 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.6 (s, 1H, NH), 9.47 (s, 1H, NH), 8.92 (t, 1H, J = 5.8 Hz, NH), 8.43 (s, 3H, NH3+), 8.17 (d, 1H, J = 8.5 Hz, ArH-3), 7.79 (d, 1H, J = 1.8 Hz, ArH-6), 7.65 (dd, 1H, J = 8.5, 1.8 Hz, ArH-4), 3.92 (d, 2H, J = 5.8 Hz, CH2), 3.66–3.71 (m, 1H, CH, overlapping with the signal for water), 3.47–3.52 (m, 1H, CH, overlapping with the signal for water), 3.36–3.42 (m, 1H, CH), 3.08–3.12 (m, 1H, CH), 2.95–3.00 (m, 1H, CH), 2.24–2.34 (m, 4H, CH, CH3), 1.96–2.06 (m, 1H, CH) ppm. Signal for COOH not seen. 13C NMR (100 MHz, DMSO-d6): δ 171.4, 165.8, 156.7, 139.3, 134.8, 129.5, 129.1, 122.9, 121.0, 119.2, 118.7, 111.2, 108.7, 55.1, 50.3, 49.0, 41.2, 29.6, 10.8 ppm. IR (ATR): ν 3348, 3235, 3017, 2922, 1733, 1636, 1602, 1509, 1410, 1317, 1042, 764, 607 cm−1. [α]25D +11.0 (c 0.100, MeOH). MS (ESI) m/z = 452.1 ([M − H]−). HRMS for C19H20Cl2N5O4: calculated 452.0892, found 452.0898. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 20–40% of acetonitrile in phosphate buffer (pH = 6.8) in 16 min, 40% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 9.252 min (99.4% at 280 nm).
4.2.2.12 4-(5-((Carboxymethyl)carbamoyl)-2-(4,5-dibromo-1H-pyrrole-2-carboxamido)phenyl)piperazin-1-ium chloride (11a). Compound 9a (50 mg, 0.079 mmol) was dissolved in 4 M HCl in 1,4-dioxane (4 mL) and THF (1 mL), and the solution was stirred at rt for 2 h. The solvents were evaporated, to the solid residue diethyl ether was added, the resulting suspension was sonicated and the solid was filtered off to give 11a (45 mg) as a beige solid. Yield 45 mg (100%); beige solid; mp 225–228 °C. 1H NMR (400 MHz, DMSO-d6): δ 13.19 (d, 1H, J = 2.8 Hz, NH), 12.52 (br s, 1H, COOH), 9.24 (br s, 2H, NH2+), 9.19 (s, 1H, NH), 8.95 (t, 1H, J = 5.8 Hz, NH), 8.06 (d, 1H, J = 9.0 Hz, ArH-3), 7.69–7.72 (m, 2H, ArH-4,6), 7.29 (d, 1H, J = 2.8 Hz, ArH), 3.93 (d, 2H, J = 5.8 Hz, CH2), 3.27–3.36 (m, 4H, 2 × CH2), 3.08–3.10 (m, 4H, 2 × CH2) ppm. 13C NMR (100 MHz, DMSO-d6): δ 171.4, 165.7, 157.0, 142.2, 134.9, 129.7, 127.5, 123.9, 122.0, 119.5, 114.0, 106.7, 98.6, 48.2, 43.0, 41.2 ppm. DEPT 45 NMR (100 MHz, DMSO-d6) δ 124.4, 122.5, 120.1, 114.5, 48.7, 43.6, 41.7 ppm. DEPT 135 NMR (100 MHz, DMSO-d6) δ 124.4, 122.4, 120.1, 114.5, 48.7 (negative), 43.6 (negative), 41.7 (negative) ppm. IR (ATR): ν 3326, 3199, 2798, 2719, 1730, 1655, 1507, 1406, 1305, 1178, 950, 746 cm−1. MS (ESI) m/z = 526.0 ([M − H]−). HRMS for C18H18Br2N5O4: calculated 525.9726, found 525.9727. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 20–40% of acetonitrile in phosphate buffer (pH = 6.8) in 16 min, 40% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 6.884 min (98.4% at 280 nm).
4.2.2.13 (S)-1-(5-((Carboxymethyl)carbamoyl)-2-(4,5-dibromo-1H-pyrrole-2-carboxamido)phenyl)pyrrolidin-3-aminium chloride (11b). Suspension of 9b (70 mg, 0.11 mmol) in 4 M HCl in 1,4-dioxane (5 mL) was stirred at rt for 2 h. The precipitate was filtered off and washed with diethyl ether and dried to give 11b (48 mg) as a grey solid. Yield 48 mg (76%); grey solid; mp 209–212 °C. 1H NMR (400 MHz, DMSO-d6): δ 13.01 (d, 1H, J = 2.4 Hz, NH), 12.38 (br s, 1H, COOH), 9.61 (s, 1H, NH), 8.84 (t, 1H, J = 5.8 Hz, NH), 8.22 (s, 3H, NH3+), 7.52–7.50 (m, 1H, ArH), 7.43–7.41 (m, 2H, 2 × ArH), 7.36 (d, 1H, J = 2.4 Hz, ArH), 3.91 (d, 2H, J = 5.8 Hz, CH2), 3.88 (s, 1H, CH, overlapping with the signal for water), 3.49–3.53 (m, 2H, 2 × CH), 3.20–3.23 (m, 1H, CH), 3.01–3.07 (m, 1H, CH), 2.20–2.29 (m, 1H, CH), 1.94–2.04 (m, 1H, CH) ppm. 13C NMR (100 MHz, DMSO-d6): δ 171.4, 166.2, 157.4, 142.6, 131.1, 129.7, 127.8, 127.0, 119.0, 115.9, 114.4, 105.8, 98.4, 53.6, 49.4, 47.7, 41.2, 29.1 ppm. IR (ATR): ν 3374, 3188, 2943, 1725, 1630, 1507, 1411, 1389, 1328, 1221, 1180, 974, 868, 755 cm−1. [α]25D −11.1 (c 0.189, MeOH). MS (ESI) m/z = 526.0 ([M − H]−). HRMS for C18H18Br2N5O4: calculated 525.9726, found 525.9726. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 20–40% of acetonitrile in phosphate buffer (pH = 6.8) in 16 min, 40% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 5.908 min (95.2% at 280 nm).
4.2.2.14 (R)-1-(5-((Carboxymethyl)carbamoyl)-2-(4,5-dibromo-1H-pyrrole-2-carboxamido)phenyl)pyrrolidin-3-aminium chloride (11c). The solution of 9c (70 mg, 0.11 mmol) in a mixture of 1,4-dioxane (10 mL) and 4 M HCl in 1,4-dioxane (5 mL) was stirred at rt for 2 h. The solvent was removed under reduced pressure, to the residue diethyl ether was added, the resulting suspension was sonicated, the precipitate was filtered off, washed with diethyl ether and dried to give 11c (62 mg) as a grey solid. Yield 62 mg (98%); grey solid; mp 209–214 °C. 1H NMR (400 MHz, DMSO-d6): δ 13.01 (d, 1H, J = 2.4 Hz, NH), 12.38 (br s, 1H, COOH), 9.61 (s, 1H, NH), 8.84 (t, 1H, J = 5.8 Hz, NH), 8.22 (s, 3H, NH3+), 7.52–7.50 (m, 1H, ArH), 7.43–7.41 (m, 2H, 2 × ArH), 7.36 (d, 1H, J = 2.4 Hz, ArH), 3.74–3.94 (m, 3H, CH, CH2 overlapping with the signal for water), 3.49–3.53 (m, 2H, 2 × CH), 3.20–3.23 (m, 1H, CH), 3.01–3.07 (m, 1H, CH), 2.20–2.29 (m, 1H, CH), 1.94–2.04 (m, 1H, CH) ppm. 13C NMR (100 MHz, DMSO-d6): δ 171.7, 167.6, 157.9, 143.0, 131.1, 129.2, 127.3, 127.2, 119.0, 115.8, 114.0, 106.4, 98.8, 53.4, 49.4, 47.9, 41.2, 28.9 ppm. DEPT 45 NMR (100 MHz, DMSO-d6) δ 127.3, 119.6, 116.5, 114.8, 54.2, 49.9, 48.3, 41.7, 29.6 ppm. DEPT 135 NMR (100 MHz, DMSO-d6) δ 127.3, 119.6, 116.5, 114.8, 54.2 (negative), 49.9, 48.3 (negative), 41.7 (negative), 29.6 (negative) ppm. IR (ATR): ν 3418, 3313, 2954, 2875, 1725, 1632, 1506, 1411, 1388, 1211, 1180, 974, 757 cm−1. [α]25D +10.8 (c 0.120, MeOH). MS (ESI) m/z = 526.0 ([M − H]−). HRMS for C18H18Br2N5O4: calculated 525.9726, found 525.9727. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 20–40% of acetonitrile in phosphate buffer (pH = 6.8) in 16 min, 40% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 5.908 min (95.1% at 280 nm).
4.2.2.15 tert-Butyl 4-(2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-((2-hydrazineyl-2-oxoethyl)carbamoyl)phenyl)piperazine-1-carboxylate (12). To the solution of compound 6c (0.589 g, 1.0 mmol) in a mixture of methanol (10 mL) and THF (10 mL) hydrazine hydrate solution (80%, 0.50 mL, 10.2 mmol) was added and the mixture was stirred under reflux for 20 h. The solvent was removed under reduced pressure, to the residue ethanol was added and the resulting suspension was sonicated, the undissolved solid was filtered off and dried to give 12 (430 mg) as a white solid. Yield 430 mg (76%); white solid; mp 156–160 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.43 (s, 1H, NH), 9.79 (s, 1H, NH), 9.23 (br s, 1H, NH), 8.71 (t, 1H, J = 5.8 Hz, NH), 8.46 (d, 1H, J = 8.6 Hz, ArH-3), 7.92 (d, 1H, J = 1.8 Hz, ArH-6), 7.75 (dd, 1H, J = 8.6, 1.8 Hz, ArH-4), 4.67 (br s, 2H, NH2), 3.84 (d, 2H, J = 5.8 Hz, CH2), 3.49–3.57 (m, 4H, 2 × CH2), 2.82–2.85 (m, 4H, 2 × CH2), 2.24 (s, 3H, CH3), 1.44 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 168.4, 165.6, 156.5, 153.7, 140.8, 136.4, 129.8, 128.9, 125.4, 121.2, 118.8, 118.4, 109.7, 108.6, 79.1, 52.1, 41.3, 28.0, 10.8 ppm. Signal for one aliphatic carbon not seen. IR (ATR): ν 3263, 1690, 1641, 1509, 1410, 1262, 1245, 1168, 1132, 1040, 713 cm−1. MS (ESI) m/z = 566.0 ([M − H]−). HRMS for C24H30Cl2N7O5: calculated 566.1685, found 566.1682. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 30–90% of acetonitrile in TFA (0.1%) in 16 min, 90% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 10.065 min (96.3% at 280 nm).
4.2.2.16 tert-Butyl 4-(2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(((5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)methyl)carbamoyl)phenyl)piperazine-1-carboxylate (13). The solution of 12 (392 mg, 0.69 mmol) and CDI (224 mg, 1.38 mmol) in a mixture of 1,4-dioxane (15 mL) and DMF (5 mL) was stirred at 101 °C for 20 h. The solvent was removed and the residue was purified with flash column chromatography using dichloromethane/methanol (10
:1) as eluent to give 13 (70 mg) as a white solid. Yield 70 mg (17%); white solid; mp 147–151 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.43 (s, 1H, NH), 12.29 (s, 1H, NH), 9.79 (s, 1H, NH), 9.03 (t, 1H, J = 5.8 Hz, NH), 8.47 (d, 1H, J = 8.6 Hz, ArH-3), 7.90 (d, 1H, J = 1.9 Hz, ArH-6), 7.76 (dd, 1H, J = 8.6, 1.9 Hz, ArH-4), 4.39 (d, 2H, J = 5.6 Hz, CH2), 3.45 (4H, 2 × CH2, overlapping with the signal for water), 2.81–2.85 (m, 4H, 2 × CH2), 2.24 (s, 3H, CH3), 1.43 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 165.5, 156.5, 154.9, 154.5, 153.7, 141.0, 136.8, 129.9, 128.1, 125.4, 121.0, 118.8, 118.6, 109.8, 108.6, 79.1, 52.0, 30.8, 28.0, 10.7 ppm. Signal for one aliphatic carbon not seen. IR (ATR): ν 3262, 2979, 2935, 1776, 1644, 1506, 1411, 1247, 1165, 1130, 1091, 1041, 762 cm−1. MS (ESI) m/z = 592.0 ([M − H]−). HRMS for C25H28Cl2N7O6: calculated 592.1478, found 592.1474. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 30–90% of acetonitrile in TFA (0.1%) in 16 min, 90% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 12.838 min (97.1% at 280 nm).
4.2.2.17 4-(2-(3,4-Dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(((5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)methyl)carbamoyl)phenyl)piperazin-1-ium chloride (14). The solution of 13 (65 mg, 0.12 mmol) in a mixture of 1,4-dioxane (10 mL) and 4 M HCl in 1,4-dioxane (6 mL) was stirred for 2 h at rt. The precipitate was filtered off and dried to give 14 (46 mg) as a grey solid. Yield 46 mg (79%); grey solid; mp 258–262 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.46 (s, 1H, NH), 12.34 (s, 1H, NH), 9.60 (s, 1H, NH), 9.18 (t, 1H, J = 5.6 Hz, NH), 9.02 (br s, 2H, NH2+), 8.46 (d, 1H, J = 8.6 Hz, ArH-3), 7.84 (d, 1H, J = 1.9 Hz, ArH-6), 7.78 (dd, 1H, J = 8.6, 1.8 Hz, ArH-4), 4.39 (d, 2H, J = 5.6 Hz, CH2), 3.24–3.31 (m, 4H, 2 × CH2), 3.06–3.12 (m, 4H, 2 × CH2), 2.24 (s, 3H, CH3) ppm. 13C NMR (100 MHz, DMSO-d6): δ 165.7, 156.6, 154.9, 154.4, 140.3, 136.6, 129.9, 128.3, 125.4, 120.9, 118.9, 118.8, 110.0, 108.7, 48.8, 43.5, 35.1, 10.7 ppm. DEPT 45 NMR (100 MHz, DMSO-d6) δ 125.9, 121.3, 119.4, 49.3, 43.9, 35.6, 11.2 ppm. DEPT 135 NMR (100 MHz, DMSO-d6) δ 125.9, 121.3, 119.4, 49.3 (negative), 43.9 (negative), 35.6 (negative) ppm. IR (ATR): ν 3245, 2956, 2797, 1773, 1639, 1507, 1460, 1309, 1258, 922, 842, 730 cm−1. MS (ESI) m/z = 492.1 ([M − H]−). HRMS for C20H20Cl2N7O4: calculated 492.0954, found 492.0959. HPLC: Agilent Eclipse Plus C18 column (5 μm, 4.6 × 150 mm); mobile phase: 20–40% of acetonitrile in phosphate buffer (pH = 6.8) in 16 min, 40% acetonitrile to 20 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 13.668 min (96.7% at 280 nm).
4.2.2.18 Methyl (S)-3-(3-((tert-butoxycarbonyl)amino)piperidin-1-yl)-4-nitrobenzoate (15c). Synthesised according to General procedure A from 1 (1.07 g, 5.39 mmol), (3S)-3-aminopiperidine 3-Boc protected (1.00 g, 5.35 mmol) and K2CO3 (0.894 g, 6.47 mmol). Yield 87% (1.78 g); orange solid; mp 120–124 °C. 1H NMR (400 MHz, CDCl3): δ 7.83 (d, 1H, J = 1.5 Hz, Ar–H-2), 7.77 (d, 1H, J = 8.4 Hz, Ar–H-5), 7.71 (dd, 1H, J = 8.4, 1.5 Hz, ArH-6), 5.05–5.15 (m, 1H, NH), 3.97 (s, 3H, CH3), 3.88–3.97 (m, 1H, CH), 3.26–3.30 (m, 1H, CH), 2.92–3.12 (m, 3H, CH, CH2), 1.84–1.93 (m, 1H, CH), 1.64–1.80 (m, 3H, CH, CH2), 1.48 (s, 9H, tBu) ppm. IR (ATR): ν 3444, 3122, 3067, 2966, 1727, 1616, 1598, 1521, 1487, 1438, 1421, 1358, 1282, 1227, 1188, 1156, 1111, 1080, 987, 917, 905, 851, 840, 799, 772, 738, 686 cm−1.
4.2.2.19 tert-Butyl 4-(2-amino-5-(methoxycarbonyl)phenyl)piperazine-1-carboxylate (16a). To the solution of compound 2a (5.44 g, 16.0 mmol) in a mixture of methanol (200 mL) and tetrahydrofuran (90 mL) under an argon atmosphere Pd–C (1.00 g) was added, the mixture was saturated with hydrogen and stirred under a hydrogen atmosphere at rt for 4 h. The catalyst was filtered off and the solvent was removed under reduced pressure to obtain 16a (4.87 g) as a white solid. Yield 90% (4.87 g); white solid; mp 135–137 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.49 (dd, 1H, J = 8.4, 1.9 Hz, ArH), 7.44 (d, 1H, J = 1.9 Hz, ArH), 6.70 (d, 1H, J = 8.4 Hz, ArH), 5.73 (br s, 2H, NH2), 3.74 (s, 3H, CH3), 3.44–3.59 (m, 4H, 2 × CH2), 2.68–2.84 (m, 4H, 2 × CH2), 1.43 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 166.3, 153.9, 147.6, 136.7, 126.8, 120.6, 116.4, 113.1, 78.8, 51.2, 50.4, 43.2, 28.0 ppm. IR (ATR): ν 3412, 3319, 2977, 2811, 1685, 1614, 1579, 1510, 1477, 1440, 1417, 1362, 1323, 1302, 1261, 1248, 1218, 1157, 1133, 1107, 1066, 1052, 1039, 993, 945, 930, 908, 870, 843, 829, 811, 769, 649, 624 cm−1. MS (ESI) m/z = 336.2 ([M + H]+). HRMS for C17H26O3N4: calculated 336.1923, found 336.1924.
4.2.2.20 tert-Butyl 4-(2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(methoxycarbonyl)phenyl)piperazine-1-carboxylate (17a). Synthesised according to General procedure B from 3,4-dichloro-5-methyl-1H-pyrrole-2-carboxylic acid (0.850 g, 4.38 mmol) and 16a (1.62 g, 4.82 mmol). During extraction, the product precipitated and was filtered off. The crude product was successively triturated with methanol and tetrahydrofuran, and the undissolved solid was filtered off and dried to give 17a (0.589 g) as a white solid. Yield 25% (0.589 g); white solid; mp 132–136 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.45 (s, 1H, NH), 9.79 (s, 1H, NH), 8.51 (d, 1H, J = 8.6 Hz, ArH), 7.78–7.89 (m, 2H, 2 × ArH), 3.84 (s, 3H, CH3), 3.52 (br s, 4H, 2 × CH2), 2.78–2.87 (m, 4H, 2 × CH2), 2.24 (s, 3H, CH3), 1.44 (s, 9H, tBu) ppm. IR (ATR): ν 3252, 2975, 2892, 2364, 1722, 1686, 1639, 1593, 1524, 1488, 1455, 1410, 1378, 1365, 1332, 1273, 1258, 1245, 1218, 1194, 1164, 1131, 1116, 1098, 1079, 1041, 998, 980, 939, 894, 860, 844, 818, 748, 740, 711, 664, 650, 632, 622, 611 cm−1. MS (ESI) m/z = 511.2 ([M + H]+). HRMS for C23H29N4O5Cl2: calculated 511.1515, found 511.1524. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Eclipse Plus C18: 5 μm, 4.6 × 150 mm): tR 16.803 min (98.3% at 280 nm).
4.2.2.21 3-(4-(tert-Butoxycarbonyl)piperazin-1-yl)-4-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzoic acid (18a). To a solution of compound 17a (201 mg, 0.391 mmol) in a mixture of methanol (10 mL) and tetrahydrofuran (7 mL) 1 M NaOH (1.56 mL, 1.56 mmol) was added and the mixture was stirred at rt for 15 h. The solvent was removed under reduced pressure, to the residue water (10 mL) was added and the mixture was acidified to pH 4 with 1 M HCl. The water phase was extracted with ethyl acetate (20 mL), the organic phase was washed with brine (10 mL), dried over Na2SO4 and the solvent was removed under reduced pressure. The crude product was triturated with diethyl ether and the undissolved solid was filtered off and dried to obtain 18a as a pale pink solid (70 mg). Yield 35% (70 mg); pale pink solid; mp 260–262 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.87 (s, 1H, COOH), 12.45 (s, 1H, NH), 9.78 (s, 1H, NH), 8.49 (d, 1H, J = 8.6 Hz, ArH), 7.77–7.88 (m, 2H, 2 × ArH), 3.42–3.63 (m, 4H, 2 × CH2), 2.75–2.91 (m, 4H, 2 × CH2), 2.24 (s, 3H, CH3), 1.44 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 166.7, 156.6, 153.8, 141.1, 137.8, 129.9, 127.3, 125.7, 122.6, 118.7, 118.7, 109.8, 108.6, 79.1, 51.9, 28.0, 10.8 ppm. One peak not seen. IR (ATR): ν 3261, 2980, 2363, 1713, 1644, 1609, 1594, 1523, 1489, 1456, 1429, 1410, 1376, 1366, 1338, 1280, 1248, 1211, 1199, 1169, 1137, 1105, 1088, 1042, 1005, 956, 897, 865, 833, 800, 765, 724, 700, 646, 629, 619, 609 cm−1. MS (ESI) m/z = 495.1 ([M − H]−). HRMS for C22H25N4O5Cl2: calculated 495.1202, found 495.1206. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Eclipse Plus C18: 5 μm, 4.6 × 150 mm): tR 14.030 min (96.3% at 280 nm).
4.2.2.22 (S)-3-(3-((tert-Butoxycarbonyl)amino)piperidin-1-yl)-4-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzoic acid (18c). To a solution of compound 17c (203 mg, 0.391 mmol) in a mixture of methanol (5 mL) and tetrahydrofuran (4 mL) 1 M NaOH (3.13 mL, 3.13 mmol) was added and the mixture was stirred at rt for 15 h. The solvent was removed under reduced pressure, to the residue water (10 mL) was added and the mixture was acidified to pH 4 with 1 M HCl. The water phase was extracted with ethyl acetate (20 mL), the organic phase was washed with brine (10 mL), dried over Na2SO4 and the solvent was removed under reduced pressure to give 18c (101 mg) as a pale-yellow solid. Yield 50% (101 mg); pale yellow solid; mp 231–233 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.89 (br s, 1H, COOH), 12.46 (s, 1H, NH), 9.83 (s, 1H, NH), 8.49 (d, 1H, J = 8.5 Hz, ArH-5), 7.75–7.91 (m, 2H, 2 × ArH), 6.80–6.99 (m, 1H, NHBoc), 3.55–3.67 (m, 1H, CH), 2.91–3.03 (m, 1H, CH), 2.78–2.88 (m, 1H, CH), 2.53–2.62 (m, 2H, CH2), 2.25 (s, 3H, CH3), 1.75–1.96 (m, 2H, CH2), 1.60–1.74 (m, 1H, CH), 1.36 (s, 9H, tBu), 1.23–1.30 (m, 1H, CH) ppm. IR (ATR): ν 3326, 2959, 2362, 2163, 2032, 2001, 1969, 1717, 1686, 1639, 1588, 1519, 1490, 1437, 1405, 1365, 1318, 1289, 1262, 1173, 1122, 1107, 1082, 1068, 1038, 1026, 997, 960, 928, 877, 864, 839, 805, 765, 731, 676, 648, 625, 608 cm−1. [α]20D = +0.44° (c 0.297, THF). MS (ESI) m/z = 511.2 ([M + H]+). HRMS for C23H29N4O5Cl2: calculated 511.1515, found 511.1512. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Eclipse Plus C18: 5 μm, 4.6 × 150 mm): tR 13.706 min (97.7% at 280 nm).
4.2.2.23 3-((1-(tert-Butoxycarbonyl)piperidin-4-yl)amino)-4-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzoic acid (18d). To a solution of 17d (40 mg, 0.076 mmol) in methanol, 1 M NaOH (0.761 mL, 0.761 mmol) was added and the reaction mixture was stirred at 60 °C for 15 h. The solvent was evaporated in vacuo and the residue was neutralised with 1 M HCl to pH 7. The resulting precipitate was filtered off and dried to obtain 18d (30 mg) as a light brown solid. Yield 77% (30 mg); light brown solid. 1H NMR (400 MHz, DMSO-d6): δ 12.60 (br s, 1H, COOH), 12.30 (s, 1H, NH), 8.93 (s, 1H, NH), 7.80 (d, 1H, J = 8.3 Hz, ArH), 7.33–7.43 (m, 2H, 2 × ArH), 5.07 (d, 1H, J = 6.9 Hz, NH), 3.86 (d, 2H, J = 13.6 Hz, CH2), 3.48 (d, 1H, J = 9.7 Hz, CH), 2.97 (s, 2H, CH2), 2.24 (s, 3H, CH3), 1.88 (d, 2H, J = 12.7 Hz, CH2), 1.40 (s, 9H, tBu), 1.25–1.38 (m, 2H, CH2) ppm.
4.2.2.24 4-(3,4-Dichloro-5-methyl-1H-pyrrole-2-carboxamido)-3-(4-phenylpiperazin-1-yl)benzoic acid (18h). To a solution of 17h (30 mg, 0.062 mmol) in methanol, 1 M NaOH (0.616 mL, 0.616 mmol) was added and the reaction mixture was stirred at 50 °C for 15 h. The solvent was evaporated in vacuo and the residue was neutralised with 1 M HCl to pH 7. The resulting precipitate was filtered off to afford 17h (28.8 mg) as a light brown solid. Yield 99% (28.8 mg); light brown solid; mp >300 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.90 (s, 1H, COOH), 12.47 (s, 1H, NH), 9.87 (s, 1H, NH), 8.51 (d, 1H, J = 8.8 Hz, ArH), 7.91 (s, 1H, ArH), 7.84 (s, 1H, ArH), 7.27 (s, 2H, 2 × ArH), 7.03 (s, 2H, 2 × ArH), 6.84 (s, 1H, ArH), 3.33 (4H, overlapped with the signal for water, 2 × CH2), 3.04 (s, 4H, 2 × CH2), 2.24 (s, 3H, CH3) ppm. IR (ATR): ν 3257, 3176, 3132, 3073, 2961, 2883, 2825, 1749, 1680, 1632, 1586, 1557, 1518, 1492, 1438, 1409, 1375, 1289, 1271, 1254, 1234, 1154, 1134, 1091, 1044, 966, 922, 883, 767 cm−1. HRMS (ESI+) m/z for C23H23Cl2N4O3 ([M + H]+): calculated 473.1142, found 473.1138. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Extend-C18 column: 3.5 μm, 4.6 × 150 mm): tR 15.437 min (95.50% at 254 nm).
4.2.2.25 3-(3-(((tert-Butoxycarbonyl)amino)methyl)piperidin-1-yl)-4-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzoic acid (18i). To a solution of compound 17i (180 mg, 0.33 mmol) in a mixture of methanol (10 mL) and tetrahydrofuran (10 mL) 1 M NaOH (1.32 mL, 1.32 mmol) was added and the mixture was stirred at rt for 15 h. The solvent was removed under reduced pressure, EtOAc (10 mL) and 1 M HCl (10 mL) were added to the residue. The phases were separated, the organic phase was washed with brine (2 × 10 mL), dried over Na2SO4, filtered and the solvent was removed under reduced pressure to give 18i (160 mg) as a white solid. Yield 92% (160 mg); white solid; mp 221–224 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.86 (br s, 1H, COOH), 12.44 (s, 1H, NH), 9.86 (s, 1H, NHAr), 8.49 (d, 1H, J = 8.4 Hz, Ar–H-5), 7.86 (d, 1H, J = 1.8 Hz, Ar–H-2), 7.78 (dd, 1H, J = 8.4, 1.8 Hz, Ar–H-6), 6.90 (t, 1H, J = 5.8 Hz, NHBoc), 2.81–2.98 (m, 3H, 3 × CH), 2.69–2.80 (m, 1H, CH), 2.59–2.69 (m, 1H, CH), 2.35–2.40 (m, 1H, CH), 2.24 (s, 3H, CH3), 1.83–1.93 (m, 1H, CH), 1.72–1.82 (m, 2H, 2 × CH), 1.59–1.71 (m, 1H, CH), 1.31 (s, 9H, tBu), 0.99–1.09 (m, 1H, CH) ppm. 13C NMR (100 MHz, DMSO-d6): δ 166.9, 156.6, 155.6, 142.3, 137.8, 129.9, 126.9, 125.7, 122.6, 118.7, 118.3, 109.9, 108.6, 77.4, 57.1, 53.3, 43.6, 37.3, 28.1, 27.6, 25.4, 10.7 ppm. IR (ATR): ν 3271, 2959, 2808, 1709, 1675, 1641, 1603, 1519, 1487, 1435, 1300, 1254, 1166, 1091, 728, 594 cm−1. MS (ESI) m/z = 525.17 ([M + H]+). HRMS for C24H31Cl2N4O5: calculated 525.1666, found 525.1662. HPLC: Agilent Zorbax 80 Å Extend-C18 (3.5 μm, 4.6 × 150 mm); mobile phase: 5% acetonitrile in 0.1% TFA to 8 min, 5–95% of acetonitrile from 8 to 15 min, 95% acetonitrile from 15 to 16 min, 95–5% of acetonitrile from 16 to 18 min, 5% of acetonitrile from 18 to 21 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 13.587 min (99.4% at 254 nm).
4.2.2.26 4-(2-(3,4-Dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(methoxycarbonyl)phenyl)piperazin-1-ium chloride (19a). Compound 17a (102 mg, 0.200 mmol) was dissolved in 1 M HCl solution in acetic acid (5 mL) and the mixture was stirred at rt for 5 h. The solvent was removed under reduced pressure, the solid residue was successively triturated with diethyl ether and water, and the undissolved solid was filtered off and dried to give 19a (71 mg) as a white solid. Yield 35% (71 mg); white solid; mp 244–248 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.51 (s, 1H, NH), 9.67 (s, 1H, NH), 9.07 (s, 2H, NH2+), 8.53 (d, 1H, J = 8.6 Hz, ArH), 7.87 (dd, 1H, J = 8.6, 1.9 Hz, ArH), 7.82 (d, 1H, J = 1.9 Hz, ArH), 3.86 (s, 3H, COOCH3), 3.22–3.31 (m, 4H, 2 × CH2), 3.05–3.14 (m, 4H, 2 × CH2), 2.25 (s, 3H, CH3) ppm. 13C NMR (100 MHz, DMSO-d6): δ 165.5, 156.7, 140.4, 138.2, 130.0, 127.6, 124.5, 122.3, 119.1, 118.7, 110.2, 108.7, 52.1, 48.7, 43.5, 10.8 ppm. DEPT 45 NMR (100 MHz, DMSO-d6) δ 128.1, 122.7, 119.6, 52.6, 49.3, 43.9, 11.2 ppm. DEPT 135 NMR (100 MHz, DMSO-d6) δ 128.1, 122.7, 119.6, 52.6, 49.3 (negative), 43.9 (negative), 11.2 ppm. IR (ATR): ν 3357, 3291, 3162, 3124, 2954, 2853, 2773, 2720, 2607, 2451, 2363, 1767, 1710, 1632, 1615, 1592, 1573, 1525, 1491, 1446, 1405, 1375, 1355, 1324, 1313, 1284, 1272, 1254, 1225, 1192, 1132, 1120, 1104, 1091, 1043, 1017, 1001, 983, 962, 947, 894, 859, 817, 761, 730, 711, 655, 639, 615 cm−1. MS (ESI) m/z = 411.1 ([M − H]−). HRMS for C18H21N4O3Cl2: calculated 411.0991, found 411.0998. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Eclipse Plus C18: 5 μm, 4.6 × 150 mm): tR 6.367 min (100% at 280 nm).
4.2.2.27 (S)-1-(2-(3,4-Dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(methoxycarbonyl)phenyl)piperidin-3-aminium chloride (19c). Compound 17c (102 mg, 0.209 mmol) was dissolved in 1 M HCl solution in acetic acid (5 mL) and the mixture was stirred at rt for 15 h. The solvent was removed under reduced pressure, the solid residue was triturated with diethyl ether, and the undissolved solid was filtered off and dried to give 19c (79 mg) as a brown solid. Yield 74% (79 mg); brown solid; mp 201–203 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.55 (s, 1H, NH), 9.64 (s, 1H, NH), 8.47 (d, J = 8.5 Hz, ArH), 8.16 (br s, 3H, NH3+), 7.79–7.90 (m, 2H, 2 × ArH), 3.86 (s, 3H, CH3), 3.26–3.36 (m, 1H, CH), 3.14–3.23 (m, 1H, CH), 2.86–2.94 (m, 1H, CH), 2.65–2.79 (m, 2H, CH2), 2.25 (s, 3H, CH3), 2.05–2.16 (m, 1H, CH), 1.85–1.92 (m, 1H, CH), 1.63–1.79 (m, 1H, CH), 1.43–1.58 (m, 1H, CH) ppm. 13C NMR (100 MHz, DMSO-d6): δ 165.6, 156.6, 141.3, 137.8, 129.9, 127.0, 124.5, 122.1, 119.3, 118.6, 110.3, 108.7, 54.7, 52.3, 52.1, 47.5, 27.5, 23.6, 10.7 ppm. IR (ATR): ν 2949, 2153, 2013, 1987, 1970, 1712, 1636, 1589, 1515, 1489, 1438, 1414, 1376, 1318, 1287, 1257, 1204, 1105, 1041, 994, 941, 873, 844, 765, 731, 657, 608 cm−1. [α]20D = −0.72° (c 0.337, THF). MS (ESI) m/z = 425.1 ([M + H]+). HRMS for C19H23N4O3Cl2: calculated 425.1147, found 425.1145. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Eclipse Plus C18: 5 μm, 4.6 × 150 mm): tR 7.370 min (98.9% at 280 nm).
4.2.2.28 (1-(2-(3,4-Dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(methoxycarbonyl)phenyl)piperidin-3-yl)methanaminium chloride (19i). Compound 17i (100 mg, 0.851 mmol) was dissolved in 4 M HCl solution in dioxane (10 mL) and the mixture was stirred for 4 h. After completion of the reaction, the solvent was removed under reduced pressure and the obtained solid was washed with diethyl ether (2 × 5 mL) to give 19i (70 mg) as a beige solid. Yield 79% (70 mg); beige solid; mp 194–198 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.53 (s, 1H, NH), 9.93 (s, 1H, NH), 8.58 (d, J = 8.4 Hz, Ar–H-3), 8.02 (br s, 3H, NH3+), 7.94 (d, J = 1.8 Hz, Ar–H-6), 7.89 (dd, J = 8.4, 1.8 Hz, Ar–H-4), 3.91 (s, 3H, COOCH3), 3.08–3.14 (m, 1H, CH), 2.89–2.98 (m, 1H, CH), 2.76–2.87 (m, 2H, 2 × CH), 2.58–2.68 (m, 2H, 2 × CH), 2.30 (s, 3H, CH3), 2.07–2.19 (m, 1H, CH), 1.90–2.00 (m, 1H, CH), 1.80–1.89 (m, 1H, CH), 1.68–1.79 (m, 1H, CH), 1.16–1.25 (m, 1H, CH) ppm. 13C NMR (100 MHz, DMSO-d6): δ 165.7, 156.6, 142.1, 138.4, 130.1, 127.1, 124.3, 122.7, 118.7, 118.5, 110.0, 108.7, 55.5, 53.5, 52.1, 41.8, 34.9, 27.1, 25.0, 10.8 ppm. DEPT 45 NMR (100 MHz, DMSO-d6) δ 127.6, 123.2, 119.0, 56.1, 54.0, 52.5, 42.3, 35.4, 27.6, 25.5, 11.3 ppm. DEPT 135 NMR (100 MHz, DMSO-d6) δ 127.6, 123.2, 119.0, 56.1 (negative), 54.0 (negative), 52.5, 42.3 (negative), 35.4, 27.6 (negative), 25.5 (negative), 11.3 ppm. IR (ATR): ν 2948, 1717, 1640, 1590, 1517, 1488, 1414, 1289, 1257, 1199, 1121, 1036, 1007, 764 cm−1. MS (ESI) m/z = 439.13 ([M + H]+). HRMS for C20H25Cl2N4O3: calculated 439.1293, found 439.1298. HPLC: Agilent Zorbax 80 Å Extend-C18 (3.5 μm, 4.6 × 150 mm); mobile phase: 5% acetonitrile in 0.1% TFA to 8 min, 5–95% of acetonitrile from 8 to 15 min, 95% acetonitrile from 15 to 16 min, 95–5% of acetonitrile from 16 to 18 min, 5% of acetonitrile from 18 to 21 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 9.373 min (95.6% at 254 nm).
4.2.2.29 4-(5-Carboxy-2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)phenyl)piperazin-1-ium chloride (20a). Compound 18a (50 mg, 0.100 mmol) was dissolved in a mixture of 1 M HCl solution in acetic acid (5 mL), tetrahydrofuran (2 mL) and dichloromethane (4 mL) and the mixture was stirred at rt for 5 h. The solvent was removed under reduced pressure, the solid residue was successively triturated with diethyl ether and water, and the undissolved solid was filtered off and dried to give 20a (16 mg) as a pale pink solid. Yield 32% (16 mg); pale pink solid; mp 258–261 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.50 (s, 1H, NH), 9.68 (s, 1H, NH), 8.50 (d, J = 8.4 Hz, ArH), 7.76–7.92 (m, 2H, 2 × ArH), 3.22–3.29 (m, 4H, 2 × CH2), 3.03–3.11 (m, 4H, 2 × CH2), 2.25 (s, 3H, CH3) ppm. Signal for NH2+ is overlapping with the signal for water. Signal for COOH proton not seen. 13C NMR (100 MHz, DMSO-d6): δ 183.4, 178.3, 166.7, 156.6, 140.4, 137.8, 129.9, 127.7, 122.4, 118.8, 110.1, 108.7, 49.0, 43.6, 10.8 ppm. IR (ATR): ν 3293, 3005, 2822, 2470, 2363, 1684, 1651, 1588, 1529, 1488, 1457, 1406, 1374, 1316, 1285, 1229, 1192, 1142, 1120, 1089, 1047, 963, 923, 885, 849, 787, 771, 726, 704, 647, 634, 606 cm−1. MS (ESI) m/z = 395.1 ([M − H]−). HRMS for C17H17N4O3Cl2: calculated 395.0678, found 395.0670. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Eclipse Plus C18: 5 μm, 4.6 × 150 mm): tR 4.769 min (98.1% at 280 nm).
4.2.2.30 (S)-1-(5-Carboxy-2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)phenyl)piperidin-3-aminium chloride (20c). Compound 18c (70 mg, 0.14 mmol) was dissolved in 1 M HCl solution in acetic acid (5 mL) and the mixture was stirred at rt for 15 h. The solvent was removed under reduced pressure, the solid residue was triturated with diethyl ether, and the undissolved solid was filtered off and dried to give 20c (64 mg) as a pale-yellow solid. Yield 91% (64 mg); pale yellow solid; mp 240–244 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.54 (br s, 1H, NH), 9.63 (s, 1H, NH), 8.45 (d, J = 9.1 Hz, ArH), 7.77–7.85 (m, 2H, 2 × ArH), 3.28–3.33 (m, 1H, CH), 3.14–3.21 (m, 1H, CH), 2.85–2.93 (m, 1H, CH), 2.64–2.78 (m, 2H, CH2), 2.25 (s, 3H, CH3), 2.04–2.14 (m, 1H, CH), 1.84–1.92 (m, 1H, CH), 1.63–1.76 (m, 1H, CH), 1.44–1.58 (m, 1H, CH) ppm. Signals for COOH and NH3+ protons not seen. 13C NMR (100 MHz, DMSO-d6): δ 166.7, 156.6, 141.2, 137.4, 129.8, 127.1, 125.8, 122.2, 119.2, 118.7, 110.3, 108.7, 54.8, 52.3, 47.6, 27.5, 23.6, 10.7 ppm. IR (ATR): ν 2926, 2154, 2050, 1992, 1682, 1634, 1589, 1518, 1490, 1406, 1322, 1249, 1201, 1125, 1107, 1090, 1043, 1017, 960, 942, 836, 766, 743, 728, 649, 608 cm−1. [α]20D = −0.55° (c 0.264, THF). MS (ESI) m/z = 411.1 ([M + H]+). HRMS for C18H21N4O3Cl2: calculated 411.0991, found 411.1002. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Eclipse Plus C18: 5 μm, 4.6 × 150 mm): tR 5.947 min (99.7% at 280 nm).
4.2.2.31 4-((5-Carboxy-2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)phenyl)amino)piperidin-1-ium chloride (20d). To a suspension of 18d (30 mg, 0.059 mmol) in 1,4-dioxane (1 mL), 4 M HCl in 1,4-dioxane (4 mL) was added and the reaction mixture was stirred at rt for 4 h. The resulting precipitate was filtered off, washed with diethyl ether and dried. Yield 94% (24.7 mg); pale yellow solid; mp 260–265 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.52 (s, 2H, NH, COOH), 9.14 (s, 1H, NH), 8.79 (s, 1H, NH), 8.65 (s, 1H, NH), 7.83 (d, 1H, J = 8.1 Hz, ArH), 7.35–7.44 (m, 2H, 2 × ArH), 3.64 (1H, overlapped with the signal for water, CH), 3.25–3.35 (m, 3H, CH, CH2), 3.05 (q, 2H, J = 10.6, 11.3 Hz, CH2), 2.25 (s, 3H, CH3), 2.05 (d, 2H, J = 13.9 Hz, CH2), 1.60–1.75 (m, 2H, CH2) ppm. Signal for one NH proton not seen. IR (ATR): ν 3117, 2955, 2786, 2323, 1722, 1657, 1620, 1581, 1519, 1500, 1471, 1402, 1371, 1335, 1311, 1282, 1218, 1179, 1135, 1117, 1090, 1076, 1055, 1031, 992, 965, 933, 901, 872, 846, 803, 763, 748, 725, 638, 606 cm−1. HRMS (ESI+) m/z for C18H21Cl2N4O3 ([M + H]+): calculated 411.0985, found 411.0980. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Extend-C18 column: 3.5 μm, 4.6 × 150 mm): tR 12.480 min (95.11% at 254 nm).
4.2.2.32 (1-(5-Carboxy-2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)phenyl)piperidin-3-yl)methanaminium chloride (20i). Compound 18i (59 mg, 0.11 mmol) was dissolved in 4 M HCl solution in 1,4-dioxane (8 mL). The mixture was stirred for 12 h. The solvent was removed under reduced pressure and the solid was washed with diethyl ether (2 × 5 mL) to give 20i (49 mg) as a white solid. Yield 96% (49 mg); white solid; mp 234–238 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.48 (s, 1H, NH), 9.87 (s, 1H, NH), 8.50 (d, J = 8.4 Hz, Ar–H-3), 7.97 (br s, 3H, NH3+), 7.88 (d, J = 1.8 Hz, Ar–H-6), 7.81 (dd, J = 8.4, 1.8 Hz, Ar–H-4), 3.03–3.10 (m, 1H, CH), 2.83–2.92 (m, 1H, CH), 2.71–2.80 (m, 2H, 2 × CH), 2.54–2.63 (m, 3H, J = 9.0 Hz, 3 × CH), 2.25 (s, 3H, CH3), 2.02–2.13 (m, 1H, CH), 1.85–1.95 (m, 1H, CH), 1.75–1.83 (m, 1H, CH), 1.63–1.73 (m, 1H, CH), 1.09–1.19 (m, 1H, CH) ppm. 13C NMR (100 MHz, DMSO-d6): δ 166.7, 156.6, 142.0, 137.9, 129.9, 127.1, 125.6, 122.8, 118.7, 118.4, 110.0, 108.6, 55.6, 53.6, 41.8, 34.8, 27.2, 25.0, 10.8 ppm. IR (ATR): ν 3446, 3070, 2888, 1722, 1635, 1605, 1521, 1422, 1378, 1219, 1201, 1140, 1041, 837, 814, 752, 730, 660, 546 cm−1. MS (ESI) m/z = 425.11 ([M + H]+). HRMS for C19H23Cl2N4O3: calculated 425.1142, found 425.1137. HPLC: Agilent Zorbax 80 Å Extend-C18 (3.5 μm, 4.6 × 150 mm); mobile phase: 5% acetonitrile in 0.1% TFA to 8 min, 5–95% of acetonitrile from 8 to 15 min, 95% acetonitrile from 15 to 16 min, 95–5% of acetonitrile from 16 to 18 min, 5% of acetonitrile from 18 to 21 min; flow rate 1.0 mL min−1; injection volume: 10 μL; tR: 8.557 min (99.2% at 254 nm).
4.2.2.33 tert-Butyl 4-(2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(hydrazinecarbonyl)phenyl)piperazine-1-carboxylate (21a). To the solution of 17a (0.71 g, 1.38 mmol) in a mixture of MeOH (20 mL) and THF (10 mL) in a high-pressure tube, hydrazine hydrate (64%, 4.71 mL, 96.8 mmol) was added. The tube was sealed and the reaction mixture was stirred at 120 °C for 15 h. The tube was cooled to rt and the precipitate was filtered off and dried to give 21a (0.579 g) as a white solid. Yield 82% (0.579 g); white solid; mp 277–280 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.42 (s, 1H, NH), 9.74 (s, 1H, NH), 9.71 (s, 1H, NH), 8.43 (d, J = 8.6 Hz, ArH), 7.84 (s, 1H, ArH), 7.70 (d, J = 8.6 Hz, ArH), 4.47 (s, 2H, NH2), 3.57–3.47 (m, 4H, 2 × CH2), 2.85–2.77 (m, 4H, 2 × CH2), 2.24 (s, 3H, CH3), 1.44 (s, 9H, tBu) ppm.
4.2.2.34 tert-Butyl 4-(2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)piperazine-1-carboxylate (22a). To a solution of compound 21a (0.500 g, 0.980 mmol) in a mixture of DMF (30 mL) and 1,4-dioxane (15 mL) 1,1′-carbonyldiimidazole (0.640 g, 3.93 mmol) was added and the reaction mixture was stirred at 101 °C for 15 h. The solvent was removed under reduced pressure, the crude product was successively triturated with acetonitrile, water and THF, the undissolved solid was filtered off and dried. The crude product was crystallized from DMF and dried to give 22a (226 mg) as a white solid. Yield 43% (226 mg); white solid; mp 260–263 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.50 (s, 1H, NH), 12.45 (s, 1H, NH), 9.73 (s, 1H, NH), 8.53 (d, J = 8.6 Hz, ArH), 7.69 (s, 1H, ArH), 7.64 (d, J = 8.5 Hz, ArH), 3.52 (s, 4H, 2 × CH2), 2.88–2.80 (m, 4H, 2 × CH2), 2.24 (s, 3H, CH3), 1.44 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 157.0, 154.9, 154.2, 154.0, 153.9, 142.3, 136.9, 130.4, 123.5, 120.1, 119.5, 119.2, 110.3, 109.1, 79.6, 52.2, 44.3, 28.5, 11.2 ppm. Peaks of two aromatic carbons overlapping. HRMS for C23H27O5N6Cl2 ([M + H]+): calculated 537.14099, found 537.14145. HPLC (0–10 min, 10–90% ACN in 0.1% TFA, 10–11 min, 90% ACN in 0.1% TFA, Waters Acquity UPLC HSS C18 column: 1.8 μm, 2.1 × 50 mm): tR 6.560 min (97.45% at 254 nm, 96.79% at 280 nm).
4.2.2.35 tert-Butyl (S)–(1-(2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)pyrrolidin-3-yl)carbamate (22b). To a solution of compound 21b (100 mg, 0.192 mmol) in a mixture of DMF (2.5 mL) and 1,4-dioxane (5 mL) 1,1′-carbonyldiimidazole (94 mg, 0.576 mmol) was added and the reaction mixture was stirred at 101 °C for 15 h. The solvent was removed under reduced pressure, the crude product was successively triturated with acetonitrile and methanol, the undissolved solid was filtered off and dried to give 22b (24 mg) as a pale-yellow solid. Yield 23% (24 mg); pale yellow solid; mp 260–263 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.40 (br s, 2H, 2 × NH), 9.50 (s, 1H, NH), 8.32 (d, J = 8.6 Hz, ArH), 7.60 (s, 1H, ArH), 7.53 (d, J = 8.6 Hz, ArH), 7.20 (d, 1H, NHBoc), 4.15–4.07 (m, 1H, CH), 3.33–3.24 (m, 1H, CH), 3.24–3.17 (m, 1H, CH), 3.12–3.04 (m, 1H, CH), 2.96–2.91 (m, 1H, CH), 2.25 (s, 3H, CH3), 2.22–2.16 (m, 1H, CH), 1.90–1.80 (m, 1H, CH), 1.40 (s, 9H, tBu) ppm. IR (ATR): ν 3342, 2980, 2850, 2361, 1721, 1681, 1643, 1620, 1602, 1512, 1491, 1416, 1366, 1329, 1275, 1216, 1171, 1118, 1086, 1037, 1003, 958, 897, 833, 761, 731, 657, 625, 608, 571 cm−1. [α]20D = 0.035 (c 0.255, DMF). HRMS for C23H27Cl2N6O5: calculated 537.1414, found 537.1416.
4.2.2.36 tert-Butyl (S)-(1-(2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)piperidin-3-yl)carbamate (22c). To a solution of compound 21c (240 mg, 0.449 mmol) in a mixture of DMF (5 mL) and 1,4-dioxane (10 mL) 1,1′-carbonyldiimidazole (219 mg, 1.35 mmol) was added and the reaction mixture was stirred at 101 °C for 15 h. The solvent was removed under reduced pressure, the crude product was triturated with a mixture of acetonitrile and methanol (20 mL, 1
:1), the undissolved solid was filtered off and dried to give 22c (202 mg) as a brown solid. Yield 84% (202 mg); brown solid; mp 260–263 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.55 (s, 1H, NH), 12.44 (s, 1H, NH), 9.76 (s, 1H, NH), 8.53 (d, J = 8.6 Hz, 1H, ArH), 7.64–7.62 (m, 2H, 2 × ArH), 6.89 (d, J = 7.5 Hz, 1H, NHBoc), 3.70–3.54 (m, 1H, CH), 3.05–2.91 (m, 1H, CH), 2.91–2.77 (m, 1H, CH), 2.70–2.54 (m, 2H, CH2), 2.24 (s, 3H, CH3), 1.99–1.75 (m, 2H, CH2), 1.74–1.58 (m, 1H, CH), 1.27–1.17 (m, 1H, CH), 1.36 (s, 9H, tBu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 156.6, 154.8, 154.4, 153.5, 142.2, 136.5, 129.9, 122.8, 119.4, 118.9, 118.7, 118.7, 108.6, 77.7, 57.4, 52.9, 47.6, 29.3, 28.2, 24.4, 10.8 ppm. IR (ATR): ν 3317, 2963, 2360, 1778, 1687, 1634,1582, 1514, 1491, 1403, 1362, 1326, 1251, 1170, 1125, 1072, 1034, 945, 913, 827, 747 cm−1. [α]20D = −0.73° (c 0.318, THF). MS (ESI) m/z = 549.1 ([M − H]−). HRMS for C24H27N6O5Cl2: calculated 549.1420, found 549.1436. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Eclipse Plus C18: 5 μm, 4.6 × 150 mm): tR 11.960 min (95.1% at 280 nm).
4.2.2.37 tert-Butyl 4-((2-(3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)amino)piperidine-1-carboxylate (22d). To a suspension of 21d (60 mg, 0.114 mmol) in a mixture of 1,4-dioxane and DMF (2
:1, 15 mL), 1,1′-carbonyldiimidazole (55.6 mg, 0.343 mmol) was added and the reaction mixture was stirred at 101 °C for 24 h. The solvent was evaporated in vacuo, the residue was suspended in acetonitrile, sonicated, filtered off and dried. Yield 56% (35 mg); yellow solid; mp 168–171 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.49 (s, 1H, NH), 12.26 (s, 1H, NH), 8.84 (s, 1H, NH), 7.78–7.74 (m, 1H, ArH), 7.19–7.14 (m, 2H, 2 × ArH), 5.19 (d, 1H, J = 7.1 Hz, NH), 3.92–3.79 (m, 2H, CH2), 3.53 (s, 1H, CH), 2.93 (m, 2H, CH2), 2.23 (s, 3H, CH3), 1.89 (d, 2H, J = 9.7 Hz, CH2), 1.40 (s, 9H, tBu), 1.37–1.25 (m, 2H, CH2) ppm. IR (ATR): ν 3347, 3328, 3265, 3122, 2974, 2931, 2852, 1767, 1660, 1590, 1533, 1496, 1478, 1417, 1366, 1322, 1241, 1212, 1175, 1143, 1093, 1074, 1043, 939, 860, 823, 734 cm−1. HRMS for C24H29Cl2N6O5: calculated 551.15710, found 551.15704.
4.2.2.38 3,4-Dichloro-5-methyl-N-(2-morpholino-4-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)-1H-pyrrole-2-carboxamide (22e). To a solution of compound 21e (400 mg, 0.970 mmol) in a mixture of DMF (20 mL) and 1,4-dioxane (20 mL) 1,1′-carbonyldiimidazole (310 mg, 1.94 mmol) was added and the reaction mixture was stirred at 101 °C for 15 h. The solvent was removed under reduced pressure, the crude product was successively triturated with acetonitrile, water and MeOH, and the undissolved solid was filtered off and dried. The crude product was crystallized from DMF and dried to obtain 22e (42.9 mg) as a white solid. Yield 11% (42.9 mg); white solid; mp 304–305 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.54 (s, 1H, NH), 12.44 (s, 1H, NH), 9.75 (s, 1H, NH), 8.54 (d, 1H, J = 8.6 Hz, ArH), 7.70 (d, 1H, J = 2.0 Hz, ArH), 7.64 (dd, 1H, J = 8.5, 1.9 Hz, ArH), 3.72–2.84 (m, 4H, 2 × CH2), 2.84–2.95 (m, 4H, 2 × CH2), 2.24 (s, 3H, CH3) ppm. 13C NMR (100 MHz, DMSO-d6): δ 157.0, 154.9, 154.0, 153.9, 142.4, 137.0, 130.4, 123.4, 120.1, 119.6, 119.2, 118.9, 109.1, 110.3, 67.0, 52.6, 11.2 ppm. HRMS for C18H18O4N5Cl2 ([M + H]+): calculated 438.07237, found 438.07304. HPLC (0–10 min, 10–90% ACN in 0.1% TFA, 10–11 min, 90% ACN in 0.1% TFA, Waters Acquity UPLC HSS C18 column: 1.8 μm, 2.1 × 50 mm): tR 5.333 min (97.38% at 254 nm, 97.45% at 280 nm).
4.2.2.39 3,4-Dichloro-5-methyl-N-(2-(2-methylmorpholino)-4-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)-1H-pyrrole-2-carboxamide (22f). To a solution of compound 21f (310 mg, 0.703 mmol) in a mixture of DMF (20 mL) and 1,4-dioxane (10 mL) 1,1′-carbonyldiimidazole (456 mg, 2.81 mmol) was added and the reaction mixture was stirred at 101 °C for 15 h. The solvent was removed under reduced pressure, the solid residue was successively triturated with acetonitrile, water and THF, and the undissolved solid was filtered off and dried. The crude product was crystallized from DMF and dried to give 22f (169 mg) as a white solid. Yield 53% (169 mg); white solid; mp 302–303 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.56 (s, 1H, NH), 12.44 (s, 1H, NH), 9.75 (s, 1H, NH), 8.55 (d, 1H, J = 8.6 Hz, ArH), 7.68 (d, 1H, J = 2.0 Hz, ArH), 7.64 (dd, 1H, J = 8.6, 2.0 Hz, ArH), 3.88–3.93 (m, 1H, CH), 3.69–3.81 (m, 2H, 2 × CH), 2.78–2.94 (m, 3H, 3 × CH), 2.55–2.61 (m, 1H, CH), 2.24 (s, 3H, CH3), 1.12 (d, 3H, J = 6.2 Hz, CH3) ppm. 13C NMR (100 MHz, DMSO-d6): δ 157.0, 154.9, 154.0, 153.9, 142.2, 137.1, 130.4, 123.4, 120.0, 119.5, 119.2, 119.0, 109.1, 110.3, 72.1, 66.7, 58.6, 52.1, 19.3, 11.2 ppm. DEPT 45 NMR (100 MHz, DMSO-d6) δ 123.4, 120.0, 119.0, 72.2, 66.7, 58.6, 52.1, 19.3, 11.2 ppm. DEPT 135 NMR (100 MHz, DMSO-d6) δ 123.4, 120.0, 119.0, 72.2, 66.7 (negative), 58.6 (negative), 52.1 (negative), 19.3, 11.2 ppm. HRMS for C19H18O4N5Cl2 ([M − H]−): calculated 450.07428, found 450.07413. HPLC (0–10 min, 10–90% ACN in 0.1% TFA, 10–11 min, 90% ACN in 0.1% TFA, Waters Acquity UPLC HSS C18 column: 1.8 μm, 2.1 × 50 mm): tR 5.703 min (95.50% at 254 nm, 95.08% at 280 nm).
4.2.2.40 3,4-Dichloro-N-(2-(2,6-dimethylmorpholino)-4-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)-5-methyl-1H-pyrrole-2-carboxamide (22g). To the solution of compound 21g (217 mg, 0.492 mmol) in a mixture of DMF (8 mL) and 1,4-dioxane (15 mL) 1,1′-carbonyldiimidazole (319 mg, 1.97 mmol) was added and the reaction mixture was stirred at 101 °C for 15 h. The solvent was removed under reduced pressure, the solid residue was successively triturated with acetonitrile, MeOH and THF, and the undissolved solid was filtered off and dried. The crude product was crystallized from DMF and dried to obtain 22g (112 mg) as a white solid. Yield 49% (112 mg); white solid; mp 301–303 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.57 (s, 1H, NH), 12.45 (s, 1H, NH), 9.79 (s, 1H, NH), 8.57 (d, 1H, J = 8.6 Hz, ArH), 7.61–7.71 (m, 2H, 2 × ArH), 3.77–3.86 (m, 2H, 2 × CH), 2.86–2.93 (m, 2H, 2 × CH), 2.43–2.48 (m, 2H, 2 × CH), 2.24 (s, 3H, CH3), 1.12 (d, 6H, J = 6.1 Hz, 2 × CH3) ppm. 13C NMR (100 MHz, DMSO-d6): δ 157.0, 154.9, 154.0, 153.9, 141.9, 137.1, 130.4, 123.5, 119.9, 119.5, 119.3, 119.1, 109.1, 110.2, 72.0, 58.1, 19.3, 11.2 ppm. HRMS for C20H22O4N5Cl2 ([M + H]+): calculated 466.10399, found 466.10434. HPLC (0–10 min, 10–90% ACN in 0.1% TFA, 10–11 min, 90% ACN in 0.1% TFA, Waters Acquity UPLC HSS C18 column: 1.8 μm, 2.1 × 50 mm): tR 6.077 min (98.33% at 254 nm, 98.27% at 280 nm).
4.2.2.41 4-(2-(3,4-Dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)piperazin-1-ium chloride (23a). To a solution of 22a (60.0 mg, 0.110 mmol) in DMF (6 mL) 4 M HCl in 1,4-dioxane (6 mL) was added and the reaction mixture was stirred at rt for 15 h. The solvent was removed under reduced pressure and the solid residue was triturated with acetonitrile, the undissolved solid was filtered off and dried to give 23a (47.4 mg) as a white solid. Yield 91% (47.4 mg); white solid; mp 287–289 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.63 (s, 1H, NH), 12.51 (s, 1H, NH), 9.60 (s, 1H, NH), 9.17 (s, 2H, NH2+), 8.53 (dd, 1H, J = 8.7, 1.3 Hz, ArH), 7.68 (d, 1H, J = 8.5 Hz, ArH), 7.62 (s, 1H, ArH), 3.23–3.31 (m, 4H, 2 × CH2), 3.08–3.15 (m, 4H, 2 × CH2), 2.25 (s, 3H, CH3) ppm. 13C NMR (100 MHz, DMSO-d6): δ 157.1, 154.9, 154.0, 153.8, 141.5, 136.9, 130.4, 123.9, 120.4, 119.6, 119.2, 118.8, 110.7, 109.2, 49.2, 43.9, 11.2 ppm. HRMS for C18H19O3N6Cl2 ([M + H]+): calculated 437.08880, found 437.08902. HPLC (0–10 min, 10–90% ACN in 0.1% TFA, 10–11 min, 90% ACN in 0.1% TFA, Waters Acquity UPLC HSS C18 column: 1.8 μm, 2.1 × 50 mm): tR 3.107 min (96.56% at 254 nm, 95.85% at 280 nm).
4.2.2.42 (S)-1-(2-(3,4-Dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)pyrrolidin-3-aminium chloride (23b). To a solution of compound 22b (24 mg, 0.045 mmol) in DMF (1 mL) 4 M HCl in 1,4-dioxane (3 mL) was added and the reaction mixture was stirred at rt for 15 h. The solvent was removed under reduced pressure and the solid residue was successively triturated with diethyl ether and acetonitrile, the undissolved solid was filtered off and dried to give 23b (13 mg) as an off-white solid. Yield 60% (13 mg); off-white solid; mp > 230 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.63 (s, 1H, NH), 12.58 (s, 1H, NH), 9.50 (s, 1H, NH), 8.32 (br s, 3H, NH3+), 8.25 (d, 1H, J = 8.5 Hz, ArH), 7.61 (d, 1H, J = 1.9 Hz, ArH), 7.55 (dd, 1H, J = 8.5, 1.9 Hz, ArH), 3.87–3.97 (s, 1H, CH), 3.50–3.57 (m, 1H, CH), 3.37–3.45 (m, 1H, CH), 3.02–3.14 (m, 2H, 2 × CH), 2.26–2.32 (m, 1H, CH), 1.98–2.06 (m, 1H, CH), 2.25 (s, 3H, CH3) ppm. IR (ATR): ν 2969, 2360, 2341, 2187, 1994, 1768, 1637, 1584, 1519, 1490, 1360, 1311, 1257, 1179, 1091, 1040, 958, 927, 832, 707 cm−1. [α]25D 1.25 (c 0.271, DMF). HRMS for C18H19Cl2N6O3: calculated 437.0890, found 437.0890. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Eclipse Plus C18: 5 μm, 4.6 × 150 mm): tR 3.523 min (96.05% at 254 nm).
4.2.2.43 (S)-1-(2-(3,4-Dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)piperidin-3-aminium chloride (23c). To a solution of compound 22c (133 mg, 0.241 mmol) in DMF (2 mL) 4 M HCl in 1,4-dioxane (6 mL) was added and the reaction mixture was stirred at rt for 15 h. The solvent was removed under reduced pressure and the solid residue was successively triturated with diethyl ether and acetonitrile, the undissolved solid was filtered off and dried to give 23c (71 mg) as an off-white solid. Yield 60% (71 mg); off-white solid; mp > 230 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.64 (s, 1H, NH), 12.57 (s, 1H, NH), 9.58 (s, 1H, NH), 8.46 (d, J = 8.4 Hz, 1H, ArH), 8.27 (br s, 3H, NH3+), 7.63–7.66 (m, 2H, 2 × ArH), 3.26–3.40 (s, 1H, CH), 3.16–3.25 (m, 1H, CH), 2.83–2.94 (m, 1H, CH), 2.65–2.81 (m, 2H, CH2), 2.25 (s, 3H, CH3), 2.05–2.13 (m, 1H, CH), 1.83–1.93 (m, 1H, CH), 1.62–1.77 (m, 1H, CH), 1.44–1.62 (m, 1H, CH) ppm. 13C NMR (100 MHz, DMSO-d6): δ 156.6, 154.4, 153.4, 141.9, 136.0, 129.8, 122.8, 120.3, 119.2, 118.7, 118.2, 110.3, 108.7, 54.7, 52.1, 47.5, 27.5, 23.6, 10.8 ppm. IR (ATR): ν 3100, 2361, 1766, 1650, 1613, 1511, 1488, 1408, 1373, 1333, 1255, 1204, 1125, 1086, 1039, 945, 919, 836, 749, 708 cm−1. MS (ESI) m/z = 449.1 ([M − H]−). HRMS for C19H19N6O3Cl2: calculated 449.0896, found 449.0890. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Eclipse Plus C18: 5 μm, 4.6 × 150 mm): tR 4.633 min (95.8% at 280 nm).
4.2.2.44 4-((2-(3,4-Dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)amino)piperidin-1-ium chloride (23d). To a suspension of 22d (25 mg, 0.045 mmol) in 1,4-dioxane (7 mL), 4 M HCl in 1,4-dioxane (3 mL) was added and the reaction mixture was stirred at rt for 3 h. The resulting precipitate was filtered off, washed with diethyl ether and dried to give 23d as a pale-yellow solid. Yield 100% (22 mg); pale yellow solid; mp 212–217 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.55 (s, 1H, NH), 12.53 (s, 1H, NH), 9.10 (s, 1H, NH), 8.72 (s, 2H, NH2+), 7.79 (d, J = 8.2 Hz, ArH), 7.17 (m, 2H, 2 × ArH), 3.67–3.72 (m, 1H, CH), 3.25–3.35 (m, 2H, CH2), 3.01–3.11 (m, 2H, CH2), 2.24 (s, 3H, CH3), 2.06 (dd, 2H, J = 13.2, 2.8 Hz, CH2), 1.65–1.72 (m, 2H, CH2) ppm. 13C NMR (100 MHz, DMSO-d6): δ 167.9, 157.6, 138.7, 130.9, 128.9, 127.7, 123.4, 120.3, 119.6, 115.3, 112.1, 109.1, 47.3, 42.1, 28.6, 11.2 ppm. HRMS (ESI+) m/z for C19H21Cl2N6O3 ([M + H]+): calculated 451.1047, found 451.1040. HPLC (0–16 min, 30–90% ACN in 0.1% TFA, 16–20 min, 90% ACN in 0.1% TFA, Agilent Extend-C18 column: 3.5 μm, 4.6 × 150 mm): tR 12.587 min (92.82% at 254 nm).
4.2.2.45 Methyl 4-(methylamino)-3-morpholinobenzoate (24). To a solution of methyl 4-amino-3-morpholinobenzoate (16e, 1.50 g, 6.35 mmol) and sodium methoxide (0.48 g, 8.89 mmol) in MeOH (35 mL) paraformaldehyde (0.48 g, 15.9 mmol) was added in one portion. The reaction mixture was heated to 40 °C and stirred for 15 h. Solid NaBH4 (0.48 g, 12.7 mmol) was added to the reaction mixture and stirred at 40 °C for 5 h. Additional 2.00 equivalents of solid NaBH4 (0.48 g, 12.7 mmol) was added and the mixture was stirred at 40 °C for 15 h. The reaction mixture was quenched with 80 mL of saturated aqueous NaHCO3 and then diluted with 80 mL of ethyl acetate. The phases were separated and the aqueous layer was washed with ethyl acetate (3 × 45 mL). The organic layers were combined, washed with brine (2 × 50 mL) and dried over Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by column chromatography using ethyl acetate/hexane (2
:3) as eluent. The crude product was purified by washing the solid with ethyl acetate to give 24 (413 mg) as a white solid. Yield 26% (413 mg); white solid; mp 125–126 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.63 (d, J = 8.5 Hz, 1H, ArH), 7.47 (s, 1H, ArH), 6.56 (d, J = 8.4 Hz, 1H, ArH), 5.86 (q, J = 4.8 Hz, 1H, NH), 3.73–3.80 (m, 7H, 2 × CH2, COOCH3), 2.81 (d, J = 4.9 Hz, 3H, NCH3), 2.72–2.79 (m, 4H, 2 × CH2) ppm.
4.2.2.46 Methyl 4-(4,5-dibromo-1H-pyrrole-2-carboxamido)-3-morpholinobenzoate (25a). To a solution of 4,5-dibromo-1H-pyrrole-2-carboxylic acid (0.500 g, 1.86 mmol) in anhydrous dichloromethane (20 mL) oxalyl chloride (0.80 mL, 9.30 mmol) was added and the mixture was stirred for 15 h at room temperature under an argon atmosphere. The solvent was evaporated under reduced pressure, anhydrous dichloromethane (15 mL), 16e (293 mg, 1.24 mmol) and pyridine (1.5 mL) were added, and the reaction mixture was stirred for 15 h under argon atmosphere at room temperature. The product was filtered off and dried to give 25a as a pale brown solid. Yield 84% (0.507 g); pale brown solid; mp 270–274 °C. 1H NMR (400 MHz, DMSO-d6): δ 13.13 (s, 1H, NH), 9.28 (s, 1H, CONH), 8.15 (d, J = 8.3 Hz, 1H, ArH), 7.71–7.80 (m, 2H, 2 × ArH), 7.15 (s, 1H, ArH), 3.85 (s, 3H, COOCH3), 3.75–3.82 (m, 4H, 2 × CH2), 2.82–2.90 (m, 4H, 2 × CH2) ppm.
4.2.2.47 4,5-Dibromo-N-(4-(hydrazinecarbonyl)-2-morpholinophenyl)-1H-pyrrole-2-carboxamide (26a). To a solution of 25a (485 mg, 1.00 mmol) in MeOH (15 mL) and THF (10 mL) in a pressure tube hydrazine monohydrate (3.40 mL, 69.8 mmol) was added and the reaction mixture was stirred at 120 °C for 15 h. The precipitate was filtered off and dried. The solvent of the mother liquor was evaporated under reduced pressure, MeOH (1 mL) was added to the crude residue, the undissolved solid was filtered off and combined with the precipitate from the first filtration. Yield 54% (263 mg); pale brown solid; mp 258–261 °C. 1H NMR (400 MHz, DMSO-d6): δ 13.08 (s, 1H, NH), 9.75 (s, 1H, NH), 9.22 (s, 1H, NH), 8.01 (d, J = 8.3 Hz, 1H, ArH), 7.68 (s, 1H, ArH), 7.61 (d, J = 8.5 Hz, 1H, ArH), 7.13 (s, 1H, ArH), 4.40–4.62 (m, 2H, NH2), 3.72–3.85 (m, 4H, 2 × CH2), 2.81–2.90 (m, 4H, 2 × CH2) ppm.
4.2.2.48 4,5-Dibromo-N-(2-morpholino-4-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)-1H-pyrrole-2-carboxamide (27a). To a solution of 26a (248 mg, 0.510 mmol) in 1,4-dioxane (10 mL) and DMF (20 mL) 1,1′-carbonyldiimidazole (336 mg, 2.07 mmol) was added, and the reaction mixture was stirred at 101 °C for 15 h. The solvent was evaporated under reduced pressure and the solid was washed successively with acetonitrile, water, and diethyl ether. The crude product was recrystallized from DMF and washed with THF to give 27a as a pale-yellow solid. Yield 51% (133 mg); pale yellow solid; mp >300 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.52 (br s, 2H, 2 × NH), 9.35 (s, 1H, NH), 8.17 (d, J = 8.3 Hz, 1H, ArH), 7.51–7.62 (m, 2H, 2 × ArH), 7.11 (s, 1H, ArH), 3.77–3.86 (m, 4H, 2 × CH2), 2.83–2.93 (m, 4H, 2 × CH2) ppm. 13C NMR (100 MHz, DMSO-d6): δ 157.4, 154.9, 154.1, 144.2, 135.4, 128.1, 123.0, 122.0, 120.4, 117.5, 114.1, 107.4, 99.1, 66.9, 52.0 ppm. HRMS for C17H16O4N5Br2 [M + H]+: calculated 511.95626, found 511.95636. HPLC (0–10 min, 10–90% ACN in 0.1% TFA, 10–11 min, 90% ACN in 0.1% TFA, Waters Acquity UPLC HSS C18 column: 1.8 μm, 2.1 × 50 mm): tR 4.900 min (95.01% at 254, 95.00% at 280 nm).
4.2.2.49 3,4-Dichloro-N,5-dimethyl-N-(2-morpholino-4-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)-1H-pyrrole-2-carboxamide (27b). To a solution of 26b (63.9 mg, 0.150 mmol) in 1,4-dioxane (3 mL) and DMF (5 mL) 1,1′-carbonyldiimidazole (73.0 g, 0.450 mmol) was added, and the reaction mixture was stirred at 101 °C for 15 h. The solvent was evaporated under reduced pressure and the solid was washed successively with acetonitrile, water, 1,4-dioxane, and methanol to give 27b as a pale-yellow solid. Yield 15% (10 mg); pale yellow solid; mp 257–259 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.52 (s, 1H, NH), 11.89 (s, 1H, NH), 7.49 (s, 2H, 2 × ArH), 7.32 (s, 1H, ArH), 3.68 (s, 4H, 2 × CH2), 3.37 (s, 3H, NCH3), 2.71–3.00 (m, 4H, 2 × CH2), 2.13 (s, 3H, CH3) ppm. 13C NMR (100 MHz, DMSO-d6): δ 162.4, 154.9, 153.8, 148.4, 140.6, 130.6, 127.6, 123.3, 121.4, 120.9, 117.1, 109.0, 107.2, 66.0, 51.5, 37.5, 11.1 ppm. HRMS for C19H18O4N5Cl2 [M − H]−: calculated 450.07394, found 450.07413. HPLC (0–10 min, 10–90% ACN in 0.1% TFA, 10–11 min, 90% ACN in 0.1% TFA, Waters Acquity UPLC HSS C18 column: 1.8 μm, 2.1 × 50 mm): tR 4.243 min (99.49% at 254, 99.82% at 280 nm).
4.2.2.50 Methyl 4-aminobenzoate (29). The solution of 28 (3.77 g, 20.8 mmol) in a mixture of MeOH and THF (100 mL) was stirred under argon for 15 min. Pd/C (0.913 g) was added, the solution was saturated with hydrogen and the reaction mixture was stirred for 3 h under hydrogen atmosphere. The catalyst was filtered off and the solvent was evaporated to give 29 (2.62 g) as a pale brown solid. Yield 2.62 g (83%); pale brown solid; mp 90–93 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.64 (d, 2H, J = 8.8 Hz, 2 × ArH), 6.57 (d, 2H, J = 8.8 Hz, 2 × ArH), 5.85 (s, 2H, NH2), 3.73 (s, 3H, CH3) ppm. IR (ATR): ν 3407, 3330, 3226, 3044, 2988, 2944, 2845, 1681, 1635, 1596, 1574, 1513, 1433, 1313, 1283, 1199, 1175, 1117, 1078, 973, 851, 842, 767, 697, 638 cm−1.
4.2.2.51 Methyl 4-(benzylamino)benzoate (30). To the solution of 29 (2.07 g, 13.7 mmol) in CH2Cl2 (70 mL) benzaldehyde (3.9 mL, 38.9 mmol) was added and the mixture was stirred at rt for 10 min, Na2SO4 (7.76 g, 54.6 mmol) was then added and the mixture was stirred at reflux for 15 h. The mixture was cooled to rt, filtered and the solvent was removed under reduced pressure. The residue was dissolved in MeOH and cooled on an ice bath, NaBH4 (1.03 g, 27.3 mmol, 2 eq.) was then added in small portions and the reaction mixture was stirred at rt for 15 h. A few mL of water were added and the solvent was removed under reduced pressure. The residue was dissolved in EtOAc (160 mL) and water (80 mL), the phases were separated and the organic phase was washed with brine (2 × 80 mL), dried over Na2SO4, filtered and the solvent evaporated. The crude product was purified with flash column chromatography using dichloromethane as the eluent. The fractions containing the product were combined, the solvent was removed under reduced pressure, the solid residue was triturated with hexane and the undissolved solid was filtered off and dried to give 30 (0.959 g) as a white solid. Yield 0.959 g (29%); white solid; mp 107–109 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.66 (d, 2H, J = 8.8 Hz, 2 × ArH), 7.29–7.39 (m, 4H, 4 × ArH), 7.20–7.28 (m, 1H, ArH), 7.14 (t, 1H, J = 6.0 Hz, NH), 6.61 (d, 2H, J = 8.8 Hz, 2 × ArH), 4.34 (d, 2H, J = 6.0 Hz, CH2), 3.72 (s, 3H, CH3) ppm. IR (ATR): ν 3416, 3027, 3005, 2952, 1686, 1598, 1569, 1529, 1494, 1456, 1432, 1419, 1344, 1312, 1275, 1236, 1186, 1172, 1111, 1064, 1027, 1003, 962, 909, 834, 767, 738, 700, 644, 618 cm−1.
4.2.2.52 Methyl 4-(N-benzyl-3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamido)benzoate (31). To a suspension of 3,4-dichloro-5-methyl-1H-pyrrole-2-carboxylic acid (0.775 g, 4.00 mmol) in anhydrous dichloromethane (55 mL), oxalyl chloride (1.39 mL, 16.0 mmol) was added dropwise and the reaction mixture was stirred at rt under an argon atmosphere for 15 h. The solvent was evaporated under reduced pressure, 30 (0.959 g, 3.97 mmol), anhydrous pyridine (8 mL) and anhydrous CH2Cl2 (30 mL) were added and the reaction mixture was stirred at rt under an argon atmosphere overnight. The solvent was removed in vacuo, and to the residue EtOAc (55 mL) and water (45 mL) were added. The phases were separated, the organic phase was washed with 1 M HCl (35 mL), saturated NaHCO3 solution (35 mL) and brine (2 × 20 mL), dried over Na2SO4, filtered and the solvent removed in vacuo. The crude product was purified with flash column chromatography using EtOAc/hexane (1
:7 to 1:3) as eluent to give 31 (0.925 g) as a brown solid. Yield 0.925 g (56%); brown solid; mp 135–140 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.19 (s, 1H, NH), 7.79 (d, J = 8.7 Hz, 2H, 2 × ArH), 7.19–7.31 (m, 5H, 5 × ArH), 7.19 (d, J = 8.7 Hz, 2H, 2 × ArH), 5.16 (s, 2H, CH2), 3.79 (s, 3H, COOCH3), 2.15 (s, 3H, pyrrole-CH3) ppm. IR (ATR): ν 3265, 3140, 3034, 2950, 1709, 1626, 1600, 1575, 1509, 1483, 1456, 1436, 1412, 1376, 1356, 1314, 1280, 1217, 1180, 1117, 1100, 1067, 1016, 977, 861, 764, 749, 733, 701, 669, 640, 613 cm−1. HRMS for C21H19O3N2Cl2 ([M + H]+): calculated 417.07672, found 417.07615.
4.2.2.53 N-Benzyl-3,4-dichloro-N-(4-(hydrazinecarbonyl)phenyl)-5-methyl-1H-pyrrole-2-carboxamide (32). To the solution of 31 (308 mg, 0.738 mmol) in a mixture of MeOH (10 mL) and THF (6 mL) in a high-pressure tube hydrazine hydrate (64%, 1.80 mL, 36.9 mmol) was added. The tube was sealed and the reaction mixture was stirred at 100 °C for 2 d. The tube was cooled to rt, the solvent was removed under reduced pressure and to the residue water was added to obtain a white suspension. The solid was filtered off and dried. The crude product was triturated with acetonitrile and the undissolved solid was filtered off and dried to give the first portion of 32 (145 mg). The filtrate was concentrated under reduced pressure and the residue was purified with flash column chromatography using CH2Cl2/MeOH (50
:1 to 25:1) as eluent to obtain the second part of 32 (125 mg). Both fractions of the product were combined. Yield 88% (270 mg); white solid; mp 144–147 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.07 (s, 1H, pyrrole-NH), 9.70 (s, 1H, NH), 7.66 (d, J = 8.7 Hz, 2H, 2 × ArH), 7.20–7.32 (m, 5H, 5 × ArH), 7.12 (d, 2H, J = 8.6 Hz, 2 × ArH), 5.14 (s, 2H, CH2), 4.47 (s, 2H, NH2), 2.14 (s, 3H, pyrrole-CH3) ppm. 13C NMR (101 MHz, DMSO-d6): δ 165.4, 161.9, 145.3, 137.7, 130.6, 128.8, 128.2, 128.1, 128.1, 127.8, 127.6, 126.0, 120.6, 110.2, 107.7, 52.8, 11.3 ppm. IR (ATR): ν 3313, 3236, 2986, 2856, 1655, 1599, 1572, 1540, 1487, 1454, 1426, 1379, 1329, 1286, 1228, 1202, 1103, 1069, 1029, 978, 861, 697, 625 cm−1. HRMS for C20H19O2N4Cl2 ([M + H]+): calculated 417.08796, found 417.08769.
4.2.2.54 N-Benzyl-3,4-dichloro-5-methyl-N-(4-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)-1H-pyrrole-2-carboxamide (33). The solution of 32 (245 mg, 0.590 mmol) and CDI (352 mg, 2.17 mmol) in 1,4-dioxane (10 mL) was stirred for 15 h at 101 °C. The solvent was removed under reduced pressure and the residue was purified with flash column chromatography starting with dichloromethane and proceeding with dichloromethane/methanol (50
:1) as eluent to give 33 (43 mg) as a pale-yellow solid. Yield 43 mg (17%); pale yellow solid; mp 202–204 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.54 (s, 1H, NH), 12.13 (s, 1H, NH), 7.64 (d, J = 8.6 Hz, 2H, 2 × ArH), 7.19–7.32 (m, 7H, 7 × ArH), 5.15 (s, 2H, CH2), 2.14 (s, 3H, pyrrole-CH3) ppm. 13C NMR (101 MHz, DMSO-d6): δ 161.8, 154.9, 153.7, 145.6, 137.6, 128.9, 128.5, 128.1, 127.7, 126.9, 126.1, 121.4, 120.4, 110.3, 107.8, 52.8, 11.3 ppm. IR (ATR): ν 3202, 2840, 1769, 1597, 1578, 1510, 1481, 1427 1407, 1381, 1351, 1275, 1209, 1182, 1075, 1031, 955, 925, 841, 750, 696, 667, 645, 613 cm−1. HRMS for C21H17O3N4Cl2 ([M + H]+): calculated 443.06722, found 443.06693. HPLC: Waters Acquity UPLC BEH C18 (1,7 μm, 2.1 × 50 mm); mobile phase: 10–90% of acetonitrile in TFA (0.1%) in 10 min; flow rate 0.4 mL min−1; injection volume: 1.75 μL; tR: 5.163 min (95.23% at 254 nm, 95.38% at 280 nm).
4.2.2.55 Methyl 3-(benzyloxy)-4-nitrobenzoate (35). To a stirred suspension of methyl 3-hydroxy-4-nitrobenzoate (34, 500 mg, 2.53 mmol) and potassium carbonate (699 mg, 5.06 mmol) in acetonitrile (10 mL) benzyl bromide (0.30 mL, 2.53 mmol) was added and the mixture was stirred at 60 °C for 3 h. The solvent was removed under reduced pressure and to the residue ethyl acetate (20 mL) and water (20 mL) were added, and separated. The organic phase was washed with brine (2 × 20 mL), dried over Na2SO4, filtered and the solvent removed under reduced pressure to give 35 (620 mg) as a yellow solid. Yield 85% (620 mg); yellow solid; mp 90–93 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.03 (d, J = 8.4 Hz, Ar–H), 7.90 (d, J = 1.2 Hz, Ar–H), 7.70 (dd, J = 8.4, 1.2 Hz, Ar–H), 7.35–7.48 (m, 5H, 5 × Ar–H), 5.41 (s, 2H, CH2), 3.92 (s, 3H, COOCH3) ppm. 13C NMR (100 MHz, DMSO-d6): δ 165.2, 150.9, 142.9, 136.1, 134.7, 129.0, 128.7, 127.9, 125.7, 122.0, 116.2, 71.2, 53.4 ppm. IR (ATR): ν cm−1. MS (ESI) m/z = 310.1 ([M + Na]+).
4.2.2.56 3-(Benzyloxy)-4-nitrobenzohydrazide (36). To a solution of 35 (4.60 g, 16.3 mmol) in MeOH (100 mL) and THF (100 mL), hydrazine monohydrate 80% solution (7.95 mL, 163 mmol) was added. The reaction mixture was stirred at 65 °C for 15 h and the solvent was removed under reduced pressure. The residue was suspended in ethanol and the flask was placed in the refrigerator for 1 h. The precipitate was filtered off, suspended in water, the suspension was sonicated, the solid filtered off and dried to give 1.44 g of 36. The ethanol from the mother liquor of the previous filtration was removed and the residue was purified with flash column chromatography (dichloromethane/methanol 10/1) to give 2.09 g of 36. Yield 77% (3.54 g). 1H NMR (400 MHz, DMSO-d6): δ 10.06 (s, 1H, NH), 7.97 (d, J = 8.4 Hz, ArH), 7.83 (d, J = 1.5 Hz, ArH), 7.54 (dd, J = 8.4, 1.6 Hz, ArH), 7.40–7.48 (m, 4H, 4 × ArH), 7.34–7.38 (m, 1H, ArH), 5.37 (s, 2H, CH2), 4.63 (s, 2H, NH2) ppm. MS (ESI) m/z = 285.9 ([M − H]−).
4.2.2.57 5-(3-(Benzyloxy)-4-nitrophenyl)-1,3,4-oxadiazol-2(3H)-one (37). To a solution of compound 30 (3.50 g, 12.2 mmol) in 1,4-dioxane (175 mL) 1,1′-carbonyldiimidazole (2.96 g, 18.3 mmol) was added and the reaction mixture was stirred at 101 °C for 15 h. The solvent was removed under reduced pressure, the residue was suspended in methanol, the suspension was sonicated, heated and the solid filtered off to give 3.22 g of 37. The mother liquor was evaporated and purified with flash column chromatography (dichloromethane/methanol 30/1). The purified product was combined with the product after filtration. Yield 98% (3.74 g). 1H NMR (400 MHz, DMSO-d6): δ 12.91 (s, 1H, NH), 8.06 (d, J = 8.4 Hz, ArH), 7.73 (d, J = 1.6 Hz, ArH), 7.54 (dd, J = 8.4, 1.6 Hz, ArH), 7.33–7.50 (m, 5H, 5 × ArH), 5.43 (s, 2H, CH2) ppm. MS (ESI) m/z = 312.0 ([M − H]−).
4.2.2.58 5-(4-Amino-3-(benzyloxy)phenyl)-1,3,4-oxadiazol-2(3H)-one (38). Compound 37 (523 mg, 1.67 mmol) was suspended in acetic acid (25 mL), iron (932 mg, 16.7 mmol) was added and the reaction mixture was stirred at rt for 90 min. Water was added and the iron was filtered through Celite. The flask was placed on an ice bath for 1 h upon which the product in the mother liquor crystalized. The product was filtered off and dried. Yield 57% (269 mg). 1H NMR (400 MHz, DMSO-d6): δ 12.22 (s, 1H, NH), 7.51 (d, J = 7.0 Hz, 2 × Ar–H), 7.40 (s, 2H, 2 × Ar–H), 7.33 (s, 1H, ArH), 7.21 (d, J = 1.8 Hz, ArH), 7.16 (dd, J = 8.2, 1.8 Hz, ArH), 6.73 (d, J = 8.2 Hz, ArH), 5.54 (s, 2H, NH2), 5.18 (s, 2H, CH2) ppm. MS (ESI) m/z = 283.9 ([M + H]+).
4.2.2.59 N-(2-(Benzyloxy)-4-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)phenyl)-3,4-dichloro-5-methyl-1H-pyrrole-2-carboxamide (39). Synthesised according to General procedure B from 3,4-dichloro-5-methyl-1H-pyrrole-2-carboxylic acid (0.123 g, 0.71 mmol) and 38 (0.150 g, 0.53 mmol). During extraction, the product precipitated and was filtered off. The crude product was successively triturated with acetonitrile, methanol and diethyl ether and the undissolved solid was filtered off. The crude product was purified by crystallization from DMF to give 39 (0.113 g) as a white solid. Yield 47% (0.113 g); white solid; mp 293–295 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.59 (s, 1H, NH), 12.42 (s, 1H, NH), 9.20 (s, 1H, NH), 8.56 (d, J = 8.5 Hz, ArH), 7.51–7.62 (m, 3H, 3 × Ar–H), 7.42–7.49 (m, 4H, 4 × Ar–H), 5.30 (s, 2H, CH2), 2.20 (s, 3H, CH3) ppm. HRMS for C21H15O4N4Cl2: calculated 457.04758, found 457.04776. HPLC (30–90% ACN in 0.1% TFA in 10 min, UPLC): tR 5.430 min (98.87% at 254 nm, 98.66% at 280 nm).
4.3. Determination of inhibitory activities on DNA gyrase and topoisomerase IV
For the determination of inhibitory activities against DNA gyrase and topoisomerase IV from E. coli, S. aureus, A. baumannii and P. aeruginosa, assay kits from Inspiralis were used according to the previously described procedures.28 Seven different inhibitor concentrations were used to determine the IC50 values, which were subsequently calculated using GraphPad Prism 6.0 software. For the most important inhibitors, IC50 values were determined based on three independent measurements, and the final results reported are mean values.4.4. Determination of inhibitory activity on human DNA topoisomerase IIα
Assay kits from Inspiralis were used for the experiments according to established protocols.38 Seven different inhibitor concentrations were used to determine the IC50 values, which were subsequently calculated using GraphPad Prism 6.0 software. For the most important inhibitors, IC50 values were determined based on three independent measurements, and the final results reported are mean values.4.5. Determination of antibacterial activity
Clinical control strains of S. aureus ATCC 29213, S. aureus ATCC 43300 (MRSA), S. aureus ATCC 700699 (VISA), E. faecalis ATCC 29212, E. coli ATCC 25922, P. aeruginosa ATCC 27853, A. baumannii ATCC 17978, K. pneumoniae ATCC 10031 and E. spp. cloacae ATCC 13047 were obtained from the University of Szeged (Hungary). Clinical control strains of E. faecium ATCC 35667, A. baumannii ATCC 19606, K. pneumoniae ATCC 700603, and K. aerogenes ATCC 13408 were obtained from Microbiologics Inc. (St. Cloud, Minnesota, USA). The antimicrobial assays were performed using the broth microdilution method in 96-well plates. The procedure complied with the Clinical and Laboratory Standards Institute guidelines37 and was performed following established methodologies.31 For selected compounds, the MIC values were determined by dose–response experiments (reported values are from at least two independent experiments, each with three replicates per concentration).4.6. Determination of frequency of resistance
To assess the spontaneous frequency of resistance, approximately 1010 cells from stationary-phase MHBII broth cultures of S. aureus ATCC 29213 and K. pneumoniae ATCC 10031 (supplied from Microbiologics) were plated on antibiotic-containing plates according to a standard protocol.39 Prior to plating, bacteria were cultured overnight in MHBII medium at 37 °C with shaking at 250 rpm, harvested by centrifugation and washed once in an equal volume of PBS to remove all by-products of bacterial metabolism from the media. After washing, the bacterial suspension was concentrated to approximately 1012 CFU mL−1 and from this concentrated cell suspension (1 mL) approximately 1010 cells were plated onto each MHBII agarose plate. Agarose was used instead of agar to reduce drug adsorption and improve assay performance. Petri dishes (145 mm) were filled with 40 mL of MHBII agarose medium containing the selective antibiotic at concentrations of 2×, 4× and 8× MIC of the respective antibiotic. Each experiment was performed in at least 3 biological replicates. The plates were incubated at 37 °C for 72 hours. Simultaneously, the total CFU was determined by plating appropriate dilutions onto antibiotic-free MHBII agar plates. The resistance frequency for each strain was calculated by dividing the number of colonies formed after 72 hours of incubation at 37 °C by the initial viable cell count.4.7. Molecular modelling
Molecular docking calculations were performed using Schrödinger Release 2023–3 (Schrödinger, LLC, New York, NY, USA, 2023). The crystal structure of E. coli DNA gyrase B in complex with a 4,5-dibromopyrrolamide-based inhibitor (PDB entry: 4ZVI)27 was prepared using the Protein Preparation Wizard with the default settings. The receptor grid was calculated for the inhibitor-binding site and the designed compounds were docked using the Glide XP protocol as implemented in Schrödinger Release 2023-3 (Glide, Schrödinger, LLC, New York, NY, USA, 2023). The highest scored docking conformation was used for analysis and presentation.4.8. Thermodynamic solubility assay
Thermodynamic water solubility was evaluated by preparing saturated solutions of selected compounds in phosphate buffered saline (PBS) with a pH of 7.4 and subsequent quantification by UPLC analysis. First, excess amounts of the compounds (1–2 mg) were weighed into Eppendorf tubes and 500 μL PBS was added to each. The resulting suspensions were shaken at 100 rpm and 25 °C. After 24 hours, the suspensions were centrifuged at 18000 rpm, and the supernatant was diluted fivefold with water/acetonitrile (1/1) to prepare the samples for UPLC analyses. In addition, 10 mM DMSO stocks of the compound were diluted in water/acetonitrile (1/1) to prepare samples for calibration curves (seven concentration points for each compound). Samples were then analysed using the Thermo Scientific Dionex Ultimate 3000 Binary Rapid Separation liquid chromatography system (Thermo Fisher Scientific, USA) equipped with a Waters C18 Acquity UPLC HSS column (1.8 μm, 2.1 × 50 mm). The mobile phase consisted of 0.1% TFA in ultrapure water (A) and acetonitrile (B), with the gradient expressed as % of B: 0–1 min 10%, 1–4 min 10–90%, 4–5 min 90%. The flow rate was set to 0.4 mL min−1 with an injection volume of 10 μL. Data processing was performed using Chromeleon CDS software (Thermo Fisher Scientific, USA), and solubilities were calculated using Excel (Microsoft, USA).Data availability
The data supporting this article have been included as part of the ESI.† The data include minimum inhibitory concentrations of compounds 6–14, inhibitory activities of 23b against DNA gyrase from A. baumannii and P. aeruginosa, spontaneous frequency of resistance table, minimum inhibitory concentrations of 23a and 23c adapted lines, synthetic procedures and analytical data, and 1H and 13C NMR spectra of the representative compounds.Author contributions
All authors conceived the project and designed the experiments. A. E. C., F. F., M. P., P. P., D. B. T., Ž. S., M. D., M. S., J. D., C. D. C., P. É. S., L. D. performed the experiments. All authors analysed the data and contributed to manuscript preparation and revision.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Slovenian Research Agency (Grant No. P1-0208, J1-3021 and J1-3031), the Research Council of Finland (Grant No. 321551) and the following research grants in Hungary: National Laboratory of Biotechnology Grant 2022-2.1.1-NL-2022-00008 (C. P.) and National Research, Development and Innovation Office K146323 (C. P.). We thank Alessia Onali, Maria Elisabetta Uda, Barbara Herlah, Una Rašić and Nika Križaj for their help with chemical synthesis. We thank Heidi Mäkkylä and Heli Parviainen for their help with the antibacterial assays.References
- Antimicrobial resistance, https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance, accessed August 18, 2023.
- J. O'Neill, Review on Antimicrobial Resistance Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations, Review on Antimicrobial Resistance, London, 2014, https://amr-review.org/sites/default/files/AMRReviewPaper-Tacklingacrisisforthehealthandwealthofnations_1.pdf, accessed August 18, 2023 Search PubMed.
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. Kashef Hamadani, E. A. P. Kumaran, B. McManigal, S. Achalapong, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F.-X. Babin, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, J. A. Berkley, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, R. Clotaire Donatien, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Duong Bich, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, C. Garcia, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, Y. Hsia, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, A. W. J. Jenney, M. Khorana, S. Khusuwan, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, K. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, A. Manoharan, F. Marks, J. May, M. Mayxay, N. Mturi, T. Munera-Huertas, P. Musicha, L. A. Musila, M. M. Mussi-Pinhata, R. N. Naidu, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, T. J. Ochoa, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, P. Ounchanum, G. D. Pak, J. L. Paredes, A. Y. Peleg, C. Perrone, T. Phe, K. Phommasone, N. Plakkal, A. Ponce-De-Leon, M. Raad, T. Ramdin, S. Rattanavong, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, V. D. Rosenthal, K. E. Rudd, N. Russell, H. S. Sader, W. Saengchan, J. Schnall, J. A. G. Scott, S. Seekaew, M. Sharland, M. Shivamallappa, J. Sifuentes-Osornio, A. J. Simpson, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Tigoi, C. Turner, P. Turner, H. R. Van Doorn, S. Velaphi, A. Vongpradith, M. Vongsouvath, H. Vu, T. Walsh, J. L. Walson, S. Waner, T. Wangrangsimakul, P. Wannapinij, T. Wozniak, T. E. M. W. Young Sharma, K. C. Yu, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- N. A. Turner, B. K. Sharma-Kuinkel, S. A. Maskarinec, E. M. Eichenberger, P. P. Shah, M. Carugati, T. L. Holland and V. G. Fowler, Nat. Rev. Microbiol., 2019, 17, 203–218 CrossRef CAS PubMed.
- World Health Organization and the European Centre for Disease Prevention and Control, Antimicrobial Resistance Surveillance in Europe 2022–2020 Data, World Health Organization and the European Centre for Disease Prevention and Control, 2022 Search PubMed.
- F. Collin, S. Karkare and A. Maxwell, Appl. Microbiol. Biotechnol., 2011, 92, 479–497 CrossRef CAS PubMed.
- T. Tomasic and L. P. Masic, Curr. Top. Med. Chem., 2014, 14, 130–151 CrossRef CAS PubMed.
- A. Nyerges, T. Tomašič, M. Durcik, T. Revesz, P. Szili, G. Draskovits, F. Bogar, Ž. Skok, N. Zidar, J. Ilaš, A. Zega, D. Kikelj, L. Daruka, B. Kintses, B. Vasarhelyi, I. Foldesi, D. Kata, M. Welin, R. Kimbung, D. Focht, L. P. Mašič and C. Pal, PLoS Biol., 2020, 18, e3000819 CrossRef CAS PubMed.
- A. M. Emmerson and A. M. Jones, J. Antimicrob. Chemother., 2003, 51, 13–20 CrossRef CAS PubMed.
- I. Laponogov, M. K. Sohi, D. A. Veselkov, X.-S. Pan, R. Sawhney, A. W. Thompson, K. E. McAuley, L. M. Fisher and M. R. Sanderson, Nat. Struct. Mol. Biol., 2009, 16, 667–669 CrossRef CAS.
- C. Alves, D. Mendes and F. B. Marques, Eur. J. Clin. Pharmacol., 2019, 75, 1431–1443 CrossRef CAS.
- European Medicines Agency, Fluoroquinolone and quinolone antibiotics: PRAC recommends restrictions on use, https://www.ema.europa.eu/en/documents/referral/quinolone-fluoroquinolone-article-31-referral-prac-recommends-restrictions-use_en.pdf, accessed August 18, 2023.
- G. S. Bisacchi and J. I. Manchester, ACS Infect. Dis., 2015, 1, 4–41 CrossRef CAS PubMed.
- S. Y. Ho, W. Wang, F. M. Ng, Y. X. Wong, Z. Y. Poh, S. W. E. Tan, S. H. Ang, S. S. Liew, Y. S. Joyner Wong, Y. Tan, A. Poulsen, V. Pendharkar, K. Sangthongpitag, J. Manchester, G. Basarab, J. Hill, T. H. Keller and J. Cherian, Eur. J. Med. Chem., 2018, 157, 610–621 CrossRef CAS PubMed.
- L. W. Tari, X. Li, M. Trzoss, D. C. Bensen, Z. Chen, T. Lam, J. Zhang, S. J. Lee, G. Hough, D. Phillipson, S. Akers-Rodriguez, M. L. Cunningham, B. P. Kwan, K. J. Nelson, A. Castellano, J. B. Locke, V. Brown-Driver, T. M. Murphy, V. S. Ong, C. M. Pillar, D. L. Shinabarger, J. Nix, F. C. Lightstone, S. E. Wong, T. B. Nguyen, K. J. Shaw and J. Finn, PLoS One, 2013, 8, e84409 CrossRef PubMed.
- A.-L. Grillot, A. L. Tiran, D. Shannon, E. Krueger, Y. Liao, H. O'Dowd, Q. Tang, S. Ronkin, T. Wang, N. Waal, P. Li, D. Lauffer, E. Sizensky, J. Tanoury, E. Perola, T. H. Grossman, T. Doyle, B. Hanzelka, S. Jones, V. Dixit, N. Ewing, S. Liao, B. Boucher, M. Jacobs, Y. Bennani and P. S. Charifson, J. Med. Chem., 2014, 57, 8792–8816 CrossRef CAS PubMed.
- T. Tomašič, S. Katsamakas, Ž. Hodnik, J. Ilaš, M. Brvar, T. Solmajer, S. Montalvão, P. Tammela, M. Banjanac, G. Ergović, M. Anderluh, L. P. Mašič and D. Kikelj, J. Med. Chem., 2015, 58, 5501–5521 CrossRef PubMed.
- M. Gjorgjieva, T. Tomašič, M. Barančokova, S. Katsamakas, J. Ilaš, P. Tammela, L. Peterlin Mašič and D. Kikelj, J. Med. Chem., 2016, 59, 8941–8954 CrossRef CAS PubMed.
- A. E. Cotman, M. Durcik, D. B. Tiz, F. Fulgheri, D. Secci, M. Sterle, S. Mozina, Z. Skok, N. Zidar, A. Zega, J. Ilas, L. P. Masic, T. Tomasic, D. Hughes, D. L. Huseby, S. Cao, L. Garoff, T. B. Fernandez, P. Giachou, L. Crone, I. Simoff, R. Svensson, B. Birnir, S. V. Korol, Z. Jin, F. Vicente, M. C. Ramos, M. de la Cruz, B. Glinghammar, L. Lenhammar, S. R. Henderson, J. E. A. Mundy, A. Maxwell, C. E. M. Stevenson, D. M. Lawson, G. V. Janssen, G. J. Sterk and D. Kikelj, J. Med. Chem., 2023, 66, 1380–1425 CrossRef CAS PubMed.
- M. Durcik, A. E. Cotman, Z. Toplak, S. Mozina, Z. Skok, P. E. Szili, M. Czikkely, E. Maharramov, T. H. Vu, M. V. Piras, N. Zidar, J. Ilas, A. Zega, J. Trontelj, L. A. Pardo, D. Hughes, D. Huseby, T. Berruga-Fernandez, S. Cao, I. Simoff, R. Svensson, S. V. Korol, Z. Jin, F. Vicente, M. C. Ramos, J. E. A. Mundy, A. Maxwell, C. E. M. Stevenson, D. M. Lawson, B. Glinghammar, E. Sjostrom, M. Bohlin, J. Oreskar, S. Alver, G. V. Janssen, G. J. Sterk, D. Kikelj, C. Pal, T. Tomasic and L. Peterlin Masic, J. Med. Chem., 2023, 66, 3968–3994 CrossRef CAS PubMed.
- M. Durcik, A. Nyerges, Z. Skok, D. G. Skledar, J. Trontelj, N. Zidar, J. Ilas, A. Zega, C. D. Cruz, P. Tammela, M. Welin, Y. R. Kimbung, D. Focht, O. Benek, T. Revesz, G. Draskovits, P. E. Szili, L. Daruka, C. Pal, D. Kikelj, L. P. Masic and T. Tomasic, Eur. J. Med. Chem., 2021, 213, 113200 CrossRef CAS PubMed.
- M. Sterle, M. Durcik, C. E. M. Stevenson, S. R. Henderson, P. E. Szili, M. Czikkely, D. M. Lawson, A. Maxwell, D. Cahard, D. Kikelj, N. Zidar, C. Pal, L. P. Masic, J. Ilas, T. Tomasic, A. E. Cotman and A. Zega, ACS Omega, 2023, 8, 24387–24395 CrossRef CAS PubMed.
- J. B. Cross, J. Zhang, Q. Yang, M. F. Mesleh, J. A. C. Romero, B. Wang, D. Bevan, K. M. Poutsiaka, F. Epie, T. Moy, A. Daniel, J. Shotwell, B. Chamberlain, N. Carter, O. Andersen, J. Barker, M. D. Ryan, C. A. Metcalf III, J. Silverman, K. Nguyen, B. Lippa and R. E. Dolle, ACS Med. Chem. Lett., 2016, 7, 374–378 CrossRef CAS PubMed.
- G. S. Basarab, P. J. Hill, C. E. Garner, K. Hull, O. Green, B. A. Sherer, P. B. Dangel, J. I. Manchester, S. Bist, S. Hauck, F. Zhou, M. Uria-Nickelsen, R. Illingworth, R. Alm, M. Rooney and A. E. Eakin, J. Med. Chem., 2014, 57, 6060–6082 CrossRef CAS PubMed.
- K. Talley Angela, A. Thurston, G. Moore, K. Gupta Vipul, M. Satterfield, E. Manyak, S. Stokes, A. Dane and D. Melnick, Antimicrob. Agents Chemother., 2021, 65(11), e01208 Search PubMed.
- A. G. Vandell, S. Inoue, J. Dennie, Y. Nagasawa, R. Gajee, J. Pav, G. Zhang, C. Zamora, N. Masuda and G. Senaldi, Antimicrob. Agents Chemother., 2018, 62(5), e02537 CrossRef CAS PubMed.
- N. Zidar, H. Macut, T. Tomašič, M. Brvar, S. Montalvão, P. Tammela, T. Solmajer, L. Peterlin Mašič, J. Ilaš and D. Kikelj, J. Med. Chem., 2015, 58, 6179–6194 CrossRef CAS PubMed.
- M. Durcik, P. Tammela, M. Barancokova, T. Tomasic, J. Ilas, D. Kikelj and N. Zidar, ChemMedChem, 2018, 13, 186–198 CrossRef CAS PubMed.
- M. Durcik, D. Lovison, Z. Skok, C. Durante Cruz, P. Tammela, T. Tomasic, D. Benedetto Tiz, G. Draskovits, A. Nyerges, C. Pal, J. Ilas, L. Peterlin Masic, D. Kikelj and N. Zidar, Eur. J. Med. Chem., 2018, 154, 117–132 CrossRef CAS PubMed.
- N. Zidar, H. Macut, T. Tomašič, L. Peterlin Mašič, J. Ilaš, A. Zega, P. Tammela and D. Kikelj, Medchemcomm, 2019, 10, 1007–1017 RSC.
- N. Zidar, T. Tomasic, H. Macut, A. Sirc, M. Brvar, S. Montalvao, P. Tammela, J. Ilas and D. Kikelj, Eur. J. Med. Chem., 2016, 117, 197–211 CrossRef CAS PubMed.
- D. B. Tiz, Z. Skok, M. Durcik, T. Tomasic, L. P. Masic, J. Ilas, A. Zega, G. Draskovits, T. Revesz, A. Nyerges, C. Pal, C. D. Cruz, P. Tammela, D. Zigon, D. Kikelj and N. Zidar, Eur. J. Med. Chem., 2019, 167, 269–290 CrossRef CAS PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 CrossRef CAS PubMed.
- M. F. Richter and P. J. Hergenrother, Ann. N. Y. Acad. Sci., 2019, 1435, 18–38 CrossRef CAS PubMed.
- D. Benedetto Tiz, D. Kikelj and N. Zidar, Expert Opin. Drug Discovery, 2018, 13, 497–507 CrossRef CAS PubMed.
- PyMOL, Delano Scientific LLC, San Francisco, CA, http://pymol.sourceforge.net.
- CLSI, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard – Ninth Edition. CLSI Document M07-A9, Wayne, PA, Clinical Standards Institute, 2012 Search PubMed.
- Ž. Skok, M. Durcik, D. Gramec Skledar, M. Barančoková, L. Peterlin Mašič, T. Tomašič, A. Zega, D. Kikelj, N. Zidar and J. Ilaš, Bioorg. Chem., 2020, 102, 104049 CrossRef PubMed.
- G. Bell and C. MacLean, Trends Microbiol., 2018, 26, 471–483 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04802d |
| ‡ Current address: Department of Life and Environmental Sciences, University of Cagliari, University Campus, S. P., Monserrato, 09042 Cagliari, Italy. |
| This journal is © The Royal Society of Chemistry 2024 |