
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Convenient syntheses of 2-acylamino-4-halothiazoles and acylated derivatives using a versatile Boc-intermediate†
Sophie Pate,
Joshua Taujanskas,
Robyn Wells,
Craig M. Robertson,
Paul M. O'Neill and
Andrew V. Stachulski *
*
Department of Chemistry, University of Liverpool, Liverpool L69 7ZD, UK. E-mail: stachuls@liv.ac.uk; Fax: +44-(0)151-794-3482; Tel: +44-(0)151-794-3482
First published on 2nd September 2024
Abstract
The 2-aminothiazole grouping is a significant feature of many series of biologically active molecules, including antibiotics, anticancer agents and NSAIDs. We have a longstanding interest in the synthesis and biological evaluation of thiazolides, viz. [2-hydroxyaroyl-N-(thiazol-2-yl)-amides] which have broad spectrum antiinfective, especially antiviral, properties. However, 2-amino-4-substituted thiazoles, especially 4-halo examples, are not easily available. We now report practical, efficient syntheses of this class from readily available pseudothiohydantoin, or 2-aminothiazol-4(5H)-one: the key intermediate was its Boc derivative, from which, under Appel-related conditions, Br, Cl and I could all be introduced at C(4). Whereas 2-amino-4-Br/4-Cl thiazoles gave low yields of mixed products on acylation, including a bis-acyl product, further acylation of the Boc intermediates, with a final mild deprotection step, afforded the desired thiazolides cleanly and in good yields. In contrast, even mild hydrolysis of 2-acetamido-4-chlorothiazole led to decomposition with fast reversion to 2-aminothiazol-4(5H)-one. We also present a correction of a claimed synthesis of 2-acetamido-4-chlorothiazole, which in fact produces its 5-chloro isomer.
Introduction
A 2-aminothiazole unit is a common feature of many biologically active molecular series,1 such as cephalosporin antibiotics, kinase inhibitor anticancer agents and non-steroidal antiinflammatory drugs, Fig. 1. It has been suggested that 2-aminothiazole substitution favourably affects both the activity profile and absorption properties.2,3 Although a thiazole unsubstituted at both C(4) and C(5) is regarded as a metabolic risk,4 this danger is readily averted by appropriate substitution, especially with electron-withdrawing substituents.5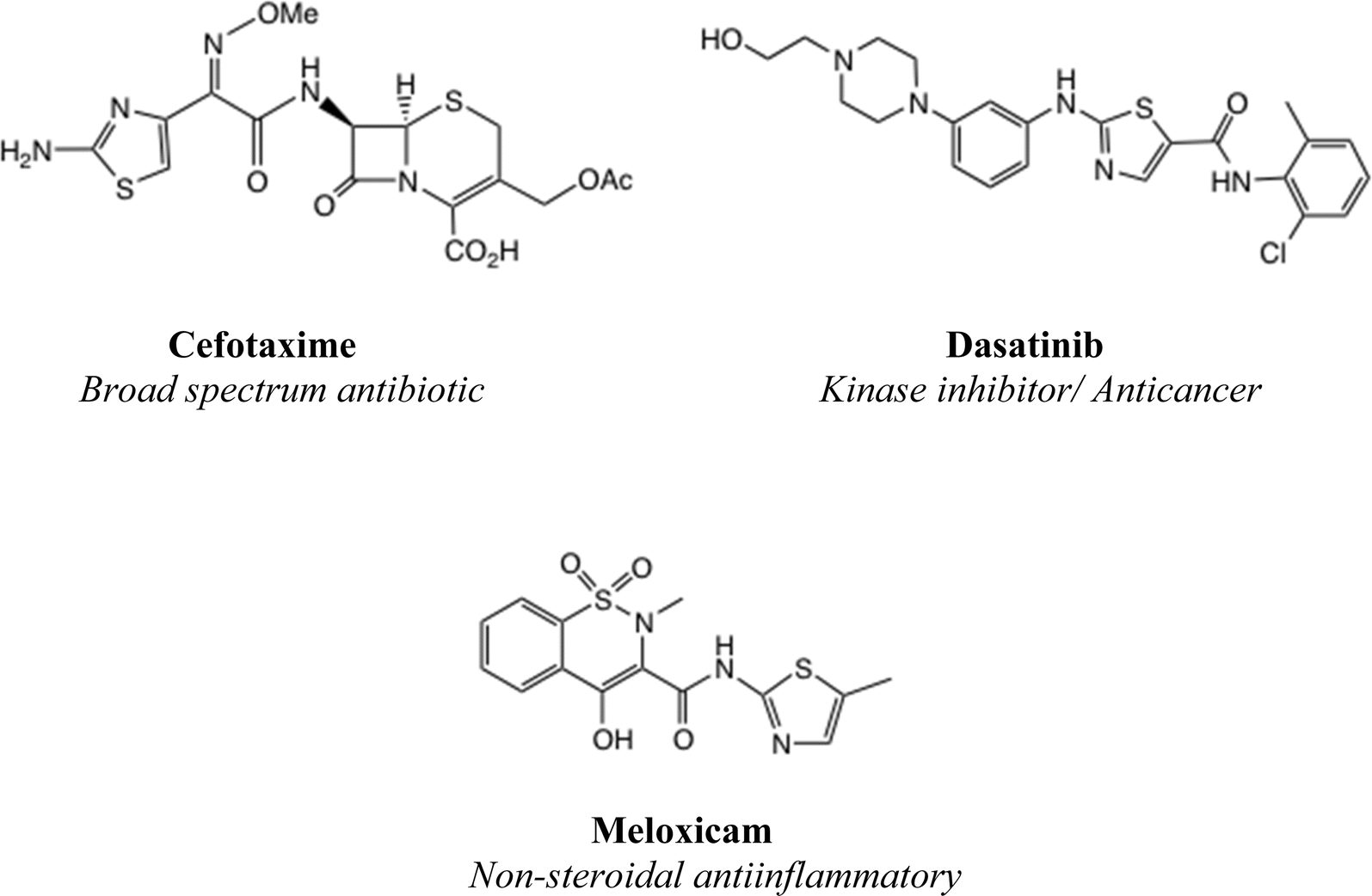 | ||
| Fig. 1 Examples of (2-aminothiazole) containing drugs. | ||
One important class of broad spectrum antiinfective 2-aminothiazole derivatives are the thiazolides, or [2-hydroxyaroyl-N-(thiazol-2-yl)-amides], typified by nitazoxanide 1a which was first reported in 1975 (Fig. 2).6 To this day 1a remains the antiparasitic agent of choice against Cryptosporidium spp.7 It was later discovered that 1a and other analogues, notably the 5-chloro analogue 1b, were broad-spectrum antiviral agents,8–10 dating from the use of 1a in treating cryptosporidiosis in AIDS patients.
 | ||
| Fig. 2 Thiazolide structures. | ||
We have described the structure–activity relationships (SAR) of a wide range of thiazolides against hepatitis B, hepatitis C and influenza A viruses.11–13 Against a typical H1N1 strain of influenza A virus, compound 1a shows IC50 = 3.3 μM and 1b shows IC50 = 3.4 μM.13b Clinical trials of 1a have been performed against rotavirus14 and acute uncomplicated influenza A.15a,b More recently, the SARS-CoV2 pandemic led to a strong resurgence of interest in small molecule antivirals, and NTZ has shown notable activity in trials against SARS-CoV2.16 The active circulating metabolites of 1a/1b in vivo are the free phenols 2a/2b, of which the phenolic acetates are prodrugs.17 Later we prepared more efficient, amino-acid ester prodrugs 3a/3b, which were shown to offer greatly improved bioavailability compared to 1a/1b.18
In general, 5-substituted thiazolides such as 1a/1b are the easiest to obtain. The natural position of electrophilic substitution of a 2-aminothiazole is at position 5, even when the 2-amine is acylated. In order to synthesise thiazolides with a 4-substituent, including 4-halo examples, various methods are possible: the 4-sulfonyl thiazolide 4 was synthesised from a thioester.13b
One approach to a 2-amino-4-bromothiazole uses the halogen dance rearrangement from a protected 5-Br thiazole, as originally described by Stangeland and Stanetty (Scheme 1),19,20 employing LiNPr2i in THF. The rearrangement of 5 to 6 is considered to proceed via the N, C(5)-dianion which is thermodynamically preferred (Scheme 1, lower). This proved a robust procedure, but on removal of the Boc group the free amine 7, Fig. 3, proved rather unstable and difficult to acylate, in contrast to 2-amino-5-bromothiazole.
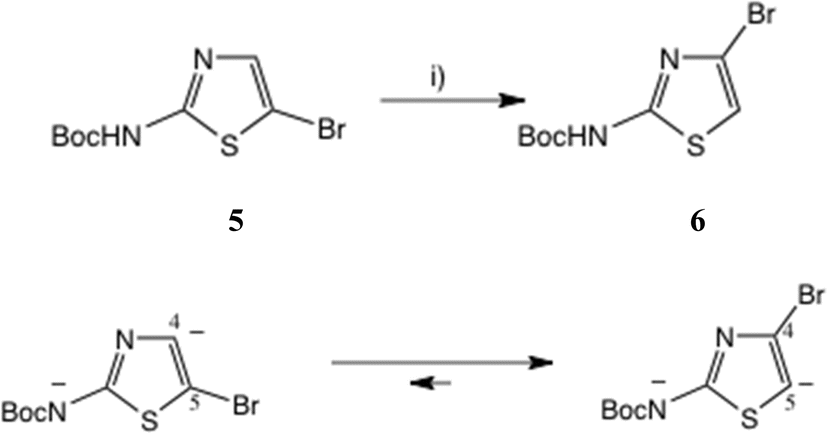 | ||
| Scheme 1 Synthesis of 2-Boc-amino, 4-bromothiazole by halogen rearrangement. Conditions: (i) LiNPr2i, THF, 0–10 °C, 20 min, 91%. | ||
 | ||
| Fig. 3 2-Amino-4-halothiazoles. | ||
The literature on 2-amino-4-chlorothiazole 8 is limited,21,22 and here again, though we were able to reproduce one synthesis of this material in very low yield,21 we found 8 was unstable as the free base and difficult to acylate, giving mixed products.
The acidity of the amide NH in thiazolides such as 1a and 1b suggested an alternative route to 4′-substituted thiazolides, viz. further acylation of N-protected versions of 7 and 8, followed by mild deprotection. We now report that t-butyl (4-oxo-4,5-dihydrothiazol-2-yl)carbamate is an ideal, versatile precursor for such derivatives.
Discussion
Acylation of 2-amino-4-bromo and 2-amino-4-chlorothiazole
Treatment of Boc derivative 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 with TFA in CH2Cl2, followed by basification with NaHCO3 and extraction, afforded the free amine 7 in 94% yield, which proved rather unstable on storage and was used immediately, Scheme 2. Reaction of 7 with O-acetylsalicyloyl chloride 9 using two-phase acylation conditions11,12 was quite unsuccessful. Instead, anhydrous acylation in THF using Et3N as base gave a slow, complex reaction. Workup after 46 h at 20 °C gave two major products by chromatography, from which the desired thiazolide 10 was isolated in 17% yield and purified by recrystallisation. A major byproduct was apparently the acetamide 11 (m/z 221, 223) though this was difficult to purify fully.
20 with TFA in CH2Cl2, followed by basification with NaHCO3 and extraction, afforded the free amine 7 in 94% yield, which proved rather unstable on storage and was used immediately, Scheme 2. Reaction of 7 with O-acetylsalicyloyl chloride 9 using two-phase acylation conditions11,12 was quite unsuccessful. Instead, anhydrous acylation in THF using Et3N as base gave a slow, complex reaction. Workup after 46 h at 20 °C gave two major products by chromatography, from which the desired thiazolide 10 was isolated in 17% yield and purified by recrystallisation. A major byproduct was apparently the acetamide 11 (m/z 221, 223) though this was difficult to purify fully.
 | ||
| Scheme 2 Acylation of 2-amino-4-bromo and 2-amino-4-chlorothiazole. Conditions: (i) THF, Et3N, 0 °C–20 °C. | ||
Similarly, anhydrous acylation of 2-amino-4-chlorothiazole 821 with 9 again gave a slow complex reaction. By chromatography, the desired thiazolide 12 was obtained in 19% yield and further purified by recrystallisation. A significant more polar product proved to be a bis-acylated derivative 13, which interestingly possessed a bis-acylamino rather than a tautomeric acylimino structure, as shown by single crystal X-ray analysis, Fig. 4. We therefore turned to alternative 2-aminothiazole intermediates.
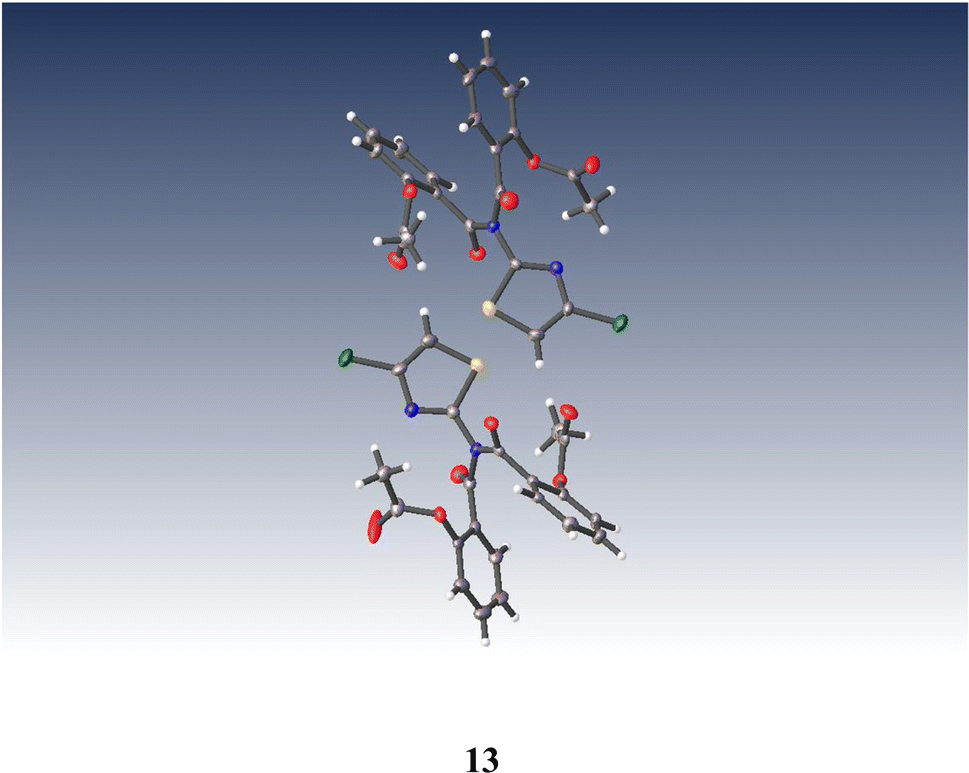 | ||
| Fig. 4 Single crystal X-ray structure of (((4-chlorothiazol-2-yl)azanediyl)bis(carbonyl))bis(2,1-phenylene) diacetate 13. | ||
Protected forms of pseudothiohydantoin
21 gave a very low yield of 2-amino-4-chlorothiazole 8.We therefore studied N-protected versions of 14, aiming first at the acetamide, Scheme 3. Heating 14 with Ac2O/AcOH24 led to a very slow reaction, even at 100–105 °C, so we switched to amine bases. Treatment of 14 with Ac2O and DMAP in THF at 20 °C gave a steady reaction and delivered very largely the previously unknown N, O-diacetate 15 in high yield; its structure was confirmed by a single crystal X-ray determination, Fig. 5, since other tautomeric products were possible. The use of Et3N gave a mixture of products including 15 and the desired monoacetamide.
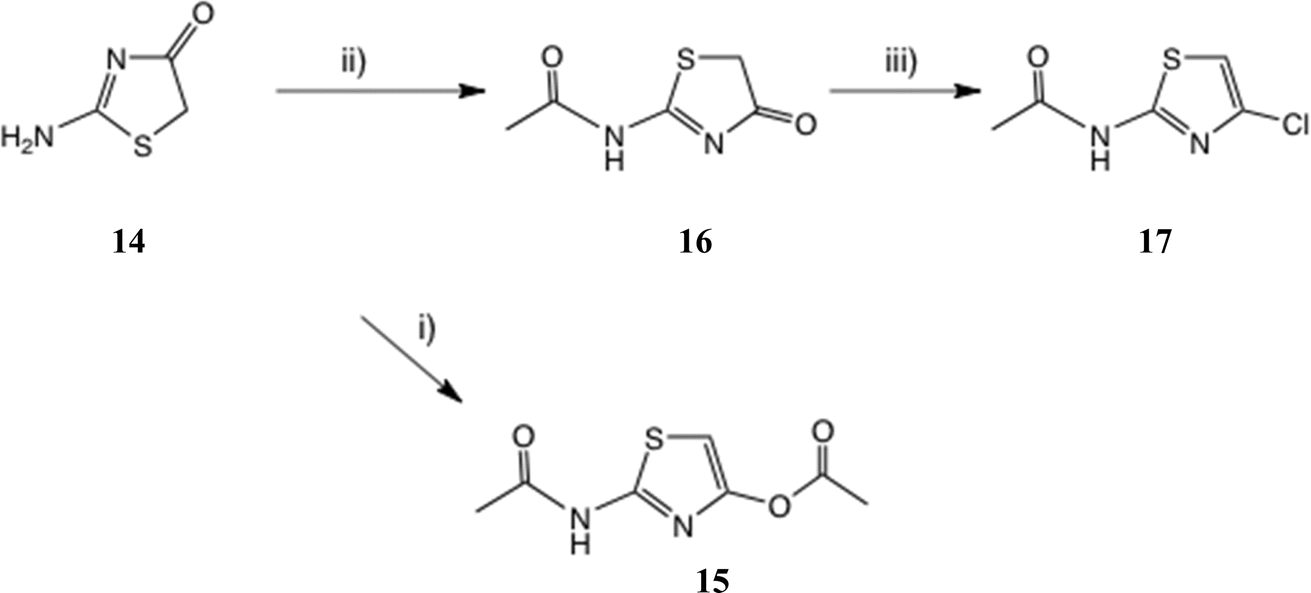 | ||
| Scheme 3 (2-Acetamido)thiazole intermediates. Conditions: (i) Ac2O, DMAP, THF, 0–20 °C, 22 h, 81%; (ii) Ac2O, N–Me morpholine, THF, 60 °C, 1.5 h, 83%; (iii) POCl3, MeCN, 50 °C, 3 h, 57% or NCS, Ph3P, MeCN, 20 °C, 5 h, 72%. | ||
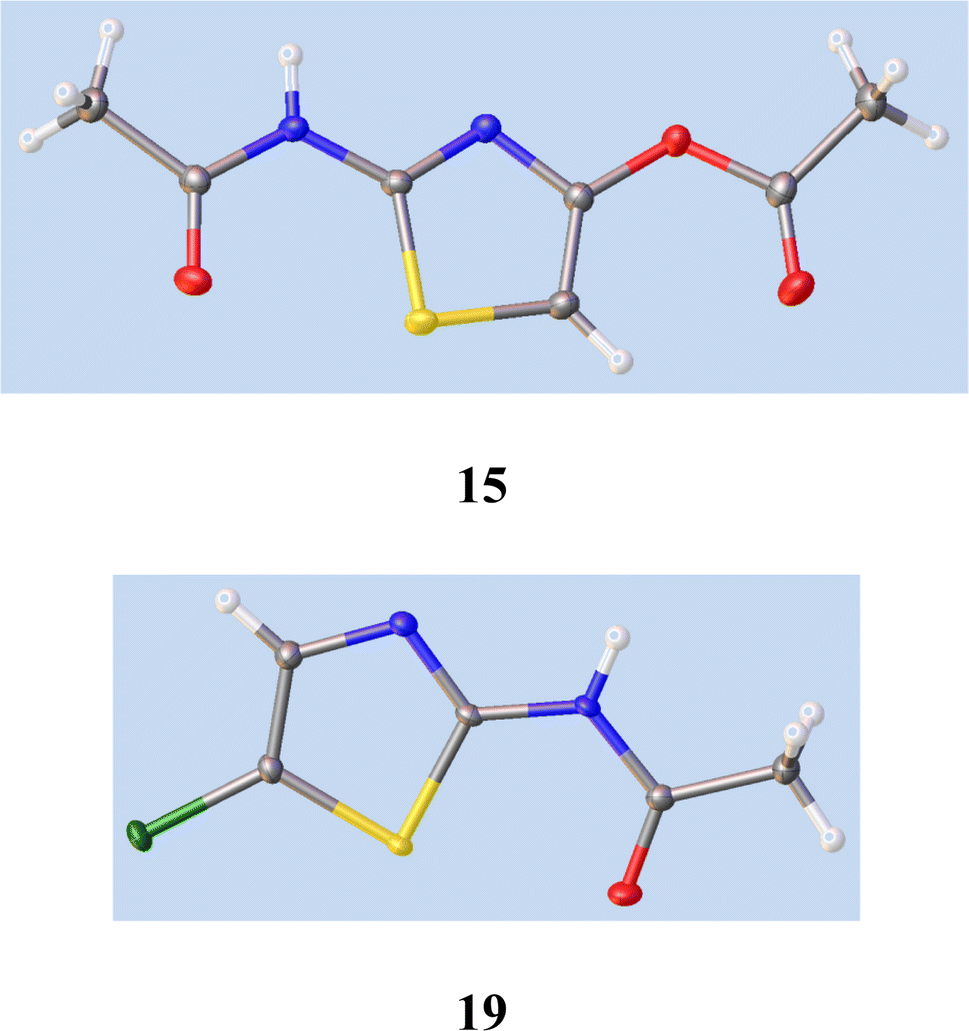 | ||
| Fig. 5 Single crystal X-ray structures of 15 and 19. See ESI† for cif file data. The syn-orientation of the S atom and carbonyl oxygen in both cases results from nonbonding overlap between the C–S σ* orbital and O lone pair electrons.26 | ||
Use of the weaker base N-methylmorpholine at 60 °C gave a controlled reaction, which generated the desired acetamide 16 in very good yield with negligible diacetylation. Treatment of 16 with POCl3 at 50 °C gave a very slow reaction until catalytic DMF was added; the 4-Cl compound 1725a was then isolated in satisfactory yield. The same product was obtained in 72% yield by reaction of 16 with Ph3P and N-chlorosuccinimide (NCS) (1.5 eq. each; cf. next section) in MeCN at 20 °C. Another route claims chlorination of 2-acetamidothiazole using ‘green’ conditions, viz. NaCl and oxone,25b but it is not clear whether the 4-Cl isomer 17 is the product since these authors' NMR data look significantly different from ours. The reaction of 2-aminothiazole with 1-chloro-1,2-benziodoxol-3-one22 was also stated to afford 17.
To seek reassurance on the regiochemical point, we studied the direct chlorination of 2-acetamidothiazole 18 with NCS in MeCN, Scheme 4. We used a similar procedure once before on a thiazolide.11 In fact this chlorination proceeded smoothly, using mild acid catalysis with Amberlyst A-15 (H+) resin, and the product, isolated in unoptimised 65% yield, was shown to be the 5-Cl isomer 19 by a single crystal X-ray determination, Fig. 5. The 1H and 13C NMR data of this material were identical with those reported25b and claimed to be the 4-Cl isomer.
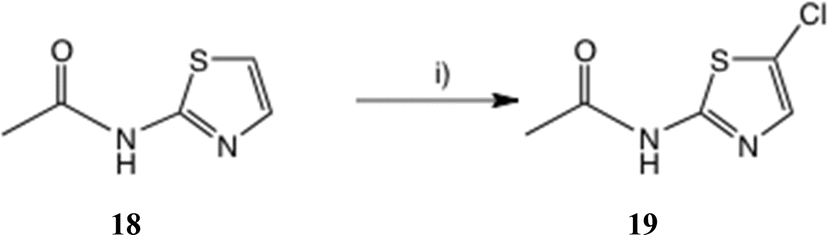 | ||
| Scheme 4 Synthesis of 2-acetamido-5-chlorothiazole. Conditions: (i) NCS, Amberlite A-15 (H+), 20 °C, 22 h, 65%. | ||
Under relatively mild conditions (HCl, aq. MeOH, 50 °C) we found that hydrolysis of 17 gave rapid decomposition with reversion to 14. This probably resulted from ring protonation at C(5) followed by attack of water at C(4).
 | ||
| Scheme 5 Conversion of pseudothiohydantoin to 2-Boc-amino-4-halothiazoles. Conditions: (i) Boc2O, aq. THF, pH10, 86%; (ii) Ph3P, Cl3CCN, CH2Cl2, 73%; (iii) Ph3P, NBS, MeCN, 63%; (iv) Ph3P, NIS, MeCN, 28%. Alternative conditions discussed in text. | ||
We anticipated that 20 would be readily enolised; hence reagents for the chlorination of other tautomeric hydroxy heterocycles such as 2-hydroxypyridine/2-pyridone under Appel-type conditions28 should be effective. More recently, variants of the original Appel method using catalytic Ph3PO29 and a sustainable procedure avoiding chlorinated solvents30 have been described. Here, Ph3P in conjunction with CCl431 (or preformed Ph3PCl232), N-chlorosuccinimide33 or trichloroacetonitrile34 all converted 20 into 21. THF, CH2Cl2 and MeCN were all adequate solvents; the best and mildest conditions proved to be Ph3P and Cl3C·CN in CH2Cl2 at 20 °C, affording 21 in 73% yield.
For the introduction of Br at C(4), as noted earlier, the Br rearrangement (‘halogen dance’)20a,b is feasible: the substrate (Scheme 1) is prepared from 2-amino-5-bromothiazole.35 Here too, however, 20 proved a highly suitable intermediate, and on treatment with Ph3P and N-bromosuccinimide33,34b 6 was readily obtained.36 Here the solvent choice was significant, with MeCN definitely superior to CH2Cl2, giving 6 in 63% yield. Another good reagent proved to be ethyl tribromoacetate,34b,37 again employing MeCN, which gave a virtually identical yield, though here purification was more difficult. It is noteworthy that MeCN often proves a superior solvent in the Appel-type halogenation reaction38 and may even divert the reaction to other products.39
2-Boc-amino-4-iodothiazole 22 was disclosed in a patent40 as a useful intermediate for Suzuki couplings, but with no preparative detail. We obtained this compound in an unoptimised 28% yield by treatment of 20 with Ph3P and N-iodosuccinimide at 0–20 °C; a little free I2 was used to initiate the reaction.41
In Scheme 6 we give a mechanism for these halogenations, using Cl3C·CN as the example donor, generating 21.
 | ||
| Scheme 6 Proposed halogenation mechanism with Cl3C·CN. | ||
We also studied the reactions of both 16 and 20 with Middleton's DAST reagent,42 hoping to gain access to 4-fluoro derivatives: currently there is no reported preparation of 2-amino-4-fluorothiazole. Neither gave useful products; the reaction of 16 gave a low yield of a complex mixture and 20 gave rapid loss of the Boc group. A 4-fluorothiazole bearing a 5-formyl substituent was recently obtained23 by displacement from a 4-chlorothiazole using SNAr reaction with anhydrous Me4N+ F−, Scheme 7.
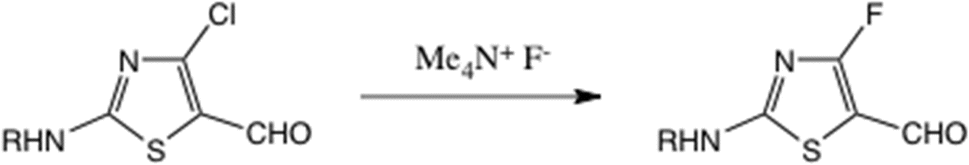 | ||
Scheme 7 Synthesis of a 4-fluorothiazole by nucleophilic halogen substitution; R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ac or Boc. Ac or Boc. | ||
Acylation of Boc intermediates and thiazolide synthesis
The NH of compounds such as 21 is considerably more acidic than a typical amide43 or even acetanilide (pKa = 13),44 and our previous experience had indeed shown that further N-acylation was possible. Using Et3N as base, acylation of 21 with O-acetylsalicyloyl chloride cleanly afforded a 70% yield of the Boc intermediate 23 (Scheme 8). Mild acidolysis (dilute CF3CO2H, CH2Cl2) then delivered thiazolide 12 in near quantitative yield, identical to the product obtained in low yield by acylation of 8, Scheme 2.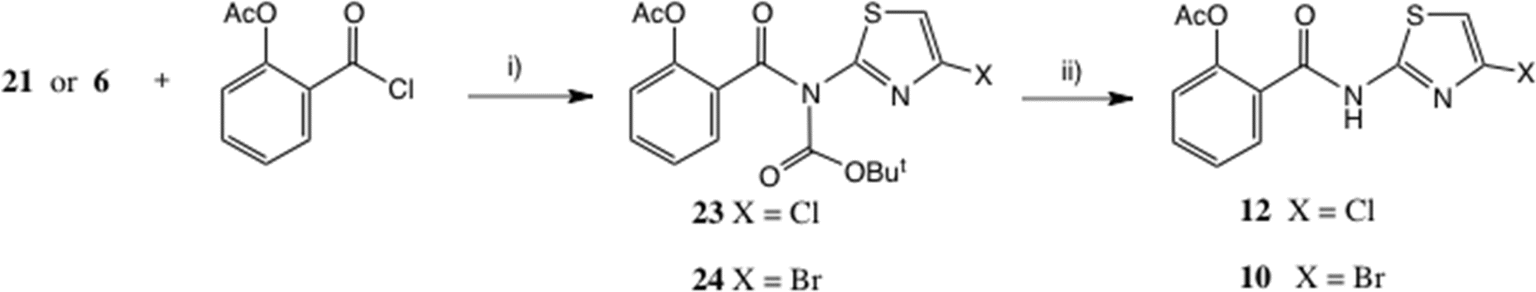 | ||
| Scheme 8 Thiazolide synthesis. Conditions: (i) Et3N, THF, 61% for 23, 70% for 24; (ii) dil. CF3CO2H, CH2Cl2, 97% for 12, 65% for 10. | ||
This sequence was equally applicable to the bromo intermediate 6, which via intermediate 24 gave 10, cf. Scheme 2. Clearly this sequence represents the method of choice for the synthesis of 10 and 12.
Conclusions
N-Boc protected forms of 2-amino-4-halothiazoles are readily available from Boc-pseudothiohydantoin, which is itself available from pseudothiohydantoin in high yield. The tendency of the heterocycle to exhibit tautomeric behaviour and to overreact with electrophiles is thus avoided. In general, N-halosuccinimides in conjunction with Ph3P under Appel-type conditions are effective reagents for the halogenation step, but Cl3CCN proved optimal for chlorination. Further acylation of these intermediates with O-acetylsalicyloyl chloride, followed by mild deprotection, offers high-yielding syntheses of 4-bromo and 4-chlorothiazolides. The relatively high acidity of amide NHs in derivatives such as 21 is significant: this bis-acylation/mild deprotection sequence may well offer good alternative syntheses for other heterocyclic amides. Direct acylation of the corresponding free amines 7 and 8, by contrast, gave low yields of mixed products.Experimental
General experimental procedures
Organic extracts were finally washed with saturated brine and dried over anhydrous Na2SO4 prior to rotary evaporation at <30 °C. Moisture sensitive reactions were carried out in anhydrous organic solvents (purchased from Sigma-Aldrich) under a N2 or Ar atmosphere. Reactions were monitored by analytical thin-layer chromatography using Merck Kieselgel 60 F254 silica plates, and were viewed under UV or by staining with KMnO4 or iodine. Preparative flash column chromatography was performed on either VWR Prolabo silica gel or Sigma-Aldrich silica gel (particle size 40–63 Å). Melting points were recorded using a Bibby-Sterlin Stuart SMP3 melting point apparatus and are uncorrected. Mass spectra were obtained in either electrospray mode (ES) with a Micromass LCT or chemical ionization (CI) mode with a Micromass Trio 1000 using ammonia. Elemental analyses were performed by Mrs Jean Ellis, University of Liverpool. 1H and 13C NMR spectra were obtained using a Bruker Avance or a Bruker DPX 400 instrument operating at 400 and 100 MHz, respectively; chemical shifts are reported in ppm (δ) relative to Me4Si. Coupling constants (J) are reported in Hz.2-Amino-4-bromothiazole 7
A solution of tert-butyl (4-bromothiazol-2-yl)carbamate 620 (0.56 g, 2 mmol) in CH2Cl2 (8 mL) was stirred at 20 °C with CF3CO2H (5 mL). After 4 h, the solution was evaporated to dryness, azeotroped with CH2Cl2 (2 × 5 mL) and the residue was partitioned between satd. aq. NaHCO3 (20 mL) and CH2Cl2 (5 × 10 mL). Evaporation gave 7 as a white solid (0.336 g, 94%) which was progressed immediately; δH (CDCl3) 5.32 (2H, br s, NH2) and 6.41 (1H, s, 5-H).
2-((4-Bromothiazol-2-yl)carbamoyl)phenyl acetate 10
Method A: a solution of 2-amino-4-bromothiazole 7 (0.42 g, 2.35 mmol) and O-acetylsalicyloyl chloride 9 (0.93 g, 4.69 mmol) in dry THF (10 mL) was stirred under N2 at 0 °C and Et3N (0.82 mL, 5.88 mmol) was added. The mixture was allowed to regain 20 °C, then after 23 h, 4- N,N-dimethylaminopyridine (0.12 g, 1 mmol) was added. After a total of 46 h, the mixture was diluted with EtOAc (20 mL) and worked up for a neutral product (0.93 g), which was chromatographed, eluting with a gradient of 30–50% EtOAc-n-hexane. Evaporation of early-eluting fractions afforded title compound 10 (0.133 g) which was recrystallised from EtOAc-n-hexane to afford pure product (0.100 g, 12.5%), m.p. 148–150 °C. Found: C, 42.3; H, 2.97; N, 7.96; S, 9.39; m/z, 362.9416. C12H9BrN2O3S requires C, 42.25; H, 2.7; N, 8.2; S, 9.4%; m/z, 362.9415 (MNa+); δH (CDCl3) 2.46 (3H, s, CH3CO), 6.92 (1H, s, thiazole 5-H), 7.40 (1H, d, Ar H), 7.63 (2H, m, Ar H), 8.04 (1H, d, Ar H) and 9.94 (1H, s, NH); δC (CDCl3) 21.3, 111.8, 121.3, 123.8, 124.4, 126.8, 130.9, 133.8, 148.5, 158.2, 162.4 and 168.3.Later column fractions were pooled and evaporated to give a white solid (0.332 g) whose spectroscopic data were consistent with the amide 11 plus other traces; δH (CDCl3) 2.33 (3H, s, CH3CO), 6.88 (1H, s, 5-H) and 10.65 (1H, br s, NH); found: m/z (CI, methane) 220.9387; C5H679BrN2OS (MH+) requires m/z, 220.9379.
Method B: a solution of Boc derivative 24 (0.292 g, 0.64 mmol, v. i.) in CH2Cl2 (2 mL) was stirred at 20 °C and CF3CO2H (0.3 mL, 4 mmol) was added dropwise. After 3 h, the solution was diluted with EtOAc (20 mL) and cautiously washed with satd. NaHCO3 (10 mL). Standard workup afforded the title compound 10 as a white solid, essentially pure (0.147 g, 65%), m.p. 148–150 °C. Analytical and spectroscopic data were identical to those obtained by Method A.
2-((4-Chlorothiazol-2-yl)carbamoyl)phenyl acetate 12 and {[(4-chlorothiazol-2-yl)azanediyl]bis(carbonyl)]bis(2,1-phenylene} diacetate 13
Method A: 2-amino-4-chlorothiazole 821 (0.135 g, 1 mmol) was dissolved in anhydrous THF (4 mL) and stirred at 20 °C with O-acetylsalicyloyl chloride 9 (0.24 g, 1.2 mmol) under N2. After addition of triethylamine (0.21 mL, 1.5 mmol), stirring was continued for 28 h then further acid chloride and triethylamine (1 mmol each) were added. After 96 h in all, the reaction was diluted with EtOAc (30 mL) and worked up for a neutral product, giving a pale orange gum (0.424 g). Chromatography, eluting with a gradient of 25–33% EtOAc in hexane, afforded firstly the mono-amide 12 (0.074 g, 25%), mp 148–149 °C. Found: C, 48.6; H, 3.0; N, 9.4; S, 10.6; m/z (ES +ve mode) 318.9918; C12H9ClN2O3S requires C, 48.6; H, 3.1; N, 9.4; S, 10.8%; C12H935ClN2O3SNa (MNa+) requires m/z, 318.9920; δH (CDCl3) 2.48 (3H, s, CH3CO), 6.79 (1H, s, thiazole 5-H), 7.26 (1H, d, ArH), 7.42 (1H, t, ArH), 7.62 (1H, t, ArH), 8.07 (1H, d, ArH) and 9.92 (1H, br s, NH); δC (CDCl3) 21.3, 108.1, 123.8, 124.3, 126.8, 131.0, 133.9, 135.4, 148.4, 157.2, 162.3 and 168.2. Later column fractions were pooled and evaporated to afford the bis-amide 13 (0.077 g, 17%), m. p. 111–112 °C. Found: C, 55.0; H, 3.3; N, 6.1; S, 6.7; m/z (ES +ve mode) 481.0230; C21H15ClN2O6S requires C, 55.0; H, 3.3; N, 6.1; S, 7.0%; C21H1535ClN2O6SNa (MNa+) requires m/z, 481.0237; δH (CDCl3) 2.35 (3H, s, CH3CO), 7.07 (1H, s, thiazole 5-H), 7.08 (1H, d, ArH), 7.16 (1H, t, ArH), 7.42 (1H, t, ArH) and 7.57 (1H, d, ArH); δC (CDCl3) 21.1 (×2), 113.7 (×2), 123.4 (×2), 125.9, 126.8, 130.3, 133.5, 136.9, 148.5, 157.7, 167.2 and 168.7. Recrystallisation of 13 gave material of excellent crystalline form suitable for single crystal X-ray determination, q. v.
Method B: the N-Boc intermediate 23 (0.171 g, 0.43 mmol, v. i.) was dissolved in CH2Cl2 (3 mL) and stirred at 20 °C, then CF3CO2H (0.5 mL) was added over 1 min. Complete reaction was observed after 1 h; the solution was diluted with EtOAc (20 mL) and washed with satd. aq. NaHCO3 (20 mL), giving an aq. pH∼8, then the organic phase was further washed with water and evaporated to give the product 12 (0.124 g, 97%) as a white solid. Analytical and spectroscopic data were identical to those obtained by Method A.
N-(4-Oxo-4,5-dihydrothiazol-2-yl)acetamide 15
A suspension of pseudothiohydantoin 14 (0.50 g, 4.31 mmol) in THF (4 mL) and Ac2O (1 mL) was stirred at 20 °C during addition of N–Me morpholine (1 mL), then heated at 65 °C for 1.5 h, when much solid had deposited. The mixture was cooled, treated with Et2O (10 mL) and stored at 0 °C for 1 h, then filtered, washed with Et2O, dried and evaporated to give essentially pure product 15 as a pale brown solid (0.565 g, 83%); an analytical sample was obtained by recrystallisation from MeOH-EtOAc. Found: C, 38.1; H, 3.8; N, 17.8; S, 20.15; m/z (CI, CH4) 159.0228. C5H6N2O2S requires C, 38.0; H, 3.8; N, 17.7; S, 20.3%; C5H7N2O2S (MH+) requires m/z, 159.0223; δH (d6-DMSO) δ 2.19 (3H, s, CH3CO), 3.85 (2H, s, CH2CO) and 12.61 (1H, br s, NH); δC (d6-DMSO) 24.3, 37.1, 173.0, 182.6 and 188.2.2-Acetamidothiazol-4-yl acetate 16
A suspension of pseudothiohydantoin 14 (0.5 g, 4.31 mmol) in THF (4 mL) and Ac2O (1 mL) was stirred at 20 °C and N,N-dimethylaminopyridine (0.61 g, 5 mmol) was added. A yellow-orange solution gradually resulted, and after 6 h the mixture was stored at 0 °C for 16 h, then partitioned between EtOAc (30 mL + 10 mL) and 7% aq. citric acid (25 mL). The combined extracts were washed with brine, dried and evaporated to give the title compound 16 as a near-white solid (0.70 g, 81%). Found: C, 42.3; H, 4.0; N, 13.6; S, 15.8; m/z (CI, CH4) 201.033; C7H8N2O3S requires C, 42.0; H, 4.0; N, 14.0; S, 16.0%; C7H9N2O3S (MH+) requires m/z, 201.0328; δH (d6-DMSO) δ 2.14, 2.26 (6H, 2s, 2xCH3CO), 6.73 (1H, 5-H) and 12.18 (1H, br s, NH); δC (d6-DMSO) 21.1, 23.0, 97.1, 149.9, 156.2, 168.7 and 169.5.N-(4-Chlorothiazol-2-yl)acetamide 17
A mixture of the acetamide 15 (0.40 g, 2.5 mmol) and POCl3 (1 mL) in MeCN (4 mL) was heated with stirring at 50 °C; reaction was initiated by addition of DMF (3 drops). After 3 h, the mixture was cooled and partitioned between EtOAc (30 mL + 10 mL) and 10% aq. Na2CO3 which was added cautiously to give a pH of 8. The combined organic extracts were washed with H2O, brine, dried and evaporated to give the title compound 17 (0.256 g, 57%) as a pale yellow solid, m. p. 144–145 °C (from EtOAc-hexane). Found: m/z (CI, CH4) 176.9892. C5H635ClN2OS (MH+) requires m/z, 176.9884; δH (d6-DMSO) δ 2.15 (3H, s, CH3CO), 7.14 (1H, s, 5-H) and 12.36 (1H, br s, NH); (CDCl3) 2.35 (3H, s, CH3CO), 6.77 (1H, s, 5-H) and 10.93 (1H, br s, NH); δC (d6-DMSO) 22.9, 108.0, 133.8, 158.7 and 169.5; (CDCl3) 23.4, 107.7, 134.1, 159.3 and 168.7.N-(5-Chlorothiazol-2-yl)acetamide 19
A solution of 2-acetamidothiazole 18 (0.28 g, 2 mmol) in acetonitrile (5 mL) was stirred with N-chlorosuccimimide (0.27 g, 2.00 mmol) over Amberlite A-15 (H+) (0.5 g) at 20 °C. After 22 h, when much white solid had been deposited, EtOAc (40 mL) was added to give a clear solution which was decanted from the resin, washed with water and brine, dried over Na2SO4 and evaporated to give 19 as a white solid (0.229 g, 65%). Recrystallisation from EtOAc-hexane afforded material suitable for a single crystal X-ray structure determination, see Fig. 1, m. p. 200–202 °C (softened ∼190 °C). Found: C, 33.9; H, 2.8; N, 15.8. C5H5ClN2OS requires C, 34.00; H, 2.85; N, 15.86%; δH 2.33 (3H, s, CH3CO), 7.27 (1H, s, 4-H) and 11.80 (1H, br s, NH); δC 22.9, 121.0, 133.5, 157.6 and 168.0.Tert-Butyl (4-oxo-4,5-dihydrothiazol-2-yl)carbamate 20
To a solution of pseudothiohydantoin 14 (1.0 g, 8.6 mmol) in 1:1 H2O:THF (15 mL) was added Boc2O (2.4 g, 11 mmol) followed by portionwise addition of NaOH (0.88 g, 22 mmol). The reaction mixture was stirred at 20 °C for 16 h, then partitioned between 7% aq. citric acid (30 mL) and CH2Cl2 (3 × 25 mL). The combined organic extracts were dried over MgSO4, filtered and evaporated to dryness, to afford the title product 20 (1.60 g, 86%) as a pale-yellow solid which was sufficiently pure to progress directly; an analytical sample was obtained by recrystallisation from EtOAc-hexane, 1:1. Found: C, 44.5; H, 5.6; N, 12.9; S, 14.9; m/z (ES +ve mode), 239.0461. C8H12N2O5S requires C, 44.4; H, 5.6; N, 12.95; S, 14.8%; C8H12N2O5SNa (MNa+) requires m/z, 239.0461; δH (CDCl3) δ 1.55 (s, 9H), 3.80 (s, 2H) and 9.61 (br s, 1H, NH); δC (CDCl3) δ 27.9, 36.4, 84.4, 153.6, 181.8 and 183.3.
tert-Butyl (4-chlorothiazol-2-yl)carbamate 21
A solution of carbamate 20 (0.54 g, 2.50 mmol) and triphenylphosphine (0.98 g, 3.75 mmol) in anhydrous CH2Cl2 (7.5 mL) was stirred at 20 °C under N2 and trichloroacetonitrile (0.38 mL, 3.75 mmol) was added over one minute. After 40 h the mixture was diluted with Et2O (30 mL), washed with H2O (2×), brine, dried and evaporated to a clear gum. Chromatography, eluting with 20%EtOAc-hexane, afforded on evaporation of appropriate fractions the title compound 21 (0.425 g, 73%) as a white solid. Found: C, 41.0; H, 4.7; N, 11.9; S, 13.7; m/z (ES +ve mode), 257.0126. C8H11ClN2O2S requires C, 40.9; H, 4.7; N, 11.9; S, 13.7%; C8H1135ClN2O2SNa (MNa+) requires m/z, 257.0122; δH (CDCl3) 1.54 (9H, s, Me3C), 6.65 (1H, s, 5-H) and 9.20 (1H, br s, NH); δC (CDCl3) 28.2, 83.0, 106.4, 134.4, 152.5 and 160.9.tert-Butyl (4-bromothiazol-2-yl)carbamate 6 (ref. 20)
A solution of N-bromosuccinimide (0.32 g, 1.77 mmol, 1.5 eq.) in anhydrous MeCN (2.0 mL) was added dropwise to a suspension of the carbamate 20 (0.25 g, 1.17 mmol) and triphenylphosphine (0.46 g, 1.74 mmol) in the same solvent (3.0 mL) with stirring under N2, then the reaction was stirred at 20 °C overnight. The reaction was quenched with H2O (20 mL) and extracted with ethyl acetate (3 × 25 mL). The combined organic extracts were washed with brine and dried over MgSO4. Evaporation followed by column chromatography (5–10% ethyl acetate/hexane) afforded the title compound 6 as an off-white solid (0.20 g, 63% yield). Found: (ES +ve mode) m/z, 300.9618. C8H1179BrN2NaO2S (MNa+) requires m/z, 300.9617; 1H NMR δH (CDCl3) 1.54 (9H, s, Me3C), 6.79 (1H, s, 5-H) and 9.50 (1H, br s, NH); δC (CDCl3) 28.2, 83.2, 110.3, 120.4, 152.2 and 161.17.tert-Butyl (4-iodothiazol-2-yl)carbamate 22
A solution of the carbamate 20 (0.22 g, 1 mmol) and Ph3P (0.39 g, 1.5 mmol) in MeCN (5 mL) was treated with N-iodosuccinimide (0.34 g, 1.5 mmol) at 0 °C and allowed to warm to 20 °C. No reaction occurred until I2 (0.25 g, 1 mmol) and further Ph3P (0.26 g, 1 mmol) were added with continued stirring at 20 °C. After a total of 22 h, EtOAc (30 mL) was added and the solution was washed with 5% aq. Na2S2O3 (20 mL), water and brine, then dried and evaporated to a near colourless residue. Chromatography using 20% EtOAc-hexane afforded on evaporation of appropriate fractions the iodo compound 22 as a white crystalline solid (0.091 g, 28%). Recrystallisation from EtOAc-hexane afforded an analytical sample. Found: C, 29.7; H, 3.4; N, 8.8; S, 9.4. m/z (ES +ve mode): 348.9479. C8H11N2O2SI requires C, 29.5; H, 3.4; N, 8.6; S, 9.8%; C8H11IN2O2SNa (MNa+) requires m/z, 348.9478; δH (CDCl3). 1.55 (9H, s, Me3C), 7.01 (1H, s, 5-H) and 8.8–9.2 (1H, br, NH); δC (CDCl3) 28.2, 83.3, 88.8, 117.6, 151.9 and 161.4.2-[(tert-Butoxycarbonyl)(4-chlorothiazol-2-yl)carbamoyl]phenyl acetate 23
Compound 21 (0.175 g, 0.75 mmol) and O-acetylsalicyloyl chloride 9 (0.15 g, 0.75 mmol) were stirred in anhydrous THF (3 mL) under N2 at 20 °C, then triethylamine (0.14 mL, 1 mmol) was added. After 22 h, when most 21 had reacted and some solid had been deposited, the mixture was diluted with EtOAc (20 mL) then washed successively with 7% aq. citric acid, satd. aq. NaHCO3 and water, then evaporated to give a sticky solid (0.295 g). Chromatography, eluting with 20% EtOAc-hexane, afforded on evaporation of appropriate fractions the title compound 23 (0.181 g, 61%) as a white solid. Found: C, 51.3; H, 4.2; N, 6.9; S, 7.4; m/z (ES +ve mode) 419.0444; C17H17ClN2O5S requires C, 51.45; H, 4.3; N, 7.1; S, 8.1%; C17H1735ClN2O5SNa (MNa+) requires m/z, 419.0439; δH (CDCl3) 1.22 (9H, s, Me3C), 2.24 (3H, s, CH3CO), 6.94 (1H, s, thiazole 5-H), 7.14 (1H, dd, aryl H), 7.26 (1H, t, ArH), 7.49 (1H, t, ArH) and 7.63 (1H, dd, ArH); δC (CDCl3) 20.9, 27.4, 85.8, 112.7, 123.3, 126.1, 127.7, 130.3, 133.3, 136.4, 148.6, 150.2, 157.6, 166.6 and 168.8.2-((4-Bromothiazol-2-yl)(tert-butoxycarbonyl)carbamoyl)phenyl acetate 24
2-(t-Butoxycarbonyl)amino-4-bromothiazole 6 (0.264 g, 0.95 mmol) was dissolved in THF (5 mL) with Et3N (0.30 mL, 2.1 mmol) and treated with O-acetylsalicyloyl chloride 9 (0.35 g, 1.80 mmol) added portionwise at 20 °C with stirring. After 16 h, the reaction mixture was diluted with EtOAc (30 mL) and worked up for a neutral product, which was purified by chromatography, eluting with a gradient of 5 to 7.5% EtOAc in hexane to afford the title compound 24 (0.292 g, 70%). Found: m/z (ES +ve mode) 462.9937; C17H17BrN2O5SNa (MNa+) requires m/z, 462.9939; δH (CDCl3) 1.32 (9H, s, Me3C), 2.34 (3H, s, CH3CO), 7.19 (1H, s, thiazole 5-H), 7.24 (1H, d, Ar H), 7.36 (1H, t, Ar H), 7.58 (1H, t, Ar H) and 7.68 (1H, d, ArH); δC (CDCl3) 20.9, 27.4, 85.8, 116.5, 122.3, 123.2, 126.1, 127.8, 130.3, 133.2, 148.5, 150.2, 158.4, 166.6 and 168.8.Crystallographic methods
Single crystals of C21H15N2O6SCl 13, C7H8N2O3S 15 and C5H5ClN2OS 19 were submitted for X-ray structural determination. A suitable crystal was selected and mounted on a MiTeGen tip using parabol oil and centred on a XtaLAB AFC12 (RCD3): Kappa single diffractometer. The crystal was kept at 100.01(10) K during data collection. Using Olex2,45 the structure was solved with the ShelXT46 structure solution program using Intrinsic Phasing and refined with the ShelXL47 refinement package using Least Squares minimisation.Crystallographic data
Cif files for compounds 13, 15 and 19 have been deposited in the CCDC database, no. CCDC 2362657, CCDC 2330479 and CCDC 2330480, respectively. The full data files have been added to ESI.†Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to the University of Liverpool for funding this work (PhD studentships to SP and JT). RW worked on this project as a final year CHEM480 student (University of Liverpool, 2018–2019).Notes and references
- Z. Jakopin, Chem.-Biol. Interact., 2020, 330, 109244 CrossRef CAS PubMed.
- H. C. Neu, Rev. Infect. Dis., 1986, 8, S237–S259 CrossRef CAS PubMed.
- P. D. R. Guay, Clin. Ther., 2002, 24, 473–489 CrossRef PubMed.
- A. S. Kalgutkar, J. Med. Chem., 2020, 63, 6276–6302 CrossRef CAS.
- R. S. Obach, A. S. Kalgutkar, T. F. Ryder and G. S. Walker, Chem. Res. Toxicol., 2008, 21, 1890–1899 Search PubMed.
- J.-F. Rossignol and R. Cavier, Chem. Abstr., 1975, 83, 28216n Search PubMed.
- (a) O. Doumbo, J.-F. Rossignol, E. Pichard, H. Traore, M. Dembele, M. Diakite, F. Traore and D. Diallo, Am. J. Trop. Med. Hyg., 1997, 56, 637–639 CrossRef CAS; (b) L. M. Fox and L. D. Saravolatz, Clin. Infect. Dis., 2005, 40, 1173–1180 CrossRef CAS.
- J.-F. Rossignol, Antiviral Res., 2014, 110, 94–103 CrossRef CAS PubMed.
- J.-F. Rossignol, Expert Opin. Drug Metab. Toxicol., 2009, 5, 667–674 CrossRef CAS PubMed.
- B. E. Korba, A. B. Montero, K. Farrar, K. Gaye, S. Mukerjee, M. S. Ayers and J.-F. Rossignol, Antiviral Res., 2008, 77, 56–63 CrossRef CAS PubMed.
- A. V. Stachulski, C. Pidathala, E. C. Row, R. Sharma, N. G. Berry, M. Iqbal, J. Bentley, S. A. Allman, G. Edwards, A. Helm, J. Hellier, B. E. Korba, J. E. Semple and J.-F. Rossignol, J. Med. Chem., 2011, 54, 4119–4132 CrossRef CAS.
- A. V. Stachulski, C. Pidathala, E. C. Row, R. Sharma, N. G. Berry, A. S. Lawrenson, S. L. Moores, M. Iqbal, J. Bentley, S. A. Allman, G. Edwards, A. Helm, J. Hellier, B. E. Korba, J. E. Semple and J.-F. Rossignol, J. Med. Chem., 2011, 54, 8670–8680 CrossRef PubMed.
- (a) J.-F. Rossignol, S. La Frazia, L. Chiappa, A. Ciucci and M. G. Santoro, J. Biol. Chem., 2009, 284, 29798–29808 CrossRef CAS PubMed; (b) A. V. Stachulski, M. G. Santoro, S. Piacentini, G. Belardo, S. La Frazia, C. Pidathala, E. C. Row, N. G. Berry, M. Iqbal, S. A. Allman, J. E. Semple, B. M Eklov, P. M O'Neill and J.-F. Rossignol, Future Med. Chem., 2018, 10, 851–862 CrossRef CAS PubMed.
- J.-F. Rossignol, M. Abu-Zekry, A. Hussein and M. G. Santoro, Lancet, 2006, 368, 124–129 CrossRef CAS PubMed.
- (a) J. Haffizula, A. Hartman, M. Hoppers, H. Resnick, S. Samudrala, C. Ginocchio, M. Bardin and J.-F. Rossignol, Lancet Infect. Dis., 2014, 14, 609–618 CrossRef PubMed; (b) NIH Trial: NCT01610245, https://clinicaltrials.gov/ct2/show/NCT01610245 Search PubMed.
- NIH Trial: NCT04486313, https://clinicaltrials.gov/ct2/show/NCT04486313, July 24, 2020; NIH Trial: NCT04406246, https://clinicaltrials.gov/ct2/show/NCT04406246, March 29 2021 Search PubMed.
- (a) J. Broekhuysen, A. Stockis, R. L. Lins, J. De Graeve and J.-F. Rossignol, Int. J. Clin. Pharmacol. Ther., 2000, 38, 387–394 CrossRef CAS PubMed; (b) A. Stockis, S. De Bruyn, C. Gengler and D. Rosillon, Int. J. Clin. Pharmacol. Ther., 2002, 40, 221–227 CrossRef CAS PubMed.
- A. V. Stachulski, K. Swift, M. Cooper, S. Reynolds, D. Norton, S. D. Slonecker and J.-F. Rossignol, Eur. J. Med. Chem., 2017, 126, 154–159 CrossRef CAS PubMed.
- E. L. Stangeland and T. Sammakia, J. Org. Chem., 2004, 69, 2381–2385 CrossRef CAS PubMed.
- (a) P. Stanetty, M. Schnurch, K. Mereiter and M. D. Mihovilovic, J. Org. Chem., 2005, 70, 567–574 CrossRef CAS; (b) B. Wang, J. Wu, Y. Wu, C. Chen, F. Zou, A. Wang, H. Wu, Z. Hu, Z. Jiang, Q. Liu, W. Wang, Y. Zhang, F. Liu, M. Zhao, J. Hu, T. Huang, J. Ge, L. Wang, T. Ren, Y. Wang, J. Liu and Q. Liu, Eur. J. Med. Chem., 2018, 158, 896–916 CrossRef CAS.
- T. Takahashi, et al., Yakugaku Zasshi, 1947, 67, 178–179 (Chem. Abstr., 1952, 112) CrossRef CAS.
- M. Wang, Y. Zhang, T. Wang, C. Wang, D. Xue and J. Xiao, Org. Lett., 2016, 18, 1976–1979 CrossRef CAS PubMed.
- This reaction has been scaled up to process levels: M. K. Hawk, S. J. Ryan, X. Zhang, P. Huang, J. Chen, C. Liu, J. Chen, P. J. Lindsay-Scott, J. Burnett, C. White, Y. Lu and J. R. Rizzo, Org. Process Res. Dev., 2021, 25, 1167–1175 CrossRef CAS.
- (a) A. A. Tsurkan, A. I. Frolova, N. I. Pospelov and L. A. Dorofeeva, Khim.-Farm. Zh., 1975, 9, 12–15 CAS; (b) S. M. Ramsh, Y. G. Basova, A. I. Ginak, N. A. Smorygo and A. A. Rodin, Khim. Geterotsikl. Soedin., 1982,(1), 30–34 CAS.
- (a) G. Caravatti, R. A. Fairhurst, P. Furet, V. Guagnaro and P. Imbach, WO2010029082, 2010; (b) V. Lakshmireddy, Y. Naga Veera, T. J. Reddy, V. J. Rao and B. China Raju, Asian J. Org. Chem., 2019, 8, 1380–1384 CrossRef CAS.
- B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. S. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS.
- (a) US Pat., 2006223843A1, Hoffmann-La Roche, 2006 Search PubMed; (b) US Pat., US2008/76771A1, Pfizer Inc., 2008 Search PubMed.
- R. Appel, Angew. Chem., Int. Ed. Engl., 1975, 14, 801–811 CrossRef.
- R. M. Denton, J. An and B. Adeniran, Chem. Commun., 2010, 46, 3025–3027 RSC.
- A. Jordan, R. M. Denton and H. F. Sneddon, ACS Sustainable Chem. Eng., 2020, 8, 2300–2309 CrossRef CAS.
- E. I. Snyder, J. Org. Chem., 1972, 37, 1466 CrossRef CAS.
- (a) G. A. Wiley, R. L. Hershkowitz, B. M. Rein and B. C. Chung, J. Am. Chem. Soc., 1964, 86, 964–965 CrossRef CAS; (b) P. J. Garegg, R. Johansson and B. Samuelsson, Synthesis, 1984, 168–170 CrossRef; (c) For a theoretical/structural study of this reagent, see: S. M. Godfrey, A. Hinchcliffe and A. Mkadmh, J. Mol. Struct.: THEOCHEM, 2005, 719, 85–88 CrossRef CAS.
- O. Sugimoto, M. Mori and K.-I. Tanji, Tetrahedron Lett., 1999, 40, 7477–7478 CrossRef CAS.
- (a) E. D. Matveeva, A. I. Yalovskaya, I. A. Cherepanov, Y. G. Bundel and A. I. Kurts, Zh. Org. Khim., 1991, 27, 1611–1618 CAS; (b) W. Kijrungphaiboon, O. Chantarasriwong and W. Chavasiri, Tetrahedron Lett., 2012, 53, 674–677 CrossRef CAS.
- B. E. Sleebs, I. P. Street, X. Bu and J. B. Baell, Synthesis, 2010, 1091–1096 CrossRef CAS.
- More recently, 13 has been made simply by Boc protection of expensive, commercially available 5: S. Havel, P. Khirsariya, N. Akavaram, K. Paruch and B. Carbain, J. Org. Chem., 2018, 83, 15380–15405 CrossRef CAS.
- P. Tonkgate, W. Pluemplanupat and W. Chavasiri, Tetrahedron Lett., 2008, 49, 1146–1148 CrossRef.
- S. G. Newman, C. S. Bryan, D. Perez and M. Lautens, Synthesis, 2011, 342–346 CAS.
- G. Burton, J. S. Elder, S. C. M. Fell and A. V. Stachulski, Tetrahedron Lett., 1988, 29, 3003–3006 CrossRef CAS.
- World Pat., WO2009106751, Sanofi, 2009 Search PubMed.
- This use of molecular I2 to ‘prime’ NIS is known in other series, e.g. J. A. Perrie, J. R. Harding, C. King, D. Sinnott and A. V. Stachulski, Org. Lett., 2003, 5, 4545–4548 CrossRef CAS PubMed.
- W. J. Middleton, J. Org. Chem., 1975, 40, 574–578 CrossRef CAS.
- The pKa of 2-acetamidothiazole is given as 10.8 by D. Suciu and Z. Gyorfi, Rev. Roum. Chim., 1974, 19, 671–677 CAS , and the presence of a 4-halogen is expected to decrease this value..
- The Merck Index, ed. M. J. O'Neil, A. Smith, P. E. Heckelman, J. R. Obenchain Jr, J. A. R. Gallipeau, M. A. D'Arecca and S. Budavari, Merck & Co. Inc., Whitehouse Station, NY, 13th edn, 2001, p. 56 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2330479, 2330480 and 2362657. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra04959d |
| This journal is © The Royal Society of Chemistry 2024 |