
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntileishmanial potential of thiourea-based derivatives: design, synthesis and biological activity†
Abdul
Hadi
abc,
Muhammad
Yaqoob
d,
Fahad
Hussain
 d,
Yasser M.S.A
Al-Kahraman
*a,
Muhammad Saeed
Jan
e,
Abid
Mahmood
f,
Thomas
Shier
c and
Umer
Rashid
*d
d,
Yasser M.S.A
Al-Kahraman
*a,
Muhammad Saeed
Jan
e,
Abid
Mahmood
f,
Thomas
Shier
c and
Umer
Rashid
*d
aDepartment of Pharmacy, COMSATS University Islamabad, Abbottabad Campus, 22060 KPK, Pakistan. E-mail: yasser@cuiatd.edu.pk
bFaculty of Pharmacy and Health Sciences, University of Balochistan, Quetta 08770, Pakistan
cDepartment of Medicinal Chemistry, University of Minnesota, Minneapolis, USA
dDepartment of Chemistry, COMSATS University Islamabad, Abbottabad Campus, 22060 KPK, Pakistan. E-mail: umerrashid@cuiatd.edu.pk
eDepartment of Pharmacy, Bacha Khan University, 24420, Charsadda, KPK, Pakistan
fDepartment of Pharmaceutical Chemistry, Government College University Faisalabad, Pakistan
First published on 19th November 2024
Abstract
Leishmaniasis is a neglected tropical disease caused by protozoan parasites and transmitted to humans by the sandfly vector. Currently, the disease has limited therapeutic alternatives. Thiourea derivatives were designed, synthesized, and screened for antileishmanial activity. The synthesized compounds 4g, 20a, and 20b demonstrated significant in vitro potency against L. major, L. tropica, and L. donovani promastigotes with IC50 values at low submicromolar concentrations. Compound 4g showed the highest activity against the amastigotes of L. major. In enzyme inhibition assays, compounds 4g, 20a, and 20b demonstrated good inhibitory potential against L. major dihydrofolate reductase (DHFR) and pteridine reductase 1 (PTR1). Reversal of the antileishmanial effect by adding folic acid revealed that the compounds 4g, 20a, and 20b act through an antifolate mechanism. Cytotoxicity data on normal human embryonic kidney cells (HEK-293) showed that the synthesized compounds displayed better safety profiles. Docking experiments on the enzymes L. major DHFR and PTR1 demonstrated the significant interactions with the active pocket residues of the target enzymes.
Introduction
The parasitic disease leishmaniasis is a neglected tropical disease (NTD) mainly caused by twenty species of Leishmania. All Leishmania infections commence with the sandfly bite, which introduces the protozoan parasite into the host's blood through the skin during its blood feeding. Every year about one million people get affected by leishmaniasis, which is becoming a social, health, and economic burden on many countries in subtropical regions. In the Middle East, Northwestern China, and Northern Africa, the main cause of cutaneous leishmaniasis is Leishmania major.1–5The symptoms of leishmaniasis depend upon the clinical form, i.e., cutaneous and visceral, and the type of the species.6 The three well-known clinical forms are cutaneous leishmaniasis abbreviated as CL, mucocutaneous leishmaniasis abbreviated as MCL and visceral leishmaniasis, also called kala-azar, abbreviated as VL.7 Post-kala-azar dermal leishmaniasis abbreviated as PKDL is the least common clinical form of the disease.8 The general symptoms are skin rashes, erythema, and papules that later become scars as seen in CL and fever, black spots on the skin, and enlargement of the spleen as seen in visceral leishmaniasis.8,9 Besides the skin, other parts of the body, such as the ears, lips, mucosa of the nose, and mouth, are also affected by leishmaniasis.10 The disease is widely distributed in the tropics as well as subtropics.11 In the US, leishmania cases are diagnosed among travellers to endemic regions, armed forces from the military, and immigrants.12 In the life cycle of Leishmania, the parasite protozoan experiences a digenetic style, i.e., promastigotes and amastigotes.13 The promastigote forms of the parasite multiply extracellularly in the vector's digestive tract or gut and are transmitted to the mammalian cells during the vector's blood-feeding meal, where they convert into amastigote forms, which are responsible for the clinical manifestation of leishmaniasis.14
The proliferation and growth of the Leishmania parasite are affected by different metabolic pathways. The metabolic pathway related to folate plays a crucial role in the biochemical processes; it depends on reduced pterin and tetrahydrofolate as essential cofactors. Hence, the most suitable targets associated with the folate metabolic pathway include pteridine reductase-1 and dihydrofolate reductase. An NADPH-dependent key enzyme called DHFR in the folate metabolism is responsible for the formation of deoxythymidine monophosphate (dTMP) from deoxy-uridine monophosphate (dUMP) and the conversion of dihydrofolate/folate to tetrahydrofolate. Thus, biosynthesis of thymidine and subsequent DNA biosynthesis can be prevented by the inhibition of DHFR, and as a result, this will disturb the parasite's folate metabolic pathway in Leishmania, which leads to the death of the parasite.5,15–18
Currently, the basis of antileishmanial treatment is chemotherapy. Different drug options are available and the choice of medication differs according to the disease state, parasite form, and geographic location.19 Drugs already available for treatment purposes are pentavalent antimonials (I.V), miltefosine (oral), pentamidine (intramuscular), liposomal amphotericin B (I.M and I.V), and paromomycin (topical). These drug candidates are in general associated with undesirable complications such as organ toxicity, teratogenicity, prolonged hospital stay, and reaction at the injection site. In addition, high costs and resistance remain the most important issues.10 Therefore, there is a need to seek alternative drug candidates and explore new therapeutic interventions to improve the existing therapy.
Several heterocyclic scaffolds such as triazole, imidazole, pyrazole, quinoline, pyrimidine, quinazoline, benzimidazole, pyrrole, and indole are being evaluated for the treatment of leishmaniasis.20,21 Moreover, N-acyl/dimeric N-arylpiperazine and halogen-rich salicylanilides have also been reported as potential antileishmanial agents.22,23 Thiourea, an organosulfur compound, is known for its therapeutic uses, including treating co-infections, acting as an antioxidant, exhibiting antibacterial activity, and showing anti-cancer properties.24 Thiourea derivatives were screened against parasitic protozoa for exploring their anti-amoebic and anti-leishmanial properties.25 Thiourea derivatives have been shown to inhibit both the wild-type and the resistant mutant forms of Plasmodium falciparum PfDHFR.26 Moreover, compounds belonging to the thiourea group (I–IV) have also been reported in the literature as antileishmanial therapeutics (Fig. 1). The cyclic propane derivatives of thiourea were screened against the promastigote form of L. major and showed significant antileishmanial potential.27 Other compounds of the thiourea class include II and III, which have also demonstrated antileishmanial potential against L. major.28 Compound IV, which is the basis of the rationale for the current study, was screened against both promastigote and amastigote forms of L. major and demonstrated efficient EC50 values with a good safety profile on mammalian cell lines.29 The compound (IV) possesses the thiourea functional group as its important constituent where only one hydrogen atom of the NH2 group of the thiourea moiety is substituted by an aromatic system.
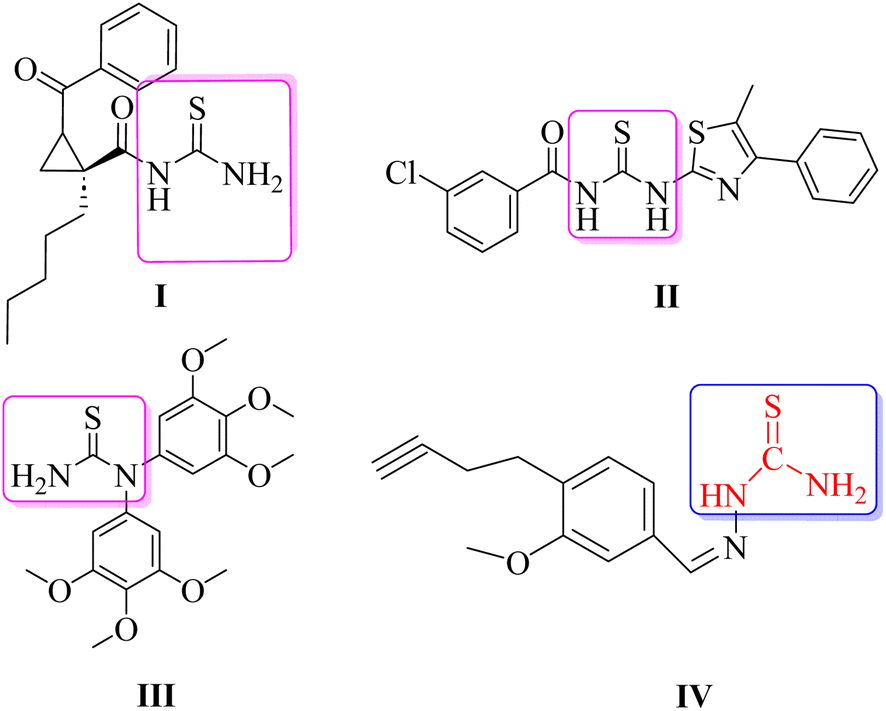 | ||
| Fig. 1 Thiourea derivatives reported against leishmaniasis. Structure IV is chosen for the rationale of the current study. | ||
Materials and methods
General information
All the chemicals and reagents were obtained from commercial sources (Sigma-Aldrich and Merck) and used without any further purification. Melting points of the compounds were determined on the Stuart SMP10 apparatus. Reactions were monitored by thin-layer chromatography (TLC) using silica gel aluminium sheets. To determine the molecular mass, high-resolution mass spectrometry was carried out using Bruker Daltonics TSQ Quantum Classic LC-MS equipment at the Chemistry Department, University of Minnesota, USA. Only a positive method was used for ionization with the polymer PEG 300 or PPG 420. Proton (1H) and carbon (13C) NMR were taken using 400 MHz and 100 MHz, spectrometer, respectively. The chemical shift values and coupling constants were presented as delta (δ) in ppm and J in Hz, respectively. The multiplicities were represented by ‘s’ for singlet, ‘d’ for doublet, ‘t’ for triplet, and ‘q’ for quintet. Analytical grade acetone, methanol, ethanol, chloroform, dimethyl sulfoxide (DMSO), and n-hexane were used to synthesize the designed thiourea derivatives. Molecular sieves of 3 Å size were heat-activated and used for the drying of acetone.Reaction procedure for the synthesis of thiourea derivatives (4a–4h and 5a–5d)
The thiourea derivatives were synthesized according to the previous method with slight modification.30 To the aromatic carboxylic acid (3 mmol) was added thionyl chloride (324 µL, 4.5 mmol). The mixture was heated to reflux along with stirring for 1 to 2 h. The resulting acid chloride was cooled to room temperature and dissolved in dry acetone. Then 3 mmol (292 mg) potassium thiocyanate was added to the solution and stirred for 2 h. Equimolar aromatic secondary amines and substituted anilines were added to the organic isothiocyanate mixture and refluxed for 3 to 4 hours to get the final product thiourea. The mixture was cooled, filtered, and washed with cold water. Finally, the product was dried and recrystallized with ethanol or methanol.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 176.5 (thiourea CS), anal. calcd for C13H12N2O3S, C, 56.51; H, 4.38; N, 10.14; O, 17.37; S, 11.60, found C, 56.49; H, 4.37; N, 10.11; O, 17.31; S, 11.57, HRMS: m/z: 299.0178 [M + Na]+.
O), 179.2 (CS), anal. calcd for C12H10N2O3S; C, 54.95; H, 3.84; N, 10.68; O, 18.30; S, 12.22, found C, 54.90; H, 3.80; N, 10.67; O, 18.27; S, 12.19, HRMS: m/z: 285.1198 [M + Na]+.
O), 178.4 (thiourea CS); anal. calcd for C12H10N2O3S; C, 54.95; H, 3.84; N, 10.68; O, 18.30; S, 12.22, found, C, 54.90; H, 3.80, N, 10.67; O, 18.27; S, 12.20, HRMS: m/z: 285.0317 [M + Na]+.
O), 168.4 (CO–OH), 178.5 (thiourea CS), anal. calcd for, C13H10N2O4S; C, 53.79; H, 3.47; N, 9.65; O, 22.05; S, 11.04, found C, 53.69; H, 3.41; N, 9.59; O, 22.09; S, 11.09. HRMS: m/z: 313.0252 [M + Na]+.
O), 178.3 (thiourea CS); anal. calcd for C13H12N2O2S2, C, 53.41; H, 4.14; N, 9.58; O, 10.94; S, 21.93, found, C, 53.39; H, 4.11; N, 9.54; O, 10.92; S, 21.90, HRMS: m/z: 315.0300 [M + Na]+.
O), 177.6 (thiourea CS), analysis calcd for C12H10N2O2S2; C, 51.78; H, 3.62; N, 10.06; O, 11.50; S, 23.04, found, C, 51.71; H, 3.58; N, 10.09; O, 11.48; S, 23.09, HRMS: m/z: 301.1078 [M + Na]+.
O), 177.6 (thiourea CS), anal. calcd for C12H10N2O2S2, C, 51.78; H, 3.62; N, 10.06; O, 11.50; S, 23.04, found, C, 51.73; H, 3.59; N, 10.01; O, 11.45; S, 23.07, HRMS: m/z: 301.1078 [M + Na]+.
S), (160.6 carbonyl–CO), 143.3, 140.0, 135.9, 133.2, 132.8, 130.7, 130.3, 130.1, 128.5, 123.9, 119.8 (Ar–C) anal. calcd for C13H10N2O3S2, C, 50.97; H, 3.29; N, 9.14; O, 15.67; S, 20.93, found, C, 50.94; H, 3.26; N, 9.10; O, 15.64; S, 20.91, HRMS: m/z: 329.0124 [M + Na]+.
S), (156.01, carbonyl CO), 147.6, 139.9, 135.0, 119.0, 117.5, 117.4, 113.2, 112.9, 31.1, Ar–C), anal. calcd for C11H9N3O2S C, 53.43; H, 3.67; N, 16.99; O, 12.94; S, 12.97, found, C, 53.39; H, 3.63; N, 16.96; O, 12.90; S, 12.94, HRMS: m/z: 270.0301 [M + Na]+.
O), 176.4 (thiourea CS), anal. calcd for C9H7N3O2S2; C, 42.68; H, 2.79; N, 16.59; O, 12.63; S, 25.31, found, C, 42.63; H, 2.75; N, 16.57; O, 12.60; S, 25.29, HRMS: m/z: 276.0191 [M + Na]+.
S), 162.7 (carbonyl CO), 147.8, 146.2, 137.4, 136.7, 136.2, 133.6, 133.2, 129.2, 123.6 (Ar–C), anal. calcd for, C11H9N3OS2, C, 44.37; H, 2.71; Cl, 11.90; N, 14.11; O, 5.37; S, 21.53, found, C, 44.31; H, 2.66; Cl, 11.88; N, 14.08; O, 5.35; S, 21.49, HRMS: m/z: 285.9771 [M + Na]+.
O), 176.6 (thiourea CS), anal. calcd for C9H7N3OS3, C, 40.13; H, 2.62; N, 15.60; O, 5.94; S, 35.71, found, C, 40.11; H, 2.59; N, 15.57; O, 5.89; S, 35.73, HRMS: m/z: 292.0660 [M + Na]+.
O), 176.5 (thiourea CS), anal. calcd for C13H12N2O3S, C, 56.51; H, 4.38; N, 10.14; O, 17.37; S, 11.60, found C, 56.49; H, 4.37; N, 10.11; O, 17.31; S, 11.57, HRMS: m/z: 299.0178 [M + Na]+.
O), 179.2 (CS), anal. calcd for C12H10N2O3S; C, 54.95; H, 3.84; N, 10.68; O, 18.30; S, 12.22, found C, 54.90; H, 3.80; N, 10.67; O, 18.27; S, 12.19, HRMS: m/z: 285.1198 [M + Na]+.
O), 178.4 (thiourea CS); anal. calcd for C12H10N2O3S; C, 54.95; H, 3.84; N, 10.68; O, 18.30; S, 12.22, found, C, 54.90; H, 3.80, N, 10.67; O, 18.27; S, 12.20, HRMS: m/z: 285.0317 [M + Na]+.
O), 168.4 (CO–OH), 178.5 (thiourea CS), anal. calcd for, C13H10N2O4S; C, 53.79; H, 3.47; N, 9.65; O, 22.05; S, 11.04, found C, 53.69; H, 3.41; N, 9.59; O, 22.09; S, 11.09. HRMS: m/z: 313.0252 [M + Na]+.
O), 178.3 (thiourea CS); anal. calcd for C13H12N2O2S2, C, 53.41; H, 4.14; N, 9.58; O, 10.94; S, 21.93, found, C, 53.39; H, 4.11; N, 9.54; O, 10.92; S, 21.90, HRMS: m/z: 315.0300 [M + Na]+.
O), 177.6 (thiourea CS), analysis calcd for C12H10N2O2S2; C, 51.78; H, 3.62; N, 10.06; O, 11.50; S, 23.04, found, C, 51.71; H, 3.58; N, 10.09; O, 11.48; S, 23.09, HRMS: m/z: 301.1078 [M + Na]+.
O), 177.6 (thiourea CS), anal. calcd for C12H10N2O2S2, C, 51.78; H, 3.62; N, 10.06; O, 11.50; S, 23.04, found, C, 51.73; H, 3.59; N, 10.01; O, 11.45; S, 23.07, HRMS: m/z: 301.1078 [M + Na]+.
S), (160.6 carbonyl–CO), 143.3, 140.0, 135.9, 133.2, 132.8, 130.7, 130.3, 130.1, 128.5, 123.9, 119.8 (Ar–C) anal. calcd for C13H10N2O3S2, C, 50.97; H, 3.29; N, 9.14; O, 15.67; S, 20.93, found, C, 50.94; H, 3.26; N, 9.10; O, 15.64; S, 20.91, HRMS: m/z: 329.0124 [M + Na]+.
S), (156.01, carbonyl CO), 147.6, 139.9, 135.0, 119.0, 117.5, 117.4, 113.2, 112.9, 31.1, Ar–C), anal. calcd for C11H9N3O2S C, 53.43; H, 3.67; N, 16.99; O, 12.94; S, 12.97, found, C, 53.39; H, 3.63; N, 16.96; O, 12.90; S, 12.94, HRMS: m/z: 270.0301 [M + Na]+.
O), 176.4 (thiourea CS), anal. calcd for C9H7N3O2S2; C, 42.68; H, 2.79; N, 16.59; O, 12.63; S, 25.31, found, C, 42.63; H, 2.75; N, 16.57; O, 12.60; S, 25.29, HRMS: m/z: 276.0191 [M + Na]+.
S), 162.7 (carbonyl CO), 147.8, 146.2, 137.4, 136.7, 136.2, 133.6, 133.2, 129.2, 123.6 (Ar–C), anal. calcd for, C11H9N3OS2, C, 44.37; H, 2.71; Cl, 11.90; N, 14.11; O, 5.37; S, 21.53, found, C, 44.31; H, 2.66; Cl, 11.88; N, 14.08; O, 5.35; S, 21.49, HRMS: m/z: 285.9771 [M + Na]+.
O), 176.6 (thiourea CS), anal. calcd for C9H7N3OS3, C, 40.13; H, 2.62; N, 15.60; O, 5.94; S, 35.71, found, C, 40.11; H, 2.59; N, 15.57; O, 5.89; S, 35.73, HRMS: m/z: 292.0660 [M + Na]+.
General procedure for the synthesis of 1-(3-oxo-butanoyl)piperidine-4-carboxylic acid (8)
Synthesis of compound 8 was carried out by using our previously reported procedure.4 A mixture of piperidine-4-carboxylic acid (7) and ethyl acetoacetate was heated under reflux in absolute ethanol as the solvent. After completion of the reaction, the reaction mixture was cooled. Ethanol was evaporated and the crude material was purified by using silica gel chromatography (n-hexane/ethyl acetate, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Transparent liquid, yield 80%, 1H NMR (400 MHz, CDCl3) δ 12.07 (s, 1H, –OH), 3.89 (s, 2H, –CH2), 3.27–3.19 (m, 4H, 2× pip-CH2), 3.09–3.04 (m, 1H, pip-CH), 2.69 (s, 3H, CH3), 2.24–2.17 (m, 2H, pip-CH2), 1.76–1.62 (m, 2H, pip-CH2).
General procedure for the synthesis of 1-(2-amino-4-methyl-6-phenylpyrimidine-5-carbonyl)piperidine-4-carboxylic acid (15)
Our previously reported multistep protocol was followed for the synthesis of 2-aminopyrimidine 15.31,32 The traditional Biginelli reaction was used to synthesize compound 12. Pyridinium chlorochromate (PCC) oxidised compound 12 into compound 13. Dihydropyrimidine (12, 5 mmol) was stirred with 30 mmol of PCC in DCM. Following the completion of the reaction (TLC, 15 hours), the solvent was removed, and silica gel chromatography (CH2Cl2/CH3OH, 9:1) was used to purify the crude product. Compound 13 (10 mmol) was then refluxed for 30 to 45 minutes in POCl3. After that, the reaction mixture was distilled to get rid of POCl3. The obtained crude product was purified using silica gel chromatography (DCM/CH3OH, 10:1). Compound 14 (10 mmol) was suspended in acetone. After adding 20 mmol of ammonium acetate, the mixture was stirred for three to four hours at RT. Compound 15 was obtained by filtering out the resultant precipitates and washing and drying them with diethyl ether.
White solid, yield 63%, m.p. 145–147 °C, 1H NMR (400 MHz, CDCl3) δ 12.07 (s, 1H, –OH), 7.37–7.28 (m, 5H, ArH), 6.69 (s, 2H, NH2), 3.78–3.70 (m, 2H, pip-CH2), 3.64–3.57 (m, 2H, pip-CH2), 3.06–3.03 (m, 1H, pip-CH), 2.56 (s, 3H, CH3), 2.39–2.30 (m, 2H, pip-CH2), 1.92–1.84 (m, 2H, pip-CH2). 13C NMR (100 MHz, CDCl3) δ 180.81, 167.52, 163.90, 163.11, 158.06, 136.09, 129.75, 129.28 (2C), 128.96 (2C), 113.20, 44.26 (2C), 40.70, 27.44 (2C), 23.46.
General procedure for the synthesis of thiourea derivatives (20a–b)
20 mmol of o-(p-tolyl)-chlorothionoformate 16 was added to a stirred solution of 2-aminopyrimidine 15 (20 mmol) in 30 mL acetone. The mixture was stirred continuously at RT for two hours. The completion of the reaction was verified using the TLC method. The precipitates obtained were filtered to obtain intermediate 17, which was used for further steps without any purification. The next step involved dissolving 10 mmol of compounds 18 and 19, 16 mmol of triethylamine, and crude intermediate 17 in 1,4-dioxane. The reaction mixture was then heated for three hours at 60 °C. After washing the resultant precipitates using diethyl ether and drying them, compounds 20a–b were obtained.33Biological screening
In the amastigote assay, serial dilutions of the synthesized compounds were made to a final concentration of 10–0.04 µg mL−1 using 50 µL culture media in 96-well flat-bottom plates. Then, to each well 50 µL suspension containing 2 × 103 cells per mL amastigotes was added. The plates were then incubated under 5% CO2 at 31 °C for 72 h. After 68 hours of incubation period, 10 µL of fluorochrome solution was added to each well and absorbance was recorded at the wavelengths of 530 nm and 590 nm for excitation and emission, respectively. The experiments were carried out in triplicate. Finally, the IC50 values were determined from the dose–response curve using GraphPad Prism 3.0. The results were expressed as mean ± SEM of the triplicate.
Docking studies
The ligand–receptor docking of the newly synthesized compounds (4g, 20a, and 20b) on LmDHFR was carried out by using our previously constructed homology model,4 while Leishmania major pteridine reductase 1 was retrieved with PDB ID: 2BFM. All the docking studies were performed on the Molecular Operating Environment, and the best-docked pose was visualized on Discovery Studio.A database of the selected synthesized compounds was prepared, and energy was minimized to obtain stabilized bond angles and geometries. The database file was saved in mdb format. The site finder option of the selected active pocket of the targeted protein was done by keeping all amino acid residues as reported in the literature.39 Furthermore, the docking procedure was validated by docking the standard drugs trimethoprim and methotrexate. The synthesized compounds were docked by retaining 20 poses for each docked ligand. The 2D interactions for the most suitable docked poses were presented, and binding energy (S) for each docked pose was noted.
Results and discussion
Design rationale
Thiourea derivatives in the current study have been designed in comparison to N,N-disubstituted thiourea derivative II (Fig. 1). The compounds 4a–h were designed by using aniline derivatives on one side of the thiourea functionality and either a furan or thiophene ring on another side, while compounds 5a–d were designed to accommodate 2-pyridine and 2-thiazole as R2 substituents. Inspired by our previous study, the dihydropyrimidine-based framework has been employed to design and synthesize a series of derivatives. Biginelli type of dihydropyrimidine-2-ones (V) emerged as potent inhibitors of L. major DHFR. In the current research, we selected 2-aminopyrimidine synthesized from DHPM-2-one. The important aspect was that we modified the C-5 ester to piperidine-4-carboxylic acid to obtain 20a–b. Piperidine-4-carboxylic acid has been reported as an important scaffold in most antileishmanial derivatives (Fig. 2).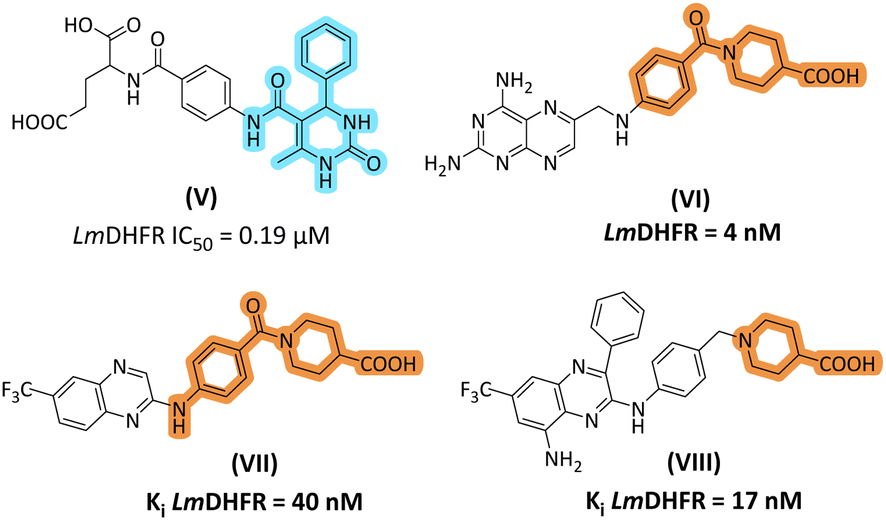 | ||
| Fig. 2 Design rationale of current study based on our previously reported DHPM-2-one based inhibitor of LmDHFR (V); piperidine-4-carboxylic acid-based inhibitors of LmDHFR (VI–VIII). | ||
Chemistry
All the furan/thiophene carboxylic acid derivatives (4a–h and 5a–d) were synthesized as per the general scheme shown in Scheme 1. The starting materials for Scheme 1 were furan carboxylic acid, thiophene carboxylic acid, and thionyl-chloride, which were refluxed for 2–3 h, leading to the formation of the respective intermediate acid chlorides. The synthesized acid chloride intermediates were further reacted with KSCN at room temperature for 2–3 h to give isothiocyanates. The final products thiourea derivatives (4a–4h and 5a–5d) were obtained by refluxing the respective isothiocyanates with substituted anilines and primary amines.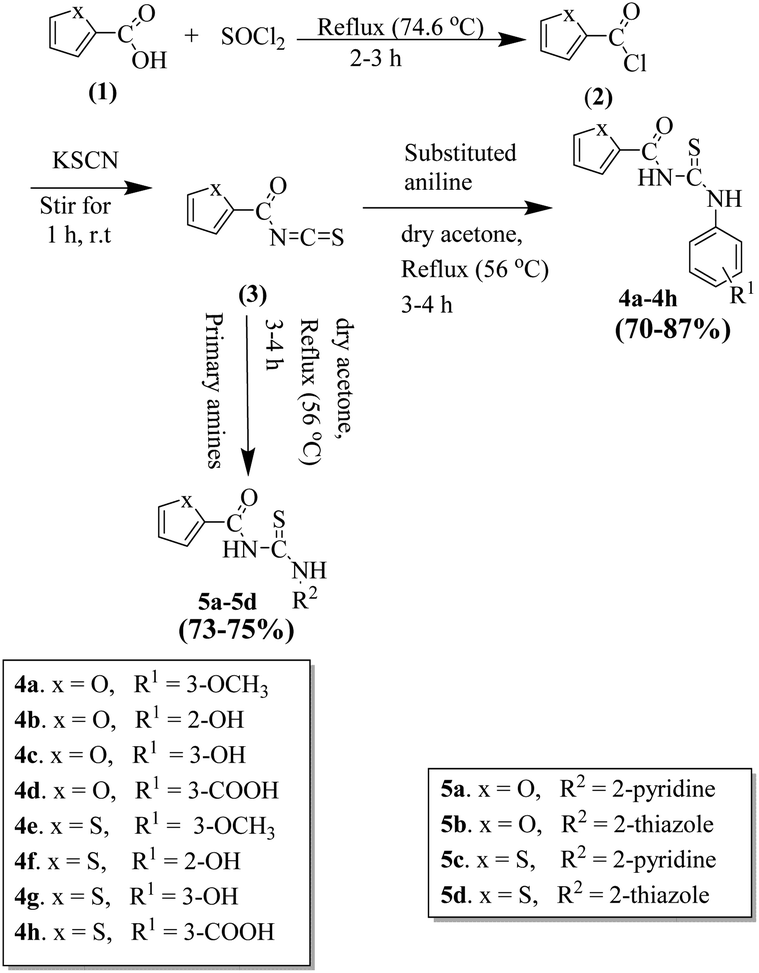 | ||
| Scheme 1 Schematic for the synthesis of thiourea derivatives (4a–4h and 5a–5d). | ||
In the next step, we synthesized pyrimidine derivatives 20a–b. For this purpose, Biginelli dihydropyrimidine derivative (12) was synthesized by using benzaldehyde (10), urea (11), and 1-(3-oxobutanoyl)piperidine-4-carboxylic acid (8, synthesized via Scheme 2). Pyridinium chlorochromate (PCC) was used to oxidize 12 to derivative 13, which was further reacted with POCl3 to yield 2-chloro derivative 14.36 2-Chloro derivative 14 was finally reacted with ammonium acetate to give the target 2-aminopyridine derivative 15 (Scheme 3).
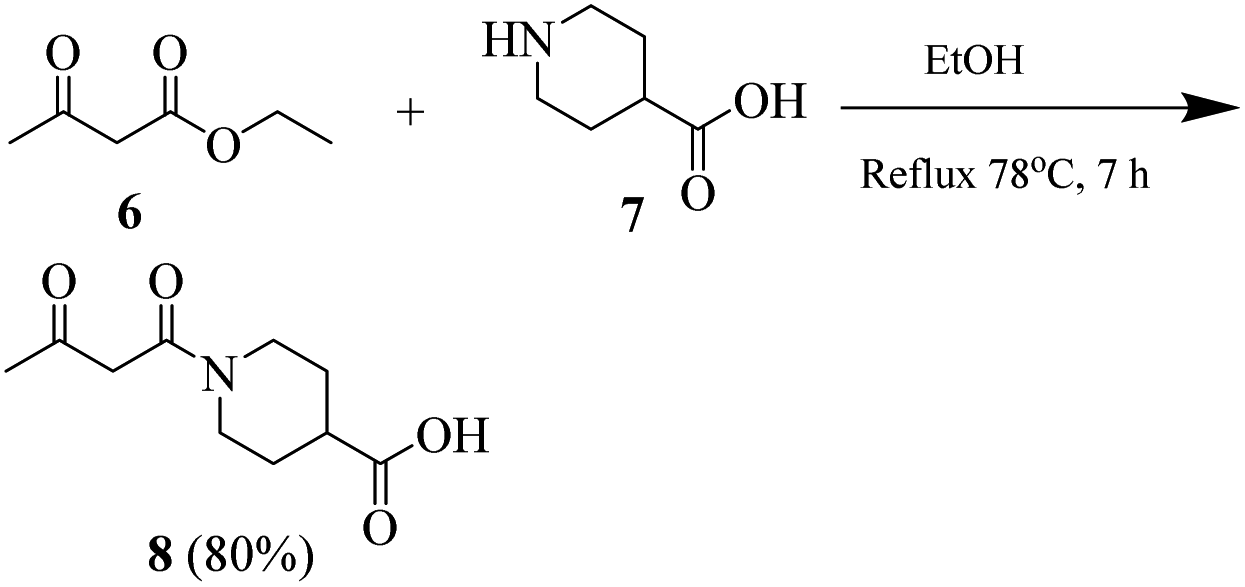 | ||
| Scheme 2 Schematic for the synthesis of 1-(3-oxobutanoyl)piperidine-4-carboxylic acid (8). | ||
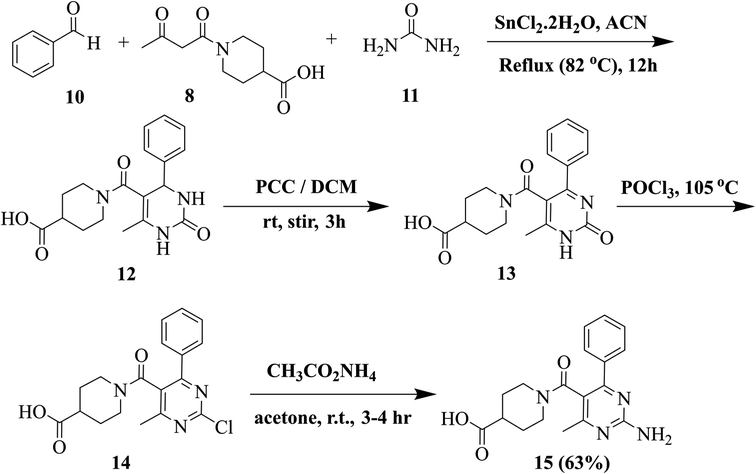 | ||
| Scheme 3 Schematic for the synthesis of 2-aminopyridine derivative 15. | ||
Finally, 2-aminopyridine-based thiourea derivatives (20a–b) were synthesized by the reaction of (15) with o-(p-tolyl)chlorothionoformate (16) in the first step and then with furan-2-ylmethanamine (18) and thiophen-2-ylmethanamine (19) using dioxane solvent to obtain the target thiourea derivatives 20a–b (Scheme 4).
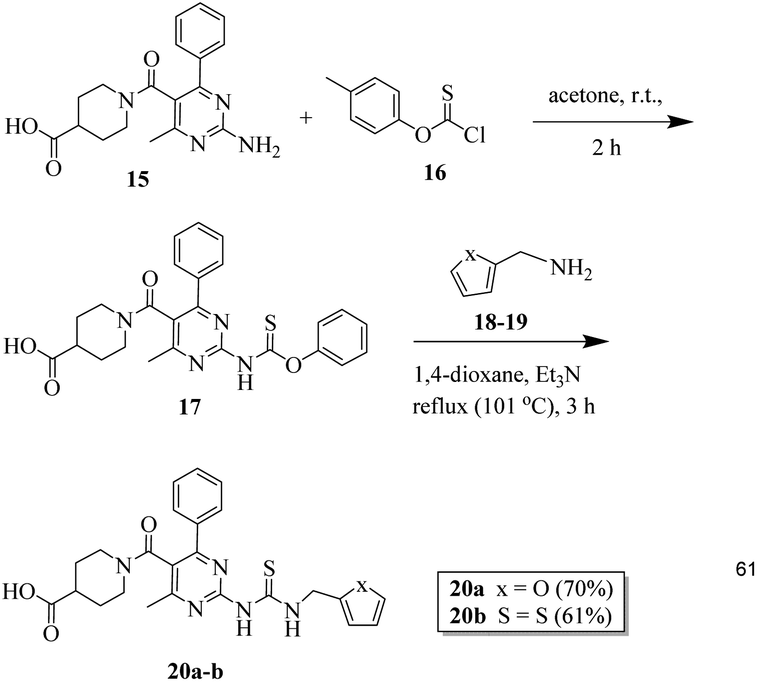 | ||
| Scheme 4 Schematic for the synthesis of 2-aminopyridine-based thiourea derivatives (20a–b). | ||
Antileishmanial assay
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Comp. no. | R1 | R2 | X | Antileishmanial activity IC50 (µM) ± SEM* | |||
| Promastigotes | Amastigotes | ||||||
| L. major | L. tropica | L. donovani | L. major | ||||
| a n = 3, SEM* = standard error of mean, n.d* = not determined. | |||||||
| 4a | 3-OCH3 | — | O | 5.34 ± 0.21 | 7.22 ± 0.61 | 10.61 ± 1.12 | n.d* |
| 4b | 2-OH | — | O | 6.08 ± 0.14 | 4.19 ± 0.10 | 2.46 ± 0.04 | n.d |
| 4c | 3-OH | — | O | 0.55 ± 0.01 | 0.68 ± 0.05 | 0.48 ± 0.04 | 0.67 ± 0.01 |
| 4d | 3-COOH | — | O | 0.83 ± 0.02 | 1.07 ± 0.08 | 1.88 ± 0.11 | 3.24 ± 0.01 |
| 4e | 3-OCH3 | — | S | 1.14 ± 0.05 | 1.23 ± 0.04 | 1.14 ± 0.02 | n.d |
| 4f | 2-OH | — | S | 0.38 ± 0.02 | 0.29 ± 0.01 | 0.41 ± 0.02 | 0.58 ± 0.01 |
| 4g | 3-OH | — | S | 0.29 ± 0.01 | 0.48 ± 0.02 | 0.24 ± 0.01 | 0.31 ± 0.01 |
| 4h | 3-COOH | — | S | 0.24 ± 0.03 | 0.33 ± 0.04 | 0.82 ± 0.04 | 3.04 ± 0.01 |
| 5a | — | 2-Pyridine | O | 0.97 ± 0.10 | 0.94 ± 0.06 | 2.30 ± 0.01 | 8.50 ± 0.01 |
| 5b | — | 2-Thiazole | O | 12.91 ± 0.42 | 12.38 ± 0.28 | 15.06 ± 0.01 | n.d |
| 5c | — | 2-Pyridine | S | 0.73 ± 0.09 | 0.79 ± 0.08 | 1.26 ± 0.01 | n.d |
| 5d | — | 2-Thiazole | S | 1.57 ± 0.04 | 1.53 ± 0.03 | 6.42 ± 0.21 | n.d |
| 20a | — | — | O | 0.18 ± 0.01 | n.d | n.d | 0.47 ± 0.01 |
| 20b | — | — | S | 0.14 ± 0.01 | n.d | n.d | 0.39 ± 0.01 |
| Miltefosine | 3.80 ± 0.16 | 2.12 ± 0.04 | 1.97 ± 0.07 | 2.89 ± 0.06 | |||
Synthesis of a small set of 2-aminopyrimidine and furan-2-yl methanamine/thiophen-2-ylmethanamine-based thiourea analogues 20a–b (Scheme 4) was also explored to evaluate their potential as antileishmanial agents. The synthesized analogues showed higher potency compared with the 4a–h and 5a–d series of compounds against L. major promastigotes. Analogues 20a and 20b also possessed good potential against the amastigotes of L. major with IC50 values of 0.47 and 0.39 µM, respectively.
In conclusion, among the 4a–h and 5a–d series of compounds, it has been observed that the OH group, its position, and the type of ring, whether furan or thiophene, largely affect the antileishmanial activity of these compounds. Therefore, we can conclude that the activity is determined not only by the position of the –OH group but also by the type of ring, whether thiophene or furan, which also plays a role in determining antileishmanial potential. However, 20a and 20b thioureas show excellent results due to the presence of an aminopyridine scaffold. Compounds 4g, 20a, and 20b were selected for further studies due to their significant antileishmanial results.
| IC50 (µM) ± SEM* | ||
|---|---|---|
| Compound no. | L. major DHFR | L. major PTR1 |
| a SEM* = standard error of the mean (n = 3). | ||
| 4g | 8.09 ± 0.01 | 0.58 ± 0.05 |
| 20a | 2.63 ± 0.01 | 15.86 ± 0.01 |
| 20b | 1.14 ± 0.01 | 19.89 ± 0.01 |
| MTX | 0.004 ± 0.000 | 0.173 ± 0.008 |
Compounds 4g, 20a, and 20b showed good inhibitory activities with IC50 values of 8.09, 2.63, and 1.14 µM, respectively, for LmDHFR and IC50 values of 0.58, 15.86, and 19.89 µM, respectively, for LmPTR1.
| Compound no. | Dose (mg kg−1 for five days) | (Mean, lesion, mm ± SD*) pre-treatment | Mean (lesion, mm ± SD*) post-treatment (after 8 weeks) | Percent-cure rate | Mice cured/mice infected | Survival time (mean, months) |
|---|---|---|---|---|---|---|
| a n = 3. Data are mean lesion size (mm) ± SD and SD* = standard deviation. | ||||||
| 4g | 30 | 0.52 ± 0.80 | 0.32 ± 0.70 | 91.51% | 6/6 | ≥2 months |
| 20a | 30 | 0.93 ± 0.22 | 0.38 ± 0.18 | 98.02% | 6/6 | ≥2 months |
| 20b | 30 | 0.80 ± 0.26 | 0.60 ± 0.54 | 86.28% | 5/6 | ≥2 months |
| Miltefosine | 15 | 0.94 ± 0.30 | 0.34 ± 0.40 | 95.10% | 6/6 | ≥2 months |
| NC | 30 | 0.71 ± 0.51 | 1.51 ± 0.51 | 0.000% | 0/6 | ≥2 months |
| Compounds | No competitor | Folic acid | |
|---|---|---|---|
| 20 µM | 100 µM | ||
| a Percentage survival is obtained by the formula = 100 − %AP, where %AP is the percent of growth inhibited by either the compound or positive control. Folate activity (through the action on DHFR and PTR1) was obtained with the compounds 20a, 20b, and 4g, along with a positive control trimethoprim. | |||
| 4g | a41% | a81% | a96% |
| 20a | a45% | a88% | a98% |
| 20b | a36% | a77% | a91% |
| Trimethoprim | a6% | — | a99% |
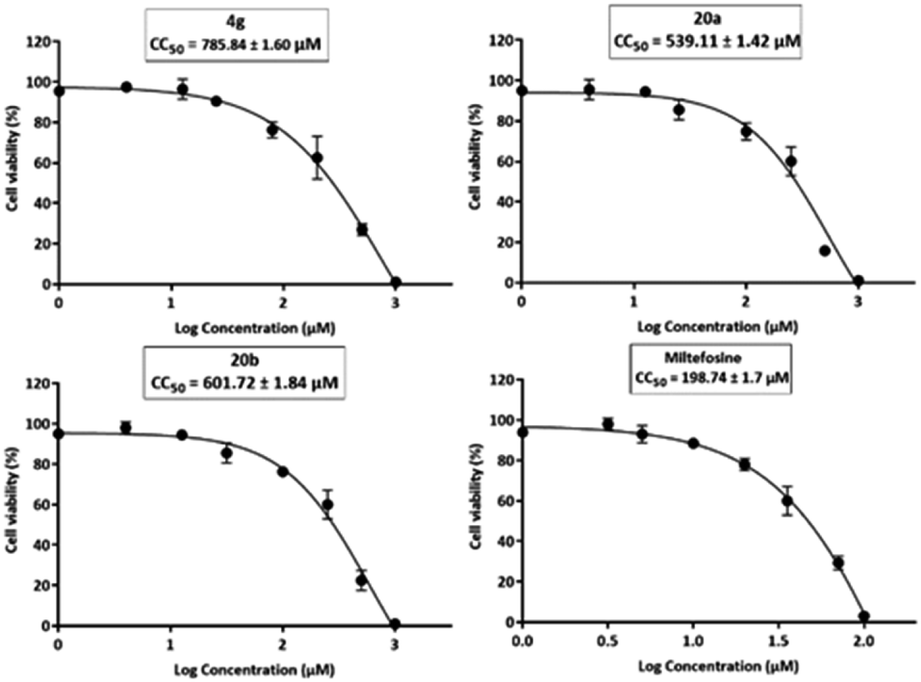 | ||
| Fig. 3 Graphs of cytotoxicity concentration 50 (CC50) of the final compounds (4g, 20a, and 20b and standard drug miltefosine) on HEK-293 cells. | ||
Evaluation of in silico studies
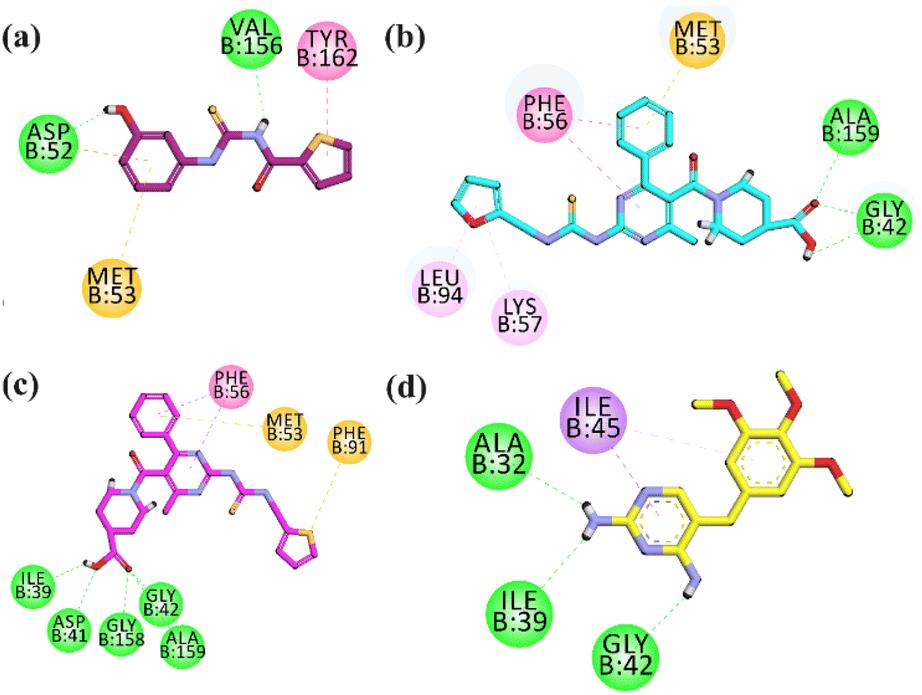 | ||
| Fig. 4 Two-dimensional (2D) interaction plots of the potent compounds (a) 4g, (b) 20a, (c) 20b and (d) standard drug trimethoprim in the binding region of homology modelled L. major DHFR. | ||
Compound 4g was a phenol derivative that established two additional hydrogen bonds with the amino acid residues of Val180 and Tyr194. In addition, two more hydrogen bonds were found between Ser111 and thiocarbamoyl functional groups as well as between Arg17 and the oxygen of the carbamoyl group. This resulted in a significantly high binding energy of −6.8573 kcal mole−1. This compound also exhibited π–sulphur and π–cation interaction with Phe113. There was also a π–alkyl interaction with Pro224, as presented in Fig. 5.
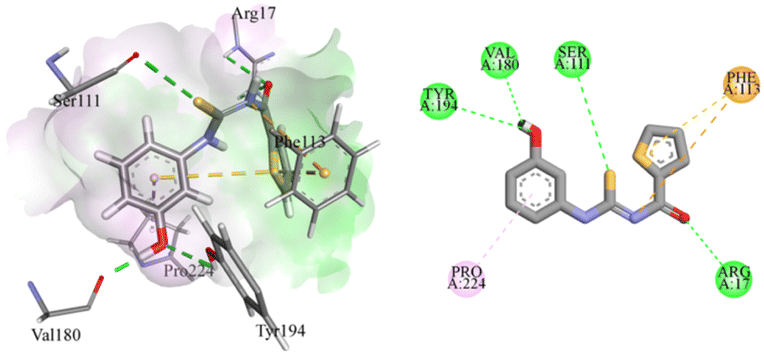 | ||
| Fig. 5 Putative binding mode of compound 4g in Leishmania major pteridine reductase 1 protein (PDB ID: 2BFM). | ||
Conclusions
In this study, thiourea derivatives were designed, synthesized, and identified as antileishmanial agents in in vitro and in vivo tests. Most of the thiourea derivatives displayed moderate to high antileishmanial activity against the promastigotes of three species L. major, L. tropica, and L. donovani, and the amastigotes of L. major as compared to the reference drug miltefosine. 2-Aminopyrimidine-piperidine-4-carboxylic acid-based thiourea hybrids demonstrated excellent in vitro inhibition potential against the promastigotes and amastigotes of L. major. The selected compounds 4g, 20a, and 20b also exhibited good in vitro results against LmDHFR and LmPTR1 enzymes. The reversal of antileishmanial activity by adding folic acid suggested that the compounds exert their action through the anti-folate pathway, which involves the inhibition of Leishmania DHFR and PTR1 enzymes. The compounds appeared safe on normal human embryonic kidney cells (HEK-293). Molecular docking studies of compound 4g on L. major PTR1 and compounds 4g and 20a–b on L. major DHFR presented significant interactions with specific amino acids of the surrounding pockets. The evaluation of the results showed that compounds 4g and 20a–b could be promising leads/hits and enrich the arsenal of antileishmanial drug development.Ethical statement
We followed the Ethical Guidelines for Animal Testing. The Ethical Committee of the Department of Pharmacy at Bacha Khan University, Pakistan via departmental ethical committee number (DECN/-2023–05) approved all the protocols. All the in vivo experiments were conducted by the Animals Bye-Laws of 2008 (Scientific Procedure, Issue-I).Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
YMSA and UR conceived, designed, and supervised this study. Both were involved in all the phases (from synthesis to pharmacological evaluation and manuscript writing/editing) that led to the completion of this manuscript. AH synthesized compounds with the help of MY. In vitro experiments were performed by AH and FH. Docking studies were performed FH and AM. Docking results were analysed and written by UR. MSJ performed in vivo studies. Compound characterization and cytotoxicity studies were performed at the Department of Medicinal Chemistry, University of Minnesota, Minneapolis, USA under the supervision of TS. AH, MY, and FH drafted the manuscript. YMSA and UR reviewed and edited the drafts. All the authors have read the manuscript and approved it for publication. The authors declare that there is no conflict of interest.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Abdul Hadi thanks the Higher Education Commission (HEC) Pakistan for supporting the research conducted under the Faculty Development Program (FDP/FMD3-001). Abdul Hadi also thanked the Medicinal Chemistry Department at the University of Minnesota, USA for conducting a portion of his PhD research.References
- O. Gupta, T. Pradhan, R. Bhatia and V. Monga, Eur. J. Med. Chem., 2021, 223, 113606 CrossRef CAS PubMed.
- K. Sweiss, A. Y. Naser, M. Samannodi and H. Alwafi, BMC Infect. Dis., 2022, 22, 398 CrossRef PubMed.
- M. Zucca and D. Savoia, Open J. Med. Chem., 2011, 5, 4 CrossRef CAS PubMed.
- M. Bibi, N. A. Qureshi, A. Sadiq, U. Farooq, A. Hassan, N. Shaheen, I. Asghar, D. Umer, A. Ullah and F. A. Khan, Eur. J. Med. Chem., 2021, 210, 112986 CrossRef CAS PubMed.
- A. Sabt, W. M. Eldehna, T. M. Ibrahim, A. A. Bekhit and R. Z. Batran, Eur. J. Med. Chem., 2023, 246, 114959 CrossRef CAS PubMed.
- S. Sasidharan and P. Saudagar, Parasitol. Res., 2021, 120, 1541–1554 CrossRef PubMed.
- M. Akhoundi, K. Kuhls, A. Cannet, J. Votýpka, P. Marty, P. Delaunay and D. Sereno, PLoS Neglected Trop. Dis., 2016, 10, e0004349 CrossRef PubMed.
- H. J. de Vries and H. D. Schallig, Am. J. Clin. Dermatol., 2022, 23, 823–840 CrossRef PubMed.
- I. Abadías-Granado, A. Diago, P. Cerro, A. Palma-Ruiz and Y. Gilaberte, Actas Dermo-Sifiliogr., 2021, 601–618 CrossRef PubMed.
- S. Pradhan, R. Schwartz, A. Patil, S. Grabbe and M. Goldust, Clin. Exp. Dermatol., 2022, 47, 516–521 CrossRef CAS PubMed.
- D. Steverding, Parasites Vectors, 2017, 10, 1–10 CrossRef PubMed.
- I. Kevric, M. A. Cappel and J. H. Keeling, Dermatol. Clin., 2015, 33, 579–593 CrossRef CAS PubMed.
- G. Volpedo, R. H. Huston, E. A. Holcomb, T. Pacheco-Fernandez, S. Gannavaram, P. Bhattacharya, H. L. Nakhasi and A. R. Satoskar, Expert Rev. Vaccines, 2021, 20, 1431–1446 CrossRef CAS PubMed.
- L. Monzote, Open Antimicrob. Agents J., 2009, 1, 9–19 CAS.
- A. H. Hassan, K. Mahmoud, T.-N. Phan, M. A. Shaldam, C. H. Lee, Y. J. Kim, S. B. Cho, W. A. Bayoumi, S. M. El-Sayed and Y. Choi, Eur. J. Med. Chem., 2023, 250, 115211 CrossRef CAS PubMed.
- H. Istanbullu, G. Bayraktar, G. Karakaya, H. Akbaba, N. E. Perk, I. Cavus, C. Podlipnik, K. Yereli, A. Ozbilgin and B. D. Butuner, Eur. J. Med. Chem., 2023, 247, 115049 CrossRef CAS PubMed.
- J. A. Shiran, B. Kaboudin, N. Panahi and N. Razzaghi-Asl, Eur. J. Med. Chem., 2024, 116396 CrossRef PubMed.
- S. Kapil, P. Singh, A. Kashyap and O. Silakari, SAR QSAR Environ. Res., 2019, 30, 919–933 CrossRef CAS PubMed.
- D. O. Santos, C. E. Coutinho, M. F. Madeira, C. G. Bottino, R. T. Vieira, S. B. Nascimento, A. Bernardino, S. C. Bourguignon, S. Corte-Real and R. T. Pinho, Parasitol. Res., 2008, 103, 1–10 CrossRef PubMed.
- N. Razzaghi-Asl, S. Sepehri, A. Ebadi, P. Karami, N. Nejatkhah and M. Johari-Ahar, Mol. Diversity, 2020, 24, 525–569 CrossRef CAS PubMed.
- R. Pal, G. Teli, M. J. Akhtar and G. S. P. Matada, Eur. J. Med. Chem., 2023, 115927 Search PubMed.
- S. B. Ansari, S. Kamboj, K. Ramalingam, R. Meena, J. Lal, R. Kant, S. K. Shukla, N. Goyal and D. N. Reddy, Bioorg. Chem., 2023, 137, 106593 CrossRef CAS PubMed.
- J. Lal, K. Ramalingam, R. Meena, S. B. Ansari, D. Saxena, S. Chopra, N. Goyal and D. N. Reddy, Eur. J. Med. Chem., 2023, 246, 114996 CrossRef CAS PubMed.
- A. Shakeel, A. A. Altaf, A. M. Qureshi and A. Badshah, J. Drug Des. Med. Chem., 2016, 2, 10 Search PubMed.
- P. W. Meireles, D. P. de Souza, M. G. Rezende, M. P. G. Borsodi, D. E. De Oliveira, L. C. R. P. da Silva, A. M. T. de Souza, G. M. Viana, C. R. Rodrigues and F. A. do Carmo, Curr. Drug Delivery, 2020, 17, 694–702 CrossRef CAS PubMed.
- M. Sharma and P. M. Chauhan, Future Med. Chem., 2012, 4, 1335–1365 CrossRef CAS PubMed.
- B. Mohammadi-Ghalehbin, J. A. Shiran, N. Gholizadeh and N. Razzaghi-Asl, Mol. Diversity, 2023, 27, 1531–1545 CrossRef CAS PubMed.
- G. M. Viana, D. C. Soares, M. V. Santana, L. H. do Amaral, P. W. Meireles, R. P. Nunes, L. C. R. P. da Silva, L. C. de Sequeira Aguiar, C. R. Rodrigues and V. P. de Sousa, Chem. Pharm. Bull., 2017, 65, 911–919 CrossRef CAS PubMed.
- M. G. Temraz, P. A. Elzahhar, A. E.-D. A. Bekhit, A. A. Bekhit, H. F. Labib and A. S. Belal, Eur. J. Med. Chem., 2018, 151, 585–600 CrossRef CAS PubMed.
- A. Mahmood, S. J. A. Shah and J. Iqbal, Eur. J. Med. Chem., 2022, 231, 114162 CrossRef CAS PubMed.
- M. A. Javed, N. Ashraf, M. Saeed Jan, M. H. Mahnashi, Y. S. Alqahtani, B. A. Alyami, A. O. Alqarni, Y. I. Asiri, M. Ikram and A. Sadiq, ACS Chem. Neurosci., 2021, 12, 4123–4143 CrossRef CAS PubMed.
- M. A. Javed, M. S. Jan, A. M. Shbeer, M. Al-Ghorbani, A. Rauf, P. Wilairatana, A. Mannan, A. Sadiq, U. Farooq and U. Rashid, Biomed. Pharmacother., 2023, 159, 114239 CrossRef CAS PubMed.
- A. Tahir, B. Mobeen, F. Hussain, A. Sadiq and U. Rashid, RSC Adv., 2024, 14, 14742–14757 RSC.
- S. N. Khattab, N. S. Haiba, A. M. Asal, A. A. Bekhit, A. A. Guemei, A. Amer and A. El-Faham, Bioorg. Med. Chem. Lett., 2017, 27, 918–921 CrossRef CAS PubMed.
- M. Teixeira, R. de Jesus Santos, R. Sampaio, L. Pontes-de-Carvalho and W. L. dos-Santos, Parasitol. Res., 2002, 88, 963–968 CrossRef PubMed.
- C. Mendoza-Martínez, N. Galindo-Sevilla, J. Correa-Basurto, V. M. Ugalde-Saldivar, R. G. Rodríguez-Delgado, J. Hernández-Pineda, C. Padierna-Mota, M. Flores-Alamo and F. Hernández-Luis, Eur. J. Med. Chem., 2015, 92, 314–331 CrossRef PubMed.
- M. Tonelli, E. Gabriele, F. Piazza, N. Basilico, S. Parapini, B. Tasso, R. Loddo, F. Sparatore and A. Sparatore, J. Enzyme Inhib. Med. Chem., 2018, 33, 210–226 CrossRef CAS PubMed.
- U. Rashid, R. Sultana, N. Shaheen, S. F. Hassan, F. Yaqoob, M. J. Ahmad, F. Iftikhar, N. Sultana, S. Asghar and M. Yasinzai, Eur. J. Med. Chem., 2016, 115, 230–244 CrossRef CAS PubMed.
- F. Hussain, A. Tahir, M. S. Jan, N. Fatima, A. Sadiq and U. Rashid, RSC Adv., 2024, 14, 10304–10321 RSC.
- E. Lebrun, Y. Tu, R. van Rapenbusch, A. R. Banijamali and W. O. Foye, Biochim. Biophys. Acta, 1990, 1034, 81–85 CrossRef CAS PubMed.
- T. J. Vickers and S. M. Beverley, Essays Biochem., 2011, 51, 63–80 CrossRef CAS PubMed.
- B. Nare, L. W. Hardy and S. M. Beverley, J. Biol. Chem., 1997, 272, 13883–13891 CrossRef CAS PubMed.
- F. H. A. Leite, T. Q. Froes, S. G. da Silva, E. I. M. de Souza, D. G. Vital-Fujii, G. H. G. Trossini, S. S. da Rocha Pita and M. S. Castilho, Eur. J. Med. Chem., 2017, 132, 322–332 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04965a |
| This journal is © The Royal Society of Chemistry 2024 |