DOI:
10.1039/D4RA05140H
(Review Article)
RSC Adv., 2024,
14, 32583-32601
Research progress of Mn-based low-temperature SCR denitrification catalysts
Received
16th July 2024
, Accepted 11th September 2024
First published on 17th October 2024
Abstract
Selective catalytic reduction (SCR) is a efficiently nitrogen oxides removal technology from stationary source flue gases. Catalysts are key component in the technology, but currently face problems including poor low-temperature activity, narrow temperature windows, low selectivity, and susceptibility to water passivation and sulphur dioxide poisoning. To develop high-efficiency low-temperature denitrification activity catalyst, manganese-based catalysts have become a focal point of research globally for low-temperature SCR denitrification catalysts. This article investigates the denitrification efficiency of unsupported manganese-based catalysts, exploring the influence of oxidation valence, preparation method, crystallinity, crystal form, and morphology structure. It examines the catalytic performance of binary and multicomponent unsupported manganese-based catalysts, focusing on the use of transition metals and rare earth metals to modify manganese oxide. Furthermore, the synergistic effect of supported manganese-based catalysts is studied, considering metal oxides, molecular sieves, carbon materials, and other materials (composite carriers and inorganic non-metallic minerals) as supports. The reaction mechanism of low-temperature denitrification by manganese-based catalysts and the mechanism of sulphur dioxide/water poisoning are analysed in detail, and the development of practical and efficient manganese-based catalysts is considered.
 Jiadong Zhang | Jiadong Zhang is a graduate student. He will start his master's degree in 2022 and his doctoral degree in 2024. He is studying at Zhejiang University, majoring in energy and power engineering. His research direction is the preparation and application of low-temperature SCR denitrification catalysts. |
 Zengyi Ma | Zengyi Ma is a professor at the School of Energy Engineering, Zhejiang University, and received his PhD in Engineering from Zhejiang University in 1998. His main research direction is clean waste incineration technology, and he has recently been engaged in the research of clean incineration technology of hazardous waste rotary kilns, clean incineration technology of high-fluorine and high-chlorine hazardous wastes, and waste incineration fly ash resource technology. |
1 Introduction
Nitrogen oxides (NOX), including nitric oxide (NO), nitrogen dioxide (NO2) and nitrous oxide (N2O), are significant air pollutants. They contribute to ecological and environmental problems such as acid rain, ozone depletion, and photochemical smog. Furthermore, NOX can cause respiratory illnesses and pose a risk to human health.1–3 NOX emissions originate from both stationary and mobile sources. Stationary sources include coal-, oil-, and gas-fired boilers and industrial furnaces, as well as waste gas pollution from petrochemical, metallurgical, and building material production processes. These emissions are typically released through exhaust stacks. Mobile sources include emissions from motor vehicles, ships and aircrafts. For thermal power plants, fossil fuel-fired industrial boilers and domestic boilers, the technologies for controlling NOX emissions encompass both combustion process control and post-combustion control. Combustion control technologies, also known as low-NOX combustion technologies, primarily include the use of low-NOX burners, staged combustion, and flue gas recirculation.4
To control NOX emissions, post-combustion control technologies are frequently employed.5 These include selective catalytic reduction (SCR) and selective non-catalytic reduction (SNCR), amongst others.6 SCR technology involves the reaction of NH3, urea, H2 or CO with NOX on the surface of catalyst to produce N2 and H2O.7 It offers advantages such as high denitrification efficiency, good product selectivity, and relatively mature technological base.8,9 The performance of the catalyst is crucial, directly impacting the denitrification effectiveness of the SCR system.10 Traditionally, SCR systems have widely utilised vanadium and tungsten-based catalysts on a TiO2 support for medium- to high-temperature applications (300–400 °C). However, these catalysts have drawbacks, including a narrow high-temperature activity window, potential for V2O5 sublimation at high temperatures, and biotoxicity concerns.11 Furthermore, SCR reactors are typically installed upstream of flue gas purification equipment, such as dust collectors and desulphurisation units. As the flue gas is not yet purified, the catalyst must withstand high dust concentrations, as well as the poisoning effects of SO2 and H2O. These factors can deactivate the catalyst, reducing denitrification efficiency and service life, leading to increased operating costs due to catalyst replacement.12 Placing the SCR reactor in a lower temperature region, such as after the flue gas purification processes and devices for dust removal and desulphurisation, could mitigate the negative impact of dust, SO2, and H2O on the catalyst. This could extend catalyst service life and reduce operating costs.13 Consequently, developing catalysts with activity in a lower temperature window holds significant practical value.
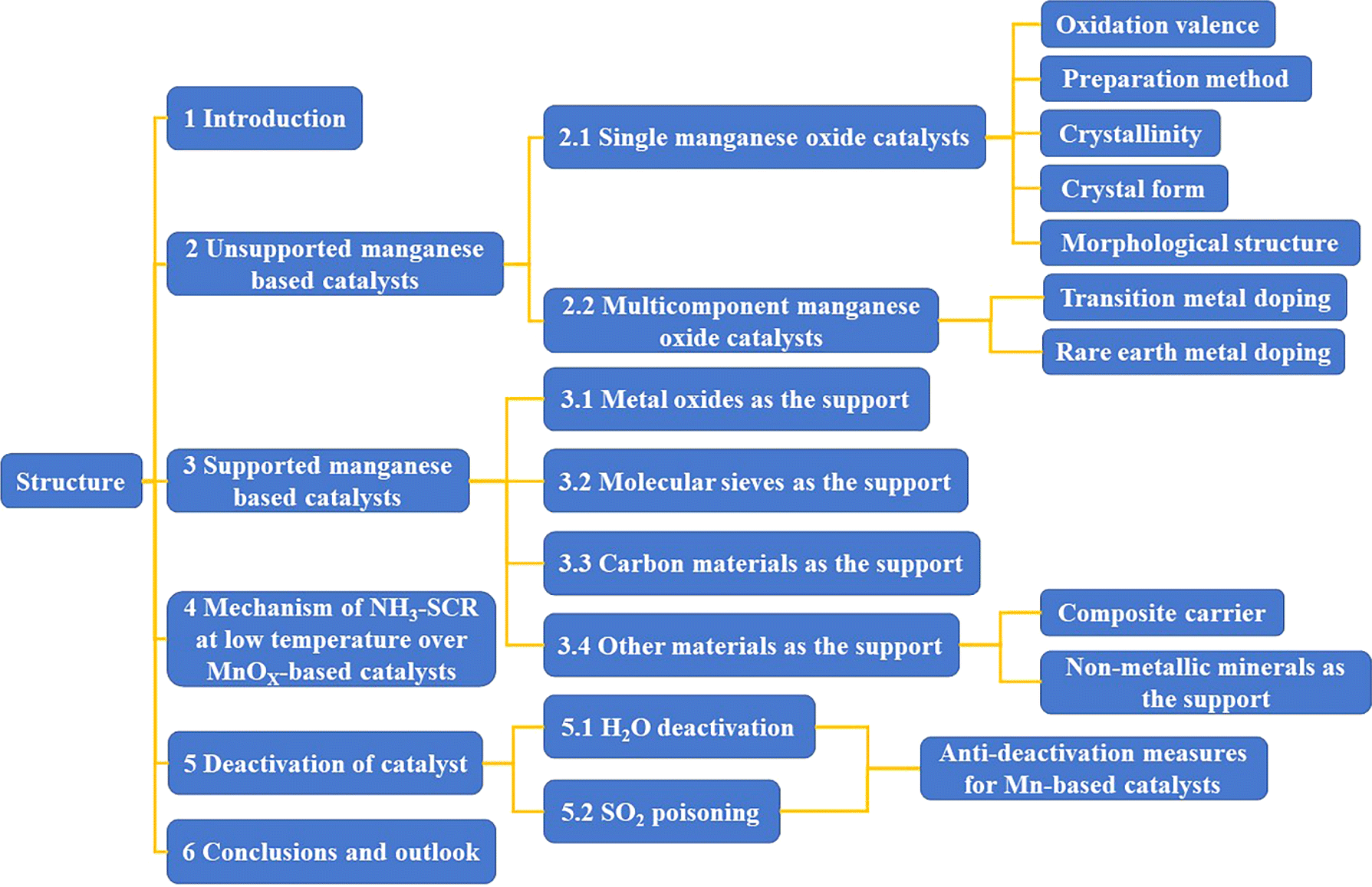
In recent years, the development and research of low-temperature catalysts has become a prominent area of focus within the field of SCR. Zeolites modified with noble metal catalysts (Pt, Pd, Ag) and transition metal ions (Cu, Fe) have been extensively investigated. Transition metal oxide catalysts (Mn, Co, Ni, Fe, Cu, etc.) exhibit excellent redox properties and promising low-temperature catalytic activity due to their facile electron gain and loss from their d-orbitals. These catalysts have emerged as a hot topic in the research of low-temperature NH3-SCR catalysts. Manganese oxides, with their abundant of multivalent states (such as Mn4+, Mn3+, Mn2+) possess strong redox capabilities, leading to excellent low-temperature denitrification activity and product (N2) selectivity. This makes them a promising candidate for industrial applications and they are considered to be the transition metal oxide catalysts with the best low-temperature SCR catalytic activity.14–17 This article reviews unsupported manganese-based catalysts, including single, binary/multicomponent manganese-based catalysts, and supported manganese-based catalysts. Supported catalysts are explored using metal oxides, molecular sieves, carbon materials, and other materials (composite carriers and inorganic non-metallic minerals) as supports. The reaction mechanisms of low-temperature denitrification using manganese-based catalysts, as well as the mechanism of SO2/H2O poisoning, are analysed and explored. The development of manganese-based catalysts with high catalytic activity, good product selectivity, and stability at low temperatures is envisioned. The structure of the article is shown in Fig. 1.
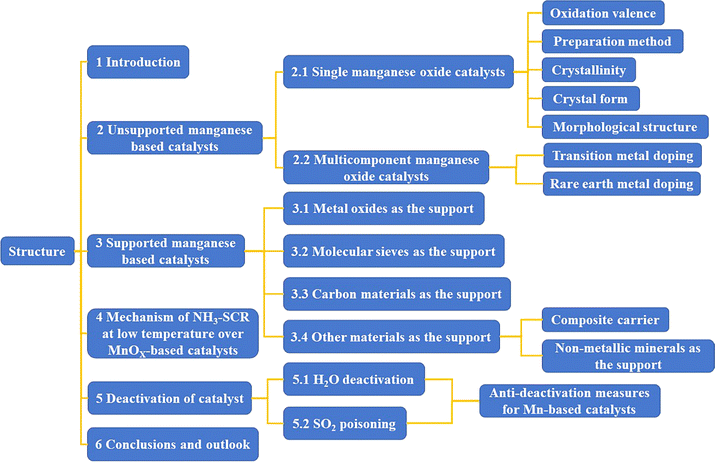 |
| | Fig. 1 The structure of article. | |
2 Unsupported manganese-based catalysts
2.1 Single manganese-based catalysts
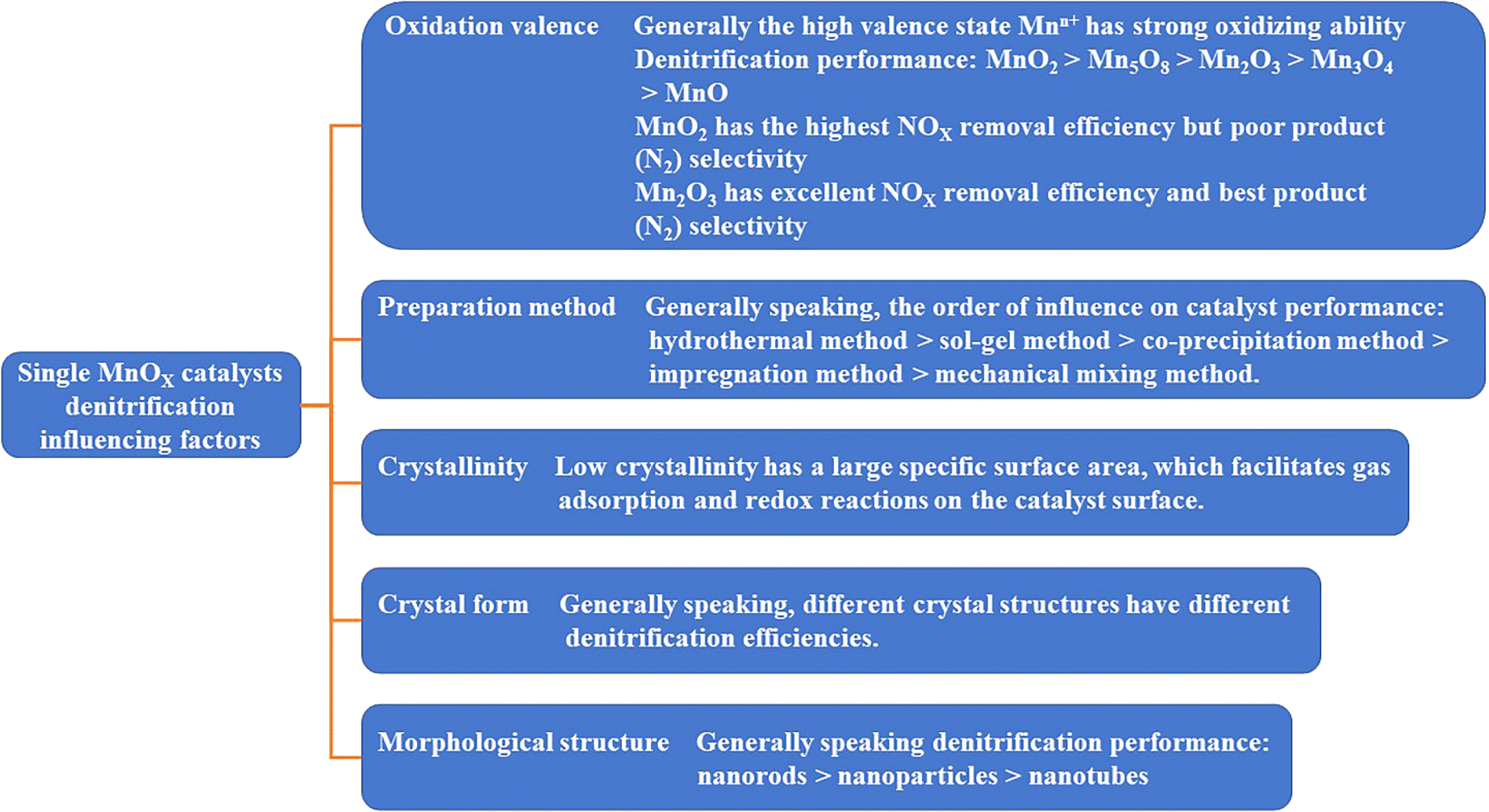
Mn has attracted extensive attention due to its abundant oxidation valence states and corresponding metal oxides, such as Mn4+, Mn3+, Mn2+.18 These different valence states contribute to its excellent redox properties.19,20 In addition, the catalytic activity of MnOX is also affected by the crystallinity, crystal crystalline structure and morphological structure.21 For example, Huang et al.22 investigated the relationship between different valence states of industrially pure MnOX compounds (MnO2, Mn2O3, Mn3O4 and MnO) under the conditions of [NO] = [NH3] = 500 ppm, [O2] = 3 vol%, N2 as the balanced gas, and GHSV = 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. The NOX conversion in the range of 120–250 °C was 100% for pure MnO2 and the NOX conversion of pure Mn2O3 reached a maximum of 92% at 160 °C. The NOX conversion of pure MnOX catalysts in the range of 50–150 °C followed the order: MnO2 > Mn2O3 > Mn3O4 > MnO.23,24 Yang et al.25 prepared manganese oxides with different valence states (MnO2, Mn2O3, and Mn3O4) by redox hydrothermal method. They conducted qualitative and quantitative investigations on the denitrification activity, NO2 generation, N2 selectivity and N2O generation (reaction condition: [NOX] = [NH3] = 500 ppm, [O2] = 11 vol%, N2 as the balanced gas and GHSV = 36000 h−1). Within the temperature range of 75–150 °C, the catalyst denitrification activity of different valence manganese oxides, the generation of NO2, and the generation of N2O increased with the increase of the temperature. However, the selectivity of N2 was in the opposite trend, decreasing as the temperature increased. The catalyst SCR of MnO2 generated more N2O, and the catalysts of Mn2O3 and Mn3O4 had better N2 selectivity than MnO2. Liu et al.26 found that due to the high oxygen instability of Mn2O3, resulting in high N2 selectivity of Mn2O3 in SCR reaction, Mn2O3 is more active than Mn3O4 for direct catalytic decomposition of NO and N2O. Kapteijn et al.15 prepared different valence states of MnOX, and the activity per unit of surface area was in the order of MnO2, Mn5O8, Mn2O3, Mn3O4 and MnO, which showed a decreasing trend in the activity per unit surface area with decreasing Mn valence, resulting in the different valence states of MnOX exhibiting different efficiencies in removing NOX ([NO] = 500 ppm, [NH3] = 550 ppm, [O2] = 2 vol%, He as the balanced gas, T = 112–302 °C and flow rate = 50 cm3 (STP) min−1). MnO2 showed the best efficiency in removing NO, and Mn2O3 showed the best selectivity of the product (N2). Tang et al.27 prepared β-MnO2 and α-Mn2O3 by redox-hydrothermal method to study the performance of NH3-SCR denitrification at 150 °C ([NO] = [NH3] = 680 ppm, [O2] = 3 vol%, He as the balanced gas and GHSV = 90000 ml g−1 h−1). MnO2 with a high valence state had higher a NO conversion and N2O generation rate than Mn2O3. MnO2 had a higher activation ability to NH3 molecules, which could break more N–H bonds in NH3 molecules, give more adsorbed nitrogen atoms, and react with gaseous NO to generate more N2O. The above studies have shown from different perspectives that different valence states of Mn lead to different NOX removal performance and product (N2) selectivity. In general, the denitrification performance of pure MnOX catalysts decreases with the decrease of the Mn valence state. Mn4+ has the highest NOX removal efficiency and poor product (N2) selectivity due to its strong oxidizing ability, whereas Mn3+ has excellent NOX removal efficiency and the best product (N2) selectivity due to its oxidation ability second only to Mn4+. The various unstable oxygen and oxidation valence states of MnOX are necessary for MnOX catalysts to complete the redox catalytic cycle.28
000 h−1. The NOX conversion in the range of 120–250 °C was 100% for pure MnO2 and the NOX conversion of pure Mn2O3 reached a maximum of 92% at 160 °C. The NOX conversion of pure MnOX catalysts in the range of 50–150 °C followed the order: MnO2 > Mn2O3 > Mn3O4 > MnO.23,24 Yang et al.25 prepared manganese oxides with different valence states (MnO2, Mn2O3, and Mn3O4) by redox hydrothermal method. They conducted qualitative and quantitative investigations on the denitrification activity, NO2 generation, N2 selectivity and N2O generation (reaction condition: [NOX] = [NH3] = 500 ppm, [O2] = 11 vol%, N2 as the balanced gas and GHSV = 36000 h−1). Within the temperature range of 75–150 °C, the catalyst denitrification activity of different valence manganese oxides, the generation of NO2, and the generation of N2O increased with the increase of the temperature. However, the selectivity of N2 was in the opposite trend, decreasing as the temperature increased. The catalyst SCR of MnO2 generated more N2O, and the catalysts of Mn2O3 and Mn3O4 had better N2 selectivity than MnO2. Liu et al.26 found that due to the high oxygen instability of Mn2O3, resulting in high N2 selectivity of Mn2O3 in SCR reaction, Mn2O3 is more active than Mn3O4 for direct catalytic decomposition of NO and N2O. Kapteijn et al.15 prepared different valence states of MnOX, and the activity per unit of surface area was in the order of MnO2, Mn5O8, Mn2O3, Mn3O4 and MnO, which showed a decreasing trend in the activity per unit surface area with decreasing Mn valence, resulting in the different valence states of MnOX exhibiting different efficiencies in removing NOX ([NO] = 500 ppm, [NH3] = 550 ppm, [O2] = 2 vol%, He as the balanced gas, T = 112–302 °C and flow rate = 50 cm3 (STP) min−1). MnO2 showed the best efficiency in removing NO, and Mn2O3 showed the best selectivity of the product (N2). Tang et al.27 prepared β-MnO2 and α-Mn2O3 by redox-hydrothermal method to study the performance of NH3-SCR denitrification at 150 °C ([NO] = [NH3] = 680 ppm, [O2] = 3 vol%, He as the balanced gas and GHSV = 90000 ml g−1 h−1). MnO2 with a high valence state had higher a NO conversion and N2O generation rate than Mn2O3. MnO2 had a higher activation ability to NH3 molecules, which could break more N–H bonds in NH3 molecules, give more adsorbed nitrogen atoms, and react with gaseous NO to generate more N2O. The above studies have shown from different perspectives that different valence states of Mn lead to different NOX removal performance and product (N2) selectivity. In general, the denitrification performance of pure MnOX catalysts decreases with the decrease of the Mn valence state. Mn4+ has the highest NOX removal efficiency and poor product (N2) selectivity due to its strong oxidizing ability, whereas Mn3+ has excellent NOX removal efficiency and the best product (N2) selectivity due to its oxidation ability second only to Mn4+. The various unstable oxygen and oxidation valence states of MnOX are necessary for MnOX catalysts to complete the redox catalytic cycle.28
Differences in the preparation methods of MnOX catalysts also affect the low-temperature denitrification activity of the catalysts. Tang et al.16 prepared amorphous unsupported manganese-oxide catalysts using three methods and investigated the catalytic activity of NH3-SCR denitrification under oxygen-rich and low-temperature conditions. They concluded that the activity of amorphous catalysts decreased in the order of MnOX (co-precipitation method), MnOX (low-temperature solid-phase reaction method), and MnOX (rheological phase reaction method) in the low-temperature range of 50–80 °C ([NO] = [NH3] = 500 ppm, [O2] = 3 vol%, N2 as the balanced gas and GHSV = 47000 h−1). Meanwhile, Tang et al.16 prepared MnOX catalysts by the low-temperature solid-phase reaction method, and examined the effects of calcination at different temperatures: 350 °C, 450 °C, 550 °C, and 650 °C. It was concluded that the crystallinity of catalysts prepared by the low-temperature solid-phase reaction method decreased with the decrease of the calcination temperature, but the catalytic activity was opposite that. The lower the crystallinity of the catalyst, the more favourable the amorphous phase is for the insertion and release of protons and promotes the chemical adsorption/desorption and redox reaction of the bulk or surface of the catalyst particles.29,30 That coincides with Andreoli et al.31 finding that catalysts with low crystallinity have better catalytic performance than crystalline catalysts. To clarify the effect of MnOX crystal crystalline structure on the denitrification efficiency of SCR reaction, Gong et al.32 prepared four different nanocrystalline structures of α-, β-, δ- and γ-MnO2, and the scanning electron microscope (SEM) images are shown in Fig. 2. The NOX redox efficiency of different crystal structures was compared as γ-MnO2 > α-MnO2 > δ-MnO2 > β-MnO2. γ-MnO2 and α-MnO2 have stronger reducing ability and stronger acidic centre, and more chemisorbed oxygen exists on the surface. In addition, the γ-MnO2 catalysts show alternating single and double bonds, which are easy to collapse and produce a large number of point-space defects and vacancies. There are more reduction/oxidation active sites in the catalyst, and γ-MnO2 has the highest catalytic activity. Experiments showed that NOX conversion reached 90% using γ-MnO2 catalyst in the temperature range of 140–200 °C ([NO] = 720 ppm, [NH3] = 800 ppm, [O2] = 3 vol%, N2 as balanced gas, and GHSV = 30000 h−1). Meanwhile, Zhao et al.33 investigated the oxidation performance of pure MnO2 catalysts concerning the crystalline structure of NOX for the most active MnO2 and found that its oxidation ability was consistent with the results of Gong et al.32
 |
| | Fig. 2 The SEM images of four different MnO2 nanocrystals: (a) α-MnO2; (b) β-MnO2; (c) δ-MnO2; (d) γ-MnO2 (reprinted from ref. 32. Copyright 2017, with permission from Elsevier). | |
The morphological structure of MnOX is also another factor affecting the denitrification activity of the catalysts. Yu et al.34 synthesized α-Mn2O3 nanocrystalline catalysts with three morphologies: octahedron (α-Mn2O3-O), truncated octahedral bipyramid (α-Mn2O3-TOB), and hexagonal nanosheets (α-Mn2O3-HN) by hydrothermal method. The influence of the crystal surface effect of α-Mn2O3 catalyst on its catalytic activity was investigated. The results showed that the exposure of the crystalline surface of the α-Mn2O3-HN catalysts {001} increased the surface density of the reactive oxygen species and enhanced the low-temperature reduction of Mn4+. Tian et al.35 prepared catalysts with different morphologies of MnO2 nanotubes, nanorods and nanoparticles by hydrothermal method. The scanning electron microscope (SEM) images are shown in Fig. 3. The results showed that the nanorod-shaped MnO2 catalysts exhibited the best denitrification performance at low temperatures (100–300 °C) and 36000 h−1 GHSV, which was mainly attributed to the low crystallinity of the nanorods, the high lattice oxygen content, the strong reducing ability and a large number of strong acid centres.
 |
| | Fig. 3 The SEM images of the MnO2 catalysts (a) nanotubes; (b) nanorods; (c) nanoparticles (reprinted from ref. 35. Copyright 2011, with permission from Elsevier). | |
Therefore, the performance and product (N2) selectivity of pure MnOX catalysts for NOX removal are not only related to the oxidation valence state of the Mn, but also closely related to the preparation method, crystallinity, crystal lattice surface, and morphological structure as well, as shown in Fig. 4.
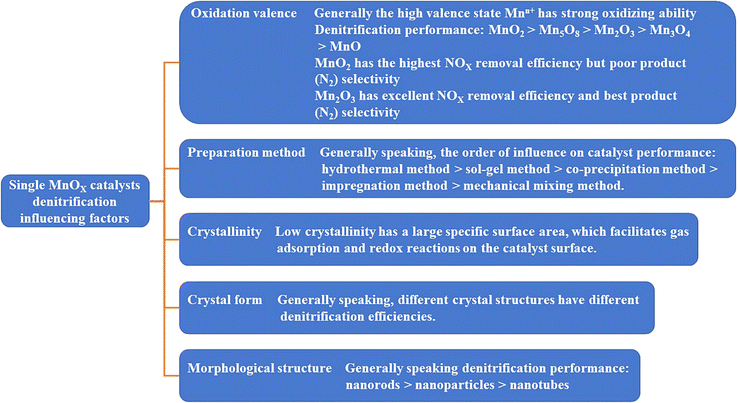 |
| | Fig. 4 Factors affecting the catalytic activity of single MnOX catalysts. | |
These factors affecting the catalytic activity of MnOX are interactive and interrelated. MnOX nanorods have lower crystallinity than MnOX nanotubes. In-depth studies are needed to determine whether the morphology or the crystallinity is the dominant factor leading to the excellent SCR catalytic activity of MnOX nanorods. While morphology, crystallinity, and specific surface area are physical properties of catalysts, chemical properties such as oxidation valence and lattice oxygen content are more indicative of the nature of catalyst performance. A single MnOX catalyst exhibits excellent low-temperature denitrification performance, but the poor stability of MnOX catalysts, resistance to H2O passivation, susceptibility to SO2 and alkali metal poisoning, and poor selectivity of the product (N2) affect its practical engineering applications.36,37 Therefore, mixing other metal oxides with MnOX for modification or doping is an important way to solve these problems.
2.2 Binary/multicomponent composite manganese-based catalysts
The introduction of other metal oxides into MnOX catalysts to form binary or multi-component composite manganese-based catalysts with the help of doping and modification can improve the reaction performance of single-component MnOX catalysts. Many researchers have introduced transition metals and rare earth metals into Mn-based catalysts for doping and modification, such as Ti,38 Fe,39,40 Cu,41 Ce,42,43 Sm,44 Co,45 etc., see Table 1.
Table 1 Part of transition metal and rare earth metal doped modified manganese based catalysts
| Catalysts |
Mental doped |
Preparation |
Reaction conditions |
Denitrification efficiency |
Ref. |
| Mn0.3Ce0.3TiOX |
Ti, Ce |
Sol–gel methode |
1000 ppm NH3, 1000 ppm NO, 3 vol% O2 |
GHSV = 40000 h−1 |
≈100% (125–350 °C) |
38 |
| MnFeOX |
Fe |
Coprecipitation method |
500 ppm NH3, 500 ppm NO, 5 vol% O2 |
GHSV = 30000 h−1 |
>80% (150–200 °C) |
39 and 40 |
| (Cu1.0Mn2.0)1−δO4 |
Cu |
Coprecipitation method |
500 ppm NH3, 500 ppm NO, 3 vol% O2 |
GHSV = 100000 h−1 |
>80% (130–240 °C) |
41 |
| MnCoCeOX |
Co, Ce |
Self-assembly, impregnation, heat treatment |
500 ppm NH3, 500 ppm NO, 5 vol% O2 |
GHSV = 24000 h−1 |
≈100% (90–240 °C) |
42 and 43 |
| MnSmOX |
Sm |
Reversed-phase precipitation |
500 ppm NH3, 500 ppm NO, 5 vol% O2 |
GHSV = 60000 h−1 |
>80% (225–325 °C) |
44 |
| MnO2–Co3O4–CeO2 |
Co, Ce |
Coprecipitation method |
1000 ppm NH3, 1000 ppm NO, 5 vol% O2 |
GHSV = 70000 h−1 |
>90% (150–175 °C) |
45 |
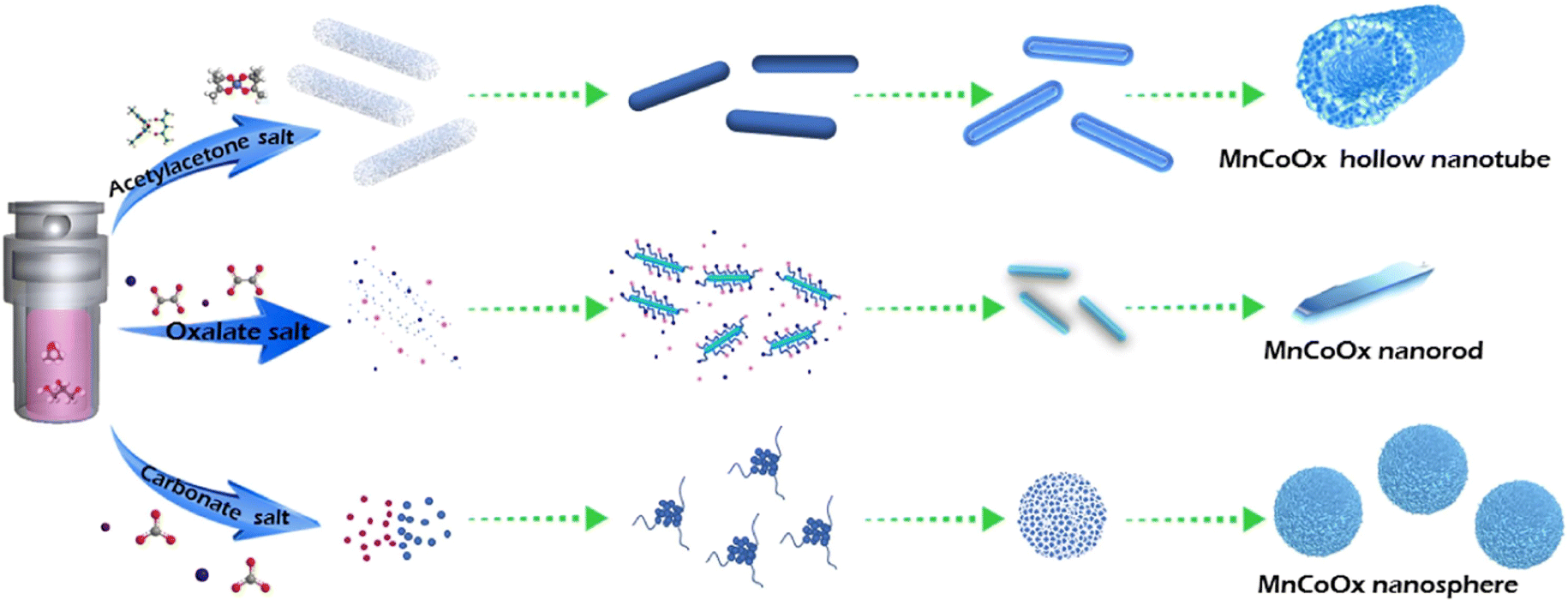
Doping transition metals can be used as structural additives to optimize the catalyst structure and enhance the stability of the catalyst, thus improving the low-temperature denitrification activity and SO2 resistance of Mn-based catalysts. Fe and Cu are often added to denitrification catalysts as additives, which resulted in a significant increase in the N2 selectivity of the SCR catalysts.46,47 Zhang et al.48 prepared Fe–Mn nanostructured oxide catalysts with NOX conversion rates exceeding 90% over the 130–300 °C range ([NO] = [NH3] = 1000 ppm, [O2] = 3 vol%, N2 as balanced gas, and GHSV = 72000 h−1), while N2 selectivity was significantly improved compared to single MnOX catalysts. The doping of FeOX made the interaction between Mnn+ and Fen+ ions stronger, which improved the denitrification ability of FeMnOX catalysts, and consequently improved the low-temperature N2 selectivity of the catalysts.49 Li et al.50 synthesized a series of MnFeOX catalysts with different Fe/Mn molar ratios by using the hydrothermal method and found that MnFe0.1OX exhibited the highest catalytic performance, with a NOX removal efficiency close to 100% at 200–350 °C ([NO] = [NH3] = 500 ppm, [O2] = 5 vol%, N2 as balanced gas, and GHSV = 28000 h−1). The addition of Fe induced the redox reaction process, increasing the oxygen concentration and acid sites for surface chemical adsorption. Gao et al.51 used the citric acid method for the preparation of Cr–Mn mixed-oxide catalysts, with high specific surface area, a large number of acidic sites and spinel structure, which exhibited excellent SCR denitrification activity with nearly 100% NOX removal efficiency and good N2 product selectivity at 100–225 °C. Yan et al.52 prepared a new class of low-temperature NH3-SCR catalysts with CuwMnyTi1−yOX using layered double hydroxide as the precursor. The Cu1Mn0.5Ti0.5OX catalyst achieved up to 90% NOX conversion at 200 °C ([NO] = [NH3] = 1000 ppm, [O2] = 5 vol%, Ar as balanced gas, and flow rate = 200 ml min−1). It was concluded that its excellent catalytic performance was mainly related to the higher specific surface area and surface acidity as well as the higher number of active MnO2 and CuO species. The catalyst exhibited significant tolerance to SO2 and H2O when CuO was introduced. Shi et al.53 synthesized nanorods, nanorods, and hollow nanotubes with the structure of the MnCoOX catalysts to investigate their low-temperature denitrification activity and tolerance to SO2 and H2O. The formation process is shown in Fig. 5. The results show that the presence of Co and Mn facilitates the improvement of redox properties, which promotes low-temperature catalytic activity, whereas the hollow nanotube-structured MnCo catalysts exhibit excellent SO2 resistance, with more than 80% NOX conversion at 150 °C even under the co-presence of H2O and SO2 ([NO] = [NH3] = 2000 ppm, [O2] = 8 vol%, [H2O] = 10 vol%, [SO2] = 200 ppm, N2 as balanced gas, and GHSV = 90000 h−1). This superior performance was attributed to the unique hollow nanotube structure, which effectively shielded the active sites on the inner surface from SO2 and alkali metal poisoning. Zhao et al.54 prepared CoMn composite oxides with layered morphology by coprecipitation. CoMn-LS-250 calcined at 250 °C showed high activity with up to 91% NOX conversion at 60 °C and good SO2 resistance at 300 °C. This was mainly attributed to the special layered structure on the surface of the catalyst, which made it rich in Lewis acid sites and strong redox capacity, as well as the high content of Mn4+, Co3+ and surface adsorbed oxygen. All these indicate that the structure of the catalyst has a certain role in the catalytic performance.
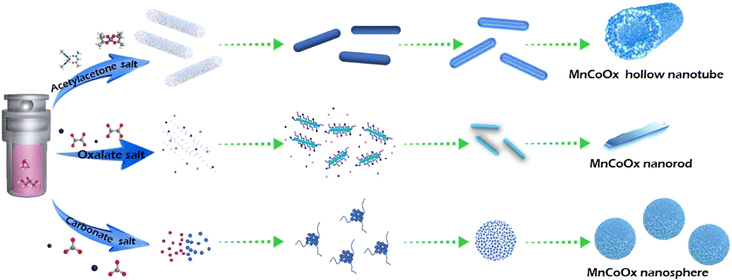 |
| | Fig. 5 The schematic illustration of the formation of the hollow nanotube, nanorod, and nanosphere of MnCoOX oxides (eeprinted from ref. 53. Copyright 2021, with permission from Elsevier). | |
Rare earth metal additives, such as Ce,42 Sm,44 Nd,55 Gd,56 etc., have good SCR denitrification performance due to their excellent oxygen storage, redox properties and good SO2 resistance. Li et al.57 prepared MnOX–CeO2 hollow binary nanotubes by a template-free method. The SEM images are presented in Fig. 6. The maximum NOX conversion was 96% at 100 °C and GHSV = 30000 h−1 with high N2 selectivity ([NO] = [NH3] = 1000 ppm, [O2] = 5 vol%, N2 as balanced gas, and GHSV = 30000 h−1). The abundant Mn4+ and Oα (surface adsorption of face), uniformly distributed active species of Mn and Ce elements, the large amount of Lewis acid and a high specific surface area brought by the hollow porous structure. The surface acidity is closely related to the activity of the catalysts,58 more acidic centres are conducive to the improvement of NH3 adsorption and low-temperature activity of the catalyst. The incorporation of Ce and the hollow porous structure reduces the possibility of SO2 occupying the surface active centres, and the doping of Ce prevents the formation of ammonium sulfate salts from blocking the active centres, thus showing good SO2 resistance. Li et al.59 prepared CeO2–MnOX catalysts with core–shell structure by chemical precipitation method. Due to the high crystallinity of α-MnO2 as well as high concentrations of Mn4+ and Ce3+, the catalyst exhibited relatively high NO conversion in the range of 110–220 °C at a molar ratio of CeO2/MnOX = 0.6. Furthermore, it demonstrated good resistance to SO2 and H2O at an air velocity of 40000 h−1, [NO] = [NH3] = 800 ppm, [O2] = 5 vol%, [SO2] = 100 ppm, [H2O] = 10 vol%, Ar as balanced gas.
 |
| | Fig. 6 The SEM images of MnOX–CeO2 hollow nanotube: (a–c). (Reprinted from ref. 57. Copyright 2018, with permission from Elsevier). | |
In addition to the CeMnOX binary catalyst, a third metal oxide was doped into the CeMnOX binary catalyst in the hope of further improving its reaction performance. Chang et al.60 investigated the denitrification performance of Cr, In, W, Ge, Sn, and Fe-doped CeMnOX, and found that the Sn-doped catalyst was modified to significantly increase the concentration of oxygen vacancies on the surface of the catalyst, improve the surface acidity and favoured the oxidation of NO to NO2. The NOX conversion rate exceeds 97% at 80 °C. Ren et al.61 investigated the performance of γ-Fe2O3 and Ce/Mn doped catalysts by quantum chemistry and density functional theory and found that SO2 and SO3 preferred to adsorb on the Ce sites, exposing more Fe active sites to participate in the NH3-SCR reaction, whereas Mn doping had little effect on the adsorption. Hao et al.62 prepared a monolithic Mn–Fe–Ce–Al–O low-temperature denitrification catalyst, which had more than 80% NO conversion in the absence of SO2 and H2O at a reaction temperature of 100 °C; the NO conversion could be maintained at about 70% at 100 °C and SO2 concentration of 200 ppm, showing excellent SO2 resistance (reaction condition: [NO] = [NH3] = 200 ppm, air balance and GHSV = 1667 h−1). Characterization analysis revealed that the presence of Ce can preferentially react with SO2 to avoid the formation of manganese sulfate, while the presence of Ce increases the amount of chemically adsorbed oxygen on the surface of the catalyst. The addition of Ce and Fe species helps to improve the catalyst's resistance to SO2 and H2O.
In summary, the doped metal oxide modification enhanced the synergistic effect between metal ions, increased the number of surface oxygen vacancies and active sites, making the active components on the catalyst surface dispersed to a higher degree, and strengthened the degree of mutual migration between electrons, thereby accelerating the NH3-SCR denitrification reaction. Manganese-based catalysts were modified by doping with one or more metal elements to improve the denitrification efficiency, and product selectivity (N2), broaden the activity temperature window, and increase the resistance to H2O and SO2.63
3 Supported manganese-based catalysts
The carrier plays an important role for low-temperature SCR catalysts,64 and the appropriate carrier is conducive to the improvement of the activity of SCR catalysts.65 Compared to unsupported catalysts, supported catalysts can not only promote the dispersion of the active components on its surface due to high specific surface area, thus preventing the catalyst from agglomeration and sintering of larger particles, but also provide more active sites for the active components dispersed on the carrier. This strengthens the synergistic effect between the active components and carrier, thereby improves the catalytic activity,66 product (N2) selectivity and anti-poisoning resistance.67–69 In the following, the performance of supported Mn-based catalysts will be investigated in terms of metal oxides, molecular sieves, carbon materials and other materials (composite carriers and inorganic non-metallic minerals) as carriers.
3.1 Metal oxide as the support
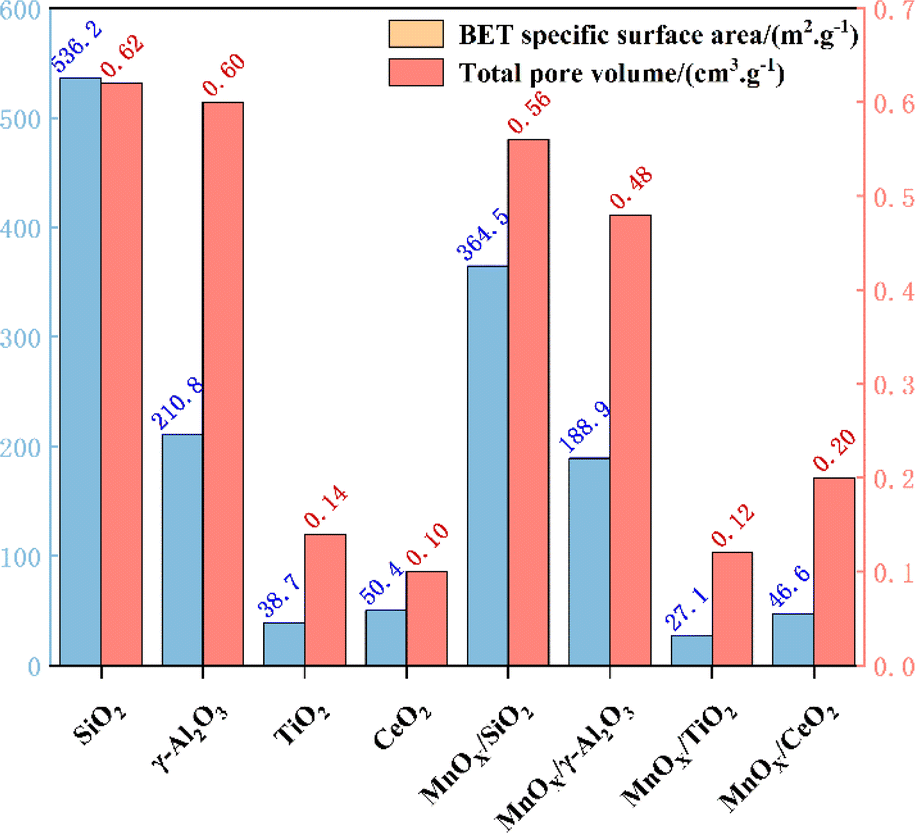
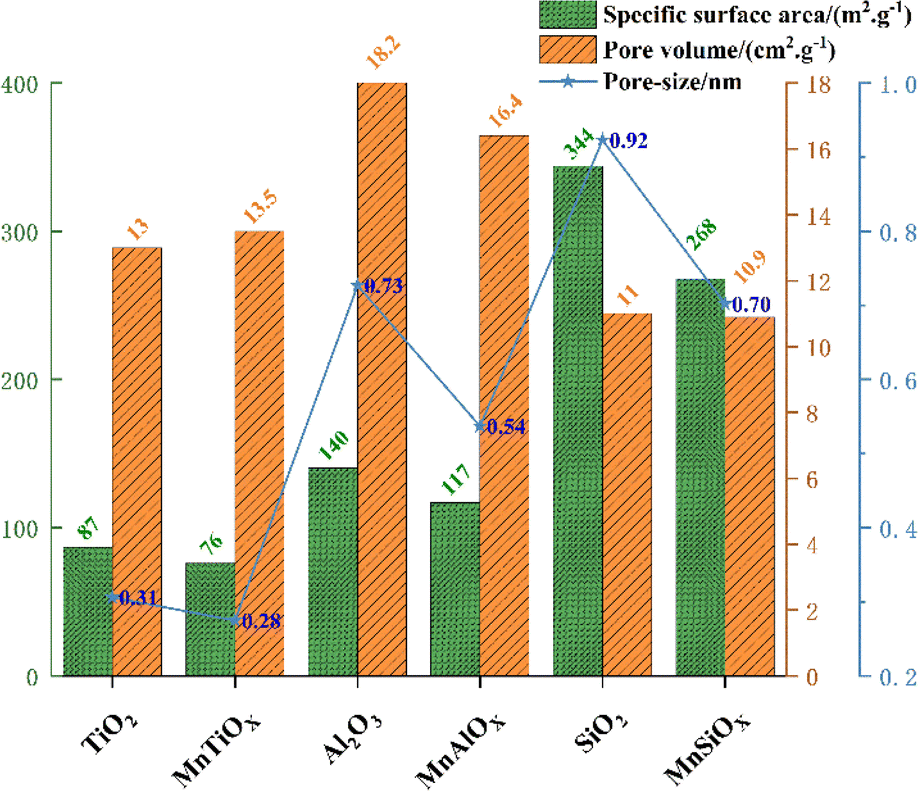
Al2O3 and TiO2 (ref. 70) as typical metal oxides, have been widely used as supports for MnOX.71 Yao et al.72 prepared MnOX/SiO2, MnOX/Al2O3, MnOX/TiO2 and MnOX/CeO2 catalysts, noting that the specific surface area of MnOX/WOy (W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si, Al, Ti, and Ce) catalysts follows the order MnOX/SiO2 > MnOX/γ-Al2O3 > MnOX/TiO2 > MnOX/CeO2 (see Fig. 7). However, in the simulated conditions: [NO] = [NH3] = 500 ppm, [O2] = 5 vol%, N2 as balanced gas and GHSV = 60000 h−1, it was found that MnOX/Al2O3 had the best performance of all the current catalysts, which was mainly related to its good dispersibility, the high number of reducing acidic sites, strong NOX adsorption capacity and abundant Mn4+ content. Liu et al.73 prepared Mn-based catalysts with γ-Al2O3, TiO2 and MCM-41 as carriers by impregnation method, and investigated their catalytic oxidation performance for NO at low temperatures (T = 80–200 °C, [NO] = 500 ppm, [O2] = 5 vol%, GHSV = 24000 h−1). The results showed that Mn/γ-Al2O3 with good Mn dispersion, excellent redox properties, moderate amount of Mn3+, Mn4+ and abundant chemically adsorbed oxygen, as well as the interaction between Mn and γ-Al2O3 carriers resulted in the strongest NO adsorption performance of Mn/γ-Al2O3, which led to its optimal catalytic activity. TiO2 can interact with active catalytic components to produce a synergistic effect and enhance catalytic activity, so it is often used as a carrier for various catalysts. Zeng et al.74 prepared MnO2/MOX (Mn/M, M = Al, Si and Ti) catalysts by impregnation method to investigate the performance of NH3-SCR reaction for NOX removal from the point of view of the supports' effect on the generation of N2O, and found that the degree of dispersion of MnO2 on MnO2/MnOX was MnSi > MnAl > MnTi (see Fig. 8). However the TiO2 support formed a stronger activity–support interaction with the impregnated MnO2, which produced a synergistic effect, thus MnTi was more strongly active than MnSi and MnAl in reducing MnO2. The strong activity–support interaction of MnTi induced the transfer of NH3 activation sites from Mn sites to Ti sites, which resulted in the separation of the activation centres of NH3 and NO + O2, and effectively suppressed the over-activation of NH3. Therefore, the generation of N2O on MnTi was much smaller than that on MnSi and MnAl, which facilitated the SCR reaction. In addition, TiO2 possesses different morphologies (anatase, rutile and brookite), and due to the different surface properties, different crystalline facets exhibit different activities. Li et al.75 employed anatase TiO2 as a support to prepare a Mn–Ce/TiO2 catalyst with varying exposed crystal faces, specifically the {001} and {101} facets. Their research revealed that preferential exposure of the anatase TiO2 {001} crystal facet significantly enhanced the catalyst's SO2 resistance and N2 selectivity. This preferential exposure effectively inhibited the formation of ammonium sulphate and ammonium bisulfate, thereby preventing the sulphation of the active Mn and Ce components. This mechanism reduces the poisoning effect of SO2 on the metal active sites.
Si, Al, Ti, and Ce) catalysts follows the order MnOX/SiO2 > MnOX/γ-Al2O3 > MnOX/TiO2 > MnOX/CeO2 (see Fig. 7). However, in the simulated conditions: [NO] = [NH3] = 500 ppm, [O2] = 5 vol%, N2 as balanced gas and GHSV = 60000 h−1, it was found that MnOX/Al2O3 had the best performance of all the current catalysts, which was mainly related to its good dispersibility, the high number of reducing acidic sites, strong NOX adsorption capacity and abundant Mn4+ content. Liu et al.73 prepared Mn-based catalysts with γ-Al2O3, TiO2 and MCM-41 as carriers by impregnation method, and investigated their catalytic oxidation performance for NO at low temperatures (T = 80–200 °C, [NO] = 500 ppm, [O2] = 5 vol%, GHSV = 24000 h−1). The results showed that Mn/γ-Al2O3 with good Mn dispersion, excellent redox properties, moderate amount of Mn3+, Mn4+ and abundant chemically adsorbed oxygen, as well as the interaction between Mn and γ-Al2O3 carriers resulted in the strongest NO adsorption performance of Mn/γ-Al2O3, which led to its optimal catalytic activity. TiO2 can interact with active catalytic components to produce a synergistic effect and enhance catalytic activity, so it is often used as a carrier for various catalysts. Zeng et al.74 prepared MnO2/MOX (Mn/M, M = Al, Si and Ti) catalysts by impregnation method to investigate the performance of NH3-SCR reaction for NOX removal from the point of view of the supports' effect on the generation of N2O, and found that the degree of dispersion of MnO2 on MnO2/MnOX was MnSi > MnAl > MnTi (see Fig. 8). However the TiO2 support formed a stronger activity–support interaction with the impregnated MnO2, which produced a synergistic effect, thus MnTi was more strongly active than MnSi and MnAl in reducing MnO2. The strong activity–support interaction of MnTi induced the transfer of NH3 activation sites from Mn sites to Ti sites, which resulted in the separation of the activation centres of NH3 and NO + O2, and effectively suppressed the over-activation of NH3. Therefore, the generation of N2O on MnTi was much smaller than that on MnSi and MnAl, which facilitated the SCR reaction. In addition, TiO2 possesses different morphologies (anatase, rutile and brookite), and due to the different surface properties, different crystalline facets exhibit different activities. Li et al.75 employed anatase TiO2 as a support to prepare a Mn–Ce/TiO2 catalyst with varying exposed crystal faces, specifically the {001} and {101} facets. Their research revealed that preferential exposure of the anatase TiO2 {001} crystal facet significantly enhanced the catalyst's SO2 resistance and N2 selectivity. This preferential exposure effectively inhibited the formation of ammonium sulphate and ammonium bisulfate, thereby preventing the sulphation of the active Mn and Ce components. This mechanism reduces the poisoning effect of SO2 on the metal active sites.
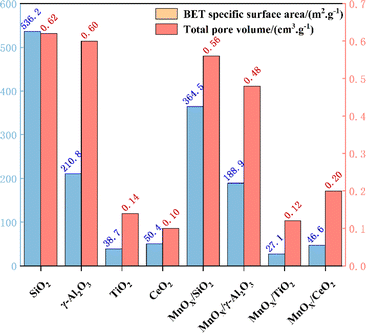 |
| | Fig. 7 BET data of different carriers and MnOX/WOy catalysts. | |
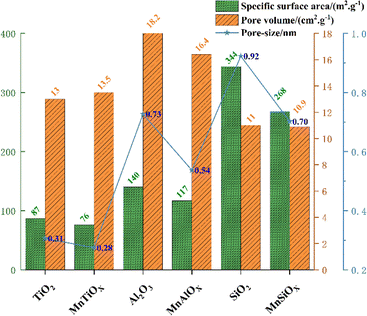 |
| | Fig. 8 BET data of different carriers and MnO2/WOy catalysts. | |
CeO2, as an active support, can form strong interactions with surface-supported components and possess abundant active oxygen to improve redox properties and thus increasing denitrification efficiency.76 Yao et al.72 prepared MnOX catalysts supported on TiO2, Al2O3, TiO2 and CeO2, investigating their H2 consumption and reduction peak temperatures. The study revealed that the H2 consumption of MnOX/SiO2, MnOX/Al2O3, and MnOX/TiO2 were broadly similar. Conversely, MnOX/CeO2 catalysts exhibited significantly greater H2 consumption compared to the above three catalysts. The reduction temperature of the MnOX/CeO2 catalyst was notably lower than the other three catalysts. From the perspective of the coordination state of Mn species, these can be attributed to the formation of a more unstable triangular biconical coordination structure between Mn and CeO2 coupled with synergistic effects between Mn and Ce. This structure facilitated the easier removal of capping O2− compared to surface lattice O2− and the subsurface lattice O2−, resulting in the superior reduction performance of MnOX/CeO2 catalysts, thereby enhancing their denitrification performance. Li et al.77 prepared a MnOX–CeO2 nanosphere catalyst with excellent low-temperature activity and SO2 resistance, and found that the MnOX–CeO2 nanosphere catalysts had long-time operation stability at 150 °C ([NO] = [NH3] = 500 ppm, [O2] = 5 vol%, N2 as the balanced gas and GHSV = 60000 h−1). The research showed that good redox properties, grain size small, high specific surface area and abundant surface Ce3+, Mn4+ and oxygen species are the main reasons for its excellent catalytic performance.
3.2 Molecular sieve as the support
Molecular sieves with unique pore structures, large specific surface areas, good adsorption, and high hydrothermal stability, such as SAPO-34,78 ZSM-5,79 β,80 zeolite (copper- and iron-zeolites),81,82 etc., are often used as supports for low-temperature NH3-SCR catalysts, which is one of the hotspot supports for Mn-based catalysts at present. Lou et al.83 prepared a series of Mn/ZSM-5 catalysts by the precipitation method and calcined the catalysts at different calcination temperatures. The results showed that MnOX existed on the catalyst surface in the form of Mn3O4 and amorphous MnO2 when calcined below 500 °C, and when the calcination temperature was 600 °C, the low-activity Mn2O3 was formed, and it became the main phase at 700 °C. The Mn concentration and specific surface area on the surface of the catalyst decreased with increasing calcination temperature. The NH3-SCR catalytic activity tests showed that the Mn/ZSM-5 catalyst calcined at 300 °C exhibited the best NO removal performance with almost 100% NO conversion in the range of 150–390 °C ([NO] = [NH3] = 600 ppm, [O2] = 4.5 vol%, N2 as the balanced gas and GHSV = 36000 h−1). Li et al.84 prepared a low-cost fly ash-derived SBA-15 molecular sieve as a support. The Fe–Mn/SBA-15 catalyst was prepared by the impregnation method. In the range of 150–250 °C, Fe–Mn/SBA-15 exhibited higher NH3-SCR activity, with synergistic effects between Mn and Fe, high dispersion on the surface of the species, suitable Mn4+/Mn3+ ratio, and the adsorption of oxygen concentration and low-temperature oxidation activity, which are favourable for the improvement of NH3-SCR catalytic activity. Xu et al.80 prepared two series of Mn/β and Mn/ZSM-5 catalysts by impregnation method using manganese nitrate, manganese acetate, and manganese chloride as the three precursors, respectively and investigated the catalytic activity of these catalysts within a reaction temperature window of 50–350 °C. In the range of 220–350 °C, the NO removal rate of Mn/β and Mn/ZSM-5 catalysts prepared by manganese acetate were above 80%, and the Mn/β prepared by manganese acetate exhibited the highest NO conversion—97.5% at 240 °C, and its activity remained above 90% in the 220–350 °C temperature window ([NO] = 1000 ppm, [NH3] = 1100 ppm, [O2] = 5 vol%, N2 as the balanced gas and GHSV = 50000 h−1). The excellent catalytic performance was attributed to the highly dispersed MnOX active phase, the appropriate amount of weak acidic centres, the higher concentration of surface Mn species, and more surface unstable oxygen groups.
3.3 Carbon materials as the support
Carbon materials with large specific surface area, porous structure, strong adsorption capacity, and high catalytic efficiency can also be used as Mn-based catalyst supports, such as activated carbon (AC),85 activated carbon fibre (ACF), graphene (GE), carbon nanotubes (CNTs), and semi-coke.86 Jiao et al.87 used a hydrothermal method for the preparation of graphene-supports manganese oxides (MnOX/GR), investigating the effect of different MnOX loadings on the catalytic activity of low-temperature NH3-SCR It was found that the catalytic activity was optimal at a Mn loading of 20% (wt), and the NO removal efficiency was greater than 90% at 190 °C, and the NO removal efficiency was close to 100% at 220 °C ([NO] = [NH3] = 600 ppm, [O2] = 3 vol%, Ar as the balanced gas and GHSV = 45000 h−1). The MnOX was dispersed as nanoparticles on the graphene surface, and it was mainly coexisted with various MnOX compounds, such as MnO, Mn3O4, and MnO2. The catalyst with a loading of 20% (wt) has high SCR activity. The reason is that it contains high-valent manganese and the surface adsorbed oxygen content increases, the redox ability is strong in the low-temperature zone, and the number of active sites is large. Zhang et al.88 prepared MnOX/CNTs catalysts with different Mn/C molar ratios by in situ precipitation. It was found that the prepared MnOX/CNTs catalysts had excellent low-temperature SCR activity with NO conversion of 57.4–89.2% for 1.2 (wt%) MnOX/CNTs catalysts in the temperature range of 80–180 °C ([NO] = [NH3] = 500 ppm, [O2] = 5 vol%, N2 as the balanced gas and GHSV = 35000 h−1). This performance was attributed to the amorphous nature of the MnOX catalysts, characterised by high Mn4+/Mn3+ and OS/(OS + OL) ratios (OS: surface adsorbed oxygen, OL: lattice oxygen). Yang et al.89 used the impregnation method to load the transition metals such as Mn, Ce, V and Fe onto nitric acid-modified biomass coke (BC) and tested for low-temperature SCR catalytic activity. The order of NO removal efficiencies in the range of 125–225 °C was Mn/BC > Ce/BC > V/BC > Fe/BC > BC, with the Mn/BC catalyst achieving the highest NO removal efficiency of 87.6% at 200 °C ([NO] = [NH3] = 600 ppm, [O2] = 11 vol%, N2 as the balanced gas and GHSV = 12000 h−1). This high performance is primarily attributed to the high specific surface area of BC support, the abundant oxygen-containing groups that provide highly active adsorption sites for NH3 and the graphite microcrystalline structure that can act as an oxidant for NO.
3.4 Other materials as the support
Composite carriers and inorganic non-metallic minerals are also often used as substrate materials for SCR reactions. Composite carriers can give full play to the advantages of different carriers relative to a single carrier, enhance the synergistic effect between active components and carriers, and improve the catalytic activity of the catalyst. He et al.90 prepared TiO2–CeO2, ZrO2–CeO2 and TiO2–ZrO2–CeO2 supports by the sol–gel method. They found that the low-temperature catalytic activity and SO2 resistance performance of MnOX/TiO2–ZrO2–CeO2 catalysts were significantly better than those of catalysts with other carriers. Additionally, the structural instability of the Mn-based catalysts was improved to reduce the temperature of the crystal formation and suppress the crystal growth. The specific surface area and pore volume of the catalysts were increased to avoid the accumulation of active components due to the high calcination temperature. Qi et al.91 found that the catalysts on the composite carriers of Al2O3 and TiO2 had better pore structure, better surface dispersion of the active substance carriers, and more active ligand NH3 in the L-acid site, and the best denitrification efficiency than the single carriers such as TiO2 or ZrO2. Li et al.65 also prepared Mn–Ce/Ti–Al–O composite carrier-type catalysts by impregnation method and found that Ti–Al–O composite carriers have larger specific surface area, pore volume and lower crystallinity than pure TiO2. When the temperature is lower than 150 °C, the Mn–Ce/TiAlOX catalysts have higher NO conversion than the Mn–Ce/TiO2 catalysts. After the passage of SO2, the NOX removal rate of the Mn–Ce/TiAlOX catalyst decreased less than that of the Mn–Ce/TiO2 catalyst, which greatly improved the SO2 resistance. This is primarily due to the improved dispersion of catalyst activity on the Ti–Al–O composite carrier, the higher concentration of Mn4+ and chemically adsorbed oxygen on the surface of the catalyst, the higher reducibility, as well as the higher adsorption capacity for NH3 and NO, thus exhibiting superior catalyst activity and sulfur resistance. Inorganic non-metallic minerals include cordierite,92 montmorillonite,93 diatomite,94 and augite. Cordierite, as a bulk silicate mineral with a honeycomb shape and regular channel structure, is usually used as a monolithic carrier to achieve better catalyst performance.95 It is one of the most widely used carriers for industrial catalysts.96 Zhao et al.97 first prepared monolithic catalysts of cordierite-supported Sm-modified Mn–Ce composite oxides by impregnation method and found that at Sm/Mn molar ratio of 0.1, Sm-MnCe/cordierite catalysts had a wide activity temperature window and the NOX removal rate was above 80% in the range of 60–270 °C ([NO] = [NH3] = 500 ppm, [O2] = 5 vol%, N2 as the balanced gas and GHSV = 20000 h−1), while maintaining a high NOX conversion within 15 h at 100 ppm SO2. The appropriate Sm content increases the specific surface area and acid sites, improves the redox environment, and enhances the Mn4+ content on the catalyst surface, which is conducive to the improvement of the catalytic activity. Attapulgite is a magnesium-aluminosilicate clay mineral with a layered chain structure, which has become a catalytic carrier for many catalytic reactions due to its unique natural one-dimensional structure, abundant surface functional groups, thermal stability and good moulding properties.98 Li et al.99 prepared Mn–Ce–Fe/attapulgite (ATP) monolithic catalysts by direct ink writing 3D printing technology to study the effect of different active components on powdered catalysts in the SCR reaction. The effect of different active components in the SCR reaction on the powder catalyst was investigated. The results showed that the Mn–Ce–Fe/ATP powder catalysts contained higher Mn4+ and adsorbed state oxygen and more reducible substances at low temperatures, and the Mn–Ce–Fe/ATP powder catalysts exhibited excellent catalytic activity (90% NO conversion and 70% N2 selectivity) in a wide temperature window range of 100–400 °C ([NO] = 1250 ppm, [NH3] = 1268 ppm, [O2] = 5 vol%, N2 as the balanced gas and GHSV = 15300 h−1). Zhang et al.100 used clay minerals (PG) as the substrate supported with Sb-modified MnOX and found that Sb doping enhanced the dispersion of Mn on the carrier surface. In the presence of SO2, Sb preferentially reacted with SO2, protecting MnOX as the active species from SO2 sulfidation. Gu et al.101 synthesized MnOX–FeOX catalysts with siliceous rock and titanium siliceous rock as supports by the wet impregnation method. They found that the latter exhibited excellent catalytic performance and H2O resistance, which was attributed to the Ti in the titanium siliceous rock carrier, resulting in more acidic sites on the surface and stronger redox capacity of the active components.
3.5 Current status of various carriers
Metal oxide carriers are excellent substrates for manganese-based catalysts because of their high specific surface area, rich distribution of acidic sites and good catalytic thermal stability. However, a single metal oxide carrier is prone to cause accumulation of the active components when the catalyst is sintered. To address this, the carriers can be composited with the help of the advantages of different carriers and composite carriers can be prepared to enhance the catalytic activity of the carrier catalysts. Molecular sieve catalyst carriers have become good catalyst carriers with their unique pore structure, large specific surface area, good adsorption and high hydrothermal stability. However, the current research on molecular sieve carriers is mainly concentrated in the medium and high-temperature zone, lacking research on the SCR low-temperature zone, and sulfur poisoning and water poisoning are also key issues hindering the development of molecular sieve-based catalysts, thereby limiting their practical application. Carbon material carriers are often used as catalyst carriers because of their strong adsorption capacity, large specific surface area, rich pore structure, and numerous oxygen-containing groups. However, the single carbon-based catalysts have the disadvantages of poor stability in long-cycle operation, easy to oxidize at low temperatures, and poor resistance to SO2 poisoning. These limitations often require surface modification to meet the demand for catalytic activity. At present, single catalyst carriers have certain defects in the catalytic process, and struggle to maintain efficient denitrification performance under the conditions of SO2 and H2O presence for a long period. Therefore, the development of efficient and stable green low-temperature denitrification catalysts is of great research significance. The composite carriers can take advantage of different carriers to carry out the composite carrier, realize the synergistic effect of “1 + 1 > 2”, and improve the catalytic activity of the catalyst. Inorganic non-metallic mineral carriers should be further tapped for stable, green, easily available, cheap and composite substances based on meeting the performance requirements.102
4 Mechanism of SCR denitrification reaction over Mn-based catalysts
So far, the NH3-SCR reaction mechanism is still controversial, as different catalyst systems having different redox and acidic capacities, producing various NHXNOy active intermediates. These intermediates, in turn, affect the reaction path and reaction efficiency. The redox property determines the low-temperature activity of the catalyst, while the surface acidity determines the high-temperature activity of the catalyst, and thus, these two components are essential for a wide activity temperature window.103,104 As shown in Fig. 9.
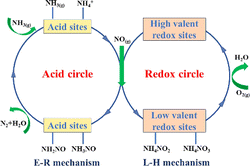 |
| | Fig. 9 Schematic diagram of NH3-SCR reaction pathway on metal oxide and zeolite catalysts (reproduced from ref. 29 with permission from ACS Publications). | |
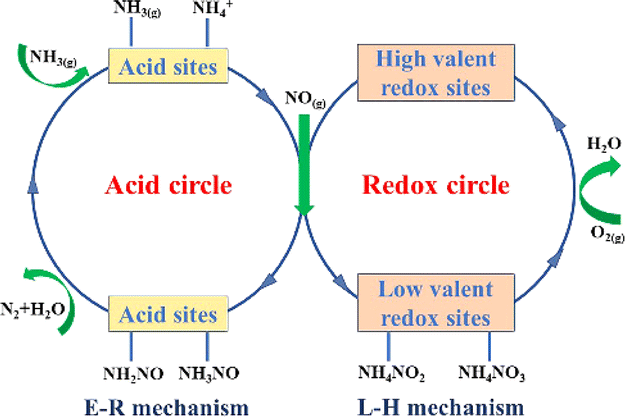
Depending on the reaction path of NO on the catalyst surface, the SCR catalytic reaction mechanism can be divided into the Langmuir–Hinshelwood (L–H) mechanism and the Eley–Rideal (E–R) mechanism.26 The L–H mechanism assumes that NH3(g) and NOX(g) (NO and NO2) are first adsorbed on the surface of the catalyst. The adsorbed NH3 then interacts with the adsorbed active sites to produce either ammonia in the coordination state (NH3-L, mainly from Lewis acid sites) or ionic ammonium (NH4+, mainly from BrØnsted acid sites). These species subsequently react with nitrates and nitrites formed from adsorbed NO to produce the transition intermediate state product NHXNOy, which is then decomposed to N2 and H2O. The E–R mechanism suggests that the adsorbed NH3 (NH3-L and NH4+) reacts with gaseous NOX to produce the transition intermediate state product NHXNOy, which then decomposes to N2 and H2O. NH3 can be adsorbed onto Lewis acid sites and BrØnsted acid sites, whereas NO is mainly physically adsorbed. The adsorption of NH3 is considered to be the first step in the catalytic reaction process because NH3 adsorbs more readily on acid sites than other reactive molecules.105,106 Generally, the activation energy required for the reacting molecules of the L–H mechanism is lower, so that the L–H mechanism is more likely to occur than the E–R mechanism at low temperatures.
According to Kapteijn, Li and Fei et al.15,107,108 on the NH3-SCR reaction mechanism, the L–H mechanism reaction pathway is approximated as, where * and (g) represent the adsorption site and gas phase:
| | |
NH4NO2* + * → N2* + 2H2O*
| (5) |
| | |
NH2*NO* + * → N2* + H2O*
| (7) |
Li et al.107 used first-principles calculations and believed that the E–R mechanism reaction pathway is:
| | |
NH3(g) + *-L → NH3* -L (Lewis acid site)
| (10) |
| | |
NH3* + * → NH2* + H*
| (11) |
| | |
NH3 + * –OH → NH4–O* (BrØnsted acid site)
| (12) |
| | |
NH2* + NO(g) → NH2*NO*
| (13) |
| | |
NH4* + NO(g) → NH4NO2*
| (14) |
| | |
NH4NO2* + * → NH2*NO* + H2O*
| (15) |
| | |
NH2*NO* + * → N2* + H2O*
| (16) |
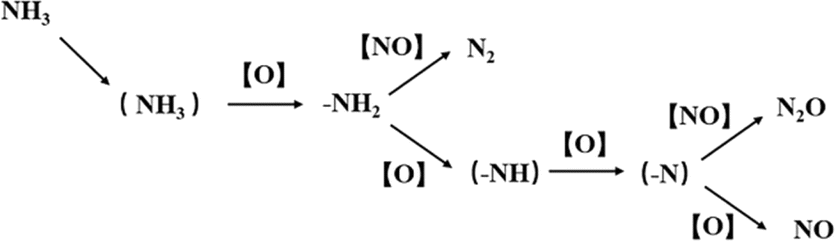
Kapteijn et al.15 believed that the denitrification pathway on pure MnOX follows the E–R mechanism, and the interaction between NO, NH3 and O2 on manganese oxides is explained by the model involves NH3 being continuously dehydrogenated by surface oxygen. As shown in Fig. 10.
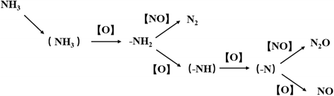 |
| | Fig. 10 NH3 activation model on MnOX (reproduced from ref. 15 with permission from ACS Publications.). | |
Ramis et al.109 conducted FT-IR research on the NH3-SCR reaction and proposed a more complete E–R reaction pathway based on the above model, as shown in Fig. 11 below.
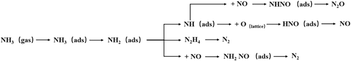 |
| | Fig. 11 Mechanism diagram of NH3 stepwise oxidation activation and NO reaction during NH3-SCR process (reproduced from ref. 109 with permission from ACS Publications.). | |
Qi et al.110 found that gaseous NH3 molecules are first adsorbed on MnOX–CeO2 catalysts to form coordinated NH3, which is then oxidized to produce NH2 and OH, and NO molecules are also adsorbed on MnOX–CeO2 catalysts, which are then oxidized to nitrates and nitrites. The reaction of NH2 and NO produces nitrosamines (NH2NO), which then decompose NH2NO to N2 and H2O. Nitrous acid is produced by the reaction in oxidation or denitrification of 2NO2 and H2O. Nitrite reacts with ammonia to produce ammonium nitrate, which is equivalent to nitrosamine hydrate (NH2NO), and both ammonium nitrite and nitrosamine are unstable intermediates, which are intermediates in numerous NH3-SCR reaction mechanisms. The reaction mechanism of NO and NH3 on the surface of the MnOX–CeO2 catalyst is as follows, where ads represents the adsorbed state:
| | |
NH3(ads) + O(ads) → NH2(ads) + OH(ads)
| (21) |
| | |
NO(g) + 1/2O2(g) → NO2(ads)
| (22) |
| | |
NH2(ads) + NO(g) → NH2NO(ads) → N2(g) + H2O(g)
| (23) |
| | |
OH(ads) + NO2(ads) → O(ads) + HNO2(ads)
| (24) |
| | |
NH3(ads) + HNO2(ads) → NH4NO2(ads) → NH2NO(ads) + H2O → N2(g) + 2H2O(g)
| (25) |
The NH3-SCR reaction process does not follow a single reaction mechanism, and many studies have shown that most of the reaction mechanisms of the current low-temperature NH3-SCR catalysts are the simultaneous existence of the L–H mechanism and the E–R mechanism, and even different temperature segments have different reaction mechanisms. For instance, at temperatures below 150 °C, the increased NO oxidation predominantly facilitates the Langmuir–Hinshelwood (L–H) mechanism on the catalyst surface. Conversely, at temperatures above 150 °C, the augmented NH3 adsorption capacity primarily promotes the Eley–Rideal (E–R) mechanism.111 Different species dominate the adsorption at acid sites on the catalyst surface depending on the temperature. Specifically, at lower temperatures, coordinated NH3 adsorbed on Lewis acid sites is predominant. As the temperature increases, NH4+ adsorbed on BrØnsted acid sites becomes the leading species.112
Gu et al.113 simulated the gas adsorption process on the Mn active centres on the MnOX/SiO2 β-cristobalite (101) surface based on density functional theory. Under anaerobic conditions, NO was more readily adsorbed on the surface of Mn2O3/SiO2 β-cristobalite (101), while NH3 was more readily adsorbed on the surface of MnO2/SiO2 β-cristobalite (101). The NO adsorption reaction mainly followed the L–H mechanism, whereas the NH3 adsorption reaction mainly followed the E–R mechanism, and the O2 adsorption processes on the Mn active centres of the two catalysts were similar. The main reason for the better catalytic activity of MnO2/SiO2 than that of Mn2O3/SiO2 is the difference in NH3 adsorption energy between the catalysts.
Wang et al.114 investigated the reaction mechanism of the synergistic effect of MnOX–CeO2 in the NH3-SCR reaction based on experimental and density-functional theory and found that the synergistic effect is to promote the catalytic activity through the formation of surface oxygen induced by the electron transfer between Ce4+ and Mn2+, and the establishment of Mn redox cycle and Ce redox cycle to activate the NH3 and O2, respectively. Firstly, owing to the oxidizing ability of Ce4+ in E-CeO2, a reaction takes place between Ce4+ and Mn2+: Ce4+ + Mn2+ → Mn3+ + Ce3+. Concurrently, surface oxygen vacancies (OV) are generated and stabilised on the surface of E-CeO2. These surface oxygen vacancies significantly enhance the adsorption and dissociation of O2, thereby oxidising Ce3+ back to Ce4+. The resultant dissociated O atoms further oxidise Ce3+ and occupy the surface oxygen vacancies, thus forming a Ce redox cycle. Gaseous NO is adsorbed onto the E-CeO2 lattice oxygen near the MnOX clusters, manifesting as nitrites and nitrates. Simultaneously, NH3 coordinates with Mn3+ species (Lewis acid sites) and is subsequently activated by the nearby Mn3+ ions. The activated NH3 then reacts with NO to yield N2 and H2O, during which a Mn3+ ion is reduced to Mn2+. After H* atoms diffuse from Mn–OH to form Ce–OH, the Mn2+ ions are oxidised back to Mn3+ by Ce4+, completing the Mn redox cycle. Fig. 12 illustrates the schematic diagram of the synergistic effect mechanism of the Mn/E-CeO2 catalyst.
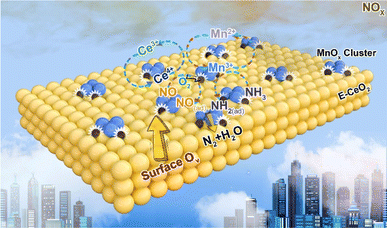 |
| | Fig. 12 Schematic reaction mechanism of MnOX–CeO2 catalyst synergistic effect (reprinted from ref. 114. Copyright 2023, with permission from Elsevier). | |
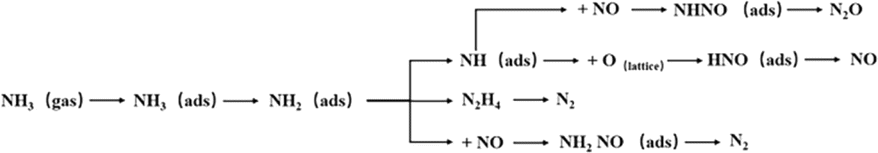
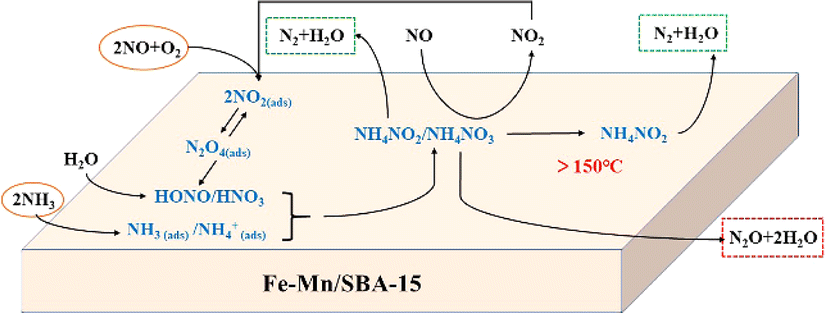
Li et al.115 investigated the mechanism of NO reduction and N2O formation, and suggested a possible mechanism on Fe–Mn/SBA-15 catalysts. The Fe–Mn/SBA-15 catalyst primarily follows the L–H mechanism at low temperatures (200 °C). However, as the temperature increases, the E–R mechanism becomes more prominent and dominates at higher temperatures (250 °C). Since Fe–Mn/SBA-15 is a strongly alkaline catalyst, the adsorption capacity of NH3 on the molecular sieve is weak, while the adsorption of NO and O2 onto the molecular sieve surface is strong. The adsorbed species form intermediates, NH4NO2 or NH4NO3, where NH4NO2 decomposes into N2O and H2O. This process aligns with the L–H mechanism. The generated intermediate NH4NO3 can react with gaseous NO to produce NH4NO2 and NO2. Furthermore, NH4NO3 can directly decompose to produce NO and H2O. The entire process adheres to the E–R mechanism. The denitrification process of the Fe–Mn/SBA-15 catalyst during the SCR reaction is illustrated in Fig. 13.
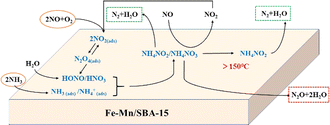 |
| | Fig. 13 Low temperature NH3-SCR reaction mechanism of Fe Mn/SBA-15 catalyst (reproduced from ref. 115 with permission from ACS Publications.). | |
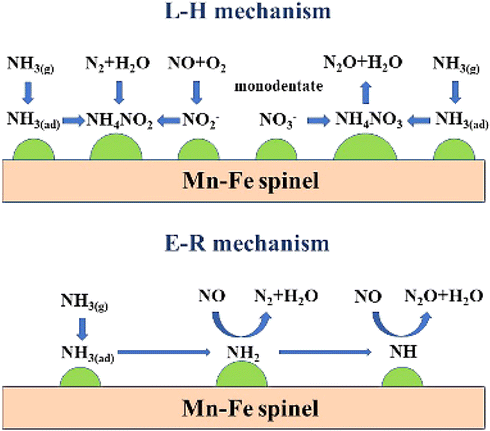
Yang et al.116 investigated the mechanism of N2O and NO generation in the low-temperature NH3-SCR process of Mn–Fe spinel using in situ diffuse reflectance Fourier transform infrared spectroscopy (in situ DRIFTS) and transient reactions, and found that the L–H mechanism and the E–R mechanism existed simultaneously in the SCR reaction. As shown in Fig. 14.
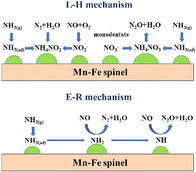 |
| | Fig. 14 Schematic diagram of the coexistence of L–H and E–R mechanisms on Mn–Fe spinel catalyst (reproduced from ref. 116 with permission from ACS Publications). | |
The L–H mechanism on Mn–Fe spinel catalysts was developed as follows:105,117
| | |
Mn4+O + NO(ads) → Mn3+–O–NO
| (28) |
| | |
Mn3+–O–NO + 1/2O2 → Mn3+–O–NO2
| (29) |
| | |
Mn3+–O–NO + 1/2O2 → Mn3+(O)2NO
| (30) |
| | |
Mn3+–O–NO + NH3(ads) → Mn3+–O–NO–NH3 → Mn3+–OH + N2 + H2O
| (31) |
| | |
Mn3+(O)2NO + NH3(ads) → Mn3+–O–NO2–NH3 → Mn3+–OH + N2O + H2O
| (32) |
| | |
Mn3+(O)2NO + NH3(ads) → Mn3+(O2)–NO–NH3
| (33) |
| | |
Mn3+–OH + 1/4O2 → Mn4+O + 1/2H2O
| (34) |
The Eley–Rideal principle on Mn–Fe spinel catalysts was as follows:105,117
| | |
NH3(ads) + Mn4+O → NH2 + Mn3+–OH
| (36) |
| | |
NH2 + NO(g) → N2 + H2O
| (37) |
| | |
NH2 + Mn4+O → NH + Mn3+–OH
| (38) |
| | |
NH + NO(g) + Mn4+O → N2O + Mn3+–OH
| (39) |
| | |
Mn3+–OH + 1/4O2 → Mn4+O + 1/2H2O
| (40) |
Zhang et al.118 prepared manganese-based SCR catalysts using homemade pyrolysis coke as the carrier, analyzing the catalytic mechanism of Mn@X/C (@ is Ce, Mo or Co) catalysts and found that the mechanism of SCR reaction was:
| | |
NH3(g) + Mn–OH → NH4+(ads)–O–Mn (BrØnsted acid site)
| (41) |
| | |
NH4+(ads) + NO(g) + Mn4+O → N2 + H2O + Mn3+–OH
| (42) |
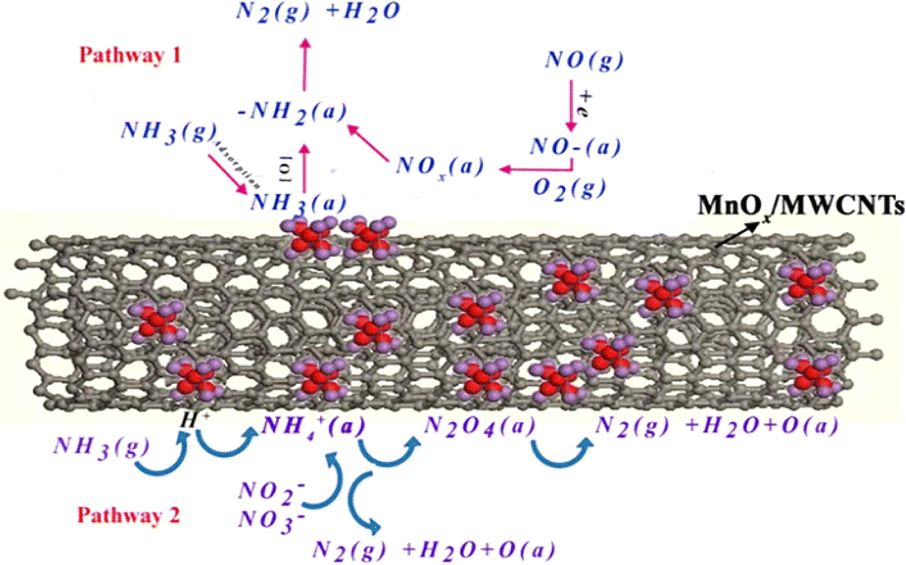
Yu et al.119 conducted a reaction mechanism study of loaded MnOX/MWCNTS using the in situ DRIFTS technique and discussed the intermediates and NH3-SCR reaction pathways during denitrification of MnOX/MWCNTS catalysts at 210 °C, and proposed two possible reaction pathways. One is the reaction of the NOX active component with NH4+ to produce NH4N2O4(a), NH4NO2(a) or NH4NO3(a) intermediates and ultimately generates N2 and H2O. The other pathway is that NH3 is first adsorbed on the active site to generate NH2, and then NH2 reacts with the NOx active component to generate the unstable intermediates NH2NO2 or NH2NO3, which then decompose into N2 and H2O. This is illustrated in Fig. 15.
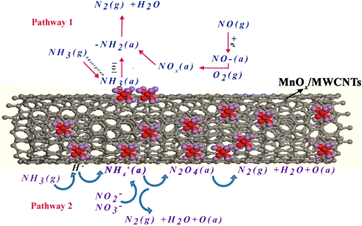 |
| | Fig. 15 Schematic diagram of two reaction pathways on MnOX/MWCNTS catalyst at 210 °C (reprinted from ref. 119. Copyright 2015, with permission from AAGR Aerosol and Air Quality Research.). | |
Although the mechanism of NH3-SCR reaction has been studied extensively, due to the complexity of the actual working flue gas conditions and the different mechanisms of Mn-based catalysts, it is necessary to further investigate the reaction mechanisms contained in NH3-SCR catalysts in-depth and to combine kinetics, solid surface chemistry, and computational chemistry to give the mechanism of NH3-SCR reaction of Mn-based catalysts.
5 Poisoning mechanism of Mn-based catalysts in NH3-SCR
The flue gas composition is complex. Even after purification by dust removal and desulfurization equipment, there will be a small amount of H2O and SO2 in the processed flue gas.120 The manganese catalyst is more sensitive to the residual deactivation substances in the flue gas. Once poisoned and deactivated, its denitrification performance will be severely impacted. Therefore, it is required that the catalyst has a high sulfur resistance and water resistance,121 so the study of the manganese catalyst poisoning mechanism is crucial for the realization of the practical application of the catalyst.
5.1 Mechanism of H2O poisoning
Water vapour will greatly reduce the catalytic activity of the catalyst in the low-temperature SCR reaction. The deactivation of catalysts by water vapour is divided into reversible and irreversible deactivation.64,122 The reversible deactivation of the catalyst by H2O vapor is usually considered to be the competitive adsorption of H2O with NO and NH3 on the catalyst surface, which occupies the active sites and leads to lower reactant adsorption reducing the NOX conversion rate. However, the inhibition will disappear gradually with the increase in temperature. Liu et al.123 by comparing the deNOX performance of β-MnO2 and Co–MnO2 catalysts in the presence of water vapour, found that water vapour will form competitive adsorption with NH3 and inhibit the adsorption and interfacial reaction of NH3 on the surface of the catalysts. This lower NH3 adsorption leads to poorer NOX conversion rate. However, H2O molecules have a reversible effect on the gas adsorption on the catalyst surface and thus have a slight impact on the NH3-SCR activity,124 which is the same as Xiong et al.125 found that the presence of water affects the effect of the SCR performance of Mn–Fe spinel. In the absence of water vapour, the NOX conversion rate of Mn–Fe spinel exceeds 80% at temperatures above 140 °C. However, the addition of 5% water vapour significantly reduces the NOX conversion rate, reaching only 40% at 140 °C ([NO] = [NH3] = 500 ppm, [O2] = 2 vol%, N2 as the balanced gas and GHSV = 120000 h−1). The presence of water forms competitive adsorption with the reacting molecules, which reduces the oxidation capacity of the catalyst and inhibits the occurrence of its interfacial reaction, resulting in a decrease in the catalytic activity. Yan et al.126 also discovered that high humidity conditions not only reduce acidity and hinder the adsorption of NH3 on the catalyst surface, but also that the dissolution of water molecules affects the structure of the catalyst, leading to a decrease in the dispersion of the active components. With the introduction of 35% water vapour, the activity of the Co–Mn–Ce/TiO2 (stearic acid) catalyst stabilises at approximately 30%. ([NO] = [NH3] = 600 ppm, [O2] = 5 vol%, N2 as the balanced gas and GHSV = 15000 h−1). But the activity of the catalyst is restored when the introduction of H2O is ceased. Similarly, Lin et al.127 and Hu et al.128 found that the adverse effect of H2O on the catalyst was reversible when the introduction of H2O was stopped. The irreversible deactivation of the catalyst by H2O vapour is typically attributed to the decomposition of H2O into hydroxyl radicals on the catalyst surface, leading to the blocking of the active sites, and resulting in a decrease in the denitrification activity and is irreversible.129 Moreover, hydroxyl radicals can only be dissociated at high temperatures (252–502 °C). After Liu et al.130 introduced water vapour into the Fe0.75Mn0.25TiOX catalyst, the hydroxyl groups produced by the decomposition of water molecules caused a transformation of some Lewis acid sites into BrØnsted acid sites. This resulted in a decrease in the intensities of the corresponding wavelengths of NH3 and NOX, leading to a reduction in the catalyst's denitrification activity. Similarly, Yan et al.126 also observed that under humid conditions, water molecules adsorbed onto the surface of the Co–Mn–Ce/TiO2 catalyst to form hydroxyl groups. These hydroxyl groups had an irreversible impact on the SCR denitrification process.131
5.2 Mechanism of SO2 poisoning
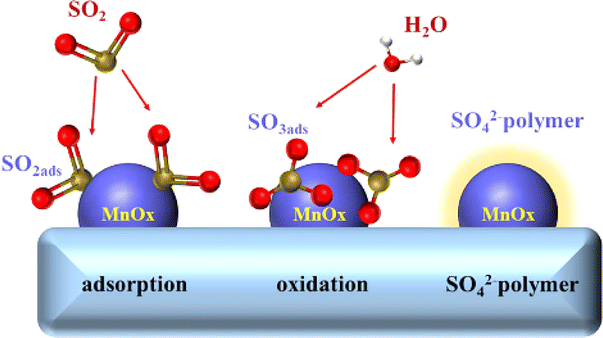
While some studies suggest that catalyst SO2 poisoning is reversible,132,133 the majority of research indicates that its effects are irreversible.134,135 The impact of SO2 on the catalyst accumulates over time, ultimately resulting in a sustained and irreversible decline in catalytic activity. The deactivation mechanism of catalyst SO2 poisoning can be categorised into three cases:136–138 Firstly, SO2 in the flue gas is oxidised to SO3 by the catalyst. This SO3 subsequently reacts with NH3 to form (NH4)2SO4 and NH4HSO4, which deposit onto the catalyst surface, blocking active sites and obstructing the pore structure. This process leads to a reduction in catalytic activity. Secondly, SO2 competes with the reactants for adsorption sites, hindering the catalytic reaction process. When both SO2 and NO are present in the flue gas, they compete for adsorption sites. SO2 preferentially occupies these sites, leading to the sulfation of the catalyst. Thirdly, SO2 can react directly with the active components of the catalyst, resulting in the sulfation of the active metal atoms. This process deactivates the catalyst and disrupts the redox cycle of the active phase. Zhang et al.139 discovered that SO2 readily oxidised to SO3 on the surface of MnOX/palygorskite (PG) catalysts, leading to the formation of polysulfuric acid. This acid encapsulated the active components and blocked the micropores, causing the initial deactivation of the MnOX/PG catalysts. The subsequent deposition of ammonium sulfate was not the primary cause of deactivation. As illustrated in Fig. 16. Xiao et al.140 proposed that the inhibition of NO conversion by SO2 was due to competitive adsorption on the active sites of the catalyst. The adsorbed SO2 was then further oxidised to inactive sulfate on the catalyst surface. Jiang et al.141 suggested that the addition of SO2 reduced the oxidation performance and surface acidity of the catalyst inhibiting NOx conversion. Xiong et al.142 found that the irreversible deactivation of the Mn3O4 spinel catalysts was primarily cause by the reaction of SO2 with Mn atoms in the active centre, resulting in the formation of MnSO4. This finding aligns with Chen et al.,143 who concluded that MnOX catalysts are poisoned by SO2, leading to the formation of MnSO4 on the surface, rather than (NH4)2SO4 and NH4HSO4. This sulfation of the active Mn atoms reduces the number of active components and reactive sites, ultimately decreasing catalytic activity.
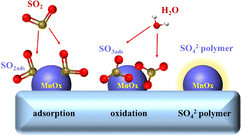 |
| | Fig. 16 SO2 poisoning process on the surface of MnOX/PG catalyst (reproduced from ref. 139 with permission from MDPI.). | |
In practice, SO2 and H2O coexist in the flue gas, and their poisoning effect on the catalyst exhibits a synergistic effect. This exacerbates the formation of sulfate and accelerates catalyst deactivation. Consequently, it is essential that the catalysts under investigation possess resistance to both sulfur and water. Common methods for enhancing catalyst performance and resistance to sulfur and water include metal modification or doping, selection of appropriate carriers, and the rational design of morphology and structure.122 Generally, manganese-based catalysts have high low-temperature catalytic activity, but their tolerance to SO2 and H2O limits their industrial applications. Therefore, researchers modify or dope them by adding metal elements to improve their resistance to sulfur and water. CeO2, with an excellent oxidative reduction ability, is often employed as a catalyst additive or carrier. Ce, acting as a sacrificial agent, preferentially reacts with SO2 to form CeSO4. This compound is easier to decompose than MnSO4, thus preventing the formation of MnSO4, which would otherwise deposit on the acidic sites of the catalyst surface and cause poisoning. By preferentially reacting with SO2, Ce effectively avoids the poisoning of the catalyst and increases the number of acidic sites on its surface.144 Yoon et al.145 synthesised Ce-doped Mn–Cr layered structure catalysts using a co-precipitation method. Their research demonstrated that Ce doping effectively inhibits the formation of manganese sulphate on the catalyst surface, reduces the decomposition temperature of ammonium sulphate, and enhances both the acidity and reducibility of the catalyst surface. These improvements contribute to a significant enhancement in the SO2 resistance of Mn catalysts. Both Sb and Ce, as rare earth metals, possess the ability to enhance sulfur and water resistance. Yan et al.146 prepared Sb-modified Mn–Ce–SbX/TiO2 catalysts using the impregnation method. Their findings indicated that the addition of Sb effectively inhibits the formation of sulphate on the catalyst surface in the presence of SO2 and H2O.147–149 Zhang et al.100 also observed that doping Sb into the MnOX/palygorskite (PG) catalyst significantly inhibited the sulphation of the active phase. Simultaneously, Sb promoted the dispersion of MnOX on the carrier surface. The preferential reaction between SbOX and SO2 effectively protected the active MnOX from sulfation by SO2, thus enhancing the catalyst's tolerance to SO2. A suitable hydrophobic carrier not only provides a large specific surface area and numerous pore structures to enhance catalyst acidity but also plays a crucial role in the synergistic effect between the carrier and the active component, significantly impacting the catalyst's resistance to sulfur water.150 Pure Al2O3, with its large specific surface area and abundant acidic sites,151,152 promotes good dispersion of the active substances. Additionally, it provides ample adsorption sites for reactants,153 facilitating the adsorption of chemically adsorbed oxygen on the catalyst surface, thereby exhibiting improved sulfur and water resistance. Liu et al.73 prepared Mn-based catalysts supported on γ-Al2O3, TiO2, and MCM-41 using an impregnation method. They observed that pure γ-Al2O3 provided abundant adsorption sites and that a strong interaction existed between Mn and γ-Al2O3. The Mn/γ-Al2O3 catalyst exhibited the strongest NO adsorption performance and good SO2 tolerance. Rational morphology and structural design are also effective measures for enhancing the sulfur and water resistance of catalysts. The unique pore structure of TiO2 nanotubes, combined with their large specific surface area, numerous acidic sites, and reactive oxygen species on the surface, makes them ideal as catalyst carriers. These properties collectively contribute to enhancing the low-temperature denitrification activity of manganese-based catalysts and improving their resistance to sulfur and water resistance.154,155 Qin et al.156 synthesised a TiO2 support using a hydrothermal method and subsequently prepared a novel flower-shaped MnCe/TiO2 catalyst. Their research revealed that the active species of the MnCe/TiO2-Flower catalyst exhibited high dispersion, abundant acid sites, a large specific surface area, and excellent redox properties. This catalyst achieved a NO conversion rate of approximately 100% at 150–250 °C, a N2 selectivity exceeding 80% at 150–350 °C ([NO] = [NH3] = 600 ppm, [O2] = 5 vol%, Ar as the balanced gas and GHSV = 108000 h−1), and demonstrated excellent tolerance to SO2 and H2O ([SO2] = 100 ppm, [H2O] = 5 vol%). In summary, the poisoning of low-temperature manganese-based catalysts by H2O and SO2 is intricately linked to the catalytic reaction process. This includes competitive adsorption of H2O and NH3 on the catalyst surface, SO2-induced acidification of active centre atoms, and the occupation of active sites by deposited (NH4)2SO4 and NH4HSO4. These phenomena are directly related to the physicochemical properties of the manganese-based catalysts. Therefore, to enhance the H2O/SO2 resistance of these catalysts during the SCR process, a top-level design approach is required, considering the catalytic mechanism. This involves strategies such as employing hydrophobic materials, constructing a unique core–shell structure, modifying the catalyst through metal doping, and integrating DFT theoretical calculations. By screening for SO2 and incorporating spatial barriers into the material design, the anti-H2O/SO2 performance of manganese-based catalysts can be significantly improved.157
6 Conclusions and outlook
Manganese-based catalysts have emerged as a research hotspot due to their excellent low-temperature denitrification performance. While single manganese-based catalysts exhibit promising NH3-SCR catalytic activity, they suffer from a narrow operating temperature window and poor water and sulfur resistance. Multicomponent manganese-based catalysts, obtained through modification by incorporating transition metals or rare earth elements, demonstrate superior catalytic activity, high denitrification efficiency, and improved water and sulfur resistance. Supported manganese-based catalysts, owing to the presence of carriers, benefit from a large surface area, strong surface acidity, and a high density of active sites. These properties contribute to their excellent low-temperature NOX removal efficiency, N2 selectivity, a wider operating temperature window, and enhanced water and sulfur resistance. Research on Mn-based denitrification catalysts have yielded significant results, highlighting the practical importance of further investigating low-temperature Mn-based denitrification catalysts with high activity and stability. These catalysts hold promise for industrial application and commercialisation. Future research should focus on the following aspects:
(1) While significant progress has been made in enhancing catalyst resistance to SO2/H2O, the long-term durability and stability of these catalysts in the continuous presence of SO2/H2O require further in-depth exploration.
(2) MnOX has been modified by introducing other metal oxides to enhance the catalytic activity of low-temperature NH3-SCR. The prevailing explanation for this enhancement is that MnOX possesses strong redox properties (high-valent Mn4+), a high density of surface defects and acidic sites, a large surface area, and significant surface chemical adsorption of oxygen. However, the underlying mechanisms requires further in-depth study.
(3) Currently, the simulated gas composition used in SCR denitrification reaction studies is simplified, leading to overly idealised experimental results. Future research should focus on conducting industrial-scale tests to verify the denitrification performance, water and sulfur resistance, and reaction mechanisms under actual operating conditions.
(4) Currently, there is limited research on the impact of catalyst forming technology on catalyst performance. In-depth studies are needed to investigate the trends in catalytic activity under different moulding process conditions. For instance, coated honeycomb catalysts and extruded honeycomb catalysts have demonstrated promising advantages in terms of high catalytic activity. Further research is warranted in this area.
(5) While the L–H and E–R mechanism of the SCR denitrification reaction have been extensively studied in the literature, the NH3-SCR reaction mechanism at low temperatures remains unclear. Techniques such as in situ DRIFTS, in situ Raman spectroscopy, and computational molecular simulation could provide valuable insights into the adsorption states of NH3, NO, O2, SO2, and H2O on the catalyst surface. By monitoring the reaction process, we can elucidate the reaction mechanism of low-temperature NOX removal and develop a comprehensive reaction mechanism that can explain the NH3-SCR reaction mechanism.
(6) Optimising catalyst structure design, extending catalyst operating cycles based on real-world operating conditions, and carefully considering the balance between catalyst cost and performance are crucial steps in developing more efficient, stable, and environmentally friendly low-temperature SCR denitrification catalysts for industrial applications.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Author contributions
Jiadong Zhang: conception, investigation, writing-review and editing; Zengyi Ma: methodology, review and editing; Ang Cao: methodology, review and editing; Jianhua Yan: methodology, review and editing; Yuelan Wang: investigation and methodology; Miao Yu: investigation and methodology; Linlin Hu: investigation and methodology; Shaojing Pan: investigation and methodology. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (2022ZFJH04), the projects of the National Natural Science Foundation (No. 52236008) and the Fundamental Reserach Funds for the Central Universities (NO. 226-2024-00225). The authors would like to thank the editor for editing the manuscript and the anonymous reviewers for providing detailed and helpful comments.
References
- Y. Shi, Y. Xia, B. Lu, N. Liu, L. Zhang, S. Li and W. Li, J. Zhejiang Univ., Sci., A, 2014, 15, 454–464 CrossRef CAS.
- L. Chen, Q. Wang, X. Wang, Q. Cong, H. Ma, T. Guo, S. Li and W. Li, Chem. Eng. J., 2020, 390, 124251 CrossRef CAS.
- Y. Shan, J. Du, Y. Yu, W. Shan, X. Shi and H. He, Appl. Catal., B, 2020, 266, 118655 CrossRef CAS.
- X. X. Cheng and X. T. T. Bi, Particuology, 2014, 16, 1–18 CrossRef CAS.
- M. Fu, C. Li, P. Lu, L. Qu, M. Zhang, Y. Zhou, M. Yu and Y. Fang, Catal. Sci. Technol., 2014, 4, 14–25 RSC.
- R. Zhang, N. Liu, Z. Lei and B. Chen, Chem. Rev., 2016, 116, 3658–3721 CrossRef CAS.
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS.
- Z. Xiong, C. Wu, Q. Hu, Y. Wang, J. Jin, C. Lu and D. Guo, Chem. Eng. J., 2016, 286, 459–466 CrossRef CAS.
- Z. Huang, X. Gu, W. Wen, P. Hu, M. Makkee, H. Lin, F. Kapteijn and X. Tang, Angew. Chem., Int. Ed., 2013, 52, 660–664 CrossRef CAS.
- D. Damma, P. R. Ettireddy, B. M. Reddy and P. G. Smirniotis, Catalysts, 2019, 9, 349 CrossRef CAS.
- Z. Liu, B. Jia, Y. Zhang and M. Haneda, Ind. Eng. Chem. Res., 2020, 59, 13916–13922 CrossRef CAS.
- S. Pan, H. Luo, L. Li, Z. Wei and B. Huang, J. Mol. Catal. A: Chem., 2013, 377, 154–161 CrossRef CAS.
- Y. Ma, F. Gao, G. Jia, S. Huang, S. Zhao, H. Yi and X. Tang, Mod. Chem. Ind., 2019, 39, 33–37 Search PubMed.
- Y. Zheng and X. Wang, Funct. Mater., 2014, 45, 11008–11012 CAS.
- F. Kapteijn, L. Singoredjo, A. Andreini and J. A. Moulijn, Appl. Catal., B, 1994, 3, 173–189 CrossRef CAS.
- X. Tang, J. Hao, W. Xu and J. Li, Catal. Commun., 2007, 8, 329–334 CrossRef CAS.
- J. Zhou, B. Wang, J. Ma, G. Li, Q. Sun, W. Xu and Y. Li, Environ. Chem., 2018, 37, 782–791 CAS.
- G. He, M. Gao, Y. Peng, Y. Yu, W. Shan and H. He, Environ. Sci. Technol., 2021, 55, 6995–7003 CrossRef CAS.
- L. Ye, P. Lu, X. Chen, P. Fang, Y. Peng, J. Li and H. Huang, Appl. Catal., B, 2020, 277, 119257 CrossRef CAS.
- H. Liu, X. Li, Q. Dai, H. Zhao, G. Chai, Y. Guo, Y. Guo, L. Wang and W. Zhan, Appl. Catal., B, 2021, 282, 119577 CrossRef CAS.
- Z. Zhang, J. Li, J. Tian, Y. H. Zhong, Z. Zou, R. Dong, S. Gao, W. B. Xu and D. L. Tan, Fuel Process. Technol., 2022, 230, 107213 CrossRef CAS.
- J. Huang, H. Huang, L. Liu and H. Jiang, Mol. Catal., 2018, 446, 49–57 CrossRef CAS.
- F. Kapteijn, J. RodriguezMirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25–64 CrossRef CAS.
- W. Shan and H. Song, Catal. Sci. Technol., 2015, 5, 4280–4288 RSC.
- J. Yang, PhD thesis, Chongqing University,China, 2022, DOI:10.27670/d.cnki.gcqdu.2022.000390.
- C. Liu, J. Shi, C. Gao and C. Niu, Appl. Catal., A, 2016, 522, 54–69 CrossRef CAS.
- X. Tang, J. Li, L. Sun and J. Hao, Appl. Catal., B, 2010, 99, 156–162 CrossRef CAS.
- C. L. Yu, B. C. Huang, L. F. Dong, F. Chen and X. Q. Liu, Catal. Today, 2017, 281, 610–620 CrossRef CAS.
- L. Han, S. Cai, M. Gao, J. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS.
- J. Liu, Y. Wei, P. Li, P. Zhang, W. Su, Y. Sun, R. Zou and Y. Zhao, ACS Catal., 2018, 8, 3865–3874 CrossRef CAS.
- S. Andreoli, F. A. Deorsola, C. Galletti and R. Pirone, Chem. Eng. J., 2015, 278, 174–182 CrossRef CAS.
- P. Gong, J. Xie, D. Fang, D. Han, F. He, F. Li and K. Qi, Chin. J. Catal., 2017, 38, 1925–1934 CrossRef CAS.
- B. Zhao, R. Ran, X. Wu and D. Weng, Appl. Catal., A, 2016, 514, 24–34 CrossRef CAS.
- Q. Yu, J. Xiong, Z. Li, X. Mei, P. Zhang, Y. Zhang, Y. Wei, Z. Zhao and J. Liu, Catal. Today, 2021, 376, 229–238 CrossRef CAS.
- W. Tian, H. Yang, X. Fan and X. Zhang, J. Hazard. Mater., 2011, 188, 105–109 CrossRef CAS.
- J. Shi, C. Gao, C. Liu, Z. Fan, G. Gao and C. Niu, J. Nanopart. Res., 2017, 19, 194 CrossRef.
- N. Husnain, E. Wang, K. Li, M. T. Anwar, A. Mehmood, M. Gul, D. Li and J. Mao, Rev. Chem. Eng., 2019, 35, 239–264 CrossRef CAS.
- X. Leng, Z. Zhang, Y. Li, T. Zhang, S. Ma, F. Yuan, X. Niu and Y. Zhu, Fuel Process. Technol., 2018, 181, 33–43 CrossRef CAS.
- Q. Huang, H. Si, S. Yu, J. Wang, T. Tao, B. Yang, Y. Zhao and M. Chen, Environ. Technol., 2020, 41, 1664–1676 CrossRef CAS.
- X. Tang, C. Wang, F. Gao, Y. Ma, H. Yi, S. Zhao and Y. Zhou, J. Environ. Chem. Eng., 2020, 8, 104399 CrossRef CAS.
- S. Xiong, Y. Peng, D. Wang, N. Huang, Q. Zhang, S. Yang, J. Chen and J. Li, Chem. Eng. J., 2020, 387, 124090 CrossRef CAS.
- X. Wang, R. Duan, W. Liu, D. Wang, B. Wang, Y. Xu, C. Niu and J. Shi, Appl. Surf. Sci., 2020, 510, 145517 CrossRef CAS.
- K. Zhu, W. Yan, S. Liu, X. Wu, S. Cui and X. Shen, Appl. Surf. Sci., 2020, 508, 145024 CrossRef CAS.
- J. Rong, W. Zhao, W. Luo, K. Kang, L. Long, Y. Chen and X. Yao, J. Rare Earths, 2023, 41, 1323–1335 CrossRef CAS.
- M. T. Le, S. Singh, M. Nguyen-Quang, A. B. Ngo, A. Bruckner and U. Armbruster, Sci. Total Environ., 2021, 784, 147394 CrossRef CAS.
- D. Ng, D. Acharya, X. Wang, C. D. Easton, J. Wang and Z. Xie, J. Chem. Technol. Biotechnol., 2021, 96, 2681–2695 CrossRef CAS.
- Y. Shi, H. Yi, F. Gao, S. Zhao, Z. Xie and X. Tang, J. Hazard. Mater., 2021, 413, 125361 CrossRef CAS.
- C. Zhang, T. Chen, H. Liu, D. Chen, B. Xu and C. Qing, Appl. Surf. Sci., 2018, 457, 1116–1125 CrossRef CAS.
- J. Jia, R. Ran, X. Guo, X. Wu, W. Chen and D. Weng, Catal. Commun., 2019, 119, 139–143 CrossRef CAS.
- Y. Li, Y. P. Li, P. Wang, W. Hu, S. Zhang, Q. Shi and S. Zhan, Chem. Eng. J., 2017, 330, 213–222 CrossRef CAS.
- F. Gao, X. Tang, H. Yi, S. Zhao, J. Wang and T. Gu, Appl. Surf. Sci., 2019, 466, 411–424 CrossRef CAS.
- Q. Yan, S. Chen, C. Zhang, Q. Wang and B. Louis, Appl. Catal., B, 2018, 238, 236–247 CrossRef CAS.
- Y. Shi, H. Yi, F. Gao, S. Zhao, Z. Xie and X. Tang, Sep. Purif. Technol., 2021, 265, 118517 CrossRef CAS.
- Q. Zhao, B. Chen, J. Li, X. Wang, M. Crocker and C. Shi, Appl. Catal., B, 2020, 277, 119215 CrossRef CAS.
- J. Huang, H. Huang, H. Jiang and L. Liu, Catal. Today, 2019, 332, 49–58 CrossRef CAS.
- Z. Fan, J. Shi, C. Gao, G. Gao, B. Wang, Y. Wang, C. He and C. Niu, Chem. Eng. J., 2018, 348, 820–830 CrossRef CAS.
- C. Li, X. Tang, H. Yi, L. Wang, X. Cui, C. Chu, J. Li, R. Zhang and Q. Yu, Appl. Surf. Sci., 2018, 428, 924–932 CrossRef CAS.
- Y. Jia, J. Jiang, R. Zheng, L. Guo, J. Yuan, S. Zhang and M. Gu, J. Hazard. Mater., 2021, 412, 125258 CrossRef CAS.
- S. Li, B. Huang and C. Yu, Catal. Commun., 2017, 98, 47–51 CrossRef CAS.
- H. Chang, X. Chen, J. Li, L. Ma, C. Wang, C. Liu, J. W. Schwank and J. Hao, Environ. Sci. Technol., 2013, 47, 5294–5301 CrossRef CAS.
- D. Ren, K. Gui and S. Gu, Appl. Surf. Sci., 2021, 561, 149847 CrossRef CAS.
- S. Hao, Y. Cai, C. Sun, J. Sun, C. Tang and L. Dong, Catalysts, 2020, 10, 1329 CrossRef CAS.
- F. Gao, X. Tang, H. Yi, J. Li, S. Zhao, J. Wang, C. Chu and C. Li, Chem. Eng. J., 2017, 317, 20–31 CrossRef CAS.
- Y. Liu, Y. Hou, X. Han, J. Wang, Y. Guo, N. Xiang, Y. Bai and Z. Huang, ChemCatChem, 2020, 12, 953–962 CrossRef CAS.
- G. Li, D. Mao, M. Chao, G. Li, J. Yu and X. Guo, J. Rare Earths, 2021, 39, 805–816 CrossRef CAS.
- A. Serrano-Lotina, M. Monte, A. Iglesias-Juez, P. Pavon-Cadierno, R. Portela and P. Avila, Appl. Catal., B, 2019, 256, 117821 CrossRef CAS.
- B. Jia, J. Guo, H. Luo, S. Shu, N. Fang and J. Li, Appl. Catal., A, 2018, 553, 82–90 CrossRef CAS.
- S. Raja, M. S. Alphin and L. Sivachandiran, Catal. Sci. Technol., 2020, 10, 7795–7813 RSC.
- Z. Wu, R. Jin, Y. Liu and H. Wang, Catal. Commun., 2008, 9, 2217–2220 CrossRef CAS.
- X. Zhang, X. Zhang, X. Yang, Y. Chen, X. Hu and X. Wu, Chem. Eng. Sci., 2021, 238, 116588 CrossRef CAS.
- W. Zhao, Y. Tang, Y. Wan, L. Li, S. Yao, X. Li, J. Gu, Y. Li and J. Shi, J. Hazard. Mater., 2014, 278, 350–359 CrossRef CAS.
- X. Yao, T. Kong, S. Yu, L. Li, F. Yang and L. Dong, Appl. Surf. Sci., 2017, 402, 208–217 CrossRef CAS.
- L. Liu, B. Shen, M. Si, P. Yuan, F. Lu, H. Gao, Y. Yao, C. Liang and H. Xu, RSC Adv., 2021, 11, 18945–18959 RSC.
- Y. Zeng, Z. Wu, L. Guo, Y. Wang, S. Zhang and Q. Zhong, Mol. Catal., 2020, 488, 110916 CrossRef CAS.
- J. Li, C. Zhang, Q. Li, T. Gao, S. Yu, P. Tan, Q. Fang and G. Chen, Chem. Eng. Sci., 2022, 251, 117438 CrossRef CAS.
- H. Yan, N. Zhang and D. Wang, Chem Catal., 2022, 2, 1594–1623 CrossRef CAS.
- L. Li, B. Sun, J. Sun, S. Yu, C. Ge, C. Tang and L. Dong, Catal. Commun., 2017, 100, 98–102 CrossRef CAS.
- X. Liu, Z. Sui, H. Chen, Y. Chen, H. Liu, P. Jiang, Z. Shen, W. S. Linghu and X. Wu, J. Environ. Sci., 2021, 104, 137–149 CrossRef CAS.
- Y. Liang, X. Wang, Q. Zhang, S. Luo and Y. Zhou, J. Fuel Chem. Technol., 2020, 48, 205–212 CAS.
- W. Xu, G. Zhang, H. Chen, G. Zhang, Y. Han, Y. Chang and P. Gong, Chin. J. Catal., 2018, 39, 118–127 CrossRef CAS.
- S. Mohan, P. Dinesha and S. Kumar, Chem. Eng. J., 2020, 384, 123253 CrossRef CAS.
- J. Chen, W. Huang, S. Bao, W. Zhang, T. Liang, S. Zheng, L. Yi, L. Guo and X. Wu, RSC Adv., 2022, 12, 27746–27765 RSC.
- X. Lou, P. Liu, J. Li, Z. Li and K. He, Appl. Surf. Sci., 2014, 307, 382–387 CrossRef CAS.
- G. Li, B. Wang, H. Wang, J. Ma, W. Xu, Y. Li, Y. Han and Q. Sun, Catal. Commun., 2018, 108, 82–87 CrossRef CAS.
- J. Yang, S. Ren, T. Zhang, Z. Su, H. Long, M. Kong and L. Yao, Chem. Eng. J., 2020, 379, 122398 CrossRef.
- L. Yao, Q. Liu, S. Mossin, D. Nielsen, M. Kong, L. Jiang, J. Yang, S. Ren and J. Wen, J. Hazard. Mater., 2020, 387, 121704 CrossRef CAS.
- J. Jiao, S. Li and B. Huang, Acta Phys.-Chim. Sin., 2015, 31, 1383–1390 CAS.
- Y. Zhang, Y. Chen, J. Huang, M. Ding, X. Li and H. Zhao, Curr. Nanosci., 2021, 17, 298–306 CrossRef CAS.
- J. Yang, J. Zhou, W. Tong, T. Zhang, M. Kong and S. Ren, J. Energy Inst., 2019, 92, 1158–1166 CrossRef CAS.
- S. He, Q. Li, Q. Zhang, Z. Zhan and L. Wang, J. Environ. Eng., 2019, 145, 04019087 CrossRef.
- Y. Qi, X. Shan, M. Wang, D. Hu, Y. Song, P. Ge and J. Wu, Water Air Soil Pollut., 2020, 231, 289 CrossRef CAS.
- S. Ding, C. Li, J. Zhang, J. Wu, Y. Yue and G. Qian, Appl. Surf. Sci., 2022, 595, 153484 CrossRef CAS.
- Z. Yang, P. Tang, C. Xu, B. Zhu, Y. He, T. Duan, J. He, G. Zhang and P. Cui, J. Energy Inst., 2023, 108, 101201 CrossRef CAS.
- J. Li, L. Shi, S. Lyu, X. Yang, X. Yan and X. Zhang, Environ. Chem., 2023, 42, 3568–3578 CAS.
- H. Zhao, H. Wang and Z. Qu, J. Environ. Sci., 2022, 112, 231–243 CrossRef CAS.
- Z. Cai, Y. Yang, Z. Feng, J. Zhang, Y. Yue and G. Qian, Colloids Surf., A, 2024, 683, 133079 CrossRef CAS.
- S. Zhao, K. Song, R. Jiang, D. Ma, H. Long and J. Shi, Catal. Today, 2023, 423, 113966 CrossRef CAS.
- W. Xie, G. Zhou, X. Zhang, J. Zhang, G. Zhang and Z. Tang, Mol. Catal., 2020, 34, 546–558 CAS.
- Y. Li, P. Jiang, J. Tian, Y. Liu, Y. Wan, K. Zhang, D. Wang, J. Dan, B. Dai, X. Wang and F. Yu, J. Environ. Chem. Eng., 2021, 9, 105753 CrossRef CAS.
- X. Zhang, S. Lv, X. Zhang, K. Xiao and X. Wu, J. Environ. Sci., 2021, 101, 1–15 CrossRef CAS.
- J. Gu, R. Duan, W. Chen, Y. Chen, L. Liu and X. D. Wang, Catalysts, 2020, 10, 566 CrossRef CAS.
- Z. Guo, W. Huo, Y. Zhang, S. Ren and J. Yang, Mater. Herald, 2021, 35, 13085–13099 Search PubMed.
- L. Lietti, J. L. Alemany, P. Forzatti, G. Busca, G. Ramis, E. Giamello and F. Bregani, Catal. Today, 1996, 29, 143–148 CrossRef CAS.
- C. Tang, H. Zhang and L. Dong, Catal. Sci. Technol., 2016, 6, 1248–1264 RSC.
- S. Yang, C. Wang, J. Li, N. Yan, L. Ma and H. Chang, Appl. Catal., B, 2011, 110, 71–80 CrossRef CAS.
- T. Chen, B. Guan, H. Lin and L. Zhu, Chin. J. Catal., 2014, 35, 294–301 CrossRef CAS.
- X. Li, Q. Li, L. Zhong, Z. Song, S. Yu, C. Zhang, Q. Fang and G. Chen, J. Phys. Chem. C, 2019, 123, 25185–25196 CrossRef CAS.
- Z. Fei, Y. Yang, M. Wang, Z. Tao, Q. Liu, X. Chen, M. Cui, Z. Zhang, J. Tang and X. Qiao, Chem. Eng. J., 2018, 353, 930–939 CrossRef CAS.
- G. Ramis, L. Yi, G. Busca, M. Turco, E. Kotur and R. J. Willey, J. Catal., 1995, 157, 523–535 CrossRef CAS.
- G. Qi, R. Yang and R. Chang, Appl. Catal., B, 2004, 51, 93–106 CrossRef CAS.
- P. Sun, R. Guo, S. Liu, S. Wang, W. Pan and M. Li, Appl. Catal., A, 2017, 531, 129–138 CrossRef CAS.
- N. Fang, J. Guo, S. Shu, H. Luo, J. Li and Y. Chu, J. Taiwan Inst. Chem. Eng., 2018, 93, 277–288 CrossRef CAS.
- S. Gu, K. Gui, D. Ren and Y. Wei, React. Kinet. Mech. Catal., 2020, 130, 741–751 CrossRef CAS.
- F. Wang, S. D. Li, R. Y. You, Z. K. Han, W. T. Yuan, B. E. Zhu, Y. Gao, H. S. Yang and Y. Wang, Appl. Surf. Sci., 2023, 638, 158124 CrossRef CAS.
- G. Li, B. Wang, Z. Wang, Z. Li, Q. Sun, W. Q. Xu and Y. Li, J. Phys. Chem. C, 2018, 122, 20210–20231 CrossRef CAS.
- S. Yang, S. Xiong, Y. Liao, X. Xiao, F. Qi, Y. Peng, Y. Fu, W. Shan and J. Li, Environ. Sci. Technol., 2014, 48, 10354–10362 CrossRef CAS.
- S. Yang, Y. Fu, Y. Liao, S. Xiong, Z. Qu, N. Yan and J. Li, Catal. Sci. Technol., 2014, 4, 224–232 RSC.
- L. Zhang, X. Wen, Z. Lei, L. Gao, X. Sha, Z. Ma, H. He, Y. Wang, Y. Jia and Y. H. Li, AIP Adv., 2018, 8, 045004 CrossRef.
- C. Yu, L. Wang and B. Huang, Aerosol Air Qual. Res., 2015, 15, 1017–1027 CrossRef CAS.
- Y. Huo, K. Liu, J. Liu and H. He, Appl. Catal., B, 2022, 301, 120784 CrossRef CAS.
- J. Tang, X. Wang, H. Li, L. Xing and M. Liu, ACS Omega, 2023, 8, 7262–7278 CrossRef CAS.
- G. Xu, X. Guo, X. Cheng, J. Yu and B. Fang, Nanoscale, 2021, 13, 7052–7080 RSC.
- Y. Liu, J. Liu, B. Zhu, J. Chen, F. Li and Y. Sun, Colloids Surf., A, 2023, 662, 130983 CrossRef CAS.
- D. An, J. Ji, Q. Cheng, X. Zhao, Y. Cai, W. Tan, Q. Tong, K. Ma, W. Zou, J. Sun, C. Tang and L. Dong, Environ. Sci. Technol., 2023, 57, 14737–14746 CrossRef CAS.
- S. Xiong, Y. Liao, X. Xiao, H. Dang and S. Yang, Catal. Sci. Technol., 2015, 5, 2132–2140 RSC.
- D. Yan, X. Hong, J. Li, Y. Wang, Y. Pan and H. Gong, J. Phys. Chem. Solids, 2024, 192, 112703 CrossRef.
- L. Lin, T. Hsieh, J. Hsu and Y. Wang, Appl. Surf. Sci., 2023, 614, 156139 CrossRef CAS.
- C. Hu, C. Liu, K. Du, C. Pang, Z. Zhuo and Q. Gao, J. Environ. Chem. Eng., 2023, 11, 110440 CrossRef CAS.
- X. Yao, T. Kong, S. Yu, L. Li, F. Yang and L. Dong, Appl. Surf. Sci., 2017, 402, 208–217 CrossRef CAS.
- F. Liu and H. He, Catal. Today, 2010, 153, 70–76 CrossRef CAS.
- R. Guo, B. Qin, L. Wei, T. Yin, J. Zhou and W. Pan, Phys. Chem. Chem. Phys., 2022, 24, 6363–6382 RSC.
- S. Liu, Y. Ji, W. Xu, J. Zhang, R. Jiang, L. Li, L. Jia, Z. Zhong, G. Xu, T. Zhu and F. Su, J. Catal., 2022, 406, 72–86 CrossRef CAS.
- Y. Pan, N. Li, S. Ran, D. Wen, Q. Luo, K. Li and Q. Zhou, Ind. Eng. Chem. Res., 2022, 61, 9991–10003 CrossRef CAS.
- L. Wang, X. Cheng, Z. Wang, C. Ma and Y. Qin, Appl. Catal., B, 2017, 201, 636–651 CrossRef CAS.
- P. Zang, J. Liu, G. Zhang, B. Jia, Y. He, Y. Wang and Y. Lv, Fuel, 2023, 331, 125800 CrossRef CAS.
- C. Sun, W. Chen, X. Jia, A. Liu, F. Gao, S. Feng and L. Dong, Chin. J. Catal., 2021, 42, 417–430 CrossRef CAS.
- Y. Wang, W. Yi, Y. Zeng and H. Chang, Environ. Sci. Technol., 2020, 54, 12612–12620 CrossRef CAS.
- L. Han, M. Gao, J. Hasegawa, S. Li, Y. Shen, H. Li, L. Shi and D. Zhang, Environ. Sci. Technol., 2019, 53, 6462–6473 CrossRef CAS.
- X. Zhang, S. Liu, K. Ma, Y. Chen, S. Jin, X. Wang and X. Wu, Catalysts, 2021, 11, 1360 CrossRef CAS.
- G. Xiao, Z. Guo, J. Li, Y. Du, Y. Zhang, T. Xiong, B. Lin, M. Fu, D. Ye and Y. Hu, Chem. Eng. J., 2022, 435, 134914 CrossRef CAS.
- W. Jiang, Y. Yu, F. Bi, P. Sun, X. Weng and Z. Wu, Environ. Sci. Technol., 2019, 53, 12657–12667 CrossRef CAS.
- S. Xiong, Y. Peng, D. Wang, N. Huang, Q. Zhang, S. Yang, J. Chen and J. Li, Chem. Eng. J., 2020, 387, 124090 CrossRef CAS.
- R. Chen, X. Fang, J. Li, Y. Zhang and Z. Liu, Chem. Eng. J., 2023, 452, 139207 CrossRef CAS.
- X. Shi, J. Guo, T. Shen, A. Fan, S. Yuan and J. Li, Chem. Eng. J., 2021, 421, 129995 CrossRef CAS.
- W. Yoon, Y. Kim, G. J. Kim, J. R. Kim, S. J. Lee, H. Y. S. Han, G. H. Park, H. J. Chae and W. B. Kim, Chem. Eng. J., 2022, 434, 134676 CrossRef CAS.
- D. Yan, J. Zhao, J. Li, G. Abbas, Z. Chen and T. Guo, Catal. Lett., 2023, 153, 2838–2852 CrossRef CAS.
- G. Yao, Y. Wei, K. Gui and X. Ling, J. Environ. Sci., 2022, 115, 126–139 CrossRef CAS.
- D. Damma, D. K. Pappas, T. Boningari and P. G. Smirniotis, Appl. Catal., B, 2021, 287, 119939 CrossRef CAS.
- X. Jia, H. Liu, Y. Zhang, W. Chen, Q. Tong, G. Piao, C. Sun and L. Dong, J. Colloid Interface Sci., 2021, 581, 427–441 CrossRef CAS.
- H. Wang, B. Huang, C. Yu, M. Lu, H. Huang and Y. Zhou, Appl. Catal., A, 2019, 588, 117207 CrossRef CAS.
- K. H. P. Reddy, B. S. Kim, S. S. Lam, S. C. Jung, J. Song and Y. K. Park, Environ. Res., 2021, 195, 110876 CrossRef CAS.
- H. W. Ryu, M. Y. Song, J. S. Park, J. M. Kim, S. C. Jung, J. Song, B. J. Kim and Y. K. Park, Environ. Res., 2019, 172, 649–657 CrossRef CAS.
- D. Wang, Q. Yao, S. Liu, S. Hui and Y. Niu, J. Energy Inst., 2019, 92, 1852–1863 CrossRef CAS.
- I. Song, H. Lee, S. W. Jeon and D. Kim, ACS Catal., 2020, 10, 12017–12030 CrossRef CAS.
- R. Cui, X. Huang, G. Zhang and Z. Tang, Ind. Eng. Chem. Res., 2024, 63, 7003–7017 CrossRef CAS.
- B. Qin, R. Guo, J. Zhou, L. Wei, T. Yin and W. Pan, Appl. Surf. Sci., 2022, 598, 153823 CrossRef CAS.
- L. Wei, R. Guo, J. Zhou, B. Qin, X. Chen, Z. Bi and W. Pan, Fuel, 2022, 316, 123438 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Ang Cao
*ab,
Ang Cao