
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTi and Zr complexes bearing guanidine-phenolate ligands: coordination chemistry and polymerization studies†
Víctor Flores-Romero ,
Jesse LeBlanc,
Zichuan Chen and
Gino G. Lavoie*
,
Jesse LeBlanc,
Zichuan Chen and
Gino G. Lavoie*
York University, 4700 Keele Street, Toronto, Ontario M3J 1P3, Canada. E-mail: glavoie@yorku.ca
First published on 16th August 2024
Abstract
A series of group 4 bis(isopropoxide) complexes M[N^O]2(OiPr)2, stabilized by guanidine-phenolate N^O ligands, have been prepared and used as catalysts for the polymerization of unpurified rac-lactide under solvent-free conditions at 130 °C. The resulting polylactic acid (PLA) presented heterotactic bias (Pr = 0.56–0.62) with molecular weights similar to those obtained in control experiments with Zr(OiPr)4·iPrOH, Ti(OiPr)4, and Sn(Oct)2. The molecular weights were lower than expected for living polymerization due to chain transfer and/or transesterification. Zr complexes were more active than the Ti homologues, with rate constants ranging from 1.17–3.21 × 10−4 s−1, comparable to that observed with the free guanidine-phenol ligands. The corresponding bis(guanidine-phenolate) titanium dichloride complexes Ti[N^O]2Cl2 were also prepared and tested in ethylene polymerization. The low activity (up to 1.1 kgPE mol−1 h−1) was associated to the strong electron-donating ability of the guanidine moiety and to the trans-N,N-cis-O,O-cis-Cl,Cl coordination mode of the guanidine-phenolate ligand.
Introduction
In recent years, polylactic acid (PLA) has attracted much interest as a biodegradable polymer that uses renewable resources, such as corn starch and sugar beet, as feedstock.1–3 Select microstructures of PLA have shown mechanical properties similar to fossil fuel-based polymers, and have found applications in consumer materials such as packaging, plastic bottles, and fibers.3–7 PLA has also been used in drug delivery systems and orthopaedic fixation devices.8,9Although PLA can be synthesized by polycondensation of lactic acid, this process is not industrially viable due to the ultrapure conditions and high conversion needed to achieve high molecular weight polymers. Thus, the industry relies on the metal-mediated ring-opening polymerization (ROP) of lactide, which exhibits more favorable kinetics for the production of high molecular weight polymers.10–13 Polylactic acid is commercially produced at elevated temperatures, in the melt phase, in the absence of solvent, with tin octanoate (Sn(Oct)2) as the most commonly used catalyst.14–17 Tin octanoate however does not impart stereoregularity to the polymer mainchain, resulting in a material with poor bulk properties, thus limiting the scope of applications of the material. Under the polymerization conditions used by the industry, Sn(Oct)2 however also catalyzes transesterification, leading to poor control of molecular weights and molecular weight distribution. Furthermore, traces of toxic tin in the polymer limit possible applications in medical devices.3,18
Catalytic systems containing nitrogen-donor ligands and less toxic metals such as alkali and alkaline earth metals,19–21 iron,22,23 and zinc24–30 have proven to be effective for the ROP of cyclic esters. In particular, group 4 metals have shown excellent activities among these non-toxic metals,31–33 polymerizing lactide by a coordination-insertion mechanism.34–37 Zirconium acetylacetonate has also proven effective in the production of PLA and copolymers by ring-opening polymerization of lactides and other cyclic esters.38 Related zirconium diketiminate complexes were found to be very active, with the substituents on the two nitrogen atoms used to modulate the sterics and electronics of the catalysts.39 Other nitrogen-donor ligands, such as phenoxy-imine, phenoxy-amine, pyrrole, and pyridine, have also drawn much interest.24,40–46 Davidson developed group 4 metal alkoxides supported by a tetradentate phenoxy-amine ligand to polymerize rac-lactide under solvent-free conditions, yielding atactic PLA.47 In contrast, zirconium catalysts of chiral phenoxy-imine ligands produced moderately heterotactic PLA under both solvent-free conditions (130 °C) and in toluene (80 °C).48
Neutral N-heterocyclic imines and the corresponding anionic iminates, especially those formally based on the corresponding N-heterocyclic carbenes (NHC), have recently drawn much attention as N-based spectator ligands due to the ability to tailor their steric and electronic contributions. These cyclic guanidines exist as two mesomeric forms, illustrating electron delocalization from the endocyclic nitrogen atoms to the exocyclic atom (Fig. 1). The electron density on this exocyclic nitrogen, and thus its electron-donating ability to metal, can be modulated by modifying the NHC scaffold itself, with its steric contributions to the system altered through the substituents on the endocyclic nitrogen atoms.
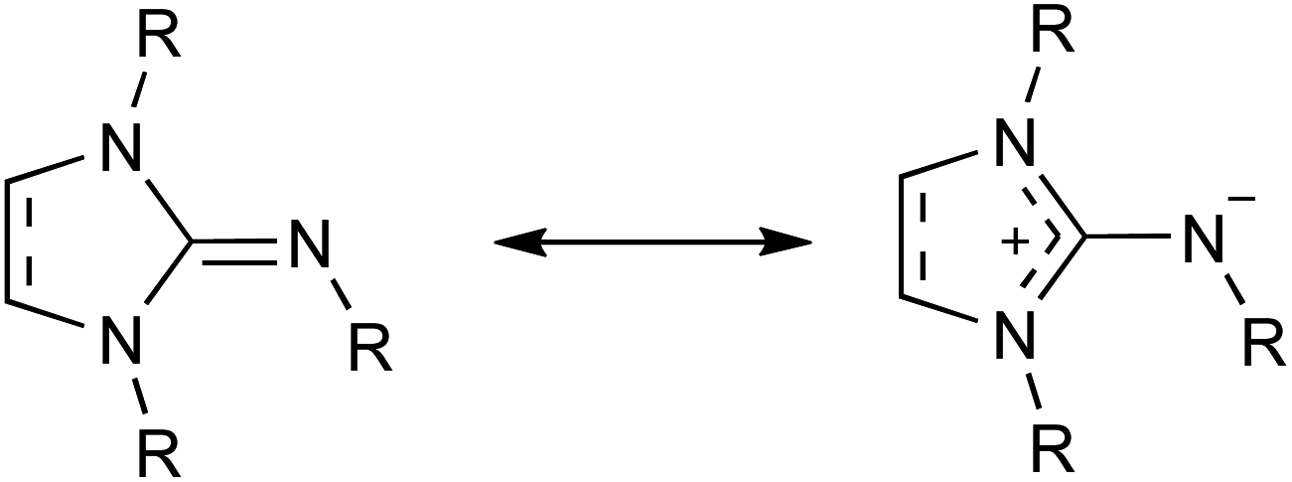 | ||
| Fig. 1 Mesomeric forms of cyclic guanidines. | ||
A decade ago, our group reported the first chelating anionic guanidine-ethenolate ligands and their coordination to group 4 metals.49 The corresponding titanium complex showed good activity in ethylene polymerization. Since then, other guanidine-based bidentate ligands, both neutral and anionic, have been reported with the metal complexes used as catalysts in both olefin and lactide polymerizations.50–53 We were especially intrigued by the guanidine-phenolate ligand systems reported by Eisen and exclusively coordinated to group 10 nickel and palladium for homo- and copolymerization of norbornene. These ligands in fact closely resemble the phenoxy-imine ligands used in group 4 metal complexes used in both olefin and lactide polymerization.54–56
On this basis, chelating ligands containing a neutral cyclic guanidine donor paired with an anionic phenolate appeared to be excellent candidates for complexation to group 4 metals and for study in both lactide and olefin polymerizations. Hence, we herein describe the synthesis and characterization of titanium and zirconium alkoxide and chloride complexes of such ligands with imidazole-, imidazolidine- and tetrahydropyrimidine-based guanidines (Fig. 2). These guanidines offer the opportunity to deconvolute the steric and electronic effects and study the catalytic performance of the corresponding metal complexes.
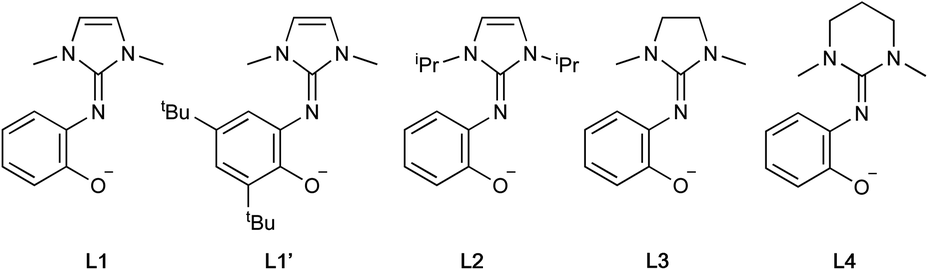 | ||
| Fig. 2 Structure of guanidine-phenolate ligands herein explored. | ||
Results and discussion
Synthesis of proligands
The guanidine-phenol hydrochloride salts LxH·HCl (wherein x = 1, 1′, 2, 3 and 4) were prepared by reacting the chloro halide salt xCl with 2-aminophenol or the corresponding tert-butyl derivative (Scheme 1).52 Deprotonation of these salts with KOH gave the corresponding guanidine-phenol LxH in 63–90% yield. Despite the formal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Nexo double bond of the guanidine, NMR spectra of both the phenol and the hydrochloride salt showed magnetically-equivalent R1 groups, consistent with free rotation about the CNexo due to electron delocalization from the Nendo lone pairs. This contrasts with restricted rotation observed in our related systems with diacylated imidazolidines, wherein the acyl groups compete for the π-electron density.50
Nexo double bond of the guanidine, NMR spectra of both the phenol and the hydrochloride salt showed magnetically-equivalent R1 groups, consistent with free rotation about the CNexo due to electron delocalization from the Nendo lone pairs. This contrasts with restricted rotation observed in our related systems with diacylated imidazolidines, wherein the acyl groups compete for the π-electron density.50
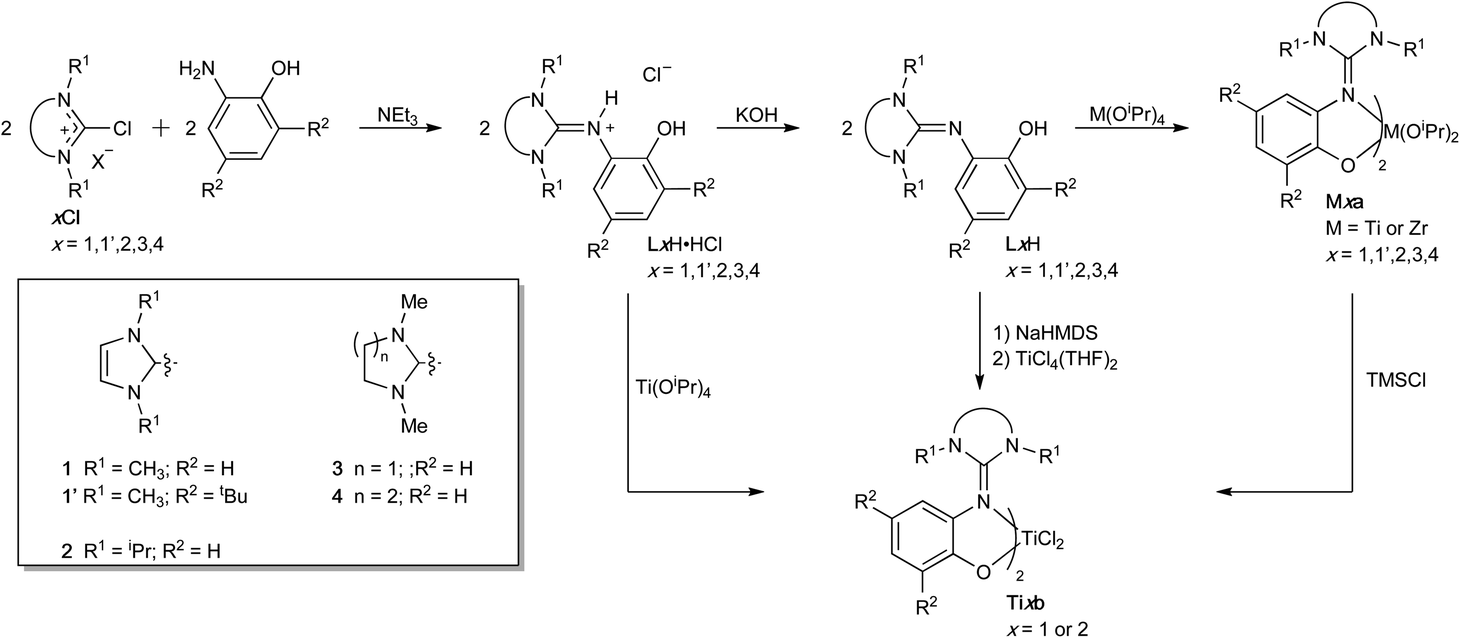 | ||
| Scheme 1 Synthesis of ligand and group 4 complexes. | ||
Synthesis of bis(guanidine-phenolate) Ti and Zr complexes
The titanium and zirconium bis(isopropoxide) complexes Mxa were respectively prepared in excellent yields (>85%) at 60 °C by addition of guanidine-phenol LxH to Ti(OiPr)4 or Zr(OiPr)4·iPrOH in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio (Scheme 1). The 1H NMR spectra of all complexes are consistent with the structure proposed and showed the generation of one single isomer. Most titanium complexes showed broad resonances for the aliphatic protons, due to a sterically crowded coordination sphere.
:1 stoichiometric ratio (Scheme 1). The 1H NMR spectra of all complexes are consistent with the structure proposed and showed the generation of one single isomer. Most titanium complexes showed broad resonances for the aliphatic protons, due to a sterically crowded coordination sphere.
The solid-state molecular structures of complexes Ti3a, Zr3a, and Ti4a are shown in Fig. 3. All complexes are six-coordinate with the isopropoxide groups in a cis arrangement. Selected bond distances and angles are given in Table 1. The guanidine-phenolate binds to the metal in a bidentate fashion to give the cis-N,N-trans-O,O diastereomer with average bite angles of 74.96° and 75.48° for Ti3a and Ti4a, respectively. As expected, a smaller bite angle of 71.79° was observed in the corresponding zirconium complex Zr3a. The O1–M–O2 bond angles deviates from linearity, at 158.35(6)°, 158.03(15) and 151.00(10)° for Ti3a, Ti4a and Zr3a, respectively. The M–OiPr bond lengths range from 1.809(3) to 1.971(3) Å, consistent with analogous group 4 complexes of phenoxy-imine ligands with the same stereo-arrangement.57 The structural parameter ρ values of 0.97–0.99 indicate extended electron delocalization within the guanidine moiety.
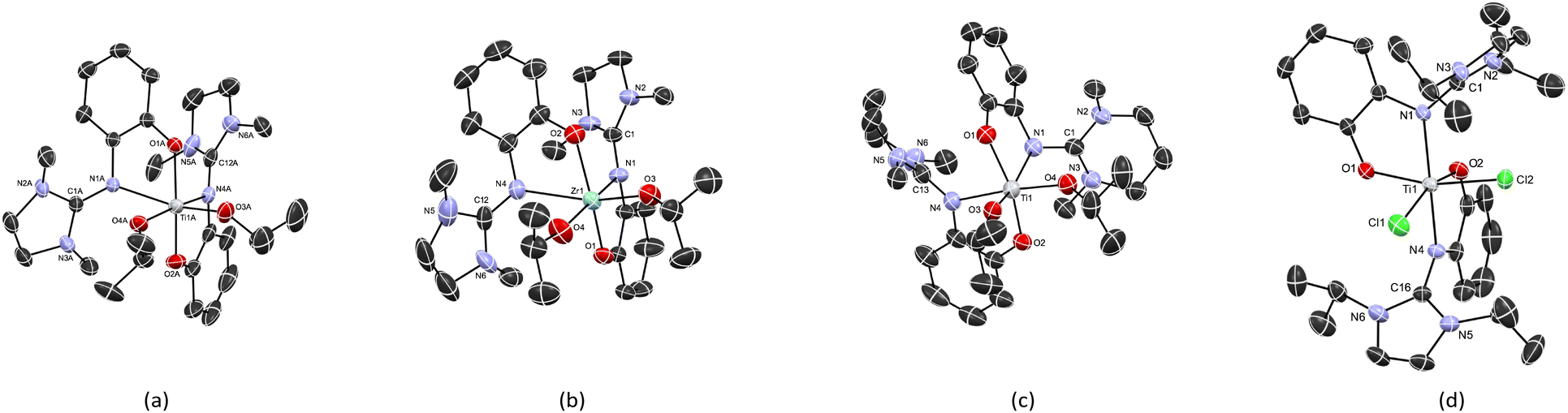 | ||
| Fig. 3 Solid-state structures of (a) Ti3a, (b) Zr3a, (c) Ti4a and (d) Ti2b. Hydrogen atoms and solvent have been omitted for clarity. ORTEP drawn at 50% probability. | ||
| Ti3a | Zr3a | Ti4a | Ti2b | ||
|---|---|---|---|---|---|
| a Averaged value of both guanidine fragments and calculated as reported in the literature.50 | |||||
| M–O1 | 1.9182(12) | 2.061(3) | 1.939(3) | Ti–O1 | 1.9098(14) |
| M–O2 | 1.9245(14) | 2.061(3) | 1.937(3) | Ti–O2 | 1.9277(14) |
| M–O3 | 1.8103(14) | 1.944(3) | 1.809(3) | Ti–Cl1 | 2.3783(6) |
| M–O4 | 1.8466(13) | 1.971(3) | 1.850(3) | Ti–Cl2 | 2.3883(6) |
| M–N1 | 2.3724(14) | 2.386(3) | 2.281(4) | Ti–N1 | 2.0664(15) |
| M–N4 | 2.2738(14) | 2.456(3) | 2.275(4) | Ti–N4 | 2.0666(14) |
| C1–N1 | 1.323(2) | 1.328(4) | 1.343(6) | C1–N1 | 1.372(2) |
| C1–N2 | 1.368(2) | 1.342(5) | 1.354(6) | C1–N2 | 1.342(2) |
| C1–N3 | 1.350(2) | 1.362(5) | 1.348(6) | C1–N3 | 1.345(2) |
| C12–N4 | 1.322(2) | 1.329(5) | 1.335(6) | C16–N4 | 1.369(2) |
| C12–N5 | 1.374(2) | 1.348(6) | 1.350(6) | C16–N5 | 1.343(2) |
| C12–N6 | 1.339(3) | 1.365(6) | 1.350(6) | C16–N6 | 1.345(2) |
| O1–M–O2 | 158.35(6) | 151.00(10) | 158.03(15) | Cl1–M–Cl2 | 87.84(2) |
| O3–M–O4 | 98.28(6) | 96.69(12) | 96.19(15) | O1–M–O2 | 94.38(6) |
| N1–M–N4 | 84.91(5) | 85.29(10) | 86.81(14) | N1–M–N4 | 167.97(6) |
| N1–M–O1 | 74.68(5) | 72.08(10) | 75.94(14) | N1–M–Cl1 | 104.59(5) |
| N4–M–O2 | 75.24(6) | 71.49(11) | 75.02(14) | N1–M–Cl2 | 85.10(6) |
| ρa | 0.97 | 0.98 | 0.99 | ρa | 1.00 |
DFT calculations on all five possible isomers of the X-ray characterized bis(isopropoxide) complexes Ti3a, Zr3a and Ti4a were performed (Fig. 4), using B3LYP-D3(BJ)/def2-SVP for geometry optimization and B3LYP-D3(BJ)/def2-TZVPP for single-point energy calculations.44. Isomer A was predicted to be the most stable isomer, consistent with the solid state structures, by 1–28 kJ mol−1 compared to the cis isopropoxide isomers B and C (Table 2), and by 35–70 kJ mol−1 for the much less stable trans isopropoxide isomers D and E (Table S1†). Considering the large unfavorable energies for the trans isopropoxide isomers D and E, computations on complexes not characterized by X-ray diffraction were then performed on only diastereomers A–C. The calculations predict the isomer A (cis-N,N-trans-O,O) to be the most stable isomer.
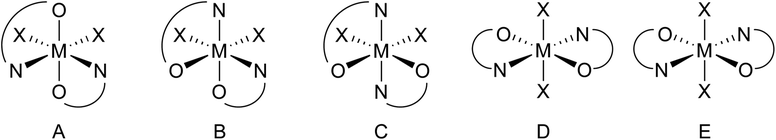 | ||
| Fig. 4 Possible isomers (excluding enantiomers) for bis(guanidine-phenolate) six-coordinate complexes. | ||
| Complex | Isomer B (cis-N,N-cis-O,O) | Isomer C (trans-N,N-cis-O,O) |
|---|---|---|
| Ti1a | 35 | 45 |
| Zr1a | 36 | 28 |
| Ti1′a | 43 | 39 |
| Zr1′a | 38 | 25 |
| Ti2a | 34 | 1 |
| Zr2a | 41 | 17 |
| Ti3a | 5 | 15 |
| Zr3a | 1 | 15 |
| Ti4a | 28 | 27 |
| Zr4a | 33 | 25 |
Replacing the methyl groups on the endocyclic nitrogen in Ti1a with larger isopropyl substituents in Ti2a resulted in an energy difference between isomers A and C to be only 1 kJ mol−1. In most cases, isomer B (cis-N,N-cis-O,O) is predicted to be the least stable diastereomer, with the exception of Ti1a, Ti3a and Zr3a, which are 5–14 kJ mol−1 more stable than isomer C. Similar results have been observed for the analogous bis(phenoxy-imine) group 4 systems adopting preferentially the N,N-cis-O,O-trans ligand arrangements (isomer A) unless bulky substituents are present.57,58
The titanium dichloride complexes Ti1b, Ti1′b, and Ti2b were prepared by reaction of 2 equivalents of the guanidinium chloride salt LxH·HCl with Ti(OiPr)4 in excellent yields (>90%) (Scheme 1). 1H NMR spectra of these complexes showcase one single set of resonances, consistent with the presence of only one isomer, with magnetically-inequivalent protons for the alkyl substituents on the endocyclic nitrogen atoms. Alternatively, these dichloride complexes could also be prepared from reaction of either the corresponding bis(isopropoxide) complexes with TMSCl or TiCl4(THF)2 with the sodium guanidine-phenolate salt, giving products that were undistinguishable spectroscopically.
The X-ray structure of Ti2b exhibits a distorted octahedral geometry, with a trans-N,N-cis-O,O ligand arrangement (isomer C; Fig. 3 and 4, Table 1). This contrasts with the cis-N,N-trans-O,O ligand arrangement (isomer A) observed for the bis(isopropoxide) complexes Ti3a, Ti4a and Zr3a and normally observed for bis(phenoxy-imine) titanium complexes. In the latter case, the trans-N,N-cis-O,O arrangement (isomer C) is limited to ligands with sterically-demanding substituents on the nitrogen.54,58 Interestingly, our earlier DFT calculations (vide supra) on the bis(isopropoxide) complexes predicted this arrangement to be the second most stable isomer. However, DFT calculations of isomers A, B and C for the dichloride complexes Ti1b, Ti1′b, and Ti2b predicted isomer C to more stable than isomers A and B by 1–18 kJ mol−1 and 21–55 kJ mol−1, respectively (Table 3). These calculations are in agreement with the solid state structure of Ti2b and further illustrate the important effect of bulky substituents on the preferred arrangement of the guanidine-phenolate (and phenoxy-imine) ligands.
| Complex | Isomer A cis-N,N-trans-O,O | Isomer B cis-N,N-cis-O,O |
|---|---|---|
| Ti1b | 1 | 36 |
| Ti1′b | 6 | 21 |
| Ti2b | 18 | 55 |
Ring-opening polymerization of rac-lactide with metal alkoxide complexes
All complexes were tested in the ring-opening polymerization of rac-lactide. Polymerizations were performed in the absence of any solvent at 130 °C with a 100:1 stoichiometric ratio of lactide to catalyst, using unpurified monomer (Table 4). All complexes gave full conversion within 6 h. The polymerization rate constants were obtained by monitoring the consumption of the monomer over time using 1H NMR spectroscopy. On average, zirconium complexes are 28% more active than the corresponding titanium homologues, with kapp average values of 1.17–1.82 × 10−4 s−1 and 1.39–3.21 × 10−4 s−1 for Ti and Zr complexes, respectively. This is consistent with other group 4 catalytic systems and attributed to an easier coordination of the monomer to the larger zirconium metal and a more nucleophilic alkoxide ligand.43 While these rate constants compare favorably to those determined with Ti(OiPr)4 (0.53 × 10−4 s−1) and Zr(OiPr)4·iPrOH (0.28 × 10−4 s−1), the activity is approximately an order of magnitude less active than that observed for Sn(Oct)2 (12.4 × 10−4 s−1).
| Entry | Catalyst | kappb (10−4 s−1) | Mnc (Da) | Mwe (Da) | Đf | Mnd (Da) | Mwd (Da) | Đg | Pr |
|---|---|---|---|---|---|---|---|---|---|
| a All polymerization were carried out at 130 °C within 6 h and a lactide-to-catalyst stoichiometric ratio of 100:1.b An average standard error of 5% was calculated for the rate constants.c Determined by 1H NMR end-group analysis assuming –OiPr as chain-end.d Determined by GPC in THF.e Determined by DOSY NMR in C6D6.f Đ = Mw DOSY ÷ Mn NMR.g Đ = Mw ÷ Mn as determined by GPC.h rac-Lactide recrystallized from toluene was employed.i 2-Propanol was employed as co-initiator 1:1 Cat:iPrOH.j Lactide/catalyst stoichiometric ratio of 50:1. |
|||||||||
| 1 | Ti1a | 1.22 | 700 | 1600 | 2.3 | 1400 | 1900 | 1.4 | 0.63 |
| 2 | Ti1′a | 1.82 | 1400 | 2400 | 1.7 | 2700 | 4000 | 1.5 | 0.62 |
| 3 | Ti2a | 1.56 | 1400 | 1900 | 1.4 | 2200 | 2900 | 1.3 | 0.56 |
| 4 | Ti3a | 1.44 | 700 | 1500 | 2.1 | 1300 | 1600 | 1.2 | 0.62 |
| 5 | Ti4a | 1.17 | 1000 | 1900 | 1.9 | 1600 | 2100 | 1.3 | 0.56 |
| 6 | Zr1a | 1.78 | 1800 | 3000 | 1.6 | 2700 | 3500 | 1.3 | 0.56 |
| 7 | Zr1′a | 2.24 | 1100 | 2100 | 1.9 | 1700 | 2400 | 1.4 | 0.58 |
| 8 | Zr2a | 2.11 | 1200 | 1900 | 1.6 | 1600 | 2100 | 1.3 | 0.58 |
| 9 | Zr2ah | 2.54 | 1100 | 2400 | 2.2 | 1700 | 2300 | 1.4 | 0.58 |
| 10 | Zr2ai | 3.21 | 1200 | 1900 | 1.6 | 1500 | 2100 | 1.4 | 0.58 |
| 11 | Zr3a | 1.63 | 900 | 1700 | 1.8 | 1500 | 1900 | 1.3 | 0.62 |
| 12 | Zr4a | 1.39 | 1500 | 2600 | 1.7 | 2300 | 2900 | 1.3 | 0.58 |
| 13 | L2Hj | 2.49 | 1400 | 3000 | 2.1 | 1700 | 2500 | 1.5 | 0.48 |
| 14 | Sn(Oct)2 | 12.4 | 2200 | 3300 | 1.5 | 3300 | 4700 | 1.4 | 0.68 |
| 15 | Ti(OiPr)4 | 0.53 | 700 | 1500 | 2.1 | 700 | 900 | 1.3 | 0.50 |
| 16 | Zr(OiPr)4·iPrOH | 0.28 | 700 | 1600 | 2.1 | 1000 | 1200 | 1.2 | 0.50 |
The number-average molecular weight (Mn) of the polymers was determined by 1H NMR end-group analysis and gel permeation chromatography (GPC) with values of 700–2200 Da and 700–3300 Da, respectively. The weight-average molecular weight (Mw) was determined by diffusion-ordered spectroscopy (DOSY) and GPC with values of, 1200–3300 Da, and 900–4700 Da, respectively (Table 4).59–61 These values are comparable to those observed with the control experiments using Ti(OiPr)4, Zr(OiPr)4·iPrOH and Sn(Oct)2 as catalysts. Surprisingly, the nature of the metal (titanium vs. zirconium) did not show a significant impact on the polymers generated. Amongst the unsubstituted phenolate-based ligands, complexes of L3 (entries 4 and 11) gave the polymers with the lowest molecular weights. Surprisingly, increasing the size of the substituent on the endocyclic nitrogen from methyl to isopropyl leads to an increase in the polymer molecular weight for titanium (entries 1 vs. 3) but a decrease for zirconium (entries 6 vs. 8). The same effect was observed when large tert-butyl substituents were installed on the phenoxide rings, with an increase in molecular weight observed with titanium (entries 1 vs. 2) and a decrease with zirconium (entries 6 and 7).
While the dispersity (Đ) of all polymers was satisfactory (1.1–2.3), the observed molecular weights were approximately 70% lower than expected based on the initial monomer-to-catalyst ratio and assuming a living polymerization. Furthermore, the polymer molecular weight remains constant with conversion and deviates from the theoretical molecular weight (Fig. S57†) thus indicating chain transfer and/or transesterification. While the activity of the catalyst improved when using purified lactide, no marked improvement was observed in the molecular weight of the polymer (entry 9). The improved rate however supports the poisoning of the catalyst by impurities present in the unpurified monomer, such as lactic acid and water.62
The 1H and 13C NMR spectra of all polymers generated showed the incorporation of an isopropyl group as a chain end, consistent with these group 4 catalysts operating through a coordination-insertion mechanism. The competing activated monomer mechanism cannot however be ruled out based on the lower-than-expected observed relative ratio for the isopropyl to the hydroxyl end groups, which is supported by the enhanced rate in the presence of two equivalents of isopropanol (entry 10). This result further supports that nucleophilic impurities in lactide can also participate as an external initiator.
The tacticity of the polymers was determined by homonuclear-decoupled 1H{1H} NMR spectroscopy and all showed a heterotactic bias (Pr = 0.56–0.62).63–65 Similar heterotactic biases were observed with the related phenoxy-imine and benzoxazole–phenoxide systems, with limited to no effect of the ligand structure.48,66 In contrast, Ti(OiPr)4 and Zr(OiPr)4·iPrOH showed no stereoselectivity, with a Pr value of 0.50, indicating that the bis(guanidine-phenolate)complexes impart some enantiomorphic-site control, albeit limited.
Given the surprisingly marginal effects of the metal and of the ligand structures on the performance of the catalysts, we became suspicious that the high temperatures used might lead to ligand dissociation. Polymerization with L2H (entry 13; Table 4) gave polylactide with a rate constant and molecular weights similar to those reported for the corresponding metal complexes. This supports the possible dissociation of the ligand from the metal complexes with the free guanidine actually catalyzing the ring-opening of the lactide, as previously reported for guanidines and cyclic esters.67,68 Complex Zr2a indeed decomposes in a first-order decay when heated in toluene at 120 °C, with an estimated half-life of 15 h, providing evidence that the ligands themselves play a role in the polymerization of lactide. Unsurprisingly, L2H does not impart any tacticity bias.
The MALDI-TOF mass spectrometry of the PLA generated by Ti2a, Zr2a, and L2H had peak separations of 72 Da, evidence of transesterification which itself explains the observed low molecular weights. In addition, the spectra provide evidence of isopropyl or hydroxyl end-capped polymer chains for both Ti2a and Zr2a as catalyst, with no evidence of cyclic oligomers, further supporting a coordination-insertion mechanism. The mass spectrum of PLA generated by L2H does not show the incorporation of the ligand into the polymer, indicating monomer activation through a hydrogen bonding mechanism.67
The reaction conditions used in large-scale productions were replicated more closely by increasing the lactide-to-catalyst ratio from 100:1 to 1000:1 for Ti2a, Zr2a and L2H. All catalysts gave polymer with the expected decrease in the pseudo first-order rate constant by approximately one order of magnitude to 1.42 × 10−5, 2.03 × 10−5 and 0.92 × 10−5 s−1, respectively (Table S2†).
Ethylene polymerization using titanium complexes
Given the structural similarities to group 4 phenoxy-imine titanium chloride complexes,54 Ti1b, Ti1′b, and Ti2b were evaluated for the polymerization of ethylene (1 atm) at room temperature with n-octyl-modified methylaluminoxane (MMAO-12) as cocatalyst. Titanium complex Ti1b and Ti2b gave comparable activities of 1.1 kgPE molTi−1 h−1 and 0.8 kgPE molTi−1 h−1 (over 1–3 hours), respectively, approximately two orders of magnitude lower than that observed with Cp2TiCl2 (over 3 min). The tert-butyl derivative Ti1′b gave only traces of polymer. This is in stark contrast with the 85-fold increase in activity reported when a tert-butyl group is installed on phenoxy-imine ligands. Fujita stated that positioning of the flanking tert-butyl groups above and below the reaction site (Cl–M–Cl), as in the cis-N,N-trans-O,O arrangement (isomer A), lowers the energy barrier for olefin insertion and mitigate side reactions.54 The DFT calculations however predicted a trans-N,N-cis-O,O arrangement (isomer C; vide supra) for the bis(guanidine-phenolate) dichloride complexes placing the tert-butyl group at the back of the reaction site. Interestingly, phenoxy-imine systems that adopt this same trans-N,N-cis-O,O arrangement also showed poor activity in ethylene polymerization.54,58,69 The low activity of the guanidine-phenolate-based catalysts might thus be attributed to both an undesirable diastereomer and a less electrophilic metal center due to the strong electron-donating guanidine fragment.Conclusions
The bis(guanidine-phenolate) group 4 alkoxide complexes were evaluated for the ring-opening polymerization of rac-lactide under solvent-free conditions and gave PLA with molecular weights and dispersity values comparable to those observed using industrial standard Sn(Oct)2 under identical conditions. The guanidine-phenol itself seemingly also plays an important role in the polymerization, with rates comparable to those observed with the corresponding metal complexes. PLA generated from these bis(alkoxide) complexes showed a stronger heterotactic bias compared to free ligand and the other catalysts used in control experiments. The related dichloride titanium complexes however showed poor activity in the polymerization of ethylene, even with the bulky tert-butyl phenolate derivative. These guanidine-phenolate ligands and the ability to further tailor its electronic and steric features offer excellent opportunities to further expand the experimental space and explore their use in other catalytic transformations. Results will be reported in due course.Experimental section
General considerations
All manipulations and materials were performed under dry argon atmosphere, using Schlenk techniques or in a nitrogen-filled MBraun glovebox. Ti(OiPr)4, Zr(OiPr)4·iPrOH, TiCl4(THF)2, Sn(Oct)2, NaHMDS, and TMSCl were purchased from Sigma-Aldrich and used without further purification. Lactide was purchased from Thermo Fisher Scientific and used as received unless otherwise stated; if needed the monomer was purified by recrystallization from toluene. 2-Amino-4,6-di-tert-butylphenol, 2-chloro-1-methylimidazole, 3Cl, 4Cl, and L3H were synthesized according to literature procedure.70,71 Deuterated solvents C6D6 and CDCl3 were purchased from Sigma-Aldrich. C6D6 and CDCl3 were dried over sodium and benzophenone, and over CaH2, respectively. Both were degassed by freeze–pump–thaw cycles, vacuum transferred to dry ampules and stored over molecular sieves prior to use.Computational details
The calculations were performed with the computational package Gaussian 16 on Shared Hierarchical Academic Research Computing Network (SHARCNET; https://www.sharcnet.ca/my/front/) and made possible by Compute Canada (https://computecanada.ca/). SHARCNET is a consortium of colleges, universities and research institutes operating a network of high-performance computer clusters across Ontario. All geometry optimizations were performed using the B3LYP functional with the Def2SVP basis set.72 Grimme's empirical dispersion correction term with Becke–Johnson damping, D3(BJ) was applied.73 Single point energies calculations were performed with B3LYP-D3(BJ)/Def2-TZVPP level of theory.Crystal structure determination
Diffraction data for complexes were collected on a Bruker APEX-II CCD diffractometer with a Mo Kα (λ = 0.71073 Å) radiation source. Crystals were selected under paratone oil and mounted under a steam of N2 and kept at 173 K during data collection. Structures were solved in Olex2 (ref. 74) using direct methods and refined with SHELXL75 refinement package using least-squares minimization.MALDI-TOF Mass Spectrometry
MALDI-TOF MS data was obtained at the AIMS (Advanced Instrumentation for Molecular Structure) laboratory of the University of Toronto on a Bruker AutoFlex Speed MALDI-TOF mass spectrometer with a 2 kHz frequency tripled Nd:YAG laser (λ = 355 nm). Samples were prepared in a dithranol THF matrix.Molecular weight determination by gel permeation chromatography
Number average molar mass (Mn), weight average molar mass (Mw), and dispersity (Đ; Mw/Mn) of samples (1 to 2 mg mL−1 in tetrahydrofuran) were determined by gel permeation chromatography (GPC) using two PLgel miniMIX-B columns (4.6 mm × 250 mm) and two PLgel miniMIX-C columns (4.6 mm × 250 mm) from Agilent on elution with tetrahydrofuran (0.4 mL min−1) at 30 °C equipped with an Agilent multi detector suite composed of an Agilent G7801A refractive index detector, an Agilent G7803A dual-angle light scattering detector and an Agilent G7802A viscometer, all of them at 30 °C. The three detectors were calibrated using a narrow polystyrene standard (PS) from Agilent (Mp = 27060 g mol−1). Columns were calibrated using eight PS standards with a conventional approach (data from refractive index detector only). Data were analyzed using the Agilent GPC software version A.02.01 software (Santa Clara, CA 95051, United States).
Synthesis
N), 149.1 (s, C–OH), 137.6 (s, C–N), 119.3 (s, CH–Ar), 119.1 (s, CH–Ar), 118.2 (s, CH–Ar), 114.6 (s, N(CH)2N), 112.4 (s, CH–Ar), 33.9 (s, NCH3). Anal. calcd for C11H13N3O (%): C, 65.01; H, 6.45; N, 20.68. Found (%): C, 65.21; H, 6.06; N, 20.92.N), 145.6 (s, C–OH), 139.7 (s, C–N), 136.6 (s, C–tBu), 132.4 (s, C–tBu), 114.3 (s, CH–Ar), 114.0 (s, N(CH)2N), 113.8 (s, CH–Ar), 34.7 (s, CMe3), 34.3 (s, NCH3), 33.9 (s, C–Me3), 31.9 (s, CMe3), 29.8 (s, CMe3). Anal. calcd for C19H29N3O (%): C, 72.34; H, 9.27; N, 13.32. Found (%): C, 72.73; H, 9.15; N, 12.99.N), 143.9 (s, C–OH), 142.4 (s, C–N), 140.0 (s, C–tBu), 125.9 (s, C–tBu), 121.7 (s, CH–Ar), 118.5 (s, N(CH)2N), 117.9 (s, CH–Ar), 35.5 (s, NCH3), 34.3 (s, C–(Me)3), 31.6 (s, C–(Me)3), 29.8 (s, C–(Me)3). Anal. calcd for C19H30ClN3O (%): C, 64.85; H, 8.59; N, 11.94. Found (%): C, 64.70; H, 8.28; N, 11.75.N), 128.6 (s, C–OH), 125.4 (s, CH–Ar), 120.4 (s, CH–Ar), 119.9 (s, CH–Ar), 118.7 (s, C–N), 114.8 (s, N(CH)2N), 50.1 (s, CH–iPr), 22.5 (s, CH–Me2). Anal. calcd for C15H22ClN3O (%): C, 60.91; H, 7.50; N, 14.21. Found (%): C, 61.29; H, 7.22, N, 14.13.N), 147.9 (s, C–OH), 138.8 (s, C–N), 119.4 (s, CH–Ar), 118.3 (s, CH–Ar), 116.0 (s, CH–Ar), 112.3 (s, CH–Ar), 110.0 (s, N(CH)2N), 46.3 (s, CH–iPr)., 22.0 (s, CH3–iPr). Anal. calcd for C15H21N3O (%): C, 69.47; H, 8.16; N, 16.20. Found (%) C, 69.35; H, 8.10; N, 16.55.N), 150.5 (s, C–OH), 126.9 (s, CH–Ar), 126.1 (s, C–N), 123.9 (s, CH–Ar), 120.4 (s, CH–Ar), 119.0 (s, CH–Ar) 48.6 (s, N–CH3), 40.5 (N–CH2), 22.4 (CH2CH2CH2). Anal. calcd for C12H18ClN3O (%): C, 56.36; H, 7.09; N, 16.43. Found (%) C, 56.61; H, 7.22; N, 16.58.N), 150.5 (s, C–OH), 138.6 (s, C–N) 120.2 (s, CH–Ar), 119.4 (s, CH–Ar), 118.7 (s, CH–Ar), 113.1 (s, CH–Ar) 48.3 (s, N–CH3), 39.0 (N–CH2), 22.1 (CH2CH2CH2) Anal. calcd for C12H17N3O (%): C, 65.73; H, 7.81; N, 19.16. Found (%) C, 65.48; H, 7.63; N, 18.95.N), 153.2 (s, C–OH), 145.2 (s, C–N), 119.0 (s, CH–Ar), 116.7 (s, CH–Ar), 115.5 (s, N(CH)2N), 113.9 (s, CH–Ar), 112.9 (s, CH–Ar), 75.6 (s, OCHMe3), 34.3 (s, NCH3), 26.5 (s, OCH–Me3) δ anal. calcd for C28H38N6O4Ti (%): C, 58.95; H, 6.71; N, 14.73. Found (%) C, 59.30; H, 6.27; N, 15.22.N), 153.8 (s, C–OH), 145.4 (s, C–N), 137.5 (s, C–C(CH3)3), 133.4 (s, C– C(CH3)3), 116.2 (s, N(CH)2N), 113.0 (s, CH–Ar), 108.0 (s, CH–Ar), 0.6 (s, OCHMe3), 35.3 (s, NCH3), 34.3 (s, C(CH3)3), 32.4 (s, C(CH3)3), 30.2 (s, C(CH3)3), 28.1 (s, OCH–Me3). Anal. calcd for C44H70N6O4Ti (%): C, 66.48; H, 8.88; N, 10.57. Found (%) C, 66.61; H, 9.13; N, 10.85.N), 150.5 (s, C–O), 145.0 (s, C–N), 117.3 (s, CH–Ar) 112.9 (s, CH–Ar), 110.9 (s, CH–Ar), 110.3 (s, N(CH)2N) 108.5 (s, CH–Ar), 71.4 (s, OCH–Me2), 45.9 (s, CH–Me2), 25.0 (s, OCH–Me2), 20.7 (s, CH–Me2), 20.6 (s, CH3–Me2). Anal. calcd for C36H54N6O4Ti (%): C, 63.33; H, 7.97; N, 12.31. Found (%) C, 63.56; H, 7.66; N, 12.60.N), 161.3 (s, C–O), 145.2 (s, C–N), 119.7 (s, CH–Ar) 116.9 (s, CH–Ar), 116.0 (s, CH–Ar), 113.6 (s, CH–Ar), 76.3 (s, OCH–Me2), 48.4 (s, NCH3), 40.4 (s, N–CH2), 26.6 (s, OCH–Me2), 22.3 (s, CH2). Anal. calcd for C28H42N6O4Ti (%): C, 58.53; H, 7.37; N, 14.63. Found (%) C, 58.92; H, 7.23; N, 14.46.N), 161.3 (s, C–O), 145.2 (s, C–N), 119.7 (s, CH–Ar) 116.9 (s, CH–Ar), 116.0 (s, CH–Ar), 113.6 (s, CH–Ar), 76.3 (s, OCH–Me2), 48.4 (s, NCH3), 40.4 (s, N–CH2) 26.6 (s, OCH–Me2), 22.3 (s, CH2) Anal. calcd for C30H46N6O4Ti (%): C, 59.80; H, 7.69; N, 13.95. Found (%) C, 60.11; H, 7.44; N, 14.19.N), 153.14.88 (s, CH–Ar) 116.2 (s, CH–Ar), 115.6 (s, CH–Ar), 115.1 (s, N(CH)2N) 114.2 (s, CH–Ar), 70.3 (s, OCH–Me2), 34.1 (s, NCH3), 27.0 (s, OCH–Me2). Anal. calcd for C28H38N6O4Zr (%): C, 54.78; H, 6.24; N, 13.69. Found (%) C, 55.14; H, 6.40; N, 13.92.N), 153.8 (s, C–OH), 145.0 (s, C–N), 136.8 (s, C– C(CH3)3), 133.4 (s, C– C(CH3)3), 116.0 (s, N(CH)2N), 113.0 (s, CH–Ar), 108.0 (s, CH–Ar), 69.6 (s, OCHMe3), 35.3 (s, NCH3), 34.3 (s, C(CH3)3), 32.4 (s, C(CH3)3), 30.2 (s, C(CH3)3), 28.1 (s, OCH–Me3). Anal. calcd for C44H70N6O4Zr (%): C, 63.04; H, 8.42; N, 10.03. Found (%) C, 63.22; H, 8.13; N, 9.88.N), 152.2 (s, C–O), 146.7 (s, C–N), 119.3 (s, CH–Ar) 115.1 (s, CH–Ar), 114.7 (s, CH–Ar), 112.2 (s, N(CH)2N) 111.3 (s, CH–Ar), 69.3 (s, OCH–Me2), 47.9 (s, CH–Me2), 27.9 (s, OCH–Me2), 23.0 (s, CH–Me2), 22.3 (s, CH3–iPr) Anal. calcd for C36H54N6O4Zr (%): C, 59.55; H, 7.50; N, 11.57. Found (%) C, 59.26; H, 7.45; N, 11.88.N), 160.2 (s, C–O), 142.7 (s, C–N), 121.3 (s, CH–Ar) 118.4 (s, CH–Ar), 116.3 (s, CH–Ar), 115.8 (s, CH–Ar), 71.0 (s, OCH–Me2), 47.8 (s, NCH3), 35.6 (s, N–CH2) 27.7 (s, OCH–Me2). Anal. calcd for C28H42N6O4Zr (%): C, 54.43; H, 6.85; N, 13.60. Found (%) C, 54.78; H, 6.95; N, 13.76.N), 160.1 (s, C–O), 144.1 (s, C–N), 120.2 (s, CH–Ar) 116.3 (s, CH–Ar), 116.2 (s, CH–Ar), 115.8 (s, CH–Ar), 70.6 (s, OCH–Me2), 48.3 (s, NCH3), 40.1 (s, N–CH2) 27.9 (s, OCH–Me2), 22.2 (s, CH2). Anal. calcd for C30H46N6O4Zr (%): C, 55.78; H, 7.18; N, 13.01. Found (%) EA C, 55.55; H, 7.03; N, 12.76.N), 152.7 (s, C–OH), 144.4 (s, C–N), 120.3 (s, CH–Ar), 120.0 (s, CH–Ar), 118.1 (s, N(CH)2N), 112.0 (s, CH–Ar), 109.2 (s, CH–Ar), 34.9 (s, NCH3), 34.7 (s, NCH3). Anal. calcd for C22H24Cl2N6O2Ti (%): C, 50.50; H, 4.62; N, 16.06. Found (%) C, 50.95; H, 5.01; N, 15.63.N), 153.4 (s, C–OH), 145.1 (s, C–N), 141.9 (s, CH–Ar), 131.6 (s, CH–Ar), 117.9 (s, N(CH)2N), 117.6 (s, N(CH)2N), 114.0 (s, CH–Ar), 104.9 (s, CH–Ar), 35.1 (s, NCH3), 34.7 (s, NCH3), 34.6 (s, C(CH3)3), 34.3 (s, C(CH3)3), 32.0 (s, C(CH3)3), 30.0 (s, C(CH3)3). Anal. calcd for C38H56Cl2N6O2Ti (%): C, 61.05; H, 7.55; N, 11.24. Found (%) C, 61.33; H, 7.40; N, 11.49.N), 150.2 (s, C–OH), 146.0 (s, C–N), 119.9 (s, CH–Ar), 119.6 (s, CH–Ar), 114.1 (s, N(CH)2N), 114.0 (s, N(CH)2N), 111.7 (s, CH–Ar), 109.0 (s, CH–Ar), 49.0 (s, CH–Me2), 48.7 (s, CH–Me2), 24.2 (s, CH–Me2), 23.3 (s, CH–Me2), 23.2 (s, CH–Me2), 23.2 (s, CH–Me2), 22.2 (s, CH–Me2). Anal. calcd for C30H40Cl2N6O2Ti (%): C, 56.70; H, 6.35; N, 13.23. Found (%) C, 56.85; H, 6.62; N, 13.44.:[Cat] ratio of 100:1. Aliquots were removed from the reaction mixture at different times to monitor the monomer conversion by 1H NMR spectroscopy. Molecular weight averages were by GPC, and by end-chain analysis (Mn) and by diffusion ordered 1H NMR spectroscopy (DOSY; Mw).:1 of MeOH and concentrated HCl. The polymer was collected by filtration and washed with water and MeOH and dried for several hours at 60 °C under vacuum.Data availability
The data supporting this article have been included as part of the “ESI”.† Crystallographic data for all four X-ray structures reported in the paper have been deposited to the CCDC with deposition numbers 2345354–2345357. All DFT calculations were performed using the computational package Gaussian 16 on the Shared Hierarchical Academic Research Computing Network (SHARCNET; https://www.sharcnet.ca/my/front/), a consortium of colleges, universities and research institutes of the province of Ontario, Canada.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. G. L. gratefully acknowledges financial support from the Natural Sciences and Engineering Research Council for a Discovery Grant. V. F. R thanks the Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCYT) of Mexico for a graduate scholarship and York University for a Carswell scholarship.References
- X. Li, Y. Lin, M. Liu, L. Meng and C. Li, J. Appl. Polym. Sci., 2023, 140, e53477 CrossRef CAS.
- K. Hamad, M. Kaseem, M. Ayyoob, J. Joo and F. Deri, Prog. Polym. Sci., 2018, 85, 83–127 CrossRef CAS.
- K. Madhavan Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef CAS PubMed.
- S. Mecking, Angew. Chem., Int. Ed., 2004, 43, 1078–1085 CrossRef CAS PubMed.
- T. A. Swetha, A. Bora, K. Mohanrasu, P. Balaji, R. Raja, K. Ponnuchamy, G. Muthusamy and A. Arun, Int. J. Biol. Macromol., 2023, 234, 123715 CrossRef CAS PubMed.
- V. H. Sangeetha, H. Deka, T. O. Varghese and S. K. Nayak, Polym. Compos., 2018, 39, 81–101 CrossRef CAS.
- A.-C. Albertsson and M. Hakkarainen, Science, 2017, 358, 872–873 CrossRef CAS PubMed.
- C.-S. Ha and J. A. Gardella, Chem. Rev., 2005, 105, 4205–4232 CrossRef CAS PubMed.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed.
- B. N. Mankaev and S. S. Karlov, Materials, 2023, 16, 6682 CrossRef CAS PubMed.
- U. Yolsal, P. J. Shaw, P. A. Lowy, R. Chambenahalli and J. A. Garden, ACS Catal., 2024, 14, 1050–1074 CrossRef CAS PubMed.
- R. P. Singh, S. Sinhababu and N. P. Mankad, ACS Catal., 2023, 13, 12519–12542 CrossRef CAS.
- Y. Hu, W. Daoud, K. Cheuk and C. Lin, Materials, 2016, 9, 133 CrossRef PubMed.
- A. Kowalski, J. Libiszowski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 1964–1971 CrossRef CAS.
- H. R. Kricheldorf, I. Kreiser-Saunders and A. Stricker, Macromolecules, 2000, 33, 702–709 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 7359–7370 CrossRef CAS.
- X. Zhang, D. A. MacDonald, M. F. A. Goosen and K. B. McAuley, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2965–2970 CrossRef CAS.
- J. Lunt, Polym. Degrad. Stab., 1998, 59, 145–152 CrossRef CAS.
- K. Devaine-Pressing, F. J. Oldenburg, J. P. Menzel, M. Springer, L. N. Dawe and C. M. Kozak, Dalton Trans., 2020, 49, 1531–1544 RSC.
- M. Fernández-Millán, P. Ortega, T. Cuenca, J. Cano and M. E. G. Mosquera, Organometallics, 2020, 39, 2278–2286 CrossRef.
- J. Hu, C. Kan, H. Wang and H. Ma, Macromolecules, 2018, 51, 5304–5312 CrossRef CAS.
- Y. Wang, X. Wang, W. Zhang and W.-H. Sun, Organometallics, 2023, 42, 1680–1692 CrossRef CAS.
- R. Duan, C. Hu, X. Li, X. Pang, Z. Sun, X. Chen and X. Wang, Macromolecules, 2017, 50, 9188–9195 CrossRef CAS.
- S. Impemba, G. Manca, I. Tozio and S. Milione, Polymers, 2023, 15, 4366 CrossRef CAS PubMed.
- W. Gruszka, L. C. Walker, M. P. Shaver and J. A. Garden, Macromolecules, 2020, 53, 4294–4302 CrossRef CAS.
- R. D. Rittinghaus, P. M. Schäfer, P. Albrecht, C. Conrads, A. Hoffmann, A. N. Ksiazkiewicz, O. Bienemann, A. Pich and S. Herres-Pawlis, ChemSusChem, 2019, 12, 2161–2165 CrossRef CAS PubMed.
- M. Shaik, J. Peterson and G. Du, Macromolecules, 2019, 52, 157–166 CrossRef CAS.
- S. Kernbichl, M. Reiter, D. H. Bucalon, P. J. Altmann, A. Kronast and B. Rieger, Inorg. Chem., 2018, 57, 9931–9940 CrossRef CAS PubMed.
- T. Ebrahimi, D. C. Aluthge, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2016, 49, 8812–8824 CrossRef CAS.
- A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS PubMed.
- S. Impemba and S. Milione, Inorg. Chim. Acta, 2024, 568, 122067 CrossRef CAS.
- A. Buchard, C. J. Chuck, M. G. Davidson, G. Gobius Du Sart, M. D. Jones, S. N. McCormick and A. D. Russell, ACS Catal., 2023, 13, 2681–2695 CrossRef CAS PubMed.
- R. Hador, M. Shuster, S. Lipstman and M. Kol, ACS Catal., 2022, 12, 4872–4879 CrossRef CAS.
- B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC.
- P. Dubois, C. Jacobs, R. Jérôme and P. Teyssié, Macromolecules, 1991, 24, 2266–2270 CrossRef CAS.
- P. Dubois, R. Jérôme and P. Teyssié, Makromol. Chem., Macromol. Symp., 1991, 42–43, 103–116 CrossRef.
- H. R. Kricheldorf, M. Berl and N. Scharnagl, Macromolecules, 1988, 21, 286–293 CrossRef CAS.
- P. Dobrzynski, J. Kasperczyk, H. Janeczek and M. Bero, Macromolecules, 2001, 34, 5090–5098 CrossRef CAS.
- I. El-Zoghbi, T. J. J. Whitehorne and F. Schaper, Dalton Trans., 2013, 42, 9376–9387 RSC.
- L.-J. Wu, W. Lee, P. Kumar Ganta, Y.-L. Chang, Y.-C. Chang and H.-Y. Chen, Coord. Chem. Rev., 2023, 475, 214847 CrossRef CAS.
- R. Dai and P. L. Diaconescu, Dalton Trans., 2019, 48, 2996–3002 RSC.
- C. B. Durr and C. K. Williams, Inorg. Chem., 2018, 57, 14240–14248 CrossRef CAS PubMed.
- A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC.
- C. Nakornkhet, T. Nanok, W. Wattanathana, P. Chuawong and P. Hormnirun, Inorg. Chem., 2022, 61, 7945–7963 CrossRef CAS PubMed.
- K. Upitak, W. Wattanathana, T. Nanok, P. Chuawong and P. Hormnirun, Dalton Trans., 2021, 50, 10964–10981 RSC.
- D. J. Gilmour, R. L. Webster, M. R. Perry and L. L. Schafer, Dalton Trans., 2015, 44, 12411–12419 RSC.
- A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Dalton Trans., 2006, 887–889 RSC.
- A. J. Chmura, D. M. Cousins, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Dalton Trans., 2008, 1437–1443 RSC.
- T. G. Larocque, S. Dastgir and G. G. Lavoie, Organometallics, 2013, 32, 4314–4320 CrossRef CAS.
- B. S. Khan, V. Flores-Romero, J. LeBlanc and G. G. Lavoie, Organometallics, 2022, 41, 2668–2677 CrossRef CAS.
- P. M. Schäfer and S. Herres-Pawlis, ChemPlusChem, 2020, 85, 1044–1052 CrossRef PubMed.
- M. Li, X. Shu, Z. Cai and M. S. Eisen, Organometallics, 2018, 37, 1172–1180 CrossRef CAS.
- M. Li, Z. Cai and M. S. Eisen, Organometallics, 2018, 37, 4753–4762 CrossRef CAS.
- H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363–2449 CrossRef CAS PubMed.
- E. F. Connor, T. R. Younkin, J. I. Henderson, A. W. Waltman and R. H. Grubbs, Chem. Commun., 2003, 2272–2273 RSC.
- T. R. Younkin, E. F. Connor, J. I. Henderson, S. K. Friedrich, R. H. Grubbs and D. A. Bansleben, Science, 2000, 287, 460–462 CrossRef CAS PubMed.
- A. L. Johnson, M. G. Davidson, M. D. Lunn and M. F. Mahon, Eur. J. Inorg. Chem., 2006, 3088–3098 CrossRef CAS.
- Z. Flisak, J. Mol. Catal. A: Chem., 2010, 316, 83–89 CrossRef CAS.
- J. L. Espartero, I. Rashkov, S. M. Li, N. Manolova and M. Vert, Macromolecules, 1996, 29, 3535–3539 CrossRef CAS.
- H. R. Kricheldorf, I. Kreiser-Saunders and C. Boettcher, Polymer, 1994, 36, 1253–1259 CrossRef.
- P. Lewinski, S. Sosnowski, S. Kazmierski and S. Penczek, Polym. Chem., 2015, 6, 4353–4357 RSC.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- M. H. Chisholm, S. S. Iyer, D. G. McCollum, M. Pagel and U. Werner-Zwanziger, Macromolecules, 1999, 32, 963–973 CrossRef CAS.
- M. T. Zell, B. E. Padden, A. J. Paterick, K. A. M. Thakur, R. T. Kean, M. A. Hillmyer and E. J. Munson, Macromolecules, 2002, 35, 7700–7707 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS PubMed.
- S. Pappuru, V. Ramkumar and D. Chakraborty, Polym. Adv. Technol., 2021, 32, 3392–3401 CrossRef CAS.
- A. Chuma, H. W. Horn, W. C. Swope, R. C. Pratt, L. Zhang, B. G. G. Lohmeijer, C. G. Wade, R. M. Waymouth, J. L. Hedrick and J. E. Rice, J. Am. Chem. Soc., 2008, 130, 6749–6754 CrossRef CAS PubMed.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- J. Strauch, T. H. Warren, G. Erker, R. Frohlich and P. Saarenketo, Inorg. Chim. Acta, 2000, 300–302, 810–821 CrossRef CAS.
- F. Bellina, M. Lessi, G. Marianetti and A. Panattoni, Tetrahedron Lett., 2015, 56, 3855–3857 CrossRef CAS.
- M. Baltrun, F. A. Watt, R. Schoch, C. Wölper, A. G. Neuba and S. Hohloch, Dalton Trans., 2019, 48, 14611–14625 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154122 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, plots to calculate kapp, and crystal and structure refinement data (PDF). CCDC 2345354–2345357. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra05146g |
| This journal is © The Royal Society of Chemistry 2024 |