
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModified Cinnabaris-stabilized Pickering emulsions loaded with the essential oil of Acorus tatarinowii Schott: preparation, characterization and in vitro evaluation†
Han Ru a,
Fei Luana,
Yajun Shia,
Xiaofei Zhanga,
Dongyan Guoa,
Bingtao Zhaia,
Jing Suna,
Dingkun Zhangb,
Liang Fengc and
Junbo Zou*a
a,
Fei Luana,
Yajun Shia,
Xiaofei Zhanga,
Dongyan Guoa,
Bingtao Zhaia,
Jing Suna,
Dingkun Zhangb,
Liang Fengc and
Junbo Zou*a
aShaanxi Province Key Laboratory of New Drugs, Chinese Medicine Foundation Research, Pharmacy College, Shaanxi University of Chinese Medicine, Xianyang 712046, PR China. E-mail: 2051078@sntcm.edu.cn; Tel: +86-153-1907-0696
bSchool of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, Sichuan, PR China
cSchool of Pharmacy, China Pharmaceutical University, Nanjing 211198, Jiangsu, PR China
First published on 2nd October 2024
Abstract
Essential oil of Acorus tatarinowii Schott (ATEO) have significant biological activity, but their physical and chemical properties are unstable and susceptible to interference by external factors, resulting in oxidation, decomposition, and isomerization of essential oils (EOs), ultimately diminishing the quality of EOs and escalating clinical risks. In this research, based on the concept of “ combination of medicine and adjuvant, ” the unsuitable stabilizer Cinnabaris in Lingzhu powder prescription was modified with a SiO2 surface to become a stabilizer suitable for Pickering emulsion. The modified Cinnabaris was synthesized, with a focus on exploring the surface modification of Cinnabaris to facilitate its role as a stabilizer in Pickering emulsion. Thermal stability studies showed that modified Cinnabaris-stabilized emulsion had higher EOs retention and lower peroxide value and hydrogen peroxide content. GC-MS analysis showed that the volatile components in the emulsion were more stable than the EOs. In vitro dissolution experiments showed that in the dissolution medium of artificial gastric juice and artificial intestinal juice, compared with the ATEO, the release in Pickering emulsion was faster within 48 h, indicating that the ATEO had been encapsulated in Pickering emulsion, which could improve the in vitro dissolution rate of EOs. This study convincingly demonstrates the potential of modified Cinnabaris-stabilized Pickering emulsion to improve the thermal stability and in vitro dissolution rate of EOs.
1 Introduction
Essential oils (EOs) and their constituents are considered to be major bioactive natural compounds, with a variety of biological activities.1–4 Pharmacological activities, including those in the cardiovascular, gastrointestinal, central nervous, and respiratory systems, have been extensively investigated in clinical research.5–7 However, due to their high irritability, poor stability, ease of volatilization at room temperature, sensitivity to air, light, and temperature, and other environmental factors, their clinical application is greatly restricted.2 The unsaturated fatty acids of EOs are easily degraded by reaction with oxygen during the storage process after opening. On the one hand, oxidation will reduce the nutritional quality of the oil; on the other hand, this process will produce an unpleasant smell and affect the odor quality of the EOs. The oxidation of EOs will produce harmful substances such as trans fatty acids and hydroperoxides, which are extremely harmful to the human body after consumption.8 Therefore, to improve the stability of EOs during the preparation process, it is necessary to control their volatility and ensure stable release.9,10Currently, the primary technique employed to improve the storage stability of EOs has been β-cyclodextrin inclusion technology.11 However, this technique presents challenges such as a low inclusion rate, limited drug loading capacity, and the need for higher dosages.12,13 Other techniques such as thin film, capsules, and liposomes have also been used to enhance the stability of EOs. However, these approaches have drawbacks including organic solvent residues, poor stability, and unknown toxic side effects.14,15 In recent years, Pickering emulsions stabilized by solid particles have gained increasing research interest due to their eco-friendly nature and high stability.16,17 Pickering emulsions are defined as the use of solid particles with appropriate surface wettability as substitutes for traditional chemical emulsifiers.18–20 These particles are able to irreversibly adsorb at the O/W interface, forming a complex and stable interfacial material that prevents coalescence and aggregation of droplets.21 In the process of physiological degradation, they provided better protection for compounds.22 In addition, compared to traditional emulsions, Pickering emulsions have numerous advantages, such as easy preparation, non-toxicity, and long-term stability.23 Therefore, Pickering emulsions have been preferred for enhancing encapsulation and improving the stability of bioactive substances.24 For example, using zein–phospholipid composite nanoparticles as emulsifiers to prepare peppermint oil emulsions has been demonstrated to enhance the stability of peppermint oil (Scheme 1).25
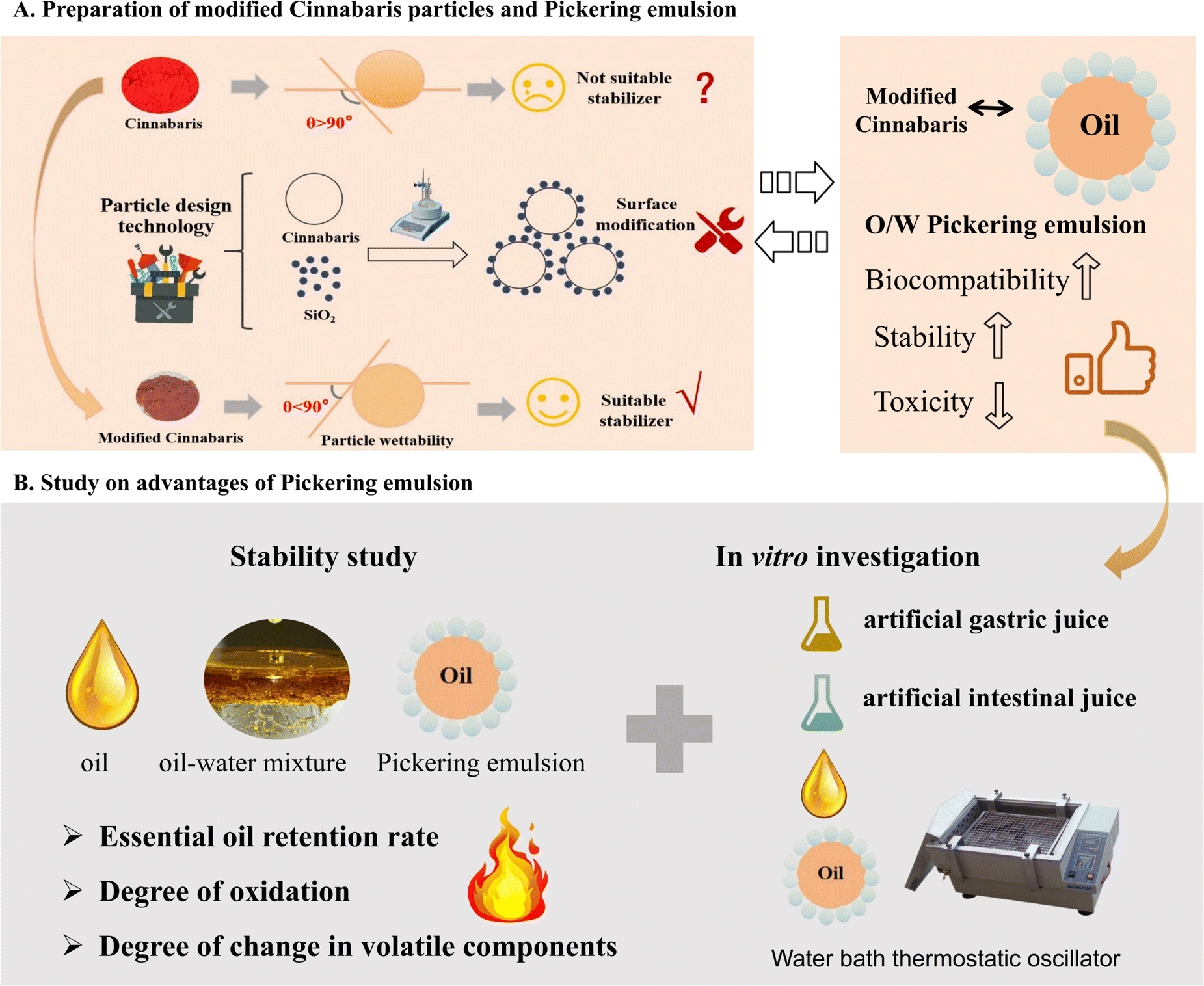 | ||
| Scheme 1 Research strategy diagram for enhancing the stability of EOs with Pickering emulsions stabilized by modified suitable Cinnabaris stabilizer. (A) Preparation of modified Cinnabaris particles and Pickering emulsions; (B) study on advantages of Pickering emulsions. | ||
Particle design technology is a cutting-edge approach that enhances the properties of materials within the field of materials science. The wettability of the particles is adjusted through surface modification to create suitable particles to stabilize the Pickering emulsions.26 Some scholars have found that the complex structure and hydrophobicity of cereal protein greatly limit its application in oil-in-water emulsion systems.27 Experimental results indicate that the θo/w of zein particles is about 114.46°, while the θo/w of gliadin is about 64.22°. With the increase of the proportion of gliadin, the θo/w of zein–gliadin complex particles (ZGCP) decreases from 104.53° to approximately 86.60°. The reason is that hydrophilic gliadin helps eliminate the hydrophobicity of zein, leading to the formation of near-neutral wettability of ZGCP. The flocculation induced by the interfacial interactions of composite particles creates a stable interface film.28 It is conducive to the formation of good viscoelastic properties and storage stability, which may help to form a stable Pickering emulsions.
Lingzhu powder, a century-old pediatric medicinal product produced by Lei Yun Shang Pharmaceuticals, functions to reduce fever and calm the nerves.29 It is commonly used in clinical practice to treat febrile convulsions in children.30 The essential oil of Acorus tatarinowii Schott (ATEO) used in the prescription is applied directly, which makes it prone to volatilization and oxidation, consequently reducing its clinical efficacy.31 Building on the research group's previous work, we revealed that the contact angle of Cinnabaris in Lingzhu powder was 124.95 ± 0.35°. When preparing the Pickering emulsions, it adhered to the water layer wall or sank to the bottom, preventing the formation of an emulsion. Thus, drawing from the concept of a “combination of medicine and adjuvant”, we prepared the Pickering emulsions by using modified Cinnabaris as a stabilizer to improve the stability of EOs in Lingzhu powder under high-temperature conditions. We carried out the in vitro dissolution experiment using the dialysis bag method to compare the in vitro release behavior of ATEO and Pickering emulsions encapsulated with ATEO in artificial gastric juice and artificial intestinal juice. Using α-asarone and β-asarone as characteristic components, we established a GC-MS method to determine the cumulative release of α-asarone and β-asarone in artificial gastric juice and artificial intestinal juice, and we drew the dissolution curve to explore the in vitro release behavior of ATEO and Pickering emulsions in artificial gastric juice and artificial intestinal juice. These results indicate that the modification of Cinnabaris by SiO2 is successful, and the reaction is a green and effective method to improve the emulsification performance of Cinnabaris. This modification can be directly used as a stabilizer to stabilize Pickering emulsions and applied to improve the stability of volatile oil in oil-containing solid preparations.
2 Materials and methods
2.1 Materials
ATEO was provided by Poli Aromatic Pharmaceutical Technology (Shanghai) Co., Ltd. Cinnabaris was provided by Lys Pharmaceutical Co., Ltd. SiO2 was provided by Shanghai Macklin Biochemical Technology Co., Ltd. In addition, 1,1,3,3-tetraethylpropane (95%) and 2-thiobarbituric acid (purity ≥98.5%) were obtained from Shanghai Yuanye Bio-Technology Co., Ltd. Nile Red and Nile Blue were provided by CSNpharm and Sigma-Aldrich. n-Docosane (99.6%) was provided by Grace Chemical Technology Co., Ltd. Pepsin and trypsin were provided by Shanghai McLean Biochemical Technology Co., Ltd. The β-asarone and α-asarone standards were provided by Shanghai Yuanye Biotechnology Co., Ltd. The 300 kDa dialysis bag was provided by SCIENTIFIC RESEARCH SPECIAL. Other commonly used chemical reagents were purchased from Tianjin Komeo Chemical Reagent Co., Ltd.2.2 Preparation and characterization of modified Cinnabaris
2.2.2.1 Scanning electron microscope and energy dispersive X-ray spectroscopy. The microstructures of the test samples were observed using a scanning electron microscope (SEM).33 A small amount of test sample particles was placed on the sample stage and underwent gold plating treatment for 20 s at a working current of 10 mA and a working voltage of 4 mV. Using an SEM, the morphology of the sample particles was observed. The imaging of the sample at a magnification of 4 μm under SEM was selected.34 The element distribution of Cinnabaris powder, SiO2, and modified Cinnabaris was analyzed by energy dispersive spectrometer (EDS).
2.2.2.2 Particle wettability. The air–water contact angles (θaw) of Cinnabaris and modified Cinnabaris were measured using an optical contact angle meter.35,36 The Cinnabaris and modified Cinnabaris samples were pressed into circular sheets with a thickness of 5 mm and a diameter of 20 mm. The image of the water droplets on the surface of the disc was taken by a high-speed camera connected to the contact angle meter to determine θaw. Subsequently, the SurfaceMeter software was used to automatically calculate the contact angle between the water droplet and the circular sheets (left and right).
2.2.2.3 Raman and Fourier transform infrared spectroscopy. Raman spectroscopy had a spectral measurement range of 2700 to 65 cm−1. Cinnabaris was derived from the sulfide mineral group, primarily composed of HgS, and its characteristic peaks were generally in the range of 400 to 80 cm−1; therefore, Raman spectroscopy was used for the determination of Cinnabaris.37 Fourier transform infrared spectroscopy (FT-IR) was performed, using a resolution of 4 cm−1, and conducting 64 scans within a test range of 4000 to 400 cm−1. SiO2 contains silicon (Si) and oxygen (O), and its characteristic peaks were generally between 1200 and 400 cm−1; thus, FT-IR was used for the determination of SiO2.38 The modified Cinnabaris contained both Cinnabaris and SiO2. Given this, both FT-IR and Raman spectroscopy were used to determine its structure.
2.3 Preparation and characterization of Pickering emulsions
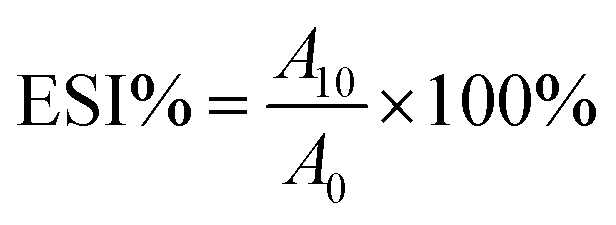 | (1) |
Following the principles of response surface design, with OD as the response value, three significant factors influencing the Pickering emulsions were selected. The independent variables were stabilizer dosage, oil phase volume fraction, and shear time. A three-factor, three-level experimental design was conducted using experimental design software with 3 replicates, resulting in a total of 17 response surface optimization experiments to determine the optimum emulsion preparation conditions.43
2.3.4.1 Measurement of particle size. The particle size of the Pickering emulsions was typically determined using the wet measurement method of the Malvern laser particle size analyzer.44 The evaluation was based on the d90 value, which represented a key parameter of the particle size distribution.
2.3.4.2 Measurement of zeta potential. The zeta potential analysis of the Pickering emulsions was performed using the zeta sizer nano ZS90 for zeta potential analysis.44 An appropriate quantity of Pickering emulsions sample, prepared according to the optimized procedure, was taken and diluted 5-fold with purified water, followed by thorough mixing. The zeta potential was then measured.
2.3.4.3 Microscopically observe. Marked the particles with Nile Blue, which showed red fluorescence under confocal microscopy. Marked the oil phase with Nile Red, which showed green fluorescence under confocal microscopy. Diluted the fluorescently labeled Pickering emulsions with distilled water at a 10-fold dilution and stirred for approximately 1 min. Then, we took 100 μL of the labeled Pickering emulsions solution, dropped it onto a glass slide, and covered it with a cover slip. This prevented the evaporation of ATEO.35 We used a confocal laser scanning microscope with a 20× oil immersion objective lens to observe the labeled Pickering emulsions.
2.3.4.4 Near-infrared spectrum. An appropriate amount of ATEO, Cinnabaris suspension, and Pickering emulsion samples were carefully placed in a 2 mm quartz cuvette. Spectra were collected at room temperature within the range of 4000 cm−1 to 1000 cm−1. The data acquisition involved 32 scans, a spectral resolution of 8 cm−1, and a gain of 1×.45 The spectra were acquired by subtracting the background using air as a reference, and each sample was repeated three times to obtain an averaged near-infrared spectrum (NIRS) measurement.
2.4 Thermal stability of Pickering emulsion
2.4.2.1 Peroxide value. Transferred 500 μL of the ATEO sample from “2.4.1” into a conical flask. Successively added 10 mL of a mixture of trichloroacetic acid (TCA) and glacial acetic acid (4
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6) and 1 mL of saturated potassium iodide solution and shook for 0.5 min. The sample was placed in a dark area for 3 min. Afterward, 30 mL of distilled water and 1 mL of 1% starch indicator were added. The mixture was titrated with a 0.001 mol per L sodium thiosulfate standard solution until the solution became colorless. The volume of sodium thiosulfate standard solution consumed during the titration was recorded and was subsequently used to calculate the peroxide value (POV).46
:6) and 1 mL of saturated potassium iodide solution and shook for 0.5 min. The sample was placed in a dark area for 3 min. Afterward, 30 mL of distilled water and 1 mL of 1% starch indicator were added. The mixture was titrated with a 0.001 mol per L sodium thiosulfate standard solution until the solution became colorless. The volume of sodium thiosulfate standard solution consumed during the titration was recorded and was subsequently used to calculate the peroxide value (POV).46
 | (2) |
2.4.2.2 Malondialdehyde. Using a precision balance, 0.1575 g of 1,1,3,3-tetraethoxypropane was accurately weighed and transferred into a 500 mL volumetric flask. The compound was then diluted with water and made up to the mark, resulting in a solution that had a concentration of 0.1 g L−1 for malondialdehyde (MDA). This solution was stored for future use. MDA standard solutions were prepared by pipetting varying volumes (0, 0.02, 0.04, 0.06, 0.08, and 0.1 mL) of the mother liquid into 10 mL volumetric flasks and diluting them to the mark with water. Consequently, these solutions had mass concentrations of 0, 0.2, 0.4, 0.6, 0.8, and 1.0 mg L−1. Each standard solution was mixed with 5.00 mL of thiobarbituric acid (TBA) solution and heated in a water bath at 90 °C for 40 min. After cooling, 5.00 mL of chloroform was added to the mixture, which was then shaken thoroughly and allowed to stand for 1 h. The supernatant was collected for subsequent analysis, and the absorbance was measured at 532 nm. The obtained regression equation was Y = 0.3001X − 0.0006, with an R2 = 0.9997.
The TCA solution was prepared by diluting 75 g of TCA and 1 g of EDTA-2Na to a final volume of 1 L with water. Transferred 500 μL of the ATEO sample from “2.4.1” into a 10.0 mL conical flask, volumizing with TCA solution. The sample and TCA mixture underwent sonication for 10 min, followed by filtration. From the filtrate, 5.0 mL was transferred to a conical flask. Subsequently, 5.0 mL of a 0.02 mol per L TBA solution was added to the flask, and the mixture was heated in a water bath at 90 °C for 40 min. After cooling for 1 h, 5.0 mL of chloroform was added, the mixture was shaken thoroughly, and allowed to stand for 1 h to separate the mixture into layers. The supernatant was collected, and its absorbance was measured at 532 nm.47
2.5 Characteristic analysis of ATEO volatile components
:1 and operated at a flow rate of 1.0 mL min−1.An electron impact ionization (EI) source was used as the ion source, with the ion source temperature set to 230 °C and the quadrupole temperature set to 150 °C. The scan range for mass analysis was set from m/z 35 to 500, and a solvent delay of 3 min was implemented.48
2.6 In vitro dissolution
:1, and the solvent delay was 3 min. The inlet temperature was 230 °C. The initial temperature was increased from 50 °C to 140 °C at a rate of 15 °C min−1, then increased to 144 °C at a rate of 0.4 °C min−1 for 5 min, and then increased to 250 °C at a rate of 10 °C min−1 for 2 min.Mass spectrometry conditions: the ionization mode was EI and the ion mode was ESI; the electron energy was set at 70 eV; and the quadrupole temperature was maintained at 150 °C, the ion source temperature was 230 °C; the scan mode was scan; and the range was m/z 35 to 500.
| Cumulate release rate% = [CnV1 + (Cn−1 + … + C1)V2]/W × 100% | (3) |
2.7 Statistical analysis
All statistical graphs were produced using GraphPad Prism software version 8.0. Results were presented as mean ± standard deviation (SD) based on at least three independent experiments. One-way analysis of variance (ANOVA) was employed to assess the significance of differences between groups, followed by post hoc comparisons using the SPSS LSD test. A p-value of P < 0.05 was considered statistically significant.3 Results and discussion
3.1 Characterization of modified Cinnabaris
 | ||
| Fig. 1 SEM of Cinnabaris (A), SiO2 (B), and modified Cinnabaris (C); EDS of Cinnabaris (D), SiO2 (E), and modified Cinnabaris (F). | ||
| Specimen | Element | Line type | Apparent concentration | K ratio | wt% | wt% Sigma |
|---|---|---|---|---|---|---|
| Cinnabaris | C | K | 2.19 | 0.02194 | 6.16 | 0.40 |
| O | K | 1.21 | 0.00407 | 0.84 | 0.12 | |
| S | K | 15.54 | 0.13387 | 10.39 | 0.31 | |
| Zr | L | 2.68 | 0.02682 | 2.37 | 0.28 | |
| Hg | M | 83.89 | 0.75983 | 74.39 | 0.83 | |
| Tl | M | 5.93 | 0.05931 | 5.85 | 0.86 | |
| Modified Cinnabaris | C | K | 5.32 | 0.05321 | 13.17 | 0.34 |
| O | K | 127.70 | 0.42972 | 50.76 | 0.27 | |
| Si | K | 53.50 | 0.42393 | 31.71 | 0.20 | |
| Pt | M | 3.92 | 0.03922 | 4.37 | 0.22 | |
| SiO2 | C | K | 2.98 | 0.02983 | 8.85 | 0.40 |
| O | K | 117.41 | 0.39509 | 49.47 | 0.32 | |
| Si | K | 58.80 | 0.46593 | 37.01 | 0.26 | |
| Pt | M | 3.87 | 0.03866 | 4.67 | 0.27 |
 | ||
| Fig. 2 The contact angle of Cinnabaris (A) and modified Cinnabais (B); Raman spectroscopy of Cinnabaris (C(a)) and modified Cinnabaris (C(b)); FT-IR spectroscopy of SiO2(D(c)) and modified Cinnabaris (D(b)). | ||
3.2 Preparation of modified Cinnabaris
3.2.1.1 Stabilizer amount. It could be seen from Fig. 3A that with the increase of the amount of modified Cinnabaris composite particle stabilizer, the contact area between the solid particles and the oil–water interface gradually increased, the overall stability score of the emulsion, and the stability of the emulsion increased, reaching the highest at 20 mg. However, as the amount of stabilizer continued to increase, the comprehensive score of the emulsion began to decrease, it suggested the presence of an optimal amount of stabilizer. The possible reason was that the amount of stabilizer was too high, and aggregation occurred at the oil–water interface, which did not participate in the formation process of Pickering emulsions, resulting in a decrease in emulsion stability. Therefore, we selected the dosage of stabilizer as 20 mg for subsequent investigation.
 | ||
| Fig. 3 One-way examination and response surface optimization experiments of Pickering emulsions preparation process. Stabilizer amount (A), ATEO phase volume fraction (B), and shear time (C) were examined; response surface plots of the interaction effect between factors (D). | ||
3.2.1.2 ATEO phase volume fraction. It could be seen from Fig. 3B that with the increase of the volume of the oil phase of AT, the comprehensive score of the emulsion increased and the stability of the emulsion was enhanced. When the volume fraction of the oil phase was 60%, the comprehensive score of the emulsion reached the highest. However, as the volume fraction of the oil phase continued to increase, the comprehensive score of the emulsion began to decrease. The possible reason was that due to too much oil phase, solid particles could only participate in the encapsulation of fixed oil volume, and excess volatile oil would remain free, resulting in a decrease in the comprehensive score of the emulsion. Therefore, the oil phase volume fraction of 60% was selected for subsequent investigation.
3.2.1.3 Shear time. It could be seen from Fig. 3C that as the shearing time increased, the comprehensive score of the emulsion increased and the stability of the emulsion increased. When the shearing time was 150 s, the comprehensive score of the emulsion reached the highest, but as the shearing time increased, the comprehensive score of the emulsion began to decrease. The possible reason was that under the action of a high-speed shearing machine, the longer the time, the more the solid particles were dispersed, resulting in decreased emulsion quality. Therefore, the shear time was selected to be 150 s.
3.2.1.4 Shear speed. It was found that the comprehensive score of the emulsion was not much different at the shear rate of 8000 to 12000 rpm, so it was not used as an index in the subsequent response surface optimization experiment. The shear speed of 10000 rpm was selected for investigation.
3.2.2.1 Analysis of variance of OD regression model of modified Cinnabaris Pickering emulsions. Design Expert 8.0.6 software was used to establish a regression model for OD data and analyze the variance. A quadratic response surface regression model was established using the following formula: Y = 92.36 + 6.27A + 5.78B − 0.100C + 3.01AB − 1.20AC − 2.96BC − 10.54A2 − 21.48B2 − 11.14C2. Analysis of variance (ANOVA) was performed on the regression equation, and the detailed results of the analysis were shown in Table S3.†
Model P < 0.001, which indicated that the difference was not significant, thus suggesting the model fitted well and the regression equation was reliable. The influences of factors A2, B2, and C2 on the response value were statistically significant (P < 0.0001). Therefore, the influence of the three factors on the response value was not a simple linear relationship, and there was a significant interaction between the selected test factors.
At the same time, the factors that had a significant impact on the experimental results were A, B, and C, indicating that the model could be used for the analysis and prediction of emulsion quality. According to the regression model, the corresponding response surface and contour maps were drawn. The slope of the response surface diagram of the interaction between stabilizer dosage and oil phase volume fraction was steeper, indicating a stronger interaction that significantly affected the comprehensive score of the emulsion. The interaction between the amount of stabilizer and the shear time had a slower response surface slope, indicating that the interaction between the two factors was weak and had little effect on the comprehensive score of the emulsion (Fig. 3D).
3.2.4.1 Measurement of particle size. By conducting three parallel experiments and recording the d90 characteristic value of the emulsion as 36.00 ± 1.26 μm (Fig. 4A), it could be concluded that the size of the modified Cinnabaris in the Pickering emulsions falls within the microscale range.
 | ||
| Fig. 4 Characterisation of the properties of Pickering emulsions. The particle size of Pickering emulsions (A); zeta potential of the Pickering emulsions (B); confocal microscope images of Pickering emulsions, bright field optical image of Pickering emulsions (C), ATEO labeled with green fluorescent dye (D), modified Cinnabaris labeled with red fluorescent dye (E) and combined confocal image of D and E (F); near-infrared spectra (G) of Cinnabaris (a), Pickering emulsions (b), and ATEO (c). | ||
3.2.4.2 Measurement of zeta potential. Each sample was measured in triplicate at a temperature of 25 °C, which resulted in a value of 33.80 ± 0.28 mV (Fig. 4B). It should have been noted that higher zeta potential values corresponded to improved emulsion stability.53
3.2.4.3 Microscopically observe. The type of emulsion formed was confirmed to be an O/W emulsion by confocal microscopy images, and the modified cinnabar particles were attached between the oil and aqueous phases, and the ATEO was completely encapsulated (Fig. 4C–F). The modified Cinnabaris, labeled with red fluorescent dye, was uniformly distributed at the boundaries of the emulsion particles (Fig. 4E). Meanwhile, EOs labeled with green fluorescent dye were observed in the inner region of the emulsion (Fig. 4D). Pickering emulsions contained smaller-sized EO droplets; the emulsions had a high content of solid particles, and the emulsion particles were spherical and well dispersed in the aqueous phase without aggregation.
3.2.4.4 Near infrared spectrum. The results in Fig. 4G indicated that the NIRS of the Pickering emulsions stabilized by modified Cinnabaris was similar to that of the modified Cinnabaris suspension. In the NIRS of the Pickering emulsions of modified Cinnabaris, only the weaker ATEO characteristic peaks were detected at 5500 to 6000 cm−1, while the ATEO characteristic peaks in general were more obvious. The NIRS results indicated that the modified Cinnabaris was encapsulated in the outer layer of ATEO in the Pickering emulsions.
3.3 Antioxidant properties of ATEO under thermal environment
As shown in Fig. 5A, the retention rate of the Pickering emulsions group was significantly higher than that of the AEO and oil–water mixture groups under the conditions of 1, 3, and 8 h at 40 °C (P < 0.01). The oxidation degree of volatile oil was reflected by the changes in POV and MDA content under an accelerated oxidation environment.54 Compared with the crude oil group, it was found that the POV and MDA contents in the volatile oil of the Pickering emulsion groups were significantly reduced. The results showed that the POV and MDA of volatile oil in the Pickering emulsion groups were the lowest, and the degree of oxidation was the lowest, indicating that it effectively removed free radicals and peroxides, prevented the oxidation reaction, and improved the stability of ATEO by reducing the formation of peroxides as well as the contents of POV and MDA. In the process of accelerated oxidation of ATEO, the Pickering emulsions prepared by modified Cinnabaris effectively delayed the oxidation degree of volatile oil, thus effectively protecting the volatile components in the EOs, as depicted in Fig. 5B and C.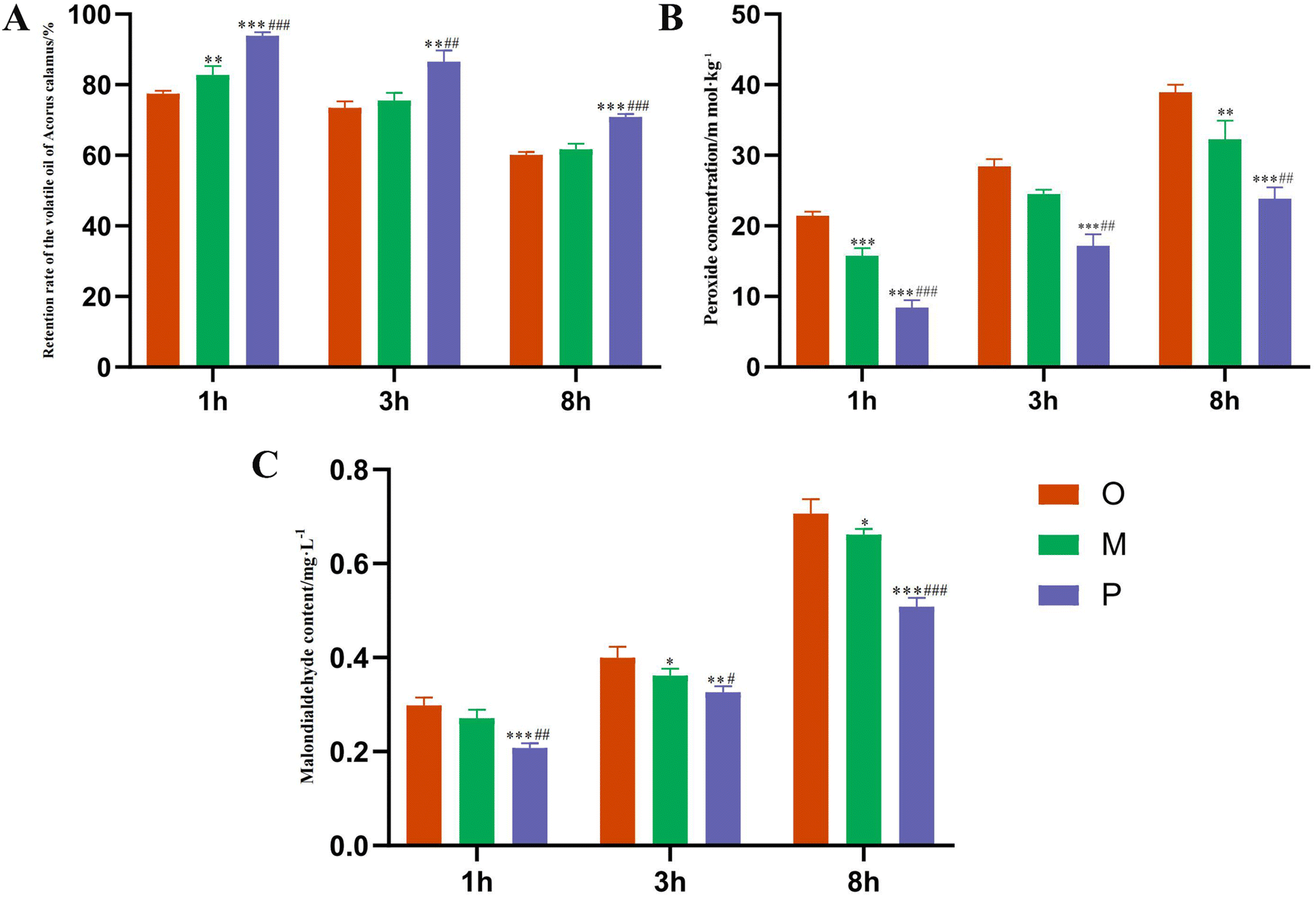 | ||
| Fig. 5 Retention rate of ATEO (A), content of POV (B), and content of MDA (C) in Pickering emulsions, oil–water mixture, and ATEO under heat treatment for 1, 3, and 8 h. (P < 0.05). *P < 0.05, **P < 0.01, ***P < 0.001 compared to the blank group. #P < 0.05, ##P < 0.01, ###P < 0.001 compared to the oil–water mixture group. | ||
3.4 Analysis of the components in ATEO
The total ion chromatogram of the components of ATEO was presented in Fig. S1A.† The results indicated that under the chromatographic conditions, the components were well separated, which met the requirements for subsequent qualitative and quantitative studies. By using the NIST 14 database, a total of 76 aromatic compounds were identified and listed in Table S4† after analyzing the total ion chromatogram.Draw the characteristic chromatogram of EO components of AT under different intervention conditions, see Fig. S1B.† The results showed that, excluding the internal standard peak, the characteristic components could be divided into seven categories: (1) 017669-14-8 and other four components, (2) 018675-33-7 and other 4 components, (3) 000469-61-4 and other 33 components, (4) 000141-27-5 and other 4 components, (5) 000586-62-9 and other 28 components, (6) 092471-23-3 and 1 component, (7) 021290-09-5 and 1 component. Among the components of category (5), as the oxidation time increased, the expression of volatile components in the Pickering emulsion group was highest during the continuous heating process at 1, 3, and 8 h, and the expression decreased slowly with the increase of heating time, while the crude oil group and oil–water mixture group decreased rapidly with the increase of heating time. In category (3), as the oxidation time increased, the expression of the crude oil group and oil–water mixture group gradually increased, while the expression of the Pickering emulsion group remained relatively consistent. It could be preliminary proved that the volatile components in Pickering emulsions were relatively stable under different heating times.
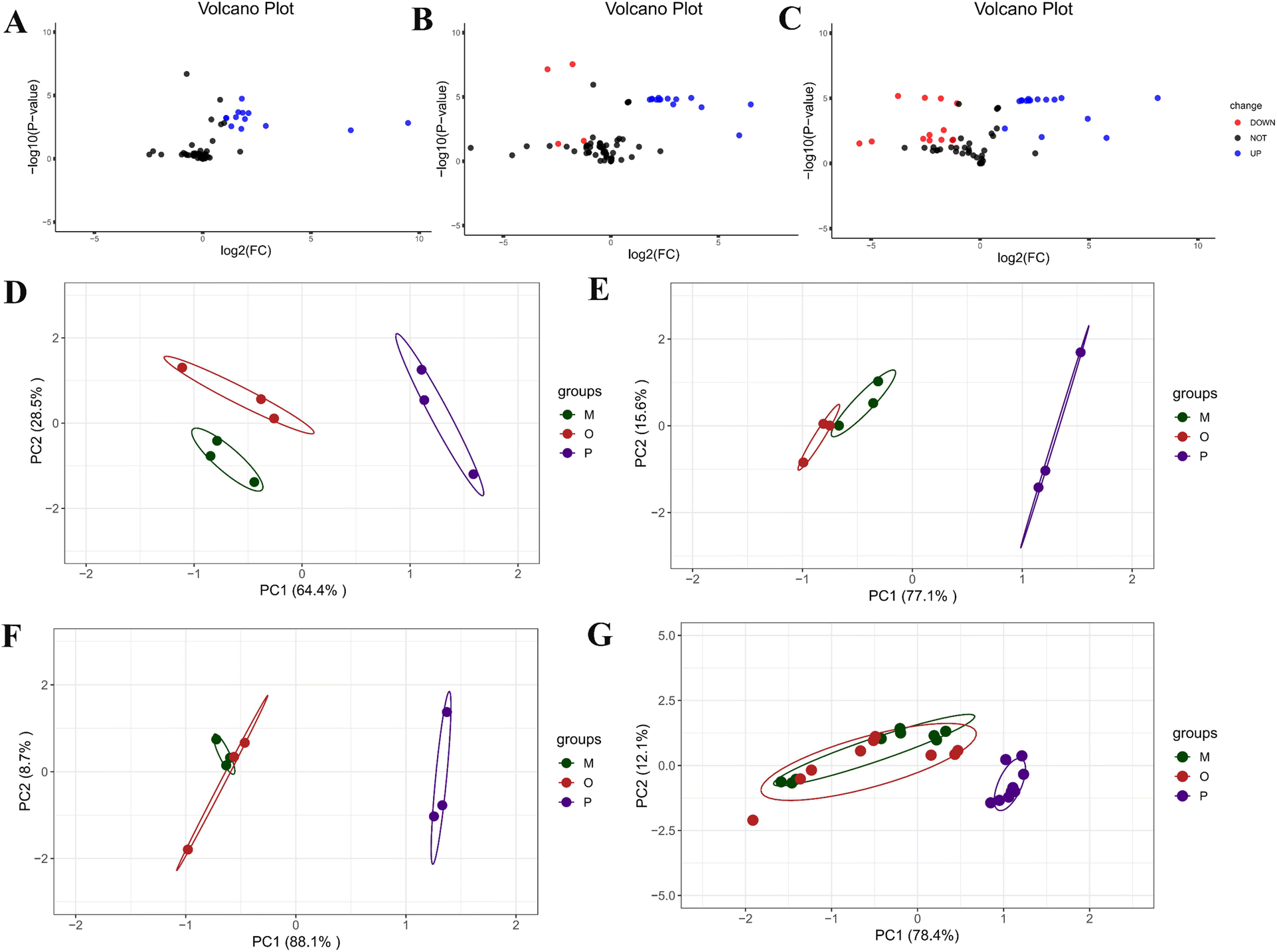 | ||
| Fig. 6 Volcano plot, PCA, and line graph analysis of volatile differential components of AT. The volcano plot of the differential components between untreated ATEO and the high temperature 1 h crude oil group (A), 3 h crude oil group (B), and 8 h crude oil group (C); the PCA plot of the differential components for the high temperature 1 h group (D), 3 h group (E), and 8 h group (F), the overall PCA plot of the differential components for different high-temperature durations (G). | ||
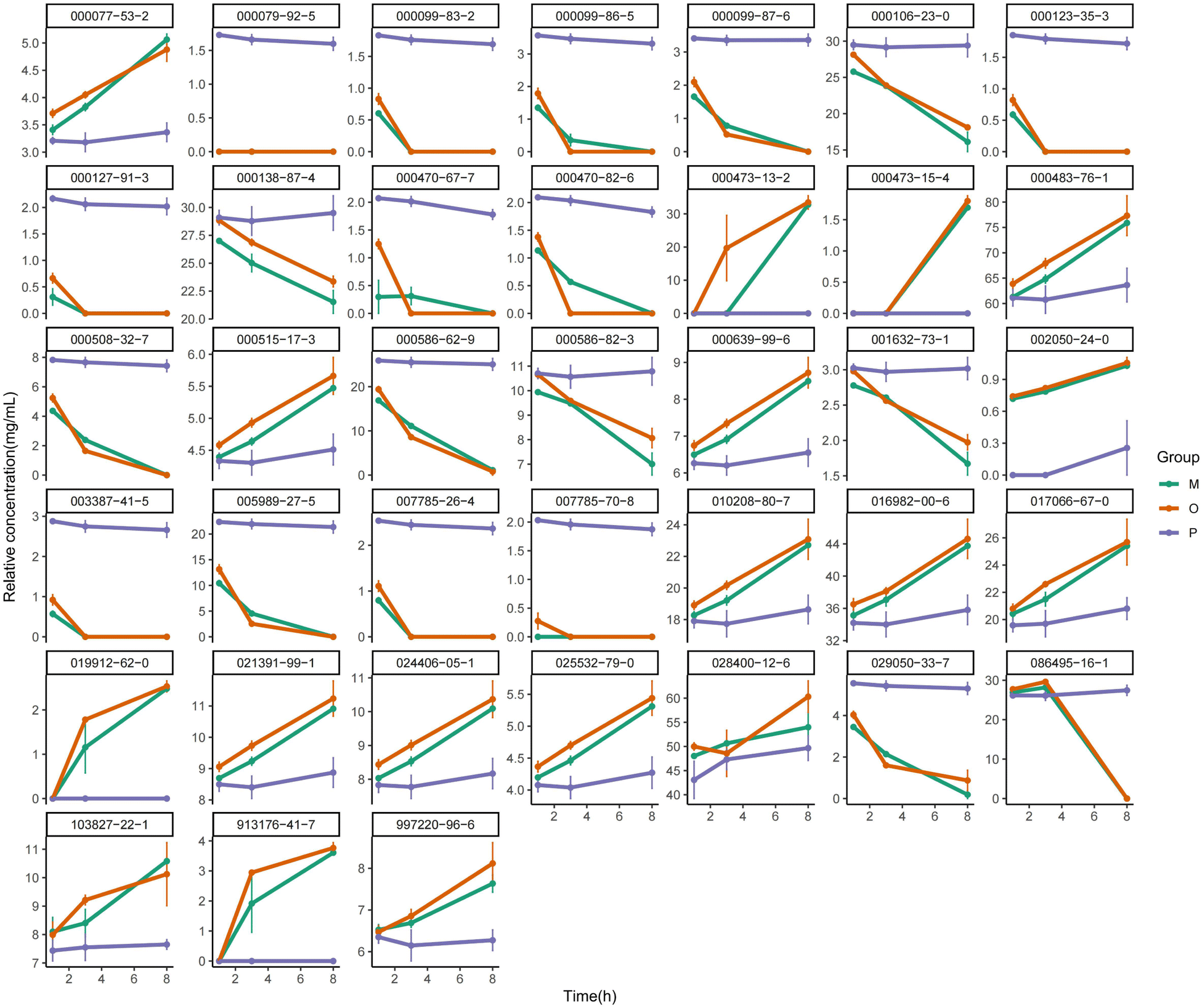 | ||
| Fig. 7 The line chart of the differential components in the Pickering emulsions, oil–water mixture, and ATEO. | ||
3.5 Quantitative changes of ATEO
The identified differential components using R language were categorized into two groups: up-regulated differential components and down-regulated differential components. For each category, the physicochemical property parameters, including relative molecular mass, melting point, boiling point, flash point, density, polarizability, surface tension, logP, and refractive index, were queried. The numerical values of these physicochemical parameters were then normalized, and the average values were subsequently calculated to assess their distribution behavior (Fig. 8A and B). Among the up-regulated differential components, the relative molecular mass, boiling point, and polarizability were the lowest, and the refractive index was the highest. Among the down-regulated differential components, the melting point was the lowest, and the boiling point, flash point, and surface tension were the highest, while the other factors were distributed in the middle. Fig. 8C showed that by using radar plots to analyze the overall physicochemical properties of up-regulated and down-regulated differential components, it was found that the relative molecular mass, boiling point, flash point, surface tension, and polarizability accounted for a larger proportion in the down-regulated components, while the distribution of up-regulated components was opposite.
 | ||
| Fig. 8 High-temperature environment affects the physical and chemical properties of the essential oil quantitative change difference components of AT. Distribution chart of the changes in physicochemical properties of up-regulated components (A), distribution chart of the changes in physicochemical properties of down-regulated components (B), radar chart of the distribution of physicochemical properties for up-regulated and down-regulated components (C), and PCA diagram of the physicochemical properties of different components affecting the quantitative variation of ATEO (D). | ||
To delve deeper into the main factors influencing the distribution behavior of components, an investigation was conducted based on various physicochemical parameters, as illustrated in Fig. 8D. Through analysis, a total of 10 principal components (PCs) were obtained. The PC1 component had a high contribution rate to the variance, accounting for 42.5% of the total variance, and was considered the main component that influenced the distribution pattern of volatile differences in AT. PC1 = 0.47 × relative molecular mass + 0.32 × melting point + 0.46 × boiling point + 0.38 × flash point + 0.24 × density − 0.31 × polarizability − 0.34 × surface tension + 0.24 × logP − 0.06 × refractive index. On the PC1 axis, the up-regulated differential components mainly exhibited a negative distribution, while the down-regulated differential components mainly exhibited a positive distribution. Relative molecular mass, melting point, boiling point, flash point, density, and logP were positively correlated, while polarizability, surface tension, and refractive index were negatively correlated. Based on the composition of the principal components, it was concluded that relative molecular mass, boiling point, flash point, polarizability, and surface tension were key physicochemical parameters that significantly influenced the distribution behavior of components. Components with higher relative molecular mass, boiling point, and flash point exhibited a propensity for distribution within the down-regulated differential component system, whereas those with lower relative molecular mass, higher surface tension, and higher refractive index were more inclined to be distributed within the up-regulated differential component system.
3.6 Qualitative changes of ATEO
A comparative analysis was conducted to examine the variations in the composition of ATEO among different groups, including untreated ATEO, heat-treated crude oil group, oil–water mixture group, and Pickering emulsion group. Based on the data presented in Fig. 9A, a total of 46 components were identified, out of which 30 were characterized as distinctive components. Analysis of the characteristic components revealed that there were 11 newly generated components, including 002050-24-0, 021290-09-5, 092471-23-3, 017699-14-8, 000473-13-2, 913176-41-7, 094535-52-1, 019912-62-0, 000473-15-4, 110983-38-5, and 997462-58-2. There were 17 disappearing components, including 007785-70-8, 000110-93-0, 000079-92-5, 000500-00-5, 000470-82-6, 000099-87-6, 005989-27-5, 000508-32-7, 086495-16-1, 003387-41-5, 000127-91-3, 000123-35-3, 000099-83-2, 007785-26-4, 000586-67-4, 997226-86-2, and 000111-84-2. In the stacked plot of newly generated components (Fig. 9B), the Pickering emulsion group, after 1, 3, and 8 h of high-temperature treatment, exhibited a higher similarity to the untreated ATEO composition compared to the crude oil group and the oil–water mixture group. This observation suggested that the volatile components in the Pickering emulsion group were less susceptible to the influence of high-temperature environments and exhibited a slower rate of oxidation. The stacked plot of disappearing components (Fig. 9C) illustrated that, in contrast to the untreated ATEO, two specific components, namely, 997226-86-2 and 000111-84-2, exhibited a more rapid rate of depletion within the Pickering emulsion group. In comparison with the crude oil group and the oil–water mixture group, the remaining components within the Pickering emulsion group exhibited a greater resemblance to the volatile components typically associated with crude oil, particularly under high-temperature conditions. This observation suggested that Pickering emulsion technology played a positive role in impeding the dispersion of ATEO and enhancing its stability. | ||
| Fig. 9 High-temperature environment affects the physical and chemical properties of the essential oil qualitative change difference components of AT. Upset plot of the changed components of ATEO (A), stacked plot of the newly generated components of ATEO (B), and stacked plot of the disappearing components of ATEO (C). Distribution chart of the physicochemical properties of newly generated components (D), distribution chart of the physicochemical properties of disappearing components (E), radar chart of the physicochemical properties distribution of newly generated and disappearing components (F), and PCA chart of physicochemical properties of different components affecting the qualitative changes of ATEO (G). | ||
The Upset plot categorizes the identified differential components into two groups: newly generated components and disappearing components. The physicochemical properties of the newly generated and disappearing components were examined, which included parameters such as relative molecular mass, melting point, boiling point, flash point, density, polarizability, surface tension, logP, and refractive index. The numerical values of various physicochemical parameters underwent normalization, followed by the calculation of average values to determine different distribution behaviors for each parameter. In terms of the newly generated components, the relative molecular mass and boiling point exhibited the lowest values, while the surface tension and refractive index displayed the highest values. Meanwhile, among the disappearing components, the relative molecular mass, boiling point, and flash point possessed the lowest values, whereas the refractive index exhibits the highest value, as depicted in Fig. 9D and E. Using radar plots to analyze the overall physicochemical properties of the newly generated and disappearing components (Fig. 9F), it was found that there were significant differences in the physicochemical properties between the newly generated and disappearing components. Further exploration was conducted through PCA analysis to investigate these differences in more depth.
Further exploration of the main components that influence the distribution behavior of the components based on different physicochemical parameters was conducted, as shown in Fig. 9G. Through analysis, a total of 10 PCs were obtained, and PC1 had a significant contribution rate to the variance, accounting for 42.9% of the total variance. PC1 was considered the main component that influenced the distribution pattern of differential components in ATEO. PC1 = 0.44 × relative molecular mass + 0.35 × melting point + 0.45 × boiling point + 0.24 × flash point − 0.08 × density − 0.40 × polarizability − 0.41 × surface tension − 0.02 × logP − 0.28 × refractive index. In the PC1 axis, the newly generated components mainly exhibited a positive distribution, while the disappearing components mainly exhibited a negative distribution. There was a positive correlation among the relative molecular mass, melting point, boiling point, and flash point; conversely, a negative correlation was observed with density, the logarithm of the partition coefficient (logP), polarizability, surface tension, and refractive index. When combined with the principal component analysis, it was apparent that the relative molecular mass, boiling point, and flash point constituted key physicochemical parameters that significantly influenced the distribution behavior of the components. Components exhibiting higher values of relative molecular mass, boiling point, and flash point were preferentially distributed within the newly generated component system, whereas those with lower corresponding values tended to align with the phasing-out component system.
3.7 In vitro dissolution
 | ||
| Fig. 10 The cumulative release curves of β-asarone and α-asarone in Pickering emulsions and ATEO. Note: artificial gastric juice (A); artificial intestinal fluid (B). | ||
The in vitro behavior of β-asarone and α-asarone, the main volatile components in Pickering emulsions and ATEO were studied by the dialysis method. The study of in vitro release behavior showed that the cumulative release of β-asarone in Pickering emulsions was 1.24 times that of ATEO, and α-asarone was 1.10 times that of ATEO when it was dissolved in artificial gastric juice for 48 h. In artificial intestinal fluid, the cumulative release of β-asarone in Pickering emulsions was 1.62 times that of ATEO at 48 h, and α-asarone was 1.91 times that of ATEO. It was found that the release rate and cumulative release of β-asarone and α-asarone in Pickering emulsions were higher than those of ATEO, and this difference was more obvious in artificial intestinal fluid. Moreover, XRD studies confirmed that the silica adsorbed on the surface of modified Cinnabaris particles usually existed in the form of molecular dissolution, that is, in an amorphous state, which could promote the in vitro dissolution of fat-soluble drugs.36
4 Conclusion
In the present work, the modified Cinnabaris stabilizer was successfully prepared. The optimum emulsion process was determined by single factor and response surface optimization experiments, and the Pickering emulsions with high stability was obtained. A series of characterizations of the Pickering emulsions showed that ATEO was tightly enveloped by the water phase. Furthermore, the alterations in the EOs were systematically investigated within the crude oil group, oil–water mixture group, and Pickering emulsion group under high-temperature conditions. Macroscopically, the escape and oxidation degree of EOs were quantified through measures such as the EOs retention rate, MDA level, and POV. Microcosmically, the changes in EOs composition and the effects of physicochemical properties on the changes in EOs composition at high temperatures were studied in terms of quantitative and qualitative changes. The Pickering emulsions was found to have significant advantages in the protection of EOs. In summary, based on the idea of “combining medicine and adjuvant,” this work provided a new idea for the preparation of mineral drug particle stabilizers, which was of great significance for improving the stability and efficacy of oil-containing solid preparations. The drug release rule showed that with the increase in dissolution time, the cumulative dissolution of the drug increased and the release rate accelerated. Compared with the ATEO, the release of essential oil in Pickering emulsions was faster within 48 h, indicating that the encapsulation of ATEO in Pickering emulsions could improve the dissolution rate of volatile oil in vitro. This may have been because the modified Cinnabaris particles, being hydrophilic solid particles, dispersed quickly in the dissolution medium, thereby facilitating the faster entry of fat-soluble substances into the dissolution medium. Secondly, the molecularly dispersed drugs also helped to improve the dissolution of volatile oil. The modified Cinnabaris could be used as a stabilizer to prepare Pickering emulsions, improving the stability and antioxidant capacity of EOs and significantly enhancing the bioavailability of active ingredients in EOs. The Pickering emulsions stabilized by modified Cinnabaris has good application potential in the field of drug delivery.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Han Ru and Fei Luan conceived the idea and recommended a structure for this review. Han Ru and Junbo Zou conducted a literature search and drafted the manuscript. Yajun Shi, Xiaofei Zhang, and Bingtao Zhai designed and created the figures. Dongyan Guo and Jing Sun finished the tables. Dingkun Zhang Liang Feng, and Junbo Zou helped to edit and revise the manuscript. All the authors listed have read and approved the final manuscript.Conflicts of interest
The authors have declared no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82274105), the Science and Technology Innovative Talent Program of Shaanxi University of Chinese Medicine (No. 2024-CXTD-03), Youth Innovation Team of Aromatic Chinese Medicine Industrialization Key Technology (No. 2022-95), the Shaanxi Province Traditional Chinese Medicine Pharmaceutical Key Discipline Funding Project (No. 303061107), the Discipline Innovation Team Project of Shaanxi University of Chinese Medicine (No. 2019-YL11), and Shaanxi Provincial Administration of Traditional Chinese Medicine Key Discipline of Traditional Chinese Medicine Pharmaceutical Engineering (No. 2017001).Notes and references
- M. Tian, T. Liu, X. Wu, Y. Hong, X. Liu, B. Lin and Y. Zhou, Nat. Prod. Res., 2019, 34, 2621–2625 CrossRef PubMed.
- G. Ren, G. Ke, R. Huang, Q. Pu, J. Zhao, Q. Zheng and M. Yang, Sci. Rep., 2022, 12, 8153 CrossRef CAS PubMed.
- S. Yogev and B. Mizrahi, ACS Appl. Energy Mater., 2020, 2, 2070–2076 CAS.
- S. Sharma, S. Barkauskaite, A. K. Jaiswal and S. Jaiswal, Food Chem., 2020, 343, 128403 CrossRef PubMed.
- F. R. Sobreira Dantas Nóbrega de Figuêiredo, Á. B. Monteiro, I. R. Alencar de Menezes, V. D. S. Sales, E. Petícia do Nascimento, C. Kelly de Souza Rodrigues, A. J. Bitu Primo, L. Paulo da Cruz, É. d. N. Amaro, G. de Araújo Delmondes, J. P. Leite de Oliveira Sobreira Nóbrega, M. J. Pereira Lopes, J. G. Martins da Costa, C. F. Bezerra Felipe and M. R. Kerntopf, Food Chem. Toxicol., 2019, 12, 27453–27462 Search PubMed.
- M. Qneibi, N. Jaradat, M. Hawash, A. N. Zaid, A.-R. Natsheh, R. Yousef and Q. AbuHasan, BioMed Res. Int., 2019, 2019, 1–11 CrossRef PubMed.
- D. Bai, X. Li, S. Wang, T. Zhang, Y. Wei, Q. Wang, W. Dong, J. Song, P. Gao, Y. Li, S. Wang and L. Dai, Front. Pharmacol, 2022, 13, 1004529 CrossRef CAS PubMed.
- B. P. Mahanta, P. K. Bora, P. Kemprai, G. Borah, M. Lal and S. Haldar, Food Res. Int., 2021, 145, 110404 CrossRef CAS PubMed.
- I. da Silva Sales, T. de Jesus Freitas, F. Barbosa Schappo, B. Aparecida Souza Machado, I. L. Nunes and C. Duarte Ferreira Ribeiro, Crit. Rev. Food Sci. Nutr., 2023, 2252067 Search PubMed.
- W. Weisany, S. Yousefi, S. P. Soufiani, D. Pashang, D. J. McClements and M. Ghasemlou, Adv. Colloid Interface Sci., 2024, 325, 103116 CrossRef CAS PubMed.
- M. Kfoury, L. Auezova, H. Greige-Gerges and S. Fourmentin, Environ. Chem. Lett., 2018, 17, 129–143 CrossRef.
- K. J. Figueroa-Lopez, D. Enescu, S. Torres-Giner, L. Cabedo, M. A. Cerqueira, L. Pastrana, P. Fuciños and J. M. Lagaron, Food Hydrocolloids, 2020, 108, 106013 CrossRef CAS.
- A. Oliveira Brito Pereira Bezerra Martins, A. G. Wanderley, I. S. Alcântara, L. B. Rodrigues, F. R. Cesário, M. R. Correia de Oliveira, F. F. Castro, T. R. Albuquerque, M. S. da Silva, J. Ribeiro-Filho, H. D. Coutinho, P. P. Menezes, L. J. Quintans-Júnior, A. A. Araújo, M. Iriti, J. R. Almeida and I. R. Menezes, Biology, 2020, 9, 1–25 CrossRef PubMed.
- H. Ali, A. R. Al-Khalifa, A. Aouf, H. Boukhebti and A. Farouk, Sci. Rep., 2020, 10, 2812 CrossRef CAS PubMed.
- V. Suvarna and S. Chippa, Curr. Drug Delivery, 2022, 20, 770–791 CrossRef PubMed.
- J. Zhang, J. Zhang, S. Wang and T. Yi, Pharmaceutics, 2018, 10, 170 CrossRef CAS PubMed.
- F. B. de Carvalho-Guimarães, K. L. Correa, T. P. de Souza, J. R. Rodríguez Amado, R. M. Ribeiro-Costa and J. O. C. Silva-Júnior, Pharmaceuticals, 2022, 15, 1413 CrossRef PubMed.
- Y. Cahyana, Y. S. E. Putri, D. S. Solihah, F. S. Lutfi, R. M. Alqurashi and H. Marta, Molecules, 2022, 28, 7872 CrossRef PubMed.
- Y. Si-Jia, H. Shu-Min, Z. Yu-Zhu, Z. Shaobo, D. Shuai and Z. Tao, Int. J. Biol. Macromol., 2023, 250, 126146 CrossRef PubMed.
- C. Zheng, Y. Huang, X. Liang, B. Shen, G. Zhang and P. Fei, Int. J. Biol. Macromol., 2023, 244, 125483 CrossRef CAS PubMed.
- L. Ming, H. Wu, A. Liu, A. Naeem, Z. Dong, Q. Fan, G. Zhang, H. Liu and Z. Li, J. Mol. Liq., 2023, 388, 122775 CrossRef CAS.
- X. Feng, Y. Sun, Y. Yang, X. Zhou, K. Cen, C. Yu, T. Xu and X. Tang, LWT–Food Sci. Technol., 2020, 122, 109029 CrossRef.
- A. Rigg, P. Champagne and M. F. Cunningham, Macromol. Rapid Commun., 2021, 43, 2100493 CrossRef PubMed.
- Y. Ma, M. Li, Z. Chen, J. Feng, R. Chen, Z. Wang, J. Chen and S. Zhang, Food Hydrocolloids, 2023, 142, 108858 CrossRef CAS.
- H. Xie, F. Ni, J. Gao, C. Liu, J. Shi, G. Ren, S. Tian, Q. Lei and W. Fang, Food Chem., 2022, 383, 132453 CrossRef CAS PubMed.
- X. Zhou, X. Zhang, D. Guo, Y. Yang, L. Liu, L. Wang, X. Zhang, J. Cheng, J. Zou and Y. Shi, Comb. Chem. High Throughput Screening, 2021, 26, 1298–1310 Search PubMed.
- H. Kumar, M. Tiwari, V. R. Dugyala and M. G. Basavaraj, Langmuir, 2024, 40, 7860–7870 CrossRef CAS PubMed.
- X. Liu, Y.-Q. Huang, X.-W. Chen, Z.-Y. Deng and X.-Q. Yang, J. Cereal. Sci., 2019, 86, 47–51 Search PubMed.
- H. Chen and S. wang, J. Nanjing Univ. Tradit. Chin. Med., 2021, 37, 290–293 Search PubMed.
- L. Sun, C. Shen, Y. Liao and J. Shan, J. Nanjing Univ. Tradit. Chin. Med., 2022, 38, 409–418 Search PubMed.
- L. Peng, L. Feng, Y. Yang, Y. Liu, X. Zhang, J. Zou and Y. Shi, Chin. Tradit. Herb. Drugs, 2023, 54, 544–552 Search PubMed.
- H. Jiang, D. Zhang, X. Han, Y. He, R. Xu, M. Yang, L. Han and Q. Tan, Chin. Pharmaceut. J., 2017, 52, 1477–1482 CAS.
- M. Koromili, A. Kapourani and P. Barmpalexis, Polymers, 2022, 15, 169 CrossRef PubMed.
- Z. Wang, B. Dai, X. Tang, Z. Che, F. Hu, C. Shen, W. Wu, B. Shen and H. Yuan, Pharmaceutics, 2022, 14, 897 CrossRef CAS PubMed.
- R. Andreia, A. M. Yaidelin, B. L. José Carlos, M. D. Madalena and B. Maria Filomena, Surf.Interfaces, 2022, 29, 101759 CrossRef.
- X. Xi, Z. Wei, Y. Xu and C. Xue, Polymers, 2023, 15, 1820 CrossRef CAS PubMed.
- J. Ming, L. Chen, B. Huang and K. Chen, Lishizhen Med. Mater. Med. Res., 2016, 27, 2423–2426 Search PubMed.
- H. Chen, Z. SUN and J. Shao, Bull. Chin. Ceram. Soc., 2011, 30, 934–937 CAS.
- Z. Yu, F. Weiting, C. Siyao, L. Hongshan, L. Jing, L. Yan and L. Bin, Int. J. Biol. Macromol., 2022, 229, 1044–1053 Search PubMed.
- Y. Li, B. Kong, Q. Liu, X. Xia and H. Chen, Process Biochem., 2017, 53, 116–124 CrossRef CAS.
- Y. Li and D. Xiang, PLoS One, 2019, 14, e0213189 CrossRef CAS PubMed.
- K. K. Agyare, K. Addo and Y. L. Xiong, Food Hydrocolloids, 2009, 23, 72–81 CrossRef CAS.
- X. Li, L. Wang and B. Wang, Food Chem., 2017, 229, 479–486 CrossRef CAS PubMed.
- Z. Chen, Y. Shi, X. Zhang, F. Luan, D. Guo, J. Sun, B. Zhai, D. Zhang and J. Zou, Heliyon, 2024, 10, e25909 CrossRef CAS PubMed.
- L. Peng, X. Zhang, D. Guo, B. Zhai, M. Wang, J. Zou and Y. Shi, RSC Adv., 2022, 12, 27453–27462 RSC.
- G 5009.227-2016 Determination of Peroxide Value in Food in National Food Safety Standards, Nation Health and Family Planning Commission of the People's Republic of China, 2016 Search PubMed.
- L. Peng, M. Wang, X. F. Zhang, D. Y. Guo, B. T. Zhai, J. B. Zou and Y. J. Shi, AAPS Open, 2023, 9, 1–12 CrossRef.
- J. Tai, J. Zou, Y. Shi, D. Guo, X. Zhang, Y. Wang, Y. Liang, J. Li, J. Cheng and M. Yang, Chin. J. Exp. Tradit. Med. Formulae, 2019, 25, 108–115 Search PubMed.
- M. Ritchie, B. Phipson, D. Wu, Y. Hu, C. Law, W. Shi and G. Smyth, Nucleic Acids Res., 2015, 43, e47 CrossRef PubMed.
- Z. Qu, Y. Shi, X. Zhang, F. Luan, D. Guo, B. Zhai, J. Sun, D. Zhang, J. Zou and M. Zhu, ACS Omega, 2024, 9, 20773–20790 CrossRef CAS PubMed.
- M. Zhu, Z. Qu, Y. Yang, R. Shi, B. Yang, Y. Shi, J. Zou and X. Jia, Pharmaceuticals, 2024, 17, 1117 CrossRef PubMed.
- Y. Liu, Z. Shi, Y. Zou, J. Yu, L. Liu and Y. Fan, Int. J. Biol. Macromol., 2023, 235, 123754 CrossRef CAS PubMed.
- A. Hajipour, Y. Heidari and G. Kozehgary, Synlett., 2016, 27, 929–933 CrossRef CAS.
- J. Tai, J. Zou, Y. Shi, D. Guo, X. Zhang, Y. Wang, Y. Liang, J. Cheng, M. Yang, F. Wang and Y. Luo, Chin. Pharmaceut. J., 2019, 54, 1237–1244 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05168h |
| This journal is © The Royal Society of Chemistry 2024 |