
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis of (−)-2-oxo epimesembranol and (+)-dihydromaritidine via a key Johnson–Claisen rearrangement†
Satyajit Majumder a,
Abhinay Yadava,
Souvik Pala,
Abhishek Mondalb and
Alakesh Bisai*ab
a,
Abhinay Yadava,
Souvik Pala,
Abhishek Mondalb and
Alakesh Bisai*ab
aDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhauri, Bhopal-462 066, Madhya Pradesh, India. E-mail: alakeshb@gmail.com
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, Nadia-741 246, West Bengal, India. E-mail: alakesh@iiserkol.ac.in
First published on 16th September 2024
Abstract
A general approach to Sceletium alkaloids of the family Aizoaceace following a key Johnson (orthoester)–Claisen rearrangement of an enantioenriched allylic alcohol has been disclosed. The tricyclic core (1c) of cis-3a-octahydroindoline skeleton was achieved via an ester-aminolysis followed by an intramolecular aza-Michael reaction with amine under elevated temperature. Utilizing aforementioned strategy, a collective total syntheses of Sceletium alkaloids, such as (−)-2-oxo-epimesembranol (1d) [the first total synthesis], (−)-6-epimesembranol (1b), and (−)-mesembrine (1a) were shown. Further this strategy was applied for total synthesis of (+)-dihydromaritidine (2c) sharing [5,11b]-ethanophenanthridine skeleton.
Introduction
Amaryllidaceae alkaloids are a structurally diverse group of plant specialized metabolites with important biological activities.1 Plants belonging to the Amaryllidaceae and Sceletium family are herbaceous perennials that grow from bulbs,2,3 More than 500 Amaryllidaceae alkaloids have been isolated, with varied biological profiles, from Amaryllidaceae plants till date.3,4 In particular, the Sceletium tortuosum, is an indigenous herb of South Africa, especially in Namaqualand, where the plant is utilized regularly as an herbal supplement in the treatment of central nervous system-related disorders for nearly 200 years,5,6 main alkaloid responsible is mesembrine (1a) and mesembranol (1b).7,8 They are also cultivated as ornamental plants for their beautiful flowers and to produce volatile oil. These alkaloids contain common core cis-3a-octahydroindoline skeleton along with a synthetically challenging benzylic all-carbon quaternary streocenter.9 Their architecture display vicinal quaternary and tertiary carbon stereocenters10 with a fused pyrrolidine ring,11 as common structural features, whose stereochemical incorporation is indeed a challenge. As a representative alkaloid of the Amaryllidaceae family with significant biological activity, maritidine12 is isolated from Pancratium maritimum, Pancratium tortuosum, and Zephyranthes genera,13 with a 5,10b-ethanophenanthridine nucleus containing dimethoxy substituents at C-8 and C-9 positions. Maritidine is of particular interest due to its cytotoxic properties14,15 and limited supplies from natural sources.It is important to note that, both antipodes of Amaryllidaceae alkaloids are naturally occurring. As for example (−)-crinine (2d) and its enantiomer (+)-vittatine (ent-2d) are isolated from different Amaryllidaceae species.5,15 Similarly, naturally occurring (−)-epi-crinine (2f) and its enantiomer (+)-epi-vittatine (ent-2f) are also isolated from different Amaryllidaceae species.5
The incorporation of sterically congested quaternary center is the critical element in the total synthesis Amaryllidaceae alkaloids sharing [5,10b]-ethanophenathridine16 and cis-3a-octahydroindoline alkaloids17,18 Although great efforts have been devoted to the development of synthetic methods to obtain maritidine type alkaloids, most of the reported approaches provided racemic products,10 and only a few asymmetric syntheses of maritidine have been reported.15,16
In this regard, the development of a general and efficient asymmetric catalytic method for the concise synthesis of Amaryllidaceae and Sceletium family of alkaloids having benzylic quaternary stereogenic center has become an important subject in organic chemistry. In this regard, in 2002, Trost and co-workers studied the direct intramolecular Pd(0)-catalyzed asymmetric decarboxylative allylic alkylation of enol carbonates and subsequently, allylation of 2-phenylcyclohexanone (88% ee using L2, 2009).19a,b However, it has been reported that the utilization of electron-rich aromatics rather very difficult. Kim et al.20a have reported that only 66% ee was obtained for 3a sharing 3,4-diOMePh as an aryl group using 5.5 mol% of L2 (Fig. 2), clearly indicating that utilization of such process with a substrate sharing electron-donating aromatic rings is indeed a considerable challenge that is worth testing. In 2018, our group also shown via an elegant Pd(0)-catalyzed Asymmetric Allylic Alkylations (AAA)20b of allyl enol carbonates (3b in 92% ee using L1) (Fig. 2). Further, Wang et al.20c has reported an elegant Pd(0)-catalyzed asymmetric allylation of α-aryl vinylogous ester 3c (84% ee) using L2 for an asymmetric total synthesis of (−)-oxomaritidine (2b) (Fig. 2).
It is clear from the literature that it is difficult to get high enantioselectivity of electron-rich enol carbonates via Pd(0)-catalyzed Asymmetric Allylic Alkylations (AAA). It is believed that a substrate having catechol methyl ether might be co-ordinating with the Pd(0) (see a proposed complex shown in Fig. 2), thereby hampering its catalytic efficiencies in terms of chemical yield (58%) as well as optical purity (66% ee). Thus, a concise catalytic asymmetric approach to the Amaryllidaceae alkaloids sharing electron-rich aromatics remains a challenge that is worth pursuing. Retrosynthetically, it was hypothesized that a Johnson–Claisen rearrangement of 3-(3,4-dimethoxyphenyl)cyclohex-2-enol can be an excellent strategic platform to install the all-carbon quaternary stereocenter required for unified strategy for Sceletium and Amaryllidaceae alkaloids shown in Fig. 1. The retrosynthetic analysis of the asymmetric total synthesis of cis-3a-octahydroindoline alkaloids is shown in Scheme 1. For a unified approach to the Scelectium alkaloids and Amaryllidaceae alkaloids, it was envisioned that a Johnson–Claisen rearrangement21 of enantioenriched allylic alcohol 9b followed by allylic oxidation (see 7b)22a and ester aminolysis (see 5) and aza-Michael reaction (6b and 1c)23 to address total synthesis of several congeners of these alkaloids.
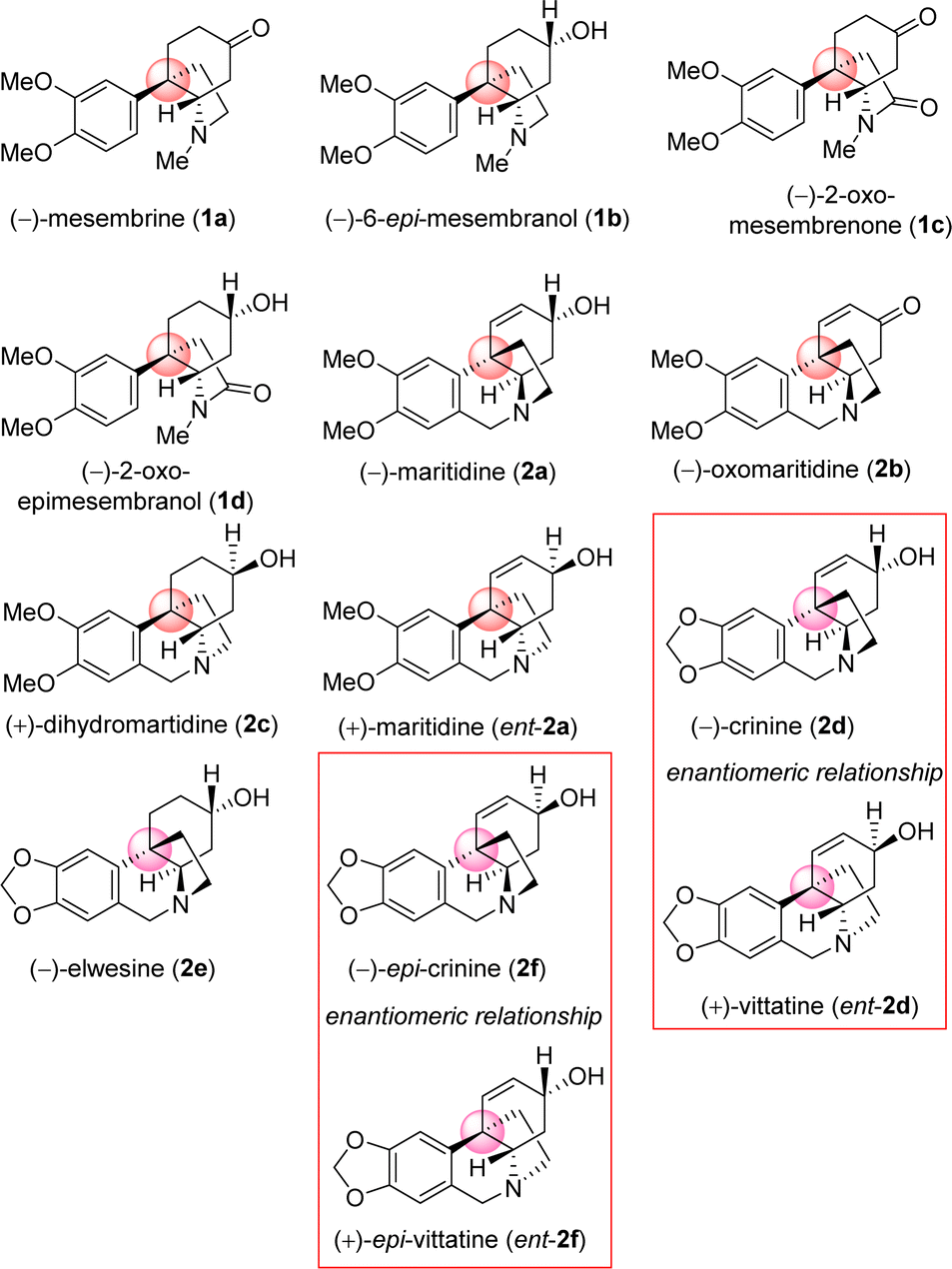 | ||
| Fig. 1 Selected naturally occurring Sceletium alkaloids and Amaryllidaceae alkaloids. | ||
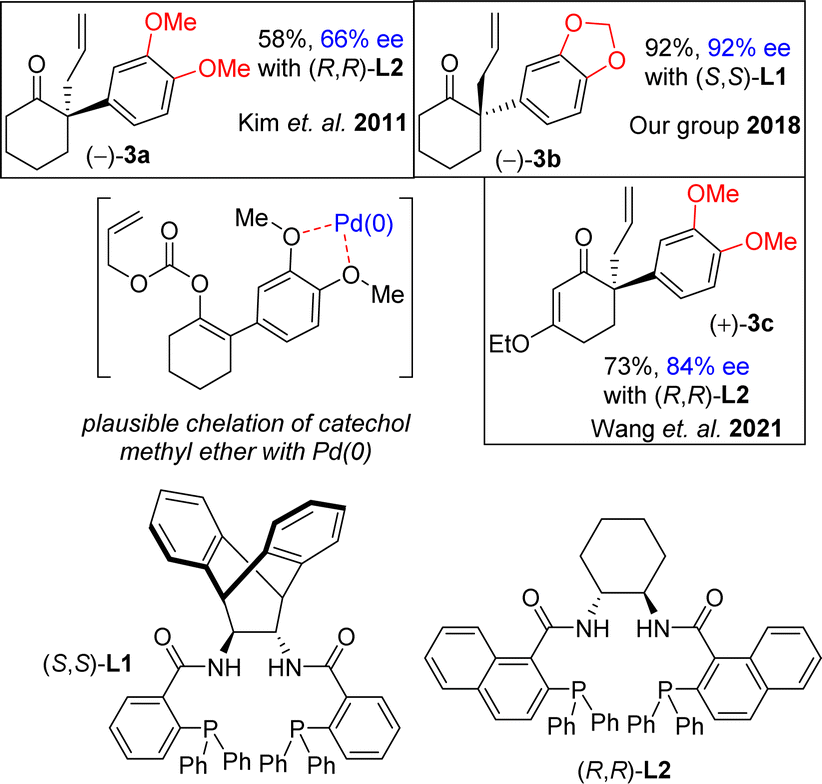 | ||
| Fig. 2 Catalytic enantioselective allylation. | ||
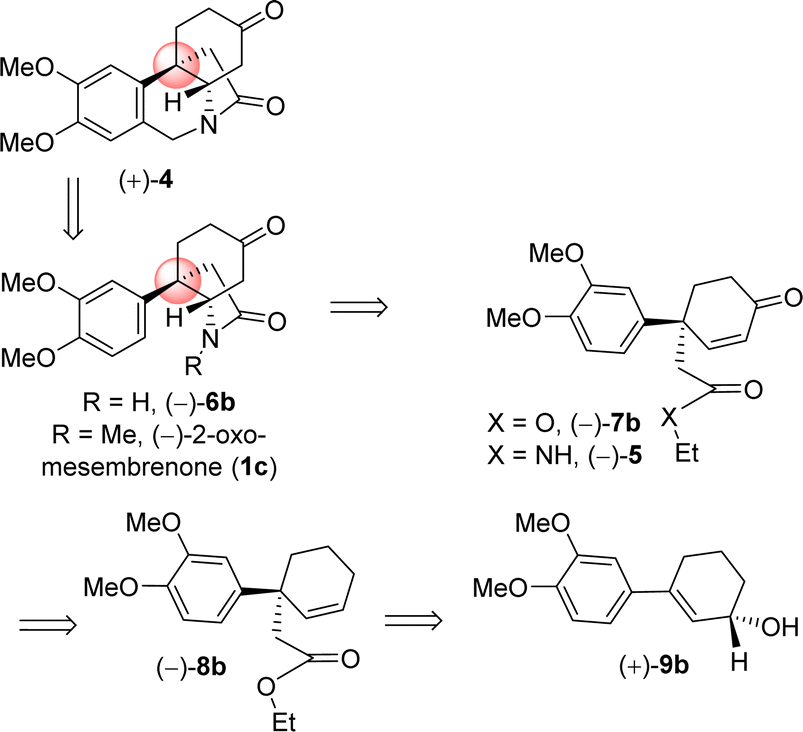 | ||
| Scheme 1 Retrosynthetic analysis. | ||
It was hypothesized that the advanced intermediate, a benzylic all-carbon quaternary stereocenter containing γ,δ-unsaturated ester 8b, could undergo direct allylic oxidation to generate the substrate for the amidation/transannular aza-conjugate addition reaction, leading to a unified pathway to access both Sceletium and Amaryllidaceae type alkaloids (Scheme 1). Enone-ester 7b can be synthesized via allylic oxidation of cyclohexene 8b, which can be obtained through the Johnson–Claisen rearrangement of allylic alcohol 9b. At this stage, it was proposed that the enantioenriched 3-(aryl)cyclohex-2-enols 9b, which can be accessed through the enantioselective CBS reduction of 3-aryl-2-cyclohexenone 11b (Scheme 2), could provide a pathway to an asymmetric synthesis. Compound 11b can be readily synthesized from vinylogous ester 10b through a well-established Stork–Danheiser sequence.
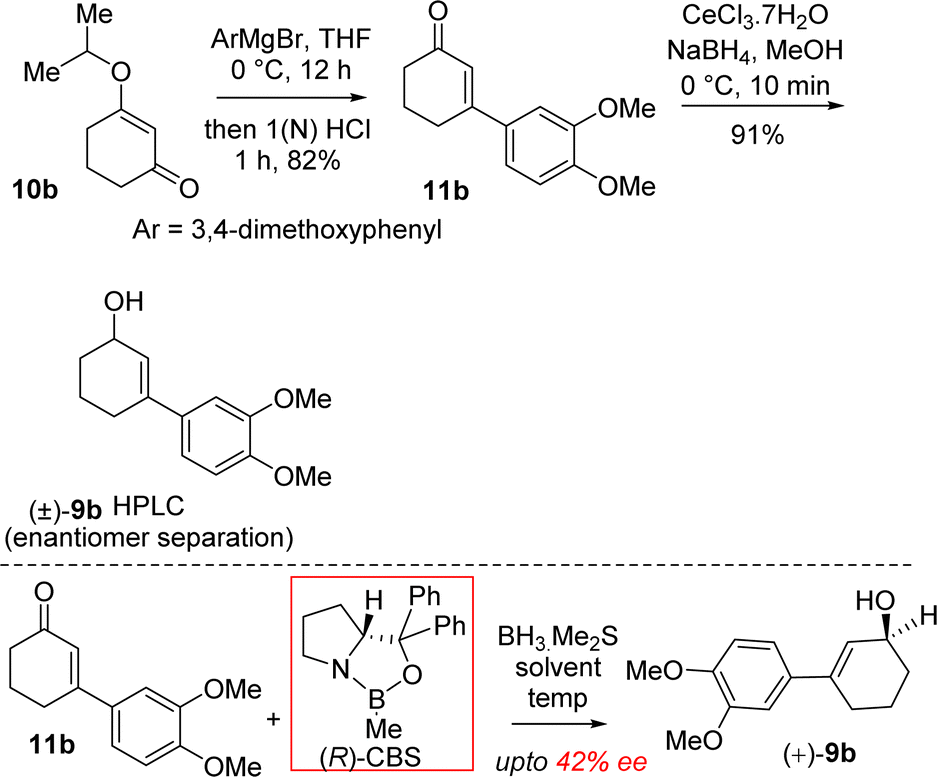 | ||
| Scheme 2 Synthesis of enantioenriched 3-(aryl)cyclohex-2-enols (+)-9b. | ||
Results and discussion
Moving forward with above proposed strategy, it was required to synthesize enantioenriched 3-(aryl)cyclohex-2-enols 9b for the orthoester Johnson–Claisen rearrangement (Scheme 2). Towards this, the Stork–Danheiser sequence on compound 10b using aryl magnesium bromide was carried out to afford 3-aryl 2-cyclohexenone 11b in 82% yields (Scheme 2). Next, it was identified that Corey–Bakshi–Shibata (CBS) reduction was reliable to reduce substituted cyclic enones in high enantioselectivity.In this regard, we first observed that coordinating polar aprotic solvent THF may reduce the acidity of BH3·Me2S results it unable to complete catalytic cycle of CBS reduction even in refluxing condition. Later on, we investigated that polar aprotic noncoordinating solvent CH2Cl2 unable to reduce acidity of BH3·Me2S results to complete the catalytic cycle of CBS reduction at rt with 20 mol% catalyst. (R)-CBS regent was used for the optimization of enantioselective reduction of 3-aryl 2-cyclohexenone 11b.22b However, our initial results in this direction were rather discouraging and it was found that even after using 100 mol% (R)-CBS regent, a maximum of 42% ee was observed.
It was argued that a sterically crowded easily removable group might help in bringing high enantioselectivity (Scheme 3). Thus, a halogen group such as bromo group that can be easily remove via reductive condition was incorporated. Therefore, Stork–Danheiser sequence bromo vinylogous ester 10a was carried out using aryl magnesium bromide to afford 2-bromo-3-aryl 2-cyclohexenone 11a in 82% yields (Scheme 4). Luche reduction of 2-bromo enone provided racemic allyl alcohol 12a for HPLC analysis.
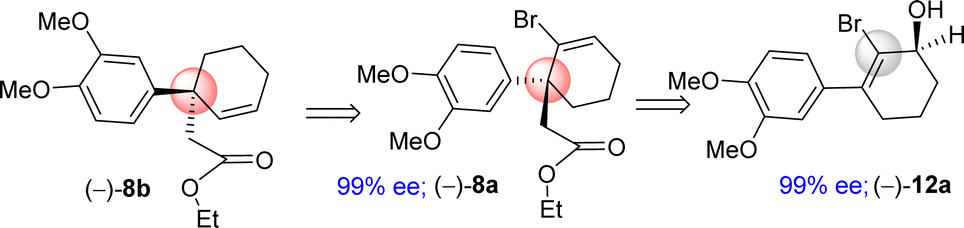 | ||
| Scheme 3 Retrosynthetic analysis. | ||
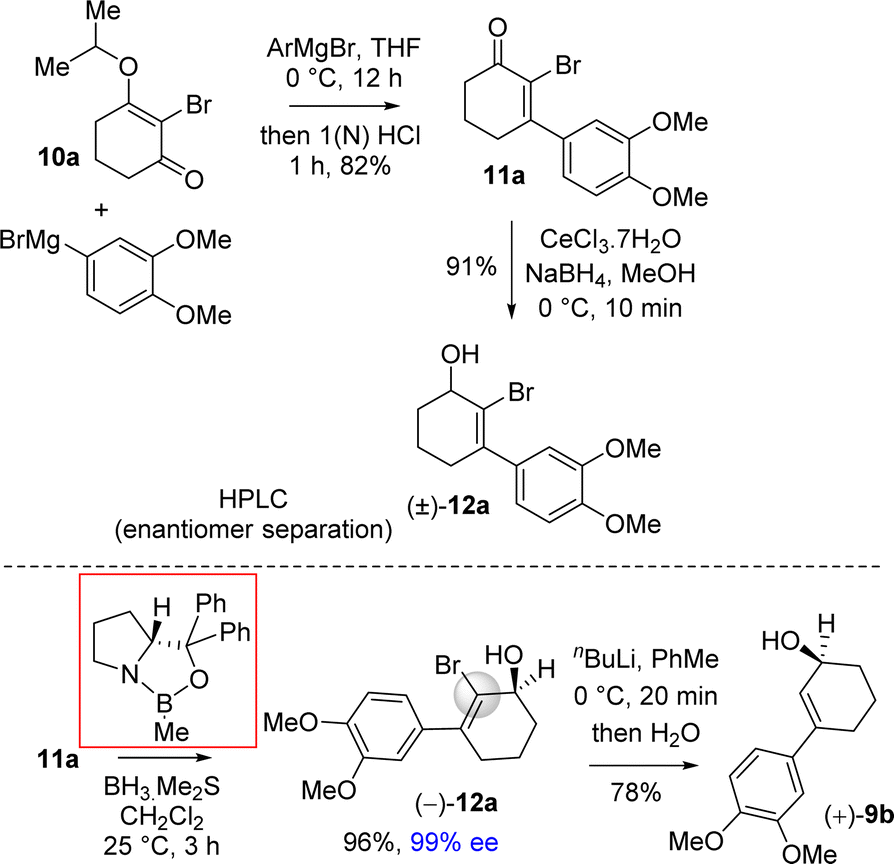 | ||
| Scheme 4 Synthesis of enantioenriched 3-(aryl)cyclohex-2-enols (+)-9b. | ||
Next, 2-bromo-3-aryl 2-cyclohexenone 11a in hand it was explored under Corey–Bakshi–Shibata (CBS) reduction and the result is summarized in Table 1. The optimization studies were conducted in different solvents such as THF and dichloromethane under different temperature using (R)-CBS regent. The initial result using 50 mol% (R)-CBS afforded allyl alcohol in 90% ee (entry 3, Table 1). Following exhaustive optimization, it was observed that dichloromethane is a better solvent for this transformation and 100 mol% of (R)-CBS in dichloromethane at room temperature afforded allyl alcohol in 97% ee (entry 7, Table 1). Gratifyingly, using 50 mol% of (R)-CBS in dichloromethane furnished product in 94% ee (entry 8, Table 1). On further decrease of catalyst loading to 20 mol% of (R)-CBS, and under slow reverse addition of bromo-enone 11a, it was found to achieve a 99% ee (entry 9, Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Entrya | (R)-CBS reagent | Solvent | Temp. | Time | Yieldb | % eec |
| a All the reactions were performed under argon atmosphere.b Yields are reported after column chromatography.c ee's were measured performing HPLC analysis by Chiralpak OD-H column.d Slow addition by syringe pump over 3 h. | ||||||
| 1 | 100 mol% | THF | 0 °C | 24 h | 0 | — |
| 2 | 100 mol% | THF | 25 °C | 0.5 h | 91% | 90% |
| 3 | 50 mol% | THF | 25 °C | 0.5 h | 50% | 90% |
| 4 | 50 mol% | THF | 60 °C | 24 h | 61% | 81% |
| 5 | 100 mol% | CH2Cl2 | 0 °C | 24 h | 10% | — |
| 6 | 20 mol% | CH2Cl2 | 25 °C | 0.5 h | 98% | 92% |
| 7 | 100 mol% | CH2Cl2 | 25 °C | 0.5 h | 93% | 97% |
| 8 | 50 mol% | CH2Cl2 | 25 °C | 2 h | 92% | 94% |
| 9d | 20 mol% | CH2Cl2 | 25 °C | 3 h | 94% | 99% |
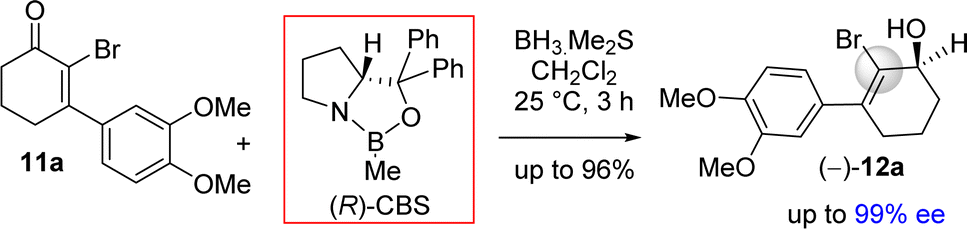
The stereochemical rationale of Corey–Bakshi–Shibata (CBS) reduction of 2-bromo-3-aryl 2-cyclohexenone 11a via transition state models are shown in Fig. 3.22c It is very clear that the bromide of bromo-enone is imparting enhanced facial bias of the ketone leading to increased enantioselectivities and is subsequently removed via a debromination step following the reduction. The asymmetric aspect of this overall transformation is therefore simplified to the preparation of an enantioenriched allylic alcohol. Thus, this account for the perfect enantioselectivity (99% ee) achieved from this reaction. In the favored transition state of CBS reduction [using (S)-CBS reagent] of 2-bromo-3-aryl 2-cyclohexenone 11a one could easily account for a ‘Si-face’ approach of hydride via a six-membered cyclic transition state in achieving optimum enantioselectivity in this process (Fig. 3). It is apparent that a bulky group such as bromo has an important role in controlling enantioselectivity, which in fact helps in retarding the ‘disfavored’ transition state and, thus, avoids ‘Re-face’ approach of hydride onto the carbonyl group (Fig. 3, see above). The transition state for the reduction of 2-bromo-3-aryl 2-cyclohexenone 11a using (R)-CBS reagent has also been shown in Fig. 3 (see above). In this case a ‘Re-face’ approach of hydride via a six-membered cyclic transition state is favored, whereas a ‘Si-face’ approach of hydride is retarded because of the steric clash (Fig. 3, see above).
 | ||
| Fig. 3 Plausible transition states. | ||
The beauty of choosing a bromo group is two-fold in our synthesis: firstly, it helps in achieving highest enantioselectivity and, secondly, this group can easily be removed by the reaction with organometallic reagents such as by the treatment with nBuLi followed by quench by water or by a reaction with n-Bu3SnH in the presence of catalytic AIBN (azo-bis isobutyronitrile) or using Pd(0)-catalyzed cross-coupling with a hydride source. In particular, the bromo group of allyl alcohol was removed by using n-BuLi at low temperature to generate vinyl carbanion which was further reacted with water to get allyl alcohol 9b in 98% ee (Scheme 4).
Further, orthoester Johnson–Claisen rearrangement of 3-(aryl)cyclohex-2-enols 9b with triethyl orthoacetate was investigated in different solvent and in presence of catalytic amount of weak acid under heating condition (Table 2). Triethyl orthoacetate was activated in presence of catalytic amount of weak acid under heating condition to produce ketene-acetal intermediate which undergoes [3,3]-sigmatropic rearrangement results γ,δ-unsaturated ester. Initially this reaction was performed in the presence of propanoic acid, pivalic acid, butyric acid and o-nitrophenol at different temperatures (Table 2). Following an exhaustive optimization, it was observed that acid catalysed orthoester Johnson–Claisen rearrangement of allyl alcohol 9b afforded product 8b with benzylic all-carbon quaternary stereogenic center in 62% isolated yield when 5 mol% of o-nitrophenol was employed as catalyst. The enantioselectivity of product 8b found to be compromised to 68% ee from 99% ee of starting allylic alcohol (entry 6, Table 2).
|
|||||
|---|---|---|---|---|---|
| Entrya | Acid | Triethyl orthoacetate | Solvent | Temp. °C | bYield% |
| a All the reactions were performed under argon atmosphere.b Yields are reported after column chromatography. | |||||
| 1 | 10 mol% propanoic acid | 10 equiv. | Toluene | 130 °C | Mixture of products |
| 2 | 5 mol% propanoic acid | 10 equiv. | Xylene | 160 °C | Mixture of products |
| 3 | 5 mol% pivalic acid | 10 equiv. | Xylene | 140 °C | Mixture of products |
| 4 | 5 mol% butyric acid | 10 equiv. | Xylene | 140 °C | Mixture of products |
| 5 | 5 mol% o-nitrophenol | 10 equiv. | Xylene | 160 °C | Mixture of products |
| 6 | 5 mol% o-nitrophenol | 10 equiv. | Xylene | 220 °C | 62% (68% ee) |
| 7 | — | 10 equiv. | DIPEA | 130 °C | Mixture of products |
| 8 | — | 10 equiv. | DIPEA | 140 °C | 20% + 36% (SM) |
| 9 | — | 10 equiv. | DIPEA | 220 °C | 73% (97% ee) |
| 10 | — | 10 equiv. | DIPEA | 160 °C | 37% + 28% (SM) |

In order to understand the loss of enantiopurity, it was thought of looking at the mechanism of orthoester Johnson–Claisen rearrangement (Scheme 5). Acid catalyzed activation of triethyl orthoacetate could form an oxocarbenium ion that could react with the allyl alcohol to generate the intermediate 16b responsible for the [3,3]-sigmatropic rearrangement via the intermediates 16b–18b (Scheme 5). Since the reaction goes through a concerted pathway, therefore, in principle, an enantioenriched allyl alcohol 9b having >99% ee should provide product 8b in >99% ee via an enantioenriched intermediate 18b (>99% ee).
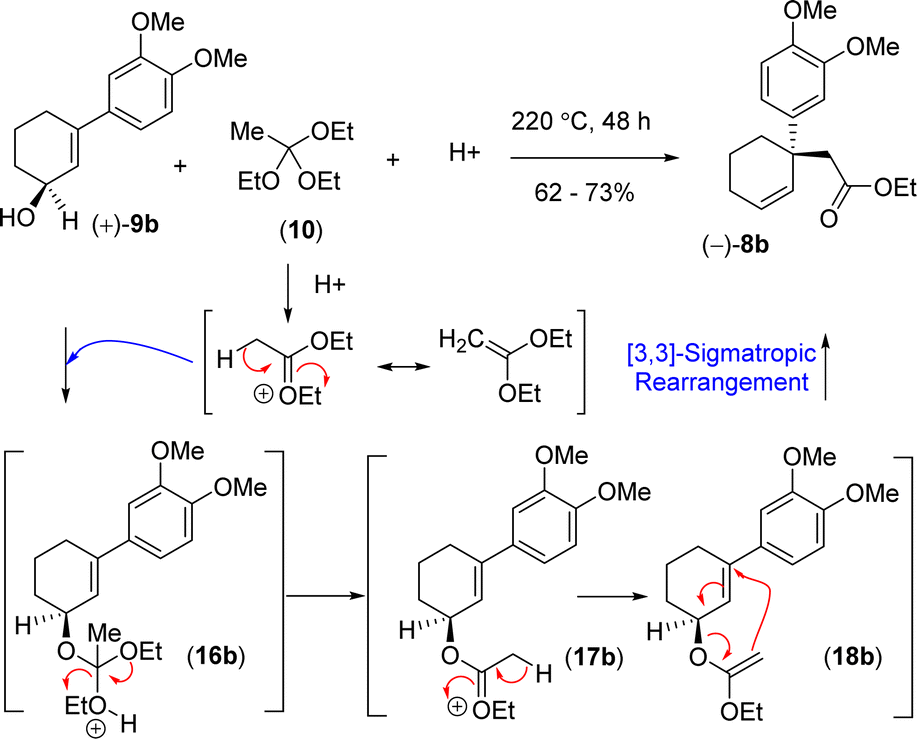 | ||
| Scheme 5 Proposed reaction mechanism of [3,3]-sigmatropic rearrangement. | ||
However, our result suggests that the electron-rich aromatics probably has a bigger role in the orthoester Johnson–Claisen rearrangement of 3-(aryl)cyclohex-2-enol 9b. It is believed that the enantioselectivity of the enantioenriched intermediate 18b is somehow compromising to 68% ee during the course of the reaction, and therefore, this account for the observed enantioselectivity of 8b as 68% ee (entry 6, Table 2). A rationale of by product formation under this type of pericyclic reaction using electron-rich aromatic ring is shown in Scheme 6.
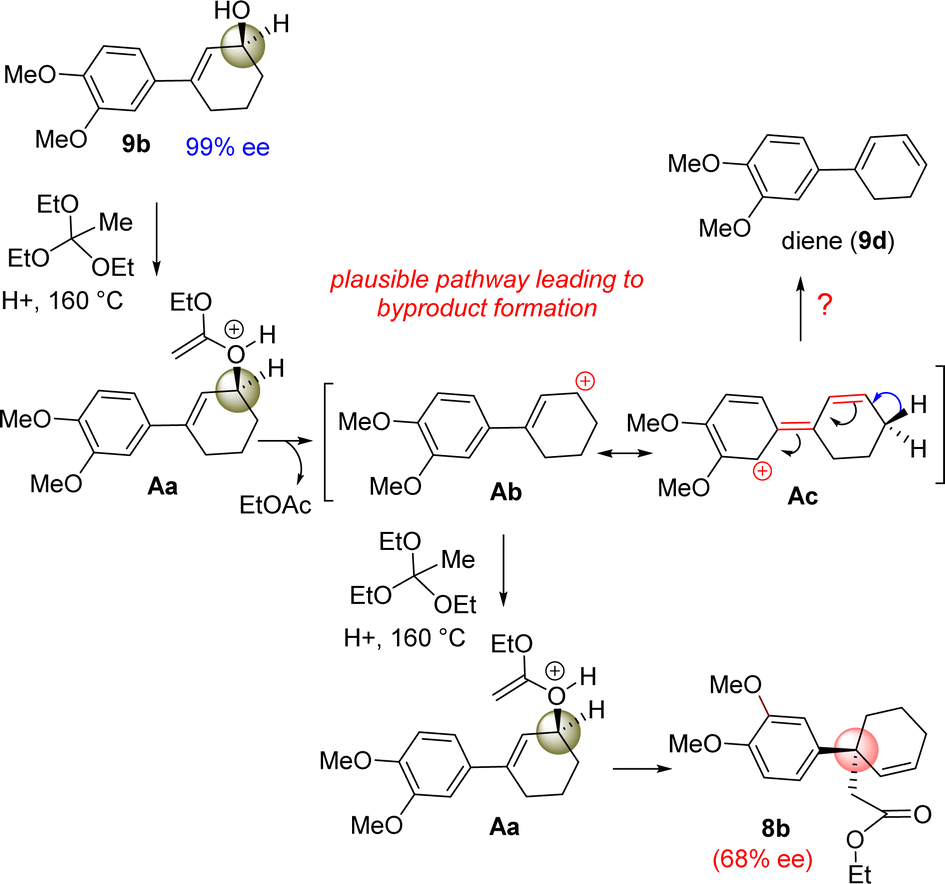 | ||
| Scheme 6 Byproduct formation under orthoester Johnson–Claisen rearrangement of (+)-9b. | ||
It is believed that 3-(aryl)cyclohex-2-enols 9b could generate some amount of an allylic carbocation such as Ab (through the intermediate Aa) in the course of reaction (Scheme 6), which is getting stabilized by the electron-donating nature of aromatic ring, in particular, 4-methoxy group of aryl ring present in the 3-position of allyl alcohol (see the formation of Ab from Ac). The allylic carbocation Ab could recombine with ethyl orthoacetate to reform intermediate Ac (68% ee) responsible for the [3,3]-sigmatropic rearrangement product.
Thus, it was thought of exploring the ortho-ester Johnson–Claisen rearrangement of 3-(aryl)cyclohex-2-enols 9b under basic condition (entries 7–10, Table 2). Under the thermal decomposition of triethyl orthoacetate one could able to generate the key intermediate responsible for [3,3]-sigmatropic rearrangement. Thus, Hünig's base i.e. N,N-diisopropylethylamine (DIPEA) was used a promoter as well as solvent. It was a pleasure to see that, after exhaustive optimization, 10 equiv. of N,N-diisopropylethylamine (DIPEA) and 10 equiv. of triethyl orthoacetate under heating condition produce γ,δ-unsaturated ester with 73% yield with 97% enantioselectivity.24 Interestingly, this reaction afforded enantioenriched [3,3]-sigmatropic rearrangement product (97% ee) with minimum loss of enantiopurity under elevated temperature in the presence of N,N-diisopropylethylamine (DIPEA) (Scheme 7).25
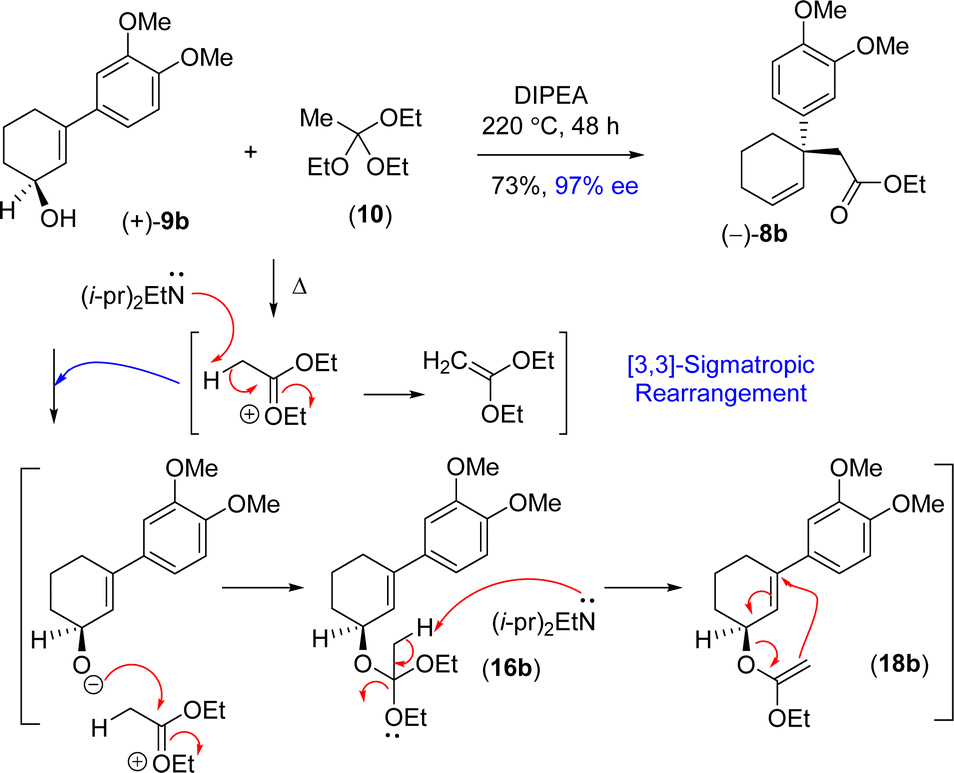 | ||
| Scheme 7 Proposed DIPEA mediated reaction mechanism of [3,3]-sigmatropic rearrangement. | ||
Next, the orthoester Johnson–Claisen rearrangement of 2-bromo 3-(aryl)cyclohex-2-enols 12b was undertaken. Since, 3-(aryl)cyclohex-2-enol having an electron-rich aromatic was difficult, it was thought of exploring the [3,3]-sigmatropic rearrangement of 2-bromo 3-(aryl)cyclohex-2-enol. The reason behind is that the substrate, 2-bromo 3-(aryl)cyclohex-2-enol, has an electron-rich aromatics and at the same time it is having an electron-withdrawing bromo functionality.
Based on this intuition, the orthoester Johnson–Claisen rearrangement was carried out under conventional approach i.e. under acid catalyzed process. Gratifyingly, it was observed that orthoester Johnson–Claisen rearrangement of 2-bromo 3-(aryl)cyclohex-2-enol was found to be very efficient and afforded product 8a in 76% yield (Scheme 8). Pleasingly, this reaction was found to be more facile under microwave condition to afford product in 79% yield (Scheme 8). Next, debromination of bromo group was conveniently done under classical condition with tri-n-butyltin hydride in the presence of AIBN. Subsequent direct conversion of 8b in to the α,β-unsaturated ketone 7b via an allylic oxidation (CrO3, 3,5-dimethylpyrazole, CH2Cl2, −10 °C to 25 °C, 16 h, 77%) set the stage for studies on its cyclization.
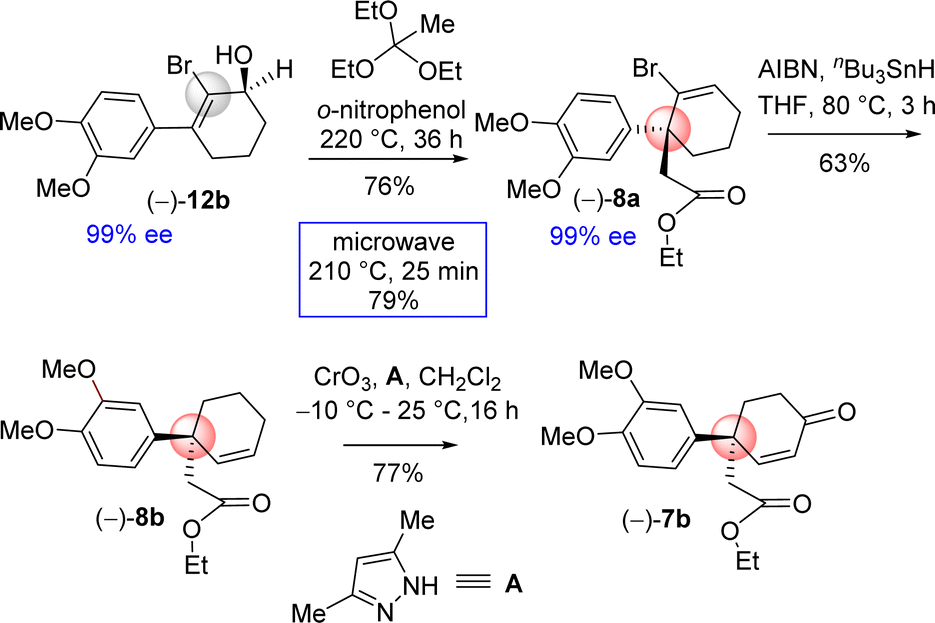 | ||
| Scheme 8 Synthesis of 4-aryl 4-alkyl 2-cyclohexenone (–)-7b. | ||
4-Aryl 4-alkyl α,β-unsaturated ketone 7b in hand, the efforts were put forward for its conversion into the advanced intermediate that is capable for concise total synthesis of cis-3a-octahydroindoline alkaloids (Scheme 9). It was hypothesized to synthesize the tricyclic core 1c from 4-aryl 4-alkyl α,β-unsaturated ketone 7b via an ester-aminolysis followed by an aza-Michael reaction with methylamine under heating (Scheme 9). Following optimization, it was observed that the treatment of 4-aryl 4-alkyl α,β-unsaturated ketone 7b with MeNH2 in THF (80 °C, 16 h) proceeded smoothly furnish bicyclic keto lactam 1c in 96% yield. A rationale of the formation of keto lactam 1c from 4-aryl 4-alkyl α,β-unsaturated ketone 7b is shown in Scheme 9.
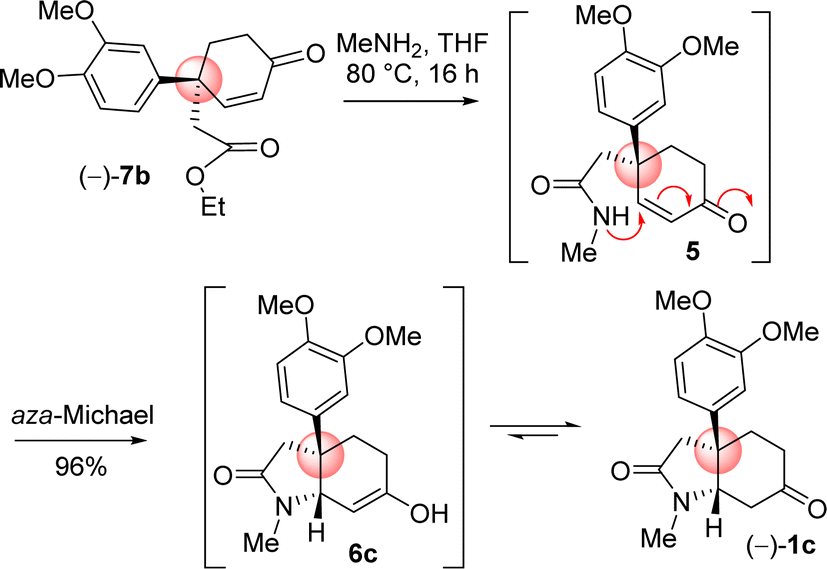 | ||
| Scheme 9 Ester aminolysis and aza-Michael cascade. | ||
Next, chemoselective reduction of keto carbonyl functionality over carboxamide carbonyl was undertaken. In this regard, a number of reducing agents were selected and reduction was performed under different condition (Table 3). Following extensive optimization using various hydride source such as NaBH4, LiAlH4, Red-Al, and K-selectride, it was found that a highly stereoselective reduction of ketone group is possible to complete the total synthesis of (–)-2-oxoepimesembranol (1d) in excellent yield (99%) and excellent diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Table 3). Thus, a two-step total synthesis of cis-3a-octahydroindoline alkaloid, (−)-2-oxoepimesembranol (1d) was accomplished from 4-aryl 4-alkyl α,β-unsaturated ketone 7b via an ester-aminolysis followed by an aza-Micheal reaction and subsequent chemoselective reduction of keto carbonyl functionality over carboxamide carbonyl.
:1) (Table 3). Thus, a two-step total synthesis of cis-3a-octahydroindoline alkaloid, (−)-2-oxoepimesembranol (1d) was accomplished from 4-aryl 4-alkyl α,β-unsaturated ketone 7b via an ester-aminolysis followed by an aza-Micheal reaction and subsequent chemoselective reduction of keto carbonyl functionality over carboxamide carbonyl.
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Reducing agent | Equivalent | Temp. | Time | Yieldb | dr |
| a All the reactions were performed under argon atmosphere.b Yields are reported after column chromatography. | ||||||
| 1 | NaBH4 (MeOH) | 1 eq. | 0 °C | 1 h | 84% | ∼8:1 |
| 2 | LiAlH4 (THF) | 1 eq. | 0 °C | 1 h | 70% | ∼5:1 |
| 3 | LiAlH4 (THF) | 1 eq. | −78 °C | 1 h | 69% | ∼10:1 |
| 4 | Red-Al (PhMe) | 1 eq. | 25 °C | 4 h | 65% | ∼5:1 |
| 5 | K-selectride (THF) | 1 eq. | −78 °C | 1 h | 99% | >20:1 |
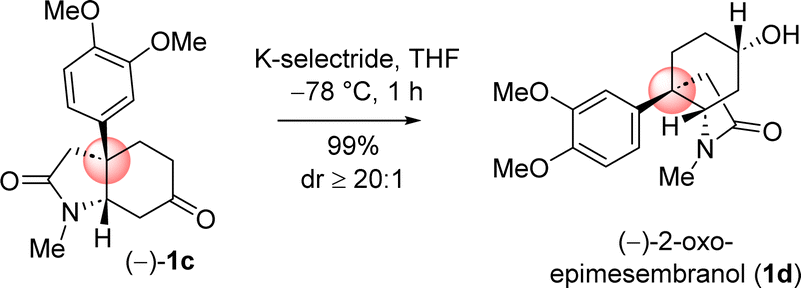
Next, the reductive removal of the lactam carbonyl of 1d upon treatment with LiAlH4 (5 equiv, THF, 70 °C reflux, 6 h) completed the total synthesis of (−)-6-epimesembranol (1b) in 92% yield (Scheme 10). Further, the treatment with IBX in DMSO (25 °C, 3 h) affords the (−)-mesembrine (1a) in 83% yield (Scheme 10). It is understood that, further utilization of this intermediate in the collective total synthesis of cis-3a-octahydroindoline alkaloid could be easily achieved with any kind of electron-rich aromatic ring present in these alkaloids. As both enantiomers [(R) as well as (S)] of CBS catalyst is commercially available, one could synthesize both antipode of the natural product only by changing the enantiomers of catalyst.
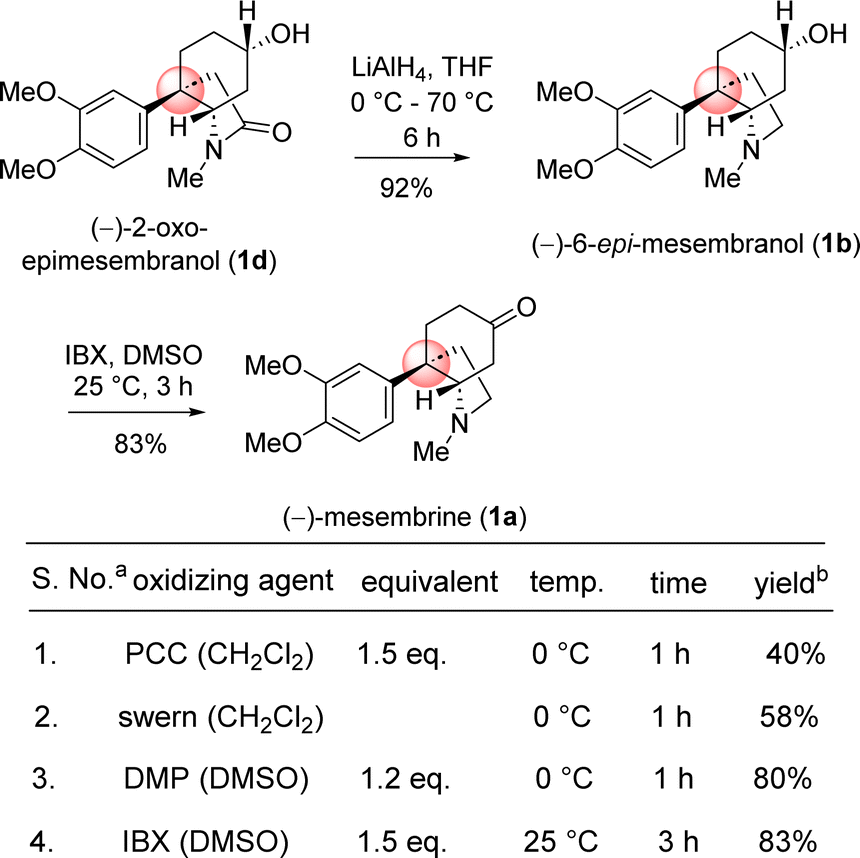 | ||
| Scheme 10 Collective total synthesis of cis-3a-octahydroindoline alkaloids. aAll the reactions were performed under argon atmosphere. bYields are reported after column chromatography. | ||
Next the total synthesis of naturally occurring dihydromaritidine was undertaken. Towards this end, it was hypothesized to synthesize the tricyclic core 6b from 4-aryl 4-alkyl α,β-unsaturated ketone 7b via an ester-aminolysis followed by an aza-Michael reaction with ammonia under heating. Following optimization, it was observed that the treatment of 4-aryl 4-alkyl α,β-unsaturated ketone 7b with NH3 in THF (70 °C, 16 h) proceeded smoothly furnish bicyclic keto lactam 6b in 94% yield (Scheme 11). Further, keto functional group of the bicyclic keto lactam 6b was protected with ethylene glycol in the presence of catalytic amount p-toluene sulfonic acid to afford 13b in 98% yield (Scheme 12). Later, lithium aluminium hydride reduction in tetrahydrofuran under reflux afforded cis-8a-octahydroindole derivative 14b in 94% yield. Other reducing agents such as Red-Al, superhydride were proved to be inefficient for this transformation. The work-up process of reduction of 13b (particularly the quench of lithium aluminum hydride) is worth mentioning, which was done under the basic condition using 1(N) NaOH. It was observed that an acidic work up using 0.5(N) HCl always afforded a mixture of spots, probably because of the deprotection of ketal functional group.
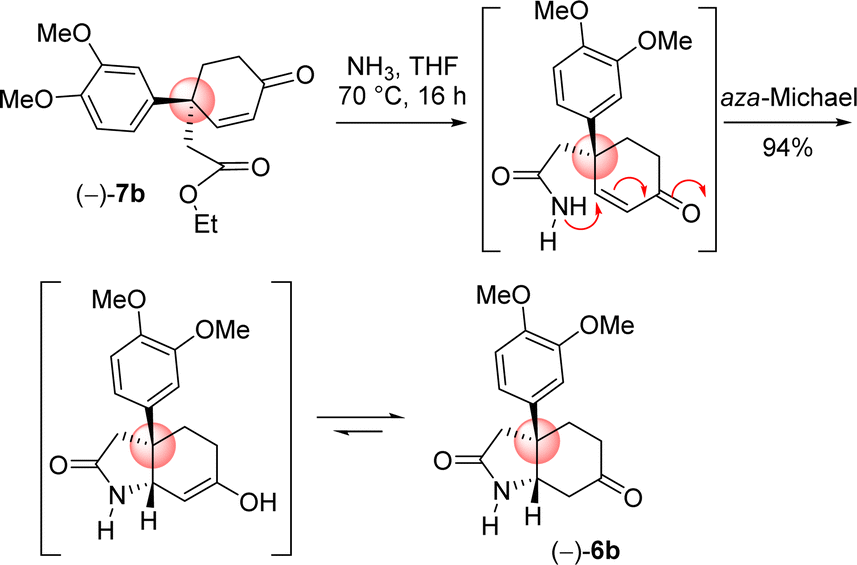 | ||
| Scheme 11 Ester aminolysis with ammonia and aza-Michael cascade. | ||
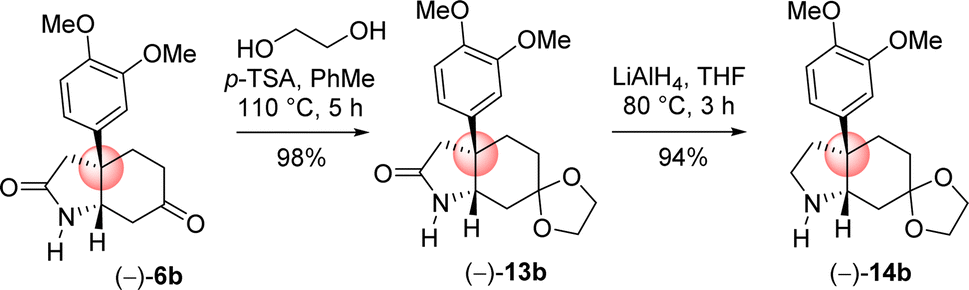 | ||
| Scheme 12 Synthesis of cis-3a-octahydroindoline skeleton. | ||
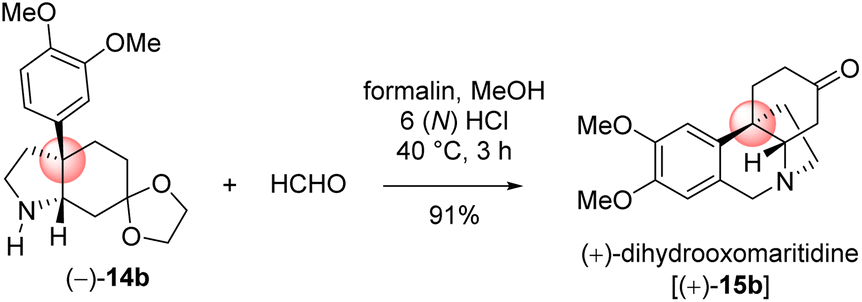
With significant quantity of cis-C-8a-octahydroindole derivative 14b in hand, the next effort was put forward to affect the Pictet–Spengler cyclization to obtain [5,11b]-ethanophenanthridine skeleton (Scheme 13). In this regard, a number of formaldehyde equivalents such as paraformaldehyde, 1,3,5-trioxane, Eschenmoser salt, and formalin were used. Following an exhaustive optimization (Table 4), it was delighted to observe that the synthesis of required [5,11b]-ethanophenanthridine skeleton, i.e. (+)-dihydrooxomaritidine (15b) could be obtained in 91% from a reaction of 14b and formalin (aqueous solution) in methanol in the presence of 6(N) HCl.
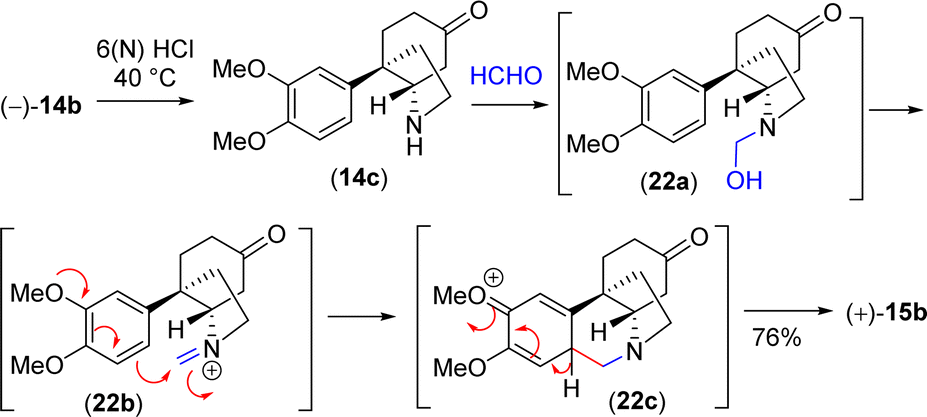 | ||
| Scheme 13 Proposed mechanism for the synthesis of [5,10b]-ethanophenanthridine skeleton. | ||
|
|||||
|---|---|---|---|---|---|
| S. no.a | Acid | HCHO equivalent | Temp. | Time | Yieldb |
| a All the reactions were performed under argon atmosphere.b Yields are reported after column chromatography. | |||||
| 1 | TFA | Paraformaldehyde | 0 °C | 8 h | 62% |
| 2 | TFA | 1,3,5-Trioxane, BF3·OEt2 | 0 °C | 8 h | 56% |
| 3 | None | Eschenmoser salt | 40 °C | 3 h | 65% |
| 4 | TFA | Formalin | 0 °C | 24 h | 71% |
| 5 | 6(N) | Formalin | 40 °C | 3 h | 91% |
A rational of the formation of Pictet–Spengler cyclization product i.e. the total synthesis of (+)-dihydrooxomaritidine (15b) from cis-3a-octahydroindole derivative 14b is shown in Scheme 13. An initial deprotection of ketal functionality in the presence of 6(N) HCl could afford ketone 14c, which on subsequent reaction with formalin could generate a hemi-aminal 22a (Scheme 13). Further activation of the hemi-aminal 22a in the presence of HCl could form iminium intermediate 22b, thereby the stage is ready for the Pictet–Spengler cyclization. Finally, the formation of [5,11b]-ethanophenanthridine skeleton, i.e. (+)-dihydrooxomaritidine (15b) would complete via an aromatic electrophilic substitution type reaction (Scheme 13).
The next effort was to synthesize the naturally occurring Amaryllidaceae alkaloids, (+)-dihydromaritidine (2c) sharing [5,11b]-ethanophenanthridine skeleton. In this regard a highly diastereoselective reduction of ketone was undertaken (Table 5). Towards this, number of reducing agents such as sodium borohydride, lithium aluminum hydride, Red-Al, and K-selectride, were employed for the total synthesis of (+)-dihydromaritidine (2c). Among various reducing agent, K-selectride was found to be the best and furnished the natural product 2c in 99% yield with >20:1 diastereoselectivity (entry 5, Table 5). Gratifyingly, a reduction using sodium borohydride at −10 °C afforded the required (+)-dihydromaritidine (2c) in 98% yield with 10:1 dr (entry 1, Table 5).
|
||||||
|---|---|---|---|---|---|---|
| S. no.a | Reducing agent | Equivalent | Temp. | Time | Yieldb | dr |
| a All the reactions were performed under argon atmosphere.b Yields are reported after column chromatography. | ||||||
| 1 | NaBH4 (MeOH) | 1 eq. | −10 °C | 1 h | 98% | ∼10:1 |
| 2 | LiAlH4 (THF) | 1 eq. | 0 °C | 1 h | 90% | ∼5:1 |
| 3 | LiAlH4 (THF) | 1 eq. | −10 °C | 5 h | 69% | ∼10:1 |
| 4 | Red-Al (PhMe) | 1 eq. | 25 °C | 4 h | 65% | ∼10:1 |
| 5 | K-selectride (THF) | 1 eq. | −78 °C | 1 h | 99% | >20:1 |

In conclusion, we describe a general approach to a number of alkaloids of Sceletium alkaloids of the family Aizoaceace following Johnson (orthoester)–Claisen rearrangement as the key step. It has been shown that acid catalysed process afforded product with compromised enantioselectivity, whereas a reaction in basic medium (such as diisopropylethylamine, DIPEA) could afford [3,3]-sigmatropic rearrangement product26 in 97% ee. Importantly, this reaction installed all carbon quaternary stereocenter at the pseudobenzylic position. The enantioenriched 3-(aryl)cyclohex-2-enol and 2-bromo 3-(aryl)cyclohex-2-enol were prepared by using catalytic enantioselective CBS reduction (up to 99% ee). Utilizing aforementioned strategy, a collective total synthesis of (−)-2-oxo-epimesembranol (1d), (−)-6-epimesembranol (1b), and (−)-mesembrine (1a) were shown. Further utilizing 7b via an ester aminolysis with ammonia followed by Pictet–Spengler cyclization leads to completion the total synthesis of (+)-dihydrooxomaritidine (15b) and (+)-dihydromaritidine (2c).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the SERB [CRG/2023/000782], [SCP/2022/000486], CSIR [02(0403)/21/EMR-II] and partial support from STARS-MoE [2023/0753] is gratefully acknowledged. SM thanks the CSIR for a research fellowship (SRF). AY and SP thank IISERB for fellowships (JRFs). AM thanks the UGC for fellowship. AB is a SERB-STAR Fellow and sincerely acknowledges the SERB [STR/2020/000061] for generous support.Notes and references
- (a) G. N. Trinadhachari, A. G. Kamat, K. R. Babu, P. D. Sanasi and K. J. Prabahar, Tetrahedron: Asymmetry, 2014, 25, 117–124 CrossRef CAS; (b) H. Kimura, T. Kawai, Y. Hamashima, H. Kawashima, K. Miura, Y. Nakaya, M. Hirasawa, K. Arimitsu, T. Kajimoto, Y. Ohmomo, M. Ono, M. Node and H. Saji, Bioorg. Med. Chem., 2014, 22, 285–291 CrossRef CAS PubMed; (c) T. Ozaki and Y. Kobayashi, Org. Chem. Front., 2015, 2, 328–335 RSC; (d) K. Geoghegan and P. Evans, J. Org. Chem., 2013, 78, 3410–3415 CrossRef CAS PubMed.
- (a) P. Lan, C. J. Jackson, M. G. Banwell and A. C. Willis, J. Org. Chem., 2014, 79, 6759–6764 CrossRef CAS PubMed; (b) J. McNulty and C. Zepeda-Velazquez, Angew. Chem., Int. Ed., 2014, 53, 8450–8454 CrossRef CAS.
- (a) N. Cortes, R. A. Posada-Duque, R. Alvarez, F. Alzate, S. Berkov, G. P. Cardona-Gomez and E. Osorio, Life Sci., 2015, 122, 42–50 CrossRef CAS PubMed; (b) W. Poet, D. Tagwireyi, H. M. Chinyanga, C. Musara, G. Nyandro, J. Chifamba and P. Nkomozepi, J. Ethnopharmacol., 2013, 148, 379–385 CrossRef PubMed; (c) V. Georgiev, I. Ivanov, S. Berkov and A. Pavlov, J. Plant Biochem. Biotechnol., 2014, 23, 389–398 CrossRef CAS.
- (a) J. J. Nair, J. Bastida, C. Codina, F. Viladomat and J. V. Staden, Nat. Prod. Commun., 2013, 8, 1335 CAS; (b) Z. Jin and X.-H. Xu, in Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes, ed. K. G. Ramawat and J. M. Merillon, Spinger-Verlag, Berlin Heidelberg, 2013, p. 497 Search PubMed; (c) Y. Ding, D. Qu, K.-M. Zhang, X.-X. Cang, Z.-N. Kou, W. Xiao and J.-B. Zhu, J. Asian Nat. Prod. Res., 2016, 12, 1 Search PubMed.
- Reviews on Amaryllidaceae alkaloids, see: (a) Z. Jin, Nat. Prod. Rep., 2009, 26, 363 RSC; (b) J. R. Lewis, Nat. Prod. Rep., 2002, 19, 223 RSC; (c) Z. Jin, Z. Li and R. Huang, Nat. Prod. Rep., 2002, 19, 454 RSC.
- The effects of Sceletium tortuosum in an in vivo model of psychological stress. M. T. Smith, N. R. Crouch, N. Gericke and M. Hrisht, J. Ethnopharmacol., 1996, 133, 119–130 CrossRef.
- (a) Pharmacological actions of the South African medicinal and functional food plant Sceletium tortuosum and its principal alkaloids. A. L. Harvey, L. C. Young, A. M. Viljoen and N. P. Gericke, J. Ethnopharmacol., 2011, 137, 1124–1129 CrossRef CAS; (b) R. E. Schultes, Hallucinogenic Plants, Golden Press, New York, 1976 Search PubMed; (c) P. W. Jeffs, in The Alkaloids, ed. Rodrigo, R. G. A., Academic Press, New York, 1981, vol. 19 Search PubMed.
- Assessing African medicinal plants for efficacy and safety: agricultural and storage practices. C. W. Fennell, M. E. Light, S. G. Sparg, G. I. Stafford and J. Van Staden, J. Ethnopharmacol., 2004, 95, 113–121 CrossRef CAS.
- K. Bodendorf and W. Krieger, Arch. Pharm., 1957, 290, 441–448 CrossRef.
- For representative racemic syntheses, see: (a) G. E. Keck and R. R Webb II, J. Am. Chem. Soc., 1981, 103, 3173 CrossRef CAS; (b) I. H. Sánchez, F. J. López, J. J. Soria, M. I. Larraza and H. J. Flores, J. Am. Chem. Soc., 1983, 105, 7640 CrossRef; (c) C. Bru, C. Thal and C. Guillou, Org. Lett., 2003, 5, 1845 CrossRef CAS PubMed; (d) N. T. Tam and C.-G. Cho, Org. Lett., 2008, 10, 601 CrossRef CAS PubMed; (e) G. Pandey, N. R. Gupta and T. M. Pimpalpalle, Org. Lett., 2009, 11, 2547 CrossRef CAS PubMed; (f) K. M. Bogle, D. J. Hirst and D. J. Dixon, Org. Lett., 2010, 12, 1252 CrossRef CAS PubMed; (g) D. A. Candito, D. Dobrovolsky and M. Lautens, J. Am. Chem. Soc., 2012, 134, 15572 CrossRef CAS PubMed; (h) M. K. Das, S. De, Shubhashish and A. Bisai, Synthesis, 2016, 48, 2093 CrossRef CAS; (i) S. Raghavan and A. Ravi, Org. Biomol. Chem., 2016, 14, 10222 RSC; (j) N. Gao, M. Banwell and A. C. Willis, Org. Lett., 2017, 19, 162 CrossRef CAS PubMed.
- (a) S. Akai, M. Kojima, S. Yamauchi, T. Kohji, Y. Nakamura and K. Sato, Asian J. Org. Chem., 2013, 2, 299–302 CrossRef CAS; (b) S. P. Chavan, S. Garai, C. Dey and R. G. Gonnade, Tetrahedron Lett., 2013, 54, 5562–5566 CrossRef CAS.
- (a) F. Sandberg and K.-H. Michel, Lloydia, 1963, 26, 78 CAS; (b) R. V. K. Rao and J. V. L. N. Sheshagiri Rao, Curr. Sci., 1979, 48, 110 CAS; (c) P. Pacheco, M. Silva, W. Steglich and W. H. Watson, Rev. Latinoam. Quim., 1978, 9, 28 CAS; (d) S. Toaima and M. Alexandria, J. Pharm. Sci., 2007, 21, 61 Search PubMed; (e) V. Zabel, W. H. Watson, P. Pacheco and M. Silva, Cryst. Struct. Commun., 1979, 8, 371 CAS.
- For biological activity of maritidine, see: (a) N. P. Gericke, and B. E. Van Wyk, WO97/46234, 1997Chem. Abstr., 1998, 12880030, (serotonin uptake inhibitor); (b) E. E. Elgorashi, G. I. Stafford, A. K. Jager and J. van Staden, Planta Med., 2006, 72, 470–473 CrossRef CAS PubMed (inhibition of [3H]citalopram binding to the rat brain serotonin transporter).; (c) G. Cea, M. Alarcon and G. Weigart, Med. Sci., 1986, 14, 90 CAS (clastogenic effect/mutagenic)..
- (a) M. Alarcon, G. Cea and G. Weigert, Environ. Contam. Toxicol., 1986, 37, 508 CrossRef CAS PubMed; (b) G. Cea, M. Alarcon and G. Weigart, Med. Sci., 1986, 14, 90 CAS (clastogenic effect/mutagenic)..
- For the total synthesis of (−)-oxomaritidine or (+)-maritidine by using a diastereoselective desymmetrization, see: (a) K. Tomioka, K. Koga and S. Yamada, Chem. Pharm. Bull., 1977, 25, 2681 CrossRef CAS; (b) P. Verma, A. Chandra and G. Pandey, J. Org. Chem., 2018, 83, 9968 CrossRef CAS PubMed; (c) For an elegant resolution of racemic oxomaritidine to (−)-oxomaritidine by Ir-catalyzed asymmetric hydrogenation, see: X.-D. Zuo, S.-M. Guo, R. Yang, J.-H. Xie and Q.-L. Zhou, Chem. Sci., 2017, 8, 6202 RSC.
- For representative asymmetric syntheses, see: (a) T. Nishimata, Y. Sato and M. Mori, J. Org. Chem., 2004, 69, 1837 CrossRef CAS; (b) K. Du, H. Yang, P. Guo, L. Feng, G. Xu, Q. Zhou, L. W. Chung and W. Tang, Chem. Sci., 2017, 8, 6247 RSC; (c) Y. R. Gao, D.-Y. Wang and Y.-Q. Wang, Org. Lett., 2017, 19, 3516 CrossRef CAS PubMed; (d) A. Yadav, S. Majumder, M. K. Das, A. Mondal and A. Bisai, Synlett, 2023, 34, 858 CrossRef CAS.
- Chiral-pool strategy from carbohydrates, see: (a) S. W. Baldwin and J. S. Debenham, Org. Lett., 2000, 2, 99 CrossRef CAS; (b) M. Bohno, H. Imase and N. Chida, Chem. Commun., 2004, 1086 RSC; (c) A. D. Findlay and M. G. Banwell, Org. Lett., 2009, 11, 3160 CrossRef CAS PubMed (chemoenzymatic resolution)..
- (a) R. E. Lyle, E. A. Kielar, J. R. Crowder and W. C. Wildman, J. Am. Chem. Soc., 1960, 82, 2625 CrossRef; (b) F. Viladomat, J. Bastida, C. Codina, W. E. Campbell and S. Mathee, Phytochemistry, 1995, 40, 307 CrossRef CAS.
- (a) B. M. Trost, G. M. Schroeder and J. Kristensen, Angew. Chem., Int. Ed., 2002, 41, 3492 CrossRef CAS; (b) B. M. Trost, J. Xu and T. Schmidt, J. Am. Chem. Soc., 2009, 131, 18343 CrossRef CAS.
- (a) J. Park, Y. K. Kim, G. Kim and B. Korean, Chem. Soc., 2011, 32, 3141 CAS; (b) M. K. Das, N. Kumar and A. Bisai, Org. Lett., 2018, 20, 4421–4424 CrossRef CAS; (c) W. Wang, J. Dai, Q. Yang, Y.-H. Deng, F. Peng and Z. Sao, Org. Lett., 2021, 23, 920–924 CrossRef CAS.
- H. Kim, H. Choi and K. Lee, Synlett, 2018, 29, 1203–1206 CrossRef CAS.
- (a) E. J. Corey and G. W. J. Fleet, Tetrahedron Lett., 1973, 14, 4499 CrossRef; (b) For CBS reduction of 2-aryl-2-cyclohexenone, see; S. Khatua, S. Niyogi and V. Bisai, Tetrahedron, 2020, 76, 130918 CrossRef; (c) A similar type of transition state has been proposed..
- (a) S. Hackett and T. Livinghouse, J. Org. Chem., 1986, 51, 1629 CrossRef CAS; (b) O. Yamada and K. Ogasawara, Tetrahedron Lett., 1998, 39, 7747 CrossRef CAS.
- E. J. Corey and C. J. Helal, Angew. Chem., Int. Ed., 1998, 37, 1986–2012 CrossRef CAS PubMed.
- S. Majumder, A. Yadav, S. Pal, A. Khatua and A. Bisai, J. Org. Chem., 2022, 87, 7786–7797 CrossRef CAS.
- V. R. Gavit, S. Kundu, S. Niyogi, N. K. Roy and A. Bisai, J. Org. Chem., 2024, 89, 1823–1835 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05275g |
| This journal is © The Royal Society of Chemistry 2024 |