
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Integrated synthesis of 3,4-carbazoquinone alkaloids N-Me-carbazoquinocin A, B and D–F†
Basavaiah Bommanaboina,
Debayan Roy and
Beeraiah Baire*
and
Beeraiah Baire*
Department of Chemistry, Indian Institute of Technology Madras, Chennai-600036, Tamil Nadu, India. E-mail: beeru@iitm.ac.in
First published on 28th August 2024
Abstract
Carbazole alkaloids carbazoquinocin A–F possessing a 1-alkyl-2-methyl-3,4-ortho-carabazoquinone framework were isolated from the microorganism Streptomyces violaceus 2448-SVT2 in 1995. Furthermore, they were found to exhibit strong inhibitory activity against lipid peroxidation. Herein, we report the integrated synthesis of N-Me-analogues of 5 members of the carbazoquinocin family of natural products, namely, N-Me-carbazoquinocin A, B and D–F. We employed an acid-catalyzed, intramolecular benzannulation of indole-appended Z-enoate propargylic alcohols, which was developed earlier in our laboratory, for the construction of the required carbazole framework. All five natural products were obtained in an overall yield of 50–60%, starting from a commercially available indole.
Introduction
Carbazoles (dibenzopyrroles) are nitrogen-based tricyclic frameworks, which are essential in medicinal chemistry1 and materials chemistry.2 They are also part of many structurally complex bioactive natural products and drug molecules.3 Among the many structurally diverse carbazole natural products, a 1-alkyl-2-methyl-carbazole framework has attracted special attention as it has been found to be present in different families of bioactive carbazole natural products such as lipocarbazoles, carbazoquinocins, and carazostatin.4 In 1995, the research group of Seto isolated six carbazoquinocins A–F, from the microorganism Streptomyces violaceus 2448-SVT2 (Fig. 1).5 These natural products share a common 1-alkyl-2-methyl-3,4-ortho-carbazoquinone framework and have been found to exhibit strong inhibitory activity against lipid peroxidation.6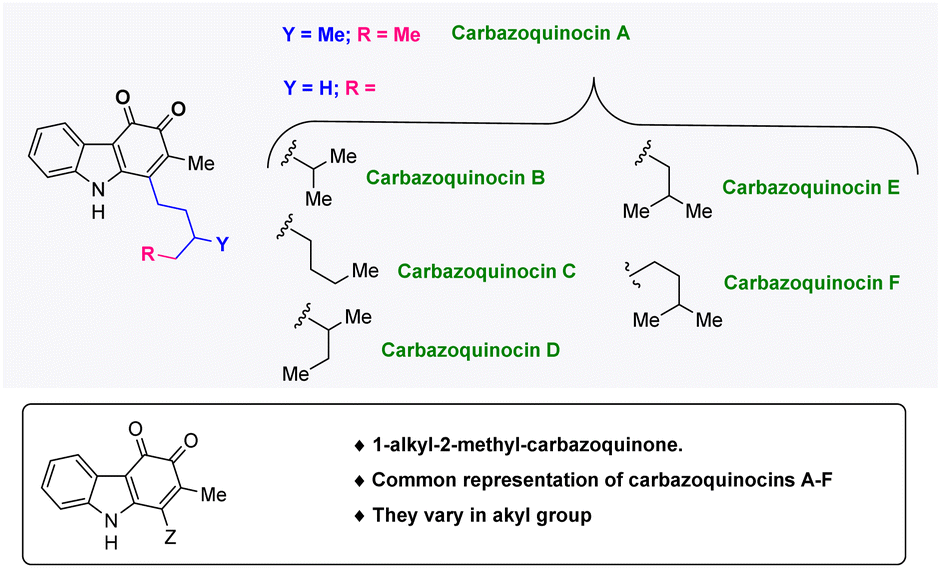 | ||
| Fig. 1 Structures of carbazoquinocin A–F natural products. | ||
Several research groups have reported the total synthesis of some of these novel alkaloids.7 For example, the total synthesis of carbazoquinocins A and D was achieved by the research group of Ogasawara in 1996.7a Alternatively, Hibino et al. reported the total synthesis of carbazoquinocins B–F.7b In 2003, Wulff and coworkers reported the total synthesis of carbazoquinocin C.7c
Recently, in 2022, we developed an approach for the total synthesis of the 1-alkyl-2-methyl-3,4-carbazoquinone natural product N-Me-carbazoquinocin C together with N-Me-carazostatin and N-Me-lipocarbazole A4.7d This approach employs a Brønsted acid-catalyzed intramolecular benzannulation of C-3-tethered indole-propargylic alcohols (1) (Scheme 1A) as the key step for the construction of a carbazole unit (2). In continuation, we envisioned an opportunity to extend this methodology for the total synthesis of other members of the carbazoquinocin family, i.e., carbazoquinocin A, B, and D–F (3–7) (Fig. 1). According to our retrosynthetic analysis (Scheme 1B), butyraldehyde present in 2 can be converted into the respective alkyl chain (as in 8) required for carbazoquinocin A, B and D–F through the Wittig olefination reaction using suitable phosphorous ylides. Further, oxidation at the C-3 and C-4 positions of the resultant carbazoles 8 to generate the natural products carbazoquinocin A, B and D–F (3–7) can be achieved through C-3-methoxylation, followed by selenium promoted oxidations.
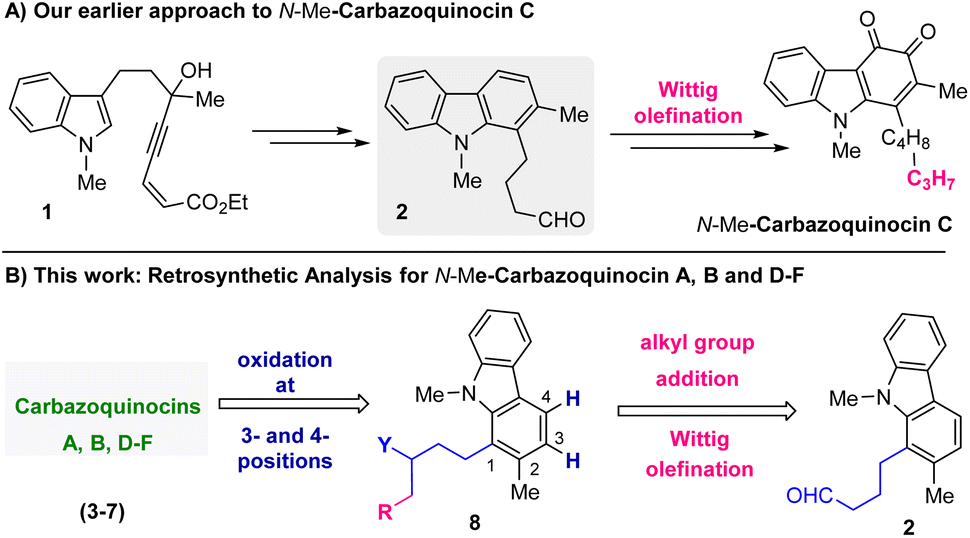 | ||
| Scheme 1 (A) Our earlier work: total synthesis of N-Me-carbazoquinocin C. (B) Retrosynthetic analysis for the total synthesis of carbazoquinocins A, B, and D–F (3–7) from aldehyde 2. | ||
Results and discussion
Our approach began with the synthesis of the required carbazole–butyraldehyde (2) from N-Me-indole, following our earlier procedure.7d Initially, we aimed at the total synthesis of N-Me-carbazoquinocin B (3) (Scheme 2, R = H). Accordingly, Wittig olefination of aldehyde 2 with phosphorous ylide (9a) at 0 °C (generated in situ from (iso-propyl)PPh3Br8 (10a) and nBuLi at 0 °C) afforded olefin (11a) in 76% yield. The hydrogenation reaction of 11a with Pd/C (10%) under an H2 atmosphere in ethyl acetate at room temperature (rt) for 6 h gave 1-isoheptylcarbazole (12a) (96% yield).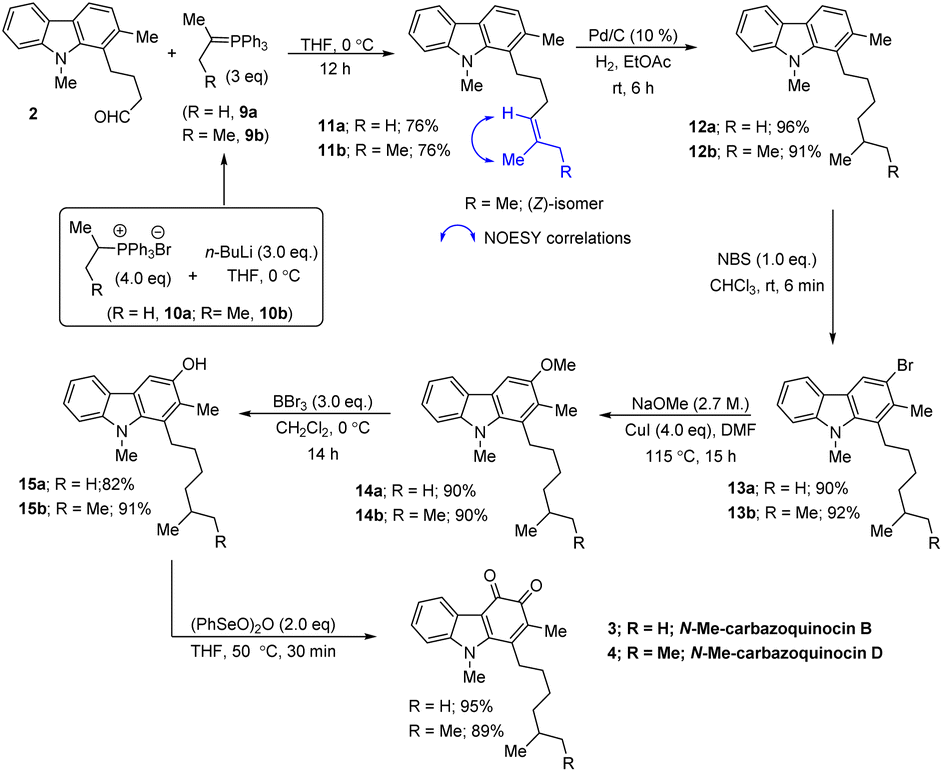 | ||
| Scheme 2 Total synthesis of N-Me-carbazoquinocin B and D. | ||
Next, the regioselective electrophilic bromination of carbazole 12a with NBS in CHCl3 at rt gave the corresponding 3-bromocarbazole (13a) (90%) within 6 min reaction time. Next, we employed a two-step strategy for the efficient conversion of bromine in 13a into a hydroxyl group. Initially, heating a DMF suspension of 13a, NaOMe and CuI at 115 °C gave 3-methoxy carbazole (14a) in 90% yield. Subsequent treatment of 14a with BBr3 in CH2Cl2 at 0 °C to rt for 14 h resulted in the formation of the phenol product (15a) in 82% yield via deprotection of the methoxy group. Finally, the synthesis of N-Me-carbazoquinocin B 3 was achieved by employing the (PhSeO)2O-promoted oxidation of 15a. Accordingly, heating (50 °C) a solution of 15a and (PhSeO)2O in THF gave N-Me-carbazoquinocin B 3 in 95% yield. After successfully completing the total synthesis of N-Me-carbazoquinocin B 3, we extended this strategy for the synthesis of N-Me-carbazoquinocin D 4 by employing the carbazole–butyraldehyde 2 as the starting point (Scheme 2, R = Me). According to our retrosynthetic analysis (Scheme 1B), the only difference between carbazoquinocin B 3 and carbazoquinocin D 4 is the incorporation of an iso-propyl group (R = H) vs. sec-butyl group (R = Me) in aldehyde 2. Therefore, olefination of aldehyde 2 using sec-butylidene phosphorous ylide 9b (generated from (sec-butyl)PPh3Br8 10b and nBuLi at 0 °C) afforded Z-olefin 11b (R = Me) in 76% yield. The geometry of the olefin in 11b was identified by using the correlations observed between ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CMe and C–H in the corresponding NOESY NMR spectrum. Further, by operating the same sequence of five functional group transformations, as depicted in Scheme 2 (R = Me), i.e., hydrogenation (12b, 91%)–bromination (13b, 92%)–methoxylation (14b, 90%)–demethylation (15b, 91%)–oxidation (4, 89%), we efficiently converted olefin 11b into N-Me-carbazoquinocin D 4, with an overall yield of 61% for five steps. Subsequently, we envisioned the extension of this methodology for the total synthesis of N-Me-carbazoquinocin E 5 and F 6 (Scheme 3). The required alkyl chains at the C1-position present in compounds 5 and 6 could be achieved by incorporating an iso-butyl group (n = 0) and iso-pentyl group (n = 1) in butyraldehyde 2 via Wittig olefination reaction, respectively. Wittig olefination of aldehyde 2 using iso-butylidene phosphorous ylide 9c (ref. 8) and iso-pentylidene phosphorous ylide 9d (ref. 8) exclusively afforded Z-olefins 11c (n = 0; 81%) and 11d (n = 1; 80%), respectively. After having both olefins 11c and 11d in hand, we employed the five-step synthetic strategy (as shown for N-Me-carbazoquinocin B 3 and D 4 in Scheme 2), i.e., hydrogenation (12c & 12d)–bromination (13c & 13d)–methoxylation (14c & 14d)–demethylation (15c & 15d)–oxidation (5 & 6), respectively. These sequential synthetic transformations efficiently converted olefins 11c & 11d into the corresponding natural products N-Me-carbazoquinocin E 5 and N-Me-carbazoquinocin F 6, respectively (Scheme 3).
CMe and C–H in the corresponding NOESY NMR spectrum. Further, by operating the same sequence of five functional group transformations, as depicted in Scheme 2 (R = Me), i.e., hydrogenation (12b, 91%)–bromination (13b, 92%)–methoxylation (14b, 90%)–demethylation (15b, 91%)–oxidation (4, 89%), we efficiently converted olefin 11b into N-Me-carbazoquinocin D 4, with an overall yield of 61% for five steps. Subsequently, we envisioned the extension of this methodology for the total synthesis of N-Me-carbazoquinocin E 5 and F 6 (Scheme 3). The required alkyl chains at the C1-position present in compounds 5 and 6 could be achieved by incorporating an iso-butyl group (n = 0) and iso-pentyl group (n = 1) in butyraldehyde 2 via Wittig olefination reaction, respectively. Wittig olefination of aldehyde 2 using iso-butylidene phosphorous ylide 9c (ref. 8) and iso-pentylidene phosphorous ylide 9d (ref. 8) exclusively afforded Z-olefins 11c (n = 0; 81%) and 11d (n = 1; 80%), respectively. After having both olefins 11c and 11d in hand, we employed the five-step synthetic strategy (as shown for N-Me-carbazoquinocin B 3 and D 4 in Scheme 2), i.e., hydrogenation (12c & 12d)–bromination (13c & 13d)–methoxylation (14c & 14d)–demethylation (15c & 15d)–oxidation (5 & 6), respectively. These sequential synthetic transformations efficiently converted olefins 11c & 11d into the corresponding natural products N-Me-carbazoquinocin E 5 and N-Me-carbazoquinocin F 6, respectively (Scheme 3).
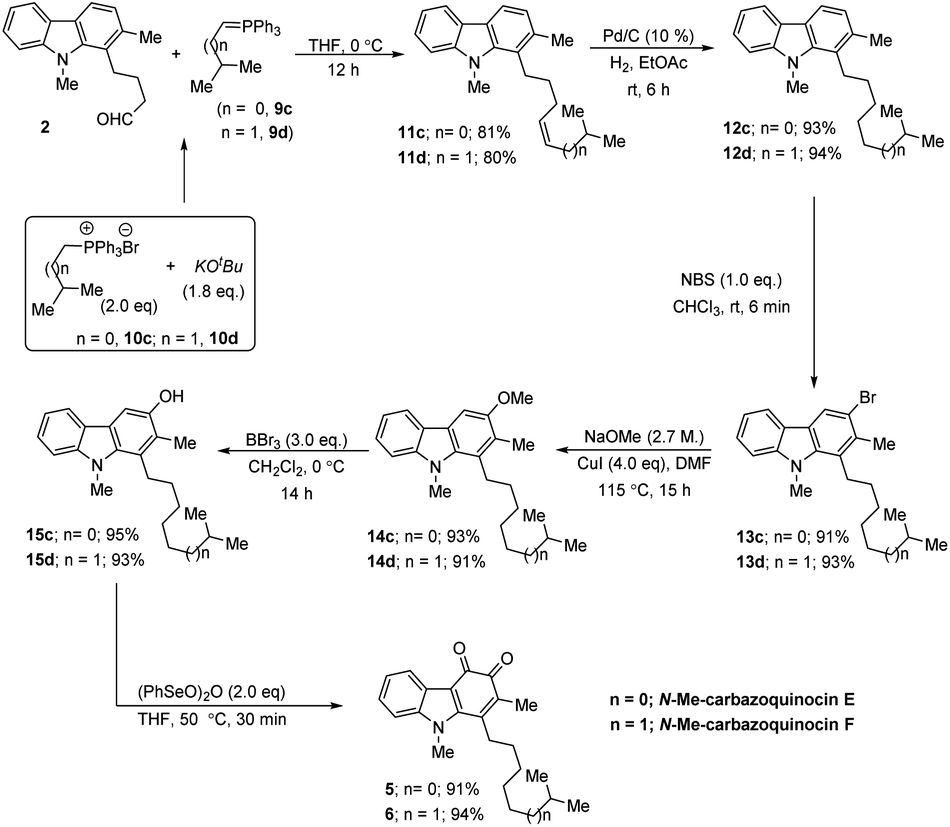 | ||
| Scheme 3 Unified total synthesis of N-Me-carbazoquinocin E and F. | ||
Finally, we also extended this strategy for the total synthesis of N-Me-carbazoquinocin A 7, which possesses a 3-methylpentyl alkyl chain at the C1-position of the carbazoquinone (Scheme 4). We envisioned carbazole ester (16) as a suitable starting material for this purpose. α-Methylation (for sec-methyl) followed by conversion of the carboxylate group into an ethyl group generates the required 3-methylpentyl chain at the C1-position. Accordingly, the LDA-mediated enolate formation of ester 16 followed by quenching with MeI gave α-methyl ester (17) in 59% yield. The reduction of 17 with LiAlH4 followed by oxidation of the resultant primary alcohol (18) with Dess–Martin periodinane (DMP) gave aldehyde (19) in 84% overall yield for two steps.
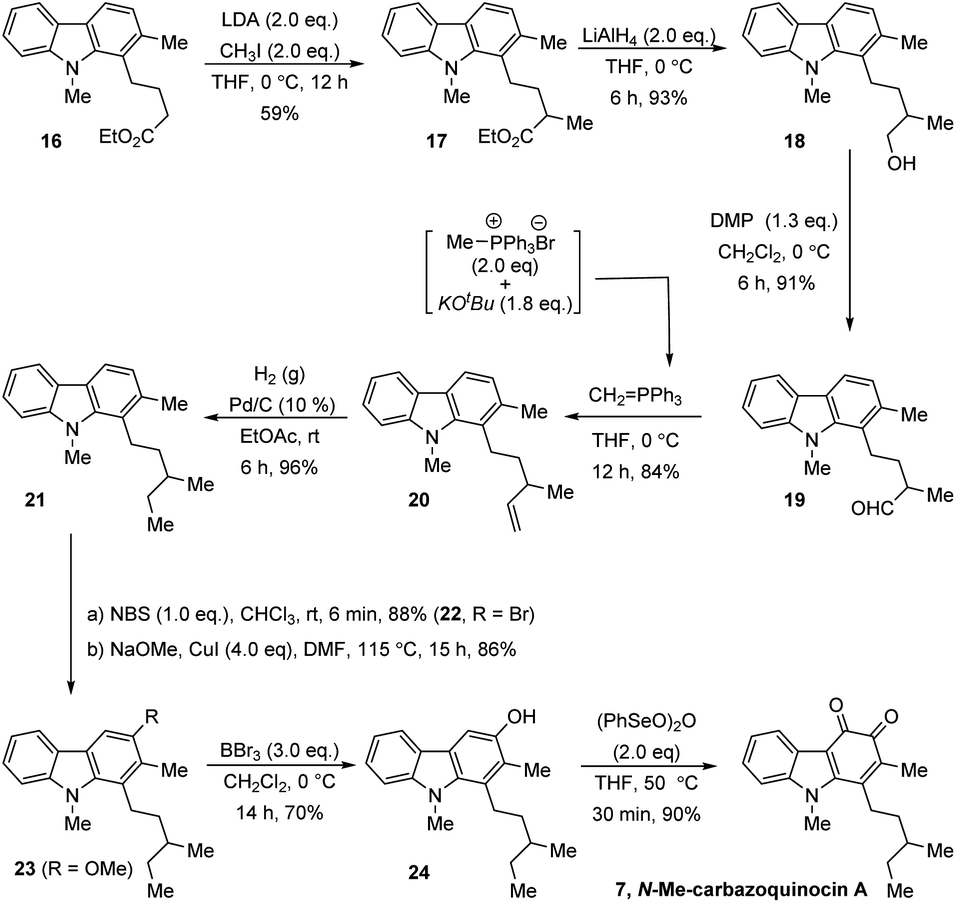 | ||
| Scheme 4 Extension to the total synthesis of N-Me-carbazoquinocin A. | ||
Further, olefination of aldehyde 19 using methylidene-phosphorous ylide, followed by hydrogenation of the resultant olefin (20) gave 1-[3-methylpentyl] carbazole (21). Subsequently, we employed a four-step synthetic strategy, which involved bromination (22)–methoxylation (23)–demethylation (24)–oxidation (7). These sequential synthetic transformations efficiently converted carbazole 21 into N-Me-carbazoquinocin A 7 with an overall yield of 48% for four steps.
Conclusions
In conclusion, we developed an efficient and practically viable synthetic approach for the unified synthesis of 3,4-carbazoquinone-based carbazole alkaloids N-Me-carbazoquinocins A, B and D–F. Our earlier methodology, an acid-catalyzed, intramolecular benzannulation of indole-appended Z-enoate propargylic alcohols, was employed for the construction of the required carbazole framework. All five natural products were obtained in an overall yield of 50–60%, starting from the commercially available indole.Experimental section
General information
All solvents were distilled prior to use and anhydrous solvents were prepared according to the standard drying procedures. All non-aqueous reactions were carried out under an atmosphere of nitrogen in flame-dried glassware. Commercially available chemicals were purchased from Sigma-Aldrich, Alfa Aesar and Spectrochem Pvt. Ltd and were used as received without further purification. Infrared (IR) spectra were recorded on a JASCO 4100 FT-IR spectrometer. 1H NMR spectra were measured on a Bruker AVANCE 400 MHz or Bruker AVANCE 500 MHz spectrometer. Chemical shifts are reported in ppm relative to solvent signals. 13C NMR spectra were recorded on a Bruker AVANCE 100 MHz or Bruker AVANCE 125 MHz spectrometer with complete proton decoupling. Chemical shifts are reported in ppm from the residual solvent as an internal standard [CDCl3 δ = 7.26 ppm for 1H, δ = 77.16 ppm for 13C or calibrated to tetramethylsilane (δ = 0.00)]. The following abbreviations are used to indicate multiplicities: s-singlet; d-doublet; t-triplet; q-quartet; quint-quintet; sext-sextet; sept-septet; m-multiplet; dd-doublet of doublet; dt-doublet of triplet; dq-doublet of quartet; td-triplet of doublet; tt-triplet of triplet; dq-doublet of quartet; br-broad; J-coupling constant in Hz. The coupling constant J (Hz) was rounded to one decimal place for all compounds. When a coupling pattern can be assigned as a combination of multiplicities, the above-mentioned abbreviations were combined to describe the observed patterns (i.e., dt, doublet of triplets). Mass spectra were recorded by the electrospray ionization (ESI) method on a Q-TOF Micro with a lock spray source. For thin layer chromatography (TLC) analysis throughout this work, E-Merck precoated TLC plates (silica gel 60 F254 grade, 0.25 mm) were used and visualized using a UV lamp (366 or 254 nm) or by using of one of the following visualization reagents: PMA: 1 g phosphomolybdic acid/10 mL ethanol; KMnO4: 0.15 g potassium permanganate, 1 g K2CO3/20 mL water. Acme (India) silica gel (100–200 mesh) was used for column chromatography.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexanes/EtOAc) provided the corresponding alkene.:1 hexanes/EtOAc) provided the corresponding alkene.:1 hexanes/EtOAc) provided the desired 3-methoxycarbazole derivatives.:1 hexane/EtOAc) provided the desired 3-hydroxycarbazole derivative.:1) as the eluent.
:1 hexanes/EtOAc) provided the corresponding alkene.:1 hexanes/EtOAc) provided the corresponding alkene.:1 hexanes/EtOAc) provided the desired 3-methoxycarbazole derivatives.:1 hexane/EtOAc) provided the desired 3-hydroxycarbazole derivative.:1) as the eluent.1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 7.7 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.44 (dd, J1 = 7.6 Hz and J2 = 7.2 Hz, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.20 (dd, J1 = 7.6 Hz and J2 = 7.2 Hz, 1H), 7.06 (d, J = 7.8 Hz, 1H), 5.26 (t, J = 7.0 Hz, 1H), 4.07 (s, 3H), 3.17–3.07 (m, 2H), 2.51 (s, 3H), 2.22 (q, J = 7.1 Hz, 2H), 1.75 (s, 3H), 1.72 (d, J = 8.6 Hz, 1H), 1.68 (s, 3H) and 1.61–1.57 (m, 1H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 142.2, 140.0, 134.6, 132.5, 125.3, 124.1, 124.1, 123.2, 122.7, 122.4, 119.6, 118.9, 117.6, 108.7, 32.6, 31.7, 28.4, 28.1, 25.9, 20.2 and 17.9 ppm.
IR (ATR): 2956, 1423, 1421, 1389, 1265 and 749 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C21H26N 292.2060; found 292.2063 (1.0 ppm).
TLC: Rf = 0.4 (19:1 hexane/EtOAc).
:1 hexanes/EtOAc) to provide the desired hydrogenation product 12a (23 mg, 0.07 mmol, 96%) as a gummy compound.1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 7.7 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.30 (dd, J1 = 7.0 Hz and J2 = 8.0 Hz, 1H), 7.21 (d, J = 8.2 Hz, 1H), 7.08 (dd, J1 = 7.0 Hz and J2 = 7.5 Hz, 1H), 6.93 (d, J = 7.8 Hz, 1H), 3.90 (s, 3H), 3.00–2.93 (m, 2H), 2.39 (s, 3H), 1.54–1.52 (m, 3H), 1.40–1.38 (m, 2H), 1.25 (q, J = 7.8 Hz, 2H) and 0.81 (d, J = 6.6 Hz, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 142.3, 140.0, 134.5, 125.3, 124.3, 123.2, 122.7, 122.5, 119.6, 118.9, 117.6, 108.7, 39.0, 32.6, 32.0, 28.5, 28.2, 27.9, 22.8 and 20.2 ppm.
IR (ATR): 3451, 2928, 2854, 1666, 1589, 1453, 1443, 1362, 1324, 1243, 1096, 826 and 738 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C21H28N 294.2216; found 294.2224 (3.0 ppm).
TLC: Rf = 0.5 (hexane).
:1 hexanes/EtOAc) provided the desired product 13a (48 mg, 0.12 mmol, 90%) as a white solid.1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 7.97 (d, J = 7.7 Hz, 1H), 7.46 (t, J = 7.6 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 7.22 (t, J = 7.3 Hz, 1H), 4.01 (s, 3H), 3.18–3.07 (m, 2H), 2.59 (s, 3H), 1.68–1.59 (m, 3H), 1.56–1.49 (m, 2H), 1.30 (d, J = 8.2 Hz, 2H) and 0.94 (d, J = 6.6 Hz, 6H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 142.5, 139.0, 132.9, 126.0, 123.7, 122.1, 121.6, 119.8, 119.3, 116.8, 108.9, 38.8, 32.7, 32.0, 29.6, 28.1, 27.8, 22.8 and 19.8 ppm.
IR (ATR): 2964, 2846, 2365, 1456, 1413, 1271, 1144, 1016, 828, 773 and 736 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C21H27NBr 372.1321; found 372.1326 (1.3 ppm).
TLC: Rf = 0.55 (hexane).
M.P.: 173–175 °C.
:1 hexanes/EtOAc) provided the desired 3-methoxycarbazole 14a (29 mg, 0.09 mmol, 90%) as a colorless solid.1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 7.7 Hz, 1H), 7.44–7.39 (m, 2H), 7.36 (d, J = 8.2 Hz, 1H), 7.17 (t, J = 7.3 Hz, 1H), 4.05 (s, 3H), 3.95 (s, 3H), 3.16–3.10 (m, 2H), 2.38 (s, 3H), 1.70–1.63 (m, 2H), 1.60 (dd, J = 13.3, 6.7 Hz, 1H), 1.52–1.31 (m, 2H), 1.29–1.26 (m, 2H) and 0.92 (d, J = 6.6 Hz, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 152.4, 142.5, 134.9, 125.8, 125.1, 124.8, 123.3, 121.9, 119.5, 118.5, 108.8, 99.3, 56.3, 39.0, 32.8, 31.9, 28.8, 28.2, 27.9, 22.8 and 12.1 ppm.
IR (ATR): 2945, 2856, 2392, 1471, 1414, 1288, 1213, 1154, 1126, 845 and 742 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C22H30NO 324.2322; found 324.2342 (6.1 ppm).
TLC: Rf = 0.2 (hexane).
M.P.: 162–164 °C.
:1 hexanes/EtOAc) provided phenol 15a (21 mg, 0.07 mmol, 82% yield) as a pale-yellow liquid.1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 7.7 Hz, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.34 (d, J = 7.8 Hz, 2H), 7.15 (t, J = 7.3 Hz, 1H), 4.03 (s, 3H), 3.16–3.08 (m, 2H), 2.40 (s, 3H), 1.70–1.58 (m, 3H), 1.53 (dd, J = 15.0, 7.2 Hz, 2H), 1.32–1.26 (m, 2H) and 0.92 (d, J = 6.6 Hz, 6H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 147.7, 142.6, 135.1, 125.7, 125.4, 122.8, 122.4, 122.2, 119.7, 118.4, 108.7, 103.1, 38.9, 32.8, 32.0, 28.8, 28.2, 27.9, 22.8 and 12.1 ppm.
IR (ATR): 3325, 2926, 2854, 2363, 2351, 1485, 1451, 1264, 1232, 902, 782 and 731 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C21H28NO 310.2165; found 310.2174 (3.0 ppm).
TLC: Rf = 0.25 (9:1 hexane/EtOAc).
:1 hexanes/EtOAc) provided the desired N-methyl carbazoquinocin B 3 (20 mg, 0.06 mmol 95% yield) as a dark-brown glittering solid.1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.18 (s, 3H), 3.80 (s, 3H), 2.68–2.55 (m, 2H), 1.82 (s, 3H), 1.49 (d, J = 5.4 Hz, 3H), 1.42 (d, J = 6.7 Hz, 2H), 1.18 (s, 2H) and 0.83 (d, J = 6.5 Hz, 6H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 183.2, 173.6, 144.9, 142.6, 139.6, 134.5, 125.7, 124.9, 121.8, 113.7, 110.9, 38.7, 33.1, 29.9, 28.6, 28.1, 27.8, 22.7 and 11.9 ppm.
IR (ATR): 2961, 2942, 2863, 1665, 1652, 1632, 1499, 1481, 1459, 1369, 1234 and 781 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C21H26NO2 324.1958; found 324.1935 (7.0 ppm).
TLC: Rf = 0.34 (4:1 hexanes/EtOAc).
M.P.: 152–154 °C.
:1) as the eluent.1H NMR (400 MHz, CDCl3) major isomer: δ 7.93 (d, J = 7.6 Hz, 1H), 7.74 (d, J = 7.7 Hz, 1H), 7.34 (dd, J1 = 7.6 Hz and J2 = 8.0 Hz, 1H), 7.27 (d, J = 8.1 Hz, 1H), 7.11 (t, J = 7.3 Hz, 1H), 6.96 (d, J = 7.7 Hz, 1H), 5.22–5.10 (m, 1H), 3.97 (s, 3H), 3.09–2.98 (m, 2H), 2.42 (s, 3H), 2.14 (d, J = 5.3 Hz, 2H), 2.06–1.90 (m, 2H), 1.65 (s, 3H), 1.59 (s, 2H) and 1.00–0.88 (m, 3H) ppm.
1H NMR (400 MHz, CDCl3) minor isomer: δ 7.93 (d, J = 7.6 Hz, 1H), 7.74 (d, J = 7.7 Hz, 1H), 7.34 (dd, J1 = 7.6 Hz and J2 = 8.0 Hz, 1H), 7.27 (d, J = 8.1 Hz, 1H), 7.11 (t, J = 7.3 Hz, 1H), 6.96 (d, J = 7.7 Hz, 1H), 5.04–4.94 (m, 1H), 3.97 (s, 3H), 3.09–2.98 (m, 2H), 2.42 (s, 3H), 2.14 (d, J = 5.3 Hz, 2H), 2.06–1.90 (m, 2H), 1.65 (s, 3H), 1.59 (s, 2H) and 1.00–0.88 (m, 3H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 142.2, 139.9, 138.2, 138.1, 134.6, 125.3, 124.1, 124.1, 123.6, 123.1, 122.6, 122.4, 119.6, 118.9, 117.6, 108.7, 32.6, 32.5, 31.9, 31.7, 28.2, 28.1, 28.0, 24.9, 23.0, 20.2, 16.2 and 13.0 ppm.
IR (ATR): 2966, 1445, 1438, 1390, 1268, 1146, 1121 and 748 cm−1.
HRMS (ESI) m/z: [M + NH4]+ calcd for C22H31N2 323.2482; found 323.2496 (4.3 ppm).
TLC: Rf = 0.8 (9:1 hexane/EtOAc).
:1 hexanes/EtOAc) to provide the desired hydrogenation product 12b (34 mg, 0.11 mmol, 91%) as a colorless liquid.1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 7.5 Hz, 1H), 7.75 (d, J = 7.7 Hz, 1H), 7.35 (t, J = 7.5 Hz, 1H), 7.27 (d, J = 8.1 Hz, 1H), 7.11 (t, J = 7.2 Hz, 1H), 6.97 (d, J = 7.7 Hz, 1H), 3.98 (s, 3H), 3.08–2.96 (m, 2H), 2.43 (s, 3H), 1.47 (m, 5H), 1.32 (m, 3H), 1.10–1.09 (m, 1H) and 0.81 (d, J = 5.8 Hz, 6H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 142.2, 139.9, 134.5, 125.3, 124.3, 123.2, 122.7, 122.4, 119.6, 118.9, 117.6, 108.7, 36.6, 34.5, 32.6, 32.0, 29.7, 28.6, 27.7, 20.3, 19.4, 11.6 ppm.
IR (ATR): 3421, 2925, 2862, 1642, 1586, 1482, 1452, 1431, 1374, 1361, 1285, 1046, 923 and 722 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C22H30N 308.2373; found 308.2382 (3.0 ppm).
TLC: Rf = 0.45 (hexane).
:1 hexanes/EA) provided the desired product 13b (34 mg, 0.08 mmol, 92%) as a gummy compound.1H NMR (400 MHz, CDCl3) δ 8.08 (s, 1H), 7.90 (d, J = 7.7 Hz, 1H), 7.40 (t, J = 7.6 Hz, 1H), 7.29 (d, J = 8.2 Hz, 1H), 7.15 (t, J = 7.1 Hz, 1H), 3.97 (s, 3H), 3.14–3.02 (m, 2H), 2.52 (s, 3H), 1.60–1.45 (m, 5H), 1.32–1.31 (m, 3H), 1.20–1.15 (m, 1H) and 0.84 (d, J = 5.7 Hz, 6H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 142.5, 139.1, 132.9, 126.0, 123.7, 122.1, 121.6, 119.8, 119.3, 116.8, 108.9, 36.5, 34.5, 32.8, 32.0, 29.6, 27.5, 19.8, 19.3, 11.6 ppm.
IR (ATR): 2934, 2868, 2376, 1468, 1402, 1282, 1149, 1016, 812, 762 and 742 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C22H29NBr 386.1478; found 386.1460 (5 ppm).
TLC: Rf = 0.5 (hexane).
:1 hexanes/EtOAc) provided the desired 3-methoxycarbazole 14b (23 mg, 0.06 mmol, 90%) as a colourless liquid.1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 7.7 Hz, 1H), 7.36–7.30 (m, 2H), 7.27 (d, J = 8.2 Hz, 1H), 7.08 (t, J = 7.3 Hz, 1H), 3.96 (s, 3H), 3.86 (s, 3H), 3.07–3.01 (m, 2H), 2.29 (s, 3H), 1.62–1.42 (m, 5H), 1.36–1.25 (m, 3H), 1.11–0.85 (m, 1H) and 0.84–0.78 (m, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 152.3, 142.5, 134.8, 125.8, 125.1, 124.8, 123.3, 121.8, 119.5, 118.5, 108.8, 99.2, 56.3, 36.6, 34.6, 32.8, 32.0, 29.7, 28.8, 27.6, 19.4, 12.1 and 11.6 ppm.
IR (ATR): 2929, 2857, 2389, 1471, 1416, 1273, 1220, 1152, 1104, 838 and 741 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C23H32NO 338.2478; found 338.2469 (3.0 ppm).
TLC: Rf = 0.18 (hexane).
:1 hexanes/EtOAc) provided the desired phenol 15b (18.2 mg, 0.05 mmol, 91% yield) as a gummy compound.1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 7.6 Hz, 1H), 7.33 (d, J = 7.4 Hz, 1H), 7.26 (s, 2H), 7.07 (t, J = 7.2 Hz, 1H), 3.95 (s, 3H), 3.08–2.96 (m, 2H), 2.31 (s, 3H), 1.63–1.40 (m, 5H), 1.28 (s, 2H) and 1.16–1.08 (m, 2H), 0.81–0.80 (m, 6H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 148.1, 141.3, 135.4, 127.9, 126.0, 124.6, 123.5, 122.3, 121.2, 111.2, 110.3, 103.1, 39.2, 32.9, 31.7, 30.3, 28.7, 28.1, 27.4, 22.8, 12.0 and 11.2 ppm.
IR (ATR): 3326, 2922, 2862, 2396, 1469, 1296, 1245, 1077 and 743 cm−1.
HRMS (ESI) m/z: [M + NH4]+ calcd for C22H33N2O 341.2587; found 341.2633 (13.4 ppm).
TLC: Rf = 0.4 (9:1, hexanes/EtOAc).
:1 hexanes/EtOAc) provided the desired N-methyl carbazoquinocin D 4 (14.2 mg, 0.04 mmol, 89% yield) as a dark-brown glittering solid.1H NMR (500 MHz, CDCl3) δ 8.12–7.99 (m, 1H), 7.21–7.14 (m, 3H), 3.82 (s, 2H), 2.63 (t, J = 8.1 Hz, 2H), 1.84 (s, 3H), 1.57–1.41 (m, 4H), 1.33–1.24 (m, 3H), 1.15–1.05 (m, 2H) and 0.88–0.74 (m, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 183.3, 173.7, 144.9, 142.6, 139.6, 134.5, 125.8, 124.9, 121.9, 113.8, 110.8, 36.4, 34.5, 33.1, 29.9, 29.6, 28.7, 27.5, 19.3, 11.9 and 11.5 ppm.
IR (ATR): 2971, 2934, 2893, 1688, 1652, 1632, 1494, 1441, 1414, 1398, 1242 and 765 cm−1.
HRMS (ESI) m/z: [M + Na]+ calcd for C22H28NO2 338.2115; found 338.2116 (0.3 ppm).
TLC: Rf = 0.3 (8:1 hexanes/EtOAc).
M.P.: 148–150 °C.
:1) as the eluent.1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 7.5 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.32 (dd, J1 = 7.0 Hz and J2 = 8.0 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 7.08 (dd, J1 = 7.5 Hz and J2 = 1.0 Hz, 1H), 6.94 (d, J = 8.0 Hz, 1H), 5.27–5.18 (m, 2H), 3.94 (s, 3H), 3.03–3.00 (m, 2H), 2.60–2.58 (m, 1H), 2.41 (s, 3H), 2.17 (q, J = 7.5 Hz, 2H), 1.65–1.62 (m, 2H) and 0.89 (q, J = 2.5 Hz, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 142.3, 140.0, 138.7, 134.6, 126.5, 125.3, 124.0, 123.2, 122.8, 122.5, 119.7, 119.0, 117.7, 108.7, 32.7, 31.7, 28.1, 27.7, 26.7, 23.3 and 20.2 ppm.
IR (ATR): 2917, 1486, 1485, 1325, 1235 and 756 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C22H28N 306.2216; found 306.2212 (1.3 ppm).
TLC: Rf = 0.4 (19:1 hexane/EtOAc).
:1 hexanes/EtOAc) provided the desired hydrogenation product 12c (37 mg, 0.12 mmol, 93%) as a colorless liquid.1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 7.5 Hz, 1H), 7.85 (dt, J1 = 7.5 Hz, J2 = 2.0 Hz, 1H), 7.45 (t, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.23–7.20 (m, 1H), 7.07 (d, J = 7.5 Hz, 1H), 4.08 (s, 3H), 3.14–3.13 (m, 2H), 2.53 (s, 3H), 1.72–1.69 (m, 2H), 1.56–1.53 (m, 1H), 1.52–1.51 (m, 2H), 1.43–1.41 (m, 2H) and 1.27–1.24 (m, 2H), 0.92–0.91 (m, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 142.3, 140.0, 134.5, 125.3, 124.3, 123.3, 122.8, 122.5, 119.6, 118.9, 117.6, 108.7, 39.2, 32.6, 31.7, 30.4, 28.5, 28.1, 27.4, 22.8 and 20.2 ppm.
IR (ATR): 3426, 2986, 2843, 1642, 1531, 1479, 1413, 1372, 1324, 1253, 1026, 866 and 732 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C22H30N 308.2373; found 308.2324 (16.0 ppm).
TLC: Rf = 0.4 (hexane).
:1 hexanes/EA) provided the desired product 13c (38 mg, 0.10 mmol, 91%) as a gummy compound.1H NMR (500 MHz, CDCl3) δ 8.14 (s, 1H), 7.96 (d, J = 7.5 Hz, 1H), 7.47 (dd, J1 = 7.5 Hz and J2 = 1.0 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.21 (dd, J1 = 8.0 Hz and J2 = 7.0 Hz, 1H), 4.00 (s, 3H), 3.14–3.11 (m, 2H), 2.58 (s, 3H), 1.67–1.65 (m, 2H), 1.58–1.52 (m, 1H), 1.51–1.49 (m, 2H), 1.42–1.38 (m, 2H) and 1.26–1.23 (m, 2H), 0.91 (d, J = 6.5 Hz, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 142.6, 139.1, 133.0, 126.0, 126.1, 123.8, 122.2, 121.6, 119.8, 119.3, 116.9, 108.9, 39.1, 32.8, 31.7, 30.3, 29.6, 28.1, 27.4, 22.8 and 19.8 ppm.
IR (ATR): 2923, 2857, 2396, 1462, 1406, 1274, 1134, 1056, 829, 762 and 746 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C22H29NBr 386.1478; found 386.1432 (12 ppm).
TLC: Rf = 0.5 (hexane).
:1 hexanes/EtOAc) provided the desired 3-methoxycarbazole 14c (31 mg, 0.09 mmol, 93%) as a yellowish liquid.1H NMR (500 MHz, CDCl3) δ 7.97 (d, J = 7.5 Hz, 1H), 7.4–7.40 (m, 2H), 7.35 (d, J = 8 Hz, 1H), 7.16 (dd, J1 = 7 Hz and J2 = 7.5 Hz, 1H), 4.04 (s, 3H), 3.94 (s, 3H), 3.12 (t, 2H), 2.37 (s, 3H), 1.55–1.51 (m, 2H), 1.52–1.50 (m, 3H), 1.39–1.31 (m, 2H), 1.26–1.21 (m, 2H) and 0.88 (d, J = 6.5 Hz, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 152.4, 142.5, 134.9, 125.8, 125.1, 124.8, 123.3, 121.8, 119.5, 118.5, 108.8, 99.3, 56.3, 39.2, 32.8, 31.7, 30.4, 28.8, 28.1, 27.4, 22.8 and 12.1 ppm.
IR (ATR): 2922, 2879, 2362, 1471, 1424, 1288, 1212, 1144, 1109, 834 and 732 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C23H32NO 338.2478; found 338.2443 (10.3 ppm).
TLC: Rf = 0.2 (hexane).
:1 hexanes/EtOAc) provided the desired phenol 15c (25.5 mg, 0.08 mmol, 95% yield) as a white gummy compound.1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 8.0 Hz, 1H), 7.41 (dd, J1 = 8.0 Hz and J2 = 7.0 Hz, 1H), 7.34–7.33 (m, 2H), 7.14 (dd, J1 = 7.5 Hz and J2 = 7.0 Hz, 1H), 4.03 (s, 3H). 3.12 (t, 2H), 2.39 (s, 3H), 1.67–1.64 (m, 2H), 1.58–157 (m, 1H), 1.54–1.50 (m, 2H), 1.39–1.37 (m, 2H), 1.26–1.21 (m, 2H) and 0.88 (d, J = 6.5 Hz, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 148.1, 141.3, 135.4, 127.9, 126.0, 124.6, 123.5, 122.3, 121.2, 111.2, 110.3, 103.1, 39.2, 32.9, 31.7, 30.3, 28.7, 28.1, 27.4, 22.8 and 12.2 ppm.
IR (ATR): 3345, 2934, 2859, 2359, 2346, 1485, 1449, 1277, 1236, 929, 782 and 734 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C22H30NO 324.2322; found 324.2278 (13.5 ppm).
TLC: Rf = 0.2 (9:1 hexane/EtOAc).
:1 hexanes/EtOAc) provided the desired N-methyl carbazoquinocin E 5 (6.6 mg, 0.02 mmol, 91% yield) as a dark-brown glittering solid.1H NMR (500 MHz, CDCl3) δ 8.09–8.08 (m, 1H), 7.20–7.20 (m, 3H), 3.84 (s, 3H), 2.66–2.64 (m, 2H), 1.88 (s, 3H), 1.58–1.52 (m, 3H), 1.47 (d, J = 6.6 Hz, 1H), 1.43–1.37 (m, 2H), 1.33–1.28 (m, 2H), 1.17–1.12 (m, 2H) and 0.81 (d, J = 6.5 Hz, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 183.4, 173.8, 145.0, 142.6, 139.7, 134.6, 125.9, 124.9, 122.0, 113.9, 110.7, 39.0, 33.1, 30.2, 30.0, 28.5, 28.1, 27.2, 22.7 and 12.0 ppm.
IR (ATR): 2952, 2934, 2863, 1658, 1647, 1635, 1482, 1463, 1434, 1372, 1234 and 762 cm−1.
HRMS (ESI) m/z: [M + Na]+ calcd for C22H27NNaO2 360.1934; found 360.1964 (8.3 ppm).
TLC: Rf = 0.4 (4:1 hexane/EtOAc).
M.P.: 156–158 °C.
:1) as the eluent.1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 7.5 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.33 (dd, J1 = 8.5 Hz and J2 = 7.0 Hz, 1H), 7.24–7.23 (m, 1H), 7.09 (dd, J1 = 7.5 Hz and J2 = 7.0 Hz, 1H), 6.95 (d, J = 8.0 Hz, 1H) 5.45–5.38 (m, 2H), 3.96 (s, 3H), 3.02 (dd, J1 = 8.5 Hz and J2 = 9.0 Hz, 2H) 2.41 (s, 3H), 2.16 (dd, J1 = 7.0 Hz and J2 = 7.0 Hz, 2H), 1.89 (t, J = 6.5 Hz, 2H), 1.70–1.61 (m, 2H), 1.60–1.51 (m, 1H) and 0.83 (d, J = 6.5 Hz, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 142.2, 139.9, 134.6, 129.8, 129.6, 125.3, 124.0, 123.2, 122.7, 122.4, 119.6, 118.9, 117.7, 108.7, 36.6, 32.7, 31.5, 28.8, 28.1, 27.7, 22.6 and 20.2 ppm.
IR (ATR): 2917, 1465, 12463, 1330, 1245, 1096, 826 and 736 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C23H30N 320.2373; found 320.2341 (10.0 ppm).
TLC: Rf = 0.4 (19:1 hexane/EtOAc).
:1 hexane/EtOAc) gave the desired hydrogenation product 12d (46 mg, 0.14 mmol, 95.4%) as a colorless gummy compound.1H NMR (500 MHz, CDCl3) δ 8.01 (d, J = 7.5 Hz, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.43 (dd, J1 = 8.0 Hz and J2 = 7.5 Hz, 1H), 7.36 (d, J = 8.5, 1H), 7.19 (dd, J1 = 7.0 Hz and J2 = 7.0 Hz, 1H), 7.05 (d, J = 8.0 Hz, 1H), 4.07 (s, 3H), 3.11 (dd, J1 = 8.0 Hz and J2 = 8.5 Hz, 2H), 2.5 (s, 3H), 1.72–1.65 (m, 2H), 1.51 (t, J = 6.7 Hz, 2H), 1.41–1.36 (m, 2H), 1.36–1.30 (m, 3H), 1.22–1.16 (m, 2H) and 0.86 (d, J = 6.5, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 142.3, 140.0, 134.5, 125.3, 124.3, 123.3, 122.8, 122.5, 119.6, 119.0, 117.6, 108.7, 39.2, 32.7, 31.7, 30.2, 29.9, 28.5, 28.1, 27.5, 22.8 and 20.2 ppm.
IR (ATR): 3413, 2928, 2855, 1648, 1595, 1429, 1413, 1372, 1324, 1256, 1049, 862 and 734 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C23H32N 322.2529; found 322.2499 (9.3 ppm).
TLC: Rf = 0.45 (hexane).
:1 hexanes/EA) provided the desired product 13d (53 mg, 0.13 mmol, 93%) as a gummy compound.1H NMR (500 MHz, CDCl3) δ 8.14 (s, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.46 (dd, J1 = 8.0 Hz and J2 = 7.0 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.22 (t, J = 7.5 Hz, 1H), 3.9 (s, 3H), 3.11 (dd, J1 = 8.0 Hz and J2 = 8.5 Hz, 2H), 2.58 (s, 3H), 1.66 (dd, J1 = 10.5 Hz and J2 = 6.0 Hz, 2H), 1.55 (dt, J1 = 14.9 Hz and J2 = 7.0 Hz, 3H), 1.40 (dd, J1 = 9.9 Hz and J2 = 4.5 Hz, 2H), 1.37–1.33 (m, 2H), 1.26–1.19 (m, 2H) and 0.91 (d, J = 6.5 Hz, 6H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 142.5, 139.1, 132.9, 126.0,126.0, 123.7, 122.1, 121.6, 119.8, 119.3, 116.2, 108.9, 39.2, 32.7, 31.7, 30.0, 29.9, 29.6, 28.1, 27.5, 22.8 and 19.8 ppm.
IR (ATR): 2961, 2856, 2377, 1462, 1402, 1276, 1132, 1012, 819, 774 and 735 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C23H31BrN 400.1634; found 400.1636 (0.5 ppm).
TLC: Rf = 0.5 (hexane).
:1 hexanes/EtOAc) provided the desired 3-methoxycarbazole 14d (32 mg, 0.09 mmol, 91%) as a colorless liquid.1H NMR (400 MHz, CDCl3) δ 8.0 (d, J = 7.6 Hz, 1H), 7.44–7.41 (m, 2H), 7.37–7.36 (m, 1H), 7.17 (dd, J1 = 7.2 Hz and J2 = 7.6 Hz, 1H), 4.0 (s, 3H), 3.95 (s, 3H), 3.13 (dd, J1 = 8.0 Hz and J2 = 8.0 Hz, 2H), 2.83 (s, 3H), 1.68 (q, J = 8.2 Hz, 2H), 1.57–1.49 (m, 3H), 1.36 (q, J = 8.3 Hz, 4H), 1.21 (t, J = 7.1 Hz, 2H) and 0.89 (d, J = 6.4, 6H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 152.2, 142.4, 134.7, 125.8, 125.1, 124.7, 123.2, 121.8, 119.5, 118.4, 108.8, 99.1, 56.2, 39.2, 32.8, 31.7, 30.1, 29.9, 28.8, 28.1, 27.5, 22.8 and 12.1 ppm.
IR (ATR): 2935, 2853, 2384, 1466, 1419, 1293, 1220, 1149, 1129, 844 and 765 cm−1.
HRMS (ESI) m/z: [M + Na]+ calcd for C24H33NNaO 374.2454; found 374.2486 (8.5 ppm).
TLC: Rf = 0.4 (19:1 hexane/EtOAc).
:1 hexanes/EtOAc) provided the desired phenol 15d (26.2 mg, 0.07 mmol, 93% yield) as a gummy compound.1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 7.6 Hz, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.34 (s, 2H), 7.14 (dd, J1 = 7.2 Hz and J2 = 7.6 Hz, 1H), 4.03 (s, 3H), 3.11 (dd, J1 = 8.0 Hz and J2 = 8.4 Hz, 2H), 2.39 (s, 3H), 1.67 (t, J = 7.8 Hz, 2H), 1.56–1.49 (m, 3H), 1.35 (dt, J1 = 16.1 Hz and J2 = 7.4 Hz, 4H), 1.22–1.17 (m, 2H) and 0.88 (d, J = 6.4, 6H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 146.6, 141.5, 133.9, 124.6, 124.2, 121.6, 121.3, 121.0, 118.5, 117.3, 107.6, 101.9, 38.0, 31.6, 30.6, 29.0, 28.7, 27.6, 26.9, 26.4, 21.6 and 11.0 ppm.
IR (ATR): 3342, 2934, 2863, 2372, 2351, 1492, 1469, 1297, 1270, 902, 776 and 734 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C23H32NO 338.2478; found 338.2485 (2.0 ppm).
TLC: Rf = 0.3 (9:1 hexane/EtOAc).
:1 hexanes/EtOAc) provided the desired N-methyl carbazoquinocin F 6 (20.6 mg, 0.06 mmol, 94% yield) as a dark-brown glittering solid.1H NMR (400 MHz, CDCl3) δ 8.06–8.05 (m, 1H), 7.18 (s, 3H), 3.82 (s, 3H), 2.62 (dd, J1 = 6.8 Hz and J2 = 8.0 Hz, 2H), 1.84 (s, 3H), 1.54–1.48 (m, 2H), 1.47–1.38 (m, 3H), 1.27 (s, 4H), 1.14–1.07 (m, 2H) and 0.80 (d, J = 6.4 Hz, 6H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 183.3, 173.6, 144.9, 142.7, 139.6, 134.5, 125.8, 124.9, 121.9, 113.8, 110.8, 77.5, 77.2, 76.8, 39.0, 33.1, 30.0, 29.9, 29.7, 28.4, 28.3, 28.1, 27.4, 22.8 and 11.9 ppm.
IR (ATR): 2961, 2934, 2846, 1661, 1655, 1642, 1489, 1472, 1424, 1368, 1262 and 742 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C23H30NO2 352.2271; found 352.2248 (6.5 ppm).
TLC: Rf = 0.4 (4:1 hexane/EtOAc).
M.P.: 169–171 °C.
1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 7.7 Hz, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.36 (t, J = 7.6 Hz, 1H), 7.29 (d, J = 8.2 Hz, 1H), 7.12 (t, J = 7.4 Hz, 1H), 6.98 (d, J = 7.8 Hz, 1H), 4.13 (q, J = 7.1 Hz, 2H), 4.00 (s, 3H), 3.16–2.97 (m, 2H), 2.59 (dd, J1 = 13.4 Hz and J2 = 7.1 Hz, 1H), 2.43 (s, 3H), 1.96–1.94 (m, 1H), 1.76–1.66 (m, 1H) and 1.28–1.18 (m, 6H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 176.5, 142.2, 139.9, 134.7, 125.4, 123.1, 123.1, 122.8, 122.5, 119.7, 119.0, 117.9, 108.7, 60.6, 40.2, 35.3, 32.7, 26.2, 20.1, 17.5 and 14.5 ppm.
IR (ATR): 2944, 1738, 1580, 1453, 1218, 1199, 1153, 1127, 1026, 925, 827 and 749 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C23H32N 324.1958; found 324.1988 (9.2 ppm).
TLC: Rf = 0.6 (9:1 hexane/EtOAc).
1H NMR (500 MHz, CDCl3) δ 7.92 (d, J = 7.7 Hz, 1H), 7.74 (d, J = 7.9 Hz, 1H), 7.34 (t, J = 7.8 Hz, 1H), 7.25 (d, J = 8.3 Hz, 1H), 7.11 (d, J = 7.5 Hz, 1H), 6.96 (d, J = 7.9 Hz, 1H), 3.95 (s, 3H), 3.53–3.41 (m, 2H), 3.12–2.94 (m, 2H), 2.41 (s, 3H), 1.76–1.56 (m, 2H), 1.52 (s, 1H), 1.42 (dt, J1 = 13.3 Hz and J2 = 5.8 Hz, 1H) and 1.00 (d, J = 6.6 Hz, 3H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 142.2, 139.9, 134.4, 125.3, 123.9, 123.1, 122.8, 122.5, 119.6, 118.9, 117.7, 108.7, 68.0, 36.5, 34.9, 32.6, 26.0 and 20.1, 16.7 ppm.
IR (ATR): 3360, 2944, 1738, 1580, 1453, 1218, 1199, 1153, 1127, 1026, 925, 827 and 749 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C19H23NO 281.1780; found 281.1792 (4.2 ppm).
TLC: Rf = 0.2 (9:1 hexane/EtOAc).
1H NMR (400 MHz, CDCl3) δ 9.57 (s, 1H), 7.89 (d, J = 7.6 Hz, 1H), 7.72 (d, J = 7.8 Hz, 1H), 7.32 (t, J = 7.5 Hz, 1H), 7.21 (d, J = 8.2 Hz, 1H), 7.09 (t, J = 7.2 Hz, 1H), 6.93 (d, J = 7.8 Hz, 1H), 3.87 (s, 3H), 3.07–2.90 (m, 2H), 2.37 (s, 4H), 1.88 (dd, J1 = 13.1 Hz and J2 = 5.6 Hz, 1H), 1.64–1.50 (m, 1H) and 1.12 (d, J = 7.1 Hz, 3H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 204.4, 142.2, 139.8, 134.5, 125.4, 123.0, 122.8, 122.5, 119.6, 119.0, 117.9, 108.7, 46.7, 32.6, 31.9, 25.7, 20.1 and 13.6 ppm.
IR (ATR): 2976, 2941, 1725, 1673, 1622, 1468, 1444, 1433, 1266, 842, 823 and 746 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C19H22NO 380.1696; found 380.1725 (8.0 ppm).
TLC: Rf = 0.4 (19:1 hexane/EtOAc).
:1) as the eluent.1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 7.6 Hz, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.30 (t, J = 7.1 Hz, 1H), 7.19 (d, J = 8.2 Hz, 1H), 7.11–7.03 (m, 1H), 6.92 (d, J = 7.6 Hz, 1H), 5.71 (dt, J1 = 17.0 Hz and J2 = 8.5 Hz, 1H), 4.96–4.62 (m, 2H), 3.86 (s, 3H), 3.04–2.82 (m, 2H), 2.36 (s, 3H), 2.29–2.16 (m, 1H), 1.53 (d, J = 6.6 Hz, 2H) and 1.01 (dd, J1 = 3.7 Hz and J2 = 2.9 Hz, 3H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 144.0, 142.2, 139.9, 134.5, 125.3, 123.9, 123.1, 122.7, 122.5, 119.6, 118.9, 117.6, 113.6, 108.6, 38.7, 38.2, 32.7, 26.2, 20.3 and 20.2 ppm.
IR (ATR): 2917, 1496, 1475, 1325, 1308, 1235, 842 and 738 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C20H26N 278.1903; found 278.1894 (3.2 ppm).
TLC: Rf = 0.4 (19:1 hexane/EtOAc).
:1 hexanes/EtOAc) provided the desired hydrogenation product 21 (44.5 mg, 0.16 mmol, 96%) as a colorless liquid.1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 7.4 Hz, 1H), 7.72 (dd, J1 = 7.8 Hz and J2 = 1.6 Hz, 1H), 7.31 (t, J = 7.6 Hz, 1H), 7.22 (d, J = 8.2 Hz, 1H), 7.08 (t, J = 6.4 Hz, 1H), 6.94 (d, J = 7.7 Hz, 1H), 3.91 (s, 3H), 3.08–2.87 (m, 2H), 2.39 (s, 3H), 1.64–1.53 (m, 1H), 1.51–1.31 (m, 3H), 1.23–1.11 (m, 1H), 0.94 (dd, J1 = 6.3 Hz and J2 = 1.6 Hz, 3H) and 0.84–0.81 (m, 3H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 142.2, 139.9, 134.4, 125.2, 124.4, 123.2, 122.7, 122.5, 119.6, 118.9, 117.5, 108.6, 38.5, 35.5, 32.6, 29.6, 26.3, 20.1, 19.3 and 11.7 ppm.
IR (ATR): 3423, 2932, 2889, 1651, 1532, 1449, 1431, 1372, 1334, 1243, 1089, 834 and 748 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C20H26N 280.2060; found 280.2063 (1.0 ppm).
TLC: Rf = 0.4 (hexane).
:1 hexanes/EtOAc) provided the corresponding 3-bromocarbazole 22 (45 mg, 0.12 mmol, 88%) as a colorless liquid.1H NMR (400 MHz, CDCl3) δ 7.99 (s, 1H), 7.82 (s, 1H), 7.31 (t, J = 7.6 Hz, 1H), 7.17 (d, J = 8.2 Hz, 1H), 7.07 (t, J = 7.4 Hz, 1H), 3.80 (s, 3H), 3.03–2.83 (m, 2H), 2.42 (s, 3H), 1.58–1.47 (m, 1H), 1.43 (dd, J1 = 12.2 Hz and J2 = 6.0 Hz, 1H), 1.37–1.27 (m, 2H), 1.15 (dt, J1 = 14.2 Hz and J2 = 7.3 Hz, 1H), 0.92 (d, J = 6.4 Hz, 3H) and 0.83 (t, J = 7.3 Hz, 3H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 142.4, 139.0, 132.8, 126.0, 126.0, 123.6, 122.1, 121.5, 119.8, 119.2, 116.8, 108.8, 38.4, 35.5, 32.7, 29.6, 27.3, 19.7, 19.2 and 11.7 ppm.
IR (ATR): 2969, 2869, 2374, 1463, 1416, 1291, 1186, 1055, 832, 768 and 749 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C20H25NBr 358.1165; found 358.1159 (2.0 ppm).
TLC: Rf = 0.5 (hexane).
:1 hexanes/EtOAc) provided the desired 3-methoxycarbazole 23 (29.6 mg, 0.09 mmol, 86%) as a pale-yellow liquid.1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 7.7 Hz, 1H), 7.32 (t, J = 7.4 Hz, 2H), 7.25 (d, J = 8.2 Hz, 1H), 7.09 (d, J = 7.3 Hz, 1H), 3.95 (s, 3H), 3.85 (s, 3H), 3.14–2.93 (m, 2H), 2.28 (s, 3H), 1.66–1.56 (m, 1H), 1.53–1.44 (m, 2H), 1.42–1.37 (m, 1H), 1.20 (dd, J1 = 13.7 Hz and J2 = 6.7 Hz, 1H), 0.97 (d, J = 6.4 Hz, 3H) and 0.86 (t, J = 7.3 Hz, 3H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 152.3, 142.4, 134.8, 125.9, 125.1, 124.6, 123.2, 121.8, 119.5, 118.4, 108.8, 99.1, 56.2, 38.4, 35.6, 32.8, 29.7, 26.6, 19.3, 11.9, and 11.7 ppm.
IR (ATR): 2935, 2862, 2388, 1476, 1413, 1278, 1229, 1164, 1122, 861 and 736 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C21H28NO 310.2165; found 310.2143 (7.0 ppm).
TLC: Rf = 0.2 (hexane).
:1 hexanes/EtOAc) provided phenol 24 (18 mg, 0.06 mmol, 70% yield) as a light-yellow gummy compound.1H NMR (500 MHz, CDCl3) δ 7.92 (d, J = 7.6 Hz, 1H), 7.42 (t, J = 7.9 Hz, 1H), 7.39–7.31 (m, 2H), 7.16 (d, J = 6.5 Hz, 1H), 4.69 (s, 1H), 4.04 (s, 3H), 3.13–3.01 (m, 2H), 2.40 (s, 3H), 1.69 (dd, J1 = 10.7 Hz and J2 = 5.6 Hz, 1H), 1.63–1.56 (m, 1H), 1.55–1.43 (m, 2H), 1.33–1.25 (m, 1H), 1.07 (dd, J1 = 6.7 Hz and J2 = 2.7 Hz, 3H) and 0.96 (t, J = 7.4 Hz, 3H) ppm.
13C[1H] NMR (125 MHz, CDCl3) δ 147.8, 142.7, 135.1, 125.8, 125.4, 122.8, 122.4, 122.2, 119.7, 118.4, 108.7, 103.1, 38.5, 35.5, 32.8, 29.7, 26.6, 19.3, 12.0 and 11.7 ppm.
IR (ATR): 3325, 2933, 2854, 2362, 2346, 1485, 1448, 1272, 1236, 921, 776 and 731 cm−1.
HRMS (ESI) m/z: [M + NH4]+ calcd for C20H29N2O 313.2274; found 313.2246 (9.0 ppm).
TLC: Rf = 0.3 (9:1 hexane/EtOAc).
:1 hexanes/EtOAc) provided the desired N-methyl carbazoquinocin A 7 (14 mg, 0.04 mmol 90% yield) as a dark-brown glittering solid.1H NMR (400 MHz, CDCl3) δ 8.13–8.11 (m, 1H), 7.25 (s, 3H), 3.88 (s, 3H), 2.81–2.57 (m, 2H), 1.90 (s, 3H), 1.50 (td, J1 = 13.2 Hz and J2 = 6.5 Hz, 2H), 1.43 (d, J = 5.2 Hz, 1H), 1.29–1.23 (m, 2H), 1.02 (d, J = 6.2 Hz, 3H) and 0.94 (t, J = 7.3 Hz, 3H) ppm.
13C[1H] NMR (100 MHz, CDCl3) δ 183.2, 173.7, 144.9, 143.0, 139.6, 134.5, 125.8, 124.9, 121.9, 113.8, 110.8, 35.4, 35.0, 33.1, 29.5, 27.9, 19.0, 11.8 and 11.6 ppm.
IR (ATR): 2966, 2957, 2853, 1662, 1658, 1636, 1492, 1471, 1454, 1379, 1235 and 776 cm−1.
HRMS (ESI) m/z: [M + H]+ calcd for C20H24NO2 310.1802; found 310.1807 (2.0 ppm).
TLC: Rf = 0.35 (4:1 hexane/EtOAc).
M.P.: 149–151 °C.
Abbreviations
| Pd/C | Palladium on carbon |
| wt% | Weight% |
| atm | Atmosphere |
| NBS | N-Bromosuccinimide |
| DIBAL-H | Diisobutylaluminium hydride |
| n-BuLi | n-Butyllithium |
| equiv. | Equivalents |
| Cat. | Catalytic amount |
| M | Molar |
| anhyd. | Anhydrous |
| mL | Microliter |
| rt | Room temperature |
| TLC | Thin layer chromatography |
| Rf | Retardation or retention factor |
| NaHCO3 | Sodium bicarbonate |
| Na2SO4 | Sodium sulfate |
| dr | Diastereomeric ratio |
| NMR | Nuclear magnetic resonance spectroscopy |
| IR | Infrared spectroscopy |
| ATR | Attenuated total reflection |
| HRMS (ESI) | High-resolution electrospray ionization mass spectrometry |
| Calcd | Calculated |
| M.P. | Melting point |
| ppm | Parts per million |
| nBu | n-Butyl group |
| atm | Atmosphere |
Data availability
The data underlying this study are available in the published article and its ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the Department of Chemistry, IIT Madras, for infrastructure and facilities. BB, and DR thanks IIT Madras for HTRA doctoral fellowship. The financial support from SERB-DST (through CRG/2023/001303 grant) is highly acknowledged.References
- (a) S. Issa, A. Prandina, N. Bedel, P. Rongved, S. Yous, M. L. Borgne and Z. Bouaziz, J. Enzyme Inhib. Med. Chem., 2019, 34, 1321–1346 CrossRef CAS PubMed; (b) R. Hegden, B. D. Emmanuel, J. Beevi and S. S. Dharan, Int. J. Pharma Sci. Res., 2020, 12, 1271–1277 Search PubMed; (c) L. S. Tsutsumi, D. Gündisch and D. Sun, Curr. Top. Med. Chem., 2016, 16, 1290–1313 CrossRef CAS PubMed; (d) A. Głuszyńska, Eur. J. Med. Chem., 2015, 94, 405–426 CrossRef PubMed; (e) A. A. Pieper, S. L. McKnight and J. M. Ready, Chem. Soc. Rev., 2014, 43, 6716–6726 RSC.
- (a) M. Manickam, P. Iqbal, M. Belloni, S. Kumar and J. A. Preece, Isr. J. Chem., 2012, 52, 917–934 CrossRef CAS; (b) K. Krzysztof and M. Lapkowski, J. Solid State Electrochem., 2015, 19, 2601–2610 CrossRef; (c) H. J. Jiang, J. Sun and J. L. Zhang, Curr. Org. Chem., 2012, 16, 2014–2025 CrossRef CAS; (d) K. Mullen and U. Scherf, Organic Light-Emitting Devices: Synthesis, Properties and Applications, Wiley-VCH, Weinheim, 2006 Search PubMed; (e) Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC; (f) J. Li and A. Grimsdale, Chem. Soc. Rev., 2010, 39, 2399–2410 RSC.
- (a) H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4428 CrossRef PubMed; (b) H.-J. Knölker, Curr. Org. Synth., 2004, 1, 309–331 CrossRef; (c) H.-J. Knölker, Chem. Lett., 2009, 38, 8–13 CrossRef; (d) W. Maneerat, T. Ritthiwigrom, S. Cheenpracha, T. Promgool, K. Yossathera, S. Deachathai, W. Phakhodee and S. Laphookhieo, J. Nat. Prod., 2012, 75, 741–746 CrossRef CAS PubMed; (e) C. Graebe and C. Glazer, Ber. Dtsch. Chem. Ges., 1872, 5, 12–15 Search PubMed; (f) A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 112, 3193–3328 CrossRef CAS PubMed.
- (a) T. Choshi, T. Sada, H. Fujimoto, C. Nagayama, E. Sugino and S. Hibino, J. Org. Chem., 1997, 62, 2535–2543 CrossRef CAS PubMed; (b) H.-J. Knölker and T. Hopfmann, Tetrahedron, 2002, 58, 8937–8945 CrossRef; (c) M. Rawat and W. D. Wulff, Org. Lett., 2004, 6, 329–332 CrossRef CAS PubMed; (d) H.-J. Knölker and W. Fröhner, Tetrahedron Lett., 1997, 38, 1535–1538 CrossRef; (e) H.-J. Knölker, K. R. Reddy and A. Wagner, Tetrahedron Lett., 1998, 39, 8267–8270 CrossRef; (f) H.-J. Knölker, W. Fröhner and K. R. Reddy, Synthesis, 2002, 557–564 CrossRef; (g) K. Schneider, J. Nachtigall, A. Hanchen, G. Nicholson, M. Goodfellow, D. R. Sussmuth and P. H. Fiedler, J. Nat. Prod., 2009, 72, 1768–1772 CrossRef CAS PubMed; (h) R. Sussmuth and A. Hanchen, Synlett, 2009, 2483–2486 CrossRef.
- For the isolation of carbazoquinocins A–F, see: M. Tanaka, K. Shinya, K. Furihata and H. Seto, J. Antibiot., 1995, 48, 326–328 CrossRef CAS PubMed.
- For recent reviews, see: (a) U. Pindur, Chimia, 1990, 44, 406–412 CrossRef CAS; (b) J. Bergman and B. Pelcman, Pure Appl. Chem., 1990, 62, 1967–1976 CrossRef CAS; (c) H.-J. Knölker, Synlett, 1992, 371–387 CrossRef; (d) T. Kawasaki and M. Sakamoto, J. Indian Chem. Soc., 1994, 71, 443–457 CAS; (e) C. J. Moody, Synlett, 1994, 681–688 CrossRef CAS; (f) H.-J. Knölker, Advances in Nitrogen Heterocycles, ed. Moody, C. J., JAI Press, Greenwich, CT, 1995, vol. 1, pp. 173–204 Search PubMed; (g) S. Hibino and E. Sugino, in Advances in Nitrogen Heterocycles, ed. Moody, C. J., JAI Press, Greenwich, CT, 1995, vol. 1, pp. 205–227 Search PubMed.
- (a) For the previous synthesis of carbazoquinocins A–F see: K. Shin and K. Ogasawara, Synlett, 1996, 922–924 CrossRef CAS; (b) T. Choshi, T. Sada, H. Fujimoto, C. Nagayama, E. Sugino and S. Hibino, J. Org. Chem., 1997, 62, 2535–2543 CrossRef CAS PubMed; (c) M. Rawat and W. D. Wulff, Org. Lett., 2004, 6, 329–332 CrossRef CAS PubMed; (d) D. Roy, P. Tharra and B. Baire, Chem. Commun., 2023, 58, 10210–10213 RSC.
- All five phosphonium salts RPPh3Br are prepared following the literature procedure. The reaction between corresponding alkyl bromides (R-Br) with triphenylphosphine (PPh3) in refluxing toluene for 48 h gave the phosphonium salts. For more details see ESI† and Homogeneous and Heterogeneous Pd-Catalyzed Selective C-P Activation and Transfer Hydrogenation for “Group-Substitution” Synthesis of Trivalent Phosphines. M. Lei, X. Chen, Y. Wang, L. Zhang, H. Zhu and Z. Wang, Org. Lett., 2022, 24, 2868–2872 CrossRef CAS PubMed.
- E. Khaskin and D. Milstein, Chem. Commun., 2015, 51, 9002–9005 RSC.
- J. Hu, T. Lan, Y. Sun, H. Chen, J. Yao and Y. Rao, Chem. Commun., 2015, 51, 14929–14932 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05347h |
| This journal is © The Royal Society of Chemistry 2024 |