DOI:
10.1039/D4RA05382F
(Paper)
RSC Adv., 2024,
14, 31954-31965
Chitosan quaternary ammonium salt-oxidized sodium alginate-glycerol-calcium ion biobased self-healing hydrogels with excellent spontaneous repair performance†
Received
25th July 2024
, Accepted 26th September 2024
First published on 8th October 2024
Abstract
Self-healing hydrogels have attracted wide attention because of their potential applications in various fields. However, the complex processes, environmental requirements, and insufficient functionality limit their practical application. Herein, we synthesized a chitosan quaternary ammonium salt-oxidized sodium alginate-glycerol-calcium ion (HACC-OSA-Gly-Ca2+) biobased hydrogel with a multi-network structure that exhibits excellent self-healing abilities. This was achieved by utilizing reversible dynamic imine bonding, electrostatic interactions, Ca2+ ions as crosslinking points, and hydrogen bonding. The oxidation of sodium alginate (SA) with sodium periodate was carried out to obtain oxidized sodium alginate (OSA) with varying oxidation degrees. The resulting OSAs were then introduced into a glycerol–water solvent system containing chitosan quaternary ammonium salt (HACC) and calcium chloride, and this reaction successfully prepared the biobased eco-friendly self-healing hydrogel. The impacts of the oxidation degree (OD) of OSA on the microscopic morphology, mechanical properties, viscoelastic properties, swelling properties, and self-healing properties of the corresponding synthetic hydrogels were investigated. The outcomes indicated that the optimal HACC-OSA-Gly-Ca2+ hydrogel possessed good mechanical properties, with a tensile stress of 0.0132 MPa and elongation at break of 551.38%. Furthermore, the multiple bond interactions led to a high self-healing ratio (100%), with an elongation at break of about 614.29%, and excellent adhesion ability (average peel strength of 6.38 kN m−1) on various substrates. Additionally, the composite hydrogels exhibited excellent water retention, thermal stability, and resilience, making them promising for various potential applications. Moreover, the properties of the composite hydrogels could be facilely and finely tuned by varying the oxidation degree of OSA and ratio of each component. Thus, the presented strategy could enrich the construction as well as application of biopolymer-based self-healing hydrogels.
Introduction
Hydrogels with flexible and suitable performance have been widely applied in sensors,1,2 drug delivery,3,4 wound healing,5,6 and so on. However, the ordinary hydrogel has obvious shortcomings; when the hydrogel is damaged or broken by external force, it cannot heal. This leads to the short service life of hydrogel materials and limits the application scenarios.7,8 Many research efforts were focused on self-healing hydrogels, which are a promising material and good substitute for the traditional hydrogel.9–11 Duan et al.12 synthesized galactomannan self-healing hydrogels and demonstrated that the self-healing properties could extend the service life of the hydrogel material. Wang et al.13 designed and synthesized a water-soluble chitosan–hyaluronic acid self-healing injectable hydrogel that can be used as a substitute for vitreous material. Research on self-healing hydrogels is generally divided into externally assisted self-healing hydrogels and intrinsically self-healing hydrogels, depending on the preparation method, with or without an additional initiator or restorer. Fang et al.14 placed repair agents (e.g., microcapsules) into the microporous structure of a hydrogel material, and when the hydrogel was broken, the repair agent was released to repair the damaged part. However, the introduction of the repair agent affects the mechanical properties of the hydrogel and does not allow the material to be repaired many times. Currently, most studies are on intrinsically self-healing hydrogels. He et al.15 prepared alginate-based cyclodextrin/azo polyacrylamide interpenetrating composite hydrogels with self-healing properties under visible or UV light irradiation by using a host–guest recognition mechanism. Wang et al.16 prepared oxidized sodium alginate–hydrazide modified polyethylene glycol hydrogels by Schiff base reaction with realistic reversible sol–gel transition under pH induction. Self-healing hydrogel materials can spontaneously repair their broken parts due to the dynamic reversible covalent bonding17–20 or non-covalent bonding interactions21,22 that are introduced inside the gel to confer reversible interactions. Common reversible dynamic covalent bonds mainly include imine bonds (Schiff base bonds), acyl hydrazone bonds, disulfide bonds, borate bonds, etc. Common reversible physical crosslinks are reversible non-covalent bonding interactions, including hydrogen, ionic, electrostatic, hydrophobic, host–guest, and other interactions.
In recent years, there has been growing interest in utilizing natural polymers to prepare hydrogel materials due to their excellent biocompatibility and biodegradability. Some commonly used natural polymers include chitosan (CS), sodium alginate (SA), guar gum, cellulose, pectin, and other polysaccharides. Additionally, proteins such as collagen and gelatin are also frequently employed as hydrogel materials. These natural polymers offer a wide range of advantages, including their availability, low cost, and ability to mimic the extracellular matrix (ECM) of biological tissues. They have shown promise in various applications, including tissue engineering, drug delivery, wound healing, and biomedical coatings.23–27 Chitosan quaternary ammonium salt (HACC) is a chitosan derivative whose quaternary ammonium salt group well solves the water solubility problem of chitosan.28,29 Sodium alginate is a natural polymer known for its excellent biocompatibility.30 One way to modify SA is by oxidizing a portion of its hydroxyl groups through a ring-opening reaction, resulting in the formation of oxidized sodium alginate (OSA) that contains aldehyde functional groups.16 The aldehyde group present in OSA can participate in a reversible dynamic covalent bond formation with the amino group of another molecule, such as chitosan.31
Single-polysaccharide hydrogels typically suffer from issues related to inadequate gel strength, poor mechanical properties, limited resilience, and inadequate self-healing properties.32 To address these issues, researchers have devised various optimization methods, such as utilizing double network (DN) structure hydrogels, interpenetrating polymer network (IPN) hydrogels, and nanocomposite (NC) hydrogels.33–35 These approaches aim to improve the mechanical properties and self-healing ability of single-polysaccharide hydrogels. DN and IPN hydrogels could achieve this by combining two or more networks with contrasting properties, thereby improving the overall properties of the hydrogel. On the other hand, NC hydrogels incorporate nanoparticles into the polysaccharide matrix, which can lead to enhanced mechanical strength and self-healing properties. Ai et al.36 designed a dual-network self-healing hydrogel based on xylan, polyvinyl ethanol (PVA), and borax (B) with double physical crosslinking. Compared to pure PVA/B hydrogel, the resulting hydrogel demonstrated significantly increased strength (approximately 81.00 kPa), greater rebound rate after compression (about 79.00%), and a faster self-healing rate (achieving an 85.80% repair rate within 30 seconds). These findings suggest the potential of this dual-network self-healing hydrogel for various applications in fields such as medicine and engineering. He et al.15 prepared a kind of hydrogel by using an interpenetrating network of cyclodextrin/azo polyacrylamide in a sodium alginate base. Based on the host–guest interaction mechanism, the hydrogel demonstrated self-healing ability under light exposure. These results highlight the potential of this hydrogel for applications in fields such as tissue engineering and drug delivery. Zhong et al.37 obtained ionic nanocomposite physical hydrogels whose mechanical properties can be easily improved through the addition of VSNP, Fe3+, monomer, and water. These findings suggest the potential of this approach for tailoring the mechanical properties of hydrogels for various applications in fields such as biomedicine and tissue engineering. Currently, the focus of research on self-healing hydrogels remains on double network structures, with few studies focusing on triple-network or multi-network structures. Therefore, this study aims to synthesize multi-network-structured self-healing hydrogels to broaden our understanding of the self-healing properties of hydrogels.
In this paper, we developed a novel method for preparing a self-healing hydrogel using chitosan quaternary ammonium salt (HACC) and oxidized sodium alginate (OSA) as raw materials. Glycerol, sodium chloride solution, and water were used as solvents, and calcium chloride was introduced to synthesize a multi-network hydrogel with spontaneous repair performance at room temperature. This hydrogel, named chitosan quaternary ammonium salt-oxidized sodium alginate-glycerol-calcium ion (HACC-OSA-Gly-Ca2+) hydrogel, exhibited excellent self-repair properties. Oxidation of sodium alginate yields OSA with an aldehyde group, which was allowed to react with the quaternary ammonium salt of chitosan with a partial amino group in a Schiff base reaction to form imine bonds. In the glycerol/water system, OSA can form multiple intermolecular hydrogen bonds. The presence of sodium chloride, which has a uniform distribution of positive and negative charges, facilitated tighter electrostatic interactions between HACC and OSA. Moreover, Ca2+ ions can form secondary crosslinks with the carboxylate ions of OSA. The structures of the hydrogels were characterized by FTIR, 1H NMR, SEM, and EDS. Additionally, the impact of different oxidation degrees (0%, 10%, 30%, 50%) of OSA on the microscopic morphology, thermal stability, mechanical strength, viscoelasticity, swelling, and self-healing ability of the composite hydrogels were evaluated. We expect to explore a new type of gentle stimuli-triggered, biopolymer-based, self-healing hydrogel with tunable mechanical strength, facile preparation, excellent stretching ability, and resilience.
Materials and methods
Materials
Sodium alginate (SA; AR, 90%) and sodium periodate (NaIO4; AR, 95%) were purchased from Shanghai Macklin Chemical Technology, Ltd (China). Chitosan quaternary ammonium salt (HACC; degree of substitution 90%) was purchased from Shanghai Yuanye Biotechnology, Ltd (China). Glycerol (AR) was purchased from Changde Bikeman Biotechnology, Ltd (China). Calcium chloride (AR) was purchased from Tianjin Baishi Chemical Technology, Ltd (China). Ethylene glycol (AR) and anhydrous ethanol (AR) were purchased from Tianjin Yongda Chemical Technology, Ltd (China). All other chemicals were AR grade and used without further purification.
Preparation of oxidized sodium alginate (OSA)
The method used for preparing oxidized sodium alginate is as follows.38 Briefly, NaIO4 was dissolved in deionized water, and then the NaIO4 aqueous solution was kept under stirring in a black bottle for 24 h. Then, 5.00 mL ethylene glycol was added under stirring for 1 h. In order to neutralize the excess periodate, the oxidated alginate was purified by adding anhydrous ethanol. The polymer was again dissolved in water, then ethylene glycol was added to reprecipitate it three times. Finally, the white polymer was isolated and dried at −20 °C under vacuum. The ratios of sodium alginate and NaIO4 are shown in Table 1.
Table 1 The ratio of sodium alginate and NaIO4
| Sample |
SA/g |
NaIO4/g |
| 10%(OD)OSA |
1.00 |
0.57 |
| 30%(OD)OSA |
1.00 |
0.86 |
| 50%(OD)OSA |
1.00 |
1.14 |
Preparation of self-healing hydrogels
First, glycerol (0.10% w/w) was added into a 5.00 mL aqueous solution containing 0.50 M NaCl and 0.10 M CaCl2, then 15.00 g of chitosan quaternary ammonium salt was dissolved into the aqueous solution. The HACC-Gly-Ca2+ aqueous solution was obtained and allowed to rest overnight in order to reduce bubbles. Next, 1.00 g of OSA with different oxidation was dissolved into the aqueous solution containing 0.50 M NaCl to prepare 10.00% wt aqueous OSA. Finally, the OSA and HACC aqueous solutions were mixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and then poured into a square mold; thus the self-healing hydrogel was obtained after 12 h.
:1 ratio and then poured into a square mold; thus the self-healing hydrogel was obtained after 12 h.
Fourier transform infrared spectroscopy
The vacuum-dried OSA, SA, HACC, and HACC-OSA samples were prepared using a KBr pellet method. The four samples were evaluated by Fourier transform infrared spectroscopy (Spectrum 100, PerkinElmer, USA). The measurements were carried out at room temperature over the wavenumbers ranging from 500 to 4000 cm−1. Besides, the hydrogel of HACC-SA-Gly-Ca2+, HACC-10%(OD)OSA-Gly-Ca2+, HACC-30%(OD)OSA-Gly-Ca2+, and HACC-50%(OD)OSA-Gly-Ca2+ were vacuum dried, and 5 mm × 5 mm thin slices were prepared for testing. The hydrogel samples were evaluated by attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR; Nicolet iS 10, Thermo Fisher, USA).
SEM analysis
To characterize the microstructure of the self-healing hydrogels, the freeze-dried hydrogel structures were observed by scanning electron microscopy (SEM). The sample was coated with gold first, and then scanned at 20.00 kV.
Nuclear magnetic resonance spectroscopy analysis
The NMR hydrogen spectra of SA, OSA, and HACC-OSA were determined using liquid NMR hydrogen spectroscopy (AVANCE 400, BRUKER Germany). The amount of test sample was greater than or equal to 50.00 mg, and it was firstly dissolved in deuterated reagent; the test conditions were set to a magnetic field of 400 MHz.
Mechanical property test
A testing machine was used to perform mechanical tests on the hydrogels with three different alginate oxidation degrees. In the compression experiment, a square hydrogel sample of 15 mm × 15 mm area and 10 mm height was used, and the compression strain was chosen to be 30%, 53%, 73%, and 80%, respectively. Three samples from each group were used in experiments to obtain an average value.
Adhesion strength test
In order to determine the adhesion strength of the hydrogel, a universal tensile test machine was used to apply four materials—wood, metal, glass and plastic—for adhesion and tensile tests, and three samples of each hydrogel type were used to obtain the average value.39
Thermogravimetric analysis
The thermal stability of HACC-SA-Gly-Ca2+, HACC-10%(OD)OSA-Gly-Ca2+, HACC-30%(OD)OSA-Gly-Ca2+, and HACC-50%(OD)OSA-Gly-Ca2+ hydrogel samples was analyzed using a comprehensive thermal analyzer (TGA5500, TA instruments, USA). The heating rate was set to 10 °C min−1, and the test temperature was 30–600 °C under nitrogen atmosphere. The TA and DTG curves were recorded synchronously.
Self-healing test
The four types of hydrogels were prepared as follows: long strips of the hydrogel samples (40 mm × 20 mm × 3 mm) were cut into two halves with a knife, and then the two halves of hydrogels were reassembled together at the cut. The samples were stored at room temperature in closed vessels (sealed with plastic bags to protect the Petri dishes) and left for a certain time (24 h/48 h/72 h) to observe the self-healing situation. The self-healed hydrogel strips were tested by tensile testing with an electronic universal testing machine. Each set of experiments was repeated for three parallel samples, and the average value was obtained. The self-healing rate of the hydrogel was calculated as R(ε) = εi/εh × 100%, where εi denotes the elongation at break of the repaired hydrogel, and εh denotes the elongation of the original hydrogel.
Swelling experiment
A certain amount of HACC-SA-Gly-Ca2+, HACC-10%(OD)OSA-Gly-Ca2+, HACC-30%(OD)OSA-Gly-Ca2+, and HACC-50%(OD)OSA-Gly-Ca2+ dry gel samples were placed in deionized water for a set period of time (2, 4, 6, 8, 12, 24, 36, 48, 60, 72, 84, 96, 120 h), and the excess water on the surface of the dry gel was suctioned off and weighed. Three samples were taken for each group of experiments to obtain the average value. The swelling ratio was calculated as follows: Sw = Mw/Md, where Mw indicates the weight of the hydrogel at a certain time, and Md indicates the weight of the dry gel.
Statistical analysis
Each experiment was performed in triplicate, and the results are expressed as mean ± standard deviation (SD). Statistical analysis was performed by using GraphPad 9 statistical software (GraphPad Software, San Diego, USA). We conducted one-way ANOVA followed by Dunnett's method to determine the significant differences (P < 0.05) between the experiment data.
Results and discussion
Preparation and structural characterization of the HACC-OSA-GLy-Ca2+ composite hydrogels
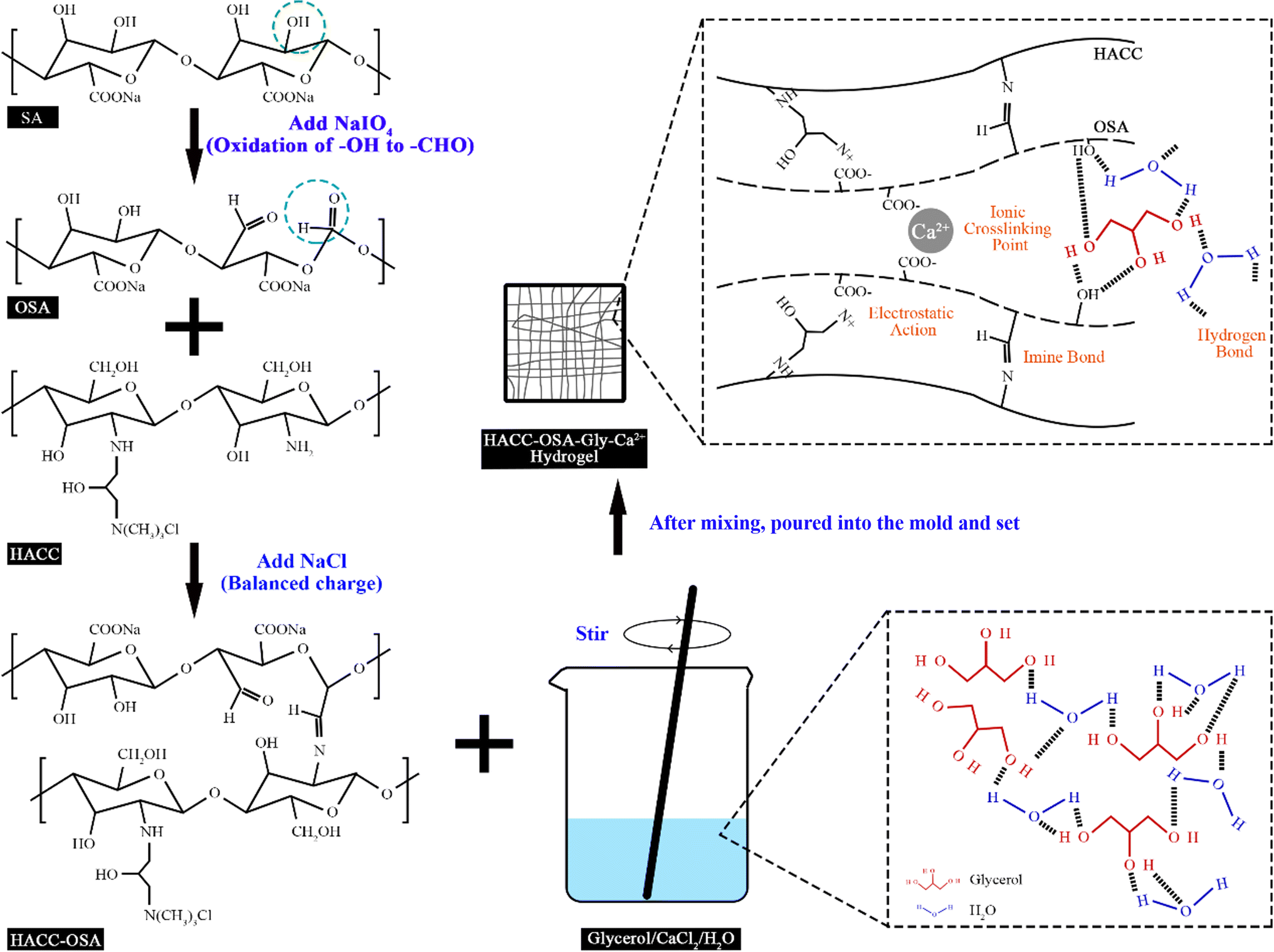
The reaction flow and mechanism of the chitosan quaternary ammonium salt-oxidized sodium alginate-glycerol-calcium ion (HACC-OSA-Gly-Ca2+) self-healing hydrogel is shown in Fig. 1. The aldehyde group was obtained after the oxidation of sodium alginate, which could react with the amino group on the chitosan quaternary ammonium salt molecular chain to form a reversible dynamic covalent bond, and there were non-covalent interactions as well. These bond interactions included the electrostatic interaction between the quaternary ammonium salt group on HACC and the carboxylic acid root of OSA, the hydrogen bond between OSA and glycerol and water, and the ionic bond cross-linking between Ca2+ and OSA. Thus, the self-healing hydrogel system was established through multiple bond interactions. This unique property inspired the design and fabrication of a biobased self-healing hydrogel with multiple network structures. Herein, it would be interesting to make use of various bond properties to integrate the automatic self-healing ability into one system to obtain multifunctional self-healing hydrogels.
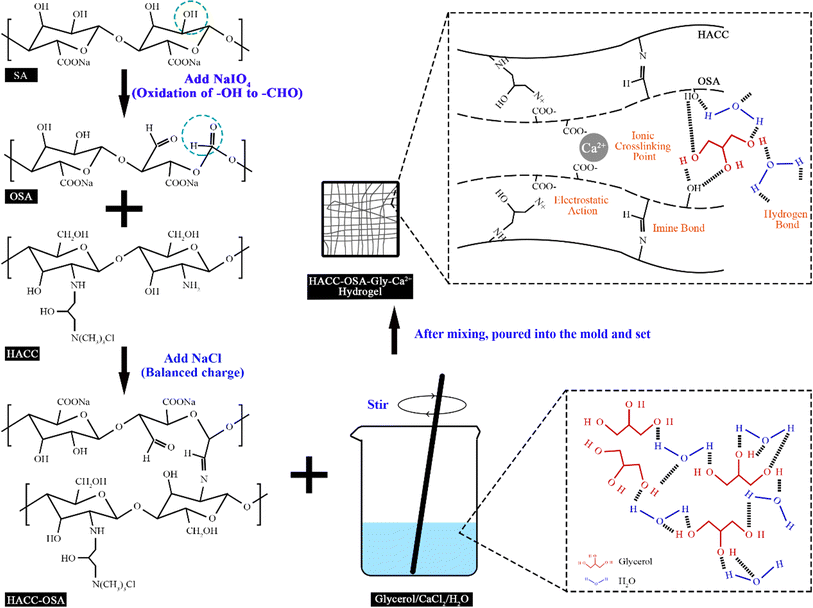 |
| | Fig. 1 Schematic illustration of the preparation process and mechanism of HACC-OSA-Gly-Ca2+ self-healing hydrogel. | |
To determine the dispersion of components in the matrix, FTIR characterization of the ingredients and hydrogels was performed (Fig. 2a–c). A broad absorption peak in the range around 3559 cm−1 was observed in Fig. 2a, which was the stretching vibration peak exhibited by the hydroxyl group (–OH) of sodium alginate (SA) and oxidized sodium alginate (OSA). Compared with the SA spectrogram, the characteristic peak at 1596 cm−1 clearly appeared in the OSA spectrum, which was the stretching vibration absorption peak of the aldehyde group (–CHO), proving the successful synthesis of oxidized sodium alginate. With the increase of oxidation degree, the characteristic peak at 1596 cm−1 became longer, which also indicated the increase of aldehyde groups formed after the oxidation reaction. As shown in Fig. 2b, for the hydrogel prepared with 50% (OD)OSA, the bending vibrational peak presented by the amino group (–NH2) on the chitosan quaternary ammonium salt disappeared at 3435 cm−1. Compared with the spectrograms of oxidized sodium alginate, the HACC-OSA, HACC-OSA-Gly, and HACC-OSA-Gly-Ca2+ at 1640 cm−1 showed the absorption peak of the stretching vibration of the imine bond, while the absorption peak of the aldehyde group at 1596 cm−1 was attenuated. Fig. 2(c) shows the FTIR spectra of the hydrogels prepared from oxidized sodium alginate with different oxidation degrees, and it was obvious that there was no difference in the characteristic peaks for these four kinds of hydrogels. To further verify the successful chemical modification, 1H NMR spectroscopy was carried out. In Fig. 2d, the proton absorption peak at 4.65 ppm for the solvent heavy water is shown as a baseline. The proton absorption peak at 3.55–3.95 ppm was observed for sodium alginate, where the primary hydroxyl group in SA was oxidized to carboxyl groups to form glyoxylate. Compared to the hydrogen spectrum of SA, the overlapping peaks at 4.10–5.20 ppm on the hydrogen spectrum of OSA were the proton absorption peaks of H6–H7 on the oxidized glyoxylate. Because of the use of acidic oxidant (sodium periodate), the aldehyde group (aldehyde) on oxidized sodium alginate and the hydroxyl group (alcohol) easily underwent nucleophilic addition reaction to form hemiacetal, so the proton absorption peaks of hemiacetal appeared at 5.37 ppm and 5.64 ppm. The combined infrared spectra and hydrogen spectra demonstrated the successful preparation of oxidized sodium alginate. As shown in Fig. 2e, the proton absorption peak at 3.55–3.95 ppm disappeared, which was the electrostatic interaction of the aldehyde glycolate on OSA with the quaternary ammonium group of HACC. The 3.16 ppm peak of the HACC-OSA spectrum was the proton absorption peak of the aldimine (R–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–R′). Taken together, this proved that the amino group of chitosan quaternary ammonium salt formed imine bonds with the aldehyde group of oxidized sodium alginate.
N–R′). Taken together, this proved that the amino group of chitosan quaternary ammonium salt formed imine bonds with the aldehyde group of oxidized sodium alginate.
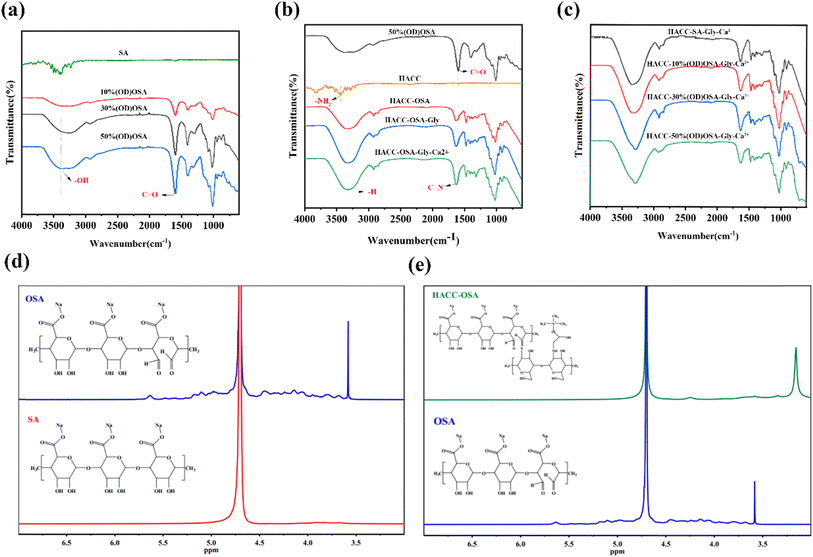 |
| | Fig. 2 (a–c) FTIR spectra of (a) SA, 10%(OD)OSA, 30%(OD)OSA, and 50%(OD)OSA; (b) hydrogels of 50(OD)%OSA, HACC, HACC-50%(OD)OSA, HACC-50%(OD)OSA-Gly, and HACC-50%(OD)OSA-Gly-Ca2+; (c) and the self-healing hydrogels of HACC-SA-Gly-Ca2+, HACC-10%(OD)OSA-Gly-Ca2+, HACC-30%(OD)OSA-Gly-Ca2+, HACC-50%(OD)OSA-Gly-Ca2+. (d) and (e) The 1H NMR spectra of (d) SA and OSA; (e) HACC-OSA and OSA. | |
The microstructures of the four dry gel samples were observed in cross section by scanning electron microscopy, and the SEM images of HACC-OSA-Gly-Ca2+ hydrogels with different oxidation degrees are shown in Fig. 3. From the SEM images, it can be seen that the HACC-OSA-Gly-Ca2+ hydrogels showed a porous reticular structure with interconnected pores. It was clearly found that the number of pores in the network of HACC-OSA-Gly-Ca2+ hydrogel gradually decreased, and the network structure became tighter, as the oxidation degree of oxidized sodium alginate increased. This was because the aldehyde group of OSA increased and the cross-linking density with HACC increased, which brought the molecular chains inside the gel closer together and appear to have thicker network lines. Therefore, after freeze-drying to remove all the water, as the oxidation degree of OSA increased, the appearance of the hydrogel appeared thicker accordingly (Fig. 3a–e). It was also observed that the pore structure of the hydrogel became more complex, thus implying a denser structure of the hydrogel, which was due to the formation of multi-network structure and the strong bonding interactions. The internal structure of the hydrogel had obvious micropores, and Ca2+ was introduced to serve as an ionic cross-linking point to cross-link its internal molecular network more tightly, as shown in Fig. 3f, where Ca2+ is small and presented a sparse distribution. Besides, Ca2+ and Na+ were detected by elemental quantitative analysis (Fig. 3g and h), which proved the successful mixing and crosslinking.
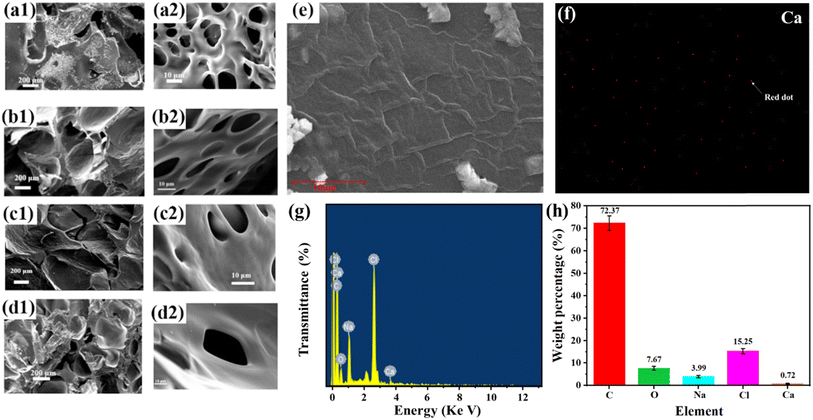 |
| | Fig. 3 SEM images of (a) HACC-SA-Gly-Ca2+, (b) HACC-10%(OD)OSA-Gly-Ca2+, (c) HACC-30%(OD)OSA-Gly-Ca2+, and (d) HACC-50%(OD)OSA-Gly-Ca2+ hydrogels; (e) SEM image with local magnification of 8000× of the HACC-50%(OD)OSA-Gly-Ca2+ hydrogel; (f) calcium distribution, indicated by red dot; (g) EDS analysis diagram of HACC-50%(OD)OSA-Gly-Ca2+ hydrogel; (h) elemental quantitative analysis. | |
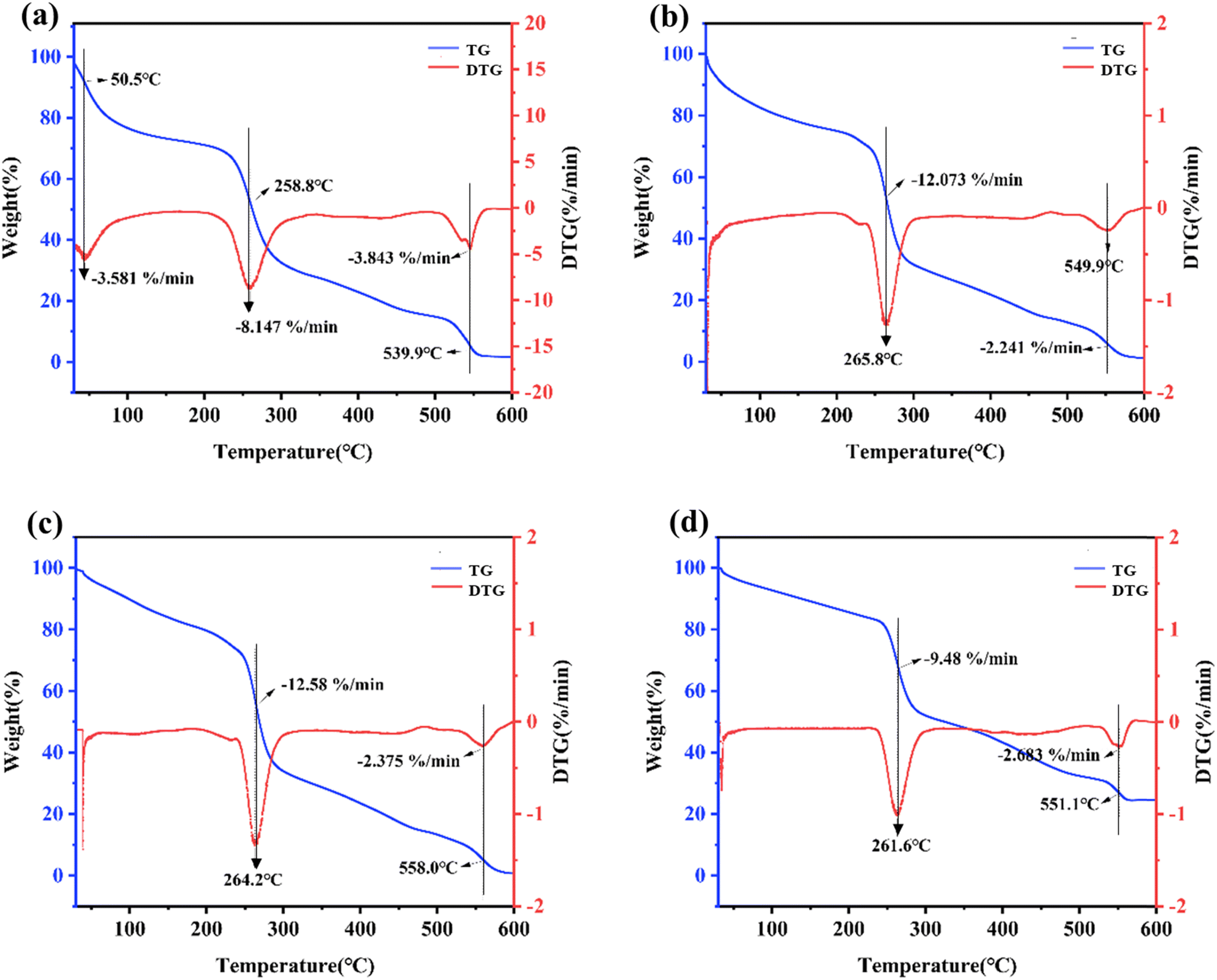
Fig. 4 depicts the TG and DTG curves of the four kinds of hydrogels. The red curve represents the thermogravimetric change of the hydrogels, while the blue curve represents the thermogravimetric differential change. The peak point indicates the temperature boundary at which the hydrogel underwent a rapid weight loss/gain step rate. As shown in Fig. 4a–d, the TG curves indicated that the residual mass of all four kinds of hydrogels at 600 °C was approximately 3% over the process from weight loss to stabilization. This was because the quaternary ammonium salt of chitosan and oxidized sodium alginate gradually decomposed and volatilized throughout the process. Glycerol had a boiling point of 290 °C, while the boiling point of sodium chloride is relatively high at 1465 °C, indicating that the remaining 3% residual mass is sodium chloride. HACC-SA-Gly-Ca2+ hydrogels exhibited three peaks within the tested temperature range, while the remaining three hydrogel types exhibited two peaks. As a result, the thermal stability of HACC-SA-Gly-Ca2+ hydrogels was inferior to that of the other three hydrogel types. At approximately 260 °C, the four hydrogel types displayed their respective peak values. Upon comparison of the TG and DTG curves, the HACC-50%(OD)OSA-Gly-Ca2+ hydrogel exhibited the lowest peak weight loss value, indicating superior thermal stability. These findings suggested that as the oxidation degree of oxidized sodium alginate increased, the crosslinking density of the hydrogel also increased, resulting in a tighter three-dimensional network structure that enhanced its thermal stability.
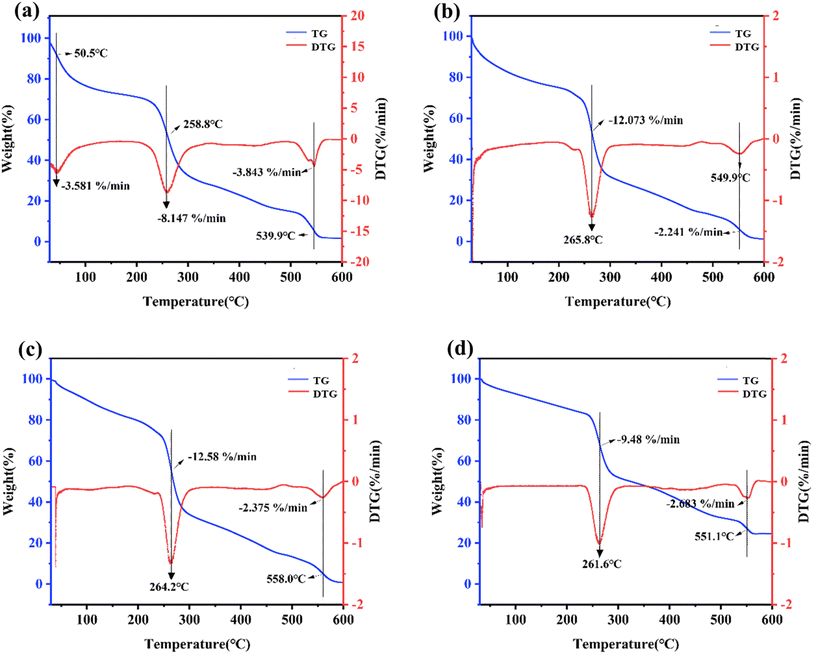 |
| | Fig. 4 TG and DTG curves of the four different kinds of hydrogels: (a) HACC-SA-Gly-Ca2+; (b) HACC-10%(OD)OSA-Gly-Ca2+; (c) HACC-30%(OD)OSA-Gly-Ca2+; (d) HACC-50%(OD)OSA-Gly-Ca2+. | |
Mechanical properties of the HACC-OSA-GLy-Ca2+ composite hydrogels
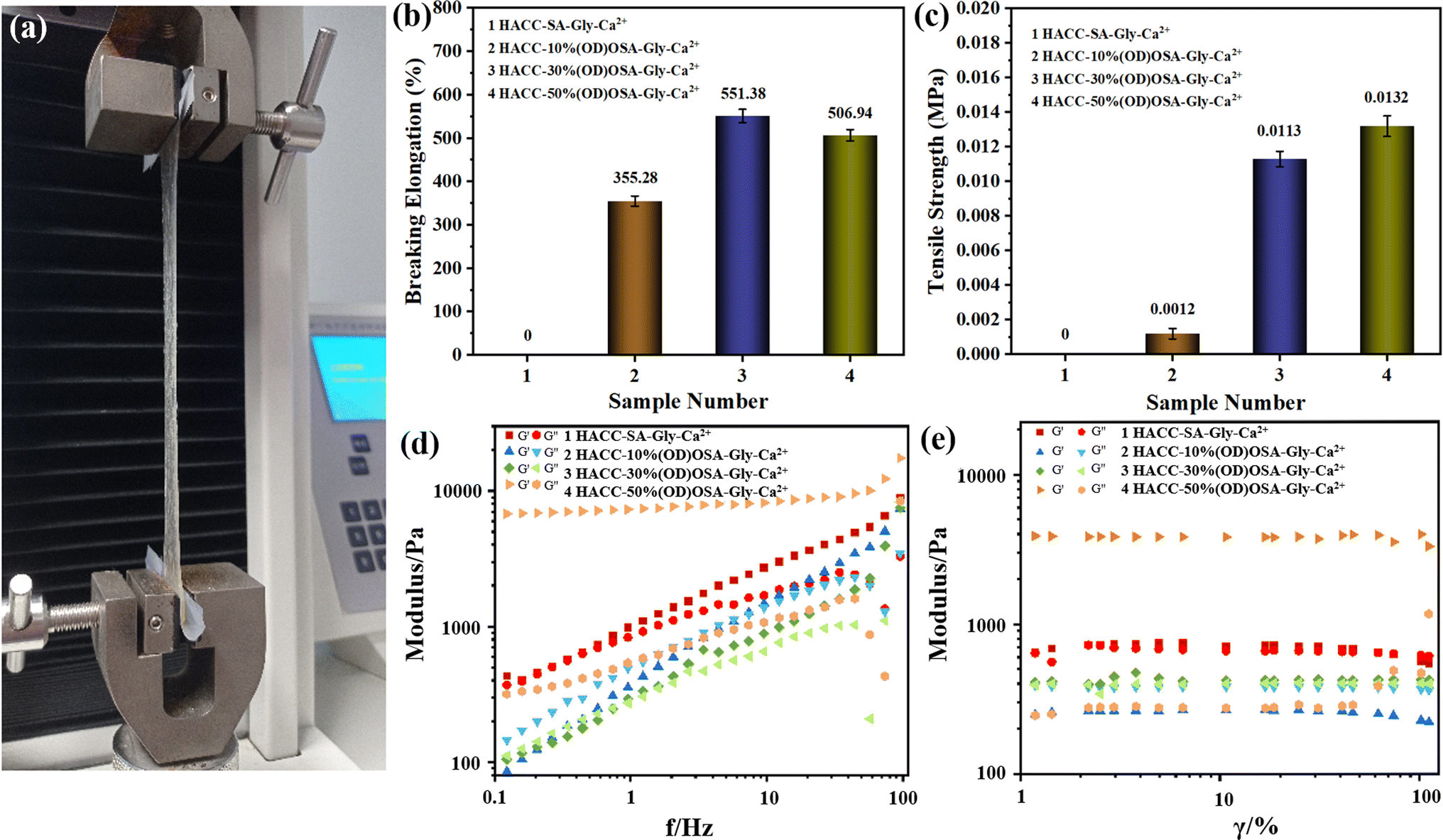
The mechanical properties of the hydrogels prepared by OSA with different oxidation degrees were tested in an electronic universal tensile testing machine (Fig. 5a). As seen in Fig. 5b, the elongation at break of sample 3 (HACC-30%(OD)OSA-Gly-Ca2+) was the largest, reaching 551.38%. It was followed by sample 4 (HACC-50%(OD)OSA-Gly-Ca2+) with 506.94%. However, because sample 1 (HACC-SA-Gly-Ca2+) was indeterminate and difficult to demold, the strength was low and difficult to stretch, and the elongation at break was 0. Fig. 5c shows the tensile strength of the hydrogels, and as the oxidation degree of OSA increased; the tensile strength of the corresponding synthesized hydrogels increased, where the hydrogel sample 4 (HACC-50%(OD)OSA-Gly-Ca2+) was the highest, being 0.0132 MPa. As shown in Fig. 5d and e, the viscoelastic properties of the self-healing hydrogels were investigated by rheological experiments. The curves show variation in the energy storage modulus (G′) and loss modulus (G′′) with strain for hydrogels prepared by OSA with different oxidation degrees. The G′ value of the HACC-SA-Gly-Ca2+ hydrogel was slightly higher than G′′ in a certain strain range, indicating that the hydrogel had a complete network structure and could withstand large strains. But when the strain reached 74%, the G′ value started to be less than G′′. The molecular structure of HACC-SA-Gly-Ca2+ hydrogel was deformed, and the hydrogel was transformed from gel-like to sol–gel-like. The G′ values of HACC-30%(OD)OSA-Gly-Ca2+ hydrogels were slightly larger than G′′ in the whole strain range, indicating the hydrogels had good elasticity. The G′ values of HACC-50%(OD)OSA-Gly-Ca2+ hydrogels were much larger than G′′, which indicated that the internal network structure was intact and the deformation was small, and the hydrogel was very elastic. On the contrary, the G′ value of HACC-10%(OD)OSA-Gly-Ca2+ hydrogel was smaller than G′′, which indicated that the hydrogel was sol–gel-like throughout the tested strain range, and there was no elasticity.
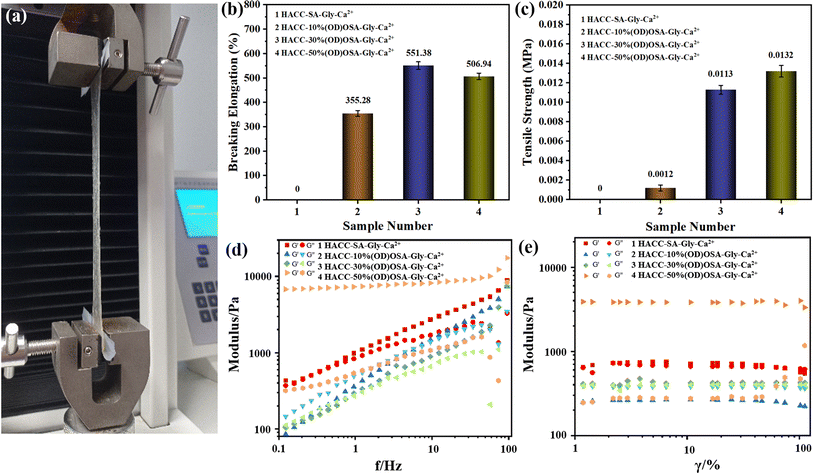 |
| | Fig. 5 (a) Test setup using the electronic universal tensile testing machine to test the mechanical properties of the four types of hydrogel: (b) elongation at break; (c) tensile strength; (d) curves of the energy storage modulus (G′) and loss modulus (G′′) with the change of frequency; (e) curves of the energy storage modulus (G′) and loss modulus (G′′) with the change of strain. | |
Self-healing ability of the HACC-OSA-GLy-Ca2+ composite hydrogels
From the macroscopic photos of the self-healing hydrogels (Fig. 6a–d), it is obvious that the spliced hydrogel, after a period of time (24 h/48 h/72 h), gradually became merged at the splice, and the color dispersed and moved to the colorless other end. The healed hydrogels were stretched vertically with an electronic universal tensile tester. As shown in Fig. 6e and f, the elongation at break and self-repair rate of the repaired HACC-30%(OD)OSA-Gly-Ca2+ and HACC-50%(OD)OSA-Gly-Ca2+ hydrogels increased and were the best as the healing time increased, and on the contrary, those for HACC-10%(OD) OSA-Gly-Ca2+ hydrogel gradually decreased. As could be seen from the data in Tables S1 and S2,† after 72 h of healing, the elongation of HACC-30%(OD) OSA-Gly-Ca2+ hydrogel was 614.29% with a self-healing rate of 100%, while the elongation of HACC-50%(OD) OSA-Gly-Ca2+ hydrogel was 417.59% with a self-healing rate of 94.41%. The self-healing rate of 100% was higher than other reports; the highest healing efficiency of sodium carboxymethyl cellulose-based hydrogel, prepared by Chen et al., was 90.50%.40 Meanwhile, the self-healing rate decreased from 92.14% at 24 h to 85.53% at 72 h for the self-healing HACC-10%(OD)OSA-Gly-Ca2+ hydrogel. To study the self-healing characteristics of the HACC-30%(OD)OSA-Gly-Ca2+ and HACC-50%(OD)OSA-Gly-Ca2+ hydrogels, these two hydrogels were subjected to alternating strain scanning (applied 1% vs. 100% strain). Fig. S1† shows that the storage modulus (G′) of the hydrogel was larger than the loss modulus (G′′) at a strain of 1%, indicating that the hydrogel is in the gel state. However, at an applied strain at 100%, G′ decreased sharply and became smaller than G′′, indicating the transition from gel-like to sol-gel-like. Upon applying 1% strain, the storage modulus (G′) was higher than the loss modulus (G′′) and changed immediately to its initial value, reforming the gel-like state, indicating that internal molecular bonds in the hydrogel were resynthesized, and the structure was reconstructed. After four cycles of low and high applied strain, the damage and healing were repeatable, which fully demonstrated that the hydrogel exhibited excellent self-healing properties. The hydrogel HACC-OSA-Gly-Ca2+ was spontaneously repaired from the fracture damage to healing, without the need for stimulation or restriction by external conditions, enabling convenient future applications.
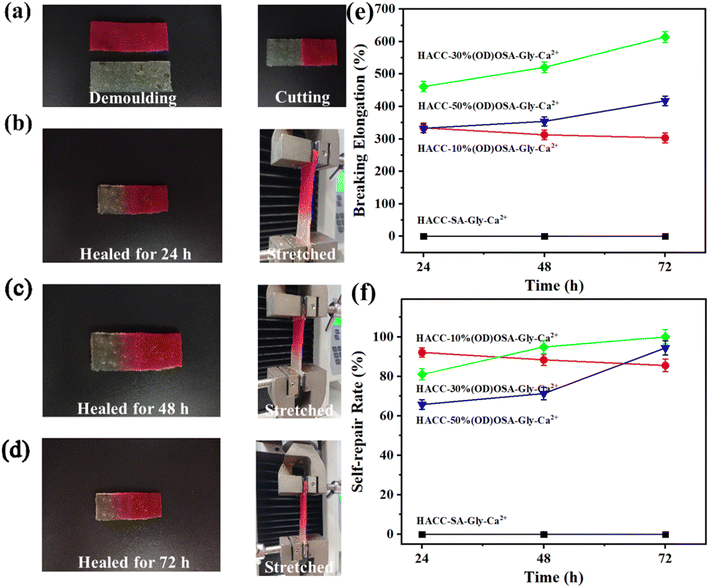 |
| | Fig. 6 (a)–(d) Self-healing process and tensile test of the HACC-50%(OD)OSA-Gly-Ca2+ hydrogel after self-healing; (e) elongation of four kinds of hydrogels after a period (72 h) of self-healing; (f) self-repair rate of four kinds of hydrogels after a period (72 h) of self-healing. | |
Swelling properties of the HACC-OSA-Gly-Ca2+ composite hydrogels
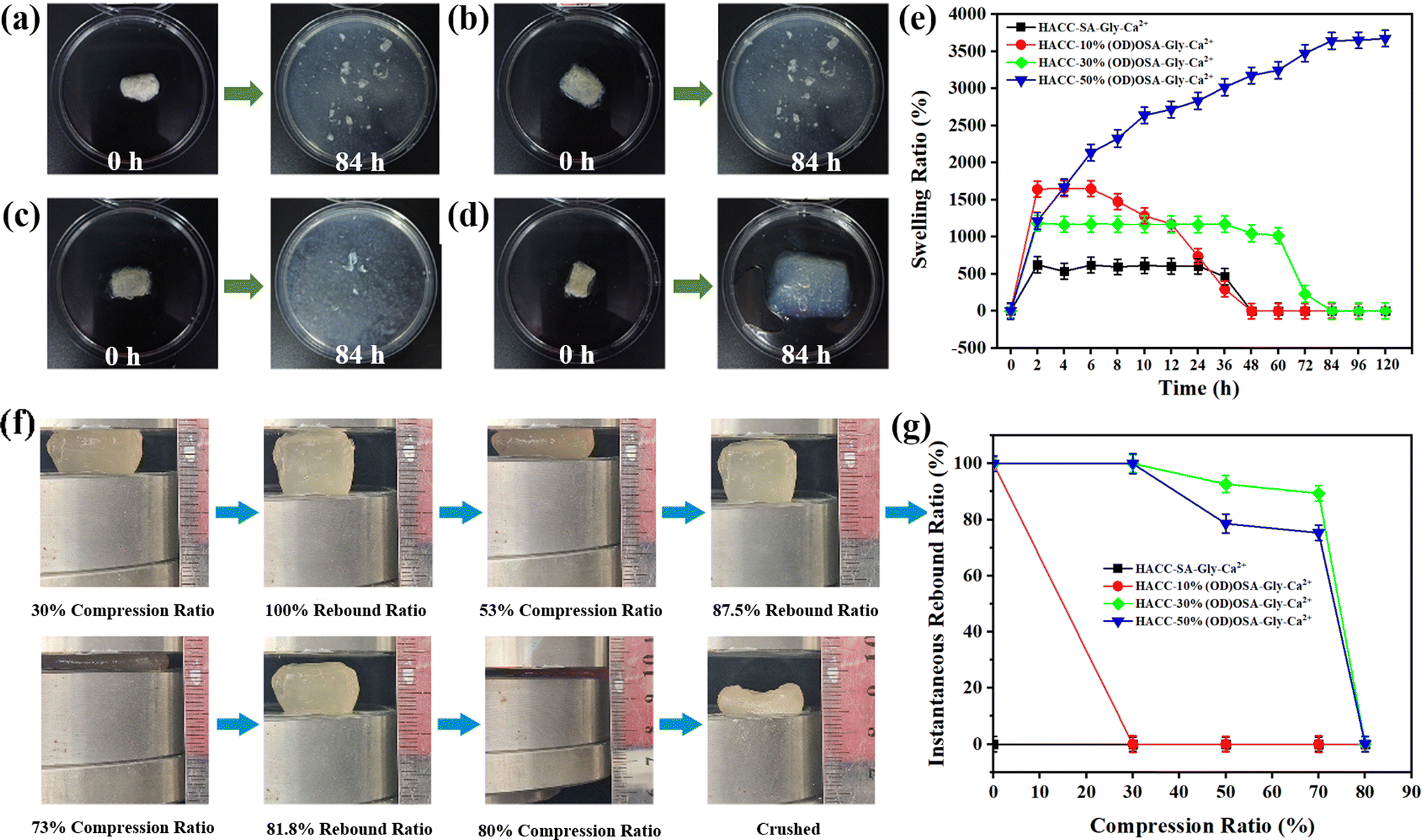
Fig. 7 illustrates the investigation into the impact of different oxidation degrees of OSA on the swelling properties of the hydrogels. Fig. 7e shows that the HACC-SA-Gly-Ca2+ and HACC-10%(OD)OSA-Gly-Ca2+ hydrogels reached their maximum swelling ratio after 2 h of immersion in water, while the HACC-30%(OD)OSA-Gly-Ca2+ hydrogel reached the maximum swelling ratio after 4 h. The swelling ratio of both HACC-SA-Gly-Ca2+ and HACC-10%(OD)OSA-Gly-Ca2+ hydrogels reached maximum at 4 h. However, at approximately 84 h, the internal structures of all three types of hydrogel were ruptured (Fig. 7a–c). In contrast, HACC-50%(OD)OSA-Gly-Ca2+ hydrogels exhibited a gradual increase in swelling ratio over time, reaching a maximum swelling ratio of 3643.00% after 84 h. An increase in the oxidation degree of oxidized sodium alginate resulted in an increase in the cross-linking degree of the gel molecular chain, which in turn led to an increase in the swelling ratio. This was because when the molecular chains were more tightly cross-linked, the pore size and voids in the structure of the hydrogel became smaller, while the network structure became thicker and stronger. As a result, the hydrogel was less likely to absorb water excessively, which reduced the chance of breakage due to swelling (Fig. 7d). To test the compression resistance of the hydrogels, we used a press to compress the hydrogels with varying oxidation degrees (Fig. 7f). The experimental results demonstrated that HACC-SA-Gly-Ca2+ and HACC-10%(OD)OSA-Gly-Ca2+ hydrogels exhibited low strength, lost their defined shape after compression, and did not rebound, indicating zero resilience and no recovery after compression. According to Fig. 7g, the HACC-30%(OD)OSA-Gly-Ca2+ hydrogel demonstrated superior resilience compared to the HACC-50%(OD)OSA-Gly-Ca2+ hydrogel when compressed by 30%, with both hydrogels exhibiting equal resilience. However, when compressed by 53%, the HACC-30%(OD)OSA-Gly-Ca2+ hydrogel achieved 87.50% resilience compared to the 75.02% resilience of HACC-50%(OD)OSA-Gly-Ca2+ hydrogel. Furthermore, when compressed by 73%, HACC-30%(OD)OSA-Gly-Ca2+ hydrogel restored 81.83% of its original length, while HACC-50%(OD)OSA-Gly-Ca2+ hydrogel only demonstrated 72.74% resilience. Both hydrogels exhibited irreversible crushing when compressed to 80%.
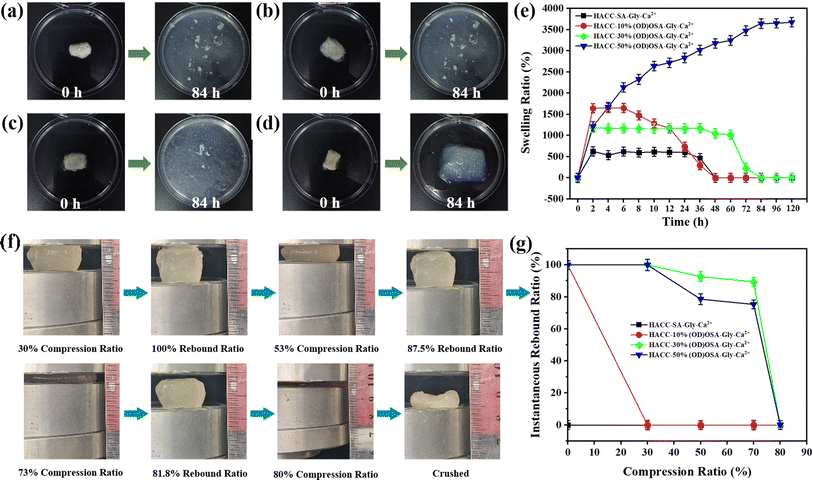 |
| | Fig. 7 (a) HACC-SA-Gly-Ca2+, (b) HACC-10%(OD)OSA-Gly-Ca2+, (c) HACC-30%(OD)OSA-Gly-Ca2+, and (d) HACC-50%(OD)OSA-Gly-Ca2+ dry gels were soaked for 84 h, respectively; (e) swelling rate curves of the four kinds of hydrogels; (f) the process of HACC-50%(OD)OSA-Gly-Ca2+ hydrogel compression experiment; (g) variation of compressive rebound of the four kinds of hydrogels. | |
Adhesion behavior of the HACC-OSA-GLy-Ca2+ composite hydrogels
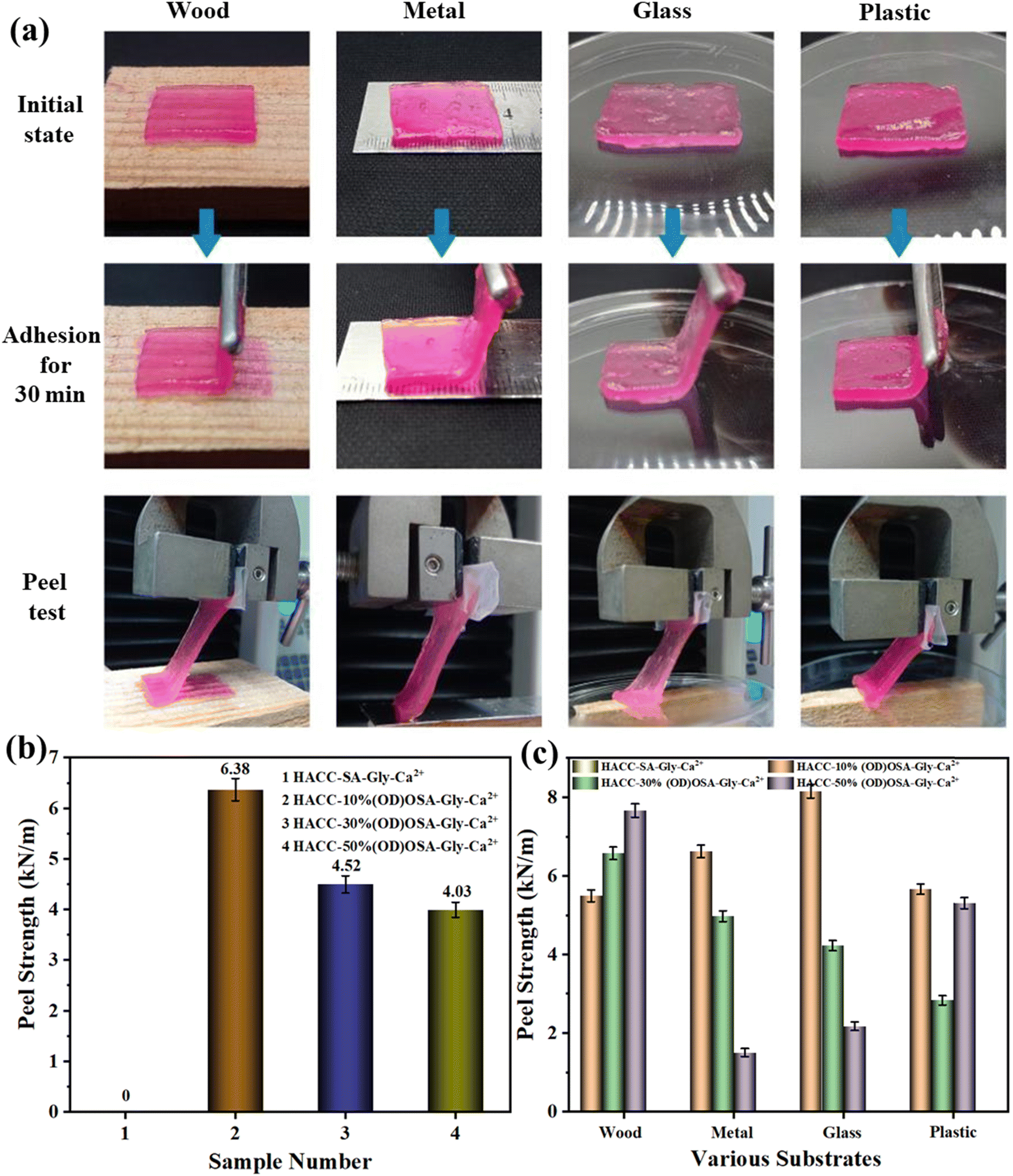
The peel strength of the hydrogel was assessed using an electronic universal testing machine, as illustrated in Fig. 8a. Due to its sol–gel state, HACC-SA-Gly-Ca2+ could not be tested for peel strength, as it was difficult to peel and tended to adhere easily. As shown in Fig. 8b, the HACC-10%(OD)OSA-Gly-Ca2+ hydrogel exhibited the highest average peel strength of 6.38 kN m−1 on wood, metal, glass, and plastic, indicating excellent adhesion performance. The peel strength of HACC-30%(OD)OSA-Gly-Ca2+ hydrogel was 4.52 kN m−1, slightly surpassing that of HACC-50%(OD)OSA-Gly-Ca2+ hydrogel at 4.03 kN m−1. Fig. 8c shows a comparison of peel strength among different kinds of hydrogels upon adhesion to various materials. All hydrogel types showed favorable adhesion performance on wood; in particular, the HACC-50%(OD)OSA-Gly-Ca2+ hydrogel demonstrated superior peel strength. HACC-10%(OD)OSA-Gly-Ca2+ hydrogel exhibited the best peel strength on metal and glass substrates. The difference in peel strength between HACC-10%(OD)OSA-Gly-Ca2+ and HACC-50%(OD)OSA-Gly-Ca2+ hydrogels on plastic was not statistically significant, measured at 5.50 kN m−1 and 5.00 kN m−1, respectively. Currently, reports about biomass-derived self-healing hydrogels with good adhesion ability are rare. The excellent adhesion properties of the hydrogels in this study are conducive to future applications in the biomedical field.
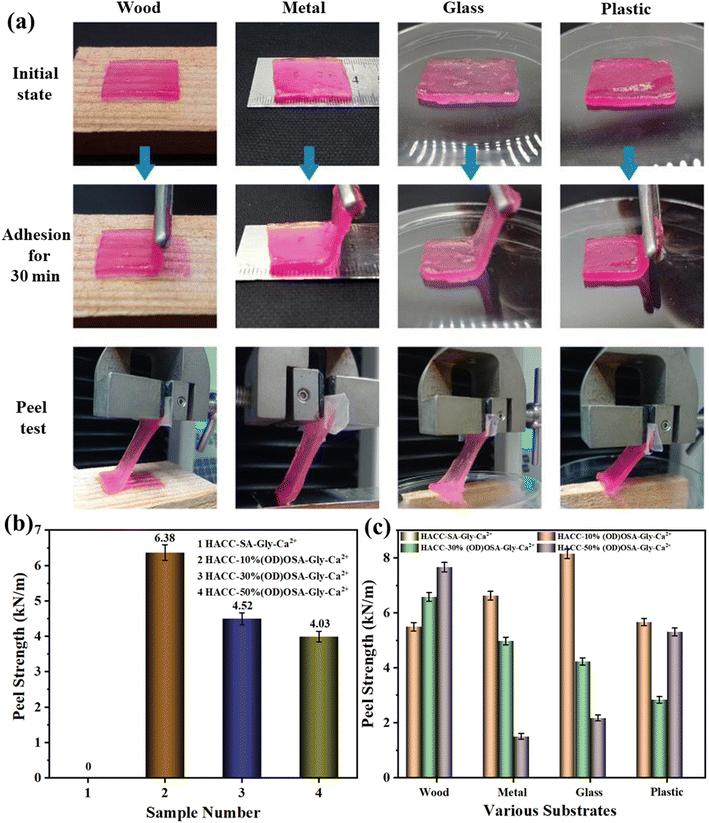 |
| | Fig. 8 Adhesion and stripping behaviors of HACC-50%(OD)OSA-Gly-Ca2+ hydrogels on wood, steel, plastic, and glass; (b) average peel strength of the four kinds of hydrogels on various substrates; (c) peel strength of the four kinds of hydrogels on various substrates. | |
Conclusion
In summary, a facile and eco-friendly method was presented for preparing self-healing hydrogels using sodium alginate and chitosan quaternary ammonium salt as raw material; glycerol, sodium chloride, and water as solvents; and Ca2+ ions as a crosslinker. Based on the experimental results, the HACC-OSA-Gly-Ca2+ hydrogel possessed good mechanical properties, water retention, thermal stability, and resilience. Significantly, the composite hydrogel exhibited super spontaneous repair ability (self-healing rate of 100%) and adhesion ability (average peel strength of 6.38 kN m−1) on various substrates. Owing to the existence of various bond interactions, such as reversible dynamic imine bonding, electrostatic interactions, and hydrogen bonding in the hydrogel system, the composite hydrogels could spontaneously repair themselves at room temperature without external stimulation after deformation and damage. The HACC-50%(OD)OSA-Gly-Ca2+ hydrogel exhibited a repair rate and elongation at break of 94.41% and 417.59%, respectively, after 72 h of self-healing, indicating impressive resilience. Moreover, the overall performance of the composite hydrogel could be tuned by adjusting the oxidation degree of OSA. These merits make the self-healing hydrogels highly attractive for researchers in the field of functional hydrogel materials. The presented strategy could broaden the list of biobased self-healing hydrogels and presents a promising candidate for a wide variety of potential applications.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Data availability
All the data and materials are accessible.
Author contributions
Le Zhong: supervision, visualization, writing – review and editing. Keli Peng: investigation, methodology. Yunqian Sun: writing – original draft, methodology, visualization. Jinxian Zhou: conceptualization, data curation. Naiyu Xiao: Methodology, project administration. Honglei Wang: data curation, validation. Xueqin Zhang: resources, formal analysis. Zheng Cheng: methodology, formal analysis, writing – review and editing.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The research is supported by the National Natural Science Foundation of China (22108086), Fundamental Research Funds from the Zhongkai University of Agriculture and Engineering (KA24YY07116), and the Science and Technology Planning Project of Guangzhou (2024A04J2301). We are grateful for the financial support.
References
- H. Qie, Z. Wang, J. Ren, S. Lü and M. Liu, Polymer, 2022, 263 Search PubMed.
- L. Xu, Z. Huang, Z. Deng, Z. Du, T. L. Sun, Z.-H. Guo and K. Yue, Adv. Mater., 2021, 33, 2105306 CrossRef CAS PubMed.
- C. Qian, T. Zhang, J. Gravesande, C. Baysah, X. Song and J. Xing, Int. J. Biol. Macromol., 2019, 123, 140–148 CrossRef CAS PubMed.
- T. Wang, G. Wang, S. Zhang, C. Qu, C. Li and Y. Gao, Mater. Rev., 2022, 36, 20060050 Search PubMed.
- Y. Yang, Y. Ma, J. Wang, L. You, R. Zhang, Q. Meng, S. Zhong, W. He and X. Cui, Carbohydr. Polym., 2023, 316, 121083 CrossRef CAS.
- Y. Liu, A. Zhou, Y. Zhang, Z. Tian, X. Cheng, Y. Gao, X. Zhou, X. Wu, K. Chen and X. Ning, Int. J. Biol. Macromol., 2023, 242, 124631 CrossRef CAS.
- M. Chen, J. Tian, Y. Liu, C. Huan, R. Li, J. Wang, J. Wu and Q. Zhang, Chem. Eng. J., 2019, 373, 413–424 CrossRef CAS.
- C. Luo, Y. Chen, Z. Huang, M. Fu, W. Ou, T. Huang and K. Yue, Adv. Funct. Mater., 2023, 33, 2304486 CrossRef CAS.
- S. Maiz-Fernandez, L. Perez-Alvarez, L. Ruiz-Rubio, J. L. Vilas-Vilela and S. Lanceros-Mendez, Polymers, 2020, 12, 2261 CrossRef CAS PubMed.
- Z. Wang, H. Cui, M. Liu, S. L. Grage, M. Hoffmann, E. Sedghamiz, W. Wenzel and P. A. Levkin, Adv. Mater., 2022, 34, 2107791 CrossRef CAS PubMed.
- Y. Shi, Y. Wang, Y. Gu, L. Zheng, S. Ma and X. Xu, Chem. Eng. J., 2020, 392, 123645 CrossRef CAS.
- J. F. Duan, Y. X. Gao, Y. R. Huang, L. Li and J. X. Jiang, Bioresources, 2019, 14, 9853–9866 CAS.
- S. Wang, J. H. Chi, Z. W. Jiang, H. W. Hu, C. Z. Yang, W. S. Liu and B. Q. Han, Carbohydr. Polym., 2021, 256, 117519 CrossRef CAS.
- Y. L. Fang, X. S. Du, Z. L. Du, H. B. Wang and X. Cheng, J. Mater. Chem. A, 2017, 5, 8010–8017 RSC.
- F. R. He, L. Z. Wang, S. J. Yang, W. Q. Qin, Y. H. Feng, Y. Y. Liu, Y. Zhou, G. B. Yu and J. C. Li, Carbohydr. Polym., 2021, 256, 117595 CrossRef CAS PubMed.
- L. Wang, W. Zhou, Q. Wang, C. Xu, Q. Tang and H. Yang, Molecules, 2018, 23 Search PubMed.
- R. H. Aguirresarobe, L. Martin, M. J. Fernandez-Berridi and L. Irusta, eXPRESS Polym. Lett., 2017, 11, 266–277 CrossRef CAS.
- C. H. Lin, D. K. Sheng, X. D. Liu, S. B. Xu, F. C. Ji, L. Dong, Y. Zhou and Y. M. Yang, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2228–2234 CrossRef CAS.
- H. Xu, N. Suzuki, A. Takahashi, T. Ohishi, R. Goseki, X. M. Xie and H. Otsuka, Sci. Technol. Adv. Mater., 2020, 21, 450–460 CrossRef CAS PubMed.
- H. Cheng, Z. Fan, Z. Wang, Z. Guo, J. Jiang and Y. Xie, Int. J. Biol. Macromol., 2023, 245, 125471 CrossRef CAS.
- A. Das, A. Sallat, F. Bohme, M. Suckow, D. Basu, S. Wiessner, K. W. Stockelhuber, B. Voit and G. Heinrich, ACS Appl. Mater. Interfaces, 2015, 7, 20623–20630 CrossRef CAS.
- S. Cho, S. Y. Hwang, D. X. Oh and J. Park, J. Mater. Chem. A, 2021, 9, 14630–14655 RSC.
- J. Cao, P. Wu, Q. Cheng, C. He, Y. Chen and J. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 24095–24105 CrossRef CAS PubMed.
- H. Yin, F. Liu, T. Abdiryim and X. Liu, ACS Mater. Lett., 2023, 5, 1787–1830 CrossRef CAS.
- S. Li, Q. Dong, X. Peng, Y. Chen, H. Yang, W. Xu, Y. Zhao, P. Xiao and Y. Zhou, ACS Nano, 2022, 16, 11346–11359 CrossRef CAS.
- Y. Wang, M. Yang and Z. Zhao, Carbohydr. Polym., 2023, 310, 120723 CrossRef CAS.
- Y. Wang, C. K. Adokoh and R. Narain, Expert Opin. Drug Delivery, 2018, 15, 77–91 CrossRef CAS PubMed.
- Y. Fu, C. Xiao and J. Liu, Environ. Technol. Innovation, 2019, 13, 340–345 CrossRef.
- X. Yin, H. Xie, R. Li, S. Yan and H. Yin, Carbohydr. Polym., 2021, 255, 117390 CrossRef CAS.
- G. Remaggi, B. Bottari, E. Bancalari, O. Catanzano, E. Neviani and L. Elviri, Int. J. Biol. Macromol., 2023, 242, 124454 CrossRef CAS.
- X. Wu, C. He, Y. Wu and X. Chen, Biomaterials, 2016, 75, 148–162 CrossRef CAS PubMed.
- D. L. Taylor and M. In Het Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS.
- X. Shang, Q. Wang, J. Li, G. Zhang, J. Zhang, P. Liu and L. Wang, Carbohydr. Polym., 2021, 257, 117626 CrossRef CAS PubMed.
- R. Yu, Z. Li, G. Pan and B. Guo, Sci. China: Chem., 2022, 65, 2238–2251 CrossRef CAS.
- I. Keiichi, T. Atsushi and O. Hideyuki, Macromolecules, 2015, 5632–5639 Search PubMed.
- J. Y. Ai, K. Li, J. B. Li, F. Yu and J. Ma, Int. J. Biol. Macromol., 2021, 172, 66–73 CrossRef CAS PubMed.
- M. Zhong, X. Y. Liu, F. K. Shi, L. Q. Zhang, X. P. Wang, A. G. Cheetham, H. G. Cui and X. M. Xie, Soft Matter, 2015, 11, 4235–4241 RSC.
- C. G. Gomez, M. Rinaudo and M. A. Villar, Carbohydr. Polym., 2007, 67, 296–304 CrossRef CAS.
- L. Han, X. Lu, K. Liu, K. Wang, L. Fang, L. T. Weng, H. Zhang, Y. Tang, F. Ren, C. Zhao, G. Sun, R. Liang and Z. Li, ACS Nano, 2017, 11, 2561–2574 CrossRef CAS PubMed.
- W. Chen, Y. Bu, D. Li, C. Liu, G. Chen, X. Wan and N. Li, Cellulose, 2020, 27, 853–865 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abcd
*abcd