
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA review of the known MTA-cooperative PRMT5 inhibitors
Mei Hu and
Xiang Chen *
*
Department of Pharmaceutical Sciences, School of Pharmacy, Southwest Medical University, 1-1 Xiangling Road, Luzhou, Sichuan 646000, People's Republic of China. E-mail: 86487165@qq.com
First published on 17th December 2024
Abstract
Protein arginine methyltransferase 5 (PRMT5), an epigenetic target with significant clinical potential, is closely associated with the occurrence and development of a range of tumours and has attracted considerable interest from the pharmaceutical industry and academic research communities. According to incomplete statistics, more than 10 PRMT5 inhibitors for cancer therapy have entered clinical trials in recent years. Among them, the second-generation PRMT5 inhibitors developed based on the synthetic lethal strategy demonstrate considerable clinical application value. This suggests that, following the precedent of poly ADP ribose polymerase (PARP), PRMT5 has the potential to become the next clinically applicable synthetic lethal target. However, due to the inherent dose-limiting toxicity of epigenetic target inhibitors, none of these PRMT5 inhibitors has been approved for marketing to date. In light of this, we have conducted a review of the design thoughts and the structure–activity relationship (SAR) of known methylthioadenosine (MTA)-cooperative PRMT5 inhibitors. Additionally, we have analysed the clinical safety of representative first- and second-generation PRMT5 inhibitors. This paper discusses the in vivo vulnerability of the aromatic amine moiety of the second-generation PRMT5 inhibitor based on its structure. It also considers the potential nitrosamine risk factors associated with the preparation process.
1 Introduction
Protein arginine methylation, as one of the post-translational modifications (PTMs), is catalysed by protein arginine methyltransferases (PRMTs).1,2 And PRMTs are known to have nine members (PRMT1-9) in human cells. Although the fact that every member of the PRMT family can catalyse the methyl transfer reaction between the cofactor S-adenosylmethionine (SAM, as the methyl group's donor) and the protein arginine residues, the resulting forms of methylated arginine catalysed by the different members of the PRMT family are not consistent with one another.3–5 Based on this, PRMTs may be further subdivided, including type I (PRMT1, 2, 3, 4, 6, 8), type II (PRMT5, 9), and type III (PRMT7).6,7 Among these, PRMT5 is responsible for catalysing the formation of omega-NG, N’G-symmetric dimethylarginines (SDMA) and omega-NG-monomethyl arginine (MMA), which are involved in various physiological multifarious physiological and biochemical processes8 such as cell cycle,9,10 the splicing of DNA repair genes,11 embryonic development and differentiation,12–14 and so on. Moreover, there is evidence that the dysregulation of PRMT5 is associated with the development of several diseases, particularly malignant tumour pathogenesis and poor patient prognosis.15–18PRMT5, as a classical SAM-binding protein, has two orthosteric sites in its C-terminal structure, namely the SAM-binding site and the substrate-binding site. A passage way, designated the Arg tunnel, exists between the SAM pockets and the substrate pockets (as shown in the Fig. 1B, PDB: 4X61).21,22 PRMT5, which has been identified as a potential target for antitumour therapy, has attracted increasing attention. Currently, according to incomplete statistics, there are more than 10 PRMT5 inhibitors entering clinical development worldwide, with the majority being in the clinical phase I. These small molecule inhibitors may be subdivided into the following several types according to their binding sites to PRMT5.
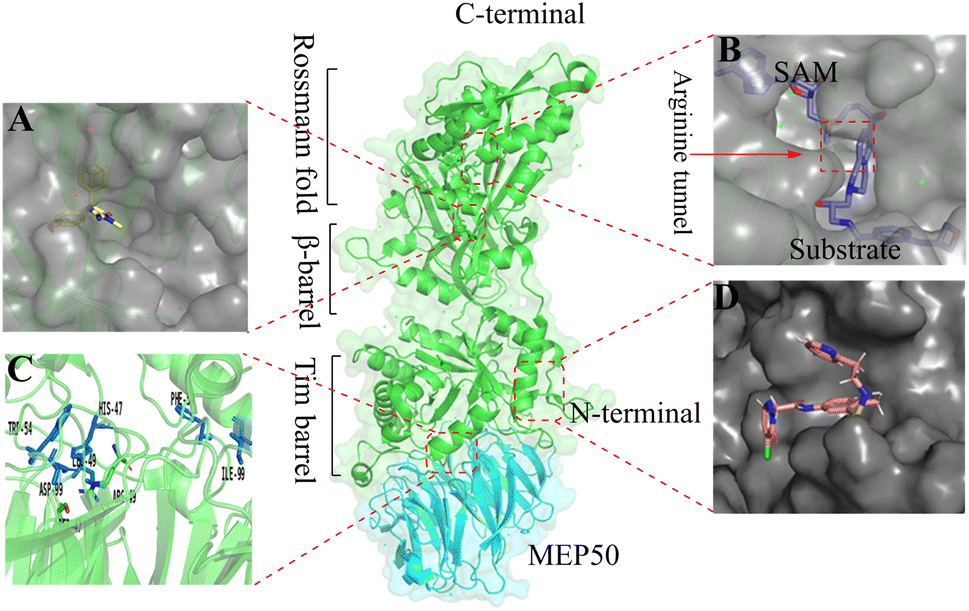 | ||
| Fig. 1 Binding sites where PRMT5 can be used for the development of pharmaceutical agents. (A) Allosteric binding site (PDB: 6uxx).19,20 (B) SAM binding site and substrate binding site. The substrate binding site and SAM-binding pocket are closely connected by the arginine tunnel (PDB: 4X61),21,22 which can be exploited by certain inhibitors, such as JNJ-64619178, to block both the SAM-binding site and substrate binding pocket simultaneously.23,24 (C) The PRMT5: MEP50 protein–protein surface interaction (PDB: 4gqb).25 (D) The PBM groove (PDB: 6U0P). The substrate adaptor proteins (SAPs) contains a highly conserved linear peptide sequence, termed the PRMT5 binding motif (PBM). The PBM peptide and PRMT5 occurs within a shallow groove (the “PBM groove”) of the TIM barrel5,26–32 | ||
(I) The SAM mimetic. The structure of this inhibitor is characterised by the adenosine scaffold as its structural core (it is highlighted in red in Fig. 2). The binding of it to PRMT5 occurs in the SAM binding pocket and competes with SAM; because of all PRMTs use the cofactor SAM, as methyl group's donor, to catalyse the arginine methylation reaction and share a highly homologous SAM-dependent methyltransferase (MTase) domain which is the catalytic domain, how to make SAM competitive PRMT5 inhibitors have the target protein selectivity, which is one of the mayor challenges researchers face. In fact, scientists obtained a lot of SAM mimetics, including but not limited to SAH, sinefungin, dehydrosinefungin and DS-437 (structure not shown here) in the early stage of PRMT5 inhibitors development. Owing to the remarkable similarity in chemical structure between SAM and these mimetics, these compounds could occupy the SAM binding pocket and show PRMT5 inhibitory activity to a certain degree, but lack of selectivity.21,42–45 Interestingly, MTA, an endogenous ligand, is a selective SAM-competitive inhibitor of PRMT5, which shows more than 100-fold selectivity for PTMT5 over all other PRMTs (however, its potency is weak, IC50 = 260 nM).46 In light of the findings of MTA research, non-covalently bound SAM competitive inhibitors, exemplified by 11 (LLy283),33 have been developed. Concurrently, covalently bound SAM competitive inhibitors with 11 (LLy283) as the parent structure were also developed such as the hemiaminal compound. Although these SAM mimetics are similar in backbone structure, their preparation routes and methods are diverse. Among the various preparation methods, the strategy of using the adenosine scaffold structure as the starting material for the synthesis of the corresponding SAM mimetic has been the subject of extensive study and has a high success rate (see Fig. 2).33–37
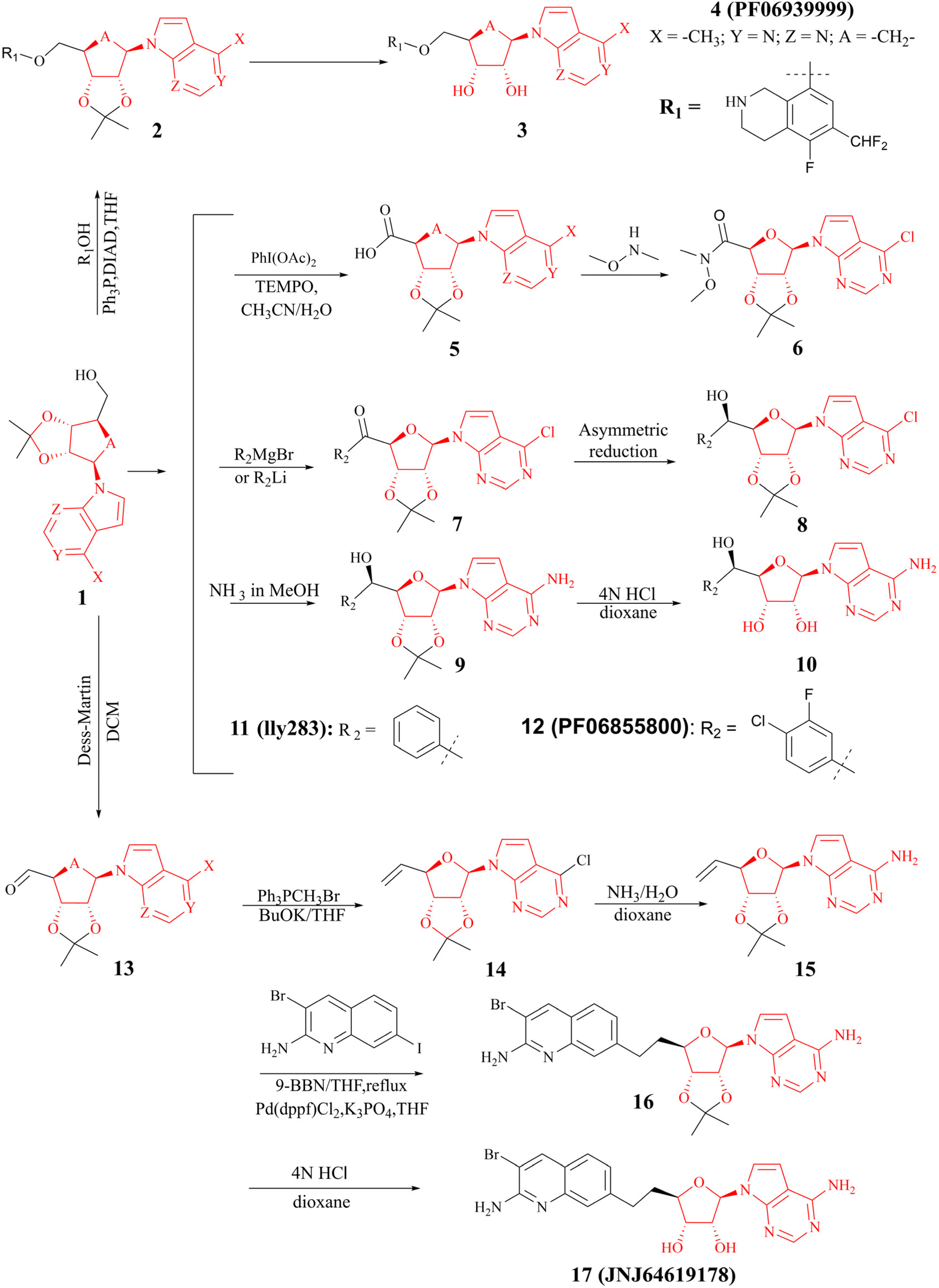 | ||
| Fig. 2 Synthesis of the SAM mimetic using the adenosine scaffold as its structural core.23,24,33–41 The adenosine scaffold is highlighted in red. | ||
(II) The dual SAM/substrate PRMT5 inhibitor. These inhibitors (such as JNJ-64619178,47,48 see the structure in Fig. 2) could potentially prohibit binding of SAM and the substrate simultaneously by occupying the SAM-binding site, and then the quinoline moiety extending into the Arg tunnel, where it interacts with the key catalytic residues Glu444 of PTMT5. Moreover, the quinoline ring forms π–π stacking with Phe327. This unique binding pattern makes it possible to have a longer retention time at the target site, thereby reducing the required dose for clinical applications. However, this tight binding pattern, along with higher efficacy, may also result in more severe toxic side effects.49–53 In terms of its chemical structure, this type of inhibitor is, in fact, a novel SAM analogue. Consequently, the most cost-effective preparation strategy is to use the adenosine scaffold structure as a starting material to synthesise the corresponding compounds. See Fig. 2.23,24,38–41
(III) The substrate competitive inhibitor. Its structure is characterised by the tetrahydroisoquinoline as its structural core (it is highlighted in red in Fig. 3). The binding site of this inhibitor with PRMT5 is in the substrate-binding pocket and competes with the substrate. Many studies have shown that the tetrahydroisoquinoline portion of this type of inhibitor forms a key cation–π interaction with the cofactor SAM, and at the same time, the aromatic ring of tetrahydroisoquinoline forms a potential π–π stacking interaction with Phe327. Phe327 is unique to PRMT5, and other PRMT enzymes at the same position are a Met. It is postulated that these factors contribute to the high selectivity and inhibitory activity of EPZ01566 against PRMT5.61–63 The preparation of this class of inhibitors is to use (R)-2-(oxiran-2-ylmethyl)-1,2,3,4-tetrahydroisoquinoline as the starting material, and the corresponding compounds are made by aminolysis and acylation reactions (see Fig. 3).54–60 And the PRMT5 degrader MS4322 with EPZ015666 as the target protein of interest (POI) ligand is also in preclinical studies.64
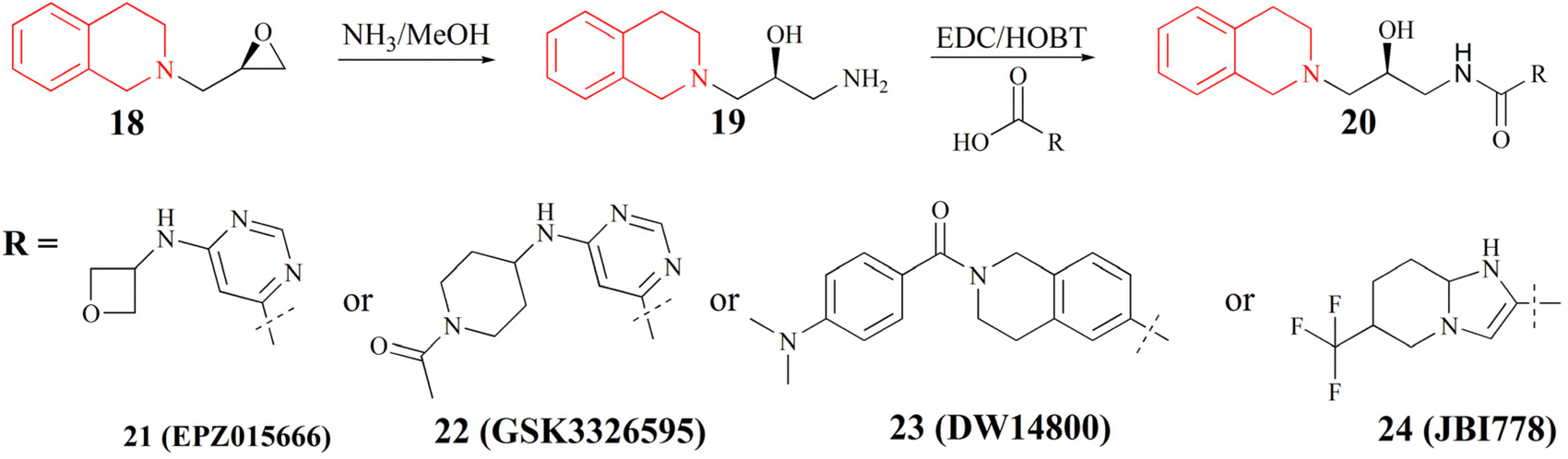 | ||
| Fig. 3 Synthesis of compounds of formula 20.54–60 | ||
(IV) Allosteric inhibitors. when the PRMT5:MEP50 complex with the inhibitor binds in a new binding site (apart from the SAM-binding site and substrate-binding pocket), the backbone structure of protein is beginning to significant structural rearrangement and further influence both the SAM-binding site and substrate-binding pocket so that none of the SAM and substrate can bind with PRMT5 in their respective binding site, such as R-1a.19,20
Although all PRMT5 inhibitors described above have shown high selectivity and inhibitory activity against PRMT5, they lack specific selectivity for tumour cells and have a narrow therapeutic window in terms of their mechanism of action. Moreover, as mentioned previously, PRMT5 plays a crucial role in normal physiological processes; PRMT5 knockdown may result in embryonic lethality42,65 and tissue-specific PRMT5 knockdown in the central nervous system,66,67 skeletal muscle,68 and haematopoietic system69 all produce substantial toxicity. These toxicities may become dose-limiting in the clinical use of the drug, potentially affecting the progress of clinical studies of drugs targeting PRMT5. In clinical experiment, the majority of such PRMT5 inhibitors also exhibit significant tumour growth inhibitory effects. However, adverse events (AEs) associated with PRMT5 inhibitor therapy, including fatigue, anaemia, nausea, alopecia and dyspeptic events, are prevalent (for a list of representative PRMT5 inhibitors in clinical trials, please refer to Table 1). For example, 4 (PF-06939999) showed significant dose-limiting toxicities including thrombocytopenia, anaemia and neutropenia in a phase I dose-escalation study. Currently, clinical development has been terminated (NCT03854227).70,71 Owing to the universality of these treatment-related AEs, the toxicity and side effects of these PRMT5 inhibitors, especially target associated haematotoxicity, need been further evaluated. Currently, the ability of these PRMT5 inhibitors to pass evaluation in further clinical trials depends on the assessment of their efficacy and safety; what is the benefit-risk ratio? Is the safety acceptable? Current clinical studies show that it is not certain. And, it may still be extremely valuable to explore other treatments with this class of drugs, such as combinations with other drugs. Hence, the study on PRMT5 inhibitors of new structures has been gradually focused on “synthetic lethality” to further higher selectivity and lower target associated toxicity.72 In order to facilitate the distinction between the various types of PRMT5 inhibitors, the above mentioned inhibitors developed on the basis of the cofactor SAM-binding site and the substrate-binding site are also collectively referred to as the first-generation PRMT5 inhibitors.
| Comp. | Ligand–protein binding mechanism | Dose range | Toxic side effect | Safety conclusion | Phase | Ref. |
|---|---|---|---|---|---|---|
| a N/A: Information not disclosed. | ||||||
 |
SAM-binding pocket. It forms a crucial interaction with Phe327 | N/A | N/A | N/A | Preclinical | 21 |
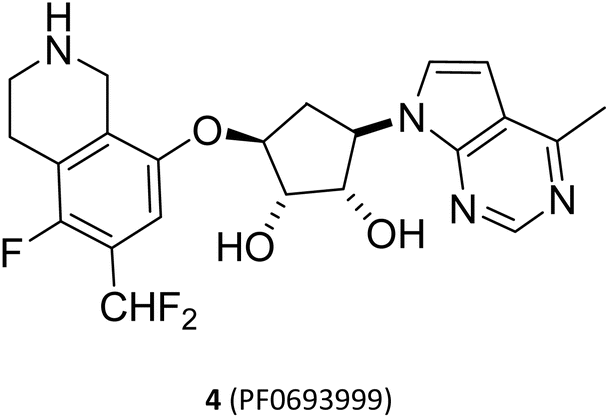 |
0.5–12 mg daily (QD or BID) dose | One patient (6 mg BID) had grade 4 treatment-related thrombocytopenia. Treatment-related adverse events occurred in 24 cases (86%) | 6 mg QD was identified as the recommended monotherapy dose for expansion | Clinical termination | 51–53 | |
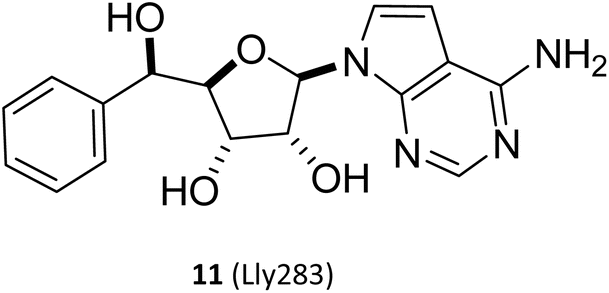 |
N/A | N/A | N/A | Preclinical | 33 | |
| Structure not disclosed SKL27969 | N/A | N/A | N/A | N/A | Phase I terminated | 24 |
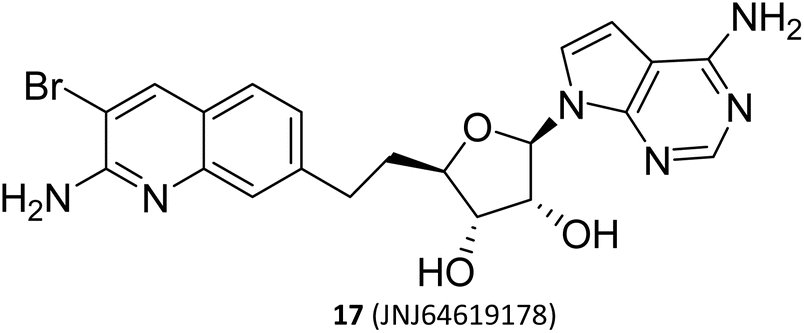 |
SAM-binding pocket and reaches the substrate-binding pocket | 2 mg QD and 3–4 mg 14 on/7 off | The dose-limiting toxicity was thrombocytopenia; 51 patients (91%) experienced a grade 3/4 trae with a treatment-related adverse event (TRAE) > 1 pt. Thirty patients (56%) experienced dose interruptions or reductions due to AEs | 0.5 mg once daily was well tolerated | Phase I | 19, 20, 65, and 66 |
| 1.5 mg intermittent and 1.0 mg once daily dosing. Demonstrated manageable dose-dependent toxicity, however limited clinical benefit other than ACC | ||||||
 |
The substrate binding pocket and competes with the substrate | 12.5 mg to 600 mg once daily (QD) and 50 mg to 200 mg twice daily | In phase I of the clinical trial for patients with solid tumour cancers, 89% of patients experienced treatment-related adverse events. Of these, 3/4 adverse events occurred | A comprehensive assessment, AE is common but manageable | NCT03614728 Terminated; NCT04676516 completed; NCT02783300 completed | 67–70 |
 |
SAM-binding pocket. It forms a crucial interaction with Phe327 | 35 mg (n = 28) or 45 mg (n = 28) | Grade 3 treatment-related adverse events were experienced by 23% of patients, no grade 4/5 treatment-related adverse events were reported | The drug was observed to be tolerated in patients with R/M ACC, but its efficacy was limited | Phase I | 38 and 39 |
| Completed | ||||||
 |
1.15–800 mg orally once daily | One patient with GBM had grade 3 thrombocytopenia. 69% (n = 31) experienced adverse events. No deaths were reported | Well tolerated, with a good safety profile under either QD or BID regimens | Phase I | 23, 40 and 41 | |
| 2.300 mg twice daily [BID] | Completed | |||||
Synthetic lethality was initially observed and defined in drosophila and yeast.73 In a nutshell, if A and B genes constitute a synthetic lethal pair, mutations in either of these two genes alone have a minimal impact on cells, but the simultaneous mutations of both genes will result in cell death.72,74–76 This concept was further elucidated in 2005, when researchers found that pharmacological inhibition of the poly ADP-ribose polymerase (PARP) may result in the death of tumour cells bearing the BRCA mutation, whereas normal cells are minimally affected due to retaining the wild-type copy. (PARP and BRCA form a synthetic lethal pair here).77,78 In 2014, the world's first synthetic lethal inhibitor olaparib, which are used in the treatment of BRCA-mutant ovarian cancer, was approved for marketing.79
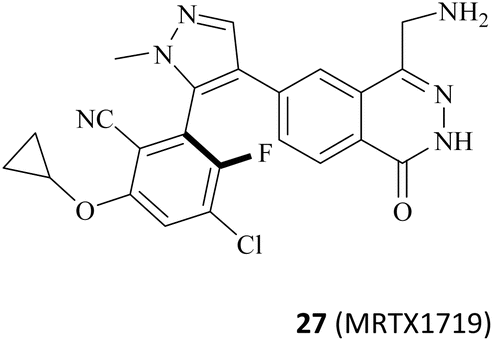
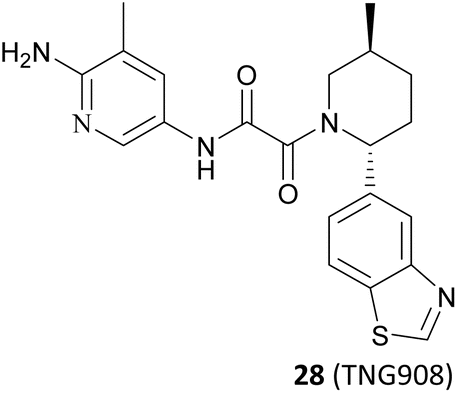
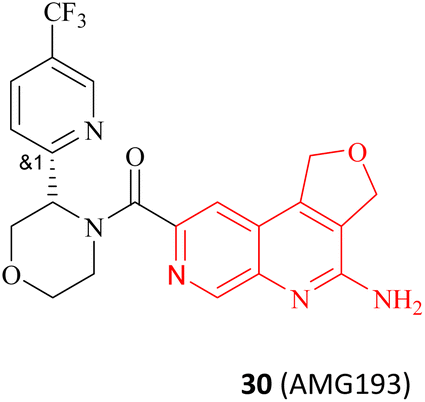
It is widely known that tumour suppressor genes CDKN2A on chromosome 9p21 (chr9p21) are homozygously deleted in multiple solid tumours. The extent of this deletion varies considerably, often leading to neighbouring genes being deleted together. Homozygous deletion of the methylthioadenosine phosphorylase (MTAP) gene takes place frequently in multiple solid tumours owing to its proximity to the tumour suppressor gene CDKN2A.80 It is noteworthy that the MTAP is in charge of methylthioadenosine (MTA) conversion in the methionine salvage pathway81,82 (see Fig. 4). As an analogue of SAM, MTA fulfils the role of a natural SAM competitive inhibitor, showing more than 100-fold selectivity for PRMT5 over all other PRMT family members. In other words, MTAP loss results in the accumulation of intracellular MTA, which produces a frail PRMT5 state that is selectively sensitive to further inhibition by PRMT5 inhibitors.42,43,46,83,84 Thus, both PRMT5 and MTAP constitute a synthetic lethal pair in this context. However, it is anticipated that the aforementioned clinical PRMT5 inhibitor will not exhibit selective inhibition of PRMT5 activity in MTAP-deleted tumours without impacting the normal cells due to the mechanism of action of these agents. Fortunately, MTA competes with SAM to generate the PRMT5-MEP50/MTA (PRMT5/MTA) complex in MTAP-deleted tumours. The researchers believe that a small molecule which could bind to and stabilise the PRMT5/MTA complex will selectively inhibit PRMT5 activity in MTAP-deleted tumour cells. Soon afterwards, several PRMT5/MTA complex inhibitors, including 28 (TNG908),85,86 27 (MRTX1719),87,88 and 30 (AMG193)89 were approved for clinical development successively.
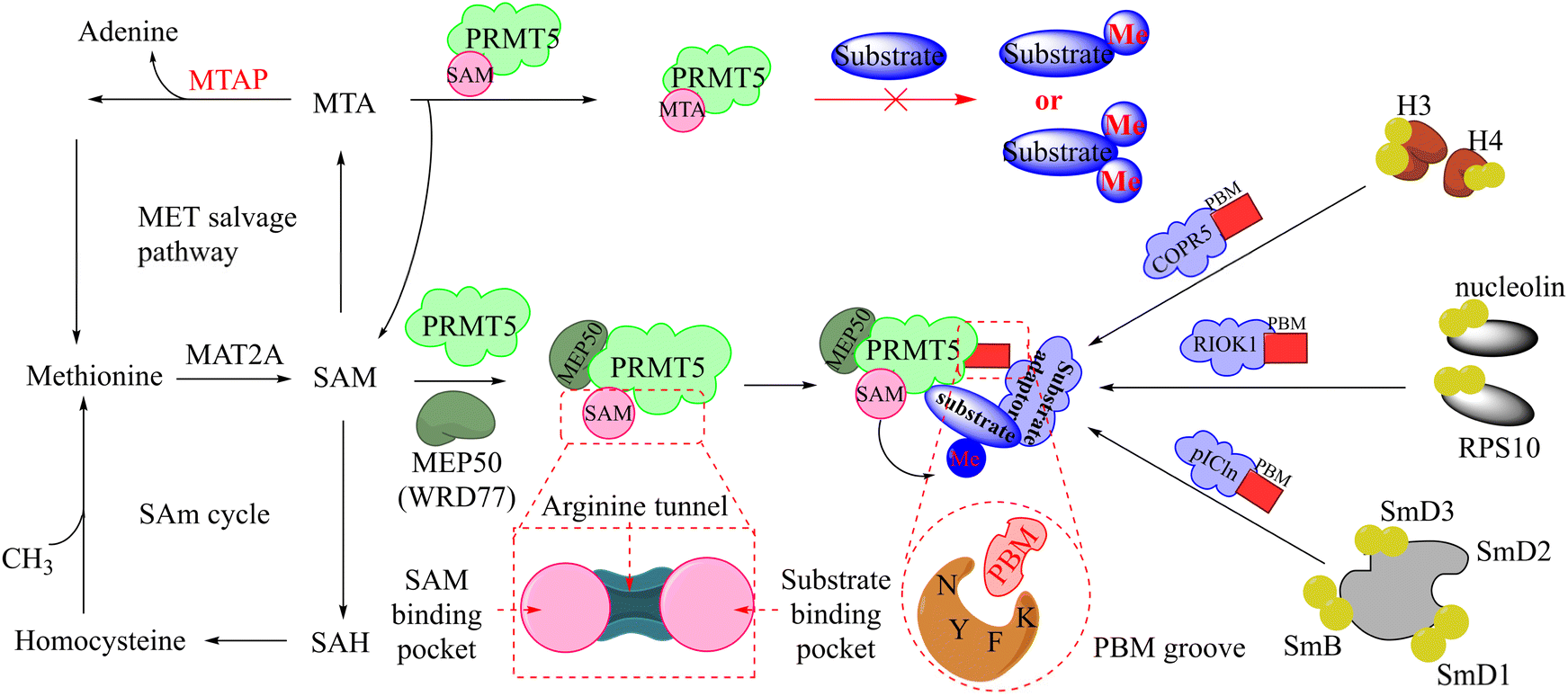 | ||
| Fig. 4 The upstream and downstream molecular regulatory mechanisms of PRMT5. The substrate binding site and SAM-binding pocket are closely connected by the arginine tunnel, which can be exploited by certain inhibitors, such as 17 (JNJ-64619178), to block both the SAM-binding site and substrate binding pocket simultaneously. Furthermore, PRMT5's activity and substrate selectivity are regulated via the formation of multisubunit protein complexes. The SAPs, including Rio domain-containing protein (RioK1), cooperator protein of PRMT5 (COPR5), and chloride channel nucleotide-sensitive protein 1A (pICln), form the multisubunit complexes with PRMT5 by the interaction between the PBM and the PBM-binding groove.5,21,26–30 | ||
In order to differentiate themselves from the aforementioned PRMT5 inhibitors, these MTA-cooperative synthetic lethal inhibitors have also been designated as second-generation PRMT5 inhibitor.
In fact, this synthetic lethality also extends to the key points of the upstream and downstream molecular regulatory mechanisms of PRMT5, including the PRMT5 complex member located downstream (see Fig. 4).26 It is known that a human PRMT5 is made up of a Rossmann fold which contains the catalytic site, a β-barrel that allows for the dimerisation of PRMT5, and a TIM barrel which houses the PBM groove and mediates interactions with different substrate adaptor proteins (SAPs).5 Owing to the positive allosteric effect of the partner protein MEP50 (WDR77), the complex composed of PRMT5 and MEP50 usually possesses a significantly higher level of MTase activity than PRMT5 alone.21,27 (Actually, a stable heterooctameric complex comprising four copies of PRMT5 and MEP50 is frequently observed within the cellular environment.)21,28 Nevertheless, the identification and recruitment of substrates for the PRMT5/MEP50 complex depend on the involvement of SAPs. It is noteworthy that the research indicates that three SAPs (RioK1, pICln, and COPR5) share a highly conserved linear peptide sequence, which has been named as the PRMT5 binding motif (PBM). And the interaction between PBM and the PBM-binding groove is responsible for mediating the methylation of over 25 substrates, including nucleolin and RPS10.29 Thus, the targeted inhibition of the protein–protein interactions (PPIs) represents another promising avenue for the development of MTA-cooperative synthetic lethal inhibitors (Fig. 4). Based on these results, the PBM-competitive inhibitor 31(BRD0639),31 the cyclic binding peptide inhibitor 32,30,32 and PRMT5:MEP50 PPI inhibitors 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 were discovered successively.
25 were discovered successively.
However, the clinical status of anti-tumour treatment with second-generation inhibitors is under pressure and it will also face with a range of potential challenges. For instance, six different malignancies, including non-small cell lung cancer and melanoma, were treated with 27 (MRTX1719) in a dose-escalating manner, and the tumour burden was reduced by approximately 30–60%. However, it is important to note that these encouraging early clinical outcomes are limited by the small sample size and more work needs to be done to verify and confirm it. Furthermore, PRMT5 is essential for the survival of all cells, especially haematopoietic cells. Despite the treatment-related adverse events (AEs) in humans were not reported at the effective dose range of 27 (MRTX1719), there was a notable reduction in normal bone marrow SDMA levels (about 52% were inhibited) in comparison to the near-total loss of SDMA in MTAP-deficient tumours.90 It has been reported that, in vitro, MTAP WT cells are capable of metabolising enriched MTA in MTAP del tumour cells. Furthermore, MTA is not enriched in human MTAP-deficient glioblastoma tissue.91
In addition, a series of studies have demonstrated that the PRMT5 enzyme is susceptible to further modulation by SAM levels in MTAP-null tumour cells, whereas its activity is either unaltered or less affected in normal tissues and in tumours where MTAP is retained. By inhibiting the upstream metabolic enzyme methionine adenosyltransferase II alpha (MAT2A) activity, SAM levels in MTAP-deleted cancers can be reduced, and selective inhibition of PRMT5 methylation activity is then possible (see Fig. 4). The MAT2A inhibitor 34 (AG-270) is currently being evaluated in clinical trials (NCT03435250).92–96 However, we will not discuss it here because the drug target MAT2A is beyond the scope of this article.
It should be noted that caution should be exercised when interpreting SAR data across references since the data presented in this article are collected from different references. In addition, all the schematic diagrams and structures in this paper are summarised from the previous research results, and were created using the drawing software Kingdraw Professional 5.0. The crystal structure data were obtained from RCSB Protein Data Bank (https://www.rcsb.org/pages/about-us/index) (https://www.rcsb.org/). The crystal structure was created by PyMol 2.1.0, a software for displaying the 3D structure of protein molecules.
2 MTA-cooperative PRMT5 inhibitors
It is well known that a lead compound with good druggability is critical to modern drug discovery. The identification of a lead compound in a timely and accurate manner has constituted a significant obstacle in the field of drug discovery. In recent years, a number of innovative drug screening techniques have emerged, including high-throughput screening (HTS), artificial intelligence-based drug molecule virtual screening (AIDD), fragment-based molecule library screening (FBDD), and structure-based molecule design (SBDD). These techniques offer potential solutions to this challenge. Among them, the concept of FBDD originated approximately 30 years ago. However, the rapid development of a wide range of experimental analytical techniques had made it an effective tool for modern drug discovery by the early 21st century. The implementation of FBDD is generally divided into three steps: the initial step is to identify a comprehensive and high-quality library of small molecules, or fragments. These small molecules should typically adhere to the Rule of Three (RO3) principle (molecular weight less than 300, no more than 3 hydrogen bond donors and acceptors, no more than 3 rotatable bonds, cLogP less than 3).97–99 Step 2, determine an appropriate high-sensitivity screening method to identify suitable fragments from the library of fragments. Of these, the most critical of these is the experimental analysis technique used to identify the binding strength of the fragments to the target protein. Currently, the main experimental techniques used in practice include differential scanning fluorometry (DSF), microscale thermophoresis (MST), surface plasmon resonance (SPR), nuclear magnetic resonance (NMR), X-ray protein crystallography (X-ray), and so on.99,100 Step 3, fragment growth and optimisation. The initial fragments obtained from FBDD screening are typically characterised by low activity. To enhance their target affinity and other properties, such as solubility, membrane permeability and bioavailability, these fragments must undergo structural extension and optimisation. In this process, SBDD is typically employed as the primary methodology to direct the expansion and enhancement of fragments.101 SBDD represents a drug design method based on the concept of molecular recognition, which employs the three-dimensional structure of ligands and targets. In comparison to the conventional HTS approach, FBDD exhibits enhanced exploration efficiency, broader chemical space coverage, more favourable binding modes, and reduced input/output. These advantageous characteristics, coupled with the insights gleaned from the crystal structure of the protein, have led to the extensive utilisation of these innovative drug screening techniques, particularly the integration of FBDD and SBDD, in the development of MTA-cooperative PRMT5 inhibitors and structure–activity relationship (SAR) exploration, with notable success.
2.1 PRMT5/MTA complex inhibitors
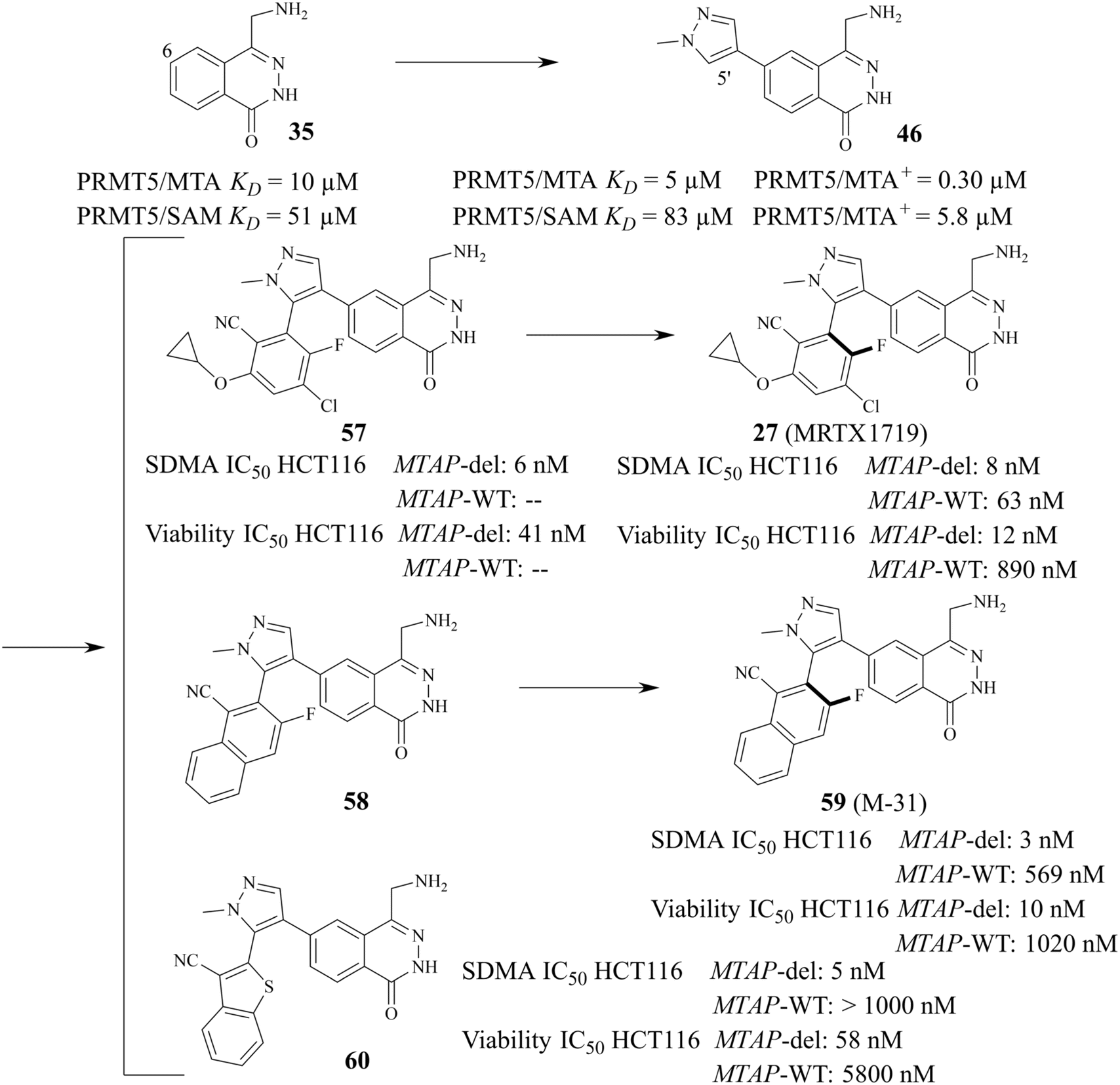
As mentioned above, PRMT5 and MTAP have been supposed to be the next potential synthetic lethal pair and several PRMT5/MTA inhibitors are currently undergoing clinical trials. Among them, 27 (MRTX-1719), as one of the most clinical application value PRMT5/MTA inhibitors, has made the fastest progress (the clinical phase I/II: NCT05245500). The 4-(aminomethyl)phthalazin-1(2H)-one fragment (35, see Fig. 5A), as the starting point for the development of 27 (MRTX1719), was confirmed via a fragment-based lead discovery approach. Firstly, SPR was used as a screening platform because of its high sensitivity, ability to detect weak binding, etc. Matthew A. Marx et al. determined the KD (KD = 27 μM) for the binding of the PRMT5-MTA complex to EPZ015666 and used it as a positive control. In the initial phase of the screening process, approximately 1000 fragments were evaluated at a single concentration of 100 μM, leading to the identification of 17 fragmented compounds. In the subsequent phase, two additional fragment libraries, namely a diverse library comprising 1692 fragments and a focused library constructed of 194 specifically selected compounds, were screened at a concentration of 500 μM, resulting in the identification of a total of 188 hit points. Subsequently, the 205 fragments obtained from the screening were subjected to PRMT5-MTA KD assay, and 100 of them exhibited saturation KD values lower than 1 mM. From these, 24 fragments were selected for further characterisation with the objective of determining whether they preferentially bind to the PRMT5/MTA complex. The results demonstrated that the binding KD of PRMT5-MTA ranged from 300 nM to 300 μM, while the selectivity for the PRMT5-SAM complex ranged from 0.2 to 35-fold, among the 24 fragments that were tested. Fragment 35, which exhibited a PRMT5-MTA KD of 10 μM and a PRMT5-SAM KD of 51 μM, was selected for further fragment growth and SAR exploration. SBDD is used as a guiding strategy.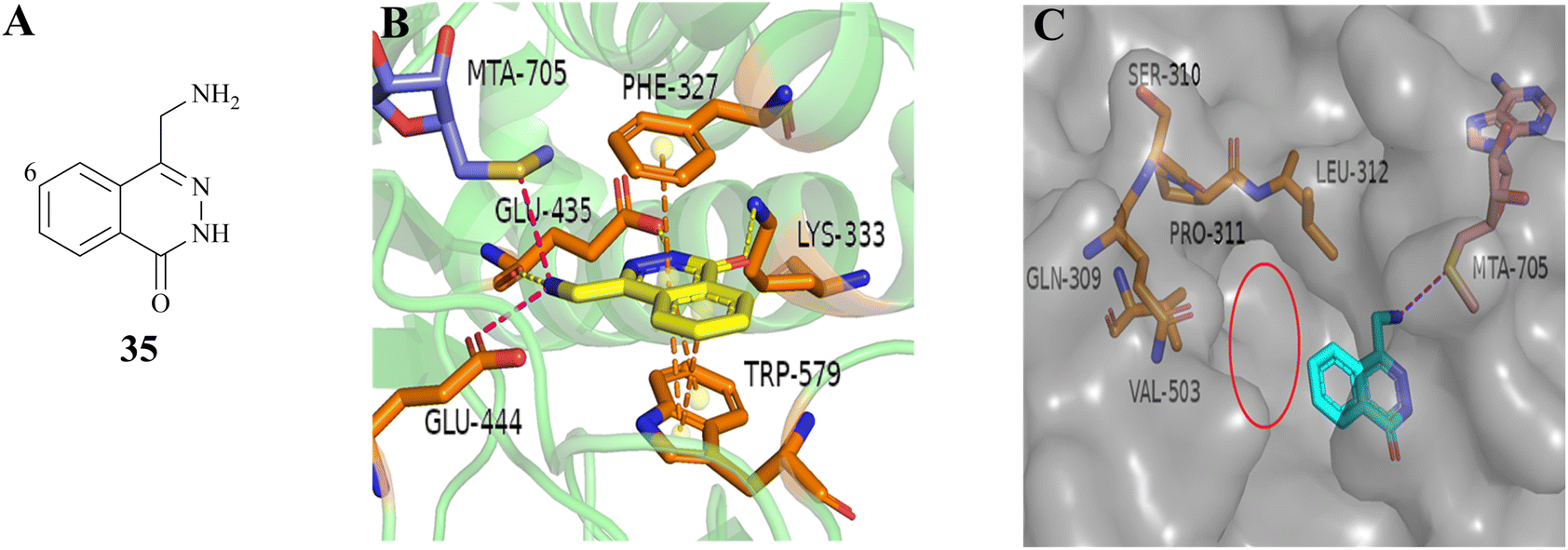 | ||
| Fig. 5 (A) Chemical structure of 35. (B) The co-crystal structure of fragment 35 with PRMT5/MTA complex (PDB ID: 7S0U). Fragment 35 forms a hydrogen bonding network with Glu435 and Lys333. Additionally, it is observed that there is an ionic interaction between the primary amine (–NH2) of 35 and Glu444, a π–stack interaction between 35 and the side chains of Phe327 and Trp579, and a van der Waals interaction between 35 and the S-atom of MTA. (C) To investigate the pocket formed by five amino acid residues including the residues Leu312, the C6-position of 35 is the most appropriate position for fragment growth.87 | ||
In the co-crystal structure (PDB ID: 7S0U, Fig. 5B), it can be observed that the fragment 35 binds in the substrate-binding pocket of PRMT5 via a series of diverse and specific mutual effects. Further analysis of the co-crystal structure revealed the presence of a pocket constituted by five amino acid residues, including the residue Leu312, at the front of the C6-position of the 35 (see Fig. 5C). Molecular modelling using MOE suggested that it can accommodate a five or six-membered heterocyclic ring, in which the heteroatom with the lone-pair electron might make a beneficial H-bond interaction with Leu312.87
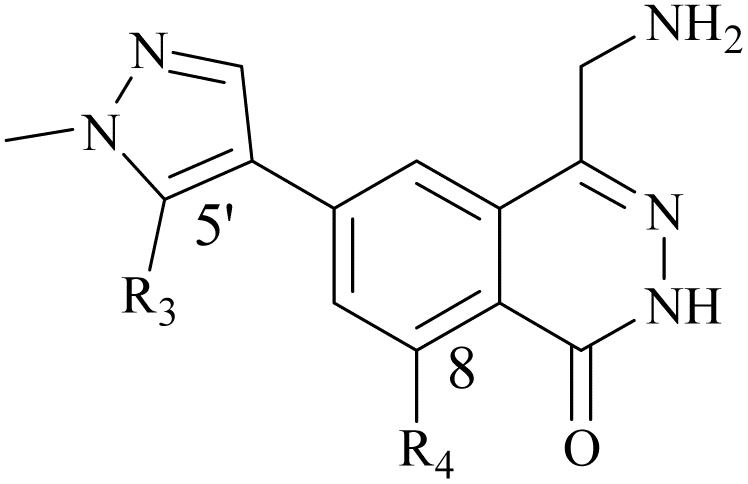
From this, a series of six-membered heterocyclic ring substituted compounds of 35 (see Table 3 for its representative compounds) were prepared. These compounds display high potency and selectivity in the PRMT5 biochemical assay, particularly compounds 39 (MTA+ IC50 = 2.3 nM, MTA− IC50 = 237 nM, selectivity ratio MTA+/MTA− = 103) and 40 (MTA+ IC50 = 3.1 nM, MTA− IC50 = 313 nM, selectivity ratio MTA+/MTA− = 100.9). It is regrettable that no further research data regarding the optimisation of this series of compounds was reported. In parallel, the five-membered heterocyclic ring substituted compounds of 35 were also prepared. Among them, those containing imidazole and N-substituted derivatives of imidazole exhibit a significant increase in potency and selectivity compared to the 35, especially compound 46 (see Table 4). The co-crystal structure of 46 with PRMT5/MTA (PDB 7S1Q, Fig. 6B)87 indicates that the N-methylpyrazole-4-yl group occupies the aforementioned pocket (Fig. 5C) and a new H-bond is observed between the backbone N–H of Leu312 and the N2 of N-methylpyrazole. Further investigation of the SAR shows that using any of the -ethyl, -propyl, -isopropyl, and -hydroxyethyl instead of H-atom of the pyrazole N1′ is also tolerated (compounds 47–50, see Table 3). Replacing the H-atom of the C5′ position of the N-methylpyrazol-4-yl group with a methyl group (compound 51) generates a 10-fold boost in inhibition compared to 46. Moreover, Phe300, Tyr304 and Val326 form the other lipophilic pocket and there is enough room to accommodate a lipophilic group (see Fig. 6C). Subsequently, compounds 56–60 were designed, synthesized, and tested (see Fig. 7 for structure and data).
| Comp. | Mechanism of action | Mechanisms of tumour cell selectivity | Dose range/toxic side effect | Safety conclusion | Phase | Ref. |
|---|---|---|---|---|---|---|
| a N/A: Information not disclosed. | ||||||
 |
Substrate competitiveness model. PRMT5/MTA complex inhibitor | Synthetic lethal | 50–800 mg orally once daily | MRTX1719 was well tolerated and no dose-limiting toxicity was observed at dose levels up to 400 mg q.d | Phase 1/2 recruiting | 90, 91, 102, and 103 |
| No dose-limiting adverse events associated with non-MTAP-selective PRMT5 inhibitors at dose levels of 400 mg q.d | ||||||
 |
N/A | N/A | Phase 1/2 recruiting | 104 | ||
 |
N/A | N/A | Phase 1 recruiting | 105 | ||
 |
40–1600 mg per day or 600 mg per day treatment-related adverse events were nausea (48.8% n = 80), fatigue (31.3%) and vomiting (30.0%). Dose-limiting toxicity was reported in 8 patients at a dose of 240 mg | The maximum tolerated dose was determined to be 1200 mg per day, and AMG 193 demonstrated a favourable safety profile without clinically significant myelosuppression | NCT06593522 phase 2 not yet recruiting | 106–111 | ||
| NCT05975073 | ||||||
| Phase 1/2 recruiting; NCT06360354 | ||||||
| Recruiting | ||||||
| NCT06333951 | ||||||
| Recruiting | ||||||
| NCT05094336 | ||||||
| Recruiting | ||||||
 |
Occupy the PRMT5 PBM groove | N/A | Poor in vivo stability and safety | Preclinical | 31 | |
 |
N/A | Poor in vivo stability and safety | Preclinical | 32 | ||
 |
Impeding PRMT5 and MEP50 protein–protein surface reactions | N/A | Poor in vivo stability and safety | Preclinical | 25 | |
 |
MTA-Cooperative MAT2A inhibition | 50 mg once daily (QD; n = 3), 100 mg QD (n = 7), 150 mg QD (n = 6), 200 mg QD (n = 11), 400 mg QD (n = 6), or 200 mg twice daily (BID; n = 6) | AG270 reduced plasma [SAM] and tumour SDMA levels at well-tolerated doses | Termination | 92 and 93 | |
| Three patients developed grade 2 and 3 diffuse erythema during the second week of administration. Reversible but dose-limiting grade 3 and 4 liver enzyme elevations in two patients treated with 200 mg of BID | ||||||
| Structure not disclosed SCR6290 (SCR6277) | N/A | Tumour-biased distribution. Intracellular enrichment concentration 50-fold greater than blood concentration | Daily doses of 10 to 160 mg | No dose-limiting toxicity (DLT) and grade 4 or 5 TRAE reported has a manageable safety profile | Phase I recruiting | 112 and 113 |
| Seventeen patients (81%) experienced treatment-related adverse events (TRAEs). six patients experienced serious adverse events (SAEs) in which chronic colitis was associated with treatment | ||||||
|
||||||
|---|---|---|---|---|---|---|
| Comp. | Ex:#a | R1 | PRMT5 biochemical IC50 (nM) | Ref. | ||
| MTA+ | MTA− | MTA+/MTA− | ||||
| a Example numbers in the table are the same as those in the cited references. | ||||||
| 36 | 3 |  |
910 | 19000 |
21 | 87 |
| 37 | 3-14 |  |
17 | 413 | 24 | 88 |
| 38 | 3-40 |  |
6 | 193 | 32 | 88 |
| 39 | 8-3 |  |
2.3 | 237 | 103 | 88 |
| 40 | 8-4 | 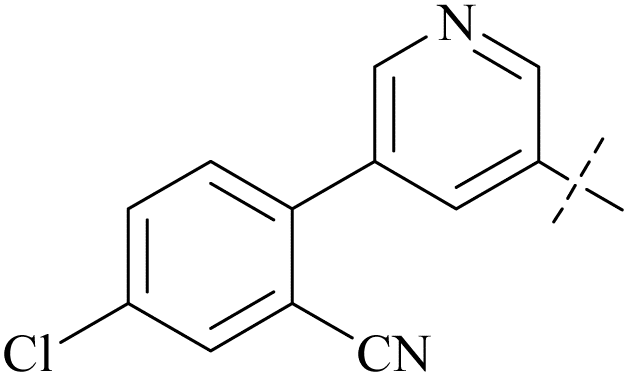 |
3.1 | 313 | 100.9 | 88 |
| 41 | 3-50 | 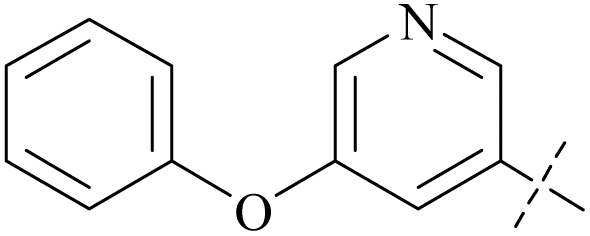 |
10 | 277 | 27.7 | 88 |
| 42 | 4-78 | 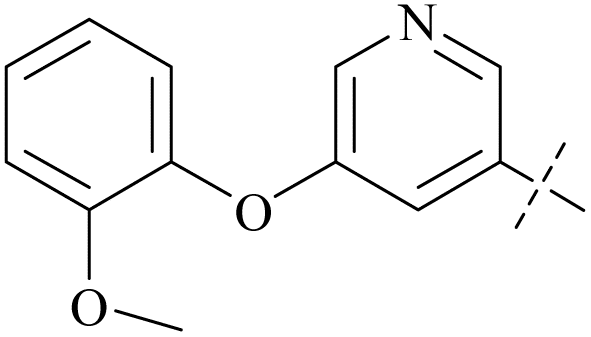 |
0.5 | 66 | 132 | 88 |
| 43 | 4-51 | 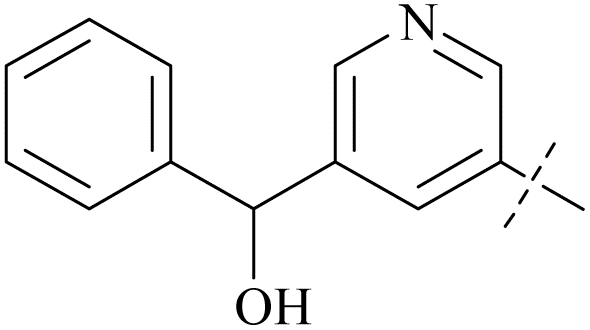 |
5.8 | 530 | 91.37 | 88 |
| 44 | 4-55 |  |
1.2 | 30 | 25 | 88 |
| 45 | 4-61 |  |
3 | 149 | 49.66 | 88 |
|
|||||||
|---|---|---|---|---|---|---|---|
| Comp. | Ex:#a | R1 | R2 | PRMT5 biochemical IC50 (μM) | Ref. | ||
| MTA+ | MTA− | MTA+/MTA− | |||||
| a Example numbers in the table are the same as those in the cited references. | |||||||
| 46 | 9 |  |
H | 0.30 | 5.8 | 19 | 87 |
| 47 | 10 | 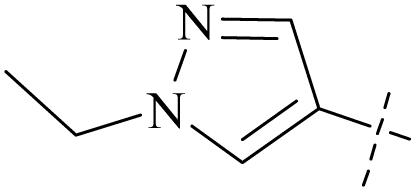 |
H | 0.51 | 10.6 | 21 | 87 |
| 48 | 11 | 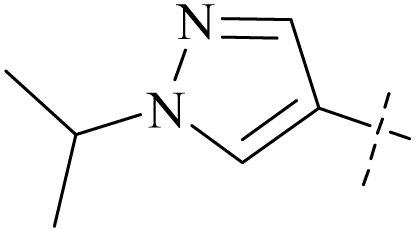 |
H | 0.67 | 11.4 | 17 | 87 |
| 49 | 12 | 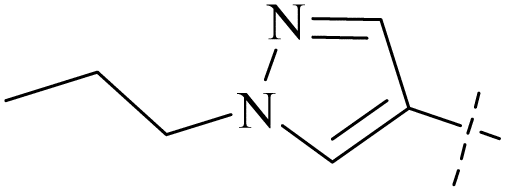 |
H | 0.32 | 7.65 | 24 | 87 |
| 50 | 13 | 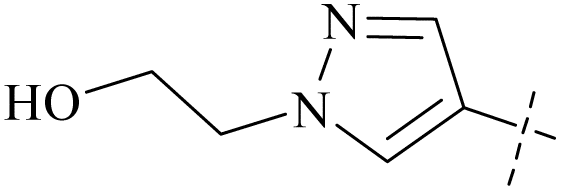 |
H | 0.71 | 11.2 | 16 | 87 |
| 51 | 14 | 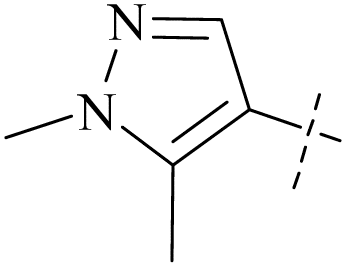 |
H | 0.05 | 1.29 | 26 | 87 |
| 52 | 6-2 | H | 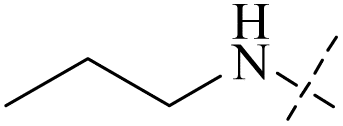 |
>102 | >102 | — | 88 |
| 53 | 5-1 | 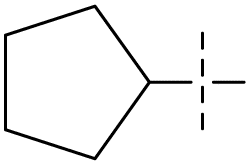 |
H | 72 | >102 | — | 88 |
| 54 | 6-5 |  |
H | >102 | >102 | — | 88 |
| 55 | 5-2 | 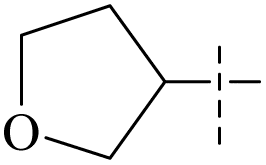 |
H | 57 | >102 | — | 88 |
| 56 | 4-48 | 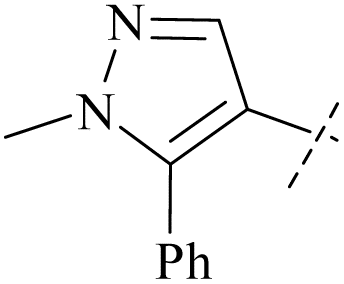 |
H | 1.6 (nM) | 0.045 | 28 | 88 |
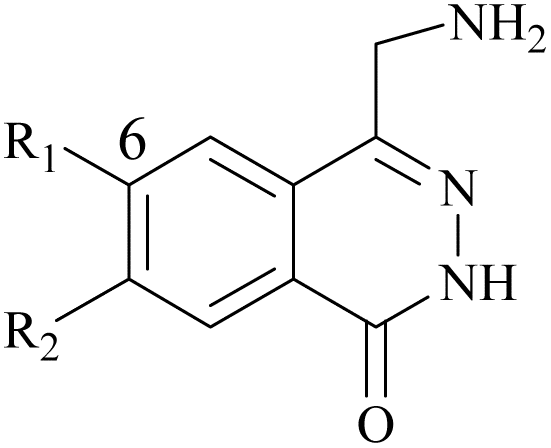
|
||||||
|---|---|---|---|---|---|---|
| Comp. | Ex: # a | R3 | R4 | SDMA IC50 HCT116 MTAP-del/MTAP-WT (nM) | Viability IC50 HCT116 MTAP-del/MTAP-WT (nM)/selectivity ratio | Ref. |
| a Example numbers in the table are the same as those in the cited references. | ||||||
| 61 | 10-1 | 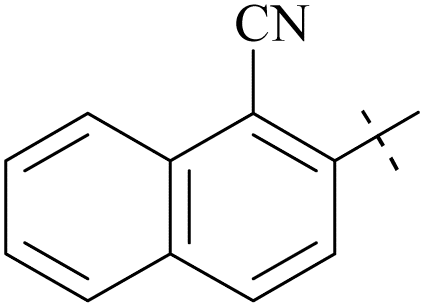 |
–Cl | 15/1123 | 36/2299/64 | 88 |
| 62 | 10-2 | 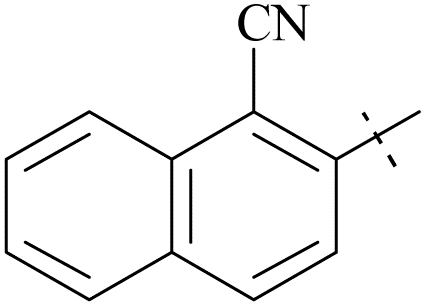 |
–F | 6/2484 | 112/3378/30 | 88 |
| 63 | 10-3 | 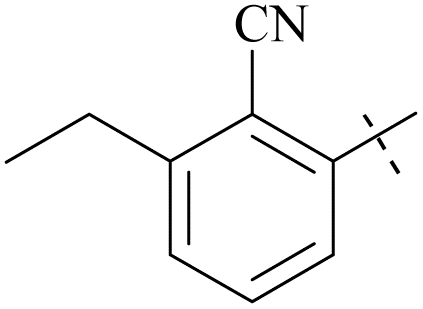 |
–Cl | 7/9215 | 128/>104/78 | 88 |
| 64 | 10-7 | 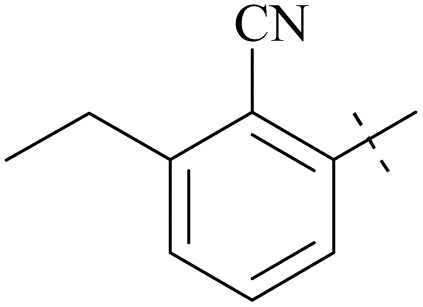 |
–CH3 | 31/1000 | 103/8386/81 | 88 |
| 65 | 2 | 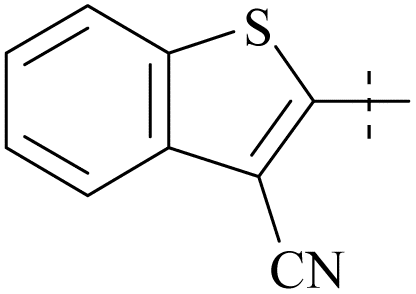 |
–H | 5/>1000 | 58/5800/100 | 114 |
| 66 | 9 |  |
Cl– | 17/>1000 | 52/734/14 | 114 |
| 67 | 10 |  |
–CH3 | 17/>1000 | 30/1470/49 | 114 |
| 68 | 11 |  |
F2HC– | 25/759 | 37/2030/55 | 114 |
| 69 | 12 |  |
–OH | 262/103 | 188/4350/23 | 114 |
| 70 | 13 |  |
–OCH2CH3 | 112/618 | 150/637/4 | 114 |
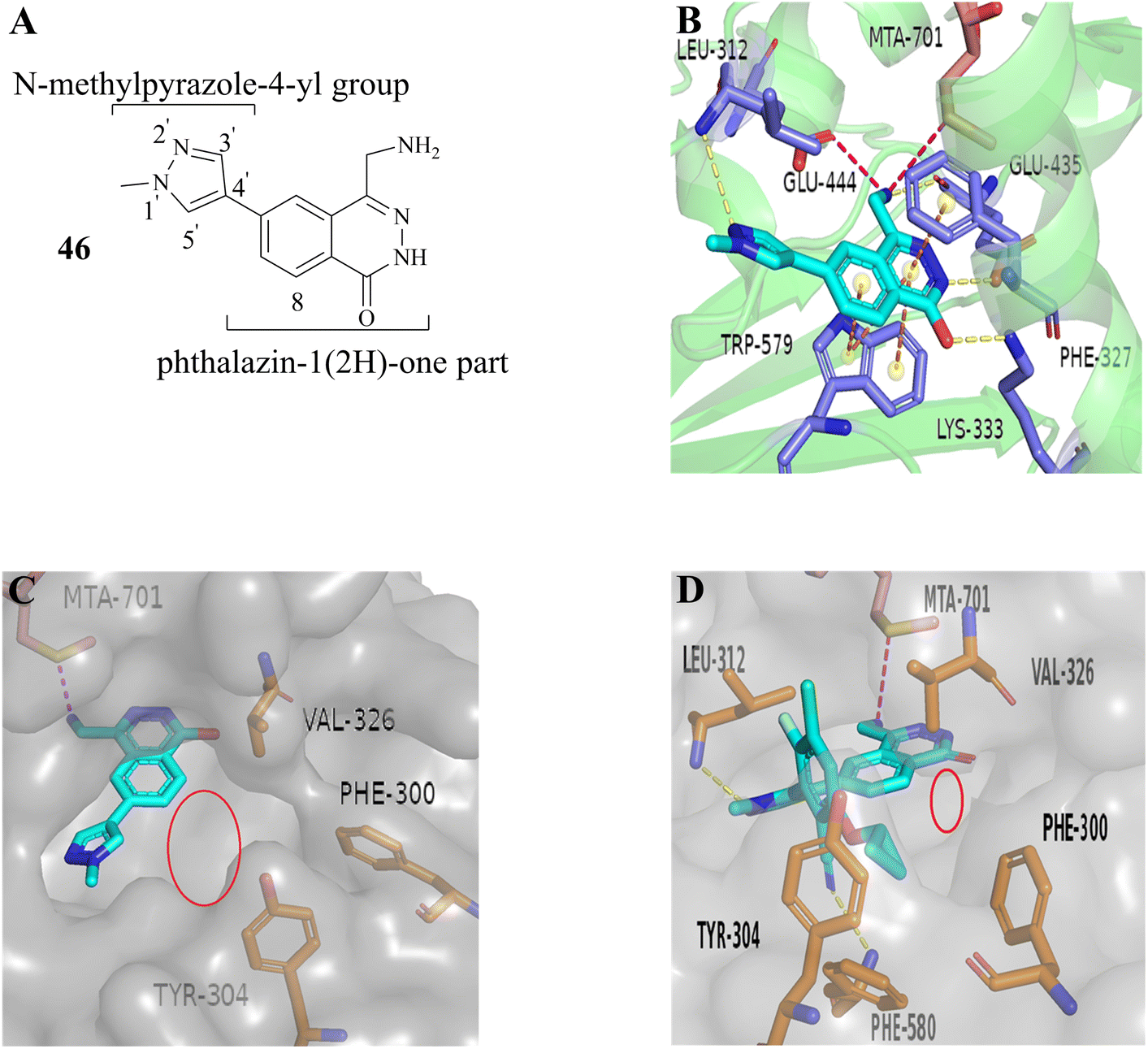 | ||
| Fig. 6 (A) Structure of 46. (B) The co-crystal structure of 46 with PRMT5/MTA (PDB 7S1Q). A new H-bond was observed between the backbone N–H of Leu312 and the N2 of N-methylpyrazole. (C) The lipophilic pocket formed by the side chains of Phe300, Tyr304 and Val326 is observed and there is enough room to accommodate a lipophilic group. (D) The co-crystal structure of 27 (MRTX1719) with PRMT5/MTA (PDB 7S1S). Additionally, there is also a smaller cavity at the side of the 8-position in the phthalazin-1(2H)-one that could accommodate a group.87 | ||
 | ||
| Fig. 7 The fragment 1 growth route of 27 (MRTX1719) and its analogues. | ||
The 1H NMR spectra of compounds 57 and 58 indicate that they might be slowly interconverting rotational isomers. Then, atropisomers 27 (MRTX1719) and (P)-27 (it is the enantiomer of 27 (MRTX1719), the structure is not shown here) are successfully separated by chira supercritical fluid chromatography (SFC). The co-crystal structure of 27 (MRTX1719) with PRMT5/MTA (PDB: 7S1S Fig. 6D) shows that the binding mode of compound 46 was maintained. The newly introduced 2-cyclopropoxy-4-chloro-5-fluoro-6-yl-benzo-nitrile substituent enters the lipophilic pocket and is oriented perpendicular to the N-methylpyrazole group. The nitrile plays a crucial role in improving the potency and selectivity of 27 (MRTX1719) due to the formation of a favourable H-bond between the nitrile and Phe580.
The F atom at the 5-position of the benzonitrile group points toward Leu312. However, using the Cl atom instead of F leads to an obvious loss of potency in the MTAP-del viability assay, and that is likely because the larger Cl-atom substituent creates a collision with the side chain of Leu312. 59 (M-31) was obtained by using the same SFC method.87,115
Based on the calculated torsion rotational energy, atropisomers are usually classified into three classes. The acquisition of 27 (MRTX1719) and 59 (M-31), which belong to class 3 (slow to no rotation), necessitates the use of chiral syntheses or kinetic racemic resolution (see Fig. 7). Significantly, the acquisition of compound 60, as a class 1 compound (free rotation), does not need the chiral syntheses or kinetic racemic resolution. It exhibits high potency and selectivity (MTAP-del IC50 = 58 nM, MTAP-WT IC50 = 5800 nM, MTAP-WT/MTAP-del = 100 fold in the HCT116 viability). However, the human hepatocyte intrinsic clearance (Clint = 41 mL min−1 kg−1) of 60 is higher than that of 27 (MRTX1719 Clint < 18 mL min−1 kg−1). After considering all cases, including but not limited to the potency, selectivity, and PK profiling in preclinical species, 27 (MRTX1719) was selected for clinical trials.87,88,114,115 In the meantime a scalable synthesis of this atropisomeric compound and an efficient isolation of the desired isomer have been established to support the clinical trials through further development of the racemic medicinal chemistry route.116,117 Additionally, there is a smaller cavity at the side of the 8-position in the phthalazin-1(2H)-one 35, which could accommodate a group (see Fig. 6C and D). And then, the SAR about the 8-position of the phthalazin-1(2H)-one 35 was also investigated (the data and structure are given in Table 5). In general, a small substituent (such as compounds 61–68) at the 8-position can show higher potency and selectivity than that of the larger substituents (such as compound 70). For the compounds in Table 5, it can be observed that –CH3-substituted compounds at this position consistently exhibit the highest potency in the HCT116 MTAP-del viability assay. Furthermore, an emerging trend suggests that –CH3 > –Cl > –CHF2 > –F > –OH. However, their selectivity ratio (viability IC50 HCT116 MTAP-del/MTAP-WT) is markedly decreased compared to –H-substituted compounds (such as compound 65). It is worth noting that the substituents of the 8-position in the 35 may have an effect on the human hepatocyte intrinsic clearance (Clint). For example, the Clint of compound 67 decreases to 27 mL min−1 kg−1 and the Clint of both (compounds 69 and 70) is below 18 mL min−1 kg−1, while compound 65 is in the high range (Clint: 41 mL min−1 kg−1). The –CHF2-substituted compound 68 (Clint: 44 mL min−1 kg−1) remains similar to 65. However, the Clint of –Cl-substituted compound (66) increases to 71 mL min−1 kg−1.
In 2023, partial details of the SAR of the 27 (MRTX1719) are further revealed by three Chinese patents (see Fig. 8. These details are highlighted in red). Among them, patent CN116178347A discloses some new information about the substituents on the benzo nitrile. Moreover, compounds 72 and 73 have similar potency as 27, possibly indicating a similar binding conformation for these three compounds (the data is not shown here).118–120
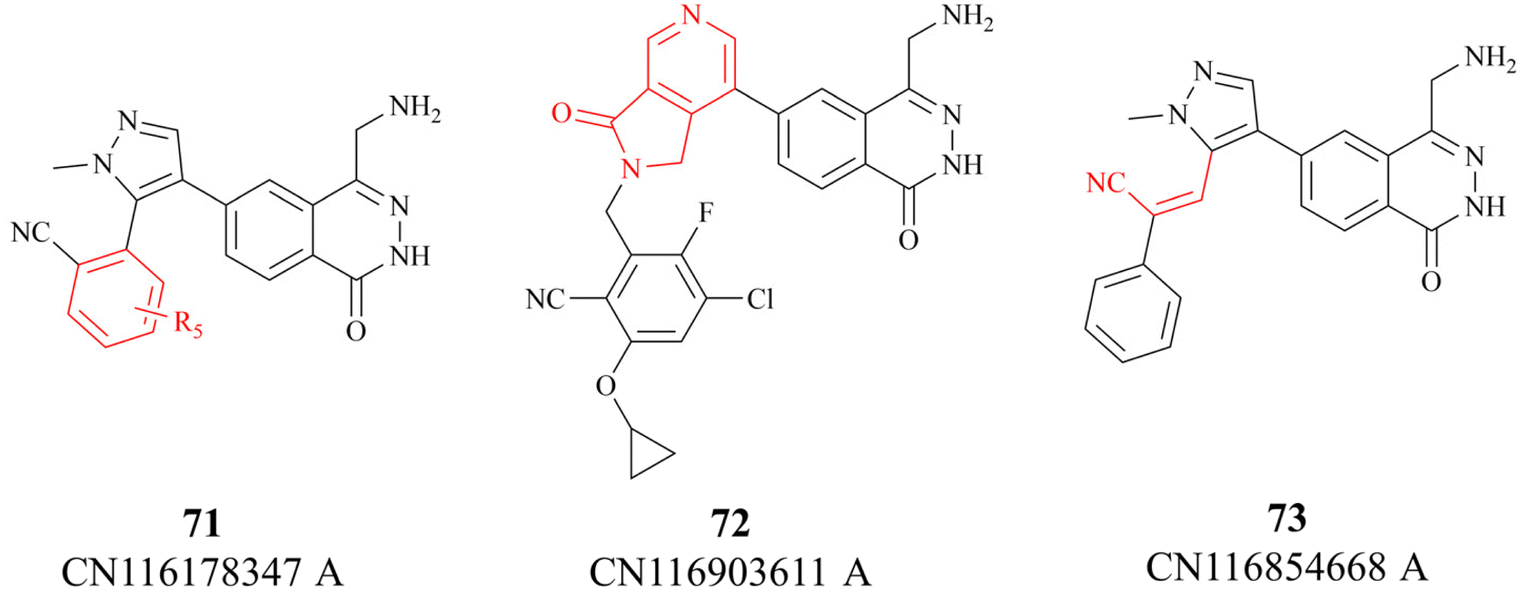 | ||
| Fig. 8 27 (MRTX1719) analogues. | ||
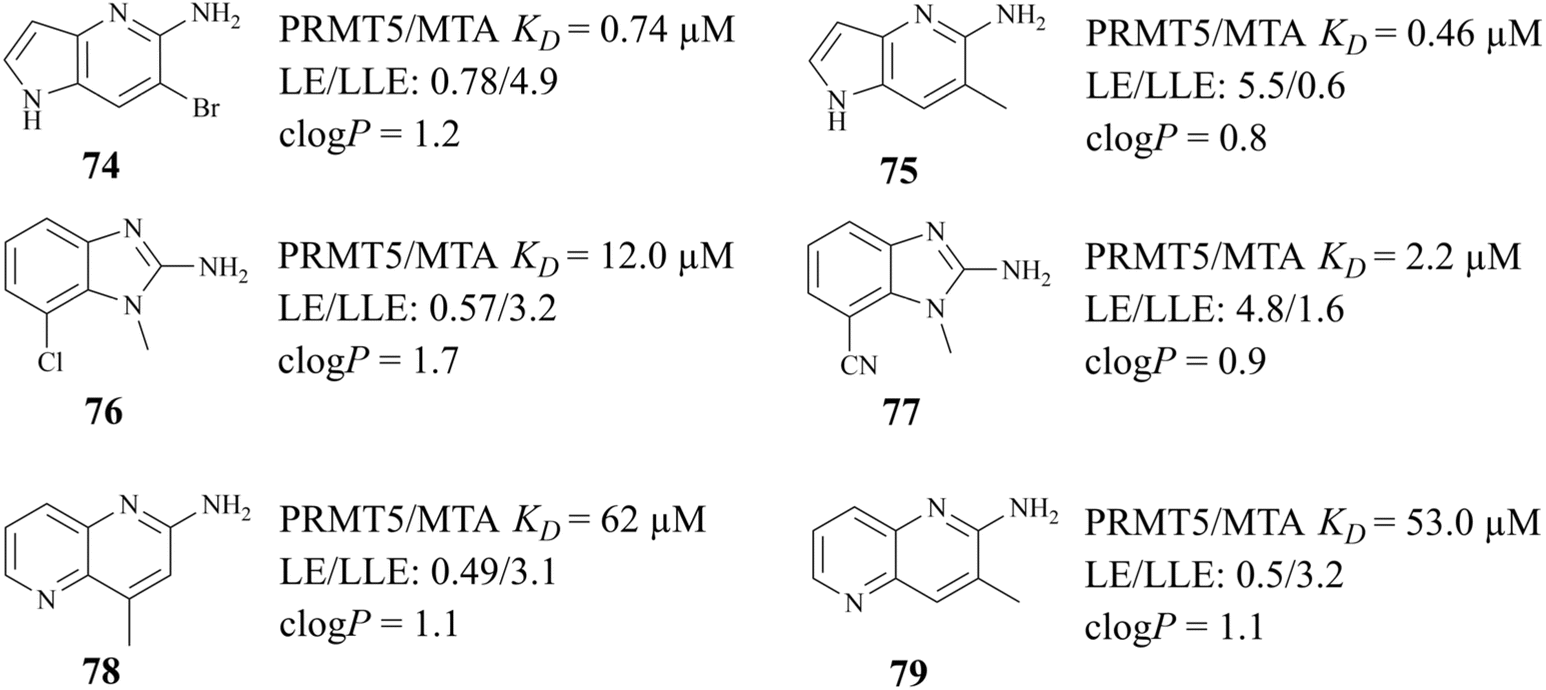
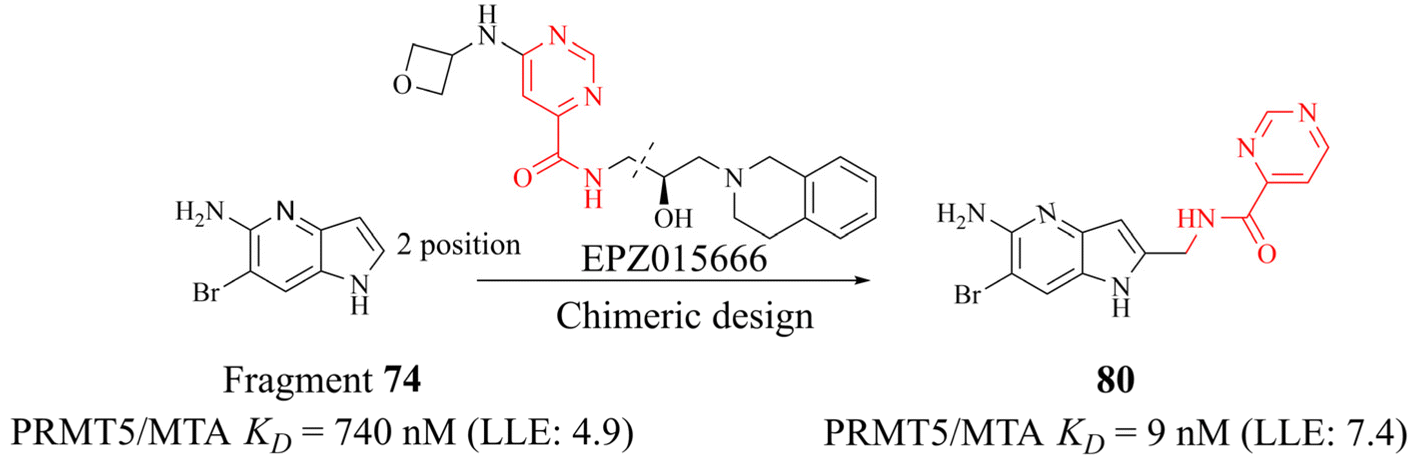
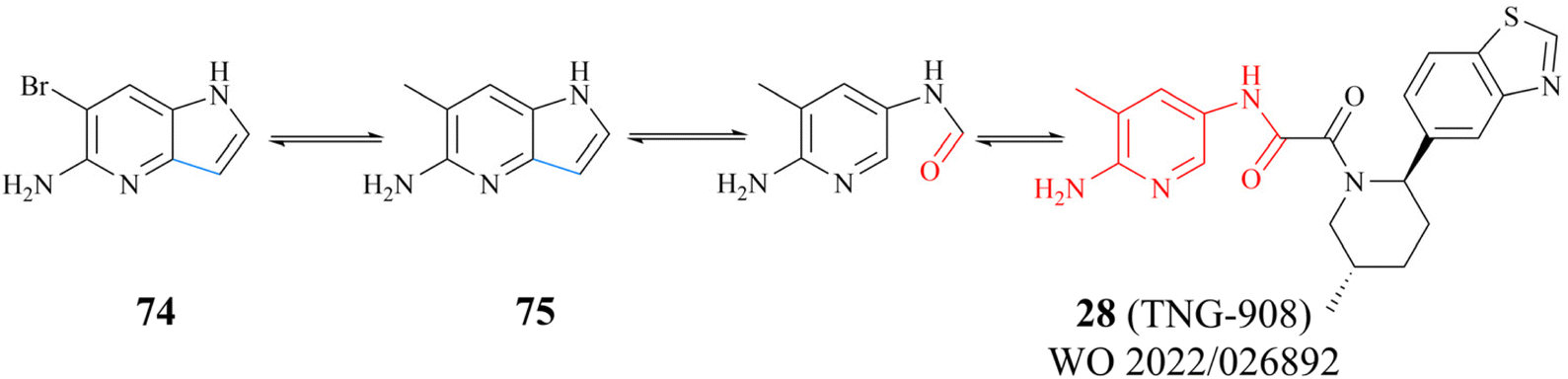
The success of 27 (MRTX1719) demonstrates that the development strategy combining the FBLD method with the co-crystal structure-based drug design (SBDD) is a highly feasible way for the discovery of PRMT5/MTA complex inhibitors. There are still several analogues of fragment 35 (fragments 74–79 shown in Fig. 9). Among them, fragments 74, 76, 78 and 79 were discovered together with the fragment 35 (PRMT5/MTA KD = 10.2 μM, LE/LLE: 0.54/4.9, clogP: 0.1) by using the FBLD approach. And fragments 75 and 77 were obtained by optimising the 74 and 76 respectively. Furthermore, each of these fragments might lead to the discovery of lead compounds just like 27 (MRTX1719). And interestingly enough, Matthew A. Marx et al. also describe another drug design strategy—chimeric design. Firstly, further analysis of the co-crystal structure of fragment 74 suggests that the 2-position of 74 is the most appropriate position for fragment growth. Subsequently, compound 80 was obtained by introducing the pyrimidine-4-carboxamide group of EPZ015666 (structure shown in red in Fig. 10) into this position. Compared to the original fragment 74 (PRMT5/MTA KD = 740 nM), the addition of the pyrimidine-4-carboxamide substituent caused an 82-fold increase in binding KD (PRMT5/MTA KD = 9 nM), and the LLE increased from 4.6 to 7.4 (ΔLLE = +2.5). These results demonstrate that it is another economically feasible way to increase the potency of a fragment by a chimeric approach with known ligands.121,122
 | ||
| Fig. 9 Analogues of fragment 35. | ||
 | ||
| Fig. 10 The chimeric design enabled optimisation of fragment 74 to obtain 80. | ||
Another useful example in the development of the PRMT5/MTA complex inhibitors is the discovery of 28 (TNG908).85,86 John P. Maxwell and colleagues initially employed a number of search strategies to identify potential lead compounds for drug discovery, including DEL and fragment screening. Among these, a high-throughput screen of a 560000-compound library at 20 μM using peptide substitution yielded a promising result. The fluorescence anisotropy (FA) method was employed to detect the displacement of a tamra-labelled histone H4 peptide from PRMT5 in the presence of 50 μM MTA. Subsequently, the compounds were subjected to further evaluation. Ultimately, 11 unique structural series, comprising 21 compounds, were obtained. Several series were eliminated due to an inability to demonstrate SAR traceability. Compound 81, one of the three remaining structural series, was selected as the lead compound and SBDD was employed as a guide for further exploration of its SAR. (Structure and physicochemical properties of 81 are presented in Fig. 11A). Preliminary SAR studies of compound 81 indicate that the 2-position of the piperidine is tolerant to a variety of different substituents, including phenyl, cyclohexyl and pyridyl. However, it is intolerant to smaller substituents such as methyl. The substitution at this position exhibits a clear stereochemical preference, with the preferred stereochemistry being R. This information was used to obtain compound 82.85
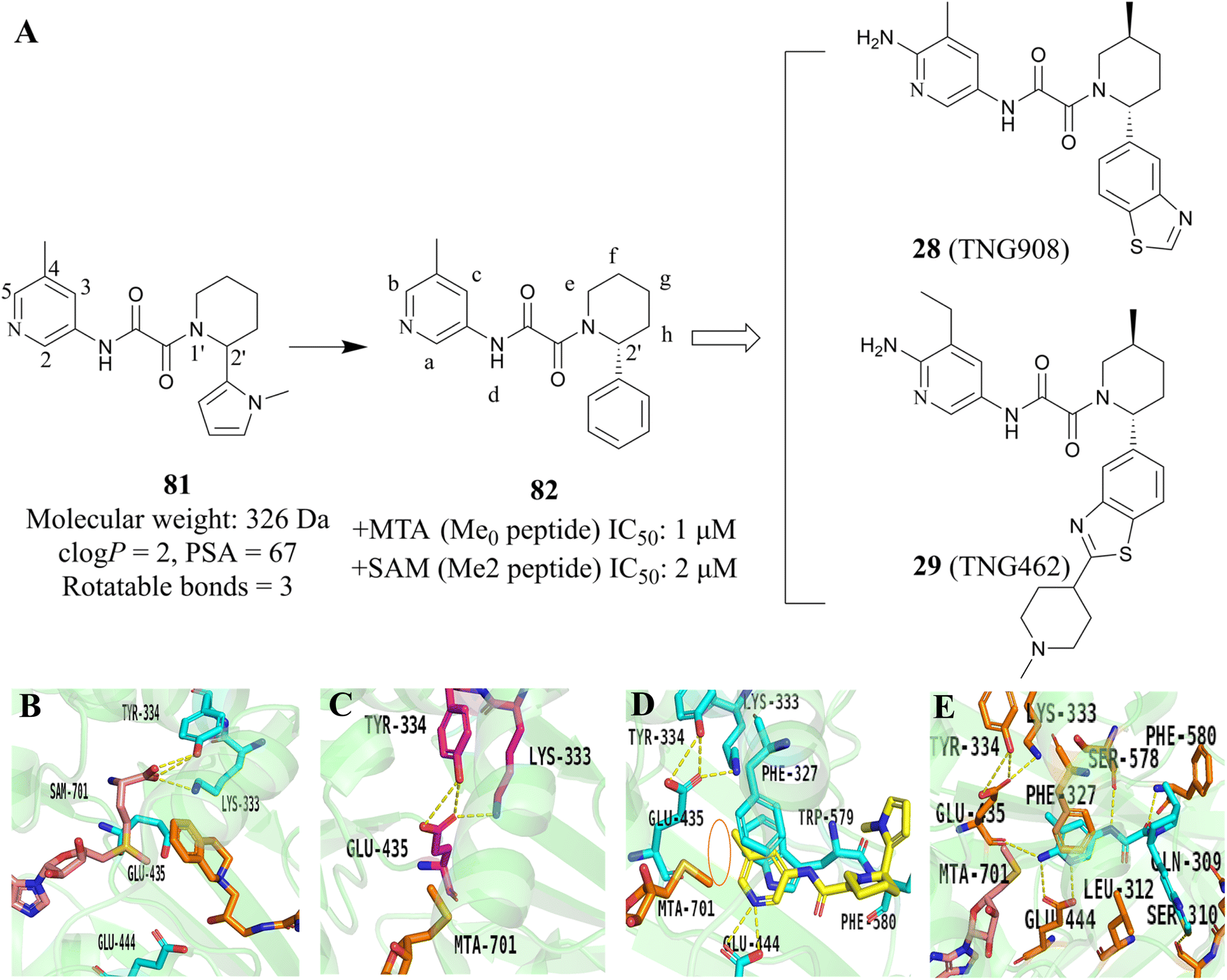 | ||
| Fig. 11 The development strategy, which combined the HTS and SBDD methodologies, yielded the 28 (TNG908).85,86 (A) The development route of 28 (TNG908) and 29 (TNG462). (B) X-ray cocrystal structure of PRMT5/SAM/substrate complex. Glu435 rotates toward substrate side chain (PDB 4X61).21,22 (C) X-ray cocrystal structure of PRMT5/MTA complex. Glu435 rotates to fill space previously filled by SAM (PDB 8VEO).21,22,85 (D) X-ray cocrystal structure of 82R (yellow) bound to PRMT5/MTA (PDB 8VET).85 (E) X-ray cocrystal structure of 28 (TNG908) bound to PRMT5/MTA (PDB 8VEY).85 | ||
Subsequently, methyl groups were introduced at each position of 82 (positions a–h, as shown in Fig. 11A) in order to further investigate the SAR of 82. The introduction of a trans methyl group at the f-position resulted in a 20-fold increase in potency while maintaining selectivity. During the SAR investigation of 82, enantiopure isomers 82R (the structure is not shown here) were obtained by chiral resolution of racemic 81 using SFC.
In order to obtain a compound that could cross the blood–brain barrier (BBB), the SBDD was employed to guide the further optimisation of compound 82. The X-ray cocrystal structure of 82R with the PRMT5/MTA complex reveals that the pyridine ring establishes van der Waals contacts with MTA and engages in π–stacking interactions with Phe327 and Trp579. Additionally, the N-atom on the pyridine ring forms a hydrogen bond with Glu444. It is noteworthy that the methyl group at the 5-position of the pyridine ring points to Lys333 and Glu435, thus locking Glu435 in the preferred spatial configuration for binding to MTA (see Fig. 11D). This prevents Glu435 from forming the rotamer necessary for binding to SAM, which helps to further explain the high selectivity of the PRMT5/MTA inhibitor (Fig. 11B and C). Furthermore, a small space near the 5- and 6-positions of the pyridine ring was observed to accommodate some small groups. The NH of oxamide forms a hydrogen bond with Ser578, and the carbonyl group of oxamide is also involved in the formation of the hydrogen bond (Fig. 11D).
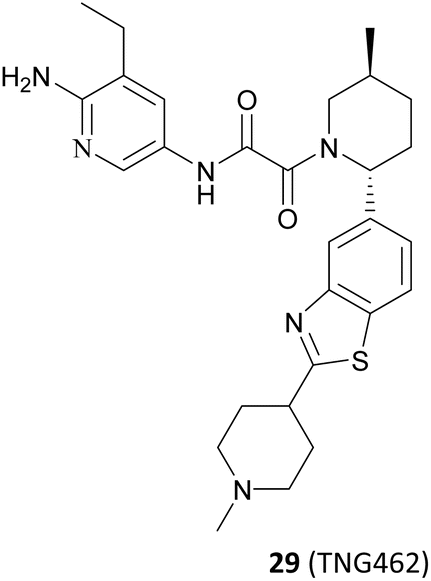
Based on these understandings, the introduction of an amino group into the 6-position of the pyridine ring of 82 results in a 200-fold increase in potency compared to compound 82, demonstrating a significant selectivity boost (HAP1 MTAP-null ICW 17 nM, HAP1 MTAP-null viability 420 nM, 14-fold viability selectivity). The substitution of the methyl group at the 5-position of the pyridine ring with the aminocarbonyl group leads to a 120-fold increase in potency relative to that of 82, while exhibiting higher selectivity (30-fold viability selectivity). Further studies of the substituent at the 2′-position of the piperidine revealed that the introduction of a hydroxyl group in the para-position of the benzene ring may form a beneficial hydrogen-bonding interaction with the carbonyl of Ser310. However, it is observed that those compounds containing more hydrogen bond donors, such as NH and OH, display a tendency to have lower penetration and higher efflux than their matched compounds without additional hydrogen bond donors. Furthermore, they exhibit lower human liver microsomes stability. (The structure of these compounds is not shown here). Interestingly, an uncommon interaction of a C–S σ* orbital and the carbonyl of Ser310 is observed when the benzothiazole is used to replace the phenyl. This interaction demonstrates that it is possible to achieve a higher valence without the addition of an additional hydrogen bond donor. In addition, rational cyclisation studies from the pyridine onto the oxamide showed that the pyridine oxamide portion and the fragments depicted in Fig. 9 such as 74 and 75 are bioisosteric groups (it is highlighted in red in Fig. 12).85,86 Nevertheless, although these cyclised compounds exhibit notable activity, they display reduced solubility and metabolic stability relative to their counterparts utilising the oxamide as a linker. By combining these SARs, 28 (TNG908),86 a potent and selective, brain penetrant PRMT5/MTA inhibitor currently undergoing phase I/II clinical trials (NCT05275478), was discovered. Another structurally similar compound 29 (TNG462),86 currently undergoing phase 1/2 clinical trials (NCT05732831), was also discovered.123
 | ||
| Fig. 12 The structural relationship between the 3-methylpyridin-2-amine pharmacophore and 74. | ||
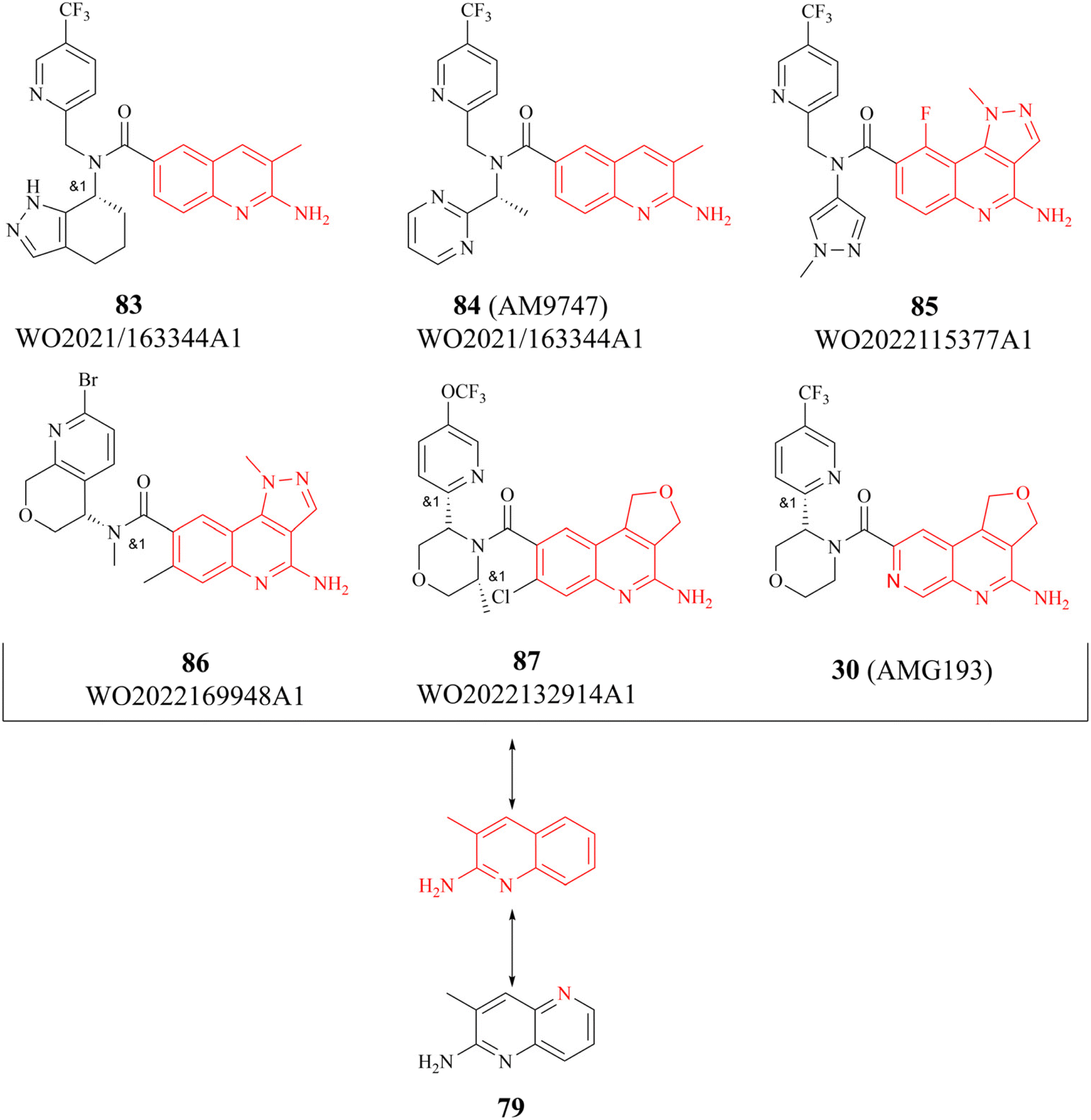
There are several other research teams developing or working in parallel on PRMT5/MTA inhibitors. Amgen Inc. has filed four patents in less than two years (2020–2022) on its PRMT5/MTA complex inhibitors, and the compounds covered by these patents possess a bicyclic (the representative compounds 83 and 84) or tricyclic (the representative compounds 85–87) quinolin-2-amine derivative scaffold, which almost all of these compounds exhibit similar biological activities as 27.124–128 The candidate compound 30 (AMG-193), as an MTA-cooperative PRMT5i, is in clinical trials (NCT06333951, NCT05094336).89
Interestingly, the quinolin-2-amine derivative scaffold is strikingly similar to that of the aforementioned fragment 79 (similar parts of their structures are highlighted in red in Fig. 13). Furthermore, the PRMT5 substrate binding site comprises the well-known double E ring (two acidic glutamate residues, Glu435 and Glu444), which facilitates the recognition of basic groups, including the protein arginine side chain.28 And it can be postulated that the known PRMT5/MTA complex inhibitors may all have a key pharmacophore. And, the pharmacophores of these known PRMT5/MTA complex inhibitors should share some common features to ensure that specific binding reactions between the pharmacophore and the PRMT5/MTA complex can be formed. For instance, an appropriate aromatic planar structure may facilitate the formation of the π–stack interaction with the side chains of Phe327 and Trp579. And the basic substituents or basic atoms (such as primary amino groups and N-atoms on fragments 35 and 74–79) on this aromatic planar structure may also be necessary for the formation of a stable specific interaction with Glu444 and MTA. Similarly, another basic or neutral substituent of appropriate size on this pharmacophore, such as the 5-methyl group on the pyridine ring of 28 (TNG908), may potentially improve target selectivity and potency.
 | ||
| Fig. 13 The heterocycles as PRMT5/MTA complex inhibitors. | ||
2.2 PRMT5-Adaptor protein interaction inhibitors (PAPII)
As previously mentioned, the PBM-binding groove is located in the TIM barrel, and a small molecule which could occupy the PBM groove would affect the methylation of more than 25 PRMT5 substrates. In 2021, Ianari et al. disclosed a series of PBM-competitive inhibitors that the 31 (BRD0639) represents.31 This research outcome remains one of the successful examples of the application of the SBDD methodologies. A competitive fluorescence polarisation (FP) analysis was employed to assess the capacity of compounds to displace the interaction between fluorophore-labelled RIOK1 PBM peptides and purified PRMT5:WDR77 heterodimers. And Ianari et al. conducted a screening of over 900K small molecule compounds with the objective of identifying inhibitors of PBM peptide interactions. Then, compound 58 (FP IC50 of 12 μM, solubility in PBS of 1.2 μM) was finally identified as the lead compound. The subsequent SAR exploration process was guided by SBDD. This process can be further divided into two phases. Initially, solubility and potency were the primary objectives, whereas in the subsequent phase, a balance between potency and glutathione (GSH) electrophilic reactivity was the key focus. The co-crystal structure of the lead compound 88 (Fig. 14A) with the PRMT5 complex shows that the PBM groove is occupied, and that cys278 makes a covalent bond with the lead compound 88 via a nucleophilic substitution reaction. In the co-crystal structure, a four-ring π–π stacking interaction (Tyr286-pyridine-arylcore-Phe243) is observed. Moreover, multiple hydrogen bonding interactions are also observed between the sulfonamide and Asn239, the sulfonamide and Lys241, the acid amides and Ser279, and the pyridazinone and Gln282 (see Fig. 14B). Further SAR studies have revealed that the replacement of the pyridyl group in the R1-portion with a six- or seven-membered nitrogen heterocyclic ring results in a loss of potency (the structure is not shown here). The pyridine isomers (compounds 89 and 90) exhibit a similar potency and stability to that of 88 (see Fig. 14C). With regard to the modification of R2, aromatic six-membered rings are tolerated such as the pyridyl ring (compound 91). Furthermore, the introduction of a methyl group at the b- or c-position is tolerated (compounds 93 and 94), while the introduction of a methyl group at the a-position results in a significant loss of potency (compounds 92). However, these modifications at the a-, b-, and c-positions may cause a reduction in solubility. And the introduction of a methyl group at the d-position is markedly less active (the structure and data are not shown here). A methyl group at the e-position is tolerated, with a very strong stereochemistry preference (the preferred stereochemistry is S such as compound 95). Meanwhile, the introduction of a methyl group at this position markedly enhances the stability of the compounds due to the steric hindrance (as exemplified by compounds 95 and 96). The electrophilic reactivity of R3 showed a negative correlation with the selectivity of the compounds. To achieve an acceptable balance between activity and reactivity, this part was further optimised. And the electrophilic reactivity of the compounds towards glutathione was used as an evaluation index of the reactivity of the compounds. As a result, the monochlorine-substituted compound 97 has been found to exhibit an optimal balance. From this, 31 (BRD0639) was obtained.31 (Fig. 14C).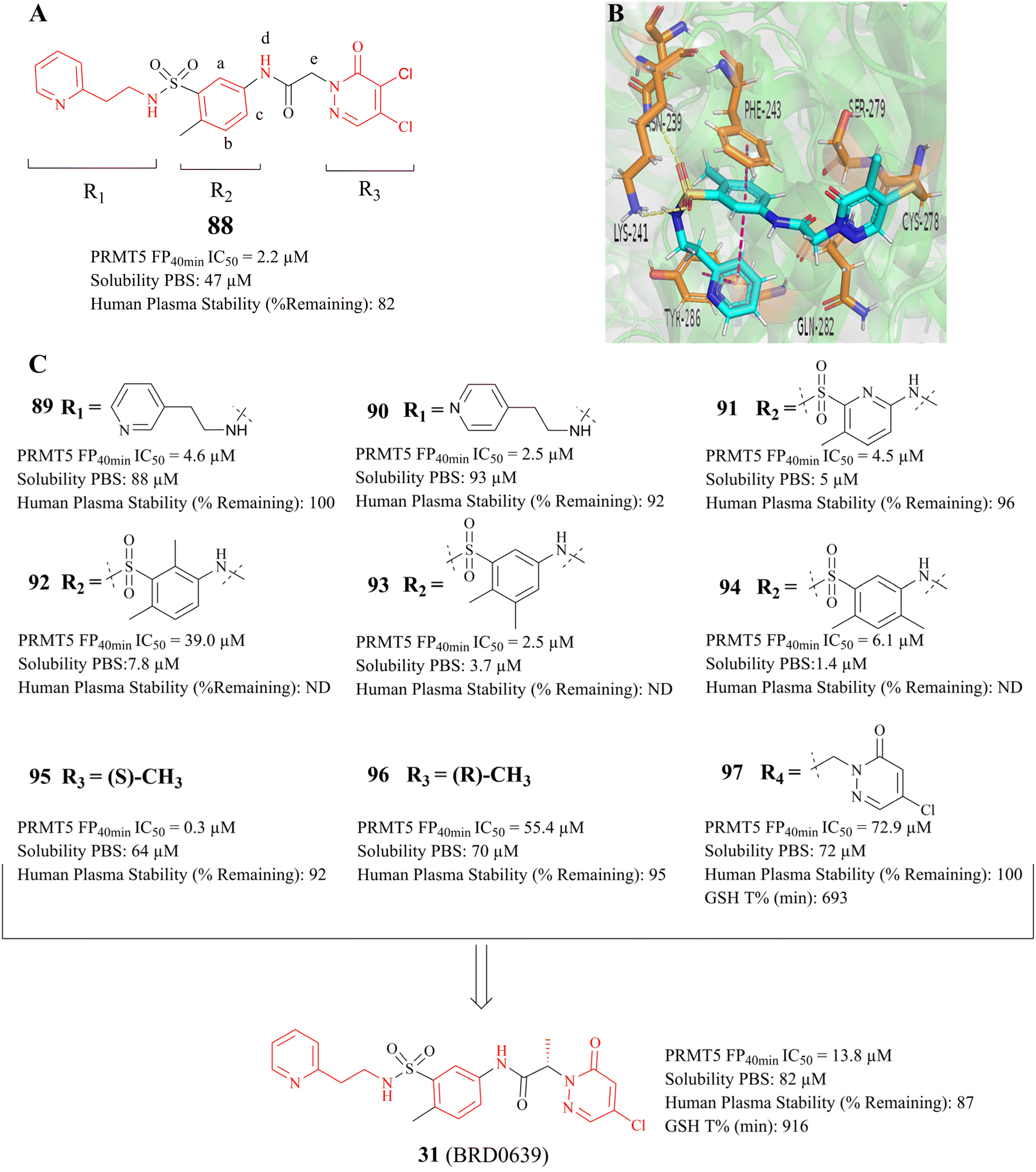 | ||
| Fig. 14 (A) Lead compound 88. (B) The cocrystal structure of compound 88 bound to the PRMT5 complex (PDB: 7M05).31 (C) Further SAR exploration of 88 results in a PBM-competitive inhibitor 31 (BRD0639). The FP is a competition fluorescence polarization assay. | ||
In 2022, Krzyzanowski and colleagues developed another type of PRMT5–adaptor protein interaction inhibitor (PAPII). The research group initially conducted a screening and identification process, whereby the RioK1-derived PBM sequence was identified as a promising candidate. It is well-known that the short amino acid chains are more easily hydrolysed by the proteolytic enzymes than those of the long chains in the physiological state. Thus, the peptide stapling technique is employed to improve the stability of the short chain. According to the co-crystal structure of RioK1-derived PBM bound to the PBM groove, Gly14 and Asp17 on the RioK1-derived PBM sequence (Fig. 15) are identified as the most ideal tether position for structural optimisation. And then these two residues may be linked through a suitable flexible chain to form a macrocycle, which would contain the key residues Gln15 and Phe16. This could maintain the spatial conformation of the short-peptide and effectively improve its enzymatic stability. Further studies have shown that this N-terminal SRV motif is not necessary for the affinity and that the N-terminal VP motif is essential for maintaining affinity (such as compounds 99–101). Owing to a potential steric effect of the attached group, the N-terminal fluorescein isothiocyanate (FITC) label (such as compound 99) may result in a reduced apparent protein binding affinity. The C-terminal FITC label (compound 100) does not interfere with binding to the PBM groove. Cyclopeptides with 5-atom-long linker are preferred such as compound 102. Then a 4-atom-long linker with a double bond is also tolerated, but only one of the isomers of the unsaturated linker will show a strong affinity for the target (the structure is not shown here). Replacing Asp20 with Gla and using Phe(4-NO2) instead of Phe16 are both beneficial in improving affinity (compounds 103–105). The combination of these modifications in the compound resulted in the cyclopeptide 32. Cyclopeptide 32 (IC50: 654 ± 476 nM) shows a comparable level of potency to that of the PBM-competitive inhibitor 31 (BRD0639) (IC50: 568 ± 284 nM) in a competitive assay. Moreover, cyclopeptide 32 does not interfere with the interaction between PRMT5 and MEP50. However, the physicochemical properties of cyclopeptide 32 reveal that its cell permeability may be poor and further optimisation is required.30,32 (Fig. 15).
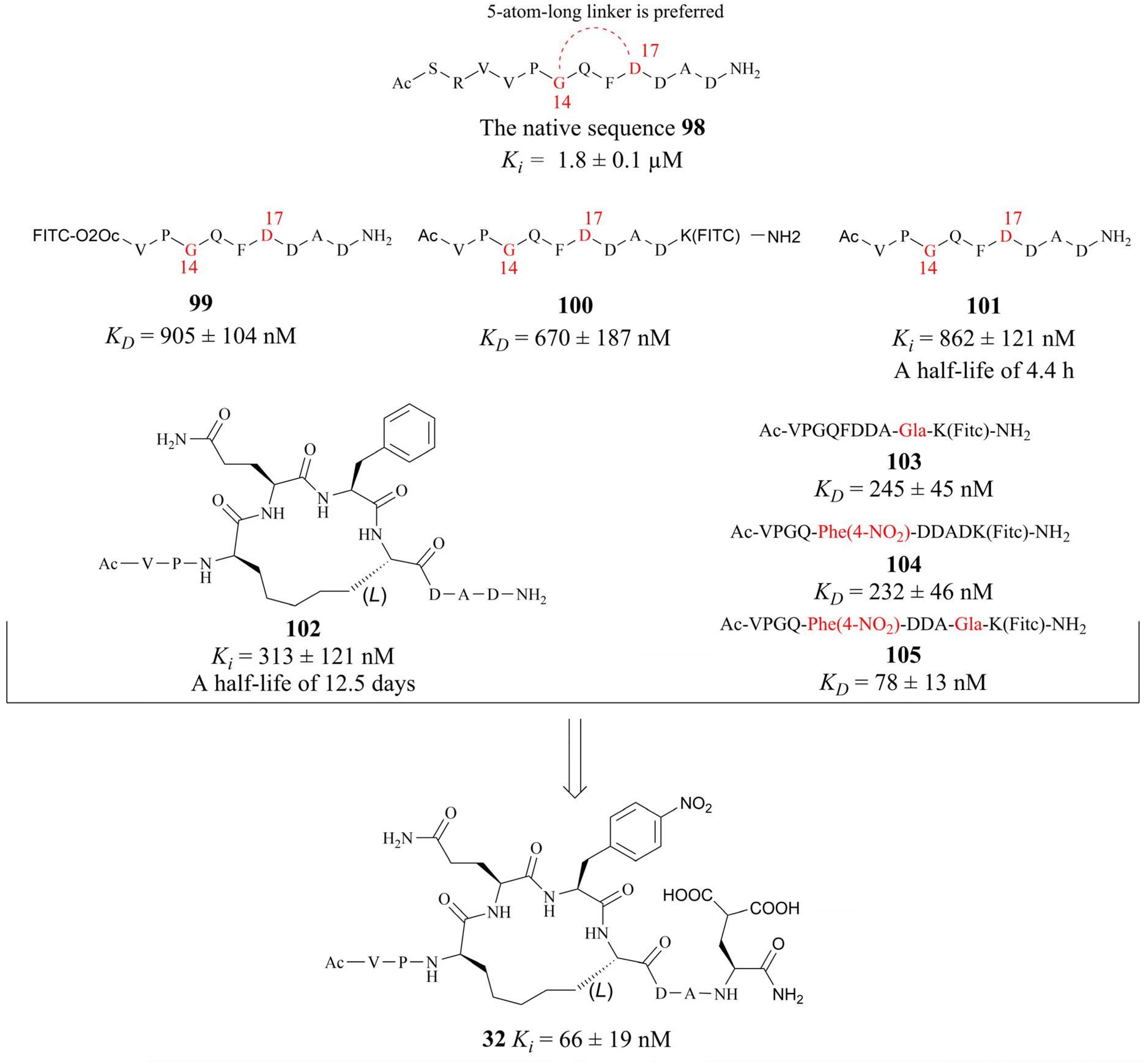 | ||
| Fig. 15 Macrocyclization of the RioK1-derived PBM sequence results in a PAPII 32. | ||
In addition, the Hu group disclosed a series of novel PRMT5:MEP50 PPI inhibitors in 2022, represented by compounds 106 and 33 (see Fig. 16). Although both 106 and 33 have almost identical chemical structures, differing only in a substituent group on the isoxazole ring (shown in red in Fig. 16), the potency of compound 33 (IC50: 430 nM) is approximately 4-times greater than that of 106 in the LNCaP cell growth inhibition assay. Nevertheless, these inhibitors, which are intended to be used as anti-cancer drugs in clinical trials, will still face significant challenges. For example, the 33 may have low solubility and its hydrazide linker does not appear to be beneficial for toxicity and stabilisation in vivo. Therefore, more work may need to be done to optimise this. Interestingly, the docking model of 106 into the TIM barrel shows that its isoxazole moiety is solvent-exposed, suggesting that it may serve as a suitable handle for structural modification.25
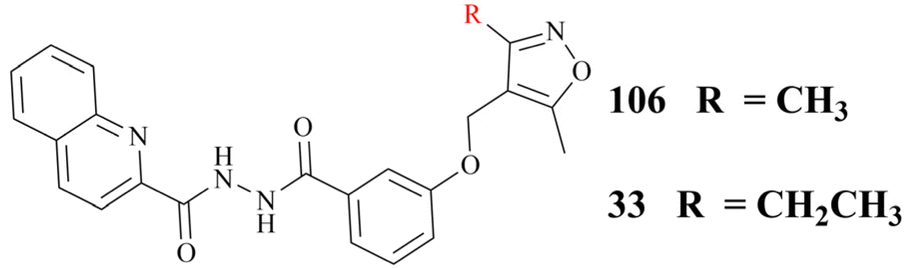 | ||
| Fig. 16 Structures of compounds 106 and 33. | ||
3 Other PRMT5 inhibitors based on the allosteric, covalent, and PROTAC modes of action
3.1 Allosteric inhibitors
In 2020, a compound 108 (R-1a) (it was originally used as the BACE inhibitor) was screened and identified by Palte et al. as a potent allosteric PRMT5 inhibitor (EC50: 108 (R-1a) = 16 nM).19,20 In the co-crystal structure (Fig. 17B, PDB: 6UXX), the adamantane moiety of 108 (R-1a) occupies a hydrophobic cage formed by five amino acid residues (Leu436, Leu437, Phe519, Phe555, and Tyr468). The iminohydantoin core forms two hydrogen bonds with Glu444. The methoxyphenyl group is surrounded by aromatic residues Phe519, Phe584, and Phe602, and makes an edge-to-face stacking interaction with Tyr613. Moreover, this group may occupy the position of Phe440 residue, thereby forcing it to move towards the SAM binding pocket. This unique binding mode deeply impacts both the SAM-binding site pocket and substrate-binding site so that none of the SAM and substrate can bind with PRMT5 in their respective binding sites. Surprisingly, the co-crystal structure of the PRMT5/MEP50 complex with 108 (PDB: 6UXX) shows that there is a narrow channel between the allosteric binding site and the substrate binding site, which appears to provide an opportunity for further structural optimisation.19 In addition, the use of other hydrophobic groups (such as a propylcyclohexane group) instead of the adamantyl moiety seems to be ok, but further screening studies on hydrophobic groups are necessary.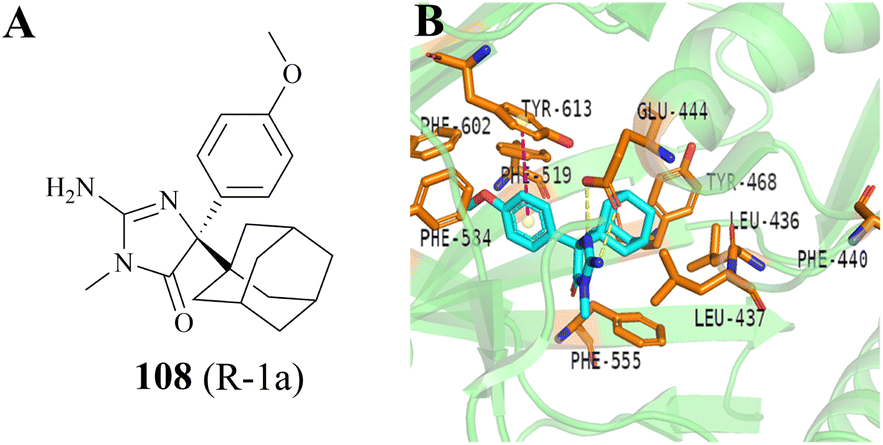 | ||
| Fig. 17 (A) The structure of 108. (B) The co-crystal structure of the PRMT5/MEP50 complex with 108 (PDB: 6UXX).19 | ||
3.2 Covalent inhibitors
In general, the binding of SAM competitive inhibitors to PRMT5 occurs by non-covalent means;33,49,129,130 However, the potency of these reversible SAM competitive inhibitors is likely to be affected by SAM levels in tumour cells.131,132 Interestingly, in the SAM binding pocket, there is a unique non-catalytic cysteine (C449), whereas other PRMTs contain a serine residue at the same position. The co-crystal structure shows that the space length between C449 and C6–NH2 (and N7 atom) of the adenine ring of MTA (or SAM) could accommodate a covalent warhead (see Fig. 18). Moreover, C449 exists in part as a thiolate ion under physiological conditions. These findings provide a foundation for the development of irreversible SAM competitive inhibitors. Based on this information, two acrylamide derivatives of MTA (see Fig. 19A compounds 109 and 110) were designed and synthesized in 2017; However, they exhibited a poor inhibitory activity (IC50: 5.01 μM and 2.38 μM, respectively) in a biochemical methylation assay.134 And then, Luengo et al. developed another series of PRMT5 covalent inhibitors using LLY-283 as the parent compound in 2019 (compounds 111–115 see Fig. 19A)34 Both compounds 112 and 113 show valuable enzymatic inhibitory potency (comparable IC50: 11 nM and 19.5 nM, respectively) in the PRMT5/MEP50 biochemical assay. Compound 112 is initially converted to 113 under the assay conditions (which closely resemble physiological conditions, with a pH of 8.0). Subsequently, a plausible trans double bond (see Fig. 19B) is formed between the S atom of Cys449 and the adenine ring C6–NH2; However, the covalent bond between Cys449 and the adenine ring of compounds 111, 114, and 115 has not been formed. This is probably due to the fact that these compounds lack the chemical reactivity with Cys449 under assay (pH: 8.0) conditions or the corresponding aldehyde does not match the active site or the potential affinity of these compounds for the SAM binding pocket is reduced. Additionally, the co-crystal structure of PRMT5/MEP50 complex with compound 113 (PDB: 6K1S) shows that, apart from the plausible trans double bond interaction, all other key binding interactions between 113 and PRMT5/MEP50 complex are similar to those of the parent compound 11 (LLY-283) (PDB: 6CKC). It should be noted that the aldehyde is very easy to be metabolized by enzymes in vivo, and further structural optimisation will be necessary.34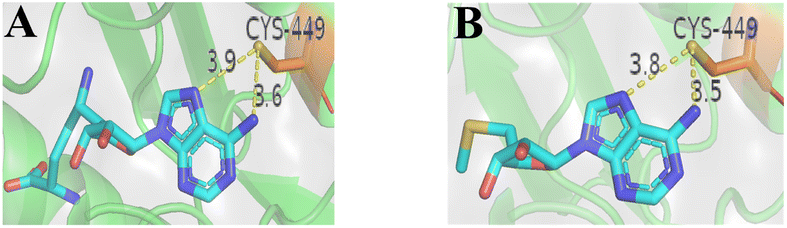 | ||
| Fig. 18 (A) The space length between C449 and C6–NH2 (and N7 atom) of the adenine ring of SAM is 3.9 (and 3.6) Å (PDB: 4GQB).25 (B) The space length between C449 and C6–NH2 (and N7 atom) of the adenine ring of MTA is 3.8 (and 3.5) Å (PDB: 5FA5).46 Note: the standard length of a carbon–carbon bond is approximately 1.54 Å.133 | ||
 | ||
| Fig. 19 (A) PRMT5 covalent inhibitors. (B) Proposed mechanism of vinyl-thio ether formation. | ||
3.3 The PRMT5 degraders based on PROTACs
Proteolysis targeting chimeras (PROTACs) have the ability to degrade a specific target protein by hijacking the E3 ubiquitin ligase machinery of the ubiquitin-proteasome system (UPS).135,136 In 2020, a selective substrate-competitive inhibitor EPZ015666 was selected as the target protein of interest (POI) ligand because Shen, et al. observed that the oxetane moiety of EPZ015666 is solvent-exposed in the co-crystal structure (PDB: 4X61 Fig. 20B). Subsequently, the first PRMT5 degrader 123 (MS4322) and two control compounds 122 (MS4370) and 121 (MS4369) were designed and prepared.64 As shown in Fig. 20C, 123 (MS4322) is composed of three parts: EPZ015666, (S, R, S)-AHPC-Me (VHL-2), and the linker. A preliminary study of the SAR indicates that the PRMT5 degraders with a relatively longer polyethylene glycol (PEG) linker have possessed stronger potency than other PRMT5 degraders which contain a short PEG linker or an all-carbon linker. For example, the PRMT5 protein level in a multitude of cell lines is clearly reduced by compound 120 at 1–5 μM (with greater efficacy observed at 5 μM), whereas none of compounds 116–119 seem to exhibit the efficacy of PRMT5 protein degradation at this concentration range. And although 123 (MS4322), 122 (MS4370), 121 (MS4369), and compound 116 share a common PEG linker, their efficacy is obviously distinct; and when is administered to MCF-7 cell lines, PRMT5 levels are significantly reduced in a time-, dose-, VHL-, and proteasome-dependent manner (DC50 = 1.1 ± 0.6 μM and Dmax = 74 ± 10%); in contrast, EPZ015666 (alone), 122 (MS4370) which contains the diastereoisomer of VHL-2, and 121 (MS4369) could not reduce PRMT5 levels in MCF7 cells at 5 μM. Compound 116 displays a weaker PRMT5 degradation activity due to the fact that VHL-2 is more effective than VHL-1 at the degrading of protein; Furthermore, 121 (MS4369) exhibits poor PRMT5 inhibition activity compared to EPZ015666 (IC50: 30 ± 3 nM), and it is probably because the 1, 2, 3, 4-tetrahydroisoquinoline moiety plays an important role in maintaining and enhancing the PRMT5 substrate-competitive inhibitors activity. These results suggest that the efficacy of the degrader 123 (MS4322) may be influenced by multiple factors, including the POI ligand, linker, and the E3 ligase ligand.64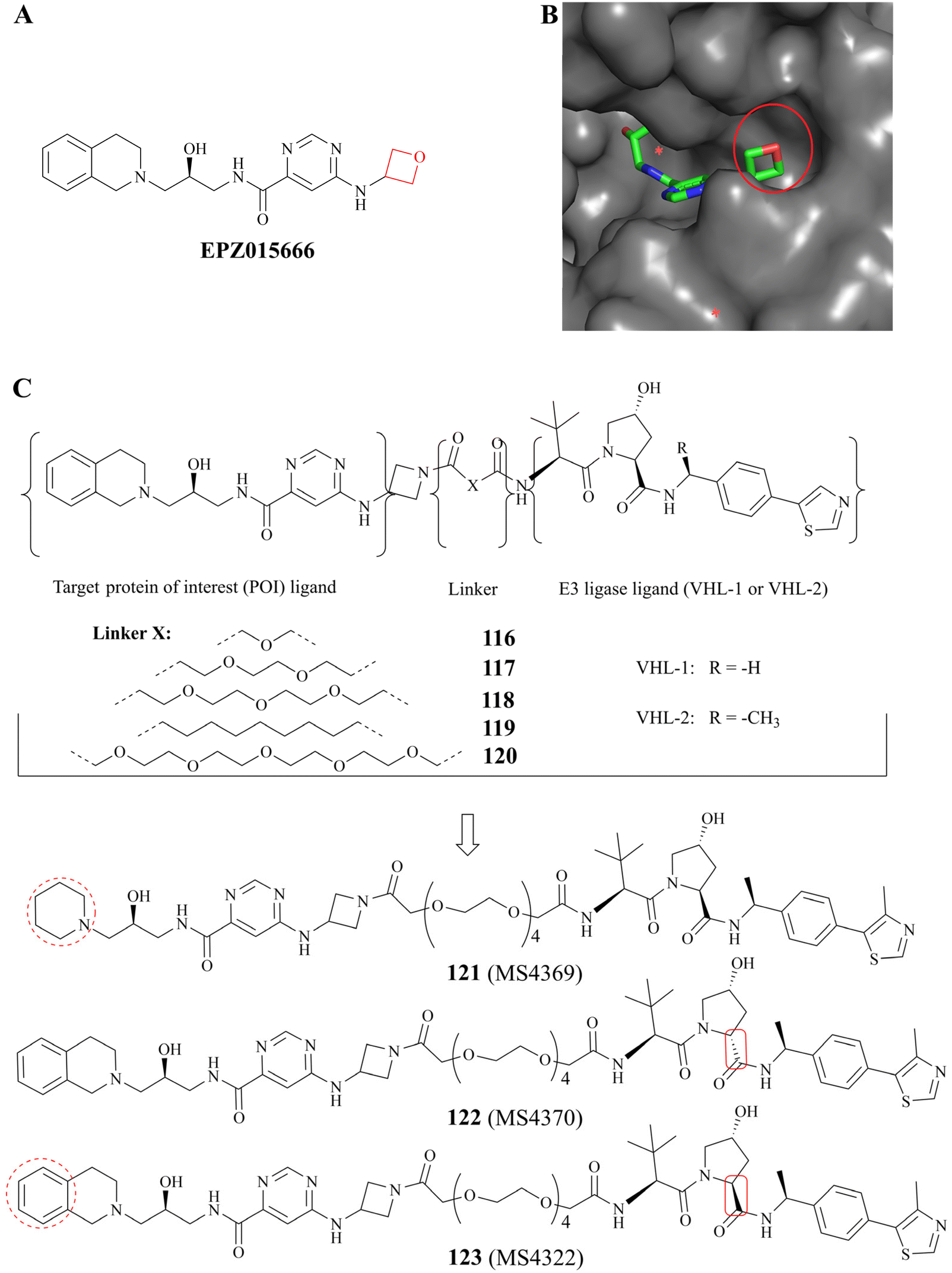 | ||
| Fig. 20 (A) EPZ015666 (the oxetane moiety is shown in red); (B) the co-crystal structure of PRMT5/MEP50 complex with EPZ015666 (PDB: 4X61). It can be observed that the oxetane moiety of EPZ015666 is solvent-exposed. (C) A brief study of the SAR of 123 (MS4322). | ||
4 The potential nitrosamine risk factors associated with the preparation process
Since the initial detection of N-nitrosodimethylamine (NDMA) in valsartan in 2018, the quality control and regulation of genotoxic impurities in the N-nitrosamine (NA) class has become a pivotal concern for pharmaceutical manufacturers and regulatory bodies. This is because they were subsequently identified in the active pharmaceutical ingredients (APIs) of other drugs, including sartan, pioglitazone, ranitidine and metformin. In consequence, drugs such as varenicline, propranolol, quinupril and orphenadrine were also recalled due to the presence of the respective nitrosamine drug-substance-related impurity (NDSRI). It is well established that the properties of amines, particularly those of secondary amines, are frequently necessary for effective binding to protein biological targets. Furthermore, amines typically exhibit a higher volume of distribution in vivo, which facilitates the enhancement of numerous pharmacokinetic parameters. However, recent studies have demonstrated that approximately 40% of active pharmaceutical ingredients (APIs) and 30% of API impurities are potential precursors of nitrosamine (NA) formation due to the presence of susceptible amine groups. If only the more reactive secondary amines are considered, there is still a potential risk for 13–15% of APIs.137–140 It is noteworthy that even in solid drug products (DPs), nitrites can form NDSRIs from levels of 1 part per million due to factors such as storage environment. Furthermore, the default Acceptable Intake (AI) for N-nitrosamines (NAs) is only 18 ng per day.The origin of nitrosamines in pharmaceuticals is generally considered to be multifactorial. Primarily, the simultaneous use of secondary amines and nitrite reagents in the synthesis routes of active pharmaceutical ingredients (APIs) (see Fig. 21) can be necessary for the formation of nitrosamines in the general assessment process. Secondly, the source of nitrosamines in pharmaceuticals can also be attributed to the use of pharmaceutical excipients or packaging materials. The formation of nitrosamines is less likely to occur with the use of tertiary amines due to the additional dealkylation step involved. In standard conditions, secondary amines are 1000 times more reactive than tertiary amines. Additionally, the generation of different cross-nitrosamine compounds may be influenced by the presence of different alkyl substituents. Furthermore, compared to secondary and tertiary amines, primary amines are challenging to produce stable nitrosamines. Typically, primary amines undergo diazotisation and subsequent amine removal to achieve this stability.141–143 Therefore, primary amines are the least likely to result in the formation of nitrosamines. Consequently, this study focuses on secondary amines in the synthesis process.
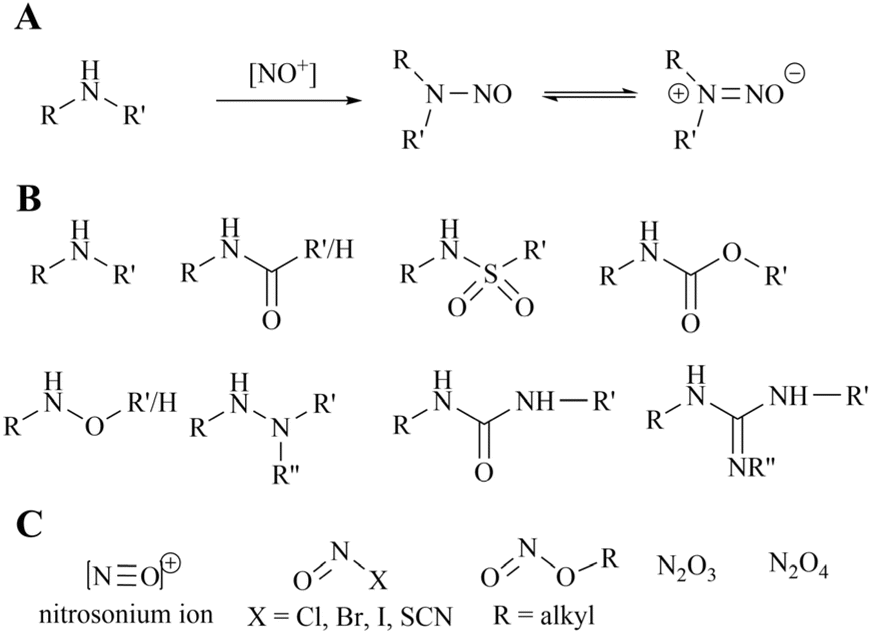 | ||
| Fig. 21 (A) Mechanism of formation of nitrosamines from secondary amines, (B) the various secondary amine structures that have the potential to form nitrosamines. (C) Common nitrosating reagents. | ||
4.1 MRTX1719
The early preparative route to 27 (MRTX1719) focused on the cross-coupling of two compounds (124 and 125) to give racemic 126. Subsequently, the pure enantiomer 27 (MRTX1719) was obtained through a separation and deprotection process utilising chiral preparative chromatography.87 However, as the clinical trials progressed, the chiral preparative chromatographic separation to obtain the highly pure (M)-enantiomers was no longer adequate for the split. In 2023, Michal Achmatowicz et al. reported an economically viable method for the production of high-purity (M)-isomers on a multi-kilogram scale. Firstly, the conventional chiral resolution method, which utilises Boc-d-phenylalanine (BDP), has replaced the enantiomeric chromatographic separation that was previously required by the original route. The efficiency of the chiral resolution process was significantly enhanced, enabling the conversion of the racemate to 98.4% d.e. MRTX1719-BDP salt 127 ((M)-4-BDP) in 75% separation yield on a current multi-kilogram scale.116 The preparation is shown in Fig. 22.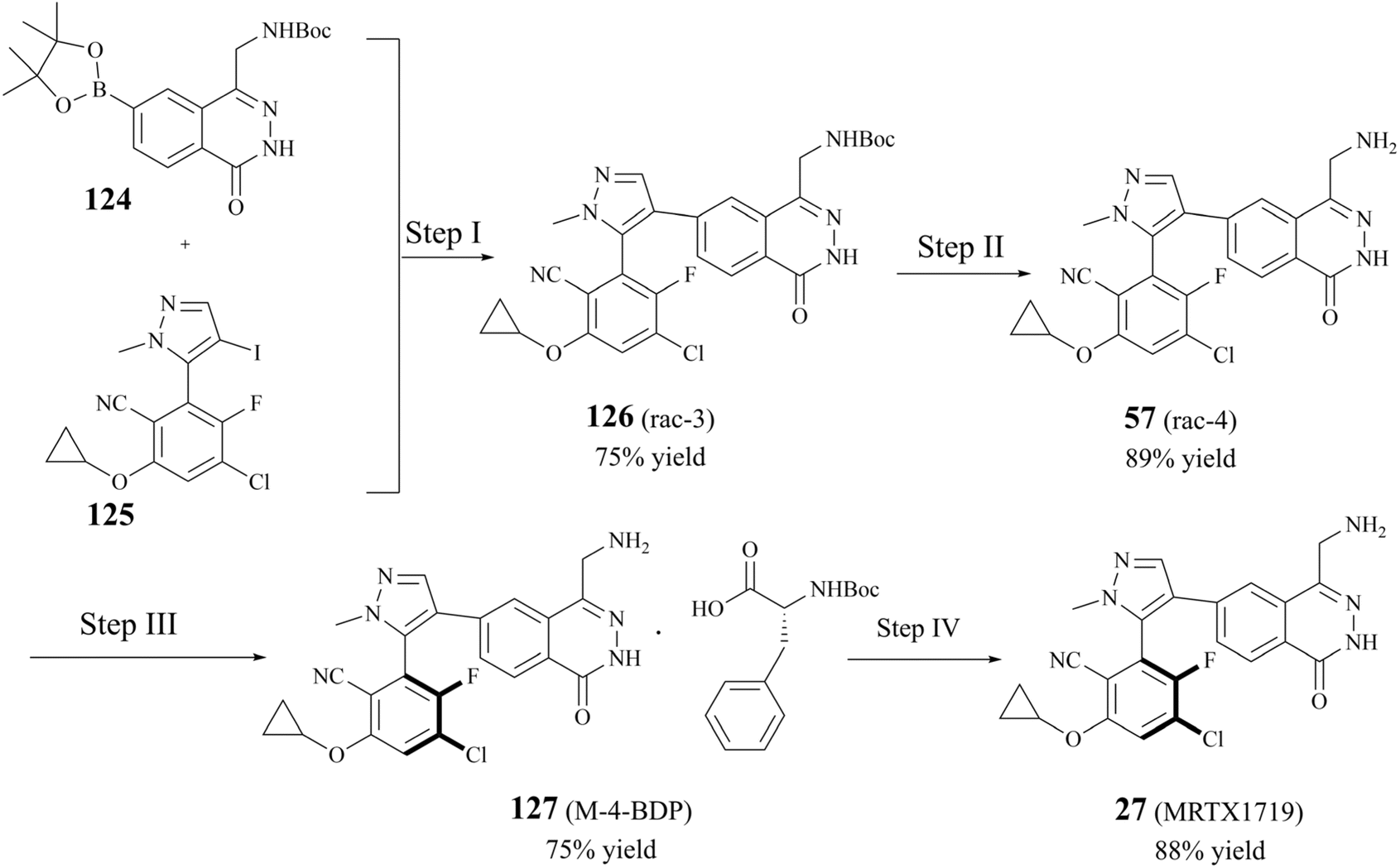 | ||
| Fig. 22 Current multikilogram GMP synthesis of 27 (MRTX1719). (Step I) 3.0 mol% Ad2nBuP-Pd-G3,3.0 eq., Cs2CO3, 12 vol. toluene, 4 vol. H2O, 55–60 °C, 18 h. (Step II) 4 M HCl/EtOAc (5 vol.), 20 vol. MeOH, 15–25 °C, 18 h; 7 M NH3/MeOH (1 vol.),10 vol. MeOH, 10–15 °C, 20 h. (Step III) 1.2 eq. Boc-D-Phe, 15 vol. EtOH/H2O 98:2, crystallizer: 20–25 °C, 16 h, racemizer: 160 °C, tR 2–4 min; 7 vol. EtOH/H2O 85:15, 15–25 °C, 16 h. (Step IV) 4.5 vol. THF, 1.5 vol. H2O, 20–25 °C; 39 vol. H2O, 1 vol. conc. aq. NH3, 20–25 °C, 6 h; 9 vol. H2O, 1 vol. IPA, 20–25 °C, 6 h. | ||
Nucleophilic reactions between compounds containing secondary amines and nitrosating reagents have the potential to produce nitrosamine impurities, as illustrated in Fig. 21. It is noteworthy that the secondary amines referred to herein encompass a range of compounds, including fatty secondary amines, amides, carbamates, hydroxylamines, hydrazines, hydrazones, ureas, and guanidines. In Fig. 22, there seems to be a structural rationale for the formation of nitrosamine-like impurities such as the presence of several intermediates with Boc-protected amide structures, including compound 124, 126 (rac 3), and BDP. Compound 124, which can be hydrolysed to boric acid under acidic conditions. Alkyl nitrites have been reported to generate nitrosamines from arylboronic acids in the presence of a copper catalyst, such as the Chan–Lam reaction. However, we carefully reviewed the conditions of the preparative process for step I–IV in the route, and did not find any obvious nitrosation reagents, and the Boc-protected amide structures exhibited a generally relatively low reactivity. It is worth noting that nitrosamines can be produced by two specific pathways, the oxidation of hydrazine and the ozonolysis of hydrazone.143 Some studies have shown that hydrazine can be oxidised in the presence of air or metal catalysts to form nitrosamines (the reaction mechanism is shown in Fig. 23A. In the presence of ozone, hydrazones can be oxidised to form nitrosamines and the reaction mechanism is shown in Fig. 23B.
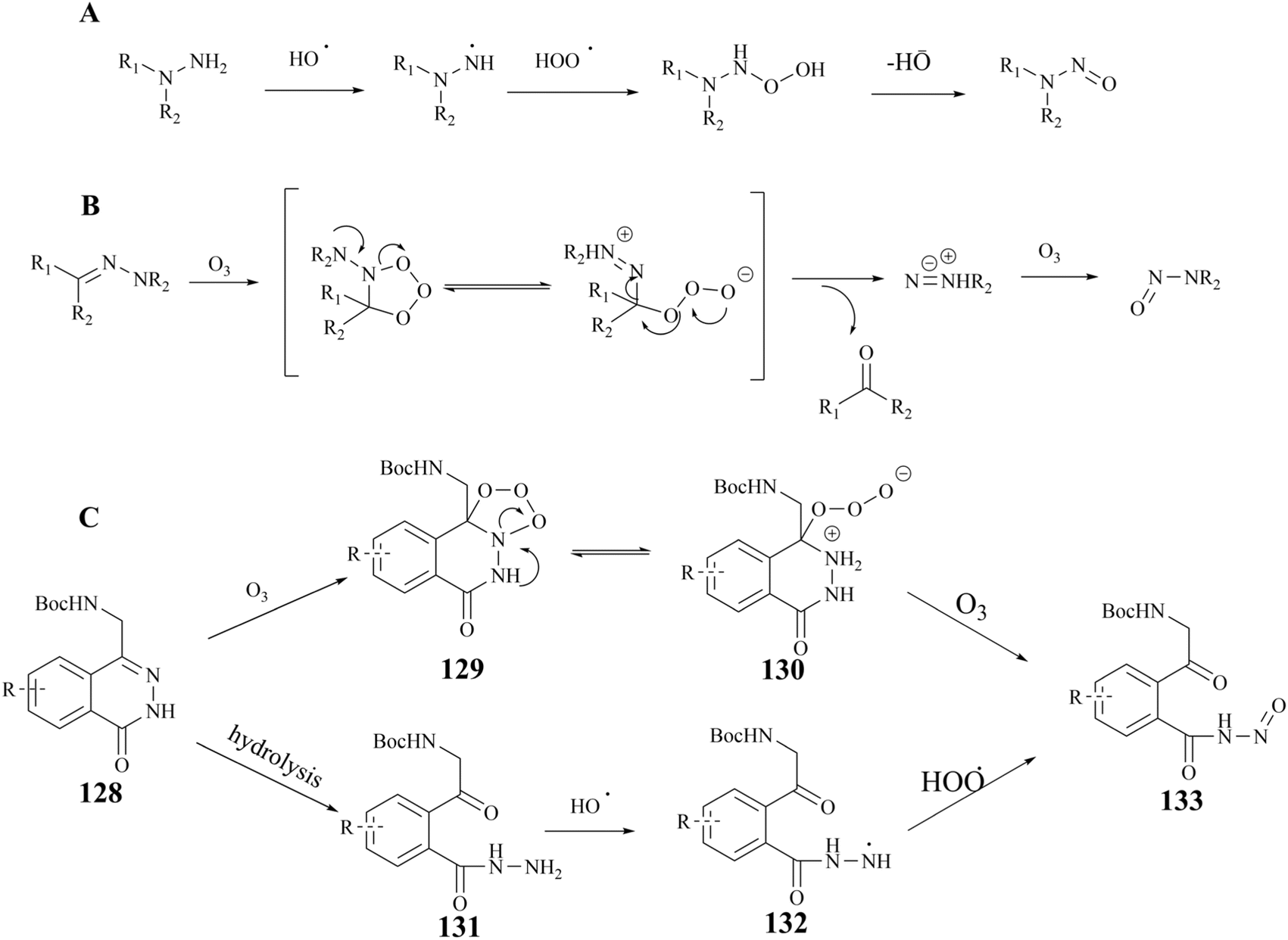 | ||
| Fig. 23 (A) The reaction mechanism of the oxidation of hydrazine. (B) The reaction mechanism of the ozonolysis of hydrazine (C) compound 133 possible production routes. | ||
4.2 AMG193
As previously stated, Amgen Inc. has patented a series of inhibitors targeting the PRMT5/MTA complex. Possibly based on synthetic convenience considerations, all of the inhibitors in this series have an aromatic pharmacodynamic core connected to another molecular group via an amide bond. Thus, the patented compounds in this series have similar preparation routes. We would like to briefly summarise them here, taking the preparation route of AMG193 as a representative. As shown in Fig. 24, the pharmacophore 4-amino-1,3-dihydrofuro [3,4-c][1,7]naphthyridine-8-carboxylic acid (Compound 136) and (S)-3-(5-(trifluoromethyl)pyridin-2-yl)morpholine (compound 138) via a condensation reaction to produce 30 (AMG193) in the presence of the peptide condensation reagent 2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluoro phosphate (HATU). Compound 136 was synthesised using methyl 5-amino-4-bromo-2-pyridinecarboxylate (133) as the initial starting material in a three-step process involving Suzuki coupling reaction, cyclisation and hydrolysis. The production of compound 138 was accomplished through a chiral splitting approach utilising the SFC method. Notably, this same chiral splitting strategy can also be employed after the synthesis of 30 (AMG193) racemates.124–127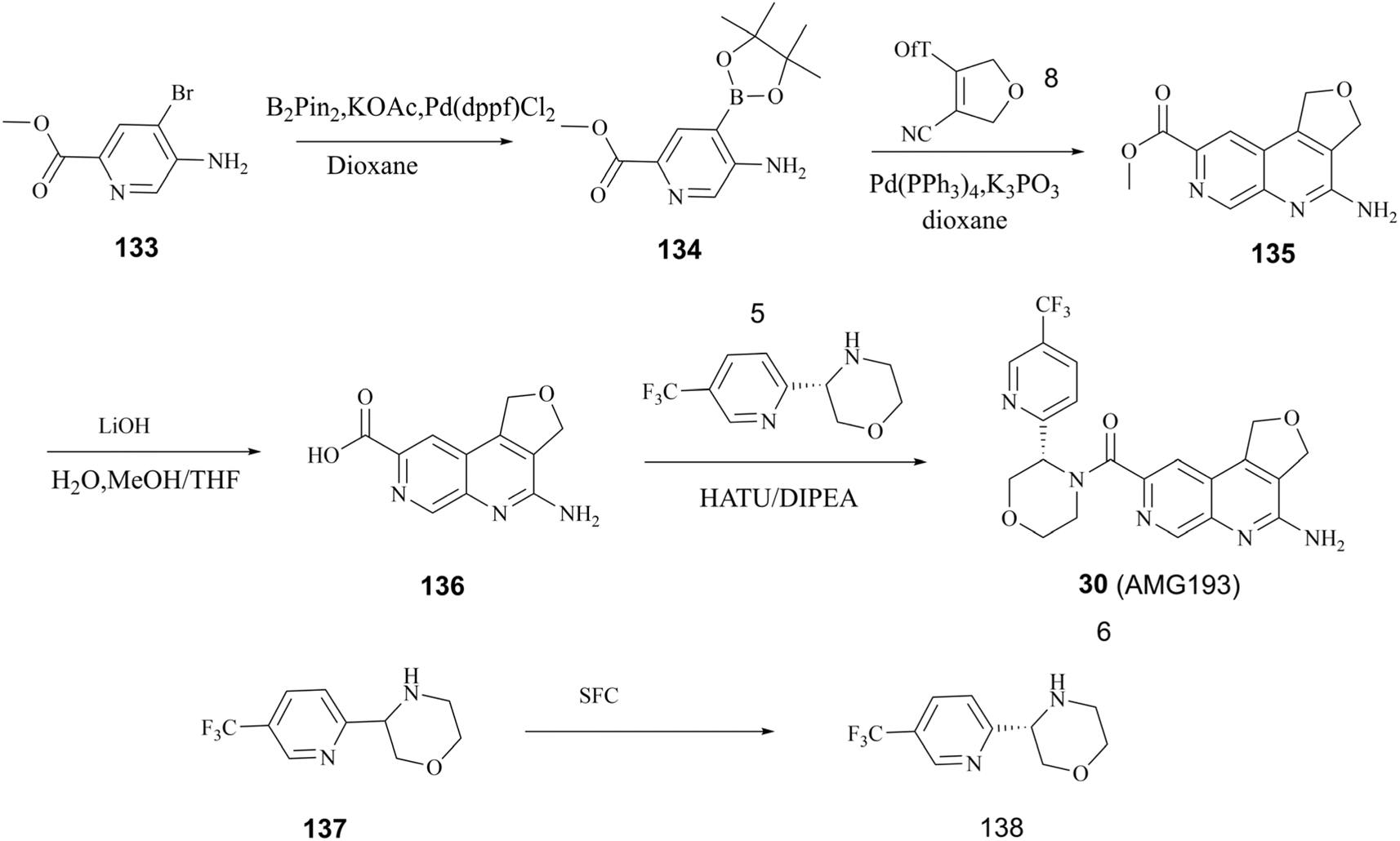 | ||
| Fig. 24 Preparation of 30 (AMG193). | ||
In additional, the morpholine moiety in compound 137 and 138 represents a cyclic secondary amine warning structure. However, of greater significance is the peptide condenser HATU. In the presence of HATU, the carboxylic acid anions within the condensation reaction system initially attack HATU, resulting in the generation of an unstable O-acyl (tetramethyl) isourea salt intermediate through an addition–elimination process. Subsequently, the OAt negative ion attacks the isourea salt, resulting in the formation of the HOAt active ester and tetramethylurea. The amine and HOAt reactive ester then undergo a reaction, forming the amide and HOAt. Therefore, tetramethylurea has the potential to serve as a source of dimethylamino group (see Fig. 25).
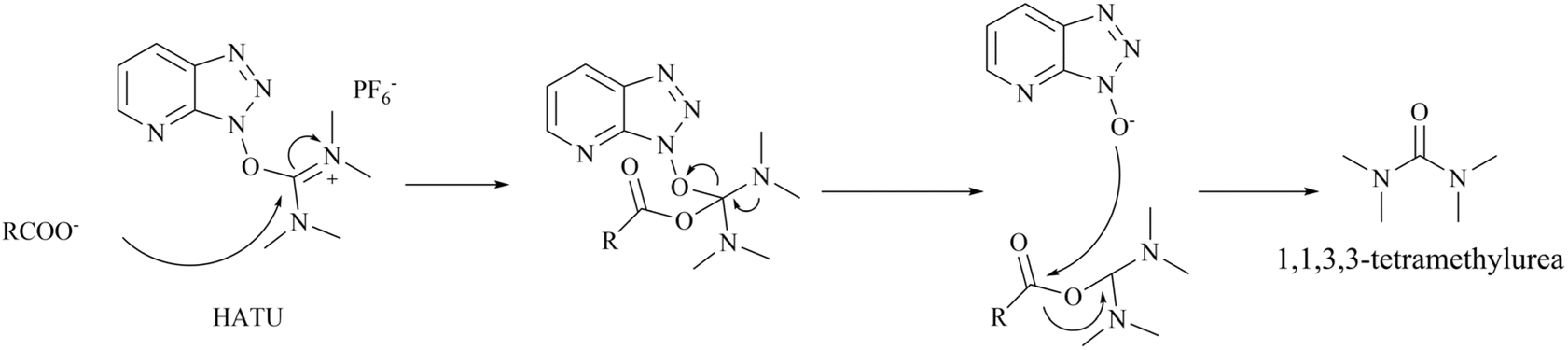 | ||
| Fig. 25 Reaction mechanism of HATU to generate 1,1,3,3-tetramethylurea.144 | ||
4.3 TNG908
The process route for the preparation of 28 (TNG908), as illustrated in Fig. 26, comprises two principal sections. In one phase of the process, pyridin-2(1H)-one (compound 139) was used as the initial reactant, which was reduced by 10% Pd/C in order to obtain racemic compound 140. Subsequently, compound 140 was protected by Boc, and in the presence of 1,1,1-trifluoro-N-phenyl-N(trifluoromethylsulfonyl)methanesulfonamide (1.25 eq., compound A), the triflate compound 141 was generated at low temperature (between −78 and −25 °C). The compound 143 was prepared by Suzuki coupling of boronates or boronic acids with Boc-protected triflate 142. Subsequently, the protection group of compound 143 was removed by treatment with trifluoroacetic acid (TFA), compound 145 is gain by a further reduction of the double bond. The other part was the preparation of compound 12 using 3-methyl-5-nitropyridine-2-amine (compound 146) as starting material. The condensation of compounds 145 and 150 was conducted in the presence of the peptide condensation reagent HATU. The Boc protecting group was then removed, and 28 (TNG908) was obtained by chiral splitting using SFC or chiral HPLC. Given that 29 (TNG462) and other derivatives possess the same fundamental backbone structure as 28 (TNG908), they can be prepared in a similar manner, and the associated details are therefore not repeated herein.85,86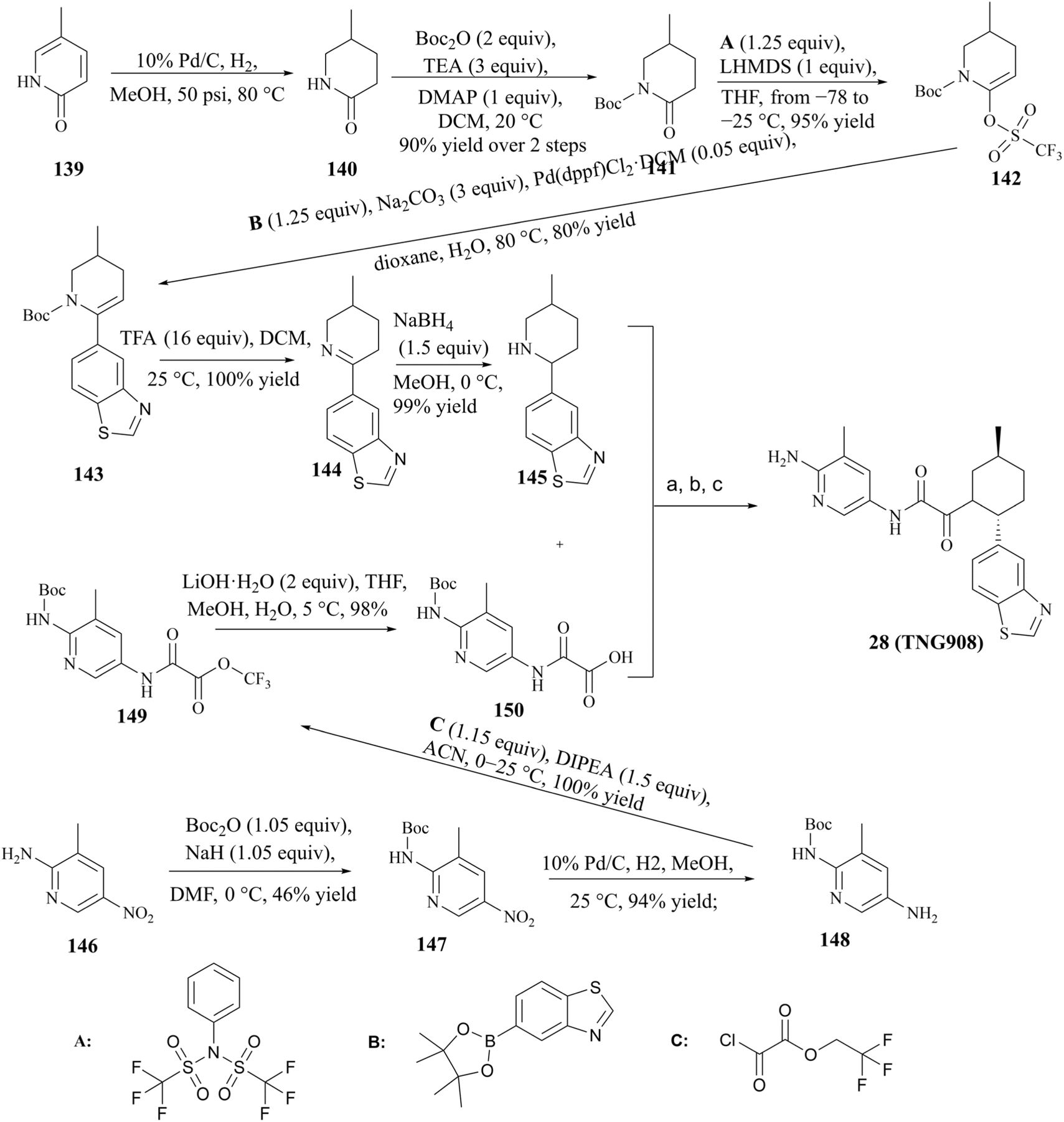 | ||
| Fig. 26 28 (TNG908) preparation route. (a) HATU (1 equiv.), TEA (6 equiv.), DMF, 25 °C; (b) 4 M HCl in dioxane, 25 °C, 59% yield over 2 steps for 28 (TNG908); (c) chiral HPLC separation of enantiomers, 48% yield for 28 (TNG908). | ||
From the perspective of compound preparation, the process route for 28 (TNG908) preparation in the initial exploratory phase of SAR incorporates the preparation of 28 (TNG908) and its derivatives, which is a highly practical process preparation route. Nevertheless, more rigorous quality control is necessary for clinical applications and GMP production environments. Firstly, with regard to the potential for the formation of nitrosamine impurities, triethylamine (TEA) and dimethylformamide (DMF) are frequently employed as reagents in the synthesis process depicted in Fig. 26. It is generally accepted that these two reagents are the source of additional secondary amines in the synthesis process. TEA may contain diethylamine, while DMF may contain dimethylamine. It is hypothesised that 1,1,1-trifluoro-N-phenyl-N(trifluoromethylsulfonyl)methanesulfonamide (compound A) may be a potential source of sulfonamide-type secondary amines. Furthermore, intermediates 140, 144, and 145 are cyclic secondary amine alert structures. In the preparative process, the nitro group of intermediate 147 was reduced by hydrogenation with 10% Pd/C, resulting in the production of an amine. This reaction is characterised by low impurities and high yield. However, the reduction reaction does in fact undergo an intermediate process of nitroso compounds, namely N-hydroxyhydroxylamine (see Fig. 27), which could potentially act as a source of nitroso groups if the process is not operated correctly or the reaction is not monitored with sufficient accuracy. Furthermore, nitroso compounds and N-hydroxyhydroxylamine can undergo a reaction to form oxoazo compounds, which can then, upon further reduction, yield intermediates such as azoic compound, 1,2-dihydrocarbonyldihydrazine, and so on. These intermediates require longer hydrogenation-catalysed reductions to be fully reduced to amine groups.145
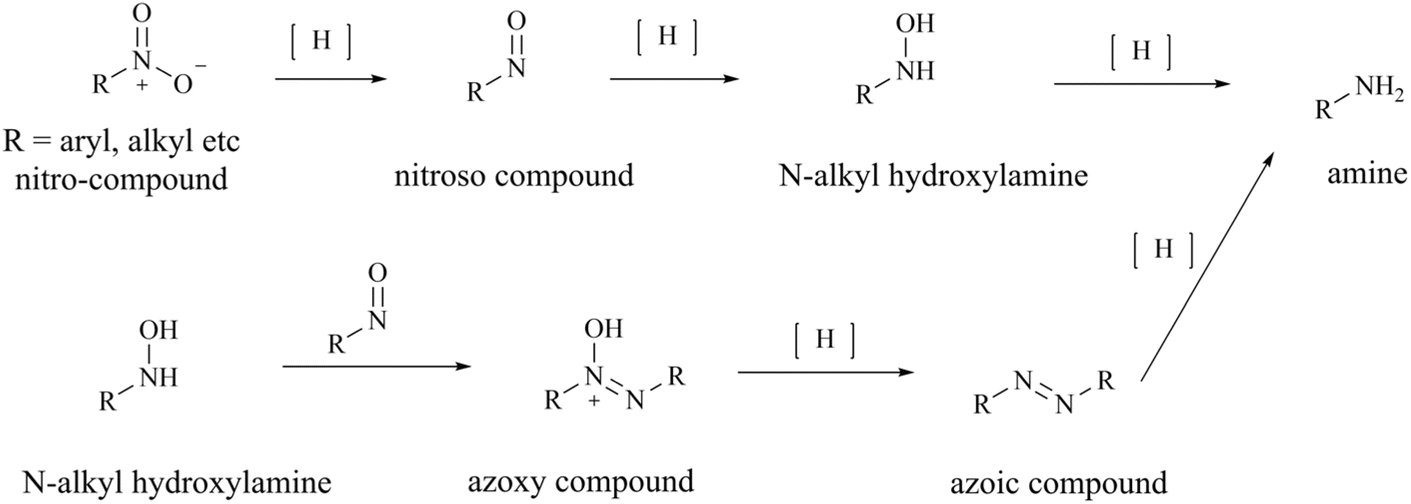 | ||
| Fig. 27 Nitrohydrogenation reduction mechanism. | ||
It should be noted that the nitrosamine risk factors discussed above are based on the chemical structures described in the current literature, which indicate the potential for the generation of nitrosamine-like impurities under certain conditions. These chemical structures must meet sufficient conditions, such as the introduction of nitrosating compounds or potentially nitrosating reagents in the process route due to unknown factors, to generate possible nitrosamine impurities. While various types of secondary amine structures are generated in our current process routes for PRMT5/MTA inhibitor preparation, in all mature processes, there are no significant nitrosation conditions. However, rigorous process operation and quality control standards remain necessary considerations based on safety concerns.
5 The vulnerability of arylamine in vivo
As previously stated, all PRMT5/MTA complex inhibitors possess a vital pharmacophoric functional group, namely the primary amine moiety (see Fig. 9). The primary amine can form H-bond interactions with the key catalytic residues Glu444 or Glu435. Additionally, it forms a specific interaction with the MTA bound in the protein complex, which serves to stabilise the PRMT5/MTA complex.Furthermore, the primary amine group is a basic group for the salt formation of these inhibitors, which facilitates the improvement of many pharmacokinetic parameters. These finds suggest that the primary amine moiety is of primary importance with regard to the efficacy, protein complex selectivity, and MTAP-deficient tumour cell selectivity of PRMT5/MTA complex inhibitors. However, the greater the importance, the greater the fragility. Small changes in primary amines inevitably result in changes in the potency and safety profile of PRMT5/MTA complex inhibitors. In the initial stages of drug discovery, this vulnerability posed significant organic synthesis challenges for structural extensions and the SAR exploration of fragments. Some promising fragments were ultimately excluded after a comprehensive evaluation.101,146,147 In vivo, exogenous drugs containing amine groups are usually metabolised by the cytochrome P450 (CYP) family of enzymes. However, arylamines can also be catalyzed by NAT enzymes to produce acetylated metabolites or conjugated with glucuronic acid to produce water-soluble products. Generally, arylamines are metabolised by cytochrome P450 (CYP) enzymes without deamination, but rather to hydroxylated metabolites (see Fig. 28A). However, many of the hydroxylated metabolites formed by exogenous arylamines are carcinogenic, such as 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP).148–152 On the basis of this information, it is reasonable to speculate that in vivo, arylamine groups may represent a significant metabolic reaction site for drugs bearing an arylamine moiety, and that these metabolic reactions are readily occurring. However, the arylamine moiety is of critical importance for the efficacy and safety of PRMT5/MTA complex inhibitors in vivo, and changes in its structure would lead to changes in its inhibitory potency and pharmacological mechanism of action, including cellular selectivity. Moreover, these inhibitors are PRMT5 substrate competitive. Hypothetically, these inhibitors such as 30 (AMG193) and 28 (TNG908) have preferential metabolism of primary amines over other metabolic sites, and there is a potential for an indirect dose-related toxicity in the case of long-term administration. It is noteworthy that the primary amine of 27 (MRTX1719) is a benzylamine group, which may offer an alternative metabolic possibility (e.g., Fig. 28B). Benzylamine molecules can be catalytically deaminated by MAO to produce aromatic aldehydes.153 However, aromatic formaldehyde is reactive in vivo and is generally further oxidised or reduced to the corresponding aromatic formic acid or aromatic methanol. These aromatic formic acids or aromatic methanols are generally more water-soluble than their parent drug. This may help to explain the lack of dose-limiting toxicity observed in vivo for MRTX1719 at a dose level of 400 mg q.d. However, it must be acknowledged that a clear picture of the dose-limiting toxicity of MRTX1719 in vivo remains elusive. Regrettably, no authoritative studies on these aspects could be identified in public databases.
 | ||
| Fig. 28 Major metabolic pathways of primary amines in vivo. (A) Major metabolic pathways of aryl amines in vivo.148–152 (B) Major metabolic pathways of benzyl amines in vivo. The vulnerability of arylamine in vivo.153 | ||
6 Conclusions and perspectives
In this paper, we have conducted a review of the design thoughts and the structure–activity relationship (SAR) of known methylthioadenosine (MTA)-cooperative PRMT5 inhibitors. We expect to elucidate the chemical-structural basis of the protein- and cell-selectivity of these inhibitors through such a comprehensive review. Additionally, we have analysed the clinical safety of representative first- and second-generation PRMT5 inhibitors. In the binding pocket, first-generation inhibitors achieve high selectivity for PRMT5 by forming a potential π–π stacking or cation–π interaction with Phe327, a specific amino acid residue of PRMT5. And the molecular structure of SAM-competitive inhibitors commonly frequently comprises an aromatic ring, which facilitates the formation of π–π stacking interaction with Phe327. However, given the critical function of PRMT5 in human cells, it is expressed in almost all cells, especially in the haematopoietic system. In vivo, this selectivity for the enzyme does not reflect the high selectivity for tissue cells, resulting in a narrow therapeutic window. Consequently, these inhibitors typically exhibit dose-limiting toxicity in clinical trials. In contrast to the first-generation PRMT5 inhibitors, the second-generation inhibitors, which were developed based on a synthetic lethality strategy, exploit the elevated levels of MTA in MTAP-deficient tumour cells, thereby achieving enhanced cellular selectivity and representing a novel therapeutic option. In the clinical phase I trial, the second-generation inhibitors demonstrated higher tolerated doses, lower dose-limiting toxicity, and higher clinical value, particularly the PRMT5/MTA complex inhibitors. The PRMT5/MTA complex inhibitors occupy the PRMT5 substrate-binding pocket and form a specific interaction with MTA bound in the SAM pocket, thereby stabilising stabilises the PRMT5/MTA complex. Moreover, the substrate-binding pocket is predominantly composed of acidic amino acids, which are more prone to interaction with basic groups. The core pharmacophore of these inhibitors is typically a nitrogen-containing aromatic ring with a spatially directed substituted amine group. Owing to the binding mechanism of these small molecule inhibitors to the PRMT5/MTA complex, minor alterations to primary amines will inevitably result in modifications to the potency and safety profile of PRMT5/MTA complex inhibitors. Consequently, the vulnerability of its aromatic amine moiety is discussed. It can be reasonably assumed that the aromatic amine moiety is, in vivo, a readily metabolisable group. A further consideration is that PRMT5/MTA complex inhibitors are substrate competitive, which may prove to be a disadvantage to their use in clinical applications.A number of second-generation inhibitors have been approved for clinical trials (Table 2 lists several representative second-generation inhibitors). Compared to the first-generation PRMT5 inhibitors, the majority of the second-generation inhibitors demonstrated satisfactory dose tolerance due to their rational pharmacological mechanism of action. For example, maximum tolerated dose of 27 (AMG193) is determined to be 1200 mg per day,111 and no dose-limiting toxicity was observed in patients treated with 30 (MRTX1719) at a dose of 400 mg once daily.103 The distinctive physicochemical characteristics of TNG908, which permit penetration of the blood–brain barrier, have facilitated promising advances in the treatment of gliomas. These results suggest that, following the precedent of poly ADP ribose polymerase (PARP), PRMT5 has the potential to become the next clinically applicable synthetic lethal target. In light of these issues, the primary challenges currently facing this field of research can be summarised as follows: firstly, it is essential to confirm the efficacy of these MTA-cooperative PRMT5 chemical inhibitors in vivo against a range of tumour types at doses that can be tolerated by patients. Secondly, it is crucial to identify any other biomarkers associated with these inhibitors. Thirdly, the mechanisms of resistance and metabolism of these inhibitors should be elucidated. It is notable that SCR6290 has been demonstrated to exhibit tumour cell selectivity through a tumour-biased distribution profile, characterised by an intracellular enrichment concentration that is 50-fold greater than that observed in the blood.112,113 This may represent a promising avenue for reducing dose-limiting toxicity of PRMT5 inhibitors. However, the exact mechanism leading to the presence of a biased tumour distribution of this inhibitor in vivo has not been disclosed.
Following comprehensive research and discussion of the design strategies and SARs of various PRMT5 inhibitors, we found that the dose-limiting toxicity of this class of compounds that this may a common problem in the drug delivery field regarding selective and effective delivery of targeted inhibitors and the development of an optimal drug delivery system has the potential to significantly address this problem and enhance the efficacy. It is well known that, in general, tumour blood vessels are much more permeable than normal blood vessels. Furthermore, the compromised lymphatic blood flow pathways allow for the preferential accumulation of certain substances in tumour tissues, where they remain for extended periods, which is known as the enhanced permeability and retention (EPR) effect.154 A nanodrug delivery strategy may be defined as the utilisation of either EPR effect-mediated passive targeting or ligand-mediated active targeting, with the objective of delivering nanodrugs to tumour tissues subsequent to systemic delivery.155 This technology allows for the precise delivery of drugs into tumours, either by targeting tumour cells directly or by targeting APC (including macrophages and DCs). In 2020, Chenggang Zhu et al. reported the development of a novel injectable in situ-forming implantation system. This system consisted of n-butyl-2-cyanoacrylate (NBCA), ethyl oleate, and a sol–gel phase transition, and was used for the delivery of protein arginine methyltransferase 1 (PRMT1) inhibitor TC-E-5003.156 In 2023, the team of Camino de Juan Romero reported the utilisation of small extracellular vesicles (EVs) isolated from diverse cell lines and encapsulated with temozolomide and EPZ015666 via direct incubation for the treatment of pancreatic cancer and glioblastoma (GBM).157,158 Nanomedicines have been developed based on active targeting nanotechnology. These include ligand-implanted nanomedicines that facilitate specific tumour delivery and enhance therapeutic efficacy. These include trastuzumab (Tra) mediated targeting of NSCLC (HER-2 positive) and substance P peptide mediated targeting of gliomas. Furthermore, Jordan J. Green and colleagues devised a bioreducible nanoparticle for the systemic delivery of siRNA into patient-derived glioblastoma cells in an in situ mouse tumour model.159 It is regrettable that, although the targeted delivery strategy may prove an effective means of enhancing the clinical value of PRMT5 inhibitors, research in this area is still in its infancy. No studies on targeted delivery systems applicable to PRMT5 inhibitors have been identified in the relevant journals or research communities.
Allostery is an inherent property of epigenetic enzymes in which topographically distinct binding sites are functionally coupled and transmissible.160 Meanwhile, due to the binding site of allosteric inhibitors being located outside of the orthosteric sites, allosteric inhibitors always show non-competitiveness with endogenous cofactors and substrates in orthosteric sites. Thus, they possess remarkable potency and safety in lower dose levels compared to orthosteric inhibitors.161 Interestingly, Palte et al. also noted that, although the cocrystal structure of the known allosteric inhibitor with PRMT5 shows that the eleven-amino acid loop (Glu435-Leu445) prevents the binding of SAM (PDB ID: 6UXY), the different conformations of this loop can be stabilized depending on the presence of allosteric inhibitors. And this suggests that the configurations in which both an allosterically binding molecule and SAM are bound seem possible.19 So, is there another possible configuration in which both an allosteric binding molecule and MTA are bound?
In addition, the MTA-cooperative PRMT5 inhibitors provide an opportunity to study the MTA-cooperative PRMT5 degraders. In general, a suitable POI ligand from the vast array of highly potent and selective inhibitors can obviously enhance the selectivity of the degraders, which may exhibit more targeted degradation selectivity than the parent inhibitor, particularly against structurally highly homologous proteins.136 A POI ligand (only EPZ015666 has been screened) is not sufficient to elucidate the profile of the PRMT5 degraders and we would like to see more extensive research in this area.
Abbreviations
| AE | adverse events |
| ADMA | omega-NG, NG-asymmetric dimethylarginines |
| AIDD | artificial intelligence-based drug molecule virtual screening |
| APIs | active pharmaceutical ingredients |
| BDP | Boc-d-phenylalanine |
| Clint | human hepatocyte intrinsic clearance |
| COPR5 | cooperator protein of PRMT5 |
| DNMTs | DNA methyltransferases |
| DSF | scanning fluorometry |
| FBDD | fragment-based molecule library screening |
| FBLD | fragment-based lead discovery |
| FP | fluorescence polarisation |
| GBM | glioblastoma |
| GSH | glutathione |
| HDACs | histone deacetylases |
| HMTs | histone methyltransferases |
| HTS | high-throughput screening |
| MEP50 | methylosome protein 50 |
| MET | methionine |
| MMA | omega-NG-monomethylarginines |
| MTA | methylthioadenosine |
| MTAP | methylthioadenosine phosphorylase gene |
| MST | microscale thermophoresis |
| NA | N-nitrosamine |
| NDSRI | nitrosamine drug-substance-related impurity |
| NDMA | N-nitrosodimethylamine |
| NMEs | new molecular entities |
| NMR | nuclear magnetic resonance |
| PARP | poly ADP ribose polymerase |
| PBM | PRMT5-binding motif |
| PPIs | protein–protein interactions |
| pICln | chloride channel nucleotide sensitive protein 1A |
| PRMTs | histone arginine methyltransferases |
| PTMs | post-translational modifications |
| PROTACs | proteolysis targeting chimeras |
| RioK1 | Rio domain-containing protein |
| TKIs | tyrosine kinase covalent inhibitors |
| SAM | S-5-adenosyl-L-methionine |
| SAH | S-adenosyl homocysteine (SAH) |
| SAR | structure–activity relationship |
| SBDD | structure-based molecule design |
| SDMA | omega-NG, N’G-symmetric dimethylarginines |
| SFC | chiral supercritical fluid chromatography |
| SPR | surface plasmon resonance |
| TRAE | treatment-related adverse event |
| UPS | ubiquitin–proteasome system |
| VHL | Von Hippel-Lindau E3 ubiquitin ligase ligand |
| WD | tryptophan-aspartate |
| X-ray | X-ray protein crystallography |
Data availability
No primary research results. Software or code have been included and no new data were generated or analysed as part of this review.Author contributions
All authors have equal sharing in all preparation steps for the review in its current final form.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was financially supported through grants from Southwest Medical University in China (No. 05/00040152).References
- R. S. Blanc and S. P. Richard, Arginine methylation: the coming of age, Mol. Cell, 2017, 65, 8–24, DOI:10.1016/j.molcel.2016.11.003.
- C. Sauter, J. Simonet, F. Guidez, B. Dumétier, B. Pernon, M. Callanan, J.-N. Bastie, R. Aucagne and L. Delva, Protein arginine methyltransferases as therapeutic targets in hematological malignancies, Cancers, 2022, 14, 5443, DOI:10.3390/cancers14215443.
- Q. Wu, M. Schapira, C. H. Arrowsmith and D. Barsyte-Lovejoy, Protein arginine methylation: from enigmatic functions to therapeutic targeting, Nat. Rev. Drug Discovery, 2021, 20, 509–530, DOI:10.1038/s41573-021-00159-8.
- W. Xiao, X.-Q. Chen, L. Liu, Y.-S. Shu, M. Zhang and Y.-C. Zhong, Role of protein arginine methyltransferase 5 in human cancers, Biomed. Pharmacother., 2019, 114, 10879, DOI:10.1016/j.biopha.2019.108790.
- Y.-Z. Yang and M. T. Bedford, Protein arginine methyltransferases and cancer, Nat. Rev. Cancer, 2013, 13, 37–50, DOI:10.1038/nrc3409.
- M. Wang, J. Fuhrmann and P. R. Thompson, Protein Arginine Methyltransferase 5 Catalyzes Substrate Dimethylation in a Distributive Fashion, Biochemistry, 2014, 53, 7884–7892, DOI:10.1021/bi501279g.
- Y.-Z. Yang, A. Hadjikyriacou, Z. Xia, S. Gayatri, D. Kim, C. Zurita-Lopez, R. Kelly, A. Guo, W. Li, S. G. Clarke and M. T. Bedford, PRMT9 is a Type II methyltransferase that methylates the splicing factor SAP145, Nat. Commun., 2015, 6, 6428, DOI:10.1038/ncomms74.
- S. K. Tewary, Y. G. Zheng and M.-C. Ho, Protein arginine methyltransferases: insights into the enzyme structure and mechanism at the atomic level, Cell. Mol. Life Sci., 2019, 76, 2917–2932, DOI:10.1007/s00018-019-03145-x.
- C. M. Koh, M. Bezzi and E. Guccione, The where and the how of PRMT5, Curr. Mol. Biol. Rep., 2015, 1, 19–28, DOI:10.1007/s40610-015-0003-5.
- D. Q. Tan, Y. Li, C. Yang, J. Li, S.-H. Tan, D. W. L. Chin, A. Nakamura-Ishizu, H. Yang and T. Suda, PRMT5 modulates splicing for genome integrity and preserves proteostasis of hematopoietic stem cells, Cell Rep., 2019, 26, 2316–2328, DOI:10.1016/j.celrep.2019.02.001.
- H. Kim and Z. A. Ronai, PRMT5 function and targeting in cancer, Cell Stress, 2020, 4, 199–215, DOI:10.15698/cst2020.08.228.
- W.-W. Tee, M. Pardo, T. W. Theunissen, L. Yu, J. S. Choudhary, P. Hajkova and M. A. Surani, Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency, Genes Dev., 2010, 24, 2772–2777, DOI:10.1101/gad.606110.
- R. V. Berrens and W. Reik, Prmt5: A guardian of the germline protects future generations, EMBO J., 2015, 34, 689–690, DOI:10.15252/embj.201591054.
- L. C. Litzler, A. Zahn, A. P. Meli, S. Hébert, A.-M. Patenaude, S. P. Methot, A. Sprumont, T. Bois, D. Kitamura, S. Costantino, I. L. King, C. L. Kleinman, S. Richard and J. M. D. Noia, PRMT5 is essential for B cell development and germinal center dynamics, Nat. Commun., 2019, 10, 22, DOI:10.1038/s41467-018-07884-6.
- Y. Lei, P. Han and D. Tian, Protein arginine methyltransferases and hepatocellular carcinoma: A review, Transl. Oncol., 2021, 14, 101194, DOI:10.1016/j.tranon.2021.101194.
- T. Wright, Y. Wang and M. T. Bedford, The Role of the PRMT5–SND1 Axis in Hepatocellular Carcinoma, Epigenomes, 2021, 5, 2, DOI:10.3390/epigenomes5010002.
- A. Motolani, M. Martin, M. Sun and T. Lu, The Structure and Functions of PRMT5 in Human Diseases, Life, 2021, 11, 1074, DOI:10.3390/life11101074.
- H. Shailesh, K. S. Siveen and S. Sif, Protein arginine methyltransferase 5 (PRMT5) activates WNT/β-catenin signalling in breast cancer cells via epigenetic silencing of DKK1 and DKK3, J. Cell. Mol. Med., 2021, 25, 1583–1600, DOI:10.1111/jcmm.16260.
- R. L. Palte, S. E. Schneider, M. D. Altman, R. P. Hayes, S. Kawamura, B. M. Lacey, M. S. Mansueto, M. Reutershan, P. Siliphaivanh, C. Sondey, H. Xu, Z. Xu, Y. Ye and M. R. Machacek, Allosteric Modulation of Protein Arginine Methyltransferase 5 (PRMT5), ACS Med. Chem. Lett., 2020, 11, 1688–1693, DOI:10.1021/acsmedchemlett.9b00525.
- M. S. Malamas, J. Erdei, I. Gunawan, J. Turner, Y. Hu, E. Wagner, K. Fan, R. Chopra, A. Olland, J. Bard and S. Jacobsen, Design and synthesis of 5, 5′-disubstituted aminohydantoins as potent and selective human β-secretase (BACE1) inhibitors, J. Med. Chem., 2010, 53, 1146–1158, DOI:10.1021/jm901414e.
- S. Antonysamy, Z. Bonday, R. M. Campbell, B. Doyle, Z. Druzina, T. Gheyi, B. Han, L. N. Jungheim, Y. Qian, C. Rauch, M. Russell, J. M. Sauder, S. R. Wasserman, K. Weichert, F. S. Willard, A. Zhang and S. Emtage, Crystal Structure of the Human PRMT5:MEP50 Complex, Proc. Natl. Acad. Sci., 2012, 109, 17960–17965, DOI:10.1073/pnas.120981410.
- L. Suna, M.-Z. Wang, Z.-Y. Lv, N. Yang, Y.-F. Liu, S. Bao, W. Gong and R.-M. Xu, Structural insights into protein arginine symmetric dimethylation by PRMT5, Proc. Natl. Acad. Sci., 2011, 108, 20538–20543, DOI:10.1073/pnas.1106946108.
- Angex Pharmaceutical Inc., Heterocyclic Compounds as PRMT5 Inhibitors, WO Pat., PCT WO2019112719 A1, 2019 Search PubMed.
- Angex Pharmaceutical Inc., Heterocyclic Compounds as PRMT5 Inhibitors, WO Pat., PCT WO 2020243178 A1, 2020 Search PubMed.
- A. M. Asberry, X. Cai, X. Deng, U. Santiago, S. Liu, H. S. Sims, W. Liang, X. Xu, J. Wan, W. Jiang, C. J. Camacho, M. Dai and C. D. Hu, Discovery and biological characterization of PRMT5:MEP50 protein-protein interaction inhibitors, J. Med. Chem., 2022, 65, 13793–13812, DOI:10.1021/acs.jmedchem.2c01000.
- A. Krzyzanowski, R. Gasper, H. Adihou, P. Hart and H. Waldmann, Biochemical investigation of the interaction of PICln, RioK1 and COPR5 with the PRMT5-MEP50 complex, ChemBioChem, 2021, 22, 1908–1914, DOI:10.1002/cbic.202100079.
- D. Musian, J. Bok, E. Massignani, L. Wu, T. Tabaglio, M. R. Ippolito, A. Cuomo, U. Ozbek, H. Zorgati, U. Ghoshdastider, R. C. Robinson, E. Guccione and T. Bonaldi, Proteomics profiling of arginine methylation defines PRMT5 substrate specificity, Sci. Signal., 2019, 12(No), eaat8388, DOI:10.1126/scisignal.aat8388.
- M.-C. Ho, C. Wilczek, J. B. Bonanno, L. Xing, J. Seznec, T. Matsui, L. G. Carter, T. Onikubo, P. R. Kumar, M. K. Chan, M. Brenowitz, R. H. Cheng, U. Reimer, S. C. Almo and D. Shechter, Structure of the Arginine Methyltransferase PRMT5-MEP50 Reveals a Mechanism for Substrate Specificity, PLoS One, 2013, 8(No), e57008 CrossRef PubMed.
- K. M. Mulvaney, C. Blomquist, N. Acharya, R. Li, M. J. Ranaghan, M. O'Keefe, D. J. Rodriguez, M. J. Young, D. Kesar, D. Pal, M. Stokes, A. J. Nelson, S. S. Jain, A. Yang, Z. M. Bernstein, J. Columbus, F. K. Bozal, A. Skepner, D. Raymond, S. LaRussa, D. C. McKinney, Y. Freyzon, Y. Baidi, D. Porter, A. J. Aguirre, A. Ianari, B. McMillan and W. R. Sellers, Molecular basis for substrate recruitment to the PRMT5 methylosome, Mol. Cell, 2021, 81, 3481–3495, DOI:10.1016/j.molcel.2021.07.019.
- H. S. Sims and M. Dai, Taming PRMT5–adaptor protein interactions, Trends Pharmacol. Sci., 2023, 44(3), 134–136, DOI:10.1016/j.tips.2023.01.002.
- D. C. McKinney, B. J. McMillan, M. J. Ranaghan, J. A. Moroco, M. Brousseau, Z. Mullin-Bernstein, M. O'Keefe, P. McCarren, M. F. Mesleh, K. M. Mulvaney, F. Robinson, R. Singh, B. Bajrami, F. F. Wagner, R. Hilgraf, M. J. Drysdale, A. J. Campbell, A. Skepner, D. E. Timm, D. Porter, V. K. Kaushik, W. R. Sellers and A. Ianari, Discovery of a first-in-class inhibitor of the PRMT5-substrate adaptor interaction, J. Med. Chem., 2021, 64, 11148–11168, DOI:10.1021/acs.jmedchem.1c00507.
- A. Krzyzanowski, L. M. Esser, A. Willaume, R. Prudent, C. Peter, P. Hart and H. Waldmann, Development of macrocyclic PRMT5-adaptor protein interaction inhibitors, J. Med. Chem., 2022, 65, 15300–15311, DOI:10.1021/acs.jmedchem.2c01273.
- Z. Q. Bonday, G. S. Cortez, M. J. Grogan, S. Antonysamy, K. Weichert, W. P. Bocchinfuso, F. Li, S. Kennedy, B. Li, M. M. Mader, C. H. Arrowsmith, P. Brown, M. S. Eram, M. M. Szewczyk, D. Barsyte-Lovejoy, M. Vedadi, E. Guccione and R. M. Campbell, LLY-283, a potent and selective inhibitor of arginine methyltransferase 5, PRMT5, with antitumor activity, ACS Med. Chem. Lett., 2018, 9(7), 612–617, DOI:10.1021/acsmedchemlett.8b00014.
- H. Lin, M. Wang, Y. W. Zhang, S. Tong, R. A. Leal, R. Shetty, K. Vaddi and J. I. Luengo, Discovery of Potent and Selective Covalent Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors, ACS Med. Chem. Lett., 2019, 10, 1033–1038, DOI:10.1021/acsmedchemlett.9b00074.
- M. Michelle, W. David, H. Chunhui, K. Shuhei, S. Sebastian and W. Murray, PRMT5 Inhibitors, WO Pat., PCT WO 2020/033285 A1, 2020 Search PubMed.
- J. Luengo, R. A. Leal, L. Hong; S. Rupa, Selective Inhibitors of Protein Arginine Methyltransferase 5 (PRMT5), WO Pat., PCT WO2018085833 A2, 2018 Search PubMed.
- J. Luengo, R. A. Leal, L. Hong and S. Rupa, Selective Inhibitors of Protein Arginine Methyltransferase 5 (PRMT5), WO Pat., PCT WO2018060824 A1, 2018 Search PubMed.
- Pfizet Inc., Substituted Carbonucleoside Derivatives Useful as Anticancer Agents, US Pat., US2017/0348313 A1, 2017 Search PubMed.
- Aligos Therapeutics Inc., Compounds Targeting PRMT5, WO Pat., PCT WO2020205867A1, 2020 Search PubMed.
- Aligos Therapeutics Inc., Compounds targeting PRMT5, WO Pat., PCT WO2021202480 A1, 2021 Search PubMed.
- Janssen Pharmaceutica Nv, Novel Monocyclic and Bicyclic Ring System Substituted Carbanucleoside Analogues for Use as PRMT5 Inhibitors, WO Pat., PCT WO2018065365 A1, 2018 Search PubMed.
- K. Marjon, M. J. Cameron, P. Quang, M. F. Clasquin, E. Mandley, K. Kunii, M. McVay, S. Choe, 1 A. Kernytsky, S. Gross, Z. Konteatis, J. Murtie, M. L. Blake, J. Travins, M. Dorsch, S. A. Biller and K. M. Marks, MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis, Cell Rep., 2016, 15, 1–14, DOI:10.1016/j.celrep.2016.03.043.
- G. V. Kryukov, F. H. Wilson, J. R. Ruth, J. Paulk, A. Tsherniak, S. E. Marlow, F. Vazquez, B. A. Weir, M. E. Fitzgerald, M. Tanaka, C. M. Bielski, J. M. Scott, C. Dennis, G. S. Cowley, J. S. Boehm, D. E. Root, T. R. Golub, C. B. Clish, J. E. Bradner, W. C. Hahn and L. A. Garraway, MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells, Science, 2016, 351, 1214–1218, DOI:10.1126/science.aad5214.
- H. G. Chin, D. Patnaik, P.-O. Este`ve, S. E. Jacobsen and S. Pradhan, Catalytic properties and kinetic mechanism of human recombinant lys-9 histone H3 methyltransferase SUV39H1: participation of the chromodomain in enzymatic catalysis, Biochemistry, 2006, 45(10), 3272–3284, DOI:10.1021/bi051997r.
- D. Smil, M. S. Eram, F. Li, S. Kennedy, M. M. Szewczyk, P. J. Brown, D. Barsyte-Lovejoy, C. H. Arrowsmith, M. Vedadi and M. Schapira, Discovery of a dual PRMT5-PRMT7 inhibitor, ACS Med. Chem. Lett., 2015, 6, 408–412, DOI:10.1021/ml500467h.
- K. J. Mavrakis, E. R. McDonald III, M. R. Schlabach, E. Billy, G. R. Hoffman, A. deWeck, D. A. Ruddy, K. Venkatesan, J. Yu, G. M. Allister, M. Stump, R. deBeaumont, S. Ho, Y. Yue, Y. Liu, Y. Y. Neale, G. Yang, F. Lin, H. Yin, H. Gao, D. R. Kipp, S. Zhao, J. T. M. Namara, E. R. Sprague, B. Zheng, Y. Lin, Y. S. Cho, J. Gu, K. Crawford, D. Ciccone, A. C. Vitari, A. Lai, V. Capka, K. Hurov, J. A. Porter, J. Tallarico, C. Mickanin, E. Lees, R. Pagliarini, N. Keen, T. Schmelzle, F. Hofmann, F. Stegmeier and W. R. Sellers, Disordered methionine metabolism in MTAP/CDKN2A deleted cancers leads to dependence on PRMT5, Science, 2016, 351, 1208–1213, DOI:10.1126/science.aad5944.
- D. Brehmer, L. Beke, T. Wu, H. J. Millar, C. Moy, W. Sun, G. Mannens, V. Pande, A. Boeckx, E. V. Heerde, T. Nys, E. M. Gustin, B. Verbist, L. Zhou, Y. Fan, V. Bhargava, P. Safabakhsh, P. Vinken, T. Verhulst, A. Gilbert, S. Rai, T. A. Graubert, F. Pastore, D. Fiore, J. Gu, A. Johnson, U. Philippar, B. Morschh€auser, D. Walker, D. D. Lange, V. Keersmaekers, M. Viellevoye, G. Diels, W. Lorenzi and S. Laquerre, Discovery and pharmacological characterization of JNJ-64619178, a novel small-molecule inhibitor of PRMT5 with potent antitumor activity, Mol. Cancer Therapeut., 2021, 20, 2317–2328, DOI:10.1158/1535-7163.MCT-21-0367.
- M. V. Haren, L. Q. V. Ufford, E. E. Moret and N. I. Martin, Synthesis and evaluation of protein arginine N-methyltransferase inhibitors designed to simultaneously occupy both substrate binding sites, Org. Biomol. Chem., 2015, 13(2), 549–560, 10.1039/C4OB01734J.
- Janssen Pharmaceutica Nv, Novel 6-6 Bicyclic Aromatic Ring Substituted Nucleoside Analogues for Use as PRMT5 Inhibitors, WO Pat., PCT WO 2017032840 A1, 2017 Search PubMed.
- Merck Sharp & Dohme Corp, PRMT5 inhibitors, WO Pat., PCT WO2020033282 A1, 2020 Search PubMed.
- R. V. Quiroz, M. H. Reutershan, S. E. Schneider, D. Sloman, B. M. Lacey, B. M. Swalm, C. S. Yeung, C. Gibeau, D. S. Spellman, D. A. Rankic, D. Chen, D. Witter, D. Linn, E. Munsell, G. Feng, H. Xu, J. M. E. Hughes, J. Lim, J. Saurí, K. Geddes, M. Wan, M. S. Mansueto, N. E. Follmer, P. S. Fier, P. Siliphaivanh, P. Daublain, R. L. Palte, R. P. Hayes, S. Lee, S. Kawamura, S. Silverman, S. Sanyal, T. J. Henderson, Y. Ye, Y. Gao, B. Nicholson and M. R. Machacek, The Discovery of Two Novel Classes of 5,5-Bicyclic Nucleoside-Derived PRMT5 Inhibitors for the Treatment of Cancer, J. Med. Chem., 2021, 64, 3911–3939, DOI:10.1021/acs.jmedchem.0c02083.
- Merck Sharp & Dohme Corp, Use of Biomarkers in Identifying Patients that Will Be Responsive to Treatment with a PRMT5 Inhibitor, WO Pat., PCT WO2021126999 A1, 2021 Search PubMed.
- Merck Sharp & Dohme Corp, PRMT5 Inhibitors, WO Pat., PCT WO2020033288 A1, 2020 Search PubMed.
- V. Saravanan, R. Sridharan, B. R. Reddy, G. Shivani, S. Dhanalakshmi, T. M. Kumar, N. Tamizharasan, S. InduN, N. Nagendra, K. Subramaniam, M. Zainuddin, S. Sayantani, S. S. Kumar and H. Prakash, Heterocyclic Compounds As PRMT5 Inhibitors, WO Pat., PCT WO 2019/102494 A1, 2019 Search PubMed.
- Aurigene Discovery Technologies Limited, Imidazolidin-2-One Compounds As Prmt5 Modulators, WO Pat., PCT WO 2019/180628 A1, 2019 Search PubMed.
- Merck Sharp & Dohme Corp, PRMT5 Inhibitors, WO Pat., PCT WO 2021/126729 A1, 2021 Search PubMed.
- Merck Sharp & Dohme Corp, PRMT5 Inhibitors, WO Pat., PCT WO 2019/094311 A1, 2019 Search PubMed.
- Merck Sharp & Dohme Corp, PRMT5 Inhibitors, WO Pat., PCT WO 2019/094312 A1, 2019 Search PubMed.
- Merck Sharp & Dohme Corp, PRMT5 Inhibitors, WO Pat., PCT WO 2021/126732 A1, 2021 Search PubMed.
- Nanjing Sanhome Pharmaceutical Research And Development Co., Ltd., Tricyclic Compound As Prmt5 Inhibitor and Application Thereof, WO Pat., PCT WO 2020/259478 A1, 2020 Search PubMed.
- K. W. Duncan, N. Rioux, P. A. Boriack-Sjodin, M. J. Munchhof, L. A. Reiter, C. R. Majer, L. Jin, L. D. Johnston, E. C. Penebre, K. G. Kuplast, M. P. Scott, R. M. Pollock, N. J. Waters, J. J. Smith, M. P. Moyer, R. A. Copeland and R. Chesworth, Structure and property guided design in the identification of PRMT5 tool compound EPZ015666, ACS Med. Chem. Lett., 2016, 7(2), 162–166, DOI:10.1021/acsmedchemlett.5b00380.
- J.-w. Shao, K.-k. Zhu, D.-H. Du, Y.-Y. Zhang, H. Tao, Z. Chen, H.-L. Jiang, K. Chen, C. Luo and W. Duan, Discovery of 2-Substituted-N-(3-(3,4-dihydroisoquinolin-2(1 H)-yl)-2-hydroxypropyl)-1,2,3,4-tetrahydroisoquinoline-6-carboxamide as Potent and Selective Protein Arginine Methyltransferases 5 Inhibitors: Design, Synthesis and Biological Evaluation, Eur. J. Med. Chem., 2019, 164, 317–333, DOI:10.1016/j.ejmech.2018.12.065.
- K.-K. Zhu, J.-L. Song, H.-R. Tao, Z.-Q. Cheng, C.-S. Jiang and H. Zhang, Discovery of new potent protein arginine methyltransferase 5 (PRMT5) inhibitors by assembly of key pharmacophores from known inhibitors, Bioorg. Med. Chem. Lett., 2018, 28, 3693–3699, DOI:10.1016/j.bmcl.2018.10.026.
- Y. Shen, G. Gao, X. Yu, H. Kim, L. Wang, L. Xie, M. Schwarz, X. Chen, E. Guccione, J. Liu, M. T. Bedford and J. Jin, Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders, J. Med. Chem., 2020, 63, 9977–9989, DOI:10.1021/acs.jmedchem.0c01111.
- X.-T. Liang, S. Duan, Q. Yang, X.-Q. Ma, Z. Li, Q. Yao, K.-Y. Wu, P. Chang, G.-Q. Feng, W. Hong, M. Cao, Q.-C. Zhou, X.-P. Zhong and H.-B. Zhao, Protein Arginine Methyltransferase 5 Is Necessary for Embryonic Development in Medaka Oryzias latipes, Fishes, 2023, 8, 19, DOI:10.3390/fishes8010019.
- M. Bezzi, S.-X. Teo, J. Muller, W. C. Mok, S. K. Sahu, L. A. Vardy, Z. Q. Bonday and E. Guccione, Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery, Genes Dev., 2013, 27, 1903–1916, DOI:10.1101/gad.219899.113.
- Y.-J. Wang, J.-B. Cao, J. Yang, T. Liu, H.-L. Yu, Z.-X. He, S.-L. Bao, X.-X. He and X.-J. Zhu, PRMT5-mediated homologous recombination repair is essential to maintain genomic integrity of neural progenitor cells, Cell. Mol. Life Sci., 2024, 81, 123, DOI:10.1007/s00018-024-05154-x.
- K. H. Kim, Z. H. Jia, M. Snyder, J. Chen, J. Qiu, S. N. Oprescu, X. Chen, S. A. Syed, F. Yue, B. T. Roseguini, A. N. Imbalzano, C. Hu and S. Kuang, PRMT5 links lipid metabolism to contractile function of skeletal muscles, EMBO Rep., 2023, 24, 1–16, DOI:10.15252/embr.202357306.
- F. Liu, G. Cheng, P. Hamard, S. Greenblatt, L. Wang, N. Man, F. Perna, H. Xu, M. Tadi, L. Luciani and S. D. Nimer, Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis, J. Clin. Invest., 2015, 125, 3532–3544, DOI:10.1172/JCI81749.
- A Dose Escalation Study Of PF-06939999 In Participants With Advanced Or Metastatic Solid Tumors, linicalTrials, NCT03854227, https://clinicaltrials.gov/study/NCT03854227cond=NCT03854227&rank=1&tab=results.
- J. Rodon, E. Rodriguez, M. L. Maitland, F. Y.-C. Tsai, M. A. Socinski, J. D. Berlin, J. S. Thomas, T. Baghdadi, I.-M. Wang, C. Guo, M. Golmakani, L. N. Clark, M. Gazdoiu, M. Li and A. W. Tolcher, A phase I study to evaluate the safety, pharmacokinetics, and pharmacodynamics of PF-06939999 (PRMT5 inhibitor) in patients with selected advanced or metastatic tumors with high incidence of splicing factor gene mutations, ESMO Open, 2024, 9(4), 101961, DOI:10.1016/j.esmoop.2024.102961.
- A. Huang, L. A. Garraway, A. Ashworth and B. Weber, Synthetic lethality as an engine for cancer drug target discovery, Nat. Rev. Drug Discovery, 2020, 19, 23–38, DOI:10.1038/s41573-019-0046-z.
- C. B. Bridges, The origin of variations in sexual and sex- limited characters, Am. Nat., 1922, 56, 51–63 CrossRef.
- T. Dobwansky, Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura, Genetics, 1946, 31, 269–290 CrossRef.
- L. H. Hartwell, P. Szankasi, C. J. Roberts, A. W. Murray and S. H. Friend, Integrating genetic approaches into the discovery of anticancer drugs, Science, 1997, 278, 1064–1068, DOI:10.1126/science.278.5340.1064.
- F. M. Behan, F. iorio, G. Picco, E. Gonçalves, C. M. Beaver, G. Migliardi, R. Santos, Y.-H. Rao, F. Sassi, M. Pinnelli, R. Ansari, S. Harper, D. A. Jackson, R. Mcrae, R. Pooley, P. Wilkinson, D. V. D. Meer, D. Dow, C. B. Doepner, A. Bertotti, L. trusolino, E. A. Stronach, J. S. Rodriguez, K. Yusa and M. J. Garnett, Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens, Nature, 2019, 568, 511–516, DOI:10.1038/s41586-019-1103-9.
- H. Farmer, N. McCabe, C. J. Lord, A. N. J. Tutt, D. A. Johnson, T. B. Richardson, M. Santarosa, K. J. Dillon, I. Hickson, C. Knights, N. M. B. Martin, S. P. Jackson, G. C. M. Smit and A. Ashworth, Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy, Nature, 2005, 434(7035), 917–921, DOI:10.1038/nature03445.
- H. E. Bryant, N. Schultz, H. D. Thomas, K. M. Parker, D. Flower, E. Lopez, S. Kyle, M. Meuth, N. J. Curtin and T. Helleday, Specific killing of BRCA2deficient tumours with inhibitors of poly(ADP-ribose) polymerase, Nature, 2005, 434(7035), 913–917, DOI:10.1038/nature03443.
- E. P. Lauraine, J. A. Ledermann, F. Selle, V. Gebski, R. T. Penson, A. M. Oza, J. Korach, T. Huzarski, A. Poveda, S. Pignata, M. Friedlander, N. Colombo, P. Harter, K. Fujiwara, I. R. Coquard, S. Banerjee, J. Liu, E. S. Lowe, R. Bloomfield, P. Pautier and the SOLO2/ENGOT-Ov21 investigators, Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a doubleblind, randomised, placebo-controlled, phase 3 trial, Lancet Oncol., 2017, 18(9), 1274–1284, DOI:10.1016/S1470-2045(17)30469-2.
- D. W. Parsons, S. Jones, X. Zhang, J. C.-H. Lin, R. J. Leary, P. Angenendt, P. Mankoo, H. Carter, I.-M. Siu, G. L. Gallia, A. Olivi, R. McLendon, B. A. Rasheed, S. Keir, T. Nikolskaya, Y. Nikolsky, D. A. Busam, H. Tekleab, L. A. D. Jr J. Hartigan, D. R. Smith, R. L. Strausberg, S. K. N. Marie, S. M. O. S. H. Yan, G. J. Riggins, D. D. Bigner, R. Karchin, N. Papadopoulos, G. Parmigiani, B. Vogelstein, V. E. Velculescu and K. W. Kinzler, An integrated genomic analysis of human glioblastoma multiforme, Science, 2008, 321, 1807–1812, DOI:10.1126/science.1164382.
- E. Guccione, M. Schwarz, F. DiTullio and S. Mzoughi, Cancer synthetic vulnerabilities to protein arginine methyltransferase inhibitors, Curr. Opin. Pharmacol., 2021, 59, 33–42, DOI:10.1016/j.coph.2021.04.004.
- M. Sauter, B. Moffatt, M. C. Saechao, R. Hell and M. Wirt, Methionine salvage and S-adenosylmethionine: essential links between sulfur, ethylene and polyamine biosynthesis, Biochem. J., 2013, 451, 145–154, DOI:10.1042/BJ20121744.
- M. Barbarino, D. Cesari, M. Bottaro, L. Luzzi, A. Namagerdi, F. M. Bertolino, C. Bellan, F. Proietti, P. Somma, M. Micheli, M. M. deSanti, R. Guazzo, L. Mutti, L. Pirtoli, P. Paladini, P. Indovina and A. Giordano, PRMT5 silencing selectively affects MTAP-deleted mesothelioma: In vitro evidence of a novel promising approach, J. Cell. Mol. Med., 2020, 00, 1–13, DOI:10.1111/jcmm.15213.
- K. Marjon, P. Kalev and K. Marks, Cancer Dependencies: PRMT5 and MAT2A in MTAP/p16-Deleted Cancers, Annu. Rev. Cancer Biol., 2021, 5, 371–390, DOI:10.1146/annurev-cancerbio-030419033444.
- K. M. Cottrell, K. J. Briggs, D. A. Whittington, H. Jahic, J. A. Ali, C. B. Davis, S. Gong, D. Gotur, L. Gu, P. McCarren, M. R. Tonini, A. Tsai, E. W. Wilker, H. Yuan, M. Zhang, W. Zhang, A. Huang and J. P. Maxwell, Discovery of TNG908: A Selective, Brain Penetrant, MTA-Cooperative PRMT5 Inhibitor That Is Synthetically Lethal with MTAP-Deleted Cancers, J. Med. Chem., 2024, 67, 6064–6080, DOI:10.1021/acs.jmedchem.4c00133.
- K. M. Cottrell and J. P. Maxwell. Piperidin-1-yl-N-pyridin-3-yl-2-oxoacetamide derivatives useful for the treatment of MTAP-deficient and/or MTA accumulating cancers and their preparation, WO Pat., PCT WO2022026892, 2022 Search PubMed.
- C. R. Smith, R. Aranda, T. P. Bobinski, D. M. Briere, A. C. Burns, J. G. Christensen, J. Clarine, L. D. Engstrom, R. J. Gunn, A. Ivetac, R. Jean-Baptiste, J. M. Ketcham, M. Kobayashi, J. Kuehler, S. Kulyk, J. D. Lawson, K. Moya, P. Olson, L. Rahbaek, N. C. Thomas, X. Wang, L. M. Waters and M. A. Marx, Fragment-Based Discovery of MRTX1719, a Synthetic Lethal Inhibitor of the PRMT5•MTA Complex for the Treatment of MTAP-Deleted Cancers, J. Med. Chem., 2022, 65, 1749–1766, DOI:10.1021/acs.jmedchem.1c01900.
- B. Thomas, S. C. Ronald, M. M. Arnold, K. J. Michael, B. A. Craig, L. J. Dawic, K. Svitlana, K. Jon and I. Anthony, MTA-cooperative PRMT5 Inhibitors, WO Pat., PCT WO2021050915A1, 2021 Search PubMed.
- B. Belmontes, K. Slemmons and S. Liu, The Discovery and Preclinical Characterization of AMG 193, a First-in-Class MTA-Cooperative PRMT5 Inhibitor with Broad Activity against MTAP-Null Cancers, Mol. Cancer Ther., 2023, 22, B177 CrossRef.
- K. M. Mulvaney, Early Clinical Success of MTA-Cooperative PRMT5 Inhibitors for the Treatment of CDKN2A/MTAP-Deleted Cancers, Cancer Discov., 2023, 13, 2310–2312, DOI:10.1158/2159-8290.CD-23-0951.
- Y. Barekatain, J. J. Ackroyd, V. C. Yan, S. Khadka, L. Wang, K.-C. Chen, A. H. Poral, T. Tran, D. K. Georgiou, K. Arthur, Y.-H. Lin, N. Satani, E. S. Ballato, E. I. Behr, A. C. deCarvalho, R. G. W. Verhaak, J. Groot, J. T. Huse, J. M. Asara, R. Kalluri and F. L. Muller, Homozygous MTAP deletion in primary human glioblastoma is not associated with elevation of methylthioadenosine, Nat. Commun., 2021, 12, 4228, DOI:10.1038/s41467-021-24240-3.
- Study of AG-270 in Participants With Advanced Solid Tumors or Lymphoma With MTAP Loss, ClinicalTrials.gov ID NCT03435250, https://clinicaltrials.gov/study/NCT03435250?cond=AG-270%26rank=1 Search PubMed.
- R. S. Heist, M. M. Gounder, S. Postel-Vinay, F. Wilson, E. Garralda, K. Do, G. I. Shapiro, P. Martin-Romano, G. Wulf, M. Cooper, C. Almon, S. Nabhan, V. Iyer, Y. Zhang, K. Marks, E. Aguado-Fraile, F. Basile, K. Flaherty and A. Howard, A phase 1 trial of AG-270 in patients with advanced solid tumors or lymphoma with homozygous MTAP deletion, Mol. Cancer Ther., 2019, 18(12_Supplement), PR03, DOI:10.1158/1535-7163.TARG-19-PR03.
- Z. Konteatis, J. Travins, S. Gross, K. Marjon, A. Barnett, E. Mandley, B. Nicolay, R. Nagaraja, Y. Chen, Y. Sun, Z. Liu, J. Yu, Z. Ye, F. Jiang, W. Wei, C. Fang, Y. Gao, P. Kalev, M. L. Hyer, B. DeLaBarre, L. Jin, A. K. Padyana, L. Dang, J. Murtie, S. A. Biller, Z. Sui and K. M. Marks, Discovery of AG-270, a First-in-Class Oral MAT2A Inhibitor for the Treatment of Tumors with Homozygous MTAP Deletion, J. Med. Chem., 2021, 64, 4430–4449, DOI:10.1021/acs.jmedchem.0c01895.
- P. Kalev, M. L. Hyer, S. Gross, Z. Konteatis, C.-C. Chen, M. Fletcher, M. Lein, E. Aguado-Fraile, V. Frank, A. Barnett, E. Mandley, J. Goldford, Y. Chen, K. Sellers, S. Hayes, K. Lizotte, P. Quang, Y. Tuncay, M. Clasquin, R. Peters, J. Weier, E. Simone, J. Murtie, W. Liu, R. Nagaraja, L. Dang, Z. Sui, S. A. Biller, J. Travins, K. M. Marks and K. Marjon, MAT2A Inhibition Blocks the Growth of MTAP-Deleted Cancer Cells by Reducing PRMT5-Dependent mRNA Splicing and Inducing DNA Damage, Cancer Cell, 2021, 39, 209–224, DOI:10.1016/j.ccell.2020.12.010.
- C. D. Fusco, M. Schimpl, U. Börjesson, T. Cheung, I. Collie, L. Evans, P. Narasimhan, C. Stubbs, M. V. Chantada, D. J. Wagner, M. Grondine, M. G. Sanders, S. Tentarelli, E. Underwood, A. Argyrou, J. M. Smith, J. T. Lynch, E. l. Chiarparin, G. Robb, S. K. Bagal and J. S. Scott, Fragment-Based Design of a Potent MAT2a Inhibitor and in Vivo Evaluation in an MTAP Null Xenograft Model, J. Med. Chem., 2021, 64, 681–6826, DOI:10.1021/acs.jmedchem.1c00067.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, A 'rule of three' for fragment-based lead discovery?, Drug Discov. Today, 2003, 8(19), 876–877, DOI:10.1016/s1359-6446(03)02831-9.
- H. Jhoti, G. Williams, D. C. Rees and C. W. Murray, The 'rule of three' for fragment-based drug discovery: where are we now?, Nat. Rev. Drug Discovery, 2013, 12, 644, DOI:10.1038/nrd3926-c1.
- G. M. Keserű, D. A. Erlanson, G. G. Ferenczy, M. M. Hann, C. W. Murray and S. D. Pickett, Design principles for fragment libraries – Maximizing the value of learnings from Pharma fragment based drug discovery (FBDD) programs for use in academia, J. Med. Chem., 2016, 59(18), 8189–8206, DOI:10.1021/acs.jmedchem.6b00197.
- M. Bissaro, M. Sturlese and S. Moro, The rise of molecular simulations in fragment-based drug design (FBDD): an overview, Drug Discovery Today, 2020, 25(9), 1693–1701, DOI:10.1016/j.drudis.2020.06.023.
- A. J. Woodhead, D. A. Erlanson, I. J. P. de Esch, R. S. Holvey, W. Jahnke and P. Pathuri, Fragment-to-Lead Medicinal Chemistry Publications in 2022, J. Med. Chem., 2024, 67(4), 2287–2304, DOI:10.1021/acs.jmedchem.3c02070.
- Phase 1/2 Study of MRTX1719 in Solid Tumors With MTAP Deletion, ClinicalTrials.gov ID NCT05245500, https://clinicaltrials.gov/study/NCT05245500?cond=MRTX1719%26rank=1 Search PubMed.
- L. D. Engstrom, R. Aranda, L. Waters, K. Moya, V. Bowcut, L. Vegar, D. Trinh, A. Hebbert, C. R. Smith, S. Kulyk, J. D. Lawson, L. He, L. D. Hover, J. Fernandez-Banet, J. Hallin, D. Vanderpool, D. M. Briere, A. Blaj, M. A. Marx, J. Rodon, M. Offin, K. C. Arbour, M. L. Johnson, D. J. Kwiatkowski, P. A. Jänne, C. L. Haddox, K. P. Papadopoulos, J. T. Henry, K. Leventakos, J. G. Christensen, R. Shazer and P. Olson, MRTX1719 is an MTA-cooperative PRMT5 inhibitor that exhibits synthetic lethality in preclinical models and patients with MTAP-deleted cancer, Cancer Discov., 2023, 13, 2412–2431, DOI:10.1158/2159-8290.CD-23-0669.
- Safety and Tolerability of TNG908 in Patients With MTAP-deleted Solid Tumors,ClinicalTrials.gov ID NCT0527547, https://clinicaltrials.gov/study/NCT05275478?cond=TNG908%26rank=1 Search PubMed.
- Safety and Tolerability of TNG462 in Patients With MTAP-deleted Solid Tumors, ClinicalTrials.gov ID NCT05732831, https://clinicaltrials.gov/study/NCT05732831?cond=TNG-462%26rank=1 Search PubMed.
- A Phase 2 Study of AMG 193 in Participants With MTAP-deleted Advanced NSCLC, ClinicalTrials.gov ID NCT06593522, https://clinicaltrials.gov/study/NCT06593522?cond=AMG193%26rank=1 Search PubMed.
- A Phase 1/2 Study of AMG 193 in Combination With IDE397 in Participants With Advanced Methylthioadenosine Phosphorylase (MTAP)-Null Solid Tumors, ClinicalTrials. gov ID NCT05975073, https://clinicaltrials.gov/study/NCT05975073?cond=AMG193%26rank=2 Search PubMed.
- A Study Evaluating AMG 193 in Combination With Other Therapies in Participants With Advanced Gastrointestinal, Biliary Tract, or Pancreatic Cancers With Homozygous Methylthioadenosine Phosphorylase (MTAP)-Deletion, ClinicalTrials.gov ID NCT06360354, https://clinicaltrials.gov/study/NCT06360354?cond=AMG193%26rank=3 Search PubMed.
- AMG 193 Alone or in Combination With Other Therapies in Subjects With Advanced Thoracic Tumors With Homozygous MTAP-deletion (Master Protocol), ClinicalTrials.gov ID NCT06333951, https://clinicaltrials.gov/study/NCT06333951?cond=AMG193%26rank=4 Search PubMed.
- A Study of AMG 193 in Subjects With Advanced MTAP-null Solid Tumors (MTAP), ClinicalTrials.gov ID NCT05094336, https://clinicaltrials.gov/study/NCT05094336?cond=AMG193%26rank=5 Search PubMed.
- J. Rodon, H. Prenen, A. Sacher, M. Villalona-Calero, N. Penel, A. El Helali, S. Rottey, N. Yamamoto, F. Ghiringhelli, M. E. Goebeler, T. Doi, S. Postel-Vinay, C.-C. Lin, C. Liu, C.-H. Chuang, K. Keyvanjah, T. Eggert and B. H. O’Nei, First-in-human study of AMG 193, an MTA-cooperative PRMT5 inhibitor, in patients with MTAP-deleted solid tumors: results from phase I dose exploration, Ann. Oncol., 2024, 35(12), 1138–1147, DOI:10.1016/j.annonc.2024.08.2339.
- A Study to Investigate the Safety and Tolerability of SCR-6920 Capsule in Patients With Advanced Malignant Tumors, ClinicalTrials.gov ID NCT05528055, https://clinicaltrials.gov/study/NCT05528055?cond=NCT05528055%26rank=1 Search PubMed.
- Y. Yu S. G. Sun, H. Q. Wang, J. Wu, X. Jiang, J. Chen, G. Yang and C. Yang, A phase I study of safety, pharmacokinetics, and pharmacodynamics of SCR-6920, a protein arginine methyltransferase 5 (PRMT5) inhibitor, in patients with advanced malignant tumors,Clinical trial identification: NCT05528055, DOI:10.1016/j.annonc.2023.09.1861.
- C. R. Smith, R. Aranda, J. G. Christensen, L. D. Engstrom, R. J. Gunn, A. Ivetac, J. M. Ketcham, J. Kuehler, J. D. Lawson, M. A. Marx, P. Olson, N. C. Thomas, X. Wang, L. M. Waters and S. Kulyk, Design and evaluation of achiral, non-atropisomeric 4-(aminomethyl) phthalazin-1(2H)-one derivatives as novel PRMT5/MTA inhibitors, Bioorg. Med. Chem., 2022, 71, 116947, DOI:10.1016/j.bmc.2022.116947.
- S. Fu, Q. Zheng, D. Zhang, C. Lin, L. Ouyang, J. Zhang and L. Chen, Medicinal chemistry strategies targeting PRMT5 for cancer therapy, Eur. J. Med. Chem., 2022, 244, 114842, DOI:10.1016/j.ejmech.2022.114842.
- M. Achmatowicz, T. Scattolin, D. R. Snead, D. J. Paymode, S. Roshandel, C. Xie, G.-h. Chen and C.-Y. Chen, Scalable Atroposelective Synthesis of MRTX1719: An Inhibitor of the PRMT5/MTA Complex, Org. Process Res. Dev., 2023, 27, 954–971, DOI:10.1021/acs.oprd.3c00072.
- J. Li and Z.-W. Wang, 4-(aminomethyl)-6-(1-methyl-1H-pyrazol-4-yl)isoquinolin-1(2H)-one derivatives as MTA-cooperative Inhibitors of PRMT5, WO Pat., PCT WO2023174250A1, 2023 Search PubMed.
- Suzhou Puhe Biopharma Co.,Led., PRMT5-MTA Inhibitors, CN Pat., CN116178347A, 2023 Search PubMed.
- X.-X. Chen, J. Li and Y.-H. Chen, The invention relates to the phthalazinone compounds and preparation method, including its composition and application, CN Pat., CN116903611A, 2023 Search PubMed.
- China Pharmaceutical University, Phthalazinone compounds including its compositions and applications, CN Pat., CN116854668A, 2023 Search PubMed.
- C. R. Smith, S. Kulyk, U. Misbha, D. Ahmad, V. Arkhipova, J. G. Christensen, R. J. Gunn, A. Ivetac, J. M. Ketcham, J. Kuehler, J. D. Lawson, N. C. Thomas, X. Wang and M. A. Marx, Fragment optimization and elaboration strategies–the discovery of two lead series of PRMT5/MTAinhibitors from five fragment hits, RSC Med. Chem., 2022, 13, 1549–1564, 10.1039/d2md00163b.
- Mirati Therapeutics Inc., Aminopyridine-based MTA-Cooperative PRMT5 Inbihitors, WO Pat., PCT WO2023278564A1, 2023 Search PubMed.
- K. M. Cottrell, Discovery of TNG462: A Highly Potent and Selective MTA-Cooperative PRMT5 Inhibitor Synthetic Lethal for MTAP Deleted Cancers, American Chemical Society National Meeting, San Francisco, CA, 2023 Search PubMed.
- J. R. Allen, A. Amegadzie, D. J. Beylkin, S. Booker, M. P. Butler, J. R. Bourbeau, M. J. Frohn, S. O. S. Glad, B. W. Husemoen, M. R. Kaller, T. J. Kohn, B. A. Lanman, K. Li, Q. Liu, P. Lopez, V. V. Ma, F. Manoni, J. Medina, A. E. Minatti, C. J. Peiro, L. Pettus, A. J. Pickrell, I. Sarvary, N. A. Tamayo and M. Vestergaard, Novel PRMT5 Inhibitors for Treating Cancer, ACS Med. Chem. Lett., 2021, 12, 1537–1538, DOI:10.1021/acsmedchemlett.1c00512.
- Amgen Inc., Tricyclic Carboxamide Derivatives as PRMT5 Inhibitors, WO Pat., PCT WO2022115377A1, 2022 Search PubMed.
- Amgen Inc., Tricyclic-amido-bicyclic PRMT5 Inhibitors, WO Pat., PCT WO2022169948A1, 2022 Search PubMed.
- J. R. Allen, A. Amegadzie, D. J. Beylkin, S. Booker, M. P. Bourbeau, J. R. Butler, M. J. Frohn, S. O. S. Glad, B. W. Husemoen, M. R. Kaller, J. R. Butler, T. J. Kohn, B. A. Lanman, K. Li, Q. Liu, P. Lopez, V. V. Ma, F. Manoni, J. Medina, A. E. Minatti, J. P. Cadahia, L. Pettus, A. J. Pickrell, I. Sarvary, N. A. Tamayo and M. Vestergaard, Novel PRMT5 Inhibitors, WO Pat., PCT WO2021163344, 2021 Search PubMed.
- Amgen Inc., PRMT5 inhibitors, WO Pat., PCT WO2022132914A1, 2022 Search PubMed.
- H. Lin and J. I. Luengo, Nucleoside Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors, Bioorg. Med. Chem. Lett., 2019, 29(11), 1264–1269, DOI:10.1016/j.bmcl.2019.03.042.
- J. H. Tatlock, I. J. McAlpine, M. B. Tran-Dube, M. J. Wyeth, E. Y. Rui, R. A. Kumpf and M. A. McTigue, Substituted Nucleoside Derivatives Useful as Anticancer Agents, US Pat., US20160244475A1, 2016 Search PubMed.
- A. Ganesan, Epigenetic drug discovery: A success story for cofactor interference, Philos. Trans. R. Soc., B, 2018, 373, 20170069, DOI:10.1098/rstb.2017.0069.
- Y. Zhang, D. Rong, B. Li and Y. Wang, Targeting Epigenetic Regulators with Covalent Small-Molecule Inhibitors, J. Med. Chem., 2021, 64(12), 7900–7925, DOI:10.1021/acs.jmedchem.0c02055.
- J. L. A. dcock, A. A. Gakh, J. L. Pollitte and C. Woods, X-ray Crystal Structure of the First Quaternary 1-Bicyclo[l.l.l]pentane Salt. The Shortest "Nonbonding" Carbon-Carbon Interaction Documented, J. Am. Chem. Soc., 1992, 114, 3981–3982, DOI:10.1021/ja00036a057.
- J. Luengo and K. Vaddi, Selective Inhibitors of Protein Arginine Methyltransferase 5 (PRMT5), WO Pat., PCT WO2017218802A1, 2017 Search PubMed.
- M. Kabir, X.-F. Yu, H. Kaniskan and J. Jin, Chemically induced degradation of epigenetic targets, Chem. Soc. Rev., 2023, 52, 4313–4342, 10.1039/d3cs00100h.
- T. Webb, C. Craigon and A. Ciulli, Targeting epigenetic modulators using PROTAC degraders: Current status and future perspective, Bioorg. Med. Chem. Lett., 2022, 63, 128653, DOI:10.1016/j.bmcl.2022.128653.
- EMA Reviewing Medicines Containing Valsartan from Zhejiang Huahai Following Detection of an Impurity: Some Valsartan Medicines Being Recalled across the EU. EMA/459276/2018, European Medicines Agency, 2018, https://www.ema.europa.eu/en/documents/press-release/ema-reviewing-medicines-containing-valsartan-zhejianghuahai-following-detection-impurity-en.pdf (accessed 2023-06-09) Search PubMed.
- FDA Provides Update on its Ongoing Investigation into ARB Drug Products; Reports on Finding of a New Nitrosamine Impurity in Certain Lots of Losartan and Product Recall, U.S. Food and Drug Administration, 2019, https://www.prnewswire.com/news-releases/fdaprovides-update-on-its-ongoing-investigation-into-arb-drug-productsreports-on-finding-of-a-new-nitrosamine-impurity-in-certain-lots-oflosartan-and-product-recall-300804982.html (accessed 2023-06-09) Search PubMed.
- R. Nudelman, G. Kocks, B. Mouton, D. J. Ponting, J. Schlingemann, S. Simon, G. F. Smith, A. Teasdale and A.-L. Werner, The Nitrosamine “Saga”: Lessons Learned from Five Years of Scrutiny, Org. Process Res. Dev., 2023, 27, 1719–1735, DOI:10.1021/acs.oprd.3c00100.
- J. Schlingemanna, M. J. Burnsb, D. J. Ponting, C. M. Avila, N. E. Romeroc, M. A. Jaywant, G. F. Smith, I. W. Ashworth, S. Simon, C. Saal and A. Wilk, The Landscape of Potential Small and Drug Substance Related Nitrosamines in Pharmaceuticals, J. Pharm. Sci., 2023, 112(5), 1287–1304, DOI:10.1016/j.xphs.2022.11.013.
- H. P. R. Vikram, T. P. Kumar, G. Kumar, N. M. Beeraka, R. Dek, S. M. Suhail, S. Jat, N. Bannimath, G. Padmanabhan, R. S. Chandan, P. Kumar and B. Gurupadayya, Nitrosamines crisis in pharmaceuticals? Insights on toxicological implications, root causes and risk assessment: A systematic review, J. Pharm. Anal., 2024, 14, 100919, DOI:10.1016/j.jpha.2023.12.009.
- N. A. Charoo, S. Dharani, M. A. Khan and Z. Rahman, Nitroso Impurities in Drug Products: An Overview of Risk Assessment, Regulatory Milieu, and Control Strategy, AAPS PharmSciTech, 2023, 24, 60, DOI:10.1208/s12249-023-02523-w.
- R. López-Rodríguez, J. A. McManus, N. S. Murphy, M. A. Ott and M. J. Burns, Pathways for N-nitroso compound formation: secondary amines and beyond, Org. Process Res. Dev., 2020, 24(9), 1558–1585, DOI:10.1021/acs.oprd.0c00323.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Large-Scale Applications of Amide Coupling Reagents for the Synthesis of Pharmaceuticals, Org. Process Res. Dev., 2016, 20(2), 140–177, DOI:10.1021/op500305s.
- M. Turáková, M. Králik, P. Lehock, L. Pikna, M. Smřcová, D. Remeteiová and A. Hudák, Influence of preparation method and palladium content on Pd/C catalysts activity in the liquid phase hydrogenation of nitrobenzene to aniline, Appl. Catal., A, 2014, 476, 103–112, DOI:10.1016/j.apcata.2014.02.025.
- G. Chessari, R. Grainger, R. S. Holvey, R. F. Ludlow, P. N. Mortenson and D. C. Rees, C–H functionalisation tolerant to polar groups could transform fragment-based drug discovery (FBDD), Chem. Sci., 2021, 12, 11976, 10.1039/d1sc03563k.
- C. W. Murray and D. C. Rees, Opportunity Knocks: Organic Chemistry for Fragment-Based Drug Discovery (FBDD), Angew. Chem., Int. Ed., 2016, 55, 488–492, DOI:10.1002/anie.201506783.
- S. T. Seger, P. Rydberg and L. Olsen, Mechanism of the N-hydroxylation of primary and secondary amines by Cytochrome P450, Chem. Res. Toxicol., 2015, 28(4), 597–603, DOI:10.1021/tx500371a.
- A. G. Siraki, Free Radical Metabolites in Arylamine Toxicity, Adv. Mod. Toxicol., 2013, 7, 39–82, DOI:10.1016/B978-0-444-62645-5.00002-X.
- J. C. Sasaki, R. S. Fellers and M. E. Colvin, Metabolic oxidation of carcinogenic arylamines by P450 monooxygenases: theoretical support for the one-electron transfer mechanism, Mutat. Res., 2002, 506–507, 79–89, DOI:10.1016/S0027-5107(02)00154-9.
- E. Sim, I. Westwood and E. Fullam, Arylamine N-acetyltransferases, Expert Opin. Drug Metab. Toxicol., 2007, 3, 2, DOI:10.1517/17425255.3.2.169.
- W. H. Schaefer, Reaction of Primary and Secondary Amines to Form Carbamic Acid Glucuronides, Curr. Drug Metab., 2006, 7, 873–881, DOI:10.2174/138920006779010629.
- K. F. Tipton and M. Strolin, Amine Oxidases and the Metabolism of Xenobiotics, 2001, p. 23, DOI:10.1002/0470846305.ch4.
- R. S. Riley, C. H. June, R. Langer and M. J. Mitchell, Delivery technologies for cancer immunotherapy, Nat. Rev. Drug Discov., 2019, 18, 175–196, DOI:10.1038/s41573-018-0006-z.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology, Adv. Drug Delivery Rev., 2014, 66, 2–25, DOI:10.1016/j.addr.2013.11.009.
- P. Zhang, H. Tao, L. Yu, L. Zhou and C. Zhu, Developing protein arginine methyltransferase 1 (PRMT1) inhibitor TC-E-5003 as an antitumor drug using INEI drug delivery systems, Drug Delivery, 2020, 27(1), 491–501, DOI:10.1080/10717544.2020.1745327.
- S. Abad, A. Manresa, E. Cãnas, M. Baile, P. -Morales, R. Mallavia, M. Saceda and C. Juan Romero, New therapy for pancreatic cancer based on extracellular vesicle, Biomed. Pharmacother., 2023, 162, 114657, DOI:10.1016/j.biopha.2023.114657.
- S. Abad, A. Manresa, E. Cañas, M. Baile, P. Morales, R. Mallavia, M. Saceda and C. Romero, Glioblastoma-Derived Small Extracellular V esicles: Nanoparticles for Glioma T reatment, Int. J. Mol. Sci., 2023, 24, 5910, DOI:10.3390/ijms24065910.
- J. Karlsson, Y. Rui, K. L. Kozielski, A. L. P. O. Choi, S. Y. Tzeng, J. Kim, J. J. Keyes, M. I. Bogorad, K. Gabrielson, H. Cazares, A. Q. Hinojosa, P. C. Searson and J. J. Green, Engineered nanoparticles for systemic siRNA delivery to malignant brain tumours, Nanoscale, 2019, 11, 20045–20057, 10.1039/c9nr04795f.
- F. Ye, J. Huang, H. Wang, C. Luoc and K. Zhao, Targeting epigenetic machinery: Emerging novel allosteric inhibitors, Pharmacol. Ther., 2019, 204, 107406, DOI:10.1016/j.pharmthera.2019.107406.
- Z. Tan, T. Li, H. Lei and X. Zhai, An update on allosteric modulators as a promising strategy targeting histone methyltransferase, Pharmacol. Res., 2021, 172, 105865, DOI:10.1016/j.phrs.2021.105865.
| This journal is © The Royal Society of Chemistry 2024 |