
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benzenesulfonohydrazide-tethered non-fused and fused heterocycles as potential anti-mycobacterial agents targeting enoyl acyl carrier protein reductase (InhA) with antibiofilm activity†
Tarfah Al-Warhia,
Ahmed Sabt b,
Małgorzata Korycka-Machalac,
Asmaa F. Kassemd,
Moataz A. Shaldame,
Hoda Atef Abdelsattar Ibrahimf,
Malwina Kawkag,
Bożena Dziadekg,
Magdalena Kuziołach,
Wagdy M. Eldehna*ei and
Jarosław Dziadek*c
b,
Małgorzata Korycka-Machalac,
Asmaa F. Kassemd,
Moataz A. Shaldame,
Hoda Atef Abdelsattar Ibrahimf,
Malwina Kawkag,
Bożena Dziadekg,
Magdalena Kuziołach,
Wagdy M. Eldehna*ei and
Jarosław Dziadek*c
aDepartment of Chemistry, College of Science, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia
bChemistry of Natural Compounds Department, Pharmaceutical and Drug Industries Research Institute, National Research Centre, Dokki, Cairo, 12622, Egypt
cLaboratory of Genetics and Physiology of Mycobacterium, Institute of Medical Biology of the Polish Academy of Sciences, Lodz, Poland. E-mail: jdziadek@cbm.pan.pl
dDepartment of Chemistry, College of Science and Humanities in Al-Kharj, Prince Sattam Bin Abdulaziz University, Al-Kharj, 11942, Saudi Arabia
eDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Kafrelsheikh University, Kafrelsheikh 33516, Egypt. E-mail: wagdy2000@gmail.com
fPediatric Department, Faculty of Medicine, Cairo University, Cairo, Egypt
gDepartment of Molecular Microbiology, Faculty of Biology and Environmental Protection, University of Lodz, Lodz, Poland
hBio-Med-Chem Doctoral School of the University of Lodz and Lodz Institutes of the Polish Academy of Sciences, Lodz, Poland
iDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Pharos University in Alexandria, Canal El Mahmoudia St., Alexandria 21648, Egypt
First published on 23rd September 2024
Abstract
Because resistant variants of the disease are always emerging, tuberculosis is a global issue that affects economies. New antitubercular medications should be developed, and this can be done by inhibiting druggable targets. Enoyl acyl carrier protein (ACP) reductase (InhA) is a crucial enzyme for the survival of Mycobacterium tuberculosis (MTB). In this study, a series of small molecules based on non-fused and fused heterocycles (pyridine, coumarin, quinoline, and indole) tethered with benzenesulfonohydrazide were prepared via an aza-Michael reaction exploiting a one-pot synthesis approach. The synthesized molecules (2–7) were evaluated for their activity against tubercle bacilli. Three analogues showed efficacy against tuberculosis, with compound 7 demonstrating a MIC value as low as 8 μg mL−1. Consequently, compounds 3 and 7 successfully hindered the growth of mycobacteria in human monocyte-derived macrophages (MDMs), demonstrating their ability to penetrate human professional phagocytes. Furthermore, they restricted the ability of mycobacteria to produce biofilms. In addition, the inhibitory effects of compounds 3 and 7 against InhA were assessed. Compound 7 exhibited the best efficacy, with an IC50 value of 0.91 μM. The findings showed that the sulfonamide and methyl ester's carbonyl functionalities were engaged in hydrogen bonding with the essential Ile194 and Tyr158 residues, respectively.
1. Introduction
The spread of infectious diseases has long been a significant worldwide health concern because of how quickly germs can be transmitted in various environments. This issue is particularly challenging in densely populated regions, where controlling the spread of diseases from person to person is a significant challenge. While numerous medications are available to treat these infections, a concerning trend is the increasing resistance of microbes to most drugs, resulting in the emergence of multidrug-resistant microorganisms. As a result, there is a pressing need to explore new therapeutic compounds to address this serious issue.1Out of all the infectious diseases, tuberculosis (TB) has been identified as the main cause of death caused by a single infectious agent, surpassing HIV/AIDS in rankings. Tuberculosis (TB), also known as ‘white plaque’, is a potentially serious infectious disease caused by the Mycobacterium tuberculosis (MTB) complex, which includes various species such as MTB itself, M. africanum, M. bovis, M. caprae, M. microti, M. pinnipedii, and M. canettii. MTB is the main pathogen that primarily affects the lung (pulmonary TB) as well as other vital organs.2 According to the WHO, approximately 2 billion individuals worldwide have a latent MTB infection that can stay dormant for many years. Around 5–10% of those infected with latent MTB eventually develop active TB. In addition, in 2022, treatment was only available to about 2 out of 5 individuals suffering from drug-resistant tuberculosis.3 Despite the effectiveness of the first-line anti-TB drugs like isoniazid (INH), rifampicin (RIF), ethambutol (EMB), and pyrazinamide (PZA) currently used to treat TB infection, they do not provide optimum results as TB threat is increasing in the developing world.4,5 Therefore, it is essential to develop novel anti-TB medicines that exhibit good tolerance and efficacy against both drug-sensitive and drug-resistant strains of MTB, minimal toxicity, and short treatment duration.6
Due to the unique cell envelope of tubercle bacilli, TB treatment is more challenging than other microbial infections. Cell envelopes consist of a defensive layer composed of mycolic acid, which is a saturated chain of β-hydroxy fatty acids with an α-alkyl side chain 7. An essential component of mycolic acid synthesis is the enoyl ACPR reductase (InhA), which reduces 2-trans enoyl-acyl carrier proteins (ACPs) in a NADH-dependent manner.8 Inhibiting the activity of the catalase-peroxidase enzyme KatG, isoniazid (INH) I inhibits InhA through its conversion into active form, INH-NADH, when it reacts with a NAD/NADH species9 (Scheme 1). The INH-NADH molecule acts as an inhibitor of the enoyl ACP reductase (InhA), which is responsible for mycolic acid production. This inhibition leads to the disruption of the cell wall and ultimately results in cell death, effectively inhibiting tuberculosis.10–12 InhA is the only essential gene encoded by inhA, but mutations in several other genes have been identified as mediating resistance to INH.13 Therefore, it is of utmost importance to note that mutations that abrogate KatG activity, which is prevalent in many clinical strains worldwide, prevent INH from being activated into active forms, preventing it from being utilized.14 It is, therefore, imperative that new compounds be developed that target InhA and can be used against multidrug-resistant strains of M. tuberculosis.15
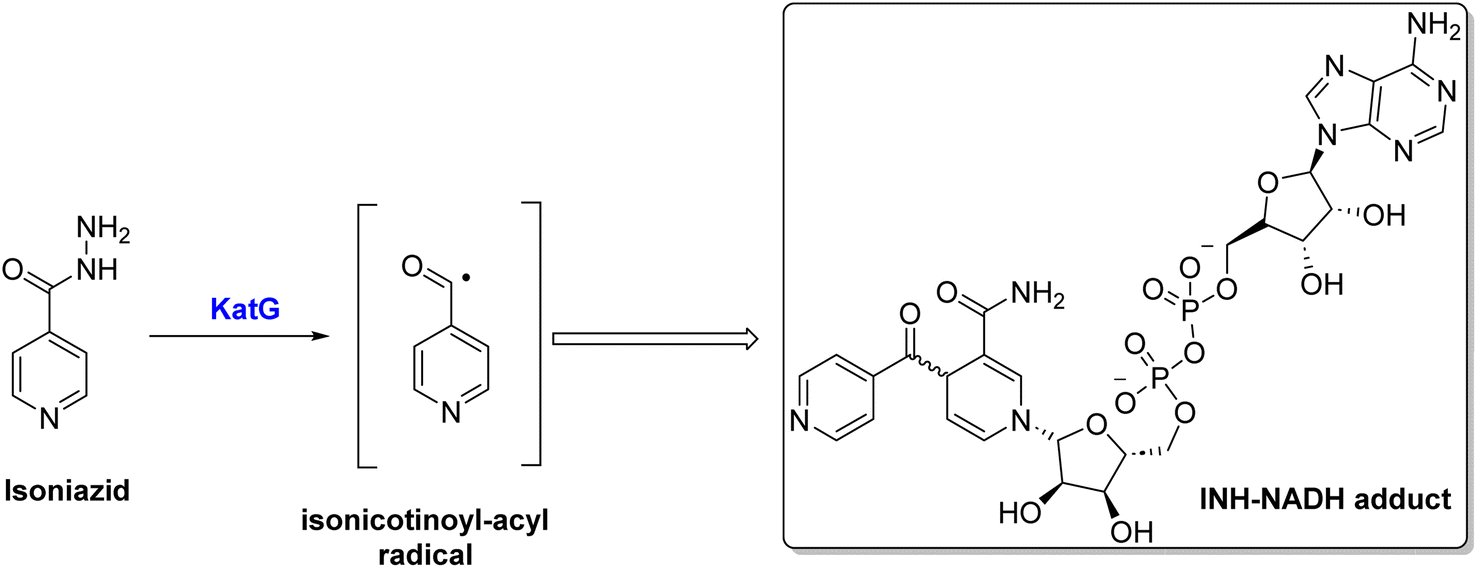 | ||
| Scheme 1 Activation of isoniazid via KatG enzyme. | ||
Three pockets are present in the InhA active site, as revealed by the binding mode of its inhibitors. As the cofactor NAD is composed of Tyr158 residues and ribose moiety, it forms its first pocket. Two hydrophobic pockets accommodate several inhibitor moieties in the second. A hydrophilic side of the third pocket forms the home for the phosphate group of NAD, while a hydrophobic side consists of Ala198 and Ile202.16
Hydrozones (HYDs) contain an azomethine group (HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and have been extensively studied for their structural properties and possible applications in medicinal chemistry.17 Many biological activities have been shown to be enhanced by sulfonyl hydrazones, including antiviral,18 antimicrobial,19 acetylcholinesterase and α-glycosidase inhibitory activities,20,21 anticancer,22 and the ability to inhibit some other enzymes.23,24 Developed aryl sulfonyl HYDs have been the focus of a lot of research recently in order to find new and more effective antitubercular agents with fewer side effects, such as compound II (Fig. 1).25 InhA is considered to be the main molecular target of N-aryl sulfonyl hydrazone derivatives. Therefore, it is crucial to undertake additional investigations to thoroughly validate this hypothesis by examining their mechanism of action.26–28
N) and have been extensively studied for their structural properties and possible applications in medicinal chemistry.17 Many biological activities have been shown to be enhanced by sulfonyl hydrazones, including antiviral,18 antimicrobial,19 acetylcholinesterase and α-glycosidase inhibitory activities,20,21 anticancer,22 and the ability to inhibit some other enzymes.23,24 Developed aryl sulfonyl HYDs have been the focus of a lot of research recently in order to find new and more effective antitubercular agents with fewer side effects, such as compound II (Fig. 1).25 InhA is considered to be the main molecular target of N-aryl sulfonyl hydrazone derivatives. Therefore, it is crucial to undertake additional investigations to thoroughly validate this hypothesis by examining their mechanism of action.26–28
 | ||
| Fig. 1 Structures of representative hybrids of bioactive cores I–VI and our newly designed compounds 2–7. | ||
On the other hand, the main moiety in heterocyclic molecules found in a variety of synthetic drugs, pharmaceuticals, bioactive natural products, and agrochemicals nowadays is either nitrogen or oxygen.29 The potency of drugs has been greatly increased by heterocyclic compounds that contain nitrogen and/or oxygen, such as coumarin, quinoline, pyridine, and/or pyridine. The pharmacological properties of these compounds span the gamut, including antibacterial, antileishmanial, anticonvulsant, anti-HIV, anticancer, and particularly anti-tuberculosis effects.30–36 Different types of these substances have been studied for their ability to fight against mycobacterial infections. A variety of coumarin compounds have become important in the search for new antimycobacterial activities,37,38 such as compound III.39 In 2020, the researchers highlighted the significance of quinoline hydrazone derivatives and their effectiveness against tuberculosis, e.g., compound IV (Fig. 1).40 Moreover, Cihan-Üstünda and coworkers41 developed and tested new indole derivatives that showed promising results in inhibiting Mycobacterium tuberculosis, with compound V being the most potent with MIC = 25 μg mL−1. In another study, a novel series of pyridine derivatives were developed and explored for in vitro anti-tubercular action.42 Compound VI (Fig. 1) exhibited the most active against Mycobacterium tuberculosis with an MIC value of 14 ± 7 μM.
On the basis of these observations and as part of our continuing interest in the search for new compounds as antitubercular agents, we have designed, synthesized, and characterized a series of heterocyclic compounds-sulfonylhydrazone hybrids, whereas the 4-methylphenyl core is expected to occupy the hydrophobic pocket and form Pi–Pi stacking with Tyr158. The sulfonylhydrazide functional group may serve as a spacer that has been modified to facilitate hydrogen bonding interactions with essential amino acid residues.
In addition, the heterocyclic scaffold can form a polar contact with the NAD cofactor (Fig. 2). The synthesized derivatives were tested for their activity toward M. tuberculosis and M. abscessus. The most potent counterparts were investigated for their inhibitory action on the Mycobacterium tuberculosis InhA enzyme. Molecular docking and molecular dynamic simulation studies were done to explore the plausible binding mode for the promising hits within the InhA active sit. Predictions have been made on the physicochemical and pharmacokinetic features of the promising candidates.
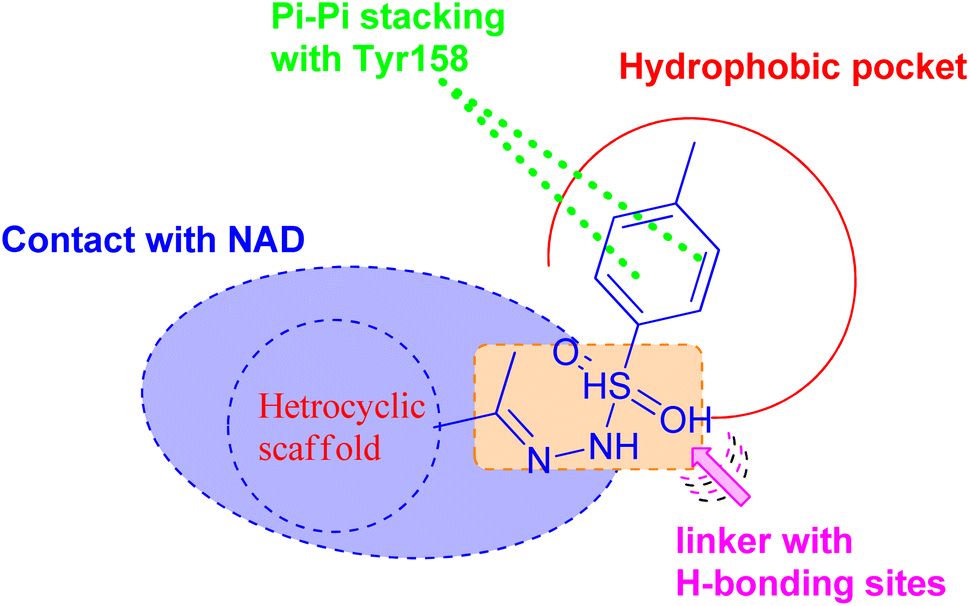 | ||
| Fig. 2 Pharmacophoric features for the most active molecules and binding interaction with the active site of InhA. | ||
The work presents a series of benzenesulfonohydrazide-tethered heterocyclic compounds designed as potential novel anti-mycobacterial candidates targeting the InhA enzyme, which is crucial for the survival of M. tuberculosis. The study emphasizes the innovative approach of synthesizing both non-fused and fused heterocycles, including pyridine, coumarin, quinoline, and indole motifs, which exhibit significant bactericidal activity against M. tuberculosis. In addition, these molecules will be evaluated for the ability to penetrate human macrophages and inhibit mycobacterial biofilm formation, addressing a critical gap in current tuberculosis treatment strategies and thereby contributing to the ongoing efforts in anti-tuberculosis drug discovery.
2. Results and discussion
2.1. Chemistry
The synthesis of target compounds 2–7 involves the preparation of key intermediates, specifically the acetyl heterocycles (9, 11, 14, and 16), as illustrated in Scheme 2. Following this, the target sulfonyl hydrazones 2–7 were synthesized through a condensation reaction between p-toluene sulfonylhydrazide and various heterocyclic compounds, including 3-acetylcoumarin (9), 4-hydroxy-3-acetylcoumarin (11), N-(4-acetylphenyl)-2-oxo-2H-chromene-6-sulfonamide (14), 1-(4-((7-chloroquinolin-4-yl)amino)phenyl)ethan-1-one (15), 3-acetylindole (17), and 2-acetylpyridine (18). This reaction was conducted in hot methanol with a small amount of hydrochloric acid, providing an efficient pathway to these sulfonyl hydrazones, which may possess significant biological activity (Scheme 3). To confirm their structures, the newly created target derivatives were analyzed using analytical and spectral techniques such as 1H NMR and 13C NMR.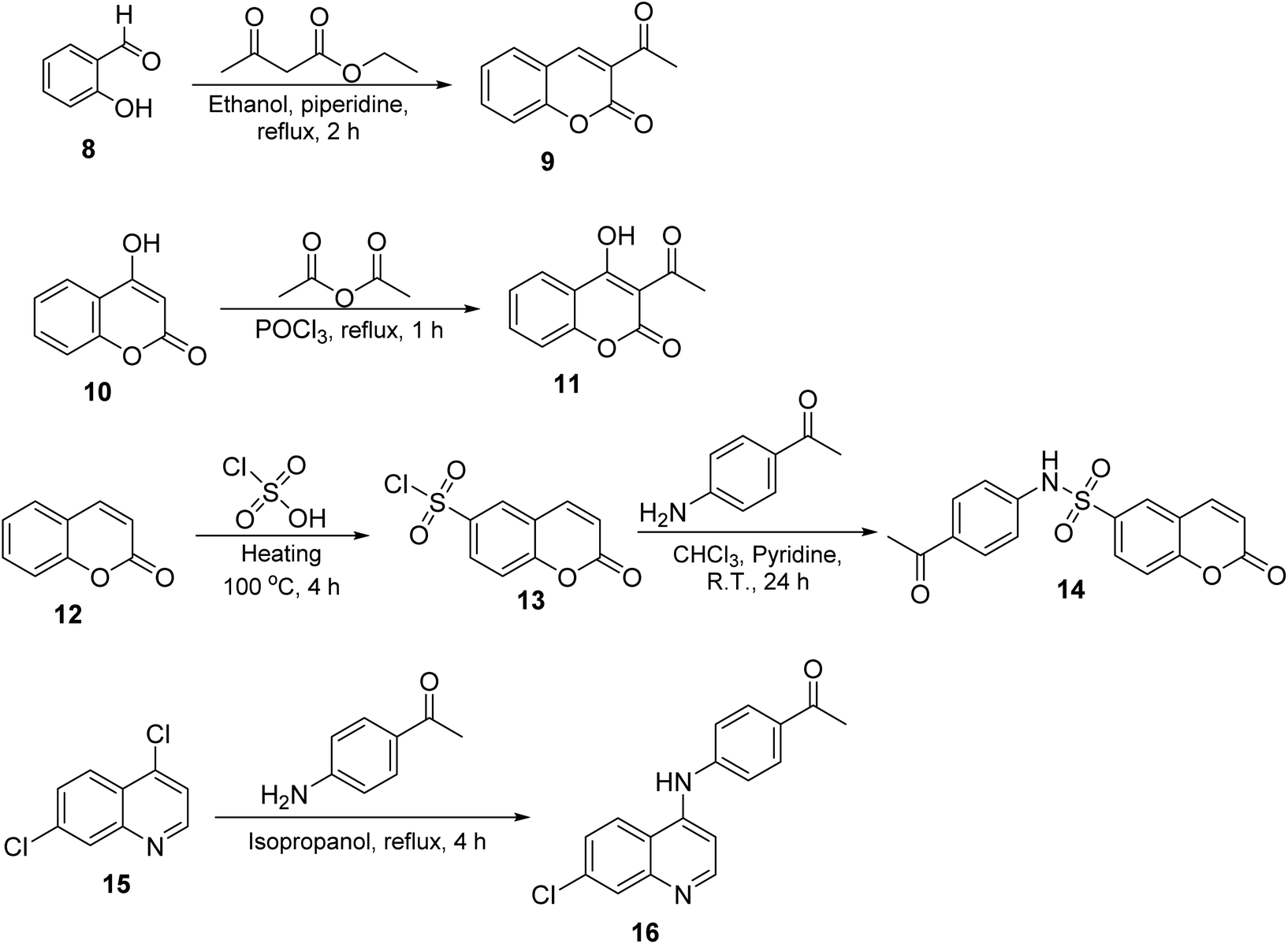 | ||
| Scheme 2 Synthesis of the target compounds 2–7. | ||
 | ||
| Scheme 3 Synthesis of the target compounds 2–7. | ||
2.2. Biological activity
| Compound | MIC [μg mL−1] | Bactericidal concentration | |
|---|---|---|---|
| M. tuberculosis | M. abscessus | ||
| a ND: not determined. | |||
| 2 | >125 | >125 | ND |
| 3 | 30 | >125 | 100 |
| 4 | >125 | >125 | ND |
| 5 | >125 | >125 | ND |
| 6 | 125 | >125 | ND |
| 7 | 8 | >125 | 60 |
| Compound | IC50 (L929 cells) | IC50/MIC (MTB) |
|---|---|---|
| 3 | 625 | 21 |
| 7 | 78 | 10 |
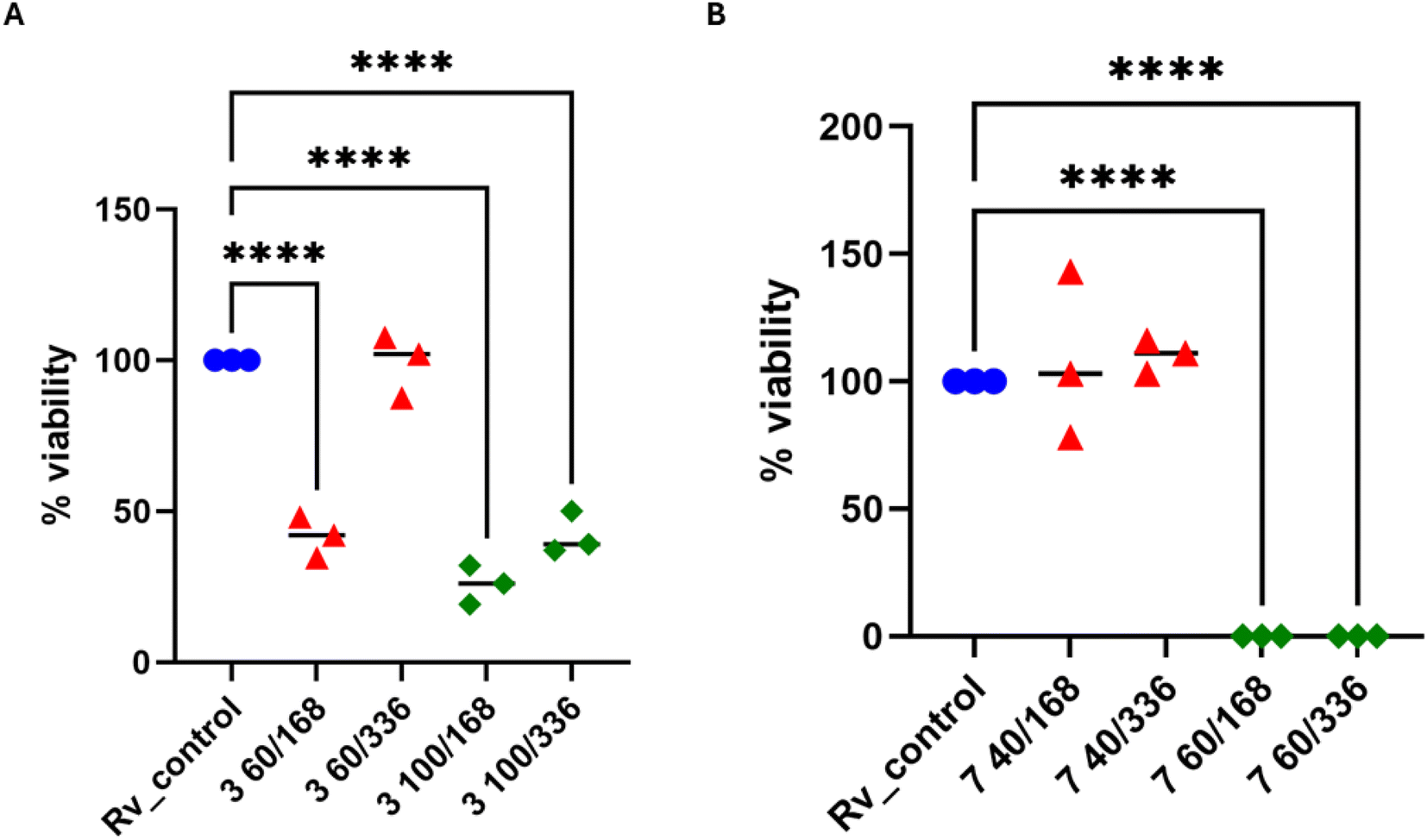 | ||
| Fig. 3 Bactericidal effect of sulfonyl hydrazone derivatives. (A) Compound 3 was used at concentrations of 60 and 100 μg mL−1 for 7 and 14 days. (B) Compound 7 was used at concentrations of 40 and 60 μg mL−1 for 7 and 14 days. The quantity of viable bacteria was evaluated through colony-forming unit (CFU) analysis. A standard one-way analysis of variance (ANOVA) test was utilized to compare the untreated M. tuberculosis control group (Rv_control) with bacilli treated with 3 and 7 compounds at specified time intervals. The corrected p-value was less than 0.0001 for paired comparisons denoted by asterisks. The statistical evaluation and visualization were conducted utilizing GraphPad Prism 9 version 9.3.1. | ||
In 2× MIC concentration, both compounds presented low cytotoxicity against MDMs. However, 4× MIC concentration of compound 3, but not 7, appeared to be toxic to the host cells. Afterward, both substances were evaluated for their effectiveness against tuberculosis bacteria present in human macrophages. To evaluate the effectiveness of the studied compound on M. tuberculosis inside human macrophages, tubercle bacilli were used to infect the MDMs at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. Following 2 hours of phagocytosis, the bacteria outside the cells were removed, and the remaining bacilli attached to the cell membrane were eliminated using gentamicin. The macrophages containing M. tuberculosis were then treated with the compounds at a concentration of twice the minimum inhibitory concentration (MIC) and left to incubate for 48 hours. The quantity of viable bacilli located inside cells was assessed by counting colony-forming units (CFU). Both compounds caused a decrease in the number of live bacteria in MDMs in comparison to the control group that did not receive any treatment. The statistical significance of the effect was observed solely in the instance of compound 7, with a p-value of 0.0358, suggesting its ability to affect the intracellular viability of tubercle bacilli (Fig. 4).
:10. Following 2 hours of phagocytosis, the bacteria outside the cells were removed, and the remaining bacilli attached to the cell membrane were eliminated using gentamicin. The macrophages containing M. tuberculosis were then treated with the compounds at a concentration of twice the minimum inhibitory concentration (MIC) and left to incubate for 48 hours. The quantity of viable bacilli located inside cells was assessed by counting colony-forming units (CFU). Both compounds caused a decrease in the number of live bacteria in MDMs in comparison to the control group that did not receive any treatment. The statistical significance of the effect was observed solely in the instance of compound 7, with a p-value of 0.0358, suggesting its ability to affect the intracellular viability of tubercle bacilli (Fig. 4).
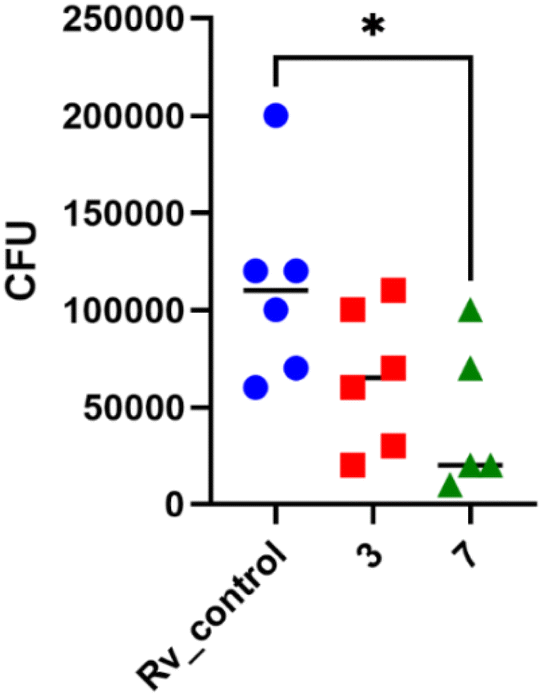 | ||
| Fig. 4 Human monocyte-derived macrophages (MDMs) were infected with M. tuberculosis and exposed to compounds 3 and 7 at a concentration of 2 times the minimum inhibitory concentration (MIC). A standard one-way analysis of variance (ANOVA) was conducted to compare the quantity of intracellular M. tuberculosis in the control group with that in the bacilli treated with compounds 3 and 7. The adjusted p-value indicated statistical significance (p = 0.0358) only for compound 7. The statistical analysis and graphical representation were performed using GraphPad Prism 9 version 9.3.1. | ||
 | ||
| Fig. 5 The impact of sulfonyl hydrazon derivatives on established biofilms consisting of M. tuberculosis was assessed. Statistical comparisons were conducted through standard one-way ANOVA analysis. Blue dots in the figures represent the absence of compound control. Detailed p values are provided in the accompanying text. The statistical analysis and graphical representation were performed using GraphPad Prism 9 version 9.3.1. | ||
| Compound | IC50 (μM) |
|---|---|
| 3 | 1.62 ± 0.69 |
| 7 | 0.91 ± 0.03 |
| Isoniazid | 0.24 ± 0.01 |
According to Table 3, compound 7 effectively inhibited the InhA enzyme at micromolar concentration, with an IC50 value of 0.91 ± 0.03 μM. Compound 3 had reduced activity against InhA, with an IC50 value of 1.62 ± 0.69 μM.
The differential antimicrobial activity of derivative 7 towards M. tuberculosis and M. abscessus could be attributed to certain factors. While both species belong to the Mycobacterium genus, their InhA enzymes share only 75–80% sequence identity, which should lead to structural differences affecting the binding of the potential inhibitors. These variations could alter the binding site's hydrophobicity, electrostatic properties, or shape, potentially resulting in different binding affinities for compound 7. Furthermore, M. abscessus is known to have a more complex and less permeable cell wall than M. tuberculosis, which may restrict the molecule's ability to reach its intracellular target. Additional factors, such as specific efflux pump systems and metabolic differences, could also potentially be responsible for the observed variance in antimicrobial activities.
2.3. Molecular docking
Molecular docking analysis was performed to enhance comprehension of the potential binding mechanisms of the potent compounds 3, 6, and 7 with InhA. The docking outcomes of the studied compounds were evaluated against the co-crystallized ligand found in the PDB ID: 4TZK,48 the reference drug utilized in this experiment has been previously demonstrated to exhibit strong inhibitory activity against InhA. During the docking procedure, the NAD cofactor was maintained in proximity to the active site. The initial stage of validating the docking process involved re-docking the co-crystallized ligand. The redocked ligand exhibits an RMSD value of 0.5699 Å, indicating a precise prediction of its correct positioning, as illustrated in Fig. 6. Analysis of the ligand interaction within the co-crystallized structure revealed the presence of multiple hydrophobic contacts and two hydrogen bonds, specifically at residues Tyr158 and Met161.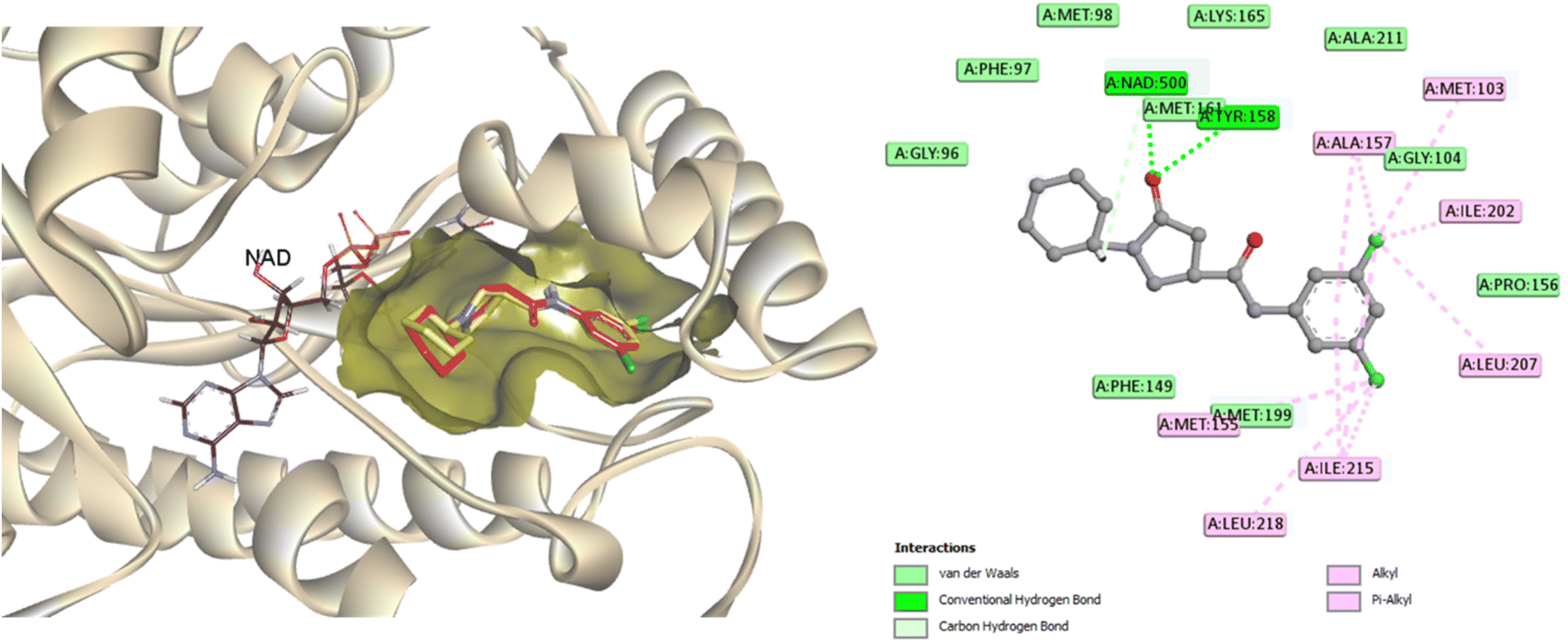 | ||
| Fig. 6 A two-dimensional interaction map and three-dimensional overlay plot were generated for the InhA protein active site (PDB ID: 4TZK)47 comparing the co-crystallized ligand (in red) with the re-docked ligand (in yellow), revealing a root mean square deviation (RMSD) value of 0.5699 Å. | ||
As seen in Fig. 7, during the tests, the active site of the NAD molecule was occupied by the compounds tested. It was observed that Tyr158 and the sulfonyl hydrazine linker of these compounds form hydrogen bond through the oxygen atom of the sulfonyl group. The phenyl ring of compounds 3 and 6 stacked in π interactions with Tyr158 and Met199. It was also discovered that there is a hydrophobic interaction between the aromatic moiety of 3 (4-hydroxycoumarin) and 6 (indole) and the active site of InhA via a hydrophobic pocket (Met103, Ala198, and Leu207). Moreover, an H-bond was formed between Met 98 and the hydroxy on coumarin for compound 3 and between Gly96 and the indole NH for compound 6.
 | ||
| Fig. 7 Molecular interactions between InhA residues and ligands 3 and 6 on a 2D and 3D scale. | ||
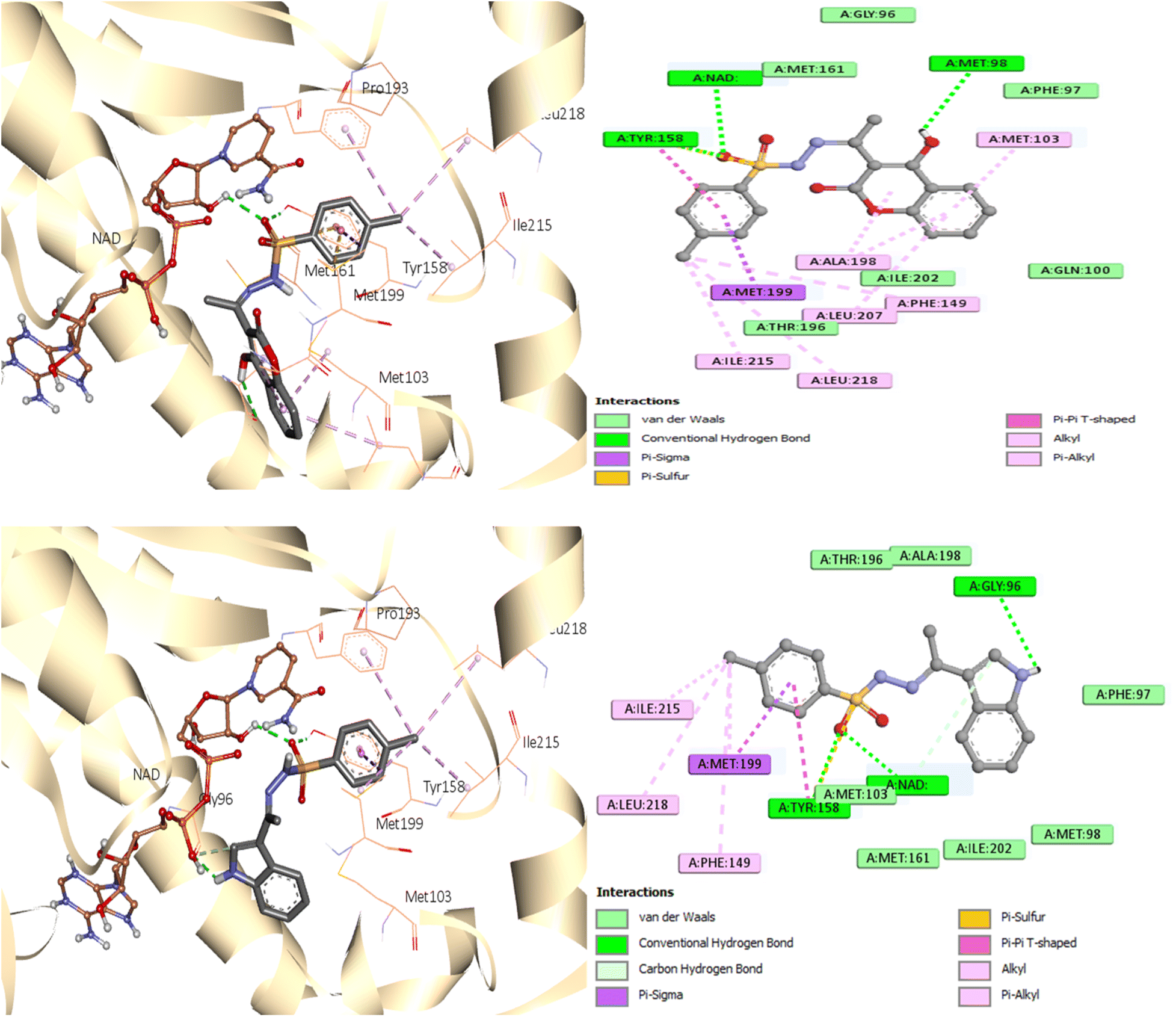
A similar reaction took place with compound 7, which occupied the NAD molecule in contact with the active site. The sulfonyl hydrazine linker forms a hydrogen bond through its oxygen with Tyr158, alongside sulfur–π interaction with its sulfur atom (Fig. 8).
 | ||
| Fig. 8 Molecular interactions between InhA residues and ligand 7 on a 2D and 3D scale. | ||
The phenyl ring showed π interactions with Met103, Met161, and Met199. It was also discovered that there is a hydrophobic interaction between the second aromatic moiety and the InhA's active site (Phe149, Pro193, Met199, and Ile215) of the hydrophobic pocket.
The most firmly bound derivatives among the tested compounds showed binding affinity of −8.3, −8.7, and −9.2 kcal mol−1, which is approximately similar to the binding affinity of the co-crystallized ligand (−9.5 kcal mol−1). By illuminating the patterns of interaction of newly synthesized compounds, these findings may provide a strong basis for the development of antituberculosis drugs.
2.4. Molecular dynamic simulations
MD study on the ideal pose for molecule 7 inside the active region of the InhA protein was carried out to examine the stability of the complex structure. Consequently, an assessment and comparison were conducted between the 7 complex system and the apoprotein using a 100 ns MD simulation conducted at ambient temperature. First, consistent temperatures, constant potential energy, and constant pressure were observed throughout the simulation, confirming the convergence of the complex system (Fig. S1†).Observing the drift of RMSD along the trajectories, the difference between the InhA protein in the presence and absence of compound 7 was examined. Because of the InhA protein's stabilization, Fig. 9 demonstrated a similar pattern between the complex RMSD values and the apoprotein one. Additionally, it was clear that the complex's SASA and radius of gyration were similar to those of the apoprotein, indicating that there was no induction of conformational changes and formation of a stable complex with 7. Furthermore, during the MD, the average distance of 7/InhA protein did not change. Additionally, the reduced draft found in the complex's RMSF relative to the free InhA protein further confirmed the stability of the 7-InhA protein complex (Fig. 9).
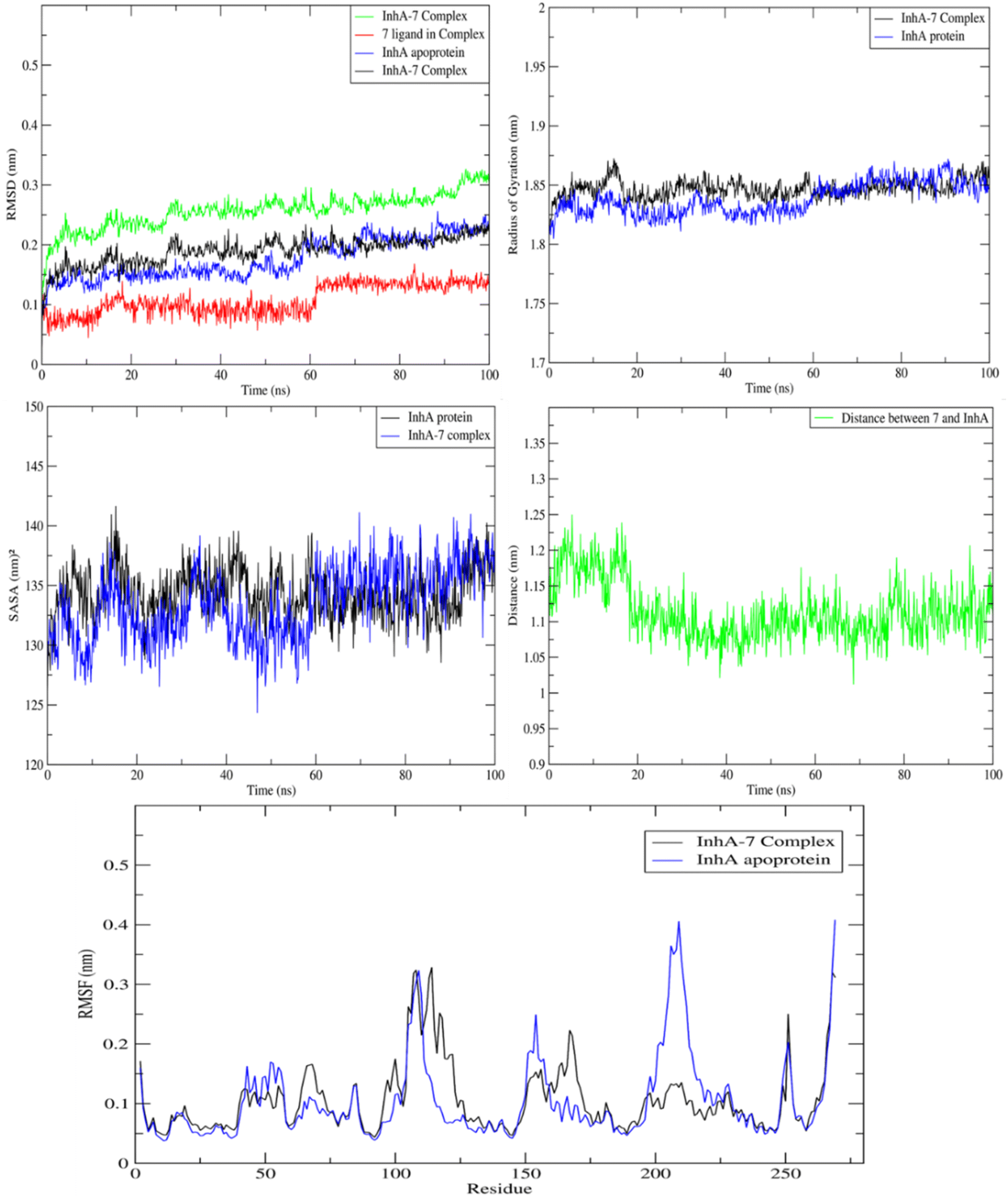 | ||
| Fig. 9 Plots of ligand-receptor distance, SASA, radius of gyration RMSD and RMSF for InhA protein with and without 7. | ||
Analysis of the MD trajectories for compound 7 simulations with the InhA receptor revealed stable binding interactions, including two robust hydrogen bonds and consistent hydrophobic interactions (Fig. 10). Specifically, two distinct hydrogen bonds were observed: one with Tyr158 and another with NAD. Additionally, intermittent π-stacking interactions were noted with Phe149 and Tyr158. Table 4 summarizes the various binding energies of the complex, calculated using the MMPBSA method. These results demonstrate how the observed interactions contribute to the overall stability of the compound–receptor complex.
 | ||
| Fig. 10 Various interacting amino acids with the sorts of interaction with 7 into InhA protein active cavity. | ||
| Binding energies | kJ mol−1 |
|---|---|
| Electrostatic energy | −23.935 ± 2.415 |
| Polar solvation energy | 54.240 ± 15.552 |
| SASA energy | −15.247 ± 0.722 |
| van der Waal energy | −121.296 ± 10.761 |
| ΔG | −106.239 ± 15.899 |
3. Conclusions
In summary, this study prepared a series of small molecules based on non-fused and fused heterocycles (pyridine, coumarin, quinoline, and indole) tethered with benzenesulfonohydrazide as potential antitubercular candidates. The developed molecules were assessed for their potential bactericidal effects against M. tuberculosis and M. abscessus. Coumarin-based derivative (3) and pyridine-based derivative (7) showed MIC values of 30 and 8 μM, respectively, against M. tuberculosis. Further tests on macrophage and antibiofilm activity were performed for the two most active compounds. Mycobacteria could not grow inside human macrophages, but there was a limited reduction in the viability of biofilm-forming mycobacteria with both compounds. A strain overexpressing InhA was also shown to be sensitive to these two compounds with IC50 values of 1.62 ± 0.69 and 0.91 ± 0.03 respectively. To rationally understand how the compounds interact with the enzyme, docking studies revealed hydrogen bond interactions with Tyr 158 to be critical for binding InhA, as well as hydrogen bonds with Met161. Based on molecular docking simulations, this compound was found to have similar binding affinities and interactions to previously reported InhA inhibitors, emphasizing the importance of hydrogen and halogen bonds, as well as other interactions that contribute to stability within the InhA active site. The MD analysis also confirmed the binding mode observed in docking studies, which supports the stability of InhA-7. The results of this study pave the way for several promising research directions and optimization opportunities. Future investigations could focus on further modifying the most potent molecules (3 and 7) to enhance their efficacy and selectivity against mycobacterial InhA. Additionally, exploring combination therapy approaches could lead to the development of more effective treatment regimens by evaluating potential synergistic effects between these compounds and existing anti-tuberculosis drugs. Moreover, conducting in vivo animal studies is a crucial next step to assess the safety profiles, pharmacokinetic properties, and therapeutic efficacy of the most promising molecules in relevant tuberculosis models. These avenues of research have the potential to advance significantly the treatment of mycobacterial infections.4. Experimental
4.1. Chemistry
The melting points of all samples were determined using the Electrothermal IA 9000 instrument without any corrections. The nuclear magnetic resonance spectra, specifically the 1HNMR (400 MHz) and 13CNMR spectra (100 MHz), were obtained using a Bruker spectrometer. TMS was used as the internal standard. The reactions were monitored using thin-layer chromatography (TLC) with silica gel and aluminum sheets 60 F254 from Merck. The eluent used was a mixture of chloroform and methanol (9.5:0.5 v/v), and the TLC plates were sprayed with an iodine-potassium iodide reagent. Compounds 3, 4, 6, and 7 were previously synthesized.49–53
N), 159.00 (CO); analysis for C18H16N2O4S, M. wt. (356.40), calcd: % C, 60.66; H, 4.53; N, 7.86; found: % C, 60.81; H, 4.49; N, 7.98.N), 154.48 (CH3–CN); analysis for C24H21ClN4O2S, M. wt. (464.97), calcd: % C, 62.00; H, 4.55; N, 12.05; found: % C, 62.19; H, 4.51; N, 12.13.4.2. Biology
4.3. Molecular modeling and molecular dynamic simulation
The M. tuberculosis InhA protein's structural coordinates were obtained in pdb format from the RCSB PDB.47 Using AutoDock Tools,61 Marvin Sketch, AutoDock Vina,62 and Discovery Studio Visualizer,63 the docking study was carried out. In addition, CHARMM-GUI solution builder,64–66 GROMACS 2020.2 software,67 VMD molecular graphics program,68 and GROMACS g_mmpbsa tool69 were exploited in molecular dynamic simulation studies. The ESI materials† provide details on all the experimental steps and protocols.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
T. A.-W.: investigation, data curation, formal analysis, funding acquisition; A. S.: conceptualization, formal analysis, methodology, resources, writing—original draft preparation; M. K.-M.: methodology, data curation; A. F. K.: methodology, investigation, formal analysis; M. A. S.: resources, formal analysis, writing—original draft preparation; H. A. A. I.: methodology, investigation, formal analysis; M. Ka.: methodology, data curation; B. D.: methodology, data curation; M. Ku.: methodology, data curation; W. M. E.: conceptualization, supervision, project administration, writing—review and editing; J. D.: conceptualization, formal analysis, supervision, writing—review and editing. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
No potential conflicts of interest was reported by the author(s).Acknowledgements
The authors extend their appreciation to the Princess Nourah bint Abdulrahman University Researchers Supporting Project number [PNURSP2024R25], Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. JD, MKu & MK-M were supported by the Ministry of Science and Higher Education, POL-OPENSCREEN, DIR/WK/2018/06 and National Science Centre, Poland UMO-2023/49/B/NZ7/01421.References
- R. V. Shingalapur, K. M. Hosamani and R. S. Keri, Synthesis and evaluation of in vitro antimicrobial and anti-tubercular activity of 2-styryl benzimidazoles, Eur. J. Med. Chem., 2009, 44, 4244–4248 CrossRef CAS PubMed.
- H. Zhang, M. Liu, W. Fan, S. Sun and X. Fan, The impact of Mycobacterium tuberculosis complex in the environment on one health approach, Front. Public Health, 2022, 10, 994745 CrossRef.
- S. S. R. Alsayed and H. Gunosewoyo, Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets, Int. J. Mol. Sci., 2023, 24(6), 5202 CrossRef CAS.
- B. W. Jhun and W. J. Koh, Treatment of Isoniazid-Resistant Pulmonary Tuberculosis, Tuberc. Respir. Dis., 2020, 83(1), 20–30 CrossRef.
- A. T. e Castro, M. Mendes, S. Freitas and P. C. Roxo, Incidence and risk factors of major toxicity associated to first-line antituberculosis drugs for latent and active tuberculosis during a period of 10 years, Rev. Port. Pneumol., 2015, 21(3), 144–150 Search PubMed.
- Y. Q. Hu, Z. Xu, S. Zhang, X. Wu, J. W. Ding, Z. S. Lv and L. S. Feng, Recent developments of coumarin-containing derivatives and their anti-tubercular activity, Eur. J. Med. Chem., 2017, 136, 122–130 CrossRef CAS PubMed.
- M. Lee, J. Lee, M. W. Carroll, H. Choi, S. Min, T. Song, L. E. Via, L. C. Goldfeder, E. Kang and B. Jin, Linezolid for treatment of chronic extensively drug-resistant tuberculosis, N. Engl. J. Med., 2012, 367, 1508–1518 CrossRef CAS.
- M. S. Prasad, R. P. Bhole, P. B. Khedekar and R. V. Chikhale, Mycobacterium enoyl acyl carrier protein reductase (InhA): A key target for antitubercular drug discovery, Bioorg. Chem., 2021, 115, 105242 CrossRef CAS PubMed.
- C. Vilchèze and Jr W. R. Jacobs, The mechanism of isoniazid killing: clarity through the scope of genetics, Annu. Rev. Microbiol., 2007, 61, 35–50 CrossRef PubMed.
- S. K. Wahan, G. Bhargava, V. Chawla and P. A. Chawla, Unlocking InhA: Novel approaches to inhibit Mycobacterium tuberculosis, Bioorg. Chem., 2024, 107250 CrossRef CAS.
- A. Khalifa, A. Khalil, M. M. Abdel-Aziz, A. Albohy and S. Mohamady, Isatin-pyrimidine hybrid derivatives as enoyl acyl carrier protein reductase (InhA) inhibitors against Mycobacterium tuberculosis, Bioorg. Chem., 2023, 138, 106591 CrossRef CAS PubMed.
- Y. Chen, Y. Ying, J. Lalsiamthara, Y. Zhao, S. Imani, X. Li, S. Liu and Q. Wang, From bacteria to biomedicine: Developing therapies exploiting NAD+ metabolism, Bioorg. Chem., 2023, 106974 Search PubMed.
- L. Liu, F. Jiang, L. Chen, B. Zhao, J. Dong, L. Sun, Y. Zhu, B. Liu, Y. Zhou, J. Yang, Y. Zhao, J. Qi and X. Zhang, The impact of combined gene mutations in inhA and ahpC genes on high levels of isoniazid resistance amongst katG non-315 in multidrug-resistant tuberculosis isolates from China, Emerging Microbes Infect., 2018, 7(1), 1–10 CrossRef CAS.
- (a) U. Theuretzbacher, S. Gottwalt, P. Beyer, M. Butler, L. Czaplewski, C. Lienhardt, L. Moja, M. Paul, S. Paulin, J. H. Rex, L. L. Silver, M. Spigelman, G. E. Thwaites and J.-D. G. Ghiano, et al., Eur. J. Med. Chem., 2020, 208, 112699 CrossRef; (b) P. Paccaud and S. Harbarth, Analysis of the clinical antibacterial and antituberculosis pipeline, Lancet Infect. Dis., 2019, 19, e40ee50 Search PubMed.
- Y. Teneva, R. Simeonova, V. Valcheva and V. T. Angelova, Recent Advances in Anti-Tuberculosis Drug Discovery Based on Hydrazide–Hydrazone and Thiadiazole Derivatives Targeting InhA, Pharmaceuticals, 2023, 16(4), 484 CrossRef CAS PubMed.
- E. F. Khaleel, A. Sabt, M. Korycka-Machala, R. M. Badi, N. T. Son, N. X. Ha, M. F. Hamissa, A. E. Elsawi, E. B. Elkaeed, B. Dziadek and W. M. Eldehna, Identification of new anti-mycobacterial agents based on quinoline-isatin hybrids targeting enoyl acyl carrier protein reductase (InhA), Bioorg. Chem., 2024, 144, 107138 CrossRef CAS PubMed.
- P. C. Sharma, D. Sharma, A. Sharma, N. Saini, R. Goyal, M. Ola, R. Chawla and V. K. Thakur, Hydrazone comprising compounds as promising anti-infective agents: Chemistry and structure-property relationship, Mater. Today Chem., 2020, 18, 100349 CrossRef CAS.
- H. Heimpel and A. Raghavachar, Hematological side effects of co-trimoxazole, Infection, 1987, 15, S248–S253 CrossRef.
- M. M. Shaaban, H. M. Ragab, K. Akaji, R. P. McGeary, A.-E. A. Bekhit, W. M. Hussein, J. L. Kurz, B. H. Elwakil, S. A. Bekhit and T. M. Ibrahim, et al., Design, synthesis, biological evaluation and in silico studies of certain aryl sulfonyl hydrazones conjugated with 1,3-diaryl pyrazoles as potent metallo-β-lactamase inhibitors, Bioorg. Chem., 2020, 105, 104386 CrossRef CAS.
- S. Murtaza, S. Shamim, N. Kousar, M. N. Tahir, M. Sirajuddin and U. A. Rana, Synthesis, biological investigation, calf thymus DNA binding and docking studies of the sulfonyl hydrazides and their derivatives, J. Mol. Struct., 2016, 1107, 99–108 CrossRef CAS.
- T. B. Fernandes, M. R. Cunha, R. P. Sakata, T. M. Candido, A. R. Baby, M. T. Tavares, E. G. Barbosa, W. P. Almeida and R. Parise-Filho, Synthesis, Molecular Modeling, and Evaluation of Novel Sulfonylhydrazones as Acetylcholinesterase Inhibitors for Alzheimer's Disease, Arch. Pharm., 2017, 350, 1700163 CrossRef.
- K. Yang, J.-Q. Yang, S.-H. Luo, W.-J. Mei, J.-Y. Lin, J.-Q. Zhan and Z.-Y. Wang, Synthesis of N-2(5H)-furanonyl sulfonyl hydrazone derivatives and their biological evaluation in vitro and in vivo activity against MCF-7 breast cancer cells, Bioorg. Chem., 2021, 107, 104518 CrossRef CAS.
- A. Queen, P. Khan, D. Idrees, A. Azam and I. Hassan, Biological evaluation of p-toluene sulphonylhydrazone as carbonic anhydrase IX inhibitors: An approach to fight hypoxia-induced tumors, Int. J. Biol. Macromol., 2018, 106, 840–850 CrossRef CAS PubMed.
- I. K. da Costa Nunes, E. T. de Souza, I. R. R. Martins, G. Barbosa, M. O. D. M. Junior, M. D. M. Medeiros, S. W. D. Silva, T. L. Balliano, B. A. da Silva and P. M. R. Silva, et al., Discovery of sulfonyl hydrazone derivative as a new selective PDE4A and PDE4D inhibitor by lead-optimization approach on the prototype LASSBio-448: In vitro and in vivo preclinical studies, Eur. J. Med. Chem., 2020, 204, 112492 CrossRef.
- V. T. Angelova, T. Pencheva, N. Vassilev, E. K-Yovkova, R. Mihaylova, B. Petrov and V. Valcheva, Development of New Antimycobacterial Sulfonyl Hydrazones and 4-Methyl-1,2,3-thiadiazole-Based Hydrazone Derivatives, Antibiotics, 2022, 11(5), 562 CrossRef CAS PubMed.
- K. N. De Oliveira, L. D. Chiaradia, P. G. A. Martins, A. Mascarello, M. N. S. Cordeiro, R. V. C. Guido, A. D. Andricopulo, R. A. Yunes, R. J. Nunes and J. Vernal, et al., Sulfonyl-hydrazones of cyclic imides derivatives as potent inhibitors of the Mycobacterium tuberculosis protein tyrosine phosphatase B (PtpB), MedChemComm, 2011, 2, 500–504 RSC.
- A. Mascarello, M. Mori, L. D. Chiaradia-Delatorre, A. C. O. Menegatti, F. D. Monache, F. Ferrari, R. A. Yunes, R. J. Nunes, H. Terenzi and B. Botta, et al., Discovery of Mycobacterium tuberculosis protein tyrosine phosphatase B (PtpB) inhibitors from natural products, PLoS One, 2013, 8, e77081 CrossRef CAS.
- S. Ghiya and Y. C. Joshi, Synthesis and antimicrobial evaluation of hydrazones derived from 4-methylbenzenesulfonohydrazide in aqueous medium, Med. Chem. Res., 2016, 25, 970–976 CrossRef CAS.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications, Molecules, 2020, 25(8), 1909 CrossRef CAS PubMed.
- A. Sabt, M. Abdelraof, M. F. Hamissa and M. A. Noamaan, Antibacterial Activity of Quinoline-Based Derivatives against Methicillin-Resistant Staphylococcus aureus and Pseudomonas aeruginosa: Design, Synthesis, DFT and Molecular Dynamic Simulations, Chem. Biodiversity, 2023, 20(11), e202300804 CrossRef CAS.
- A. Sabt, W. M. Eldehna, T. M. Ibrahim, A. A. Bekhit and R. Z. Batran, New antileishmanial quinoline linked isatin derivatives targeting DHFR-TS and PTR1: Design, synthesis, and molecular modeling studies, Eur. J. Med. Chem., 2023, 246, 114959 CrossRef CAS PubMed.
- A. Sabt, M. H. Abdulla, M. S. Ebaid, J. Pawełczyk, H. A. Abd El Salam, N. T. Son, N. X. Ha, M. A. Vaali Mohammed, T. Traiki, A. E. Elsawi and B. Dziadek, Identification of 2-(N-aryl-1,2,3-triazol-4-yl) quinoline derivatives as antitubercular agents endowed with InhA inhibitory activity, Front. Chem., 2024, 12, 1424017 CrossRef.
- R. Z. Batran, A. Sabt, M. A. Khedr, A. K. Allayeh, C. Pannecouque and A. F. Kassem, 4-Phenylcoumarin derivatives as new HIV-1 NNRTIs: Design, synthesis, biological activities, and computational studies, Bioorg. Chem., 2023, 141, 106918 CrossRef CAS PubMed.
- W. M. Eldehna, S. T. Mahmoud, E. R. Elshnawey, Z. M. Elsayed, T. A. Majrashi, M. K. El-Ashrey, M. Rashed, L. R. Hemeda, A. A. Shoun, E. B. Elkaeed and M. A. El Hassab, Novel indolinone-tethered benzothiophenes as anti-tubercular agents against MDR/XDR M. tuberculosis: Design, synthesis, biological evaluation and in vivo pharmacokinetic study, Bioorg. Chem., 2024, 143, 107009 CrossRef CAS PubMed.
- Z. M. Elsayed, W. M. Eldehna, M. M. Abdel-Aziz, M. A. El Hassab, E. B. Elkaeed, T. Al-Warhi, H. A. Abdel-Aziz, S. M. Abou-Seri and E. R. Mohammed, Development of novel isatin–nicotinohydrazide hybrids with potent activity against susceptible/resistant Mycobacterium tuberculosis and bronchitis causing–bacteria, J. Enzyme Inhib. Med. Chem., 2021, 36(1), 384–392 CrossRef CAS PubMed.
- E. B. Elkaeed, M. S. Taghour, H. A. Mahdy, W. M. Eldehna, N. M. El-Deeb, A. M. Kenawy, B. A. Alsfouk, M. A. Dahab, A. M. Metwaly, I. H. Eissa and M. A. El-Zahabi, New quinoline and isatin derivatives as apoptotic VEGFR-2 inhibitors: design, synthesis, anti-proliferative activity, docking, ADMET, toxicity, and MD simulation studies, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 2191–2205 CrossRef CAS PubMed.
- V. T. Angelova, V. Valcheva, N. G. Vassilev, R. Buyukliev, G. Momekov, I. Dimitrov, L. Saso, M. Djukic and B. Shivachev, Antimycobacterial activity of novel hydrazide-hydrazone derivatives with 2H-chromene and coumarin scaffold, Bioorg. Med. Chem. Lett., 2017, 27, 223–227 CrossRef CAS.
- V. T. Angelova, V. Valcheva, T. Pencheva, Y. Voynikov, N. Vassilev, R. Mihaylova, G. Momekov and B. Shivachev, Synthesis, antimycobacterial activity and docking study of 2-aroyl-[1]benzopyrano[4,3-c]pyrazol-4(1H)-one derivatives and related hydrazide-hydrazones, Bioorg. Med. Chem. Lett., 2017, 27, 2996–3002 CrossRef CAS.
- V. T. Angelova, V. Valcheva, T. Pencheva, Y. Voynikov, N. Vassilev, R. Mihaylova, G. Momekov and B. Shivachev, Synthesis, antimycobacterial activity and docking study of 2-aroyl-[1]benzopyrano[4,3-c]pyrazol-4(1H)-one derivatives and related hydrazide-hydrazones, Bioorg. Med. Chem. Lett., 2017, 27, 2996–3002 CrossRef CAS PubMed.
- G. S. T., S. Subramanian and S. Eswaran, Design, Synthesis and Study of Antibacterial and Antitubercular Activity of Quinoline Hydrazone Hybrids, Heterocycl. Commun., 2020, 26, 137–147 CrossRef.
- G. Cihan-Üstündağ, D. Şatana, G. Özhan and G. Çapan, Indole-based hydrazide-hydrazones and 4-thiazolidinones: Synthesis and evaluation as antitubercular and anticancer agents, J. Enzyme Inhib. Med. Chem., 2016, 31, 369–380 Search PubMed.
- S. A. Bonnett, D. Dennison, M. Files, A. Bajpai and T. Parish, A class of hydrazones are active against non-replicating Mycobacterium tuberculosis, PLoS One, 2018, 13, e0198059 CrossRef PubMed.
- M. D. Johansen, J. L. Herrmann and L. Kremer, Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus, Nat. Rev. Microbiol., 2020, 18(7), 392–407 CrossRef CAS PubMed.
- A. Bekier, M. Kawka, J. Lach, J. Dziadek, A. Paneth, J. Gatkowska, K. Dzitko and B. Dziadek, Imidazole-Thiosemicarbazide Derivatives as Potent Anti-Mycobacterium tuberculosis Compounds with Antibiofilm Activity, Cells, 2021, 10(12), 3476 CrossRef CAS PubMed.
- J. Esteban and M. García-Coca, Mycobacterium Biofilms, Front. Microbiol., 2018, 8, 2651 CrossRef.
- A. Dokic, E. Peterson, M. L. Arrieta-Ortiz, M. Pan, A. Di Maio, N. Baliga and A. Bhatt, Mycobacterium abscessus biofilms produce an extracellular matrix and have a distinct mycolic acid profile, Cell Surf., 2021, 7, 100051 CrossRef CAS PubMed.
- A. K. Ojha, A. D. Baughn, D. Sambandan, T. Hsu, X. Trivelli, Y. Guerardel, A. Alahari, L. Kremer, Jr W. R. Jacobs and G. F. Hatfull, Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria, Mol. Microbiol., 2008, 69(1), 164–174 CrossRef CAS PubMed.
- X. He, A. Alian, R. Stroud and R. Paul, Ortiz de Montellano, Pyrrolidine Carboxamides as a Novel Class of Inhibitors of Enoyl Acyl Carrier Protein Reductase from Mycobacterium tuberculosis, J. Med. Chem., 2006, 49, 6308–6323 CrossRef CAS.
- R. Z. Batran, M. A. Khedr, N. A. Latif, A. A. Abd El Aty and A. N. Shehata, Synthesis, homology modeling, molecular docking, dynamics, and antifungal screening of new 4-hydroxycoumarin derivatives as potential chitinase inhibitors, J. Mol. Struct., 2019, 1180, 260–271 CrossRef CAS.
- P. Govindaiah, N. Dumala, I. Mattan, P. Grover and M. J. Prakash, Design, synthesis, biological and in silico evaluation of coumarin-hydrazone derivatives as tubulin targeted antiproliferative agents, Bioorg. Chem., 2019, 91, 103143 CrossRef CAS PubMed.
- C. Lei, Y. J. Yip and J. S. Zhou, Nickel-catalyzed direct synthesis of aryl olefins from ketones and organoboron reagents under neutral conditions, J. Am. Chem. Soc., 2017, 139(17), 6086–6089 CrossRef CAS.
- S. M. Sondhi, M. Dinodia and A. Kumar, Synthesis, anti-inflammatory and analgesic activity evaluation of some amidine and hydrazone derivatives, Bioorg. Med. Chem., 2006, 14(13), 4657–4663 CrossRef CAS PubMed.
- A. Sabt, O. M. Abdelhafez, R. S. El-Haggar, H. M. Madkour, W. M. Eldehna, E. E. El-Khrisy, M. A. Abdel-Rahman and L. A. Rashed, Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: synthesis, in vitro biological evaluation, and QSAR studies, J. Enzyme Inhib. Med. Chem., 2018, 33(1), 1095–1107 CrossRef CAS.
- S. G. Franzblau, R. S. Witzig, J. C. McLaughlin, P. Torres, G. Madico, A. Hernandez, M. T. Degnan, M. B. Cook, V. K. Quenzer, R. M. Ferguson and R. H. Gilman, Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay, J. Clin. Microbiol., 1998, 36(2), 362–366 CrossRef CAS PubMed.
- M. Korycka-Machała, M. Kawka, J. Lach, R. Płocińska, A. Bekier, B. Dziadek, A. Brzostek, P. Płociński, D. Strapagiel, M. Szczesio, K. Gobis and J. Dziadek, 2,4-Disubstituted pyridine derivatives are effective against intracellular and biofilm-forming tubercle bacilli, Front. Pharmacol, 2022, 13, 1004632 CrossRef.
- M. Korycka-Machała, A. Viljoen, J. Pawełczyk, P. Borówka, B. Dziadek, K. Gobis, A. Brzostek, M. Kawka, M. Blaise, D. Strapagiel, L. Kremer and J. Dziadek, 1H-Benzo[d]Imidazole Derivatives Affect MmpL3 in Mycobacterium tuberculosis, Antimicrob. Agents Chemother., 2019, 63(10), 1–13 CrossRef.
- M. Kawka, A. Brzostek, K. Dzitko, J. Kryczka, R. Bednarek, R. Płocińska, P. Płociński, D. Strapagiel, J. Gatkowska, J. Dziadek and B. Dziadek, Mycobacterium tuberculosis Binds Human Serum Amyloid A, and the Interaction Modulates the Colonization of Human Macrophages and the Transcriptional Response of the Pathogen, Cells, 2021, 10(5), 1264 CrossRef CAS PubMed.
- M. Korycka-Machała, A. Brzostek, B. Dziadek, M. Kawka, T. Popławski, Z. J. Witczak and J. Dziadek, Evaluation of the Mycobactericidal Effect of Thio-functionalized Carbohydrate Derivatives, Molecules, 2017, 22(5), 812 CrossRef.
- M. Korycka-Machała, J. Pawełczyk, P. Borówka, B. Dziadek, A. Brzostek, M. Kawka, A. Bekier, S. Rykowski, A. B. Olejniczak, D. Strapagiel, Z. Witczak and J. Dziadek, PPE51 Is Involved in the Uptake of Disaccharides by Mycobacterium tuberculosis, Cells, 2020, 9(3), 603 CrossRef.
- X. He, A. Alian, R. Stroud, R. Paul and O. de Montellano, Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis, J. Med. Chem., 2006, 49(21), 6308–6323 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- E. Dall and H. Brandstetter, Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation, Proc. Natl. Acad. Sci., 2013, 110, 10940–10945 CrossRef CAS.
- J. Lee, X. Cheng, J. M. Swails, M. Sun Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. Brooks III, A. D. MacKerell Jr. and J. B. Klauda, Wonpil Im, CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell Jr, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York, and M. Karplus, CHARMM: The biomolecular simulation program, 2009, vol. 30, pp. 1545–1614 Search PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, Jr., CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields, J. Comput. Chem., 2010, 31, 671–690 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14(33–38), 27–38 Search PubMed.
- R. Kumari, R. Kumar and A. Lynn, g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05616g |
| This journal is © The Royal Society of Chemistry 2024 |