DOI:
10.1039/D4RA05646A
(Review Article)
RSC Adv., 2024,
14, 33536-33567
Accessing the synthesis of natural products and their analogues enabled by the Barbier reaction: a review
Received
4th August 2024
, Accepted 12th October 2024
First published on 22nd October 2024
Abstract
The Barbier reaction is significantly referred to as one of the efficient carbon–carbon bond forming reactions which involves the treatment of haloalkanes and carbonyl compounds by utilizing the catalytic role of a diverse range of metals and metalloids. The Barbier reaction is tolerant to a variety of functional groups, allowing a broad substrate scope with the employment of lanthanides, transition metals, amphoteric elements or alkaline earth metals. This reaction is also water-resistant, thereby overcoming the challenges posed by moisture sensitive organometallic species involving C–C bond formation reactions. The Barbier reaction has significantly found its applicability towards the synthesis of intricate and naturally occurring organic compounds. Our review provides an outlook on the synthetic applications of the Barbier reaction and its variants to accomplish the preparation of several natural products, reported since 2020.
1 Introduction
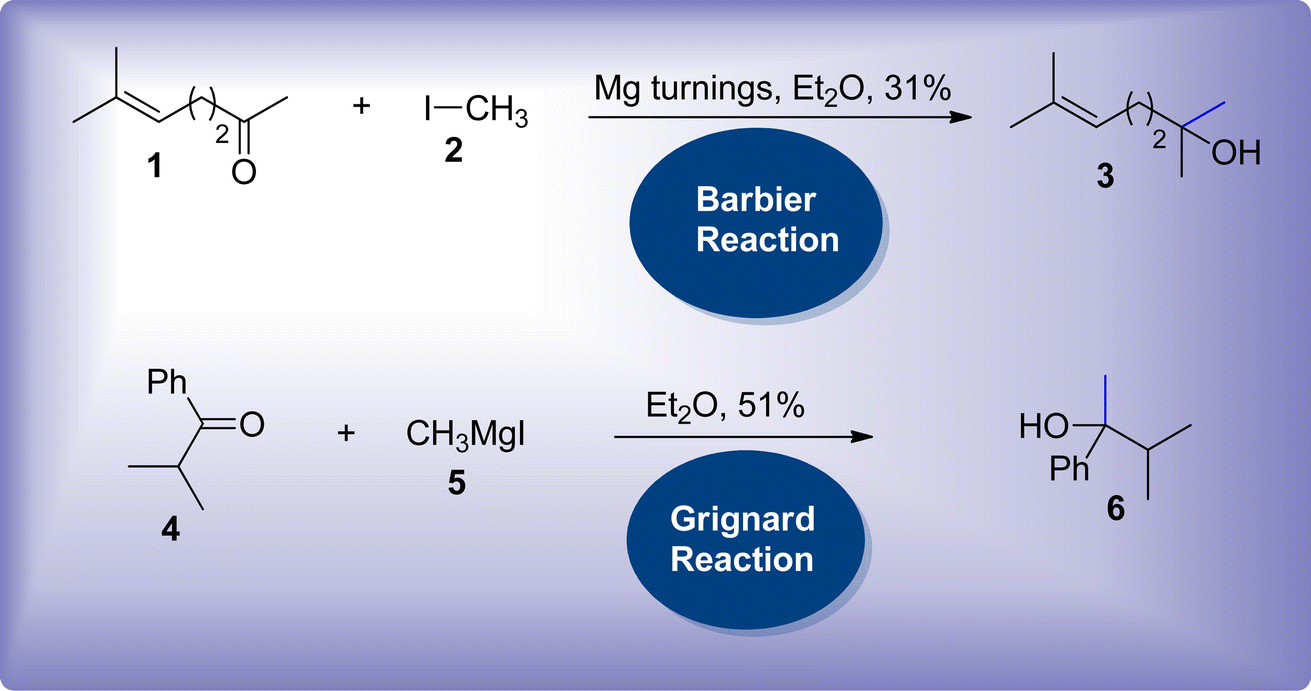
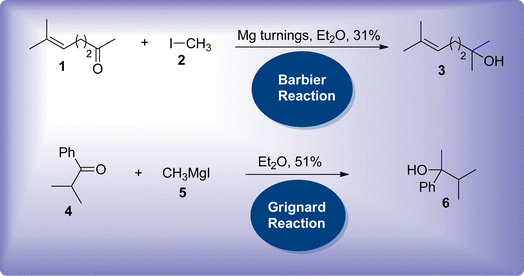
Carbon–carbon bond forming reactions are of substantial interest in organic chemistry to afford the functionalization of chains along with their branching and elongation.1,2 These reactions have garnered considerable attention in synthetic organic chemistry, thereby leading to the synthesis of intricate molecules.3,4 Until the end of the 19th century, zinc metal was predominantly employed in several organic reactions.5,6 P. Barbier and V. Grignard introduced the utilization of magnesium metal in the addition reactions of carbonyl compounds.7,8 This employment led towards the improvisation of carbon–carbon bond forming reactions, thus exploring the use of other metals in these reactions. Initially, the Barbier reaction reported the utilization of magnesium metal-based turnings in the treatment of methylheptenone 1 with methyl iodide 2 to afford the synthesis of 2,6-dimethylheptenol 3. The attainment of the target molecule in moderate yield with limited replicability hindered the exploration of the Barbier reaction (Scheme 1).7
 |
| | Scheme 1 Synthesis of tertiary alcohol from Barbier and Grignard reaction. | |
Later, Barbier's pupil Grignard performed this reaction by modifying the protocol, where he isolated the generated nucleophile i.e., organomagnesium intermediate, rather than exploiting a one-pot reaction. The synthesized nucleophile 5 was then further treated with carbonyl compound 4 resulting in relatively higher yield with consistent replicability (Scheme 1).8 Ever since 1900, the Grignard reaction has held vital importance in synthetic organic chemistry, owing to its facile pathway towards the procurement of various organic molecules.9–11 Based on his significant contribution in synthetic chemistry, Grignard was awarded with the Nobel prize in 1912.
Both Grignard and Barbier reactions are regarded as nucleophilic addition reactions of carbonyl compounds by treating them with alkyl halides in the presence of metal or its salts, thus resulting in the synthesis of higher order alcohols. However, they mainly vary on the way, the nucleophilic specie (organometallic specie) is produced followed by its attack on the electrophilic centre of carbonyl functionality.12,13 Though, Grignard reaction was more frequently employed in past years, most recent modifications that sustain the original one-pot characteristics of Barbier reaction are referred as “Barbier-type reactions”.14 They have been found to be even more favourable than Grignard reactions as they surpass the certain limitations imposed by Grignard reactions which involve the requirement of anhydrous conditions, withdrawal and handling of organometallic specie. This issue is resolved by substituting Mg metal with some other metal or amphoteric element that are resistant to moisture.15,16 Thus, there has been continuous advancement regarding the usage of variety of metals, bimetallic system, zero valent metals and benign metal slats in the Barbier reaction. Moreover, water has been employed as solvent in several methodologies, eliminating the use of toxic and expensive solvents.17
Besides the methodological development, substrate scope of Barbier reaction has also been explored and thoroughly investigated in several protocols to include the treatment of various vinyl, alkyl, propargyl and allyl halides in combination with imines, esters, ketones, nitriles, aldehydes and several azo compounds. The broad range of substrate scope has been analyzed in Barbier reaction to produce a diverse range of complicated products.18–20
Stereoselectivity and regioselectivity are some other considerable factors in Barbier reaction, which arise under certain conditions. The α or γ-adduct emerges as a result of treating γ-substituted allyl compounds. The nature of regioselective product is dependent upon the solvent, γ-substrate bulkiness, utilized additive and metal promoter species. γ-Adduct is usually obtained predominantly by employing In,21–23 Sn,24–26 Zn27,28 or Ti29,30 metals in Barbier reaction. However, α-adduct is selectively produced by the introduction of praseodymium31 and neodymium32 metals in Barbier reaction utilizing γ-substituted allyl compounds as precursors (Scheme 2).
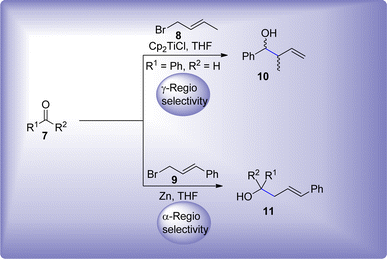 |
| | Scheme 2 Layout for γ-regioselective and α-regioselective Ti & Zn-mediated Barbier allylation reaction. | |
Furthermore, in order to achieve highly enantioselective Barbier product, chiral additives or ligands i.e., (1S,2R)-2-amino-1,2-diphenylethanol,33 R-pybim34 and β-cyclodextrin35 are significantly employed (Scheme 3). γ-Regioselectivity approach of Barbier reaction has the probability of leading towards two optically active centers in the target molecule, whose relative stereochemistry is defined by the ability of utilized metal towards the formation of cyclic transition state.
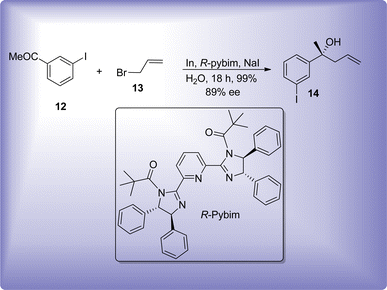 |
| | Scheme 3 Layout for an enantioselective In-mediated Barbier allylation reaction. | |
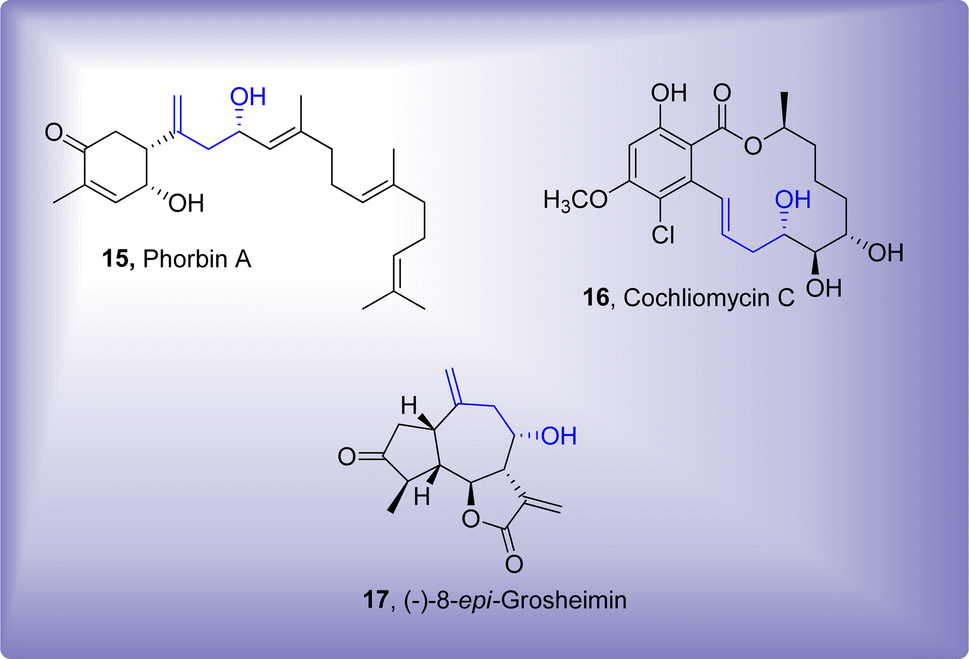
Utility of Barbier reaction has also been explored in polymer chemistry where benzoyl group and organic halide constituting monomers were added via one-pot A2, AB and B2 Barbier polyaddition to attain a catalog of polymethanol substituted polymers.36 Several one-pot reactions are utilized to accomplish the synthesis of complex organic scaffolds.37–41 There has been significant advancement towards the synthesis of several biologically active naturally occurring organic compounds.42–44 Barbier reaction has also found its significance in this specific domain, as it has been found to involve as one of the main reaction steps leading towards the procurement of natural products.45,46 Phorbin A,47 Cochliomycin C48 and (−)-8-epi-grosheimin49 are some of the naturally occurring compounds endowed with homolallylic alcohol functionality, introduced via Barbier allylation reaction (Fig. 1).
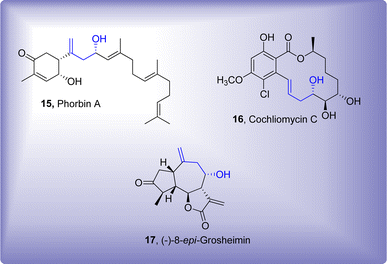 |
| | Fig. 1 Synthesis of some naturally occurring organic compounds by employing Barbier allylation reaction as a key step. | |
Considering the wide synthetic utility of Barbier reaction and its variants, significant research groups have contributed to summarize its scope, merits and remarkable applications.50–56 However, very few reports have been published (till now) to emphasize the utilization of Barbier reaction towards the procurement of natural products. Recently, in 2022, Petrides and Georgiades reported a specific overview of stereocontrolled Barbier allylation and its applications towards the synthesis of natural products.57 The objective of our review is to highlight the latest applications of Barbier and Barbier-type reactions towards the synthesis of a variety of natural products, reported within 2020–2024.
2 Literature review
2.1. Synthesis of alkaloids-based natural products
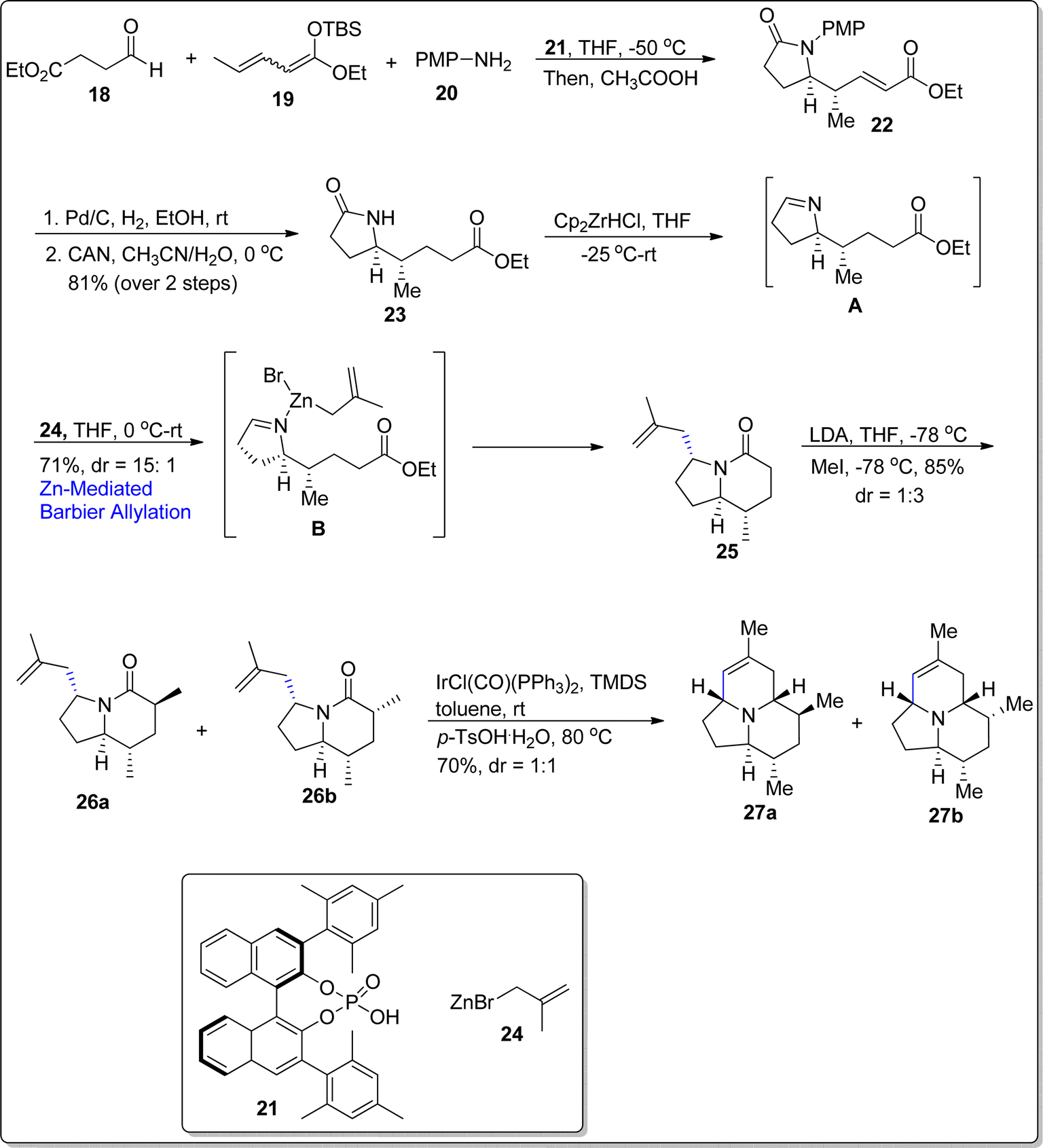
The alkaloid (–)-205B is a medicinally active and lipid-soluble natural product, which was first came across by Daly and colleagues.58 The alkaloid (–)-205B is structurally composed of azaacenonaphthylene moiety with five chiral centers.59 The total synthesis of this naturally occurring alkaloid has been performed by several research groups via diverse synthetic and organic transformations. Taking into consideration the medicinal weightage and intricate structure of this alkaloid, Tripathy and Schneider60 attempted its total synthesis by involving various name reactions i.e., Mukaiyama–Mannich reaction, aza-Prins annulation and zinc promoted Barbier allylation as key steps. Their developed briefest synthetic strategy initiated with vinylogous Mukaiyama–Mannich reaction between aldehyde moiety 18, tethered ester group 19 and amine 20 in the presence of compound 21 followed by the addition of acetic acid to generate the lactam ring 22 in 73% yield with 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 enantiomeric ratio and more than 20:1 diastereoslectivity ratio. The γ-lactam 23 was easily derived from compound 22 in 81% yield over two steps. In the next step, the lactam 23 was initially reduced to the respective imine A on treatment with Cp2ZrHCl in tetrahydrofuran at −25 °C, which was then further subjected to zinc promoted Barbier allylation using methallylbromide 24 to afford indolizidinone 25 in 71% yield with 15:1 diastereomeric ratio, thereby resulting in substrate control optically active center. It was observed that indium promoted Barbier allylation reaction resulted in decreased yield and stereoselectivity of indolizidinone. The next step involved the introduction of methyl group at 6th position of indolizidinone on reaction with lithium diisopropyl amide and tetrahydrofuran to generate the corresponding enolate which was further made to react with methyl iodide in THF at −78 °C. As a result, diastereomeric mixture of methylated compounds 26a & 26b was attained in 1:3 diastereomeric ratio with 85% yield. The diastereomeric mixture was then subjected to aza-Prins annulation reaction in the presence of Vaska's complex IrCl(CO)(PPh3)2 and TMDS followed by the treatment with p-TsOH.H2O to afford the target natural product 27a with its 6-epimer 27b (via elimination reaction) in 1:1 diastereomeric ratio. The chromatography technique was carried out to separate the resulting isomers, thereby resulting in the isolation of alkaloid (–)-205B 27a in 35% yield (overall 12% yield) (Scheme 4).
:2 enantiomeric ratio and more than 20:1 diastereoslectivity ratio. The γ-lactam 23 was easily derived from compound 22 in 81% yield over two steps. In the next step, the lactam 23 was initially reduced to the respective imine A on treatment with Cp2ZrHCl in tetrahydrofuran at −25 °C, which was then further subjected to zinc promoted Barbier allylation using methallylbromide 24 to afford indolizidinone 25 in 71% yield with 15:1 diastereomeric ratio, thereby resulting in substrate control optically active center. It was observed that indium promoted Barbier allylation reaction resulted in decreased yield and stereoselectivity of indolizidinone. The next step involved the introduction of methyl group at 6th position of indolizidinone on reaction with lithium diisopropyl amide and tetrahydrofuran to generate the corresponding enolate which was further made to react with methyl iodide in THF at −78 °C. As a result, diastereomeric mixture of methylated compounds 26a & 26b was attained in 1:3 diastereomeric ratio with 85% yield. The diastereomeric mixture was then subjected to aza-Prins annulation reaction in the presence of Vaska's complex IrCl(CO)(PPh3)2 and TMDS followed by the treatment with p-TsOH.H2O to afford the target natural product 27a with its 6-epimer 27b (via elimination reaction) in 1:1 diastereomeric ratio. The chromatography technique was carried out to separate the resulting isomers, thereby resulting in the isolation of alkaloid (–)-205B 27a in 35% yield (overall 12% yield) (Scheme 4).
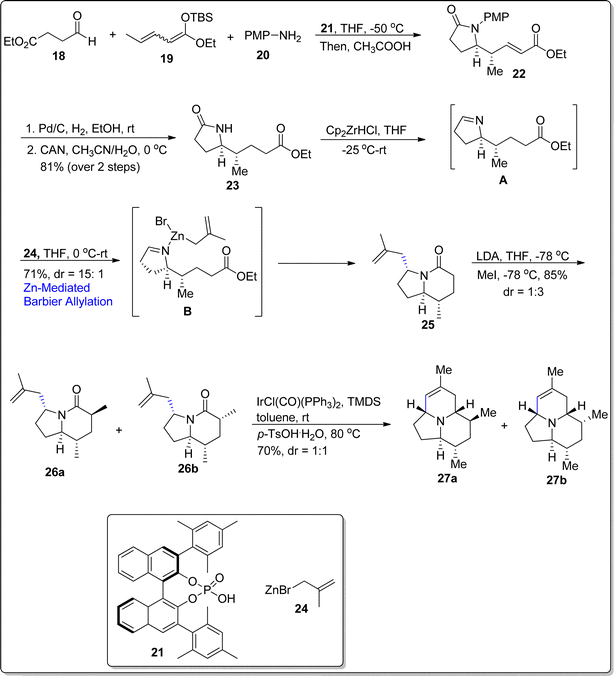 |
| | Scheme 4 Synthesis of the alkaloid (–)-205B 27a. | |
Indole alkaloids belong to the biologically active class of natural products, which are utilized in several organic transformations.61 Synthesis of indole alkaloids constituting aza-quaternary optically active center i.e., subincanadine A, arborisidine and taberdivamine B, encounter severe challenges due to their convoluted structural framework. Arborsidine is composed of 5-membered cyclic structure with two chiral centers. It has been found to be potent against gastric cancer cell lines in coordination with pimelautide. Cancer is a deadly disease and researchers are continually making efforts to develop and synthesize anti-cancer agents.62,63 Till now, two successful total synthesis of arborisidine have been reported.64,65 In 2021, Wang and Jiao66 established an efficient asymmetric total synthesis of arborisidine by employing Barbier-type addition reaction as a key step. In order to achieve the asymmetric synthesis of this natural product, Barbier type addition reaction was performed between harmalane 28 and allyl bromide 29 by involving chiral indium reagent in THF/DMF at 35 °C followed by protection of nitrogen with Boc to afford compound (±)-30 in 51% yield. In the next step, compound (±)-30 was subjected to palladium mediated intramolecular dearomative asymmetric allylic alkylation (AAA) exploiting diaminocyclohexane (DACH)-phenyl Trost ligand (R,R)-L1 (via parallel kinetic resolution (PKR) regime), which resulted in asymmetric synthesis of compound (±)-31 & (±)-32. The compound (±)-31 was further treated with benzene sulfenylchloride followed by reaction with 2,4,6-coolidine/tetrabutylammonium chloride involving elimination reaction to forge phenyl sulfide substituted compound (±)-33. The compound (±)-33 then underwent N-Boc deprotection with subsequent alkylation to afford corresponding alcohol (−)-34 in 34% overall yield (as a single isomer). The proceeding step involved the iodination of compound (−)-34 followed by 5-exo-trig radical cyclization to result in compound (−)-35 in 74% yield. Later, the compound (−)-35 was transformed over few steps to assemble the naturally occurring (−)-arborisidine (−)-36 in fine yield with efficient diastereoselectivity (Scheme 5).
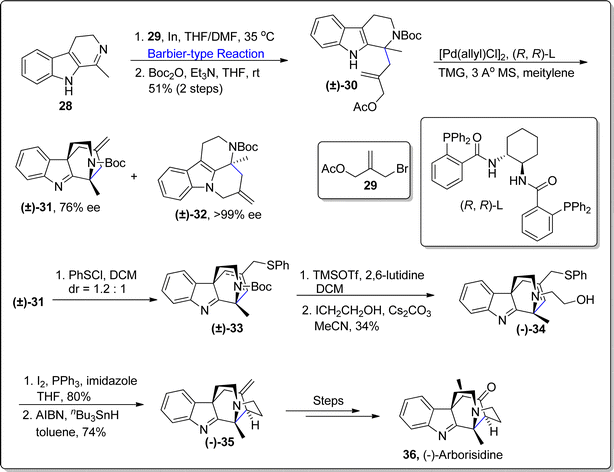 |
| | Scheme 5 Synthesis of (−)-arborsidine (−)-36. | |
Casuarine is a natural product that is extracted from Eugenia jambolana Lam and Casuarina equisetifolia L.67 Casuarine is composed of pyrrolizidine ring with 6 chiral centers, thus it exists in almost 64 stereoisomers. It also acts as an efficient isomaltase, maltase (rat instetinal) and α-glucosidase inhibitor (rice). Moreover, it has also been found to exhibit the properties of competitive inhibitor of C. riparius trehalase, NtMGAM (human N-terminal subunit of maltase-glucoamylase) and A. niger amyloglucosidase. In 2021, Li et al.68 reported the synthesis of naturally occurring casuarine 41 and its natural stereoisomer 6-epi-casuarine 42 by applying Barbier conditions in one of the key steps. Their synthetic route initiated with the reaction of nitrone 37 (obtained from D-arabinose) with allylbromide in the presence of zinc (Barbier-type reaction) in tetrahydrofuran and ammonium chloride followed by subsequent reaction with carboxybenzyl chloride (CBzCl) in the presence of sodium carbonate to afford compound 38 in 75% yield. In the following step, compound 38 was subjected to SeO2 promoted allylic oxidation by employing t-BuOOH in dichloromethane to give corresponding alcohol 39 in 53% yield (as a single isomer). Here, the Barbier-type reaction involving pathway led to the relatively higher yield of alcoholic isomer 39 in comparison to Grignard reagent involving protocol. In the next step, –OH group of compound 39 was protected with MOM ether followed by diastereoselective dihydroxylation by employing osmium tetroxide and NMO in acetone to furnish the mixture of triols 40a and 40b in 84% yield. The isomers 40a and 40b were treated individually over few steps to accomplish the synthesis of casuarine 41 and 6-epi-casuarine 42 respectively (Scheme 6).
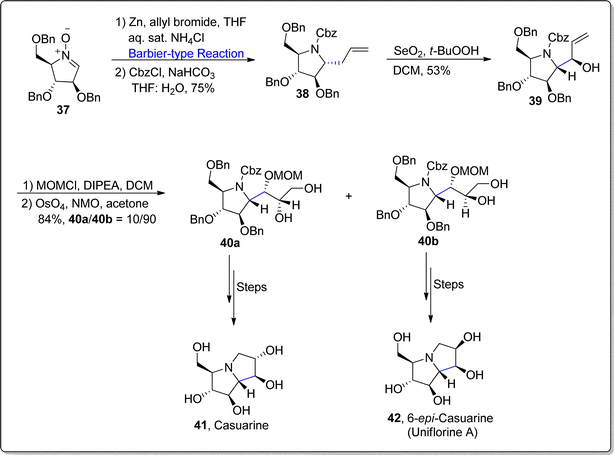 |
| | Scheme 6 Synthesis of casuarina 41 and 6-epi-casuarine 42. | |
Most of the biologically active natural products consist of spirocyclic frameworks.69 In 2021, two diastereomeric naturally occurring isoindolinone alkaloids i.e., spirocollequins A and B have been extracted from Colletotrichum boninense AM-12-2 (an endophytic fungus).70 They are composed of a furan ring alongwith isoindolinone skeleton within their spirocyclic skeleton.71 These isoindolinone alkaloids have the potency to exhibit anti-plasmodial activity. In 2022, Ichikawa and coworkers72 carried out the first total synthesis of these alkaloids involving Barbier reaction as a key step. Their synthetic endeavor initiated with the reaction of phthalic anhydride 43 with 2-aminoethanol in the presence of triethylamine, which was proceeded by the protection of hydroxyl groups with tert-butyl silyl group to furnish compound 44 in 86% yield. In the next step, the compound 44 was subjected to magnesium induced Barbier allylation reaction with allylbromide, resulting in the synthesis of isoindolinone 45 in 82% yield, as a single product. The allylic functionality in compound 45 was further subjected to cross-metathesis reaction on treatment with compound 46, utilizing Grubb's 2nd generation catalyst with copper iodide in diethyl ether. This reaction was followed by osmium tetroxide NMO induced dihydroxylation followed by treatment with PPTS (pyridinium p-toluenesulfonate) to result in diastereomeric mixture of 47a and 47b (40% isolated yield). The compound 47a (34%) was purified via selective TBS protection of its hydroxyl functionality. The reaction was proceeded by the deprotection strategy followed by subsequent prenylation reaction to afford spirocollequins A 49a in 89% yield. On the similar ground, 47b was converted to spirocollequins B 49b in 93% yield. As a result, the foremost total synthesis of (±)-spirocollequins B 49b and A 49a was achieved in 24% & 19% overall yields, correspondingly (Scheme 7).
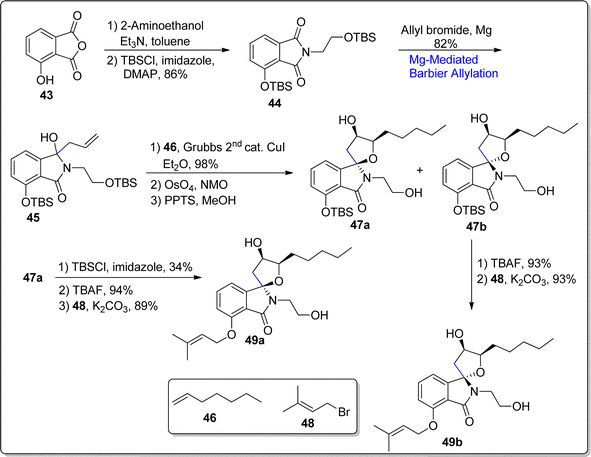 |
| | Scheme 7 Synthesis of (±)-spirocollequins A 49a & B 49b. | |
2.2. Synthesis of lactones-based natural products
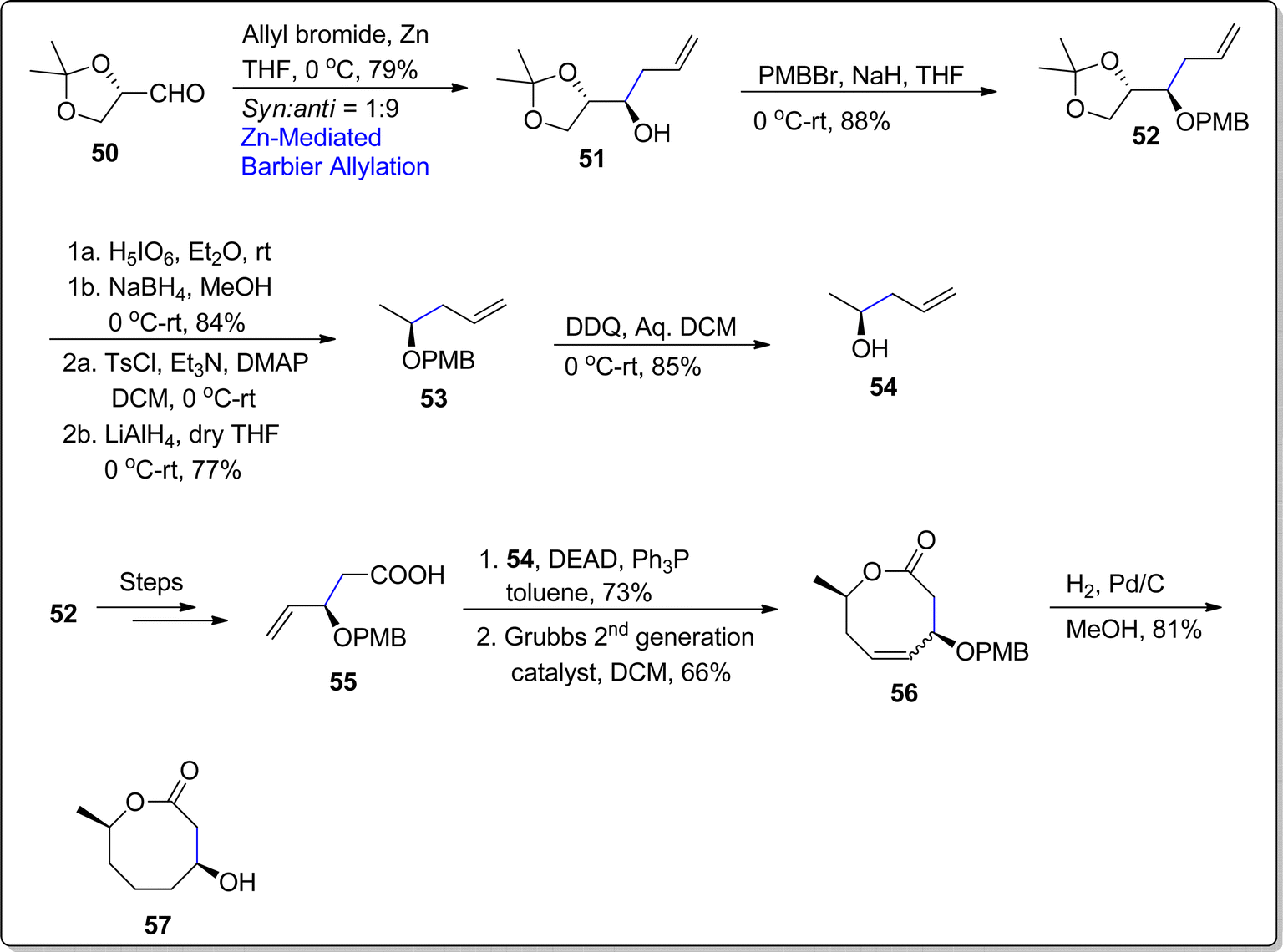
Cephalosporolides are of huge significance owing to their unique structural and medicinal attributes. (−)-Cephalosporolide D is a naturally occurring eight membered lactone which is extracted from Cephalosporium aphidicola (fermentation fungus).73 In 2000, Shiina and coworkers reported the first total synthesis of this naturally occurring lactone along with the ascertainment of its (absolute and relative) stereochemistry.74 Since then, there have been several reports concerning the synthesis of this bioactive natural product. Being persuaded by the gripping structural and pharmaceutical features of (−)-Cephalosporolide D, Kalavakuntla et al.75 in 2022, carried out an efficient and compact route towards the its total synthesis by employing Barbier allylation reaction, Mitsunobu esterification and RCM (Ring Closing Metathesis) as key steps. Their synthetic approach commenced by treating optically active aldehyde 50 with allylbromide via zinc promoted Barbier allylation in tetrahydrofuran at 0 °C to afford carbinols (51a & 51b) in a divisible mixture (syn and anti = 1:9) with 79% yield. The next step involved the protection of alcoholic functionality to ether 52 (in 88% yield) on treatment with p-methoxybenzyl bromide by utilizing sodium hydride as base in tetrahydrofuran. PMB ether 52 was then subjected to react with periodic acid followed by sodium borohydride mediated reduction to yield alcohol in 84% yield. The tosyl group was then installed on alcohol proceeded by its subsequent reaction with lithium aluminum hydride in dry tetrahydrofuran to access compound 53 in 77% yield. In the next step, alcoholic compound 54 was obtained (in 85% yield) as a result of deprotection of PMB ether by treating compound 53 with 2,3-dichloro-5,6-cyano-1,4-benzoquinone (DDQ) in aqueous DCM. PMB ether 52 was also transformed to corresponding acid 55 over few steps. The resulting acid 55 then underwent Mitsunobu esterification with already prepared alcohol 54 followed by ring closing metathesis (RCM) reaction (exploiting G-II catalyst) to forge the eight membered heterocycle 56 in 66% yield. Finally, the compound 56 was subjected to Pd/C mediated hydrogenation to accomplish the synthesis of targeted compound 57 in 81% yield (Scheme 8).
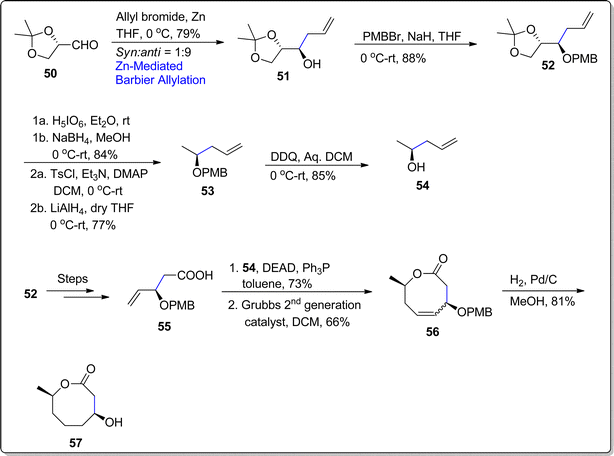 |
| | Scheme 8 Synthesis of Cephalosporolide D 57. | |
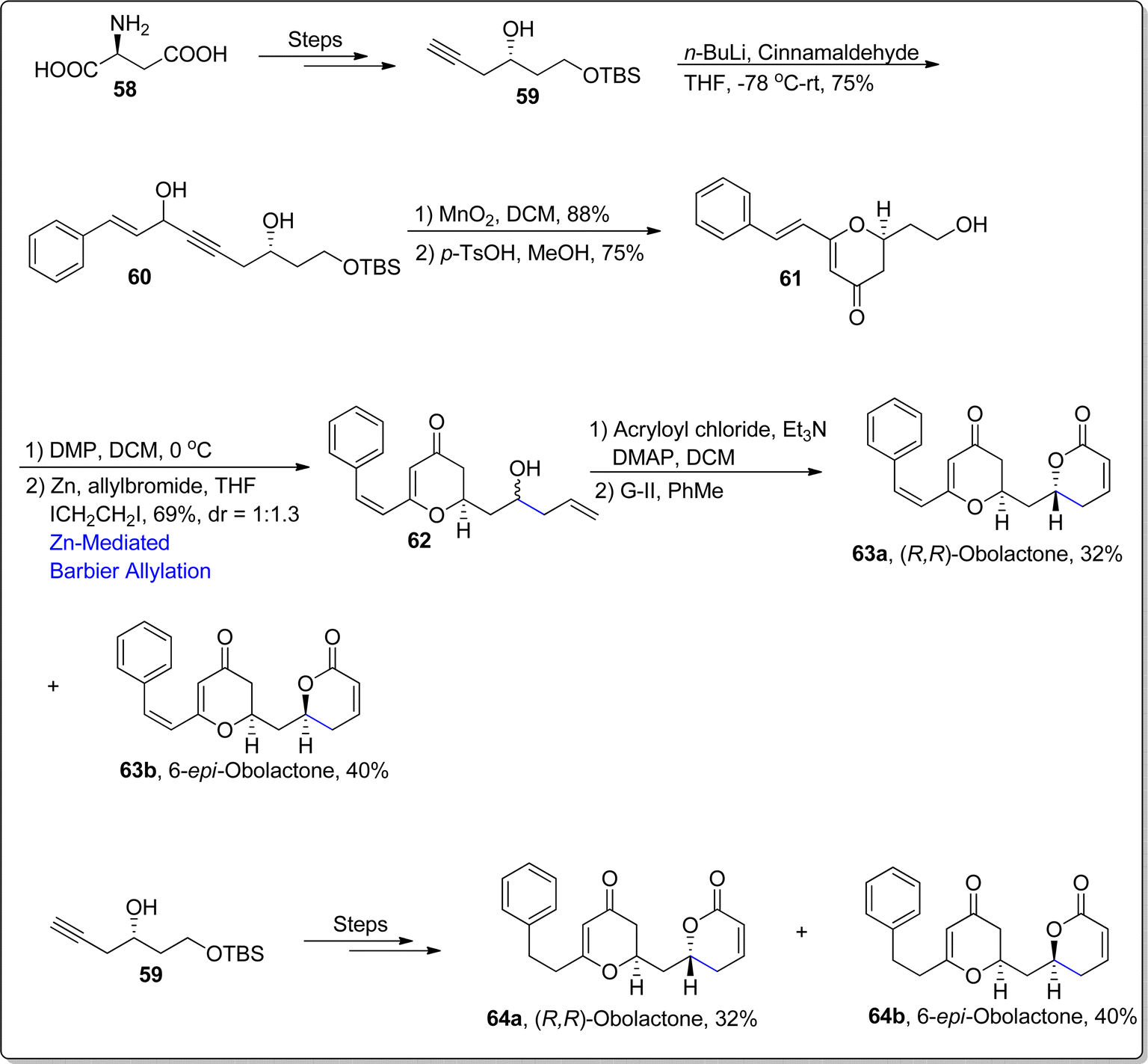
The 5,6-dihydro-pyrone functionalities are copiously found in several medicinally active natural products as they illustrate efficient potency against various deadly diseases and a wide range of cancer cells. Obolactone and dihydroobolactone are natural products, whose structural formulas are incorporated with 5,6-dihydro-pyrone moieties. Cryptocaria obovate is the main source of obolactones, which are known to exhibit potent efficacy against Trypanosoma brucei 5.3 μM nasopharyngeal cancer cells with IC50 = 3 μM.76,77 In 2021, Saini et al.78 accomplished the total synthesis of these naturally occurring and pharmaceutically significant organic compounds. Their synthetic pathway initiated with the transformation of L-aspartic acid 58 to alkyne 59 over few steps. In the next step, alkyne 59 was subjected to nucleophilic addition reaction to install the alkynol functionality 60 in 75% yield. Later, compound 60 underwent MnO2 mediated oxidation followed by p-toluene sulfonic acid promoted rearrangement to forge the 2,3-dihydro-pyranone moiety 61 in 75% yield. The alcoholic functionality in compound 61 was subjected to Dess–Martin periodinane oxidation to transform it into aldehyde which further underwent treatment with allylbromide via zinc promoted Barbier allylation to result in the synthesis of compound 62 in 69% yield with dr = 1:1.3. The substrate directed Barbier approach was chosen to induce chirality within allylation reaction to afford the synthesis of natural products' both diastereomers. The Barbier allylated product 62 was further transformed into naturally occurring (R,R)-obolactone 63a (32%) and its separable isomer i.e., 6-epi-obolactone 63b (40%) via esterification reaction with ensuing G–II mediated ring closing metathesis (RCM). In the similar manner, alkyne 59 was also subjected to react with 3-phenylpropanal followed by the same series of reactions to afford the synthesis of naturally occurring 7′,8′-dihydroobolactone 64a and its separable isomer i.e., 6-epi-7′,8′-dihydroobolactone 64b in 33% and 39% yield respectively (Scheme 9).
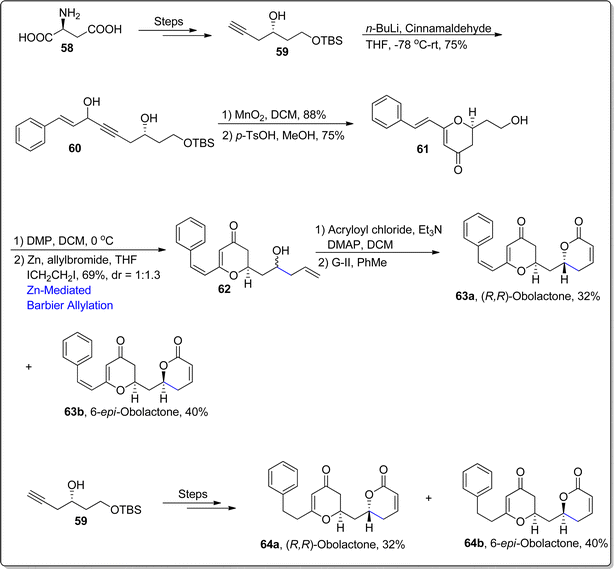 |
| | Scheme 9 Synthesis of (R,R)-obolactone 63a, 6-epi-obolactone 63b, 7′,8′-dihydroobolactone 64a and 6-epi-7′,8′dihydroobolactone 64b. | |
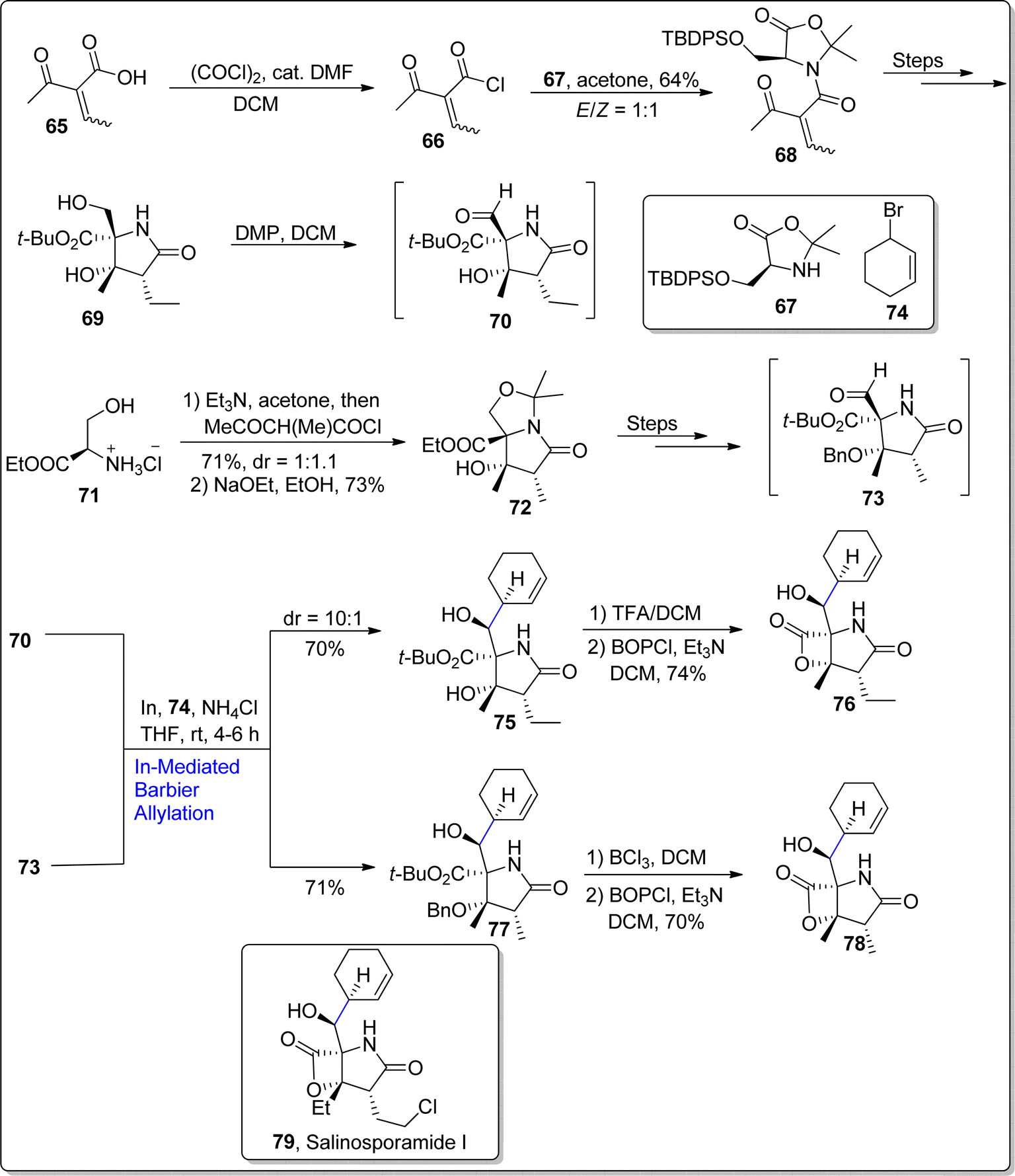
Marine bacteria (of Salinispora genus) are involved in the production of salinosporamides based natural products. They are structurally composed of bicyclic framework having γ-lactam-β-lactone functionality. Considering the unique characteristics of salinosporamides, Park et al. in 2022,79 unveiled the synthesis of salinosporamide B, which was initially extracted from the S. tropica. Their established protocol involved the use of Barbier allylation as one of the key steps. In the foremost step, β-ketoacid 65 was transformed to compound 66 as a result of reaction with (COCl)2 followed by its condensation reaction with oxazolidinone 67 in acetone to generate required substrate 68 in 64% yield with E/Z = 1:1. Later, the compound 68 was treated over several steps to result in compound 69. In the next step, alcoholic functionality in compound 69 was subjected to Dess–Martin periodinane mediated oxidation to convert it into aldehyde intermediate 70. The aldehyde 70 was then further subjected to react with bromo cyclohexene 74 via indium catalyzed Barbier allylation reaction in the presence of ammonium chloride (additive) and tetrahydrofuran at room temperature to afford the addition product 75 in 70% yield with 10:1 diastereoselective ratio. It was observed that the Barbier reaction streamlined the synthetic pathway, thereby affording the product 75 in efficient yield with higher diastereoelectivity. Addition product 75 was then subjected to tert-butyl ester hydrolysis via TFA proceeded by exposure with BOP-Cl to fulfil the synthesis of salinosporamide B 76 in 74% yield. Recently, in 2023, Park et al.80 also utilized indium promoted Barbier allylation approach to afford the first chiral synthesis of salinosporamide D 78 in eleven steps with 12% overall yield. To start with, D-serine based ester 71 was made to react with triethylamine and acetone followed by coupling with β-ketoacid and reaction with sodium ethoxide in ethanol to procure aldol compound 72 in 73% yield with more than 99% ee (as a single diastereomer). The synthesized aldol compound 72 was converted to corresponding aldehyde over several steps. The aldehyde 73 was then treated further via similar In-mediated Barbier allylation to afford compound 77 in 71% yield (as a single isomer). The resulting compound 77 was then made to react with BCl3 to carry out the removal of benzyl and tert-butyl groups proceeded by exposure to BOPCl (bis(2-oxo-3-oxazolidinyl)phosphinic chloride) to furnish the total synthesis of salinosporamide D 78 in 70% yield. Similarly, in-promoted Barbier reaction involving allylation approach was employed to synthesize salinosporamide I 79 (Scheme 10).
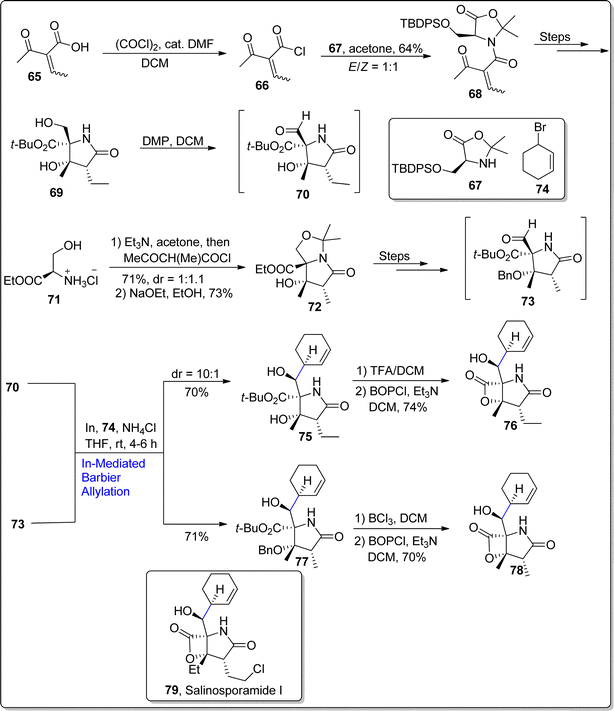 |
| | Scheme 10 Synthesis of salinosporamide B 76, salinosporamide D 78 and salinosporamide I 79. | |
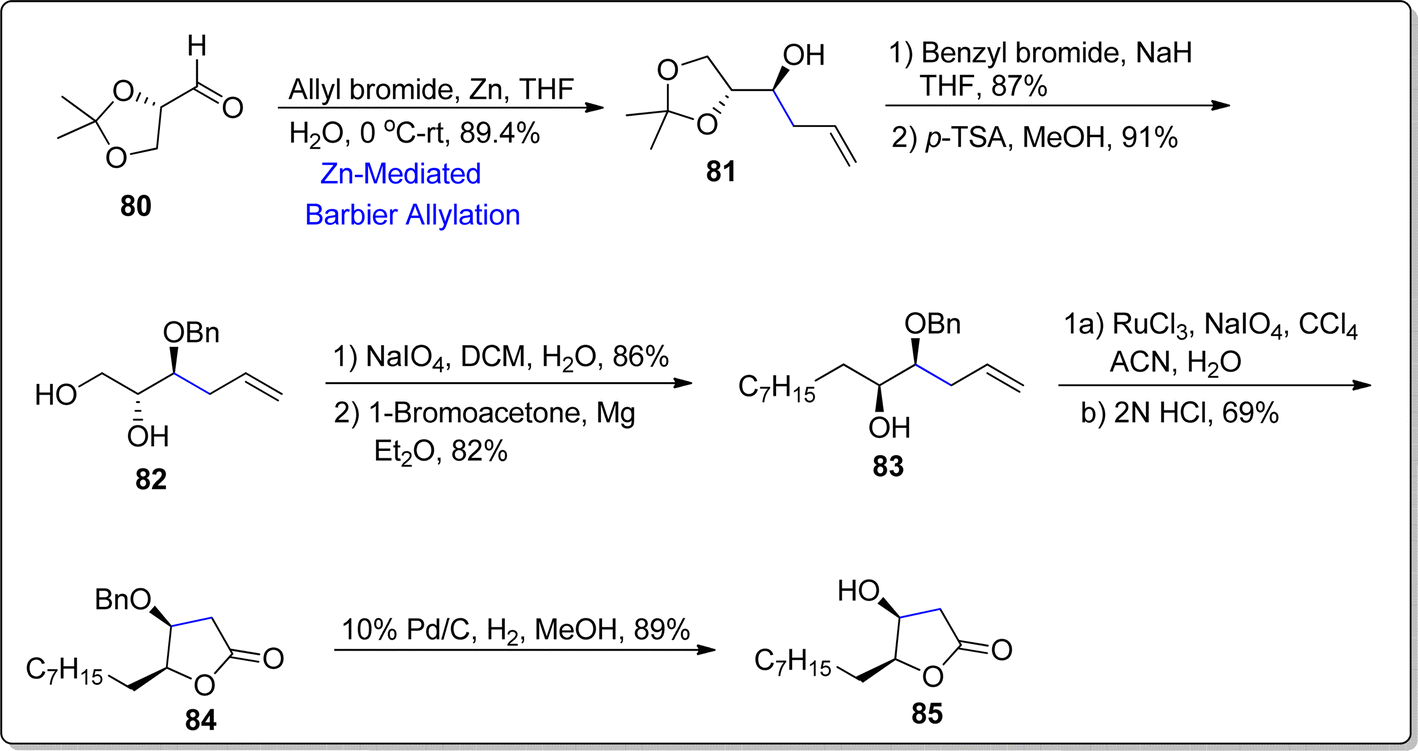
Almost 10% of natural products constitute γ-butyrolactone functionalities, indicating the importance of these organic moieties. The medicinal and pharmaceutical attributes of these butyrolactones have not been studied due to their extraction (from natural sources) in minute quantity. Thus, the need to synthesize these naturally occurring butyrolactones in efficient yields is enhancing continuously. In 2022, Gayke et al.81 adopted a stereoselective route towards the asymmetric synthesis of (4S,5S)-4-hydroxy-γ-decalactone 85, which was first isolated alongwith its major isomer ((4S,5R)-4-hydroxy-γ-decalactone) by Raspotnig group from the secretion of harvestmen Egaenus convexus ejection. In arachnids, these butyrolactones act as defensive agents by releasing exocrine secretions against hazardous chemicals and perpetrators. Gayke and coworkers initiated the synthesis by treating aldehyde 80 with allyl bromide via zinc promoted Barbier allylation reaction in tetrahydrofuran and water to generate homoallyl alcohol 81 in 89.4%. The hydroxyl group in compound 81 was protected on reaction with benzyl bromide using sodium hydride as base, followed by removal of acetonide on treatment with p-toluene sulfonic acid to forge respective diol 82 in 91% yield. The diol 82 then underwent oxidative cleavage by exploiting sodium periodate and dichloromethane followed by Grignard reaction with 1-bromoacetone using magnesium in diethyl ether to synthesize compound 83 in 82% yield. The compound 83 was treated further to result in lactone 84 (with 69% yield) on exposure with oxidizing agents (RuCl3 and NaIO4) proceeded by acidic treatment. Finally, deprotection of hydroxyl group furnished the synthesis of naturally occurring minor isomer of (4S,5S)-4-hydroxy-γ-decalactone 85 in 89% yield (Scheme 11).
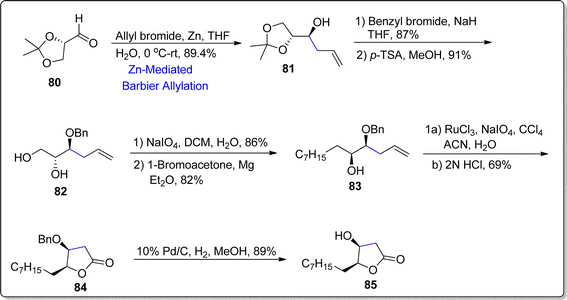 |
| | Scheme 11 Synthesis of (4S,5S)-4-hydroxy-γ-decalactone 85. | |
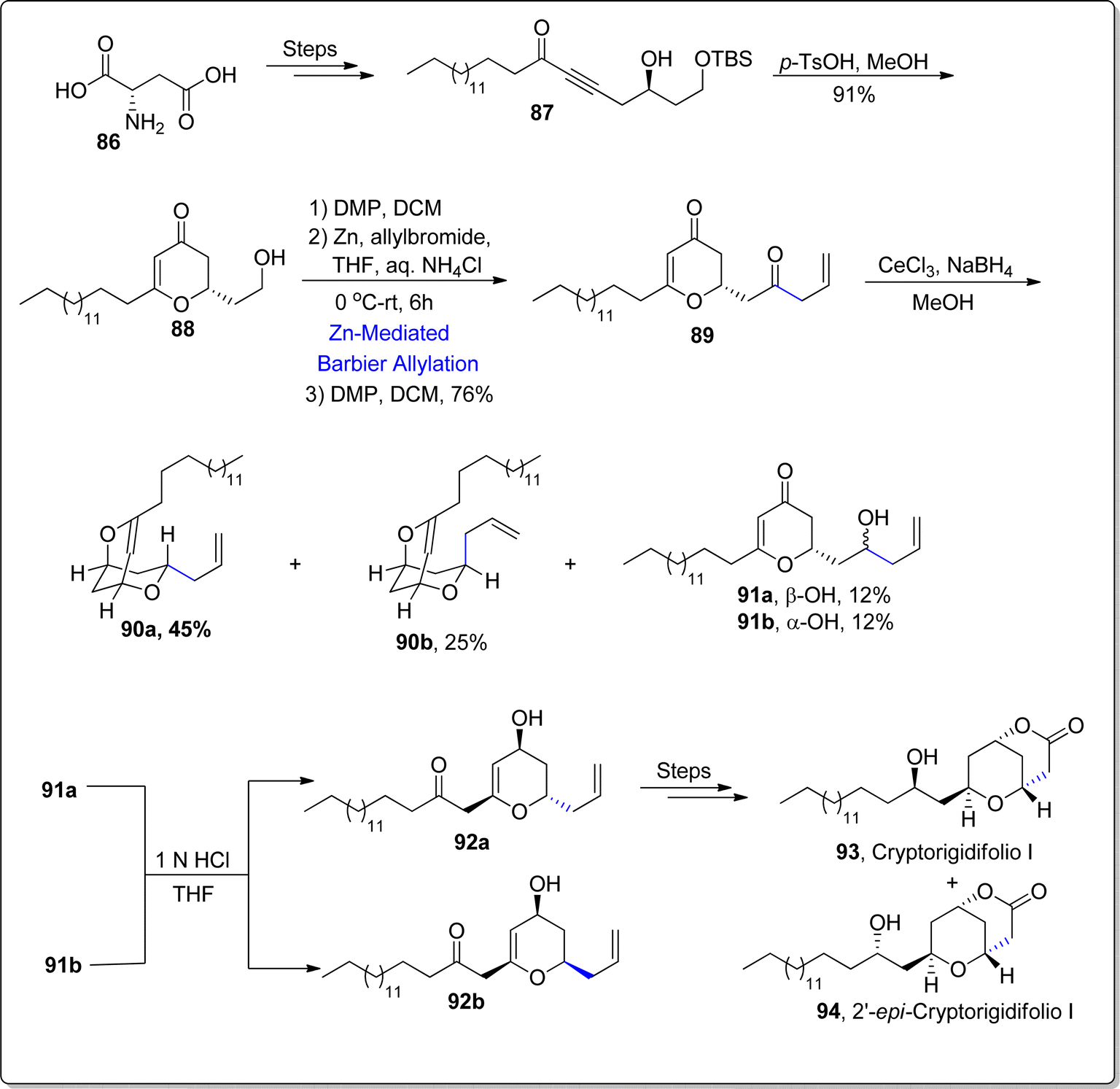
Cryptorigidifoliol is a tetrahydrone pyrone constituting oxygenated natural product, which was obtained as a result of extraction from the Cryptocarya rigidifolia (in 2015).82 Cryptorigidifoliol along with its other class members tend to showcase a broad spectrum of medicinal applications, thereby acting as efficient anti-fungal, anti-cancer, anti-inflammatory and anti-bacterial agents.83,84 Besides its utilization as efficacious analgesics, this natural product has been found to demonstrate cytotoxic activity against specific human cancer cell lines and the antimalarial activity against Plasmodium falciparum. Till now, a single report has been documented focusing the synthesis of cryptorigidifoliol I in 10.8% overall yield by Mohapatra and coworkers. Recently, in 2024, Jangid and Fernandes85 established a successful route to access this naturally occurring compound by employing Barbier allylation as one of the central steps. Their synthetic journey initiated with the procurement of δ-hydroxyalkynone 87 by subjecting L-aspartic acid 86 to a series of consecutive reactions. The δ-hydroxyalkynone intermediate 87 was then transformed into dihydropyranone 88 (in 91% yield) via p-TsOH mediated rearrangement reaction. The resulting dihydropyranone was then exposed to DMP promoted oxidation in continuation with zinc promoted Barbier allylation reaction with allyl bromide in tetrahydrofuran to afford diastereomeric mixture. The diastereomeric products were feasibly separated via column chromatography. The subsequent oxidation step resulted in the synthesis of diketone 89 (within 76% yield). Furthermore, the Luche reduction of diketone 89 furnished diastereomeric mixture of bis-ethers 90a & 90b (dr about 2:1), alongwith reduction products 91a & 91b. The acidic hydrolysis of these ethers rendered diospongin analogues 92a and 92b respectively. The diospongin analogue 92a was then made to react over several steps including sequence to accomplish the total synthesis of cryptorigidifoliol I 93 alongwith its epimer 94 (Scheme 12).
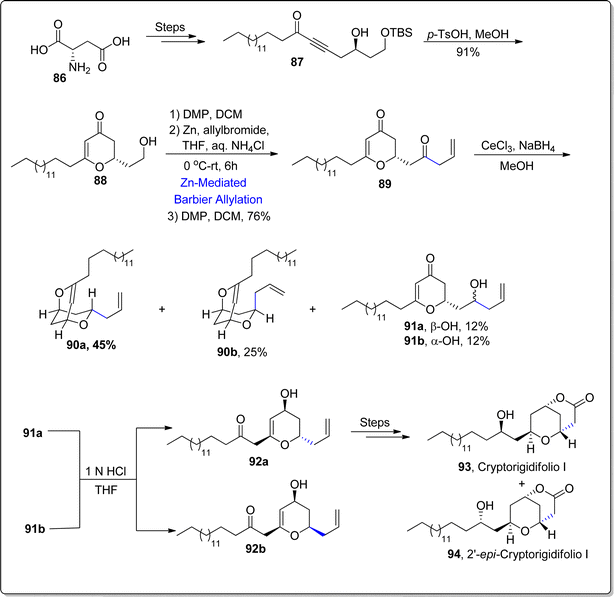 |
| | Scheme 12 Synthesis of cryptorigidifoliol I 93 and 2′-epi-cryptorigidifoliol I 94. | |
2.2.1. Synthesis of macrolides-based natural products. Thuggacins A–C are natural products, which were withdrawn from the myxobacterium i.e., Sorangium cellulosum.86 Their structural skeleton is composed of α,β-unsaturated macrolide alongwith thiazole ring and conjugated diene framework. They exhibit potent activity against a diverse variety of microorganisms. They have been found to affect the respiratory chain of Mycobacterium tuberculosis, thereby acting as therapeutic agent against tuberculosis. Thuggacin cmc-A (which is composed of 17 members) was isolated by Jansen and colleagues from myxobacterium Chondromyces crocatus Cmc5. In 2021, Tsutsumi et al.87 provided the first total synthesis of thuggacin cmc A 108 by applying indium-mediated Barbier propargylation as one of the central steps. Initially, L-malic acid 95 was treated over a number of steps to afford C1–C12 fragment of thuggacin cmc-A 96. In order to access C13–C25 fragment, optically active oxazolidinone 97 was treated with sodium hexamethyldisilazide followed by reaction with BrCH2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CTMS and sodium borohydride mediated reduction to attain corresponding alcohol 98 in 97% yield. The alcohol 98 was then transformed to terminal alkyne 99 over few steps. The reaction of terminal alkyne 99 with (tributylstannyl)methylmagnesium (which was generated within the reaction) utilizing copper cyanide as catalyst proceeded by the addition of methyl group and iodine afforded iodoalkene 100 in 56% yield. Iodoalkene 100 was further made to undergo Pd(PPh3)4 catalyzed Suzuki–Miyaura coupling with Boronic acid 101 along with subsequent (TPAP-promoted) oxidation to furnish aldehyde 102 in 85% yield. The aldehyde 102 was next subjected to react with aminoalcohol 103 and allybromide Singaram's developed indium promoted Barbier propargylation to afford the required compound 104 in 91% yield. The Barbier reaction involving propargylation resulted in excellent yield (91%) of a single required isomer in comparison to the addition of Grignard reagent which afforded the product in unsatisfactorily diastereomeric ratio. The hydroxyl group in compound 104 was further protected with tert-butyl silyl group to generate fragment C13–C25 105 in 100% yield. The fragment C1–C12 96 and C13–C25 105 were further coupled via Sonogashira coupling in the presence of copper iodide, palladium chloride using triethylamine as base to achieve alkyne 106 in 90% yield. The removal of TBS group in alkyne 106 was carried out via tert-butylammonium fluoride to generate –OH substituted alkyne 107, which was then reacted over several steps to give rise to thuggacin cmc-A 108 (Scheme 13).
CTMS and sodium borohydride mediated reduction to attain corresponding alcohol 98 in 97% yield. The alcohol 98 was then transformed to terminal alkyne 99 over few steps. The reaction of terminal alkyne 99 with (tributylstannyl)methylmagnesium (which was generated within the reaction) utilizing copper cyanide as catalyst proceeded by the addition of methyl group and iodine afforded iodoalkene 100 in 56% yield. Iodoalkene 100 was further made to undergo Pd(PPh3)4 catalyzed Suzuki–Miyaura coupling with Boronic acid 101 along with subsequent (TPAP-promoted) oxidation to furnish aldehyde 102 in 85% yield. The aldehyde 102 was next subjected to react with aminoalcohol 103 and allybromide Singaram's developed indium promoted Barbier propargylation to afford the required compound 104 in 91% yield. The Barbier reaction involving propargylation resulted in excellent yield (91%) of a single required isomer in comparison to the addition of Grignard reagent which afforded the product in unsatisfactorily diastereomeric ratio. The hydroxyl group in compound 104 was further protected with tert-butyl silyl group to generate fragment C13–C25 105 in 100% yield. The fragment C1–C12 96 and C13–C25 105 were further coupled via Sonogashira coupling in the presence of copper iodide, palladium chloride using triethylamine as base to achieve alkyne 106 in 90% yield. The removal of TBS group in alkyne 106 was carried out via tert-butylammonium fluoride to generate –OH substituted alkyne 107, which was then reacted over several steps to give rise to thuggacin cmc-A 108 (Scheme 13).
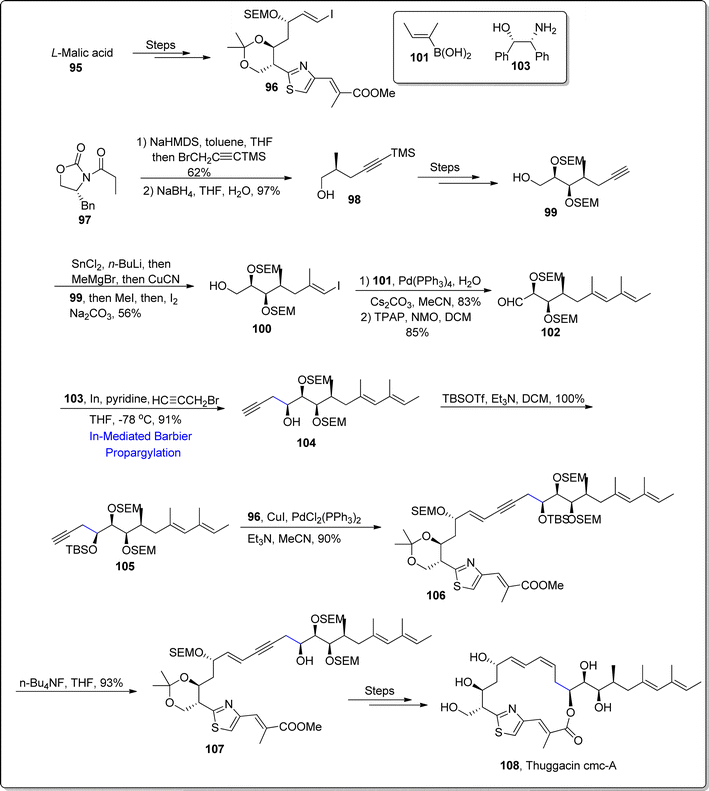 |
| | Scheme 13 Synthesis of thuggacin cmc-A 108. | |
Resorcylic acid lactones are natural occurring compounds, constituting 14-membered macrolides in their structural framework.88 Ever since the extraction of radiciol, this particular class of natural products has been investigated keenly. It has been found to demonstrate the efficient potency as anti-bacterial, anti-fungal and anti-viral agents. There is continuous progress towards the development of efficient anti-microbial agents.89,90 In 2012, Chen and colleagues isolated naturally occurring resorcyclic acid lactones i.e., Paecilomycins A–F.91 In propagation of their strives towards the total synthesis of various Paecilomycins, Gurram and coworkers92 performed the synthesis of Pecilomycin F by employing Barbier-type allylation as a key step. Initially, they treated epoxide 109 over several steps involving synthetic pathway to forge the isopropylidine based ester 110. The synthesized ester 110 was then reduced via DIBAL-H in dichloromethane proceeded by zinc mediated Barbier allylation reaction with allylbromide in the presence of ammonium chloride and tetrahydrofuran to afford non-separable diastereomeric mixture of homo-allylated alcohol 111 in 78% yield with 10:1 dr. The hydroxyl functionality in compound 111 was protected on treatment with MOMCl and DIPEA in dichloromethane with subsequent removal of silyl group by using tetrabutyl ammonium fluoride to furnish the alcoholic fragment 112 in 83% yield. Furthermore, tri-hydroxy substituted benzoic acid 113 was reacted over a number of steps to result in required acid fragment 114. In the next step, synthesized alcoholic and acid fragments (112 & 114) were joined via Steglich esterification to forge ester, which was subjected to Grubbs II catalyst promoted ring closing metathesis to accomplish the synthesis of Paecilomycin F 115 (85% yield) (Scheme 14).
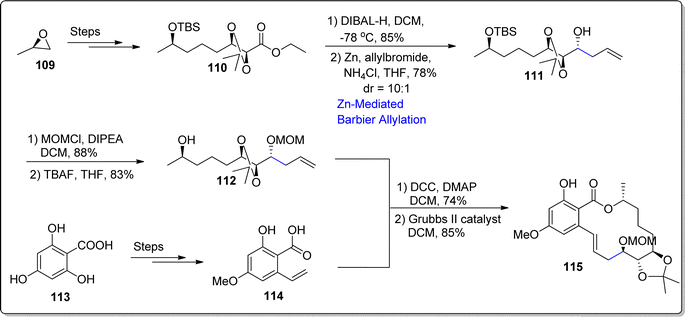 |
| | Scheme 14 Synthesis of Paecilomycin F 115. | |
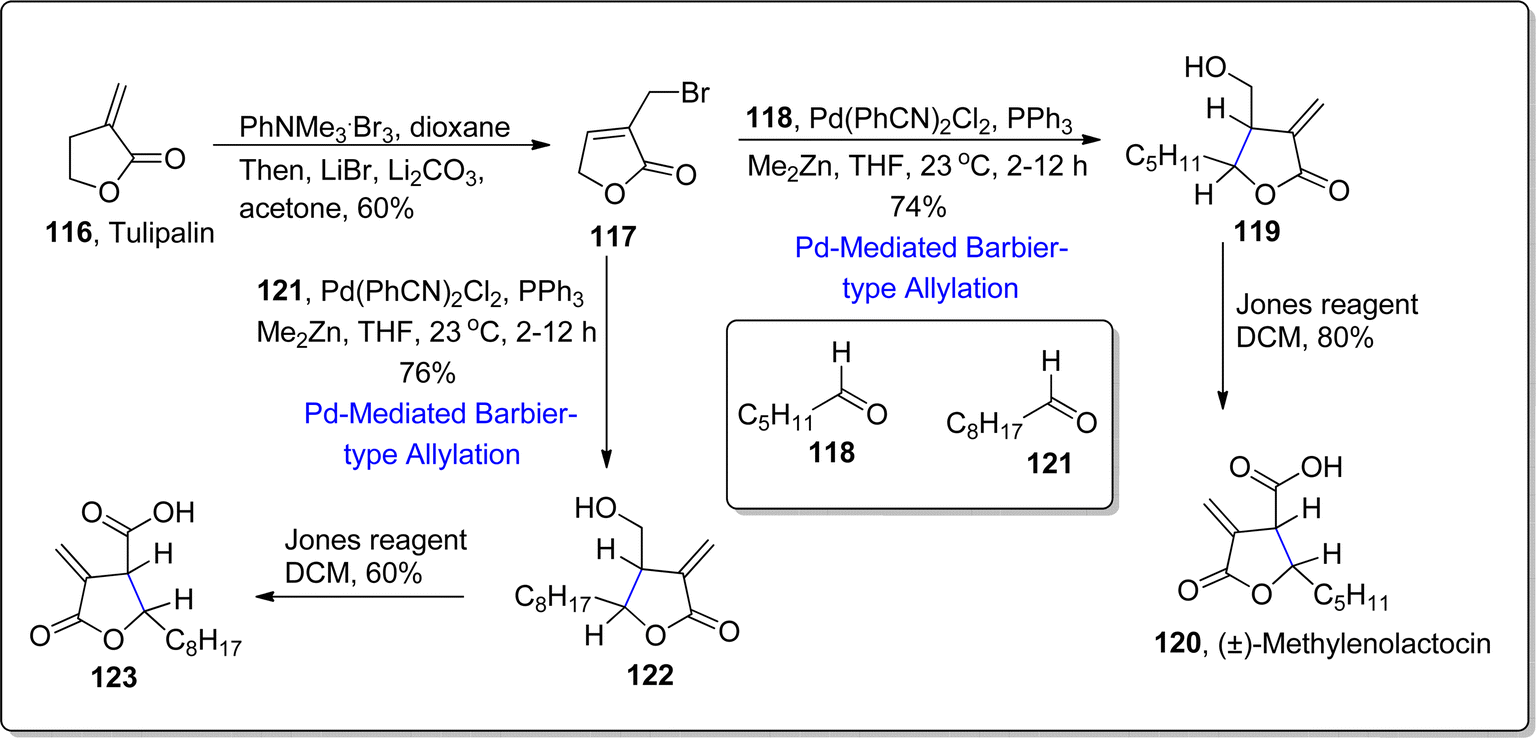
2.2.2. Synthesis of sesquiterpene lactones-based natural products. Sesquiterpene lactones are incorporated with α-exo-methylene-γ-butyrolactone which exhibit promising biological activities i.e., deoxyelephantopin, helenalin and arglabin.93 Structural framework of paraconic acids is also endowed with this bioactive skeleton. The previously employed allylation routes to access bioactive skeleton resulted in anti-relationship within substituents, thus another route was required to access the syn-relationship. For this purpose, considering the utility of palladium metal in organic synthesis,94,95 palladium promoted Barbier-type allylation was employed by involving dimethyl zinc as reducing agent. In 2021, Liu et al.96 developed Barbier-type allylation with subsequent translactonization reaction by using Pd(PhCN)2Cl2 as catalyst with triphenylphosphine ligand in the presence of dimethyl zinc (reducing agent) in THF/toluene to afford the diastereoselective mixture of β,γ-disubstituted α-exo-methylene-γ-butyrolactones. Dimethyl zinc was observed to play crucial role in both steps by acting as a nucleophile (for polarity inversion of π-allylpalladium) and Lewis acid respectively. This developed approach was then exploited further to achieve the synthesis of two paraconic acids and two 1,10-seco-guaianolides. In this regard, market ready tulipalin A 116 was transformed to bromolactone 117 in 60%. In the next step, bromolactone 117 was subjected to treat with hexanal 118 via developed dimethylzinc involving Barbier-type allylation and translactonization sequence in tetrahydrofuran to attain the resulting addition product 119 in 74% yield. The addition product was subjected to Jones oxidation to accomplish the synthesis of (±)-methylenolactocin 120 in 80% yield. In another route, bromolactone 117 was made to react with nonanal 121 via palladium promoted Barbier allylation and subsequent lactonization to furnish compound 122 in 76% yield. In the final step, Jones oxidation of compound 122 resulted in the synthesis of second paraconic acid i.e., (+)-C75 123 in 60% yield (Scheme 15).
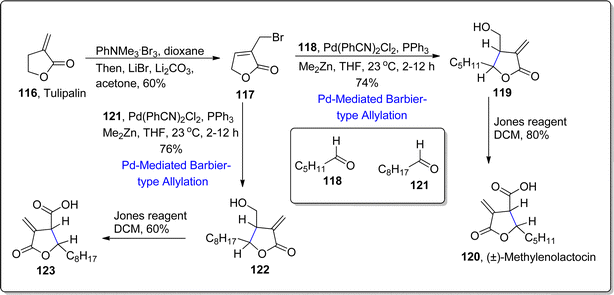 |
| | Scheme 15 Synthesis of (±)-methylenolactocin 120 and (+)-C75 123. | |
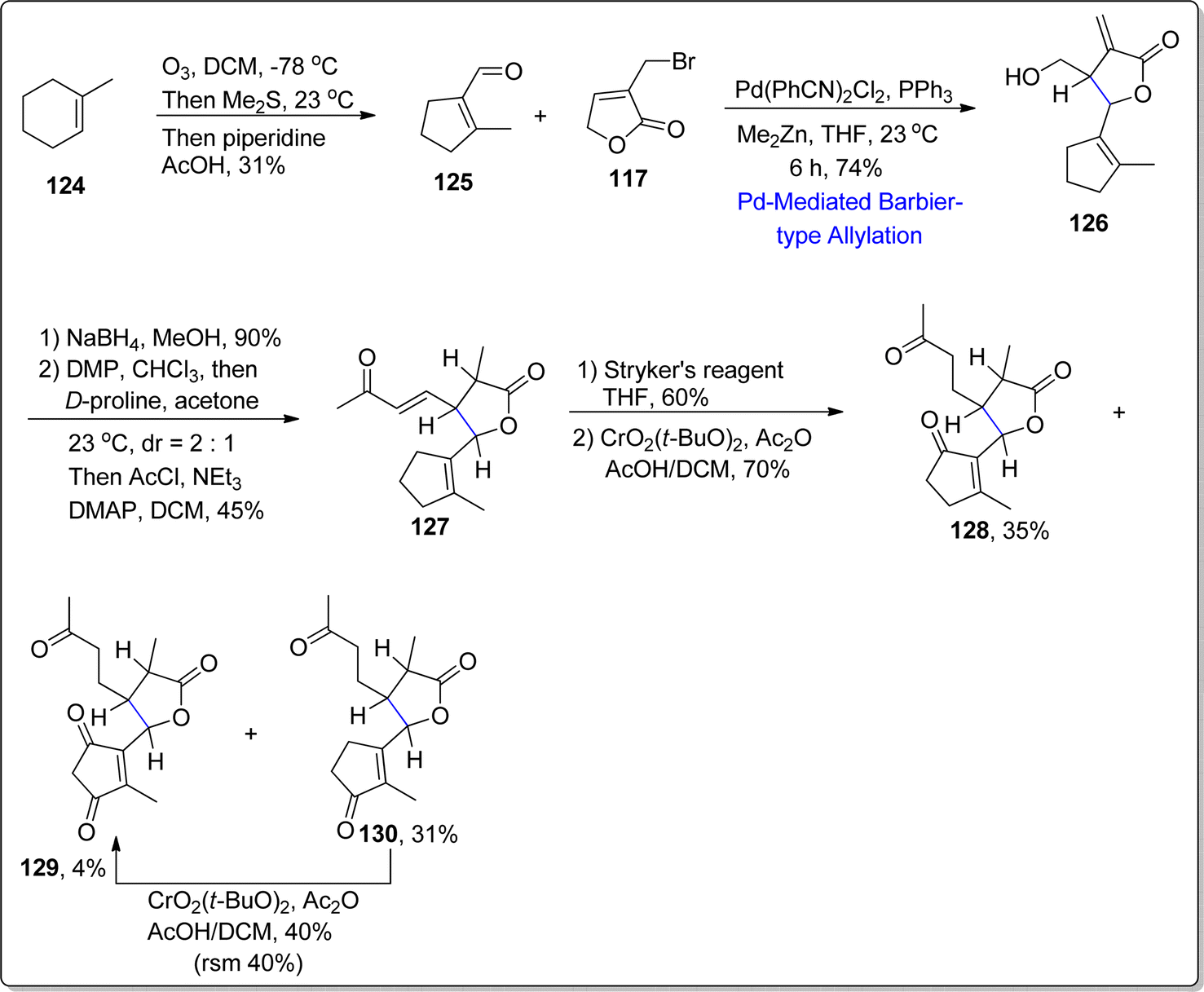
Further Barbier-type allylation sequence was harnessed for the synthesis of 1,10-seco-guaianolides 3-deshydroxy-iso-seco-tanapartholide 128 and 1,10-dioxo-1,10-deoxy-1,10-secogorgonolide 129 which are extracted from Achillea ligustica and Artemisia gorgonum respectively. Initially, alkene 124 was treated over few steps to synthesize corresponding aldehyde 125, which was further treated with bromolactone 117 via developed Barbier-type allylation and translactonization sequence to acquire addition product 126 in 74% yield. The compound 126 was subjected to reduction and oxidation followed by proline mediated aldol condensation and acylation to afford conjugated ketone 127 in 45% yield. The conjugated ketone 127 further resulted in the synthesis of mixture of three compounds 128–130 as a result of Stryker's reagent induced reduction (of α,β-unsaturated double bond) followed by allylic oxidation with the addition of CrO2(Ot-Bu)2. In this way, two naturally occurring 1,10-seco-guaianolides 128 and 129 were obtained in 35% and 4% yield respectively. Other non-natural compound 130 (from the resulting mixture) was subjected to similar allylic oxidation to transform it into naturally occurring 1,10-seco-guaianolide 129 (Scheme 16).
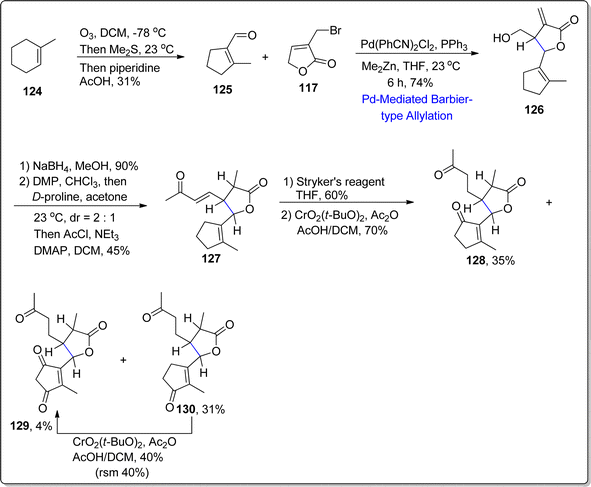 |
| | Scheme 16 Synthesis of 1,10-seco-guaianolides 128 and 129. | |
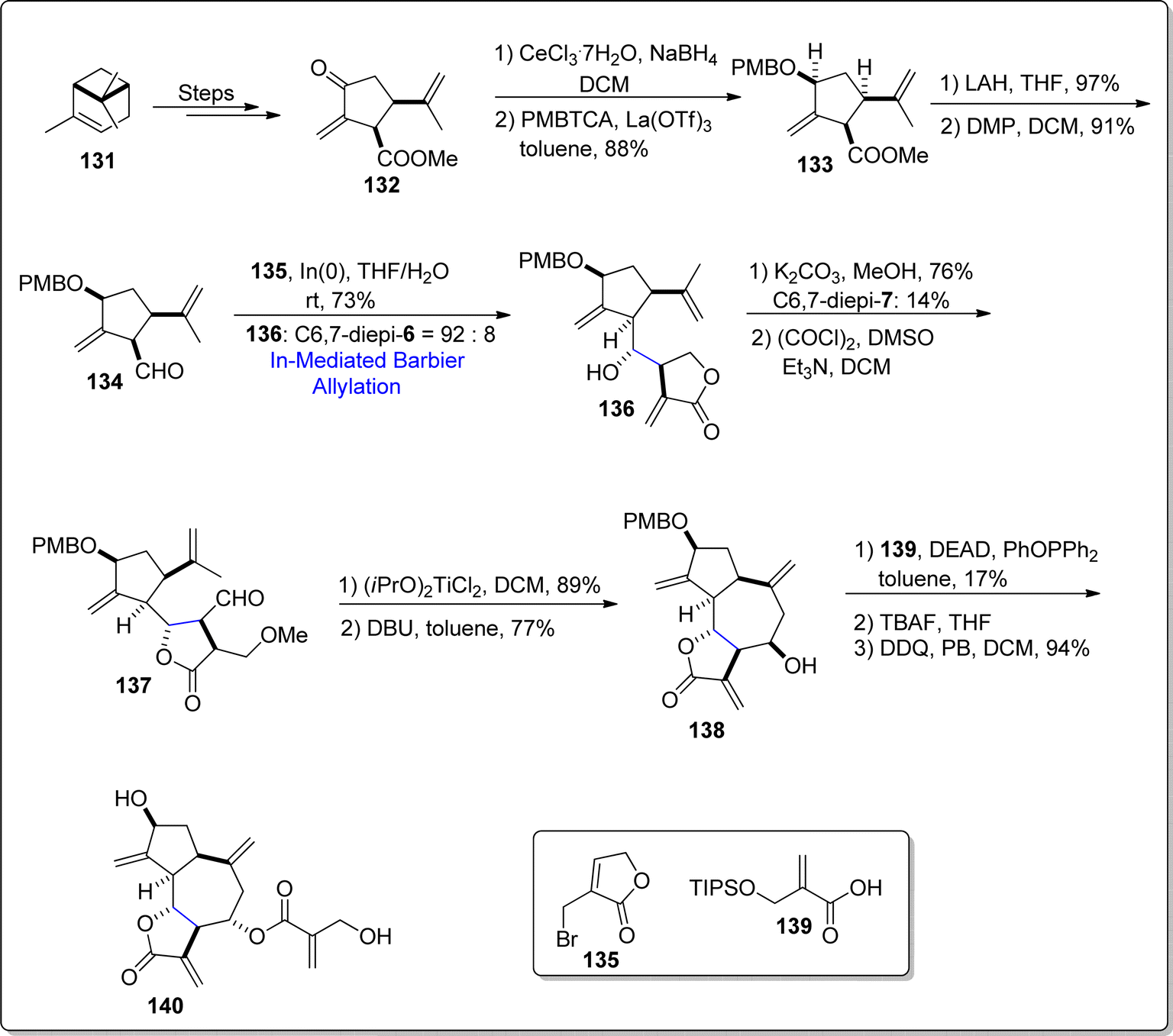
Cynaropicrin is a naturally occurring sesquiterpene lactone, obtained from Cynara scolymus (artichoke). This particular natural product is composed of complex structural framework, consisting of three fused cyclic rings, four exo double bonds with six stereogenic centers. Cynaropicrin has been discovered to play the role of anti-trypanosomal, NF-κB activation inhibitors and anti-hepatitis agents. In 2021, Nakamura et al.97 carried out the first total synthesis of (+)-cynaropicrin 140 by employing Favorskii rearrangement and indium mediated diastereoselective Barbier reaction as major steps. Initially, easily accessible natural precursor i.e., (S)-α-pinene 131 was made to undergo several steps to generate corresponding lactone 132. Lactone 132 then underwent Luche reduction proceeded by hydroxyl group protection with p-methoxy benzyl to furnish compound 133 in 88% yield. The ester functionality in compound 133 was reduced by utilizing lithium aluminium hydride followed by Dess–Martin periodinane mediated oxidation to procure aldehyde 134 in 91% yield. The aldehyde 134 was then made to react with compound 135 via indium catalyzed diastereoselective Barbier allylation reaction in tetrahydrofuran/water at room temperature, which gave diastereomeric mixture of compound 136 in 92:8. It was observed that indium promoted Barbier allylation resulted in better yield with higher diastereomeric ratio as compared to zinc mediated Barbier approach. Compound 136 was then subjected to trans-lactonization in step sequence with Swern oxidation to attain aldehyde 137. The aldehyde 137 was further converted to tri-cyclic compound 138 (in 77% yield) via intramolecular ene reaction succeeded by DBU promoted elimination of methanol. In the next step, Mitsunobu–Mukaiyama inversion reaction between compound 138 and acid 139 installed the side chain to fused tricyclic ring skeleton followed by TBAF and DDQ induced deprotection of hydroxyl group to furnish the desired natural product 140 in 94% yield (Scheme 17).
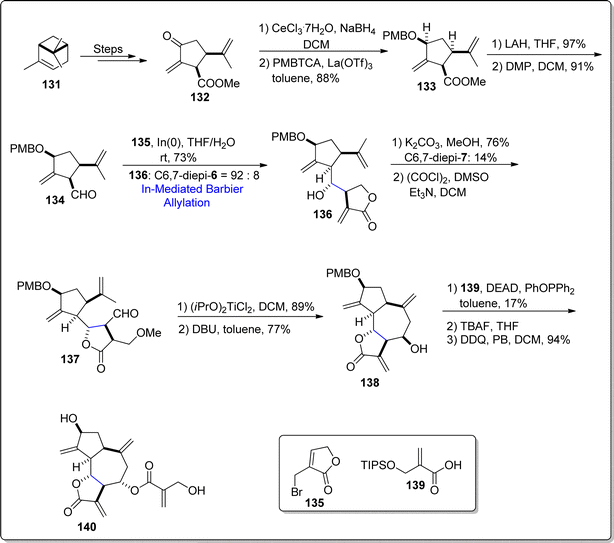 |
| | Scheme 17 Synthesis of 1,10-seco-guaianolides 138 and 140. | |
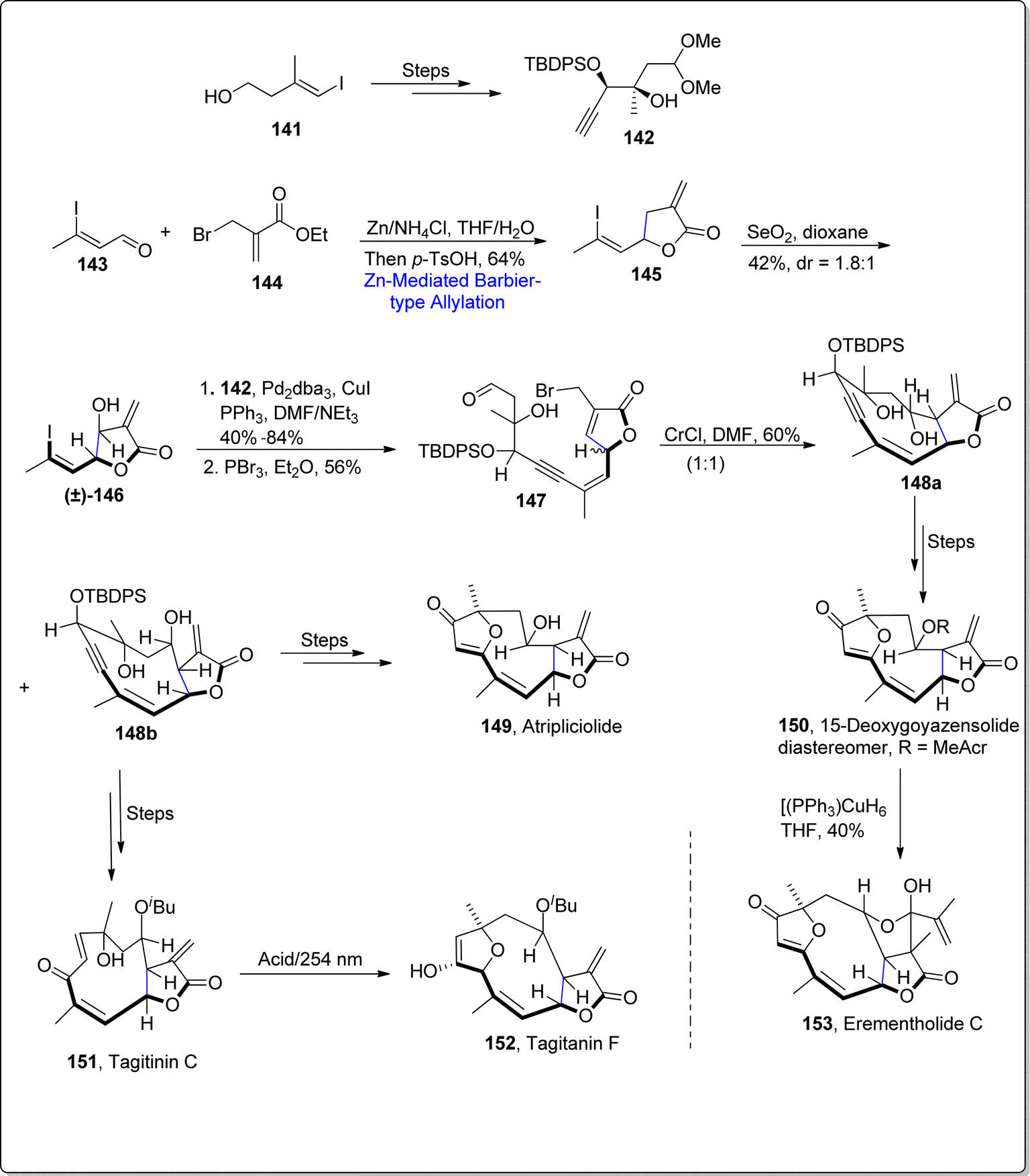
Sesquiterpenes are naturally occurring organic compounds which have been observed to display a significant array of biological activities. As, 15-deoxygoyazensolide has unveiled itself as potent cytotoxic agents in comparison to parent i.e., goyazensolide, which indicates the improvement in pharmaceutical activity on slight structural modifications.98 In 2021, Liu et al.99 reported the synthesis of naturally occurring sesquiterpenes by carrying out zinc promoted Barbier-type allylation as a key step. To achieve the required task, they initiated the synthetic pathway by treating vinyl alcohol 141 over few steps involving route to procure alkyne 142. Then, zinc mediated Barbier-type allylation was carried out by reacting aldehyde 143 with allyl bromide 144 in tetrahydrofuran/water using ammonium chloride followed by lactonization to give the substituted iodoalkene 145 in 64% yield. The utilization of Barbier reaction within allylation approach led towards the installation of α-exo-methylene-γ-butyrolactone. The synthesized alkene 145 further experienced serium oxide (SeO2) allylic oxidation to furnish vinyl iodide 146 in diastereomeric ratio of dr = 1:8.1 with 42% yield. In the next step, vinyl iodide was made to react with initially generated alkyne 142 via Sonogashira coupling reaction proceeded by reaction with PBr3 in diethyl ether, which resulted in the attainment of compound 147. The compound 147 was further converted into two diastereomers 148a and 148b on reaction with CrCl2 in DMF. The diastereomer 148b acted as an essential synthetic intermediate to afford the total synthesis of various naturally occurring sesquiterpenes i.e., atripliciolide 149 and tagitinic C 151 (via several steps involving synthetic pathway). Exposure of acidic conditions to tagitanin C 151 led to the synthesis of tagitanin F 152. Similarly, diastereomer 148a acted as an essential precursor to attain 15-deoxygoyazensolide 150 via several steps sequence, which was then subjected to Stryker's reagent to furnish eremantholide C 153 in 40% yield (Scheme 18).
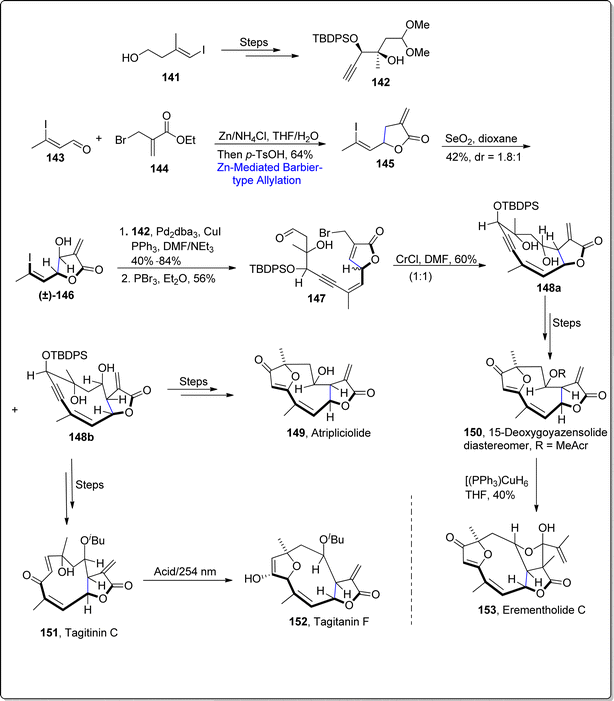 |
| | Scheme 18 Synthesis of naturally occurring sesquiterpene lactones 149–153. | |
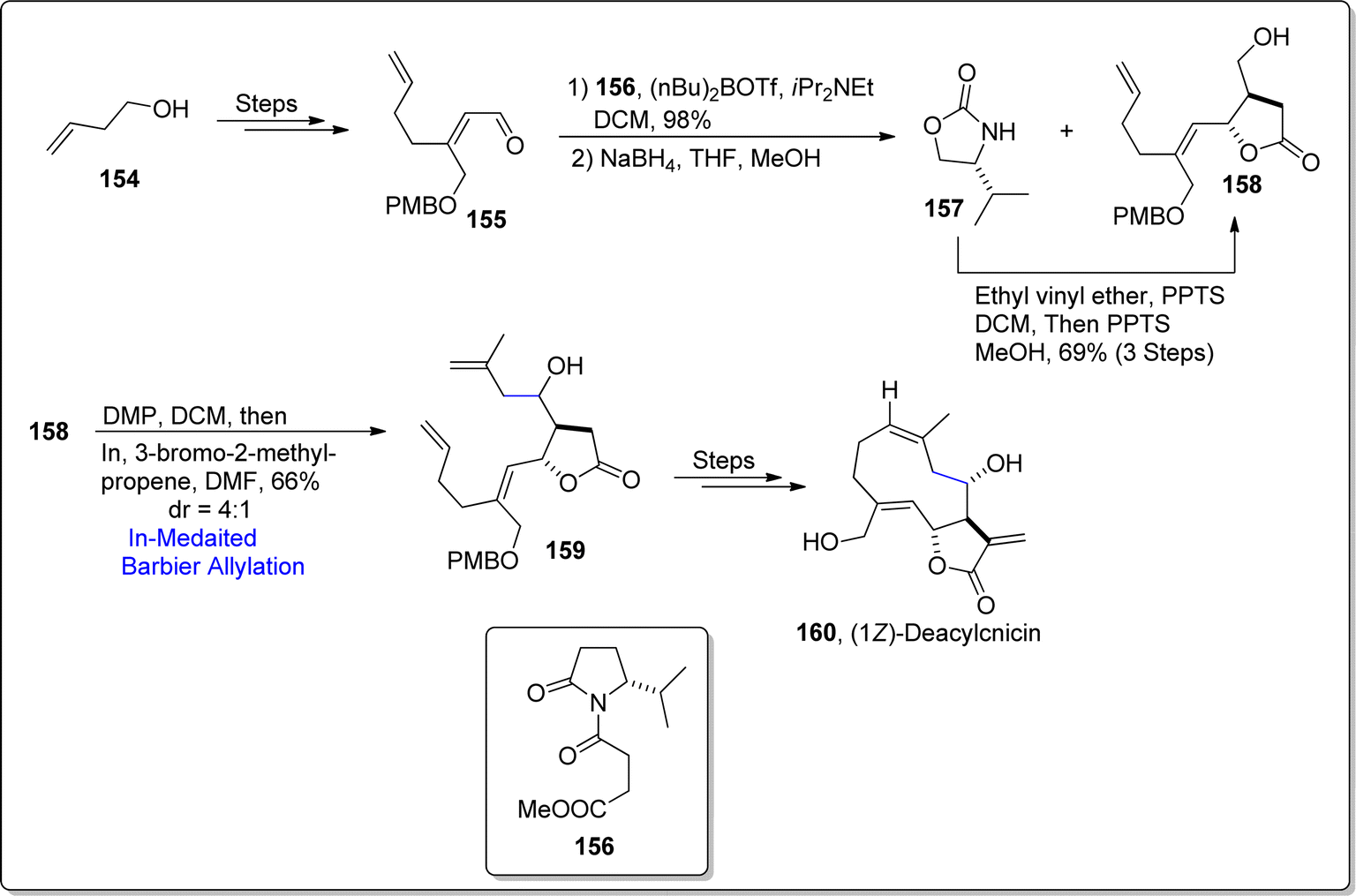
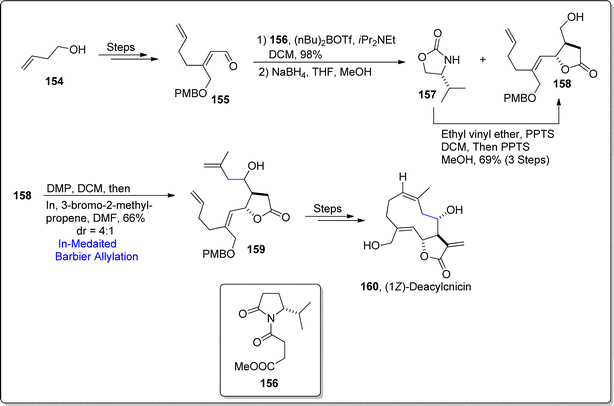
A germacranolide sesquiterpene based lactone i.e., cnicin was initially extracted by Šorm and colleagues in 1959, from the Cnicus benedictus's leaves.100 Cnicin is structurally composed of a diene constrained in a ten-membered ring joined with a lactone ring (5-membered). This naturally occurring sesquiterpene lactone exhibits allelopathic, anti-myeloma, cytostatic, anti-bacterial and anti-trypanosoma brucei activities.101 Its striking pharmacological potential urged the Kimura and Usuki102 (in 2022) to develop an efficient route to access cnicin, exploiting Barbier allylation as a key step. Their synthetic pathway involved the procurement of aldehyde 155 by subjecting alcohol 154 to several steps involving sequence. The synthesized aldehyde 155 was then joined with Evans aldol substrate 156 via 1,2-addition reaction in the presence of (n-Bu)2OTf and diisopropyl ethylamine in dichloromethane in sequence with sodium borohydride mediated reduction to afford the inseparable mixture of compounds 157 and 158. However, treatment of compound 157 with ethyl vinyl ether and PPTS resulted in the synthesis of pure alcoholic fragment 158 in 69% yield (via three steps). The alcoholic functionality in compound 158 was oxidized using Dess–Martin Periodinane followed by indium promoted Barbier allylation with 3-bromo-2-methyl propene, thereby yielding diastereomeric mixture of coupled product 159 in 66% yield (dr = 4:1). The resulting product 159 was treated over a number of steps (which also involve RCM) to attain the synthesis of cnicin analogue i.e., (1Z)-deacylcnicin 160. The inability of RCM to furnish the 1E-form as major product hindered the total synthesis of cnicin, thus paved pathway to result in cnicin analogue (Scheme 19).
 |
| | Scheme 19 Synthesis of (1Z)-deacylcnicin 160. | |
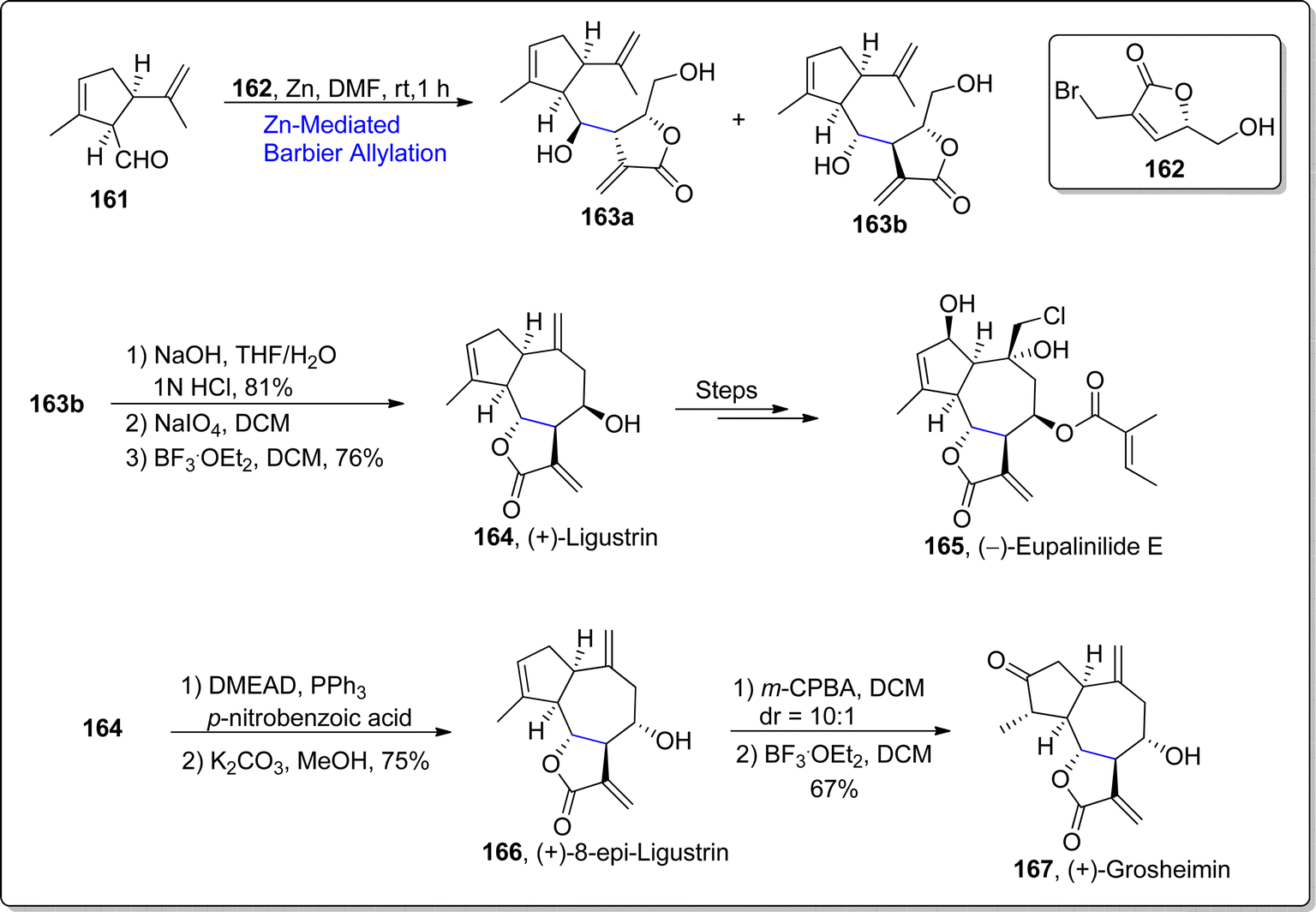
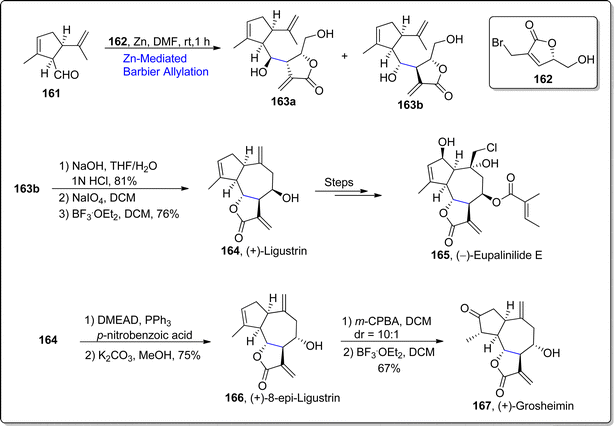
γ-Butyrolactones are abundantly found in the structural skeleton of diverse naturally occurring compounds.103 These scaffolds have been observed to display a significant array of potent biological activities which include cytotoxic, anti-bacterial, anti-viral, anti-inflammatory and anti-fungal activities.104 Guaianolides i.e., (+)-ligustrin, (+)-8-epi-ligustrin, (+)-grosheimin, (+)-8-epi-grosheimin & (−)-eupalinilide are naturally occurring sesquiterpene lactones whose structural constituents involve tricyclic ring system framework with γ-butyrolactone and hydroazulene moieties. These natural products demonstrate remarkable potencies as biological agents. In 2022, Fernandes and Ramakarishna105 carried out the total synthesis of these sesquiterpene lactones by employing diastereoselective Barbier allylation and translactonization as key steps. The first step of synthetic journey involved the treatment of allyl substituted chiral bromolactone 162 with aldehyde 161 via diastereoselective Barbier allylation in the presence of zinc in dimethylformamide to afford the diastereomeric inseparable mixture of three-stereocenters involving compound 163a and 163b (dr = 1:7.5), which was subjected to column chromatography to obtain dr = 8:92 of the resulting mixture. The compound 163b was further translactonized followed by two steps involving aldehyde-ene reaction, which ultimately furnished the (+)-ligustrin 164 in 76% yield. (+)-ligustrin 164 was treated over few steps involving synthetic route to furnish the synthesis of (−)-eupalinilide E 165. Furthermore, (+)-ligustrin 164 was transformed to (+)-8-epi-ligustrin 166 (in 75% yield) via Mitsunobu conditions. Next, epoxidation of (+)-8-epi-ligustrin 166 was proceeded by ring opening rearrangement with boron trifluoride diethyl etherate to accomplish the synthesis of (+)-grosheimin 167 in 67% yield (Scheme 20).
 |
| | Scheme 20 Synthesis of naturally occurring sesquiterpene lactone 164–167. | |
2.3. Synthesis of terpenoids-based natural products
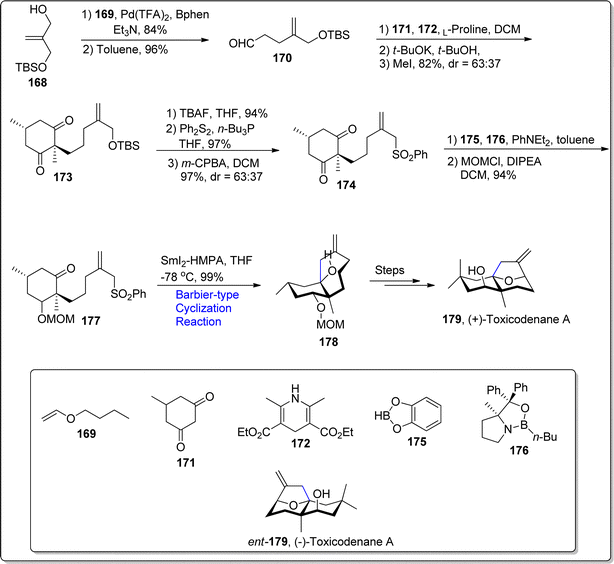
2.3.1. Synthesis of sesquiterpenoids-based natural products. Toxicodenane A is a sesquiterpenoid which consists of three fused cyclic rings anlogwith exocyclic double bond and bridged ether functionality. In 2013, it was first withdrawn from Toxicodendron vernicifluum by Cheng and coworkers.106 Racemic isolation of toxicodenane A urged the researchers to carry out its enantioselective synthesis, to evaluate its biological applications. In 2021, Han and colleagues107 demonstrated the first enantioselective synthesis of toxicodenane A by carrying out Lewis acid catalyzed transacetalation and Prins cascade reaction. In 2022, Nishikawa et al.108 reported an efficient route to achieve the enantioselective synthesis of toxicodenane A followed by the biological assessment of both enantiomers against lipotoxicity. They accomplished the asymmetric synthesis of toxicodenane A by implementing SmI2 mediated Barbier-type cyclization, allylic oxidation and dehydration induced cyclization as key steps. The synthesis began with the vinylation of silyl ether 168 by involving Pd(TFA)2 and Bphen (bathophenanthroline) followed by Claisen rearrangement to afford required aldehyde 170 in 96% yield. The resulting aldehyde 170 was then subjected to react with cyclohexanedione 171 and compound 172 via reductive Knoevenagel condensation which resulted in diastereomeric mixture of 173 in 82% yield (with dr = 63:37). The silyloxy group in compound 173 was then transformed to sulfonyl group 174, which on further asymmetric desymmetrization and protection of hydroxyl group with methoxymethyl gave enantiopure compound 177 in 94% yield. In the next step, compound 177 was subjected to samarium iodide (SmI2-HMPA) promoted Barbier-type annulation in tetrahydrofuran at −78 °C to afford compound 178 stereo selectively in 99% yield with cis-configuration. The cyclized product 178 was then treated over several steps to forge (+)-toxicodenane A 179. Similarly, the synthesis of (−)-toxicodenane A ent-179 was procured by treating compound 174 with boronic acid 175 and compound ent-176 via similar mentioned (Barbier-type annulation involving) protocol. Both the enantiomers were found to protect the cells against lipotoxicity promoted inflammatory effects (Scheme 21).
 |
| | Scheme 21 Synthesis of (−)-toxicodenane A 179 and (−)-toxicodenane A ent-179. | |
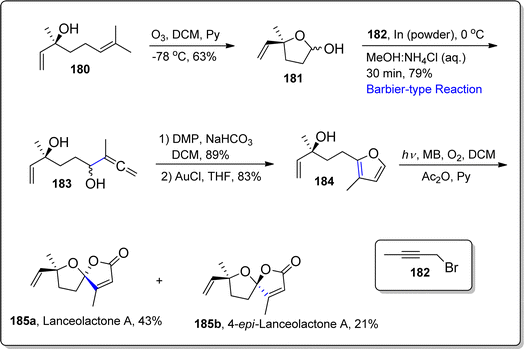
Oxaspirolactone constituting terpenoids are known to exhibit remarkable biological role as they are efficiently employed against several deadly diseases.108,109 In 2015, Kubo et al. first time isolated the oxaspirolactone based tetranorsesquiterpenoids i.e., lanceolactone A and B from the Illicium lanceolatum's leaves.110 These naturally occurring sequiterpenoids were observed to illustrate the potent anti-microbial activity against teeth pathogen i.e., Porphyromonas gingivalis. Nanda's group, in 2018,111 carried out the initial report on the asymmetric total synthesis of lanceolactone in 16.2% overall yield by employing various name reactions as key steps. Being interested in the chemistry of oxaspirolactones, in 2022, Borade and Kontham112 established a facile synthetic route to achieve the synthesis of lanceolactone A and its possible stereoisomers by exploiting chiral precursors without installation of any protecting group. They accomplished the synthesis of target natural products by carrying out ozonolysis, Barbier-type addition reaction, gold-promoted cycloisomerization and dye-sensitized photooxidation as significant steps. In the first step, (S)-linalool 180 was made to undergo ozonolysis in the presence of pyridine and DCM to afford lactol 181 in 63% yield, without incorporating any protecting group. The synthesized lactol 181 was then subjected to indium powder promoted Barbier-type addition reaction with propargyl bromide 182 in MeOH:NH4Cl, which gave intermediate i.e., allene-diol 183 in 79% yield. The resulting allene-diol 183 was further subjected to Dess–Martin Periodinane mediated oxidation followed by treatment with AuCl in tetrahydrofuran to synthesize hydroxyalkyl-joined-furan 184 in 83% yield. Compound 184 was further exposed to MB (Mitsunobu–Vassilikogiannakis's) dye-sensitized photo-oxidation reaction, which afforded lanceolactone A 185a and its epimer 185b in 43% and 21% yield respectively (Scheme 22).
 |
| | Scheme 22 Synthesis of lanceolactone A 185a and 4-epi-lanceolactone 185b. | |
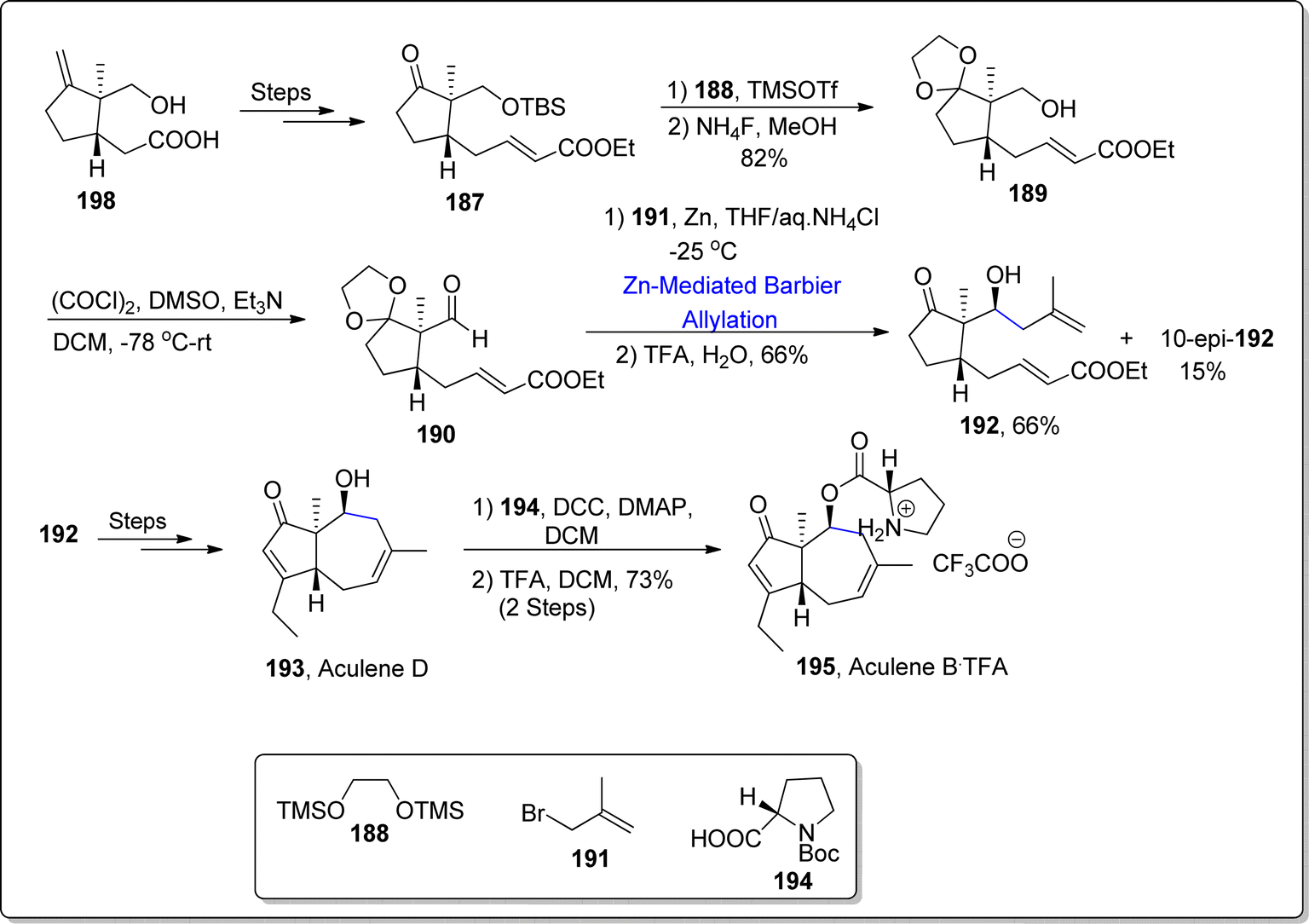
Aculenes A–E are affiliated with bicyclic norsesquiterpene based L-proline associated keto esters or alcohols, which were extracted from Aspergillus aculeatus culture media and from Penicillium sp. XWS02F62 and SCS-KFD08.113 These norsesquiterpenes are fungal metabolites endowed with nordaucane framework. Among them, aculenes C, D and E perform the inhibition of bacterial communication (quorum sensing restrictive potency) opposed to Chromobacterium violaceum CV026 bacterium.114 Thus, being persuaded by the efficient pharmacological role of aculenes, Yokokawa et al.115 in 2023, reported a significant pathway leading towards the synthesis of aculene B and D. Their synthetic journey involved the utilization of zinc promoted Barbier allylation as one of the essential steps. The synthesis commenced with the transformation of easily available precursor i.e., carboxylic acid 186 into α,β-unsaturated ester 187 via several steps. In the next step, ketone functionality in compound 187 was subjected to acetalization reaction followed by the treatment with tetra ammonium fluoride, thereby giving acetal 189 in 82% yield. The obtained acetal was then subjected to undergo Swern oxidation to afford aldehyde 190. The aldehyde 190 was then made to undergo zinc-mediated Barbier allylation reaction with allylbromide 191, thereby giving the required diastereomer 192 in major quantity (66% yield) on subsequent removal of acetal group via acidic hydrolysis. The diastereomer 192 was made to react over a number of steps to furnish the naturally occurring aculene D 193. Aculene D 193 was treated further to acquire aculene B 195 in 73% yield as a result of its Steglich esterification with Boc protected L-proline 194 proceeded by the deprotection of amine (Scheme 23).
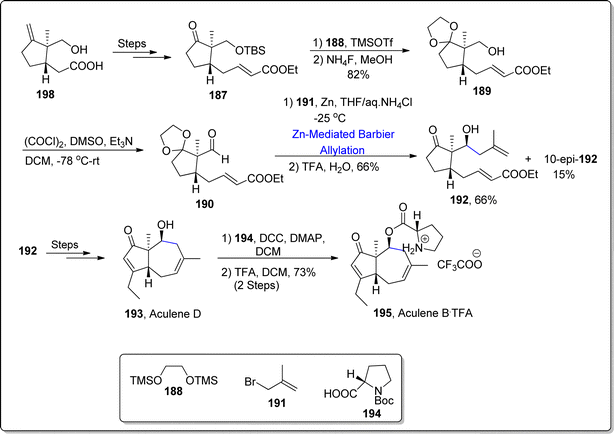 |
| | Scheme 23 Synthesis of aculene D 193 aculene B.TFA 195. | |
2.3.2. Synthesis of diterpenoids-based natural products. Havellockate belongs to the cembranoid family, which was first obtained from Sinularia granosa. Its structure is composed of fused tricyclic ring framework with β-hydroxybutanolide ring constituting overall eight chiral centers.116 Till now, no successful synthesis of havellockate has been presented. Considering the challenges towards its total synthesis, in 2022, Hafeman et al.117 performed the total synthesis of havellockate by utilizing Julia–Kocienski olefination, Diels–Alder cascade reaction, zinc catalyzed Barbier allylation alongwith copper mediated aerobic oxidation as major steps. The synthesis kickstarted with the synthesis of required aldehyde 197 and sulfone 199 individually by treating enone 196 and acyl oxazolidinone 198 respectively over few steps. The aldehyde 197 and sulfone 199 were then coupled via KHMDS mediated Julia–Kocienski olefination followed by desilylation to afford diol 200 in 57% yield with >20:1 E:Z. The resulting diol 200 was further made to underwent Steglich esterification with propiolic acid followed by [4 + 2] cycloaddition reaction to furnish Diels–Alder adduct 201 in 51% yield with >20:1 dr. The adduct 201 was later treated over several steps to synthesize enone 202, which was subjected to transesterification reaction to result in ester 203 in 70% yield. The ester 203 further underwent zinc promoted Barbier allylation reaction with substituted allyl bromide 204 in tetrahydrofuran followed by subsequent deacetylation to forge triol 205 in 25% with dr = 1:1. The triol compound 205 was later transformed to tetrol 206 in 34% yield as a result of iridium promoted anti-Markonikov hyrdrosilylation alongwith Tamao–Fleming oxidation. In the final step, synthesis of havellockate 207 was achieved in 80% by treating compound 206 via copper mediated (aerobic) oxidation/cyclization reaction (Scheme 24).
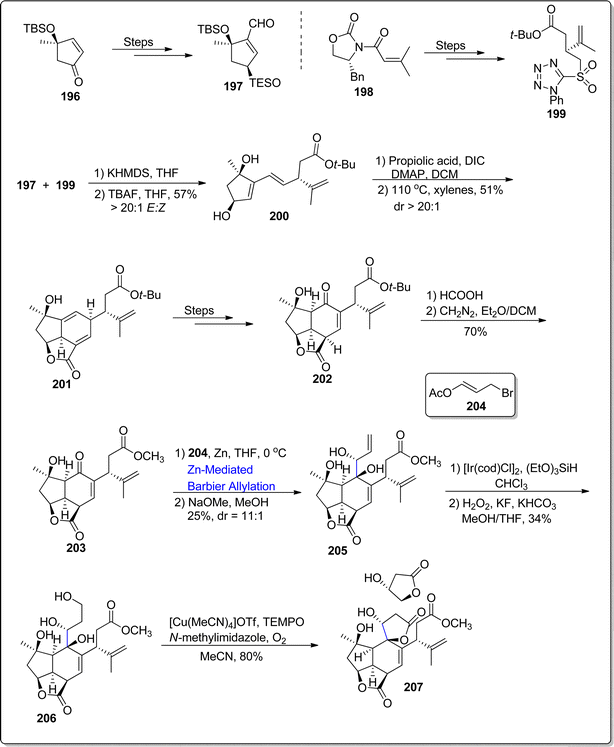 |
| | Scheme 24 Synthesis of havellockate 207. | |
Synthesis of naturally occurring organic compounds have taken frontseat owing to their applicability as efficient drugs. Makassaric acid and fascioquinol B are meroditerpenoids obtained from sponges i.e., Acanthodendrilla and Fasciospongia sp. respectively. Makassaric acid acts an efficient inhibitor of MAPKAP kinase 2 (MK2) enzyme,118 thus acting as potent anti-inflammatory agent. Similarly, fascioquinol B has been known to exhibit remarkable anti-bacterial potency. In 2022, Rosales et al.119 presented a synthetic layout for the accomplishment of these naturally occurring meroterpenoids. For the synthesis of Makassaric acid derivative, initially, Grignard derivative 208 was added to isoprene monoxide by exploiting the catalytic efficiency of CuBr.DMS, to result in 92% of allylic alcohol 210. The resulting alcohol was then converted to aldehyde 211 via oxidation using Dess–Martin periodinane. The synthesized aldehyde 211 was further subjected to exposed to compound 212 via Cp2TiCl mediated Barbier-allylation reaction, which furnished polyene 213 in 51% yield. This catalytic strategy is essential for the formation of diversely substituted integrated polyenes. The hydroxyl group in ploynene 213 was acetylated to afford compound 214 in 99% yield, which was ultimately treated over several steps to procure the naturally occurring makassaric acid's derivative 215. The similar strategy was adopted by treating 2-bromo-1,4-dimethoxybenzene as a precursor, which followed the above synthetic protocol to furnish the synthesis of a close derivative of fascioquinol B 216 (Scheme 25).
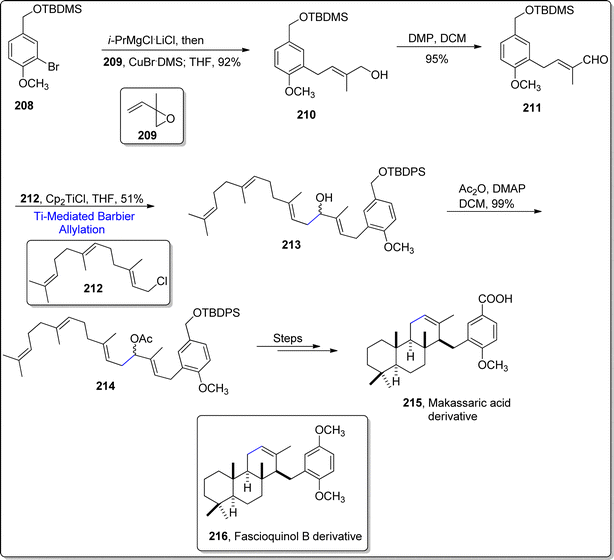 |
| | Scheme 25 Synthesis of makassaric acid's derivative 215 & fascioquinol derivative 216. | |
The most enormous class of plants based secondary metabolites generally constitute diterpenoids (subclass of terpenoids).120 C20 diterpenoids have been structurally classified into same main categories which include ent-beyerane, ent-kaurane, ent-trachylobane, ent-gibberellane and ent-atisane etc. The main reason for the origin of these C8-ethano bridging diterpenoids is the series of cyclization reaction of geranylgeranyl pyrophosphate which gives rise to ent-beyeranyl cation I.121 This cation plays a key role in enzyme-mediated conversions to synthesize a variety of diterpenoids. Taking into consideration the medicinal applications and structural diversity of these diterpenoids, in 2023, Fan group122 unveiled a unified strategy by exploiting late stage conversions of ent-trachylobane and ent-kaurane scaffolds to achieve the synthesis of 9 ethano-bridged diterpenoids. The synthetic pathway commenced with the preparation of corresponding aldehyde 218 from optically active cyclohexenone 217. Meanwhile, Appel substitution reaction of γ-cyclogeraniol 219 afforded optically active allylic bromide 220 in 85% yield. The two resulting monoterpene fragments were then subjected to lithium-mediated Barbier allylation reaction by utilizing lithium/naphthalene in tetrahydrofuran followed by acidic exposure to furnish the diastereomeric mixture of 221a and 221b in about 1:1 diastereoselectivity ratio. Next, enonols 223α and 223β were individually attained as a result of MnO2 promoted allylic oxidation of 221a and 221b respectively. The earlier obtained diastereomeric mixture (221a and 221b) was also exposed to IBX and Dess-Martin periodinane mediated oxidation to procure 91% yield of keto-enone 222. Enonols 223α and 223β were individually subjected to optimized hydrogen atom transfer cyclization (MHAT) to result in the generation of trans,trans-C2 224a and tran,trans-C1 224b along with cis,cis-C1 224c. These precursors were obtained in 36% yield with 75% brsm affording a diastereomeric ratio of 7:1. The synthesized trans,trans C2 224a was treated over several steps thereby furnishing the synthesis of atisirene 226, which was subjected to Muakiyama hydration reaction condition to result in ent-atisan-16apha-ol 227 in 76% yield. Similarly, compound 224a was transformed into epi-canadol A 225a within 71% yield (ent-kaurane type diterpenoid) via Wolff–Kishner–Huang reaction conditions. Similarly, the trans,trans-C1 224b was transformed into sicanadiol 228 over several steps. This diastereomer 224b resulted in the synthesis of canadol A 225b (in 69% yield) via Wolff–Kishner–Huang reduction. The resulting canadol A 225b underwent cobalt mediated Mukaiyama hydration to accomplish the synthesis of powerol 229 in more than 20:1 diastereoselective ratio with 71% yield (Scheme 26).
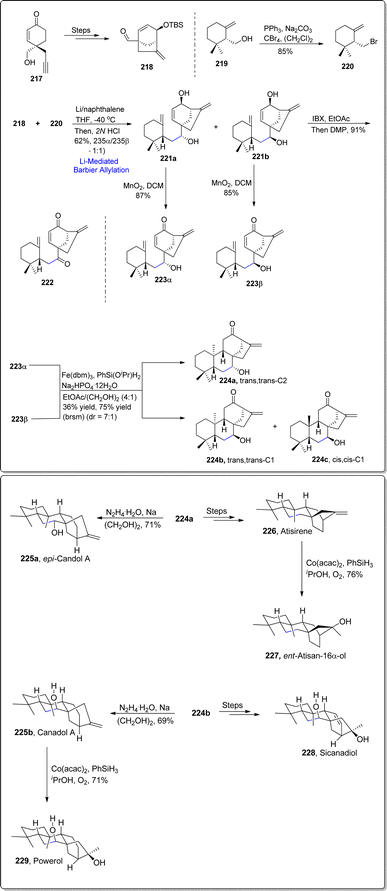 |
| | Scheme 26 Synthesis of C20 diterpenoids 224–229. | |
The initially synthezsized keto-enone 222 was also subjected to MHAT cyclization to give rise to trans,trans-C3-CP 230, which was treated over numerous steps to achieve naturally occurring trachinol 231a and epi-trachinol 231b. Similarly, Wolff–Kishner reduction of trans,trans-C3-CP 230 generated ent-beyerane 232 in 62% yield (Scheme 27).
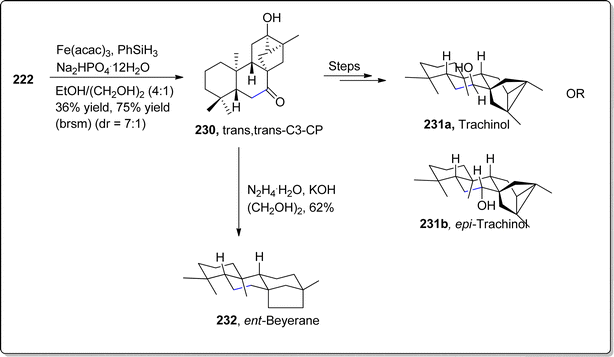 |
| | Scheme 27 Synthesis of C20 diterpenoids 231a, 231b and 232. | |
2.4. Synthesis of lignans-based natural products
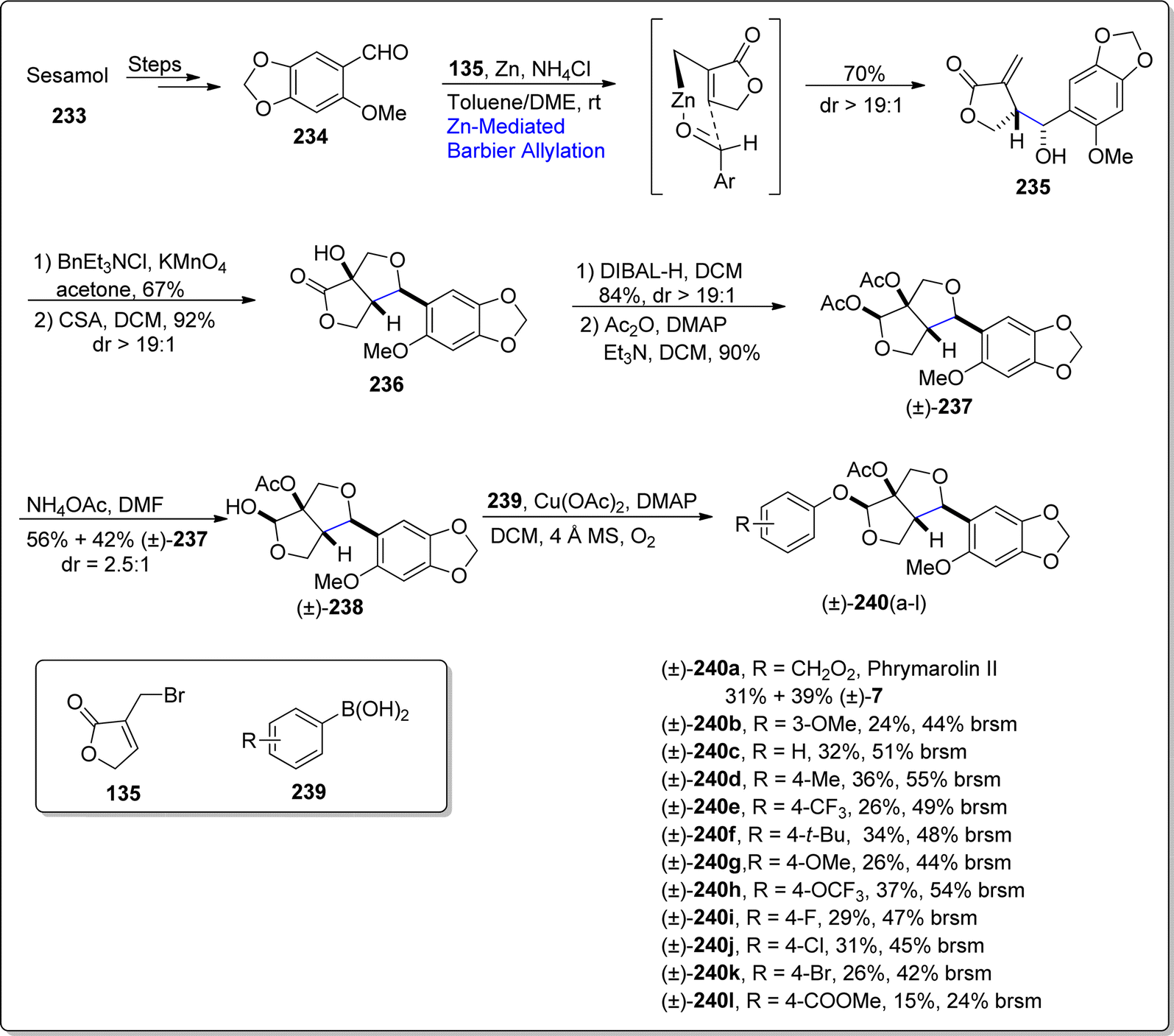
Phrymarolin II is a 3,7-dioxabicyclooctane framework featuring furofuran lignan, which has been extracted from renowned chinese’ traditional medicinal plant i.e., Phryma leptostachya L.123 This plant's roots have been determined to depict the angiogenesis, pesticidal, anti-bacterial and anti-inflammatory activities. There have been several reports concerning the total synthesis of phrymarolin II and its derivatives in literature, however they were observed to usually suffer from non-economic and low-yielding outcomes. Thus, in order to establish more convenient pathway for the synthesis of naturally occurring lignan, Chi and coworkers in 2021,124 endeavored an efficient synthetic pathway to access phrymarolin II and its analogues. They utilized the market-ready precursor i.e., sesamol to accomplish the synthesis of this natural product (with overall 5.7% yield) in total nine steps, exploiting Barbier allylation reaction and anomeric O-arylation as significant steps. Initially, sesamol 233 was subjected to react over reported two steps to afford carbaldehyde 234. The resulting carbaldehyde was then made to react with 3-bromomethyl substituted furanone 135 via zinc promoted Barbier allylation reaction with the addition of ammonium chloride in toluene/dimethylether, thus procuring compound 235 in 70% yield with more than 19:1 diastereomeric ratio. The Barbier allylated product 235 was then exposed to dihydroxylation conditions proceeded by camphor sulfonic acid (CSA) mediated cyclization to forge tetrahydrofuran 236 in 92% yield. In the ongoing step, tetrahydrofuran was subjected to DIBAL-H promoted reduction in sequence with treatment of resulting lactol with acetic anhydride using dimethyl aminopyridine and triethylamine in dichloromethane, thus giving diacetate 237 in 90% yield. Further, deacetylation reaction by employing ammonium acetate resulted in diastereomeric mixture of compound 238 and starting diacetate 237 in 2.5:1 dr. Finally, compound 238 was further made to undergo copper acetate promoted Chan–Lam–Evan cross coupling reaction with substituted aryl boronic acids 239 to afford the total synthesis of phrymarolin II 240 and its various analogues. The in vivo studies interpret the good to excellent potential of these analogues against the activity of tobbaco mosaic virus, in comparison with ningnanmycin (Scheme 28).
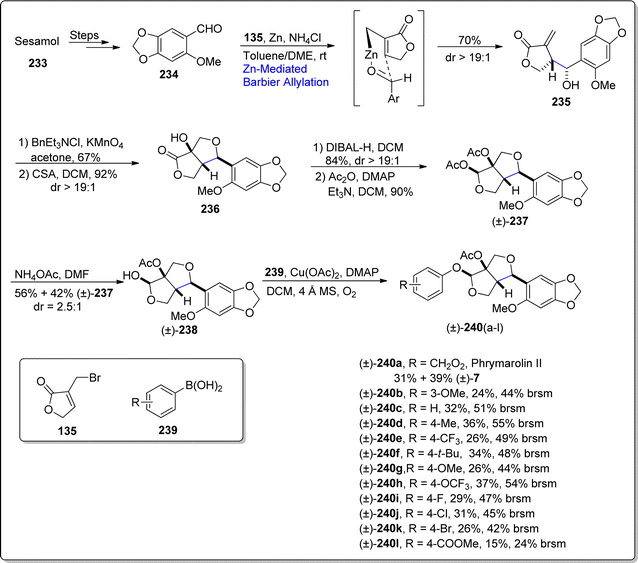 |
| | Scheme 28 Synthesis of phrymarolin II 240 and its various analogues. | |
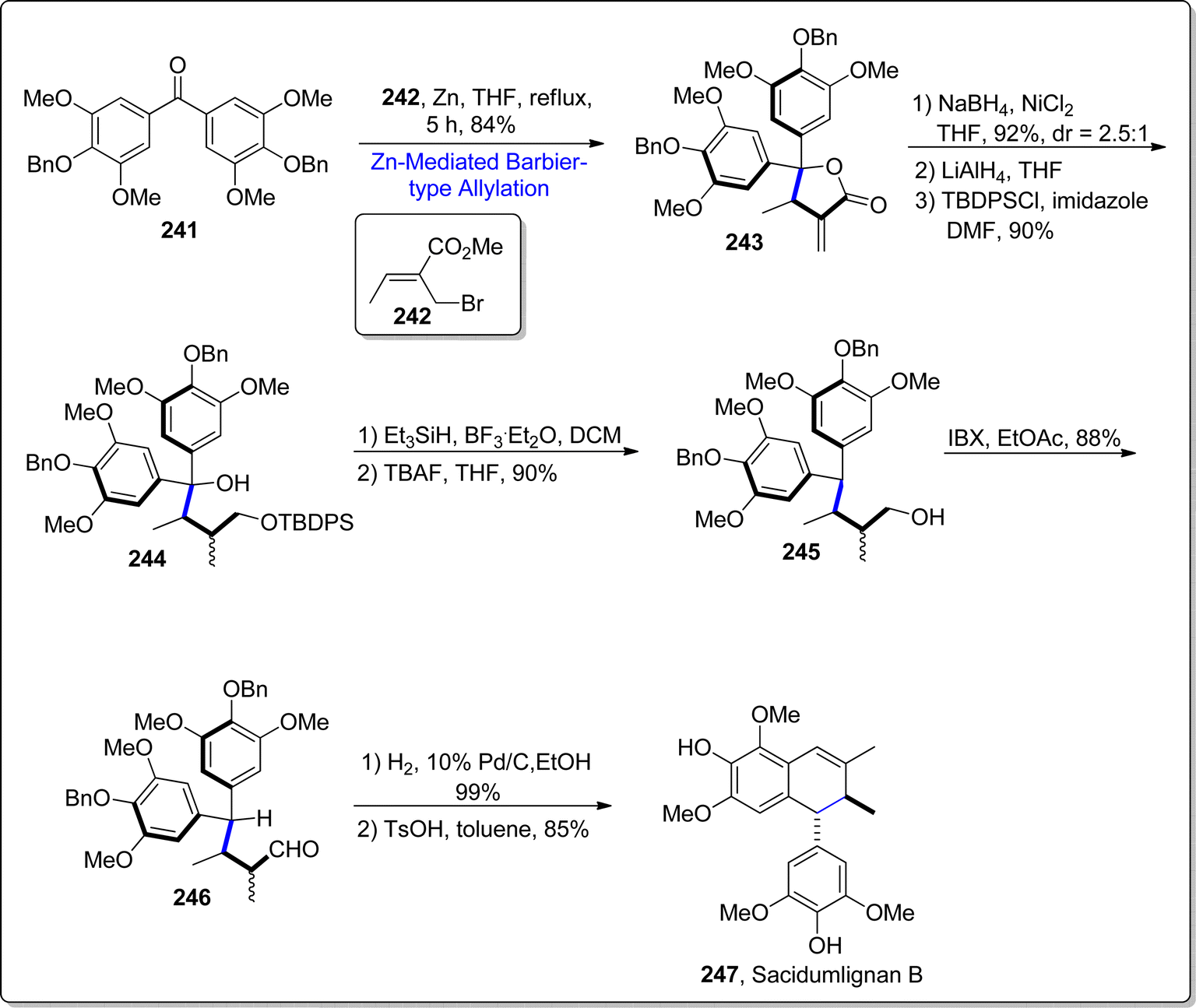
Sacidmulignans A–D are naturally occurring compounds, which have been extracted from the Sarcostemma acidum (Roxb.) located in the China's Hainan island.125 Efforts towards the total synthesis of sacidmulignans A–C (which constitute of 2,7′ cyclolignans structurally) have gained significant momentum in synthetic organic chemistry.126 Attempts towards the synthesis of sacidmulignan B have been reported by Ramana and Peng group individually, which resulted in 10% and 3% overall yield of this natural product respectively.127,128 In 2022, Zhuang et al.129 carried out the first racemic total synthesis of sacidmulignan B by exploiting zinc promoted Barbier-type allylation as a key step. Initially, diaryl ketone 241 was subjected to react with crotyl bromide 242 via zinc mediated Barbier-type allylation reaction in tetrahydrofuran to furnish the lactone 243 in 84% yield. The resulting lactone 243 experienced two reduction reactions consecutively followed by the selective protection of hydroxyl group to acquire monoalcohol 244 in 90% yield. The resulting tertiary alcohol 244 was subjected to Et3SiH and boron trifluoride diethyl ether assisted reduction conditions proceeded by the deprotection conditions leading to the removal of tert-butyldiphenylsilyl group, which afforded primary alcohol 245 (in 90% yield). The exposure of IBX promoted oxidation conditions to generated alcohol 245 gave the corresponding aldehyde 246 in 88% yield. The compound 246 was further subjected to hydrogenation to deprotect the benzyl protected alcoholic functionality proceeded by the p-TsOH mediated (Friedel–Crafts) cyclization, thus procuring sacidmulignan B 247 in 85% yield (Scheme 29).
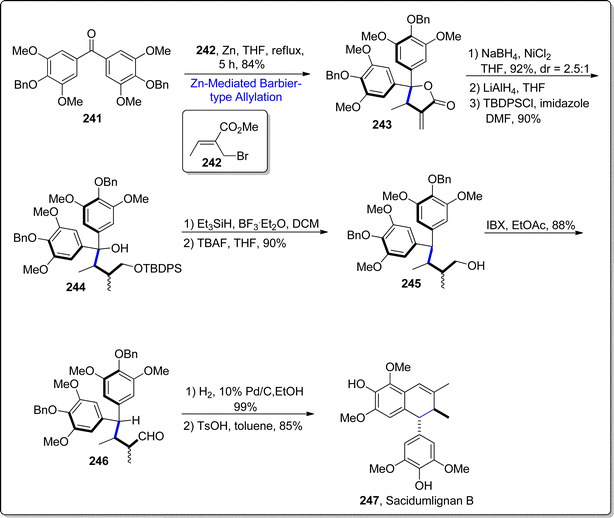 |
| | Scheme 29 Synthesis of sacidmulignan 247. | |
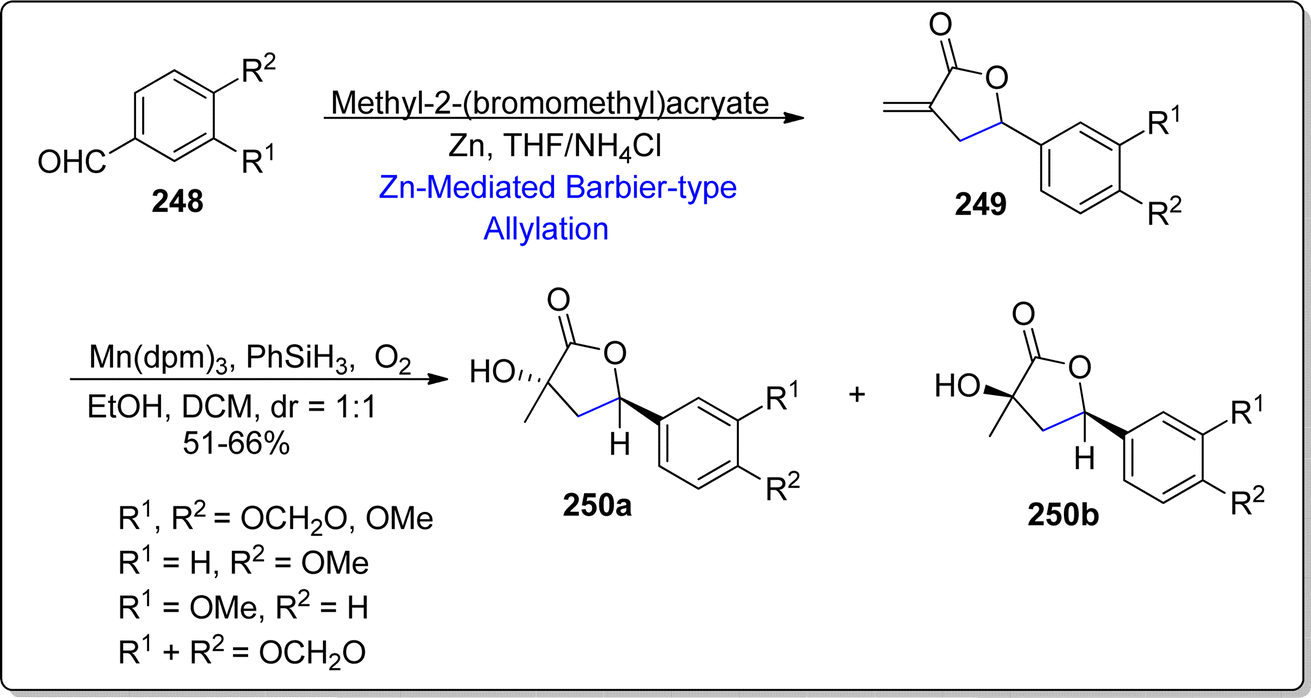
Tobacco mosaic virus is one the highly afflicting disease of plants, which is responsible for the detrimental and hazardous effects on almost 125 plant species.130 Considering the availaibility of very few anti-viral agents such as ningnanmycin, ribavirin and dufulin, there is a constant urge to discover and develop efficient anti-viral compounds. Naturally occurring norlignan i.e., nicotlactone A, which was extracted from Nicotiana tabacum, identified by Hu and colleagues.131 The anti-viral potency of this naturally occurring product was unveiled to be more efficient as compared to commercially utilized standard i.e., ningnanmycin. These results indicate the capability of nicotlactone A as efficacious lead compound against tobacco mosaic virus. In 2022, He et al.132 procured the synthesis of natural product (±)-8-demethylnicotlactone A and its analogues by utilizing zinc mediated Barbier-type allylation reaction. The substituted benzaldehydes 248 were treated with methyl-2-(bromoethyl)acrylate via zinc promoted Barbier-type allylation reaction in tetrahydrofuran and ammonium chloride to afford the corresponding lactones 249. The synthetic pathway was then proceeded by the Mn(dpm)3 promoted hydration of synthesized lactones to attain the diastereomeric mixture of compound 250a & 250b in 1:1 diastereomeric ratio (Scheme 30).
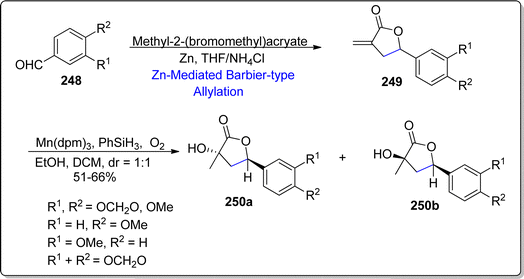 |
| | Scheme 30 Synthesis of (±)-8-demethylnicotlactone 250a and its analogues. | |
2.5. Synthesis of miscellaneous natural products
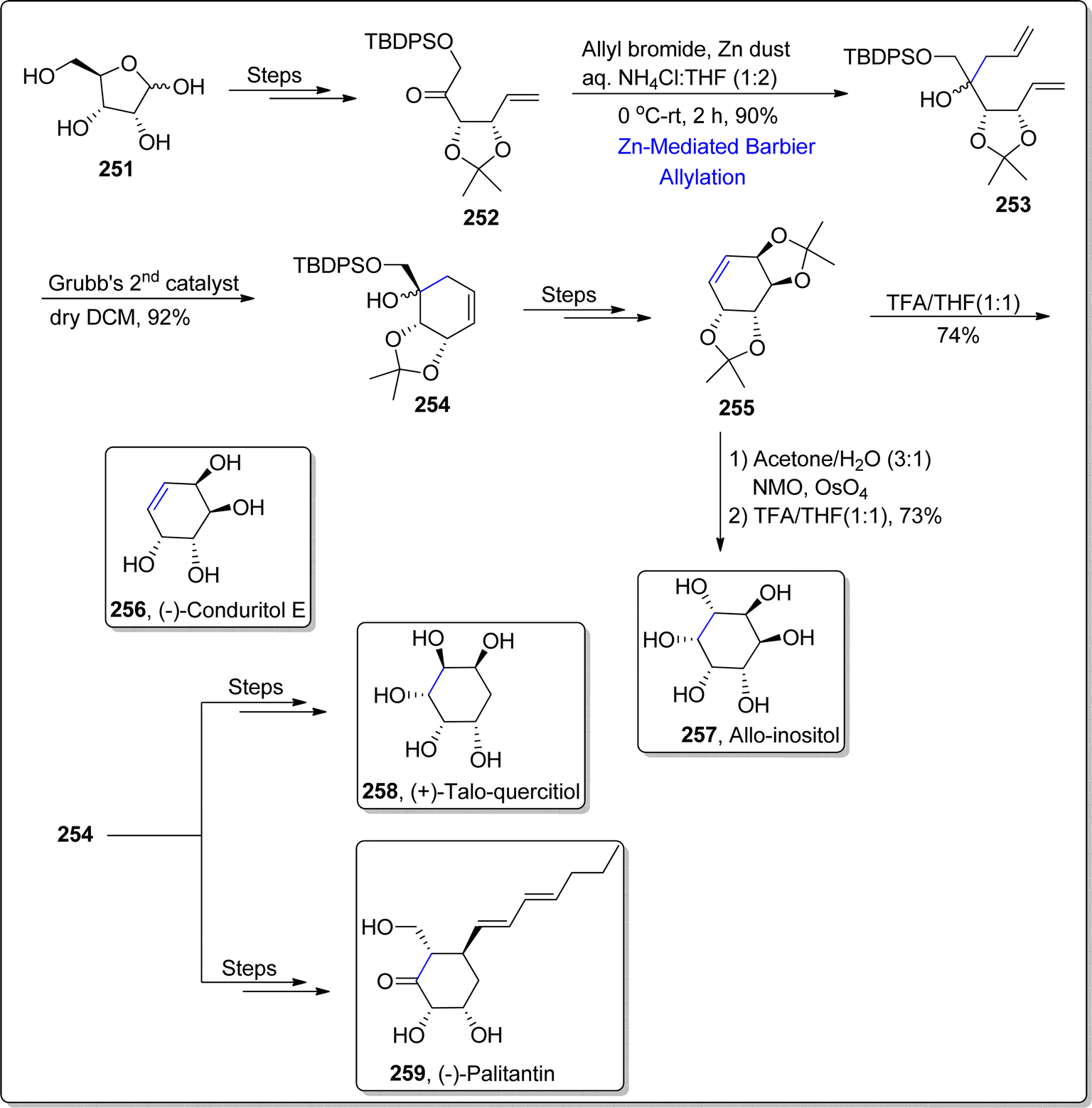
Various natural products can be synthesized in a feasible manner by employing carbohydrates as precursors. Carbohydrates endowed with several hydroxyl group functionalities bear significant weightage owing to their enhanced water solubility and bio-availability. Synthesis of several natural products by employing a common intermediate is an efficient divergent route, exploited widely in past. On the similar ground, Sivakarishna et al.133 in 2022, carried out the total synthesis of various polyhydroxylated natural products by employing an efficient divergent synthetic route. They demonstrated the synthesis of common intermediate by implementing Barbier allylation and ring closing metathesis as major steps. At first, D-Ribose was transformed to ketone 251 over several steps involving synthetic pathway. In the next step, the resulting ketone 252 was made to react with allylbromide via zinc dust promoted Barbier allylation in ammonium chloride:tetrahydrofuran (1:2) at 0 °C to room temperature, thereby furnishing the allylation product i.e., diene 253 in 90% yield. The zinc metal was exploited within Babier reaction considering its inexpensive feature. The diene 253 was further subjected to ring closing metathesis (RCM) in the presence of Grubb's 2nd generation catalyst in dichloromethane to afford cyclohexenyl derivative 254 in (inseparable) diastereomeric mixture (92%). The cyclohexenyl derivative 254 was then utilized as a common intermediate to accomplish the synthesis of various natural products. This common intermediate 254 was treated over several steps to generate alkene 255. The alkene 255 was made to undergo syn-dihydroxylation via NMO and osmium tetroxide followed by subsequent trifluoroacetic acid mediated acid hydrolysis, which ultimately resulted in the synthesis of allo-inositol 257 in 73% yield. On the other hand, conduritol E 256 was easily accessed in 74% yield (overall 10% yield) via acid hydrolysis of alkene 255. The cyclohexenyl derivative 254 was also utilized to attain the asymmetric synthesis of (−)-palitantin 259 and (+)-talo-quercitol 258 (Scheme 31).
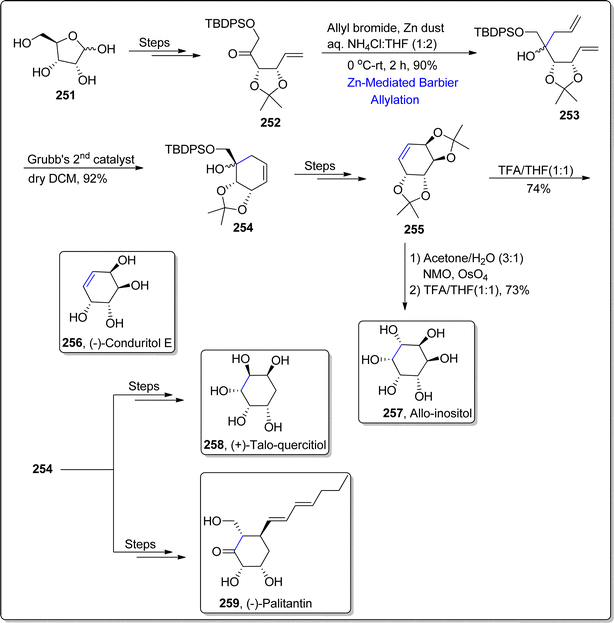 |
| | Scheme 31 Synthesis of (−)-conduritol E 256, allo-inositol 257, (−)-palitantin 259 and (+)-talo-quercitol 258. | |
Azimic acid is a trisubstituted piperidine constituting natural product, extracted from the Azima tetrucuntha Lam. This hydrolyzed form of azimine alkaloid has been observed to manifest promising medicinal applications. In 2023, Gharpure et al.134 developed a novel approach by subjecting enynyl amines to Lewis acid i.e., TMSOTf (5/6-endo-dig) reductive hydroamination in the absence of metal to access piperidne and pyrrolidine based natural products. They conducted the formal synthesis of naturally occurring (+)-azimic acid by employing the beneficial aspects of the developed 5/6-endo-dig reductive hydroamination and zinc mediated Barbier reaction as key steps. In the first step, aldehyde 259 was made to undergo zinc promoted Barbier propargylation reaction with alkynyl bromide 260 in the presence of ammonium chloride and tetrahydrofuran:water to give alkynylamine 261 in 80% yield (dr = 1:1). The alcoholic functionality in compound 261 was then acylated with acetic anhydride followed by the Sonogashira coupling of resulting alkyne with vinyliodide 262 to give enynylamine 263 in 60% yield (syn isomer = 20%). In the next step, generated enynylamine 263 was made to undergo developed reductive hydroamination reaction by using TMSOTf, Et3SiH in DCM to furnish diastereomeric mixture (dr = 1.5:1) of piperidine derivative in 63% yield. Enynylamine 263 was subjected to react with TMSOTf and Et3SiH in dichloromethane followed by potassium hydroxide involving hydrolysis in methanol to furnish piperidine derivative 264. The removal of tosyl group from piperidine derivative 264 was carried out in the presence of magnesium in methanol to result in azimic acid 265 (Scheme 32).
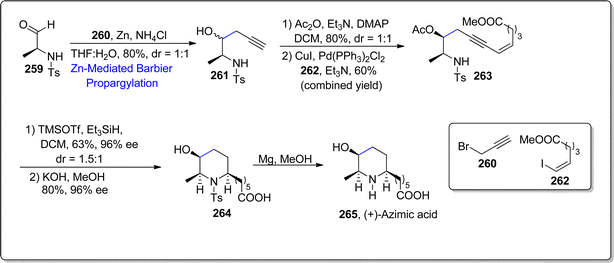 |
| | Scheme 32 Synthesis of (+)-azimic acid 265. | |
Ambergis is a naturally occurring compound, generally produced within the digestive cavity of sperms whales. It is known to constitute (–)-cis-α-ambrinol, which is a naturally occurring product and is highly acclaimed in perfumes industry. In 2023, Martinez et al.135 reported the two pathways for the facile generation of (–)-cis-α-ambrinol 269a (in 35% over all yield) from α-ionone/α-dihydroionone. In pathway A, market-ready α-ionone 266 was transformed to dihydroionone via reduction over raney-nickle, followed by m-CPBA mediated oxidation to afford the epoxide containing oxidized compound 267. Epoxide ring opening reactions result in the synthesis of diverse and useful organic compounds.136–138 The epoxide ring in compound 267 was treated with acid followed by the formation of carbonate on reaction with ethyl chloroformate. The synthesized carbonate 268 was subjected to Cp2TiCl2 mediated intramolecular Barbier allylation reaction to result in cis-α-ambrinol 269a alongwith trans-α-ambrinol 269b and compound 270 (Scheme 33).
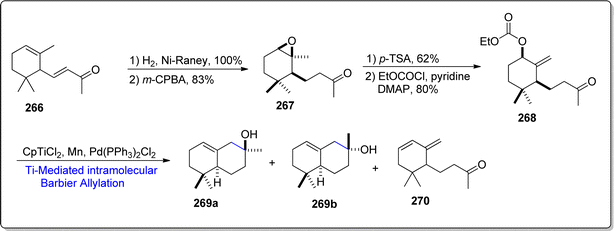 |
| | Scheme 33 Synthesis of (–)-cis-α-Ambrinol 269a. | |
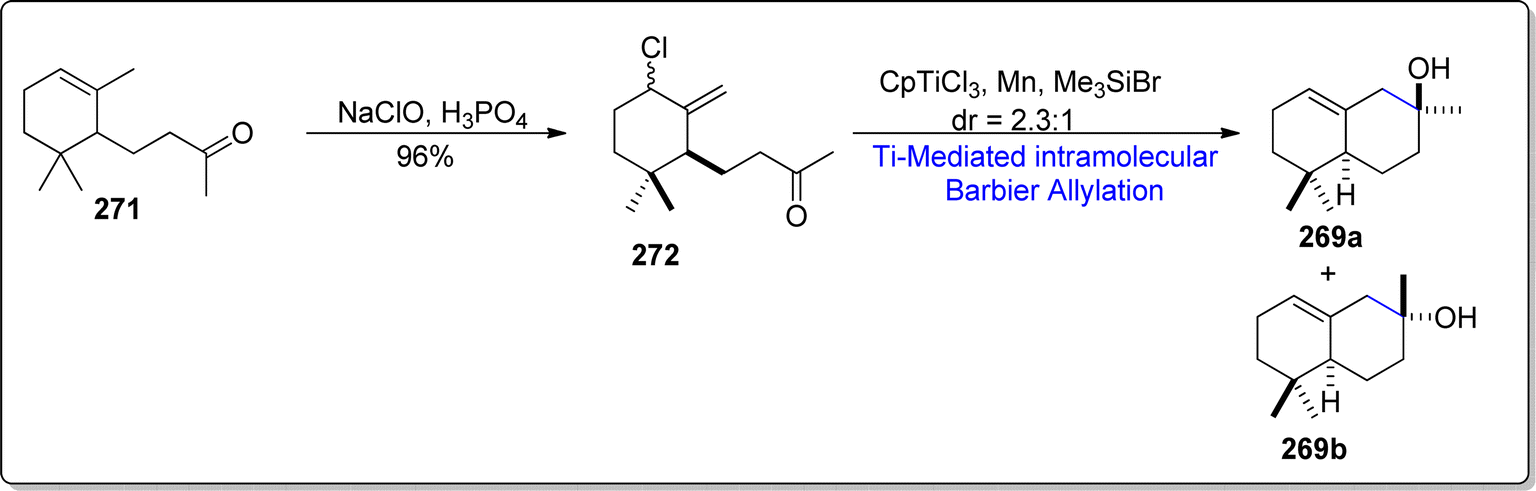
The second synthetic strategy for the synthesis of cis-α-ambrinol 269a involved the chlorination of α-dihydroionone 271 with the addition of sodium perchlorate and phosphoric acid, thereby yielding allylic chloride 272 in 96% yield. The synthesized allylic chloride 272 was then exposed to CpTiCl2 (generated within the reaction as a result of reduction of CpTiCl3 with manganese) promoted intramolecular Barbier allylation in the presence of trimethyl silylbromide to afford the separable diastereomeric mixture of cis-α-ambrinol 269a and trans-α-ambrinol 269b in 2.3:1. (46% overall yield) (Scheme 34).
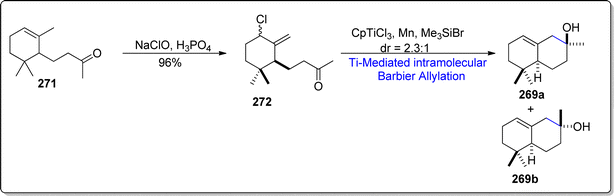 |
| | Scheme 34 Synthesis of (–)-cis-α-ambrinol 269a. | |
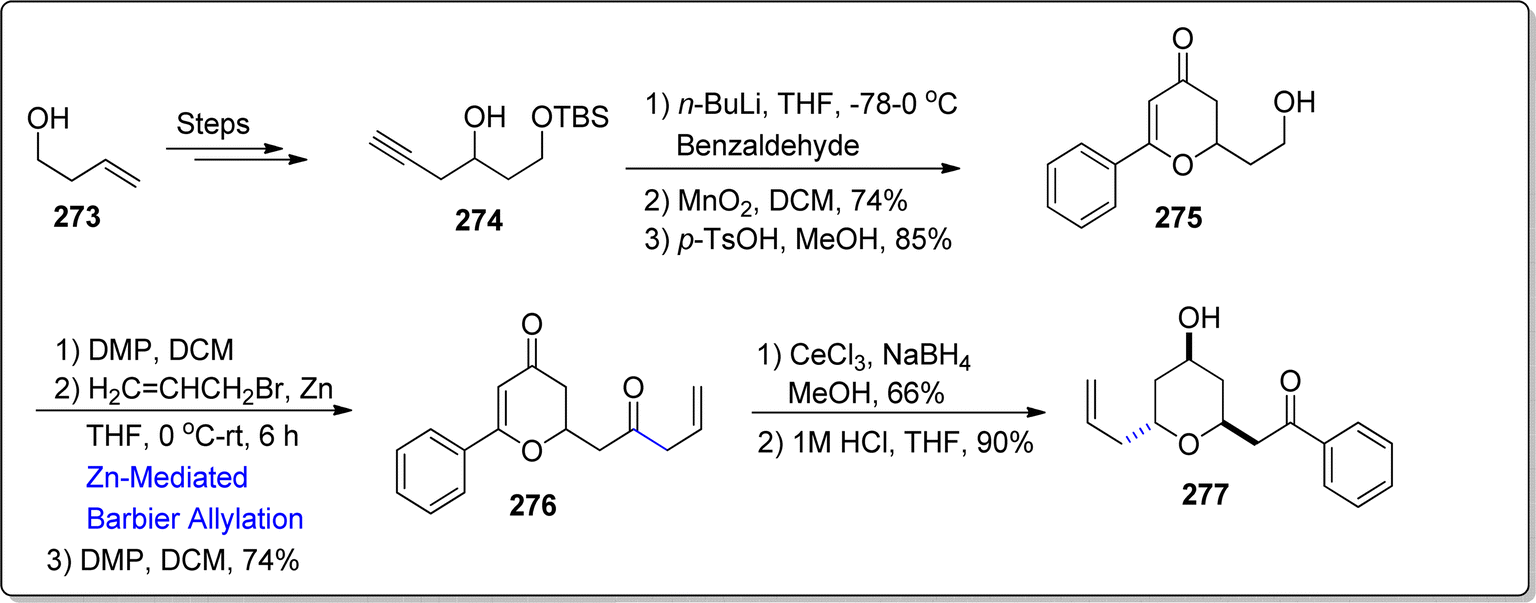
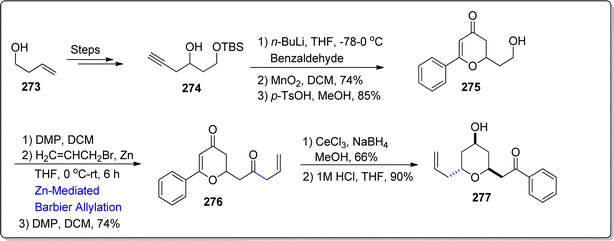
Diospongin B, a disubstituted tetrahydropyran (THP) based natural product, was isolated from Dioscorea spongiosa's rhizomes by Kadota and colleagues.139 It has been known to demonstrate anti-osteoporotic activity. Recently, in 2024, Fernandes et al.140 attempted the synthesis of Diospongin B analogue by utilizing Barbier reaction and Luche reduction as significant steps. Their synthetic route initiated with the preparation of alkynol 274 by treating alkenol 273 over few steps. The synthesized alkynol 274 was then added to 4-chlorobenzaldeyde with subsequent oxidation reaction and deprotection conditions to yield 2,3-dihydro-4H-pyran-4-one 275 in 85% yield. The hydroxyl group in tetrahydropyan compound 275 was subjected to oxidation proceeded by zinc mediated Barbier allylation reaction with allylbromide in tetrahydrofuran to afford diastereomeric mixture of corresponding alcohols. The Barbier product was further oxidized to afford diketone 276 in 74% yield. The diketone 276 was then transformed into diospongin B analogue 277 (in 90% yield) via Luche reduction in sequence with acidic hydrolysis (Scheme 35).
 |
| | Scheme 35 Synthesis of diospongin B analogue 277. | |
3 Conclusion
To conclude, a summary of recent applications of Barbier reaction and its variants to achieve a variety of natural products have been provided in this review. Barbier reaction is one of the renowned C–C bond forming reactions, which possibly results in the formation of optically active center, leading to the stereoselectivity in organic synthesis. This reaction has found several applications in synthetic organic chemistry, most significantly towards the total synthesis of a variety of natural products. In this regard, synthetic layouts of several alkaloids, lactones, terpenoids and lignans etc., harnessing Barbier reaction as one the significant steps, have been overviewed here, documented within 2020–2024. For the synthesis of each natural product, utility of corresponding Barbier methodology has also been highlighted. We envision that this communication will stimulate the synthetic chemists to further delve within the Barbier reaction to unveil its potential beneficiary roles in organic synthesis.
Data availability
No new data has been generated and all data is contained in the manuscript.
Author contributions
A. M. wrote the manuscript. M. N. A., S. G. K., N. A. and U. N. did literature survey and data analysis. A. M., H. A., A. R. C. and A. I. did data analysis and acquired funding. A. F. Z. supervised the project. All authors reviewed and edited the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
A. Irfan extends his appreciation to the Deanship of Research and graduate studies at King Khalid University for funding this work through Large Groups Research Project under grant number (RGP2/146/45). A. R. Chaudhry is thankful to the Deanship of Graduate Studies and Scientific Research at the University of Bisha, for supporting this work through the Fast-Track Research Support Program.
References
- H. Mayr, B. Kempf and A. R. Ofial, π-Nucleophilicity in carbon–carbon bond-forming reactions, Acc. Chem. Res., 2003, 36, 66–77 CrossRef CAS PubMed.
- D. Ravelli, S. Protti and M. Fagnoni, Carbon–carbon bond forming reactions via photogenerated intermediates, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed.
- G. Brahmachari, Design for carbon–carbon bond forming reactions under ambient conditions, RSC Adv., 2016, 6, 64676–64725 RSC.
- V. Nair, S. Vellalath and B. P. Babu, Recent advances in carbon–carbon bond-forming reactions involving homoenolates generated by NHC catalysis, Chem. Soc. Rev., 2008, 37, 2691–2698 RSC.
- X. F. Wu and H. Neumann, Zinc-Catalyzed Organic Synthesis: C-C, C-N, C-O Bond Formation Reactions, Adv. Synth. Catal., 2012, 354, 3141–3160 CrossRef CAS.
- A. Guijarro, D. M. Rosenberg and R. D. Rieke, The reaction of active zinc with organic bromides, J. Am. Chem. Soc., 1999, 121, 4155–4167 CrossRef CAS.
- P. Barbier, Synthèse du dioxyde d'éthylène et de ses dérivés (Synthesis of ethylene oxide and its derivatives), C. R. Hebd. Seances Acad. Sci., 1899, 128, 110–111 CAS.
- V. Grignard, Sur une nouvelle méthode de synthèse d'organomagnésiens" (On a new method of synthesis of organomagnesium compounds), C. R. Hebd. Seances Acad. Sci., 1900, 130, 1322–1324 CAS.
- M. Westerhausen, A. Koch, H. Görls and S. Krieck, Heavy Grignard reagents: synthesis, physical and structural properties, chemical behavior, and reactivity, Chem.–Eur. J., 2017, 23, 1456–1483 CrossRef CAS PubMed.
- M. Haroon, S. Ahmad, A. F. Zahoor, S. Javed, M. N. Ahmad, S. G. Khan, A. A. Al-Mutairi, A. Irfan, S. A. Al-Hussain and M. E. Zaki, Grignard Reaction: An ‘Old-Yet-Gold’synthetic gadget toward the synthesis of natural Products: A review, Arabian J. Chem., 2024, 17, 105715 CrossRef CAS.
- F. Sato, The preparation of Grignard reagents via the hydromagnesation reaction and their uses in organic synthesis, J. Organomet. Chem., 1985, 285, 53–64 CrossRef CAS.
- K. Maruyama and T. Katagiri, Mechanism of the Grignard reaction, J. Phys. Org. Chem., 1989, 2, 205–213 CrossRef CAS.
- R. M. Peltzer, J. Gauss, O. Eisenstein and M. Cascella, The Grignard reaction–unraveling a chemical puzzle, J. Am. Chem. Soc., 2020, 142, 2984–2994 CrossRef CAS PubMed.
- P. Bauer and G. Molle, The barbier reaction: No organometallic pathway in a one-step alternative grignard reaction, Tetrahedron Lett., 1978, 19, 4853–4856 CrossRef.
- R. D. Rieke, Use of activated metals in organic and organometallic synthesis, in Organic Syntheses, Springer Berlin Heidelberg, 2006, pp. 1–31 Search PubMed.
- R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, J. Jiang, S. Maeda, H. Takaya, K. Kubota and H. Ito, Mechanochemical synthesis of magnesium-based carbon nucleophiles in air and their use in organic synthesis, Nat. Commun., 2021, 12, 6691 CrossRef CAS PubMed.
- M. B. Gawande, V. D. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Benign by design: catalyst-free in-water, on-water green chemical methodologies in organic synthesis, Chem. Soc. Rev., 2013, 42, 5522–5551 RSC.
- H. Y. Kang and S. E. Song, Barbier-type reactions of nitriles and alkyl iodides mediated by samarium (II) iodide in the presence of catalytic nickel (II) iodide, Tetrahedron Lett., 2000, 41, 937–939 CrossRef CAS.
- S. H. Kim, H. S. Lee, K. H. Kim and J. N. Kim, An expedient synthesis of poly-substituted 1-arylisoquinolines from δ-ketonitriles via indium-mediated Barbier reaction protocol, Tetrahedron Lett., 2009, 50, 6476–6479 CrossRef CAS.
- A. F. Zahoor and U. Kazmaier, A Straightforward Approach towards Functionalized γ-Hydroxy and Heterocyclic Amino Acids, Synthesis, 2011, 18, 3020–3026 Search PubMed.
- T. P. Loh, J. R. Zhou and Z. Yin, A highly enantioselective indium-mediated allylation reaction of aldehydes, Org. Lett., 1999, 1, 1855–1857 CrossRef CAS.
- Z. Zha, Y. Wang, G. Yang, L. Zhang and Z. Wang, Efficient Barbier reaction of carbonyl compounds improved by a phase transfer catalyst in water, Green Chem., 2002, 4, 578–580 RSC.
- J. W. Lim, K. H. Kim, B. R. Park and J. N. Kim, Facile synthesis of γ-alkenylbutenolides from Baylis–Hillman adducts: consecutive In-mediated Barbier allylation, PCC oxidation, isomerization, and Zn-mediated Barbier allylation, Tetrahedron Lett., 2011, 52, 6545–6549 CrossRef CAS.
- Y. Zhou, Z. Zha, Y. Zhang and Z. Wang, Sn/I2 Mediated allylation of carbonyl compounds with allyl (crotyl) halide in water, Arkivoc, 2008, 11, 142–153 Search PubMed.
- Z. Wang, Z. Zha and C. Zhou, Application of tin and nanometer tin in allylation of carbonyl compounds in tap water, Org. Lett., 2002, 4, 1683–1685 CrossRef CAS PubMed.
- Y. Masuyama, M. Kishida and Y. Kurusu, γ-Syn-selective carbonyl allylation by 1-Bromo-2-butene with tin (II) iodide and tetrabutylammonium bromide, Tetrahedron Lett., 1996, 37, 7103–7106 CrossRef CAS.
- Z. Zha, Z. Xie, C. Zhou, M. Chang and Z. Wang, High regio-and stereoselective Barbier reaction of carbonyl compounds mediated by NaBF 4/Zn (Sn) in water, New J. Chem., 2003, 27, 1297–1300 RSC.
- K. I. Takao, T. Miyashita, N. Akiyama, T. Kurisu, K. Tsunoda and K. I. Tadano, Construction of all-carbon quaternary stereocenters by zinc-mediated barbier-type allylation in aqueous media, Heterocycles, 2012, 86, 147–153 CrossRef CAS PubMed.
- Z. Wang, Z. Zha and C. Zhou, Application of tin and nanometer tin in allylation of carbonyl compounds in tap water, Org. Lett., 2002, 4, 1683–1685 CrossRef CAS PubMed.
- S. Jana, C. Guin and S. C. Roy, Mild and efficient allylation of aldehydes mediated by titanium (III) chloride, Tetrahedron Lett., 2004, 45, 6575–6577 CrossRef CAS.
- S. Wu, Y. Li and S. Zhang, α-Regioselective barbier reaction of carbonyl compounds and allyl halides mediated by praseodymium, J. Org. Chem., 2016, 81, 8070–8076 CrossRef CAS PubMed.
- F. Zhang, R. Wang, S. Wu, P. Wang and S. Zhang, Highly α-regioselective neodymium-mediated allylation of diaryl ketones, RSC Adv., 2016, 6, 87710–87718 RSC.
- L. C. Hirayama, S. Gamsey, D. Knueppel, D. Steiner, K. DeLaTorre and B. Singaram, Indium-mediated Barbier-type allylation of aldehydes as a convenient method for the highly enantioselective synthesis of homoallylic alcohols, Tetrahedron Lett., 2005, 46, 2315–2318 CrossRef CAS.
- S. Nakamura, Y. Hara, T. Furukawa and T. Hirashita, Enantioselective Barbier-type allylation of ketones using allyl halide and indium in water, RSC Adv., 2017, 7, 15582–15585 RSC.
- H. R. Appelt, J. B. Limberger, M. Weber, O. E. Rodrigues, J. S. Oliveira, D. S. Lüdtke and A. L. Braga, Carbohydrates in asymmetric synthesis: enantioselective allylation of aldehydes, Tetrahedron Lett., 2008, 49, 4956–4957 CrossRef CAS.
- X. L. Sun, D. M. Liu, D. Tian, X. Y. Zhang, W. Wu and W. M. Wan, The introduction of the Barbier reaction into polymer chemistry, Nat. Commun., 2017, 8, 1210 CrossRef PubMed.
- S. I. Bhat, One-Pot Construction of Bis-Heterocycles through Isocyanide Based Multicomponent Reactions, ChemistrySelect, 2020, 5, 8040–8061 CrossRef.
- J. Lachheb, Barium Hydroxyapatite Ba10 (PO4) 6 (OH) 2 and Barium Nitrate: New Recyclable Catalysts for the Synthesis of 3, 4-dihydropyrimidin-2 (1H)-ones/thiones, J. Chem. Soc. Pak., 2023, 45, 453 CAS.
- S. ben Moussa, A. Mehri and B. Badraoui, Magnesium modified calcium hydroxyapatite: An efficient and recyclable
catalyst for the one-pot Biginelli condensation, J. Mol. Struct., 2020, 1200, 127111 CrossRef.
- M. K. M. A. Lathiff, R. Suresh, R. Senthamarai, S. Annadurai, R. R. Bhandare and A. B. Shaik, Exploring substituted 3, 4-dihydropyrimidinone and thione derivatives as anti-prostate cancer agents: Computational screening, synthesis, characterization, and in vitro efficacy assessment, J. Saudi Chem. Soc., 2024, 28, 101798 CrossRef.
- V. K. Sharma, A. Barde and S. Rattan, One-pot sequential synthesis of quinazolin-8-ol derivatives employing heterogeneous catalyst for Suzuki–Miyaura coupling, Synth. Commun., 2020, 50, 2962–2968 CrossRef CAS.
- B. B. Toure and D. G. Hall, Natural product synthesis using multicomponent reaction strategies, Chem. Rev., 2009, 109, 4439–4486 CrossRef CAS PubMed.
- S. Munawar, A. F. Zahoor, S. Ali, S. Javed, M. Irfan, A. Irfan, K. Kotwica-Mojzych and M. Mojzych, Mitsunobu reaction: a powerful tool for the synthesis of natural products: a review, Molecules, 2022, 27, 6953 CrossRef CAS PubMed.
- P. Tang and Y. Qin, Recent applications of cyclopropane-based strategies to natural product synthesis, Synthesis, 2012, 44, 2969–2984 CrossRef CAS.
- J. Jang, L. Zhang, H. J. Kim, S. R. Lee and S. H. Kim, Silver (I)-catalyzed hydroamination of (3S, 4R)-4-acetoxy-3-[(R)-1-tert-butyldimethylsiloxy) ethyl] azetidine-2-one derivatives for the synthesis of carbapenem skeleton, Tetrahedron Lett., 2022, 94, 153712 CrossRef CAS.
- Z. Xin, H. Wang, H. He, X. Zhao and S. Gao, Asymmetric total synthesis of norzoanthamine, Angew. Chem., Int. Ed., 2021, 60, 12807–12812 CrossRef CAS PubMed.
- J. G. Hubert, D. P. Furkert and M. A. Brimble, Preparation of cis-γ-Hydroxycarvone Derivatives for Synthesis of Sesterterpenoid Natural Products: Total Synthesis of Phorbin A, J. Org. Chem., 2015, 80, 2231–2239 CrossRef CAS PubMed.
- P. Pal, J. Chakraborty, A. Mali and S. Nanda, Asymmetric total synthesis of paecilomycin F, cochliomycin C, zeaenol, 5-bromo-zeaenol and 3, 5-dibromo-zeaenol by Heck coupling and late stage macrolactonization approach, Tetrahedron, 2016, 72, 2336–2348 CrossRef CAS.
- B. M. Fraga, Natural sesquiterpenoids, Nat. Prod. Rep., 2012, 29, 1334–1366 RSC.
- C. J. Li, Aqueous Barbier-Grignard type reaction: scope, mechanism, and synthetic applications, Tetrahedron, 1996, 52, 5643–5668 CrossRef CAS.
- C. J. Li, Organic reactions in aqueous media with a focus on carbon− carbon bond formations: a decade update, Chem. Rev., 2005, 105, 3095–3166 CrossRef CAS PubMed.
- R. G. Soengas and A. M. Estevez, Applications of Barbier type reactions in carbohydrate chemistry, Curr. Org. Synth., 2013, 10, 183–209 CrossRef CAS.
- E. Erdik and M. Koçoğlu, A brief survey on the copper-catalyzed allylation of alkylzinc and Grignard reagents under Barbier conditions, Appl. Organomet. Chem., 2006, 20, 290–294 CrossRef CAS.
- W. J. Bowyer, B. Singaram and A. M. Sessler, Nature of the intermediates formed during indium mediated allylation under Barbier conditions. Spectroscopic and experimental data on allylindium species, Tetrahedron, 2011, 67, 7449–7460 CrossRef CAS.
- C. Blomberg, The Barbier Reaction and Related One-step Processes, Springer, Verlag Berlin Heidelberg, 2012 Search PubMed.
- Y. J. Chen, L. T. Wu, H. Xiao, X. L. Sun and W. M. Wan, Recent advances and challenges in barbier polymerization, ChemPlusChem, 2023, 88, e202200388, DOI:10.1002/cplu.202200388.
- S. Petrides and S. N. Georgiades, Stereocontrolled Barbier reactions for generation of homoallylic alcohols: New applications in the synthesis of natural products, Trends Org. Chem., 2022, 23, 1–32 Search PubMed.
- J. W. Daly, T. F. Spande and H. M. Garraffo, Alkaloids from amphibian skin: a tabulation of over eight-hundred compounds, J. Nat. Prod., 2005, 68, 1556–1575 CrossRef CAS PubMed.
- N. Toyooka, A. Fukutome, H. Shinoda and H. Nemoto, Total synthesis of the antipode of alkaloid 205 B, Angew. Chem., Int. Ed., 2003, 42, 3808–3810 CrossRef CAS PubMed.
- M. Tripathy and C. Schneider, Short synthesis of alkaloid (−)-205B, J. Org. Chem., 2020, 85, 12724–12730 CrossRef CAS PubMed.
- A. J. Kochanowska-Karamyan and M. T. Hamann, Marine indole alkaloids: potential new drug leads for the control of depression and anxiety, Chem. Rev., 2010,(110), 4489–4497 CrossRef CAS PubMed.
- I. Shahzadi, A. F. Zahoor, B. Tüzün, A. Mansha, M. N. Anjum, A. Rasul, A. Irfan, K. Kotwica-Mojzych and M. Mojzych, Repositioning of acefylline as anti-cancer drug: Synthesis, anticancer and computational studies of azomethines derived from acefylline tethered 4-amino-3-mercapto-1, 2, 4-triazole, PLoS One, 2022, 17, e0278027, DOI:10.1371/journal.pone.0278027.
- R. Akhtar, A. F. Zahoor, A. Rasul, M. Ahmad, M. N. Anjum, M. Ajmal and Z. Raza, Design, synthesis, in-silico study and anticancer potential of novel n-4-piperazinyl-ciprofloxacin-aniline hybrids, Pak. J. Pharm. Sci., 2019, 32, 2215–2222 Search PubMed.
- Z. Zhou, A. X. Gao and S. A. Snyder, Total synthesis of (+)-arborisidine, J. Am. Chem. Soc., 2019, 141, 7715–7720, DOI:10.1021/jacs.9b03248.
- R. Andres, Q. Wang and J. Zhu, Asymmetric total synthesis of (−)-arborisidine and (−)-19-epi-arborisidine enabled by a catalytic enantioselective Pictet–Spengler reaction, J. Am. Chem. Soc., 2020, 142, 14276–14285 CrossRef CAS PubMed.
- F.-Y. Wang and L. Jiao, Total Synthesis of (_)-Arborisidine, Angew. Chem., Int. Ed., 2021, 60, 1–6 CrossRef.
- R. J. Nash, P. I. Thomas, R. D. Waigh, G. W. Fleet, M. R. Wormald, P. M. D. Q. Lilley and D. J. Watkin, Casuarine: a very highly oxygenated pyrrolizidine alkaloid, Tetrahedron Lett., 1994, 35, 7849–7852 CrossRef CAS.
- Y. X. Li, J. Z. Wang, A. Kato, Y. Shimadate, M. Kise, Y. M. Jia, G. W. J. Fleet and C. Y. Yu, Stereocomplementary synthesis of casuarine and its 6-epi-, 7-epi-, and 6, 7-di epi-stereoisomers, Org. Biomol. Chem., 2021, 19, 9410–9420 RSC.
- K. Babar, A. F. Zahoor, S. Ahmad and R. Akhtar, Recent synthetic strategies toward the synthesis of spirocyclic compounds comprising six-membered carbocyclic/heterocyclic ring systems, Mol. Diversity, 2021, 25, 2487–2532 CrossRef CAS PubMed.
- N. R. Ariefta, T. Koseki, Y. Nishikawa and Y. Shiono, Spirocollequins A and B, new alkaloids featuring a spirocyclic isoindolinone core, from Colletotrichum boninense AM-12-2, Tetrahedron Lett., 2021, 64, 152736 CrossRef CAS.
- S. P. Upadhyay, P. Thapa, R. Sharma and M. Sharma, 1-Isoindolinone scaffold-based natural products with a promising diverse bioactivity, Fitoterapia, 2020, 146, 104722 CrossRef CAS PubMed.
- K. Ichikawa, T. Inuzuka, H. Yoda and T. Sengoku, Total synthesis and structural confirmation of (±)-spirocollequins A and B, Tetrahedron Lett., 2022, 107, 154109 CrossRef CAS.
- M. J. Ackland, J. R. Hanson, P. B. Hitchcock and A. H. Ratcliffe, Structures of the cephalosporolides B–F, a group of C 10 lactones from Cephalosporium aphidicola, J. Chem. Soc., Perkin Trans. 1, 1985, 1, 843–847 RSC.
- I. Shiina, An effective method for the synthesis of carboxylic esters and lactones using substituted benzoic anhydrides with Lewis acid catalysts, Tetrahedron, 2004, 60, 1587–1599 CrossRef CAS.
- C. Kalavakuntla, V. B. Kummari and J. S. Yadav, Total synthesis of (–)-cephalosporolide D, Nat. Prod. Res., 2021, 36, 4021–4025 CrossRef PubMed.
- S. M. Siegel, Inhibitory activity of the phenolic glucoside psilotin and its reversal by gibberellic acid and thiols, Phytochemistry, 1976, 15, 566–567 CrossRef CAS.
- J. B. Tunac, B. D. Graham and W. E. Dobson, Novel Antitumor Agents CI-920, PD 113, 270 and PD 113, 271 I. Taxonomy, Fermentation and Biological Properties, J. Antibiot., 1983, 36, 1595–1600 CrossRef CAS PubMed.
- D. Saini, P. Kumar and R. A. Fernandes, Stereoselective total synthesis of obolactones and 7′, 8′-dihydroobolactones, New J. Chem., 2021, 45, 18976–18982 RSC.
- S. Park, J. Lee, J. H. Kim, Y. Jeong, S. Lee, S. W. Lee and S. Kim, Evolution of a Strategy for Concise Enantioselective Total Synthesis of the Salinosporamide Family of Natural Products, Angew. Chem., 2022, 134, e202210317, DOI:10.1002/ange.202210317.
- S. Park, Y. Jeong, C. Lim and S. Kim, First Asymmetric Total Synthesis of Salinosporamides D and I Using Memory of Chirality and Dynamic Kinetic Resolution, Eur. J. Org Chem., 2023, 26, e202300774, DOI:10.1002/ejoc.202300774.
- M. Gayke, H. Narode, R. S. Bhosale and J. S. Yadav, Stereoselective total synthesis of arachnid harvestmen natural product:(4 S, 5 S)-4-hydroxy-γ-decalactone, Nat. Prod. Res., 2024, 38, 1168–1176 CrossRef CAS PubMed.
- Y. Liu, L. H. Rakotondraibe, P. J. Brodie, J. D. Wiley, M. B. Cassera, J. S. Miller, F. Ratovoson, E. Rakotobe, V. E. Rasamison and D. G. I. Kingston, Antimalarial 5, 6-dihydro-α-pyrones from Cryptocarya rigidifolia: related bicyclic tetrahydro-α-pyrones are artifacts1, J. Nat. Prod., 2015, 78, 1330–1338 CrossRef CAS PubMed.
- S. E. Drewes, B. M. Sehlapelo, M. M. Horn, R. Scott-Shaw and P. Sandor, 5, 6-Dihydro-α-pyrones and two bicyclic tetrahydro-α-pyrone derivatives from Cryptocarya latifolia, Phytochemistry, 1995, 38, 1427–1430 CrossRef CAS.
- X. P. Fang, J. E. Anderson, C. J. Chang, P. E. Fanwick and J. L. McLaughlin, Novel bioactive styryl-lactones: goniofufurone, goniopypyrone, and 8-acetylgoniotriol from Goniothalamus giganteus (annonaceae). X-Ray molecular structure of goniofufurone and of goniopypyrone, J. Chem. Soc., Perkin Trans. 1, 1990, 1, 1655–1661 RSC.
- R. A. Fernandes and D. Jangid, Asymmetric Total Synthesis of Cryptorigidifoliol, J. Org. Chem., 2024, 89, 5207–5214 CrossRef CAS PubMed.
- G. Höfle, N. Bedorf, H. Steinmetz, D. Schomburg, K. Gerth and H. Reichenbach, Epothilone A and B—Novel 16-Membered Macrolides with Cytotoxic Activity: Isolation, Crystal Structure, and Conformation in Solution, Angew. Chem., Int. Ed. Engl., 1996, 35(13-14), 1567–1569 CrossRef.
- T. Tsutsumi, M. Matsumoto, H. Iwasaki, K. Tomisawa, K. Komine, H. Fukuda, J. Eustache, R. Janesan and S. H. J. Ishihara, Total Synthesis of Thuggacin cmc-A and Its Structure Determination, Org. Lett., 2021, 23, 5208–5212 CrossRef CAS PubMed.
- M. Stob, R. S. Baldwin, J. Tuite, F. N. Andrews and K. G. Gillette, Isolation of an anabolic, uterotrophic compound from corn infected with Gibberella zeae, Nature, 1962, 196, 1318 CrossRef CAS PubMed.
- M. Maqbool and G. M. Ishaq, Recent promising advances in development of antimicrobial agents: A review, J. Drug Delivery Ther., 2018, 8, 82–86 CrossRef CAS.
- A. F. Zahoor, M. Yousaf, R. Siddique, S. Ahmad, S. A. R. Naqvi and S. M. A, RizviSynthetic strategies toward the synthesis of enoxacin-, levofloxacin-, and gatifloxacin-based compounds: A review, Synth. Commun., 2017, 47, 1021–1039 CrossRef CAS.
- L. Xu, Z. He, J. Xue, X. Chen and X. Wei, β-Resorcylic acid lactones from a Paecilomyces fungus, J. Nat. Prod., 2010, 73, 885–889 CrossRef CAS PubMed.
- R. Gurram and S. Pabbaraja, A Convergent Approach for Protected (+)-Paecilomycin F, Lett. Org. Chem., 2023, 20, 185–192 CrossRef CAS.
- Y. Ren, J. Yu and A. Douglas Kinghorn, Development of anticancer agents from plant-derived sesquiterpene lactones, Curr. Med. Chem., 2016, 23, 2397–2420 CrossRef CAS PubMed.
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Pd metal catalysts for cross-couplings and related reactions in the
21st century: a critical review, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- S. McCarthy, D. C. Braddock and J. D. Wilton-Ely, Strategies for sustainable palladium catalysis, Coord. Chem. Rev., 2021, 442, 213925 CrossRef CAS.
- W. Liu, Z. Yu and N. Winssinger, Total Syntheses of Paraconic Acids and 1, 10-seco-Guaianolides via a Barbier Allylation/Translactonization Cascade of 3-(Bromomethyl)-2 (5 H)-furanone, Org. Lett., 2021, 23, 969–973 CrossRef CAS PubMed.
- T. Nakamura, D. B. Pitna, K. Kimura, Y. Yoshimoto, T. Uchiyama, T. Mori, R. Kondo, S. Hara, Y. Egoshi, S. Yamaguchi, N. Suzuki, Y. Suzuki and T. Usuki, Total synthesis of cynaropicrin, Org. Biomol. Chem., 2021, 19, 6038–6044 RSC.
- Y. L. Ren, J. C. Gallucci, X. X. Li, L. C. Chen, J. H. Yu and A. D. Kinghorn, Crystal Structures and Human Leukemia Cell Apoptosis Inducible Activities of Parthenolide Analogues Isolated from Piptocoma rufescens, J. Nat. Prod., 2018, 81, 554–561 CrossRef CAS PubMed.
- W. Liu, R. Patouret, S. Barluenga, M. Plank, R. Loewith and N. Winssinger, Identification of a covalent importin-5 inhibitor, goyazensolide, from a collective synthesis of furanoheliangolides, ACS Cent. Sci., 2021, 7, 954–962 CrossRef CAS PubMed.
- M. Suchý, V. Benešová, V. Herout and F. Šorm, Contribution on the structure of cnicin, the bitter principle from cnicus benedictus L, Tetrahedron Lett., 1959, 1, 5–9 CrossRef.
- R. G. Kelsey and L. J. Locken, Phytotoxic properties of cnicin, a sesquiterpene lactone from Centaurea maculosa (spotted knapweed), J. Chem. Ecol., 1987, 13, 19–33 CrossRef CAS PubMed.
- K. Kimura and T. Usuki, Synthesis of (1Z)-deacylcnicin, Tetrahedron Lett., 2022, 107, 154102 CrossRef CAS.
- H. M. R. Hoffmann and J. Rabe, Synthesis and biological activity of α-methylene-γ-butyrolactones, Angew. Chem., Int. Ed. Engl., 1985, 24, 94–110 CrossRef.
- B. M. Fraga, Natural sesquiterpenoids, Nat. Prod. Rep., 2010, 27, 1681–1708 RSC.
- R. A. Fernandes and G. V. Ramakrishna, Diastereoselective Allylation in the Divergent Total Syntheses of Guaianolides (+)-Ligustrin and (+)-Grosheimin and the Formal Synthesis of (−)-Eupalinilide E, J. Org. Chem., 2022, 89, 815–824 CrossRef PubMed.
- J. B. He, J. Luo, L. Zhang, Y. M. Yan and Y. X. Cheng, Sesquiterpenoids with new carbon skeletons from the resin of Toxicodendron vernicifluum as new types of extracellular matrix inhibitors, Org. Lett., 2013, 15, 3602–3605 CrossRef CAS PubMed.
- X. L. Qin, G. J. Wu and F. S. Han, Enantioselective Total Synthesis and Absolute Configuration Assignment of (+)-Toxicodenane A, Org. Lett., 2021, 23, 8570–8574 CrossRef CAS PubMed.
- K. Nishikawa, K. Kikuta, T. Tsuruta, H. Nakatsukasa, S. Sugahara, S. Kume and Y. Morimoto, Asymmetric Total Synthesis of Toxicodenane A by Samarium-Iodide-Induced Barbier-Type Cyclization and Its Cell-Protective Effect against Lipotoxicity, Org. Lett., 2022, 24, 531–535 CrossRef CAS PubMed.
- J. Degenhardt, T. G. Köllner and J. Gershenzon, Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants, Phytochemistry, 2009, 70, 1621–1637 CrossRef CAS PubMed.
- M. Kubo, Y. Nishikawa, K. Harada, M. Oda, J.-M. Huang, H. Domon, Y. Terao and Y. Fukuyama, Tetranorsesquiterpenoids and Santalane-Type Sesquiterpenoids from Illicium lanceolatum and Their Antimicrobial Activity against the Oral Pathogen Porphyromonas gingivalis, J. Nat. Prod., 2015, 78, 1466–1469 CrossRef CAS PubMed.
- R. K. Acharyya and S. Nanda, Asymmetric total synthesis of naturally occurring spirocyclic tetranorsesquiterpenoid lanceolactone A, Org. Biomol. Chem., 2018, 16, 5027–5035 RSC.
- B. R. Borade and R. Kontham, Concise total synthesis of (+)-lanceolactone A: revision
of absolute stereochemistry, J. Org. Chem., 2022, 87, 12867–12876 CrossRef CAS PubMed.
- L. M. Petersen, C. Hoeck, J. C. Frisvad, C. H. Gotfredsen and T. O. Larsen, Dereplication guided discovery of secondary metabolites of mixed biosynthetic origin from Aspergillus aculeatus, Molecules, 2014, 19, 10898–10921 CrossRef PubMed.
- F. D. Kong, L. M. Zhou, Q. Y. Ma, S. Z. Huang, P. Wang, H. F. Dai and Y. X. Zhao, Metabolites with Gram-negative bacteria quorum sensing inhibitory activity from the marine animal endogenic fungus Penicillium sp. SCS-KFD08, Arch. Pharmacal Res., 2017, 40, 25–31 CrossRef CAS PubMed.
- H. Yokokawa, S. Ishizawa, K. Saito, Y. Meguro, S. Kuwahara and M. Enomoto, Total synthesis of aculenes B and D, Eur. J. Org Chem., 2023, 26, e202201482, DOI:10.1002/ejoc.202201482.
- A. S. R. Anjaneyulu, M. J. R. V. Venugopal, P. Sarada, J. Clardy and E. Lobkovsky, Havellockate, A Novel Seco and Spiro Lactone Diterpenoid from the Indian Ocean Soft Coral Sinularia granosa, Tetrahedron Lett., 1998, 39, 139–142 CrossRef CAS.
- N. J. Hafeman, M. Chan, T. J. Fulton, E. J. Alexy, S. A. Loskot, S. C. Virgil and B. M. Stoltz, Asymmetric total synthesis of havellockate, J. Am. Chem. Soc., 2022, 144, 20232–20236 CrossRef CAS PubMed.
- A. Kotlyarov, A. Neininger, C. Schubert, R. Eckert, C. Birchmeier, H. D. Volk and M. Gaestel, MAPKAP kinase 2 is essential for LPS-induced TNF-α biosynthesis, Nat. Cell Biol., 1999, 1, 94–97 CrossRef CAS PubMed.
- J. Rosales, G. Cabrera and J. Justicia, Exploring Short and Efficient Synthetic Routes Using Titanocene (III)-Catalyzed Reactions: Total Synthesis of Natural Meroterpenes with Trisubstituted Unsaturations, Molecules, 2022, 27, 2400 CrossRef CAS PubMed.
- H. D. Sun, S. X. Huang and Q. B. Han, Diterpenoids from Isodon species and their biological activities, Nat. Prod. Rep., 2006, 23, 673–698 RSC.
- H. Oikawa, K. Nakamura, H. Toshima, T. Toyomasu and T. Sassa, Proposed mechanism for the reaction catalyzed by a diterpene cyclase, aphidicolan-16β-ol synthase: experimental results on biomimetic cyclization and examination of the cyclization pathway by ab initio calculations, J. Am. Chem. Soc., 2002, 124, 9145–9153 CrossRef CAS PubMed.
- X. H. Zhao, L. L. Meng, X. T. Liu, P. F. Shu, C. Yuan, X.-T. An, T.-X. Jia, Q.-Q. Yang, X. Zhen and C. A. Fan, Asymmetric divergent synthesis of ent-kaurane-, ent-atisane-, ent-beyerane-, ent-trachylobane-, and ent-gibberellane-type diterpenoids, J. Am. Chem. Soc., 2022, 145, 311–321 CrossRef PubMed.
- I. K. Park, S. C. Shin, C. S. Kim, H. J. Lee, W. S. Choi and Y. J. Ahn, Larvicidal activity of lignans identified in Phryma leptostachya Var. asiatica roots against three mosquito species, J. Agric. Food Chem., 2005, 53, 969–972 CrossRef CAS PubMed.
- Y. Chi, H. Zhou, H. W. He, Y. D. Ma, B. Li, D. Xu, J.-M. Gao and G. Xu, Total synthesis and anti-tobacco mosaic virus activity of the furofuran lignan (±)-phrymarolin II and its analogues, J. Nat. Prod., 2021, 84, 2937–2944 CrossRef CAS PubMed.
- L. S. Gan, S. P. Yang, C. Q. Fan and J. M. Yue, Lignans and Their Degraded Derivatives from Sarcostemma a cidum, J. Nat. Prod., 2005, 68, 221–225 CrossRef CAS PubMed.
- H. Q. Zhang, C. X. Yan, J. Xiao, Y. W. Wang and Y. Peng, Recent advances in the total synthesis of 2, 7′-cyclolignans, Org. Biomol. Chem., 2022, 20, 1623–1636 RSC.
- J. K. Rout and C. V. Ramana, Total synthesis of (−)-sacidumlignans B and D, J. Org. Chem., 2012, 77, 1566–1571 CrossRef CAS PubMed.
- J.-J. Zhang, C.-S. Yan, Y. Peng, Z.-B. Luo, X.-B. Xu and Y.-W. Wang, Total synthesis of (±)-sacidumlignans D and A through Ueno–Stork radical cyclization reaction, Org. Biomol. Chem., 2013, 11, 2498–2513 RSC.
- Z. Zhuang, Z. Luo, S. Yao, Y. Wang and Y. Peng, A concise synthesis of sacidumlignan B, Molecules, 2022, 27, 5775 CrossRef CAS PubMed.
- W. Islam, M. Qasim, N. Ali, M. Tayyab, S. Chen and L. Wang, Management of tobacco mosaic virus through natural metabolites, Rec. Nat. Prod., 2018, 12, 403 CrossRef CAS.
- X. M. Gao, X. S. Li, X. Z. Yang, H. X. Mu, Y. K. Chen, G. Y. Yang and Q. F. Hu, Lignan derivatives from the leaves Nicotiana tabacum and their activities, Heterocycles, 2012, 85, 147–153 CrossRef CAS.
- H. W. He, Y. Chi, C. Y. Chen, F. Y. Wang, J. X. Wang, D. Xu, H. Zhou and G. Xu, Synthesis and structure–activity relationship studies of nicotlactone analogues as Anti-TMV agents, Synthesis, 2022, 54, 3642–3650 CrossRef CAS.
- B. Sivakrishna, S. Sahoo, A. Kumar and S. Pal, Development of a Divergent Synthetic Avenue towards Conduritol-E, allo-Inositol, talo-Quercitol and Palitantin from D-Ribose, ChemistrySelect, 2022, 7, e202203346, DOI:10.1002/slct.202203346.
- S. J. Gharpure, R. K. Patel and K. S. Gupta, Total Synthesis of Pyrrolidine and Piperidine Natural Products via TMSOTf-Mediated “5/6-endo-dig” Reductive Hydroamination of Enynyl Amines, Org. Lett., 2023, 25, 5850–5855 CrossRef CAS PubMed.
- J. L. López-Martínez, I. Torres-García, I. Moreno-Gutiérrez, P. Oña-Burgos, A. Rosales Martínez, M. Muñoz-Dorado, M. Álvarez-Corral and I. Rodríguez-García, A Concise Diastereoselective Total Synthesis of α-Ambrinol, Mar. Drugs, 2023, 21, 230 CrossRef PubMed.
- M. Thirumalaikumar, Ring opening reactions of epoxides. A review, Org. Prep. Proced. Int., 2022, 54, 1–39 CrossRef CAS.
- S. Faiz and A. F. Zahoor, Ring opening of epoxides with C-nucleophiles, Mol. Diversity, 2016, 20, 969–987 CrossRef CAS PubMed.
- S. Ahmad, A. F. Zahoor, S. A. R. Naqvi and M. Akash, Recent trends in ring opening of epoxides with sulfur nucleophiles, Mol. Diversity, 2018, 22, 191–205 CrossRef CAS PubMed.
- J. Yin, K. Kouda, Y. Tezuka, Q. Le Tran, T. Miyahara, Y. Chen and S. Kadota, New diarylheptanoids from the rhizomes of Dioscorea spongiosa and their antiosteoporotic activity, Planta Med., 2004, 70, 54–58 CrossRef CAS PubMed.
- R. A. Fernandes, D. Jangid and D. A. Gorve, Unanticipated Bicyclic Etherification in Luche Reduction: Asymmetric Total Syntheses of Diospongins A and B and Tetraketide, Org. Lett., 2024, 26, 433–437 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Mirza Nadeem Ahmadb,
Samreen Gul Khana,
Naheed Akhterc,
Usman Nazeerd,
Asim Manshaa,
Hamad Ahmade,
Aijaz Rasool Chaudhryf and
Ahmad Irfang
*a,
Mirza Nadeem Ahmadb,
Samreen Gul Khana,
Naheed Akhterc,
Usman Nazeerd,
Asim Manshaa,
Hamad Ahmade,
Aijaz Rasool Chaudhryf and
Ahmad Irfang