
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimised syntheses and purifications of 3-aryl/heterocyclic dihydrobis- and hydrotris-(pyrazolyl)borate ligands as their alkali salts†
Jarrod R. Thomas ,
Jonathan T. Mifsud and
Scott A. Sulway*
,
Jonathan T. Mifsud and
Scott A. Sulway*
School of Chemistry, The University of New South Wales (UNSW), Kensington, Sydney, 2052, Australia. E-mail: s.sulway@unsw.edu.au
First published on 26th September 2024
Abstract
Though poly(pyrazolyl)borate ligands, namely dihydrobis(pyrazolyl)borate (Bp) and hydrotris(pyrazolyl)borate (Tp), have been used in coordination chemistry for decades, their synthesis and purification are of great importance, when targeting high purity and yield of metal complexes. As borohydride substitutions is temperature and pyrazolyl dependent, determining the reaction conditions for each ligand is non-trivial and does not always result in a temperature where only the desired products form. Herein we report the purification of two known TpR (R = phenyl, 2′-thienyl), as their tris-substituted temperatures coincide with their tetrakis-temperatures via fraction crystallisation from MeCN, and an optimised safe procedure for 3-aryl/heterocyclic BpR ligands (R = 2′-pyridyl, 2′-furyl, 2′-thineyl). These novel techniques allow for the safe isolation of alkali salts of poly(pyrazolyl)borate ligands in high yields and purities without the use of highly toxic thallium(I) salts.
Introduction
Dihydrobis(pyrazolyl)borate (Bp) and hydrotris(pyrazolyl)borate (Tp, known as scorpionate) ligands were first introduced in the late 1960's by Trofimenko and featured a new synthetic route yielding a B–N bond.1 Since their introduction these poly(pyrazolyl)borate ligands have been used extensively throughout coordination chemistry. First generation poly(pyrazolyl)borate featured small alkyl and/or halide substituents on the heterocyclic pyrazolyl groups in the 3rd, 4th and 5th positions.1c Coordination of first generation scorpionates to transition metals and lanthanides resulted in the formation of bis-capped and tris-capped metal complexes, of general form [M(TpR)2]1b,c and [Ln(Tp)3],2 respectively, regardless of stoichiometry due to high formation constants of the final complexes.1b,c To synthesis mono-scorpionate metal complexes the use of bulkier substituents were utilized, with first examples of such complexes including [Co(TptBu)(SCN)] (TptBu = hydrotris(3-tert-buytlpyrazol-1-yl)borate) and [Co(TpPh)(SCN)(THF)] (TpPh = hydrotris(3-phenylpyrazol-1-yl)borate).3 Fine tuning of scorpionate ligands has since become a vast section of coordination chemistry and thus optimising the synthesis of these ligands is of utmost importance. Dihydrobis(pyrazolyl)borate ligands were initially coordinated in a similar matter, forming complexes of the generally form [M(Bp)2],1 before pyrazole substitution were used to synthesis a range of different metal complexes.3The synthesis of poly(pyrazolyl)borate ligands has remained relatively unchanged, following the Trofimenko procedure which entails heating pyrazolyl reagents in the presence of alkali, or highly toxic thallium(I), borohydrides (Scheme 1).1,3–6 Post transmetalation of alkali to thallium(I) salts is another synthetic method used largely for purification of alkali poly(pyrazolyl)borate and the solubility of thallium(I) based scorpionates in organic solvents.5 For pyrazolyl substituents with high steric demand some of the syntheses of scorpionate ligands moved to solution-based procedures, using high boiling point solvents, reacting said pyrazole with previously prepared dihydrobis(pyrazolyl)borate.3 The two-step processes can seem tedious and for the most part results in low yields of desired products. Recent reporting of the previously inaccessible poly(pyrazolyl)borates has been achieved by in situ reaction of sodium pyrazolylides with dichloroborane dimethylsulfide, though isolation of the sodium salts is far from optimised, and instead, transmetalate to the thallium(I) salts for purification.6
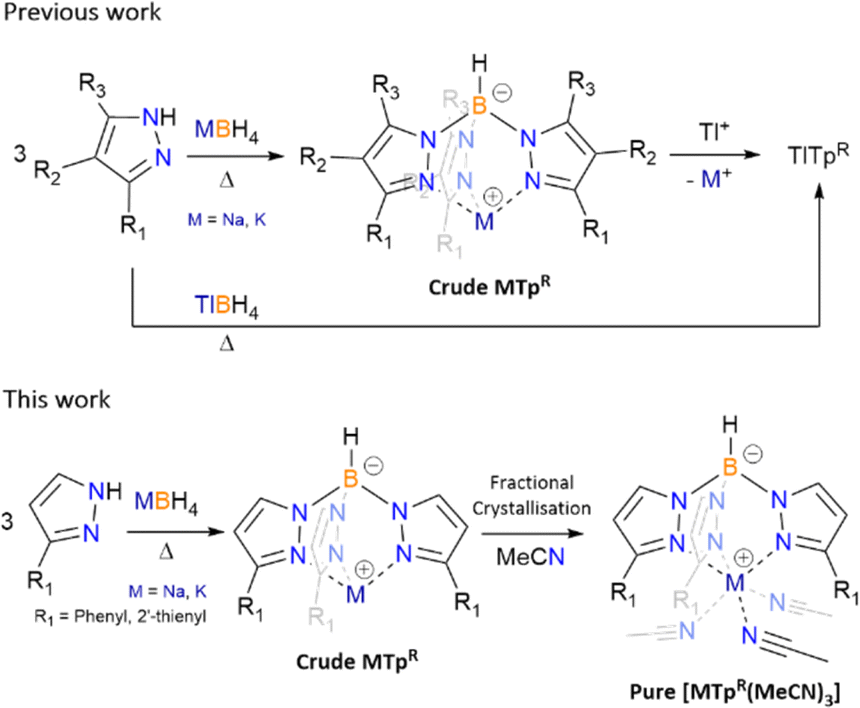 | ||
| Scheme 1 Previous synthesis of scorpionate ligands and new fractional crystallisation of acetonitrile (MeCN) adducts for TpPh (R1 = phenyl) and Tp2-Fu (R1 = 2′-thienyl). | ||
Due to safety concerns, we restrict the use of thallium(I) salts in our laboratory, thus we targeted alkali salt poly(pyrazolyl)borates as they can be considered a readily safe and useable source of the ligands, and we would also assume the same is true for many other groups. Thus the synthetic routes that allow for the isolation of poly(pyrazolyl)borate ligands, in high purities and moderate yields, is important for further (salt) metathesis reactions. From our previous work, the synthesis of scorpionates that employ 3-R substituted pyrazoles, e.g. hydrotris(3-(2′-pyridyl)-pyrazol-1-yl)borate (Tp2-py) and hydrotris(3-(2′-furyl)-pyrazol-1-yl)borate (Tp2-Fu), as both their sodium and potassium salts can be readily achieved using Trofimenko's procedure (melts at ca. 200 °C for 20 hours with a slight excess of pyrazole), resulting in high yields and purity of desired tris-products.7–9 However, not all pyrazole derivatives are made alike, likely owing to their pKa's, as reagents such as 3-phenylpyrazole and 3-(2′-thienyl)-pyrazole do not have optimal temperatures where tris-products exclusively form. From initial syntheses, which are discussed vide infra and as reported previously with KTpPh,7 once a certain temperature is met the formation of both tris- and tetrakis-products occurs for the aforementioned pyrazoles. Herein we discuss a new purification technique for scorpionates that encounter oversubstituted contaminates through the isolation of [M(TpR)(MeCN)3] (1-Na, M = Na, R = phenyl; 2-Na, M = Na, R = 2′-thienyl (Tp2-Th = hydrotris(3-(2′-thienyl)-pyrazol-1-yl)borate); 2-K, M = K, R = 2′-thienyl), and previous isolation of [K(TpPh)(MeCN)3] (1-K),7 via fractional crystallisation from MeCN.
The same temperature dilemma occurs during the synthesis of dihydrobis(pyrazolyl)borate ligands. Optimal temperatures for the synthesis of Bp ligands using 3-R substituted pyrazolyl reagents employing Trofimenko's method can result in the isolation of crude Bp salts that are contaminated with tris-, and in some cases, tetrakis-products. The use of these crude products can result in impure products and unwanted side-reactions in subsequent metathetical reactions. Thus, an alternate method for the synthesis of pure MBpR (3-K, M = K, R = 2′-pyridyl (Bp2-py = dihydrobis(3-(2′-pyridyl)-pyrazol-1-yl)borate); 3-Na, M = Na, R = 2′-pyridyl; 3-Li, M = Li, R = 2′-pyridyl; 4, M = K, R = 2′-furyl (Bp2-Fu = dihydrobis(3-(2′-furyl)-pyrazol-1-yl)borate); 5, M = K, R = 2′-thienyl (Bp2-Th = dihydrobis(3-(2′-thienyl)-pyrazol-1-yl)borate)) is illustrated, without the use of purification by thallium(I) abstraction.6
Results and discussion
Synthesis and spectroscopic characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :tetrakis and NMR Data for 1-Na, 1-K, 2-Na and 2-K
:tetrakis and NMR Data for 1-Na, 1-K, 2-Na and 2-K
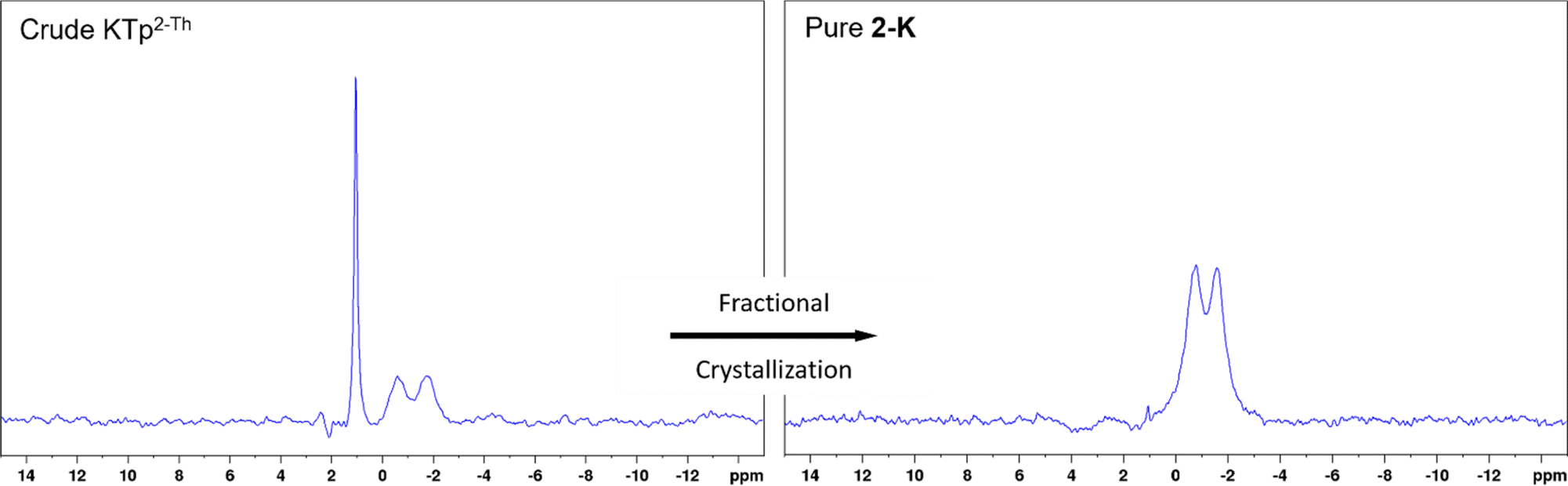 | ||
| Fig. 1 11B NMR spectra of the crude and purified product 2-K where pre-purification the presence of tris- and tetrakis-products is observed but post fractional crystallisation the desired tris-product is solely observed. Baseline corrections have been made to remove undesired borosilicate peak from glassware. | ||
Once crude material is isolate by the parent procedure, NMR spectroscopy can confirm the presence of described products (Fig. 1, ESI Fig. S1–S3†). Many attempts were made to purify the crude material by column chromatography, but no separation was observed between tris- and tetrakis-products. In trial reactions with crude KTp2-Th, it was discovered that 2-K can be fractionally crystallised from MeCN as single crystal X-ray diffraction analysis on these repeated reaction resulted in the repeated isolation of 2-K. Thus for reactions producing NaTpPh, KTpPh, NaTp2-Th and KTp2-Th, fractional crystallisations were performed with these crude scorpionates from MeCN yielding crystals of 1-Na, 1-K, 2-Na and 2-K, respectively, over a 2 day period. Yields of the pure MeCN adducts are between 40–60% (from metal borohydride), which are slightly lower than other scorpionates where over substitution is not a problem. Both 1H and 11B NMR spectra on 1-Na, 1-K, 2-Na and 2-K show only one borate species with the correct multiplicity (Table 1, Fig. 1 and ESI Fig. S4–S7†), and the addition of an MeCN peak at the correct chemical shift and integration. Identification of νB–H and νC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N is visible in FTIR spectra for the above compounds alongside microanalyses reflecting the correct compositions of tris-MeCN adducts.
N is visible in FTIR spectra for the above compounds alongside microanalyses reflecting the correct compositions of tris-MeCN adducts.
We feel that this new purification technique is highly important for further transmetalation reaction incorporating these ligands, or any non-trivial scorpionate, to increase yields and purity of metal complexes. It has been stated previously by others that scorpionates can be hard to purify but given their high denticity they will form the desired complexes when crude material is used.10 We have had similar purification issues particularly when optimal temperatures cannot be found for purely tris-borate formation. Thus the clean and efficient synthesis of 1-M and 2-M present two cases where scorpionate synthesis is not selective but purification is possible.
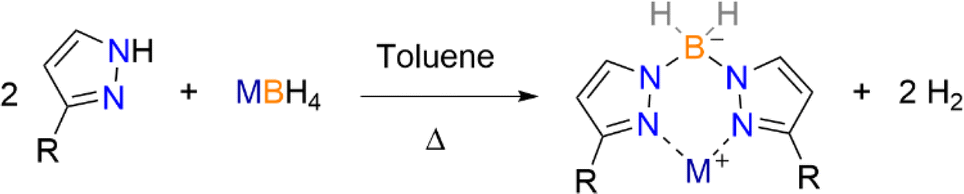 | ||
| Scheme 2 Synthetic route to 3-M (M = Li, Na, K), 4 and 5. | ||
It is worth noting that attempting the same reactions with other alkali borohydrides, namely sodium and lithium, did not yield the same single isolation of preferred bis-products for most pyrazole reagents used herein. The single case that resulted in the isolation of bis-products for other alkali metals, in moderate yields, was with 3-(2′-pyridyl)-pyrazole yielding LiBp2-py (3-Li) and NaBp2-py (3-Na). There were subtle differences in the workups for said reactions compared to the potassium salts as these Bp salts are soluble in toluene; a reduction of the solvent and washing with Et2O to remove unreacted pyrazole reagents yields desired bis-products in moderate to high yields (Table 2, ESI Fig. S8 and S9†). Both 3-Li and 3-Na were recrystallized by slow diffusion of Et2O from CH2Cl2, which results in the formation of the dimeric species [M(μ-Bp2-py)]2 (3-Li2, M = Li; 3-Na2, M = Na), which have been analysed vide infra. These recrystallizations must be performed in anhydrous conditions as the use of ‘wet’ solvents results in the isolation of the water bridged dimers [{M(μ-Bp2-py)}2(μ-OH2)] (see Fig. S29† showcasing the solid-state structure for when M = Na).
Solid-state characterisation
Single crystal X-ray diffraction was performed on the fractionally crystallised 1-Na, 2-Na, 2-K and optimised toluene refluxed products 3-Li2, 3-Na2 and 4 (X-ray crystallographic data are summarised in Table S1,† molecular structures of these compounds are shown in Fig. 2 and 3 and S25,† asymmetric units are shown within the ESI Fig. S22–S27†), where diffractions studies on 1-K have been previously reported.7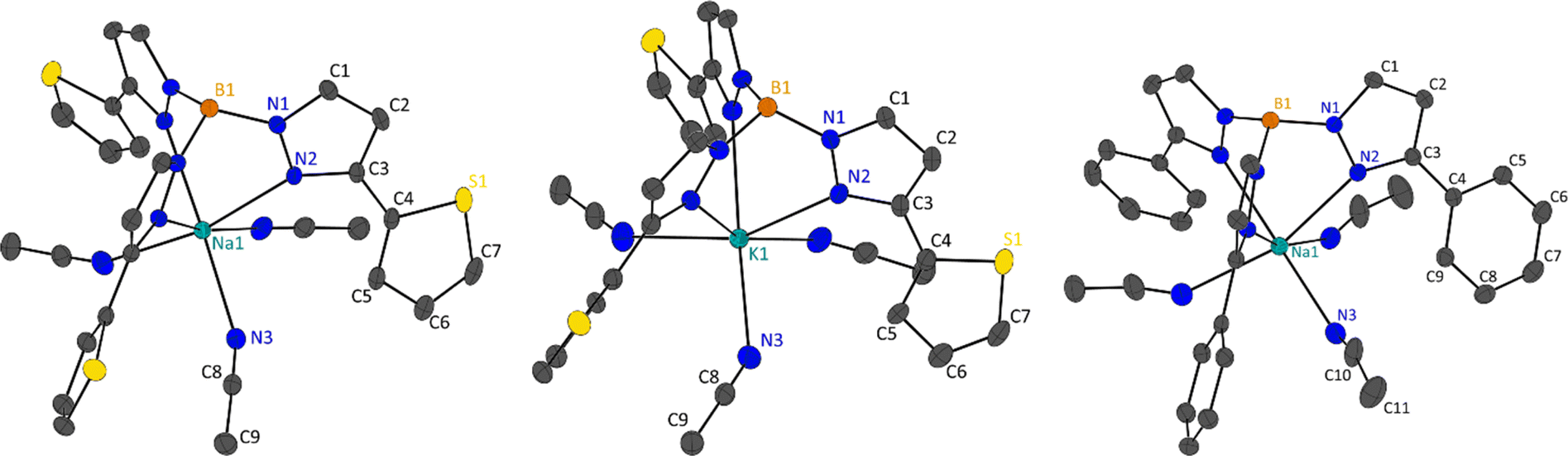 | ||
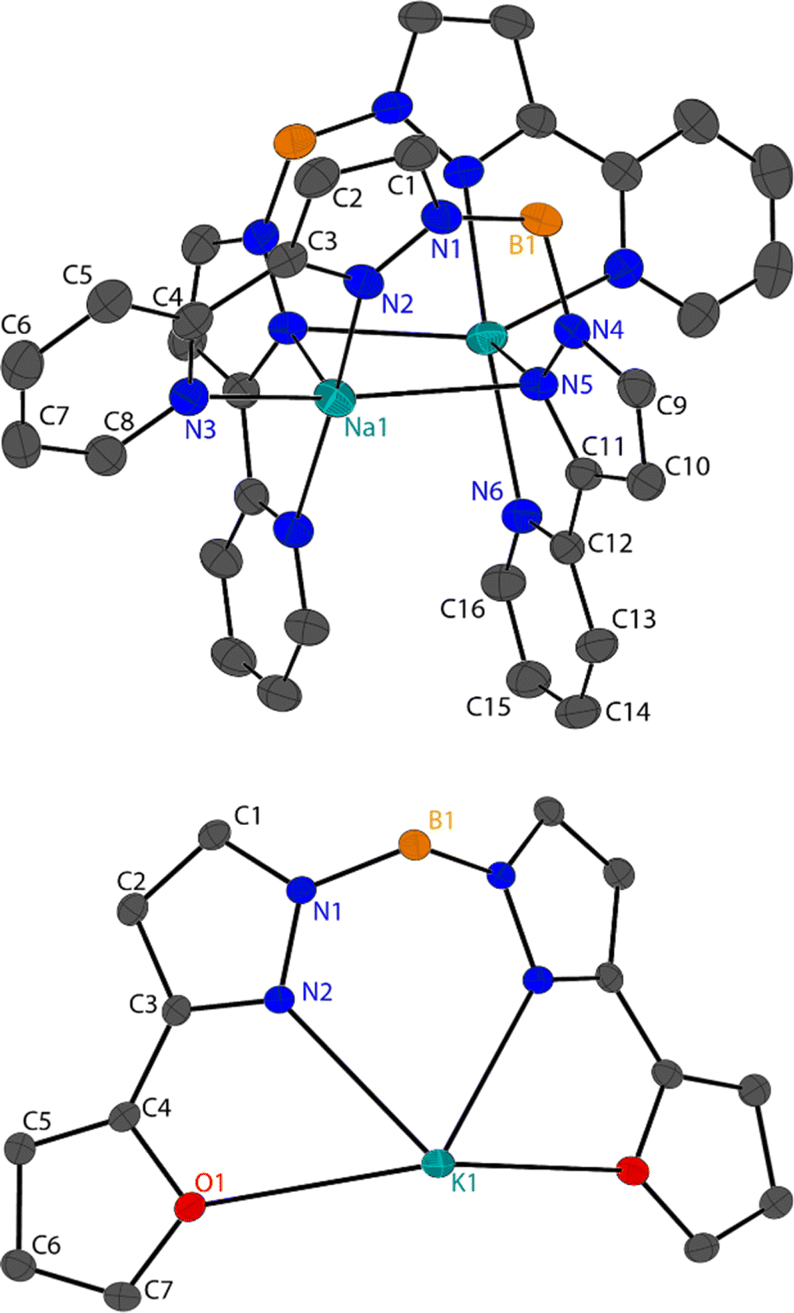
| Fig. 2 Molecular structure of 2-Na (left), 2-K (middle) and 1-Na (right), shown with 50% ellipsoids, hydrogen atoms have been omitted for clarity. For 2-Na and 2-K molecular structures are grown by (1 + Y − X, 1 − X, +Z); (1 − Y, +X − Y, +Z) and for 1-Na by (1 − X, +X − Y, +Z); (1 + X − Y, 1 − X, +Z). | ||
 | ||
| Fig. 3 Molecular solid-state structures for 3-M2 (top, where M = Na) and 4 (bottom), shown with 50% ellipsoids, hydrogen atoms have been omitted for clarity. | ||
| 1-Na | 1-K a | 2-Na | 2-K | |
|---|---|---|---|---|
| a Data taken from literature.7 | ||||
| M1···B1/Å | 3.383(3) | 3.738(3) | 3.416(5) | 3.774(9) |
| M1–N2/Å | 2.5355(13) | 2.8325(15) | 2.533(2) | 2.816(4) |
| M1–N3/Å | 2.5935(14) | 2.870(3) | 2.535(3) | 2.836(4) |
| M1⋯B1⋯N2/° | 48.01(6) | 49.2(1) | 47.50(8) | 48.2(1) |
| N2–C3–C4–C5/° | 163.88(12) | 163.88(17) | ||
| N2–C3–C4–S1/° | 159.15(19) | 161.4(3) | ||
| CShM (Oh) | 1.390 | 2.575 | 0.644 | 1.943 |
1-Na packs in the trigonal P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) and isomorphous to our previous reporting for 1-K.7 As described, 1/3 of the molecule comprises the asymmetric in 1-Na with one pyrazolyl substituent and one MeCN ligand, and the sodium and boron atoms lying on the C3 axis, which extends in the c-axis direction. Coordination bonds (M1–N2 and M1–N3) are shorter in 1-Na then in 1-K owing to ionic size (Table 3). Within the 1-M structures the scorpionate ligands align down the c-direction in columns, all in the same direction with complete overlap, where the adjacent columns are in an antiparallel arrangement (opposing directions). The ‘antiparallel’ arrangement of the columns is likely due to the edge-to-face π-stacking of phenyl rings in adjacent molecules (Fig. S30†) where the distance between the hydrogen atom and the centroid of the phenyl rings is 2.661 Å in 1-Na and 2.832 Å in 1-K.
and isomorphous to our previous reporting for 1-K.7 As described, 1/3 of the molecule comprises the asymmetric in 1-Na with one pyrazolyl substituent and one MeCN ligand, and the sodium and boron atoms lying on the C3 axis, which extends in the c-axis direction. Coordination bonds (M1–N2 and M1–N3) are shorter in 1-Na then in 1-K owing to ionic size (Table 3). Within the 1-M structures the scorpionate ligands align down the c-direction in columns, all in the same direction with complete overlap, where the adjacent columns are in an antiparallel arrangement (opposing directions). The ‘antiparallel’ arrangement of the columns is likely due to the edge-to-face π-stacking of phenyl rings in adjacent molecules (Fig. S30†) where the distance between the hydrogen atom and the centroid of the phenyl rings is 2.661 Å in 1-Na and 2.832 Å in 1-K.
2-Na and 2-K are also isomorphic, packing in the acentric trigonal space group R3c. Like 1-M, 2-M structures have 1/3 of the molecular structure in the asymmetric unit with one pyrazolyl substituent and one MeCN ligand, where the C3 rotational axes pass through the alkali metal and boron atoms. Similarly, due to the ionic radius of the alkali metal used, 2-Na has shorter coordination bonds than 2-K (Table 3). Similar to 1-M, 2-M also form columns of molecules down the c-direction, where all columns are ‘parallel’ to each other. Molecules within these columns are not align and instead are generated by glide planes that are parallel with the C3 axes. Interestingly, the S1 atoms do not coordinate to the metal centres in 2-M, rending the coordination mode of the scorpionate to κ3, and instead the 2′-thienyl substituent are at torsion angles of 159.15(19)° in 2-Na and 161.4(3)° in 2-K (torsion is taken from N2–C3–C4–S1). In transition metal complexes employing 3-heterocyclic substituted pyrazoles with coordinating atoms, the heterocyclic ring generally have similar torsion angles, however in lanthanide and actinide complexes these functional groups coordinate.8–10,12,13 Looking at the supramolecular packing of 2-M, the 2′-thienyl groups are not directed towards anything of chemical significance, however, there is an edge-on π interaction of the heterocyclic ring with MeCN ligands of adjacent molecules. The distances between the terminal carbon of the MeCN ligands and the centroids of the 2′-thienyl substituents are 3.380 Å in 2-Na and 3.328 Å in 2-K (Fig. S31†).
| 3-Li2 | 3-Na2 | |
|---|---|---|
| a Molecular geometry grown via (1 − X, Y, ½ − Z). | ||
| M1⋯M1a/Å | 3.303(8) | 3.2724(16) |
| M1⋯B1/Å | 3.676(5) | 3.786(3) |
| M1a⋯B1/Å | 3.772(5) | 3.955(2) |
| M1–N2/Å | 2.032(4) | 2.3374(17) |
| M1–N3/Å | 2.130(4) | 2.5028(19) |
| M1–N5/Å | 2.869(4) | 2.7345(19) |
| M1a–N5/Å | 2.053(4) | 2.4114(18) |
| M1a–N6/Å | 2.121(4) | 2.4793(18) |
| N5–M1–N5a | 96.86(15) | 100.76(6) |
| M1–N5–M1a | 82.49(15) | 78.70(5) |
| N2–C3–C4–N3 | 1.8(3) | 0.0(3) |
| N5–C11–C12–N6 | 14.8(3) | 11.9(3) |
| CShM (SP) | 4.829 | 5.785 |
Unlike 1-M and 2-M, 3-M2 have the heterocyclic 3-R-pyrazolyl substituents coordinating to the metal centres which leads to small torsion angles of said heterocycles with pyrazolyl rings (<15°). The M1–N5 and M11–N5 in 3-M2 produce a rectangle that allow for short M1⋯M11 distances of 3.303(8) Å in 3-Li2 and 3.2724(16) Å in 3-Na2. Two pyridyl groups in 3-M2 from different Bp2-py anions within the dimer display π–π interaction, with centroid–centroid distances of 3.465 and 3.935 Å from 3-Li2 and 3-Na2, respectively (Fig. S32†), with a similar packing seen in adjacent molecules though the centroid distances are much larger (ca. 3.5–4.0 Å, Fig. S33†).
Compound 4 packs in the orthorhombic space group Pnma and forms a 2-D coordination polymer where one Bp2-Fu anion coordinates to three potassium ions. One half of the KBp2-Fu ion pair in 4 compromises the asymmetric where the boron (including bound hydrogen atoms) and potassium atoms lie on a mirror plane and have half occupancies in the asymmetric unit. The typical coordination bonds from the pyrazolyl substituents are K1–O1 = 3.1369(9) Å and K1–N2 = 2.7997(11) Å. The 2-D nature of the solid-state structures derives from both hydrides bridging to a neighbouring potassium centre which extends the network in the a-direction, with a boron–potassium distance of 3.2431(19) Å (Fig. S34†), which is much closer than the K1⋯B1 distance of 3.883(2) Å in the asymmetric unit (see Fig. 3). The network extends in the c-direction due to the N1 atoms coordinating to a different adjacent potassium cation, with a coordination distance of 3.3132(11) Å (Fig. S34†).
Conclusions
The purification of scorpionate ligands that suffer from over substitution at tris-substituted activation temperatures, i.e. TpPh and Tp2-Th, by fractional crystallisation has been exemplified through the isolation of 1-Na, 1-K, 2-Na and 2-K of the general formula [M(TpR)(MeCN)3]. These tris-MeCN adducts have been characterised by FTIR, NMR and X-ray diffraction showing a high purity in bulk materials. Alongside these, the safe and straightforward syntheses of the 3-heteroclylic dihydrobis(pyrazolyl)borate ligands 3-M (M = Li, Na, K) 4 and 5 are exemplified. We implore other groups to use these novel techniques for the purification of other poly(pyrazolyl)borate ligands that suffer from the same over substitution problem, without resorting to the use of highly toxic thallium(I) salts.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the University of New South Wales (UNSW) for their funding and the Mark Wainwright Analytical Centre (MWAC) for the use of their instrumentation for the collection of single crystal X-ray diffraction, NMR Data and the XRF laboratory for elemental analysis.Notes and references
-
(a) S. Trofimenko, J. Am. Chem. Soc., 1966, 88(8), 1844 CrossRef
; (b) S. Trofimenko, J. Am. Chem. Soc., 1967, 89(13), 3170–3177 CrossRef CAS
- K. W. Bagnall, A. C. Tempest, J. Takats and A. P. Masino, Inorg. Nucl. Chem. Lett., 1976, 12, 555–557 CrossRef CAS
-
(a) J. C. Calabrese, S. Trofimenko and J. S. Thompson, J. Chem. Soc., Chem. Commun., 1986, 1122–1123 RSC
- M. Kitamura, Y. Takenaka, T. Okuno, R. Holl and B. Wunsch, Eur. J. Inorg. Chem., 2008, 8, 1188–1192 CrossRef
- S. Trofimenko, Chem. Rev., 1993, 93, 943–980 CrossRef CAS
- M. M. Melero, Z. Klosek, C. R. Arellano and A. Olmos, J. Org. Chem., 2023, 88, 9130–9135 CrossRef CAS PubMed
- J. R. Thomas and S. A. Sulway, RSC Adv., 2021, 11, 16158–16160 RSC
-
(a) J. R. Thomas, M. J. Giansiracusa, R. A. Mole and S. A. Sulway, Cryst. Growth Des., 2024, 24, 573–583 CrossRef CAS
- J. R. Thomas and S. A. Sulway, Acta Crystall., 2024, C80, 153–158 Search PubMed
- N. Armaroli, G. Accorsi, F. Barigelletti, S. M. Couchman, J. S. Fleming, N. C. Harden, J. C. Jeffery, K. L. V. Mann, J. A. McCleverty, L. H. Rees, S. R. Starling and M. D. Ward, Inorg. Chem., 1999, 38(25), 5769–5776 CrossRef CAS
-
(a) S. Ossinger, C. Nather, A. Buchholz, M. Schmidtmann, S. Mangelsen, R. Beckhaus, W. Plass and F. Tuczek, Inorg. Chem., 2020, 59(12), 7966–7979 CrossRef CAS PubMed
-
(a) H. Adams, S. R. Batten, G. M. Davies, M. B. Duriska, J. C. Jeffery, P. Jensen, J. Lu, G. R. Motson, S. J. Coles, M. B. Hursthouse and M. D. Ward, Dalton Trans., 2005, 1910–1923 RSC
-
(a) D. A. Bardwell, J. C. Jeffery, P. L. Jones, J. A. McCleverty, E. Psillakis, Z. Reeves and M. D. Ward, J. Chem. Soc., Dalton Trans., 1997, 2079–2086 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2357156–2357161. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra05723f |
| This journal is © The Royal Society of Chemistry 2024 |