
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChitosan supported ionic liquid, a multifaceted catalyst for streamlined and efficient synthesis of carboxylic, amino acid and carbohydrate esters†
Praachi Kakati and
Satish Kumar Awasthi*
and
Satish Kumar Awasthi*
Department of Chemistry, Chemical Biology Laboratory, University of Delhi, Delhi 110007, India. E-mail: satishpna@gmail.com
First published on 11th November 2024
Abstract
This work presents a sustainable approach for synthesizing esters from carboxylic acids, amino acids and carbohydrates using a robust and eco-friendly chitosan-incorporated ionic liquid under solvent-free conditions. Ionic liquids with carbon chain lengths ranging from 3 to 8 were integrated into the chitosan molecule, resulting in a heterogeneous catalyst optimized for esterification reactions. Among these, the 6-carbon chain ionic liquid demonstrated superior catalytic activity and substrate tolerance. The catalyst's effectiveness was confirmed using advanced analytical techniques. The acidity of the ionic liquid was assessed by observing the interaction between the synthesized IL6 (1,4-bis(5-carboxypentyl)pyrazine-1,4-diium ([BCPPD][Br])) and p-nitroaniline via UV-Vis studies. Chitosan-IL6, an ionic liquid supported on chitosan, functions as a heterogeneous catalytic system that can be easily removed from reaction mixtures through simple filtration. It also exhibits excellent reusability, maintaining high catalytic activity and structural integrity over 10 catalytic cycles. Moreover, the methodology was successfully scaled up for the gram-scale synthesis of key compounds such as diisopropyl azodicarboxylate, methyl nicotinate, methyl cysteinate, and glucose pentaacetate, highlighting its practical viability.
Introduction
Ester linkages are one of nature's most rudimentary and coveted chemical bonds in lieu of the fact that esters are suggested to achieve profound application in chemical and pharmaceutical industries, being the source of natural products, medicines, chemicals, polymers, flavours and fragrances, etc.1 Amino acid esters are value added intermediates in organic synthesis, which have been put to various use in areas such as peptide synthesis,2 medicinal chemistry, chiral sources,3,4 polymer materials5 and various other fields. Similarly, mono- or oligosaccharide esters show intriguing surface activity as well as potentially effective anticancer and plant growth-inhibiting activities.6 Having properties of good biodegradability and low toxicity, they have been widely used in food, detergents, cosmetics and biomedical industries mainly due to their benign nature.7 Because of these diverse properties, esters and their feasible and green synthesis over their conventional methods are worthy of investigation.Traditional methodologies of ester synthesis using condensation reactions of carboxylic acids and alcohols require strong mineral acids (H2SO4, HCl, etc.) (Fig. 1a)8–10 as catalysts. This makes reaction conditions corrosive and extremely vigorous resulting in low yields due to unwanted by-products. Another drawback of these conventional synthetic routes is their reversible nature as they hydrolyse the ester product in presence of moisture or traces of acid. This shifting of the equilibrium further adds complications to the purification. Rachele et al.11 reported esterification of amino acid using aqueous HCl eliminating the use of dry solvent or any gaseous HCl. Several halogenated compounds such as SOCl2,12,13 triphosgene14 and trimethylchlorosilane2 were also used for esterification as acid substitutes. Additionally, enzymatic15–18 reaction is another common approach that is frequently used for esterification of acids in an enantioselective manner (Fig. 1b). Carbohydrates and their derivatives are also esterified using enzymes such as Candida rugosa lipase,19,20 Yarrowia lipolitica,21 Candida rugosa,22 Chromobacterium viscosum7 lipases and various others, albeit with lower yields. With the advancement in metal catalysis, great efforts have been made to synthesize esters in recent years. Itami23 and Baudoin24 reported palladium based catalysts used for esterification of acids with aryl halides. Zhang25 and co-workers proposed a metal organic framework (MOF) as a nanocatalyst to produce biodiesel by esterifying lauric acid. Peiber26 and group demonstrated a photocatalysis approach using nickel as semi-heterogeneous catalyst, wherein they used a homogeneous nickel catalyst combined with a heterogeneous metal-free carbon nitride semiconductor for dual catalysis. Despite their high yields these metal-based catalysts are still not practiced for pharmaceutical purposes. Even an inconspicuous amount of it in any step of esterification may cause serious health hazards (Table S1†).
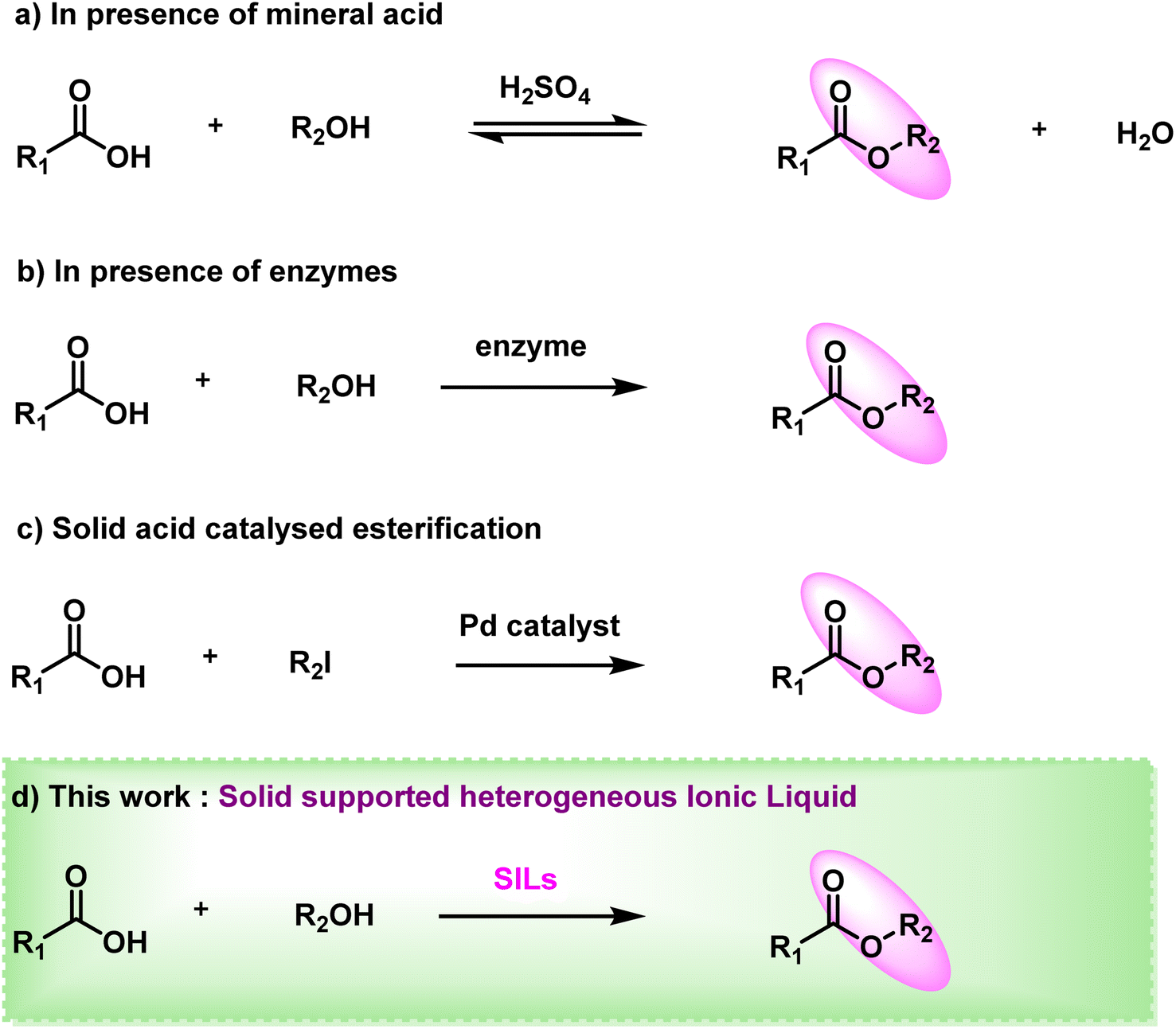 | ||
| Fig. 1 Previous work and current method. | ||
The use of solid acid catalysts27,28 for the esterification reaction has been the subject of recent reports29 (Fig. 1c). Highly stable magnetic solid acid catalyst was reported by Liu et al.30 with a total acid site content of 2.57 mmol g−1. It was synthesised using sawdust as the biomass and magnetic porous carbon nanomaterials as the solid acid catalyst, with the addition of Iron. Chellappan et al.31 also developed a solid acid catalyst from cassava peel in their research. For biodiesel production aided by microwaves, the catalyst demonstrated good stability and reusability. This is due to its advanced features, which include a high specific area, thermal stability, large pore volumes and, most crucially, the ease with which the catalyst can be recovered and the product purified. In heterogeneous catalytic esterification systems, various kinds of mesoporous silica,32 such as zeolite, SBA family members and sulfonic acid-functionalized inorganic frameworks29,33 have been utilised.
Recent studies have reported chitosan as a promising material for use as a support for catalytic applications in heterogeneous molecular catalysis, due to its hydrophilicity, chemical reactivity, unique three-dimensional structure, hydroxyl and amino groups, excellent chelating properties and mechanical properties.34 Chitosan is a biopolymer derived from chitin by deacetylation.35 Due to the presence of alcohol and amine functional groups in the chemical structure of chitosan, it is possible to modify this polysaccharide through a wide variety of chemical reactions. And because it can be broken down by microorganisms in both soil and water, chitosan is also more ecologically benign and efficient. Chitin and chitosan have attracted significant attention from the scientific and industrial communities for a wide variety of uses, including those in the pharmaceutical, waste water treatment, cosmetics, drug delivery, heavy metal chelation and heterogeneous catalysts industries.
Organic solvents are volatile liquids that due to their evaporation into the atmosphere lead to adverse effects on the environment and human health. Moreover, their flammability and low boiling point lead to further adversities. To successfully address these issues, we sought a greener approach for this condensation reaction that did not involve the use of any hazardous solvents and conditions, as well as minimizing the usage of corrosive reagents.
Ionic liquids have gained overwhelming interest over the past few decades because of their potential applications as catalysts, co-catalyst and green solvents36 in several chemical processes.37 ILs are a class of molecules with unique and tuneable physical and chemical properties that makes them ideal green solvents for a variety of organic reactions due to their ability to control product distribution,38 enhance rates39 and/or reactivity,40 facilitate product recovery,41 immobilise catalysts.42 They have negligible vapour pressure and due to their thermal and chemical stability they are preferred as solvents in most chemical reactions relieving the use of hazardous and polluting organic solvents.43 Furthermore, due to their structural diversity, ease of separation and recyclable nature they are environmentally benign and is classified as a green catalyst.44 The excessive use of ILs as a catalyst45 or a solvent, which results in a significant amount of waste materials, is one of the main downsides of homogeneous catalysis utilising ILs apart from high temperature and larger reaction time. Furthermore, it is quite challenging to dispose of these waste products, making the entire process unsatisfactory from an environmental standpoint.46 Different ILs have been immobilised on solid supports to combine the benefits of ILs and heterogeneous support materials, avoiding the drawbacks of homogeneous catalysis involving ILs. So, in this paper, we report solid support ionic liquids (SILs),47 which use insoluble supports to create heterogeneous acid catalyst systems which underpin sustainable chemical processes. The solid support furnishes a specially designed environment around the catalytic site,48 allowing this heterogenous catalyst49 to outperform its homogeneous equivalents in terms of size and positional selectivity.
In this work, we have designed and characterised chitosan incorporated ionic liquid, Chitosan-IL6 by synthesizing six atom carbon chain ionic liquid 1,4-bis(5-carboxypentyl)pyrazine-1,4-diium ([BCPPD][Br]) and incorporating it in chitosan as the solid support (Scheme 1). This ‘designer catalyst’ was then facilitated for the synthesis of esters (Fig. 1d) with excellent yield and without showing any significant forfeit in catalytic activity up to 10 catalytic cycles (Fig. 5, and S4†).
 | ||
| Scheme 1 Schematic representation of (a) synthesis of 6 atom carbon chain ionic liquid IL6 1,4-bis(5-carboxypentyl)pyrazine-1,4-diium ([BCPPD][Br]) (b) incorporating of IL6 in an insoluble solid support chitosan to form the desired catalyst Chitosan-IL6. | ||
Results and discussion
To initiate the process, we selected the archetypical substrates of benzoic acid A1 (1 mmol) and ethanol B1 (1 mmol) in presence of several organic solvents such as water, chloroform, acetonitrile and ethanol (entry 1–4, Table 1) and refluxed it at 100 °C for 6 h which resulted in negligible yield. Following that 10 mg (Table S2†) of the catalyst Chitosan-IL6 was added to the reaction mixture and refluxed at 100 °C for 3 h which resulted in 67% yield (entry 6, Table 1) of ethyl benzoate C1. It was also observed that the use of solvent (or ethanol) in excess has no discernible change in the reaction yield (entry 5 and 6, Table 1). This manifests that the reaction proceeds particularly in the presence of Chitosan-IL6. Further optimising the reaction conditions of temperature and time, by decreasing temperature to 50 °C and RT and time to 3 h, 1 h and 30 min (entry 7–11, Table 1) optimal yields of 96% were obtained when the reaction was carried out at RT for 30 min (entry 11, Table 1). Following that, modified catalysts having different chain lengths of ionic liquid were used in the reaction that fabricated somewhat similar yields (entry 12), with the highest yield being obtained with Chitosan IL6 and hence it was selected as the catalyst for the reaction. The reason being that the alkyl chain length with 6 carbon chain provides the optimum environment for the reaction to proceed. Moreover, according to Table 1 (entry 11, 13 and 14) optimizing the reaction in presence of IL6, chitosan and Chitosan-IL6 procured yields indicating that the presence of chitosan has no effect on the reaction and acts only as a heterogeneous media. Thus, the optimal reaction conditions for the esterification of benzoic acid A1 (1 mmol) and ethanol B1 (1 mmol) is under solvent free conditions catalysed by 10 mg of Chitosan-IL6 for 30 min at room temperature.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst | Temperature (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: benzoic acid A1 (1 mmol), ethanol B1 (1 mmol), catalyst (10 mg), at appropriate temperature and time.b Isolated yield. IL: ionic liquid, RT: room temperature, CHCl3: chloroform, CH3CN: acetonitrile, EtOH: ethanol, NR: no reaction. | |||||
| 1 | Water | — | 100 | 6 | NR |
| 2 | CHCl3 | — | 100 | 6 | NR |
| 3 | CH3CN | — | 100 | 6 | NR |
| 4 | EtOH | — | 100 | 6 | Trace |
| 5 | EtOH | Chitosan-IL6 | 100 | 3 | 68 |
| 6 | — | Chitosan-IL6 | 100 | 3 | 67 |
| 7 | — | Chitosan-IL6 | 50 | 3 | 70 |
| 8 | — | Chitosan-IL6 | RT | 1 | 90 |
| 9 | — | Chitosan-IL6 | 100 | 0.5 | 86 |
| 10 | — | Chitosan-IL6 | 50 | 0.5 | 96 |
| 11 | — | Chitosan-IL6 | RT | 0.5 | 96 |
| 12 | — | Chitosan-IL3–8 | RT | 0.5 | ∼90 |
| 13 | — | IL6 | RT | 0.5 | 96 |
| 14 | — | Chitosan | RT | 0.5 | 45 |
The optimized reaction conditions were subsequently employed for the synthesis of other derivatives of esters (Schemes 2–4). As illustrated in Scheme 2, the substrate scope was generated by taking ethanol, methanol or isopropyl alcohol (B) and substituted benzoic acid having both electron donating as well withdrawing groups. It was perceived that electron withdrawing groups (C5–C12) have superior yields (96–99%) than electron donating groups (C2–C4). Furthermore, it was observed that heterocyclic derivatives (C13–C15) have considerable good yields of (89–93%). Moreover, the aliphatic substrates (C16–C29) yielded their corresponding esters in good yields (84–92%). It was observed that the presence of ethyl or methyl groups in the alcohol moiety has no discernible effect on the yield of the corresponding ester product (C21, C27).
 | ||
| Scheme 2 Substrate scope for Chitosan-IL6 catalyzed selective esterification of carboxylic acids to esters C1–C29.aReaction conditions: carboxylic acid (1 mmol), alcohol (1 mmol), Chitosan-IL6 (10 mg) under neat conditions at RT for appropriate time.bIsolated yield. | ||
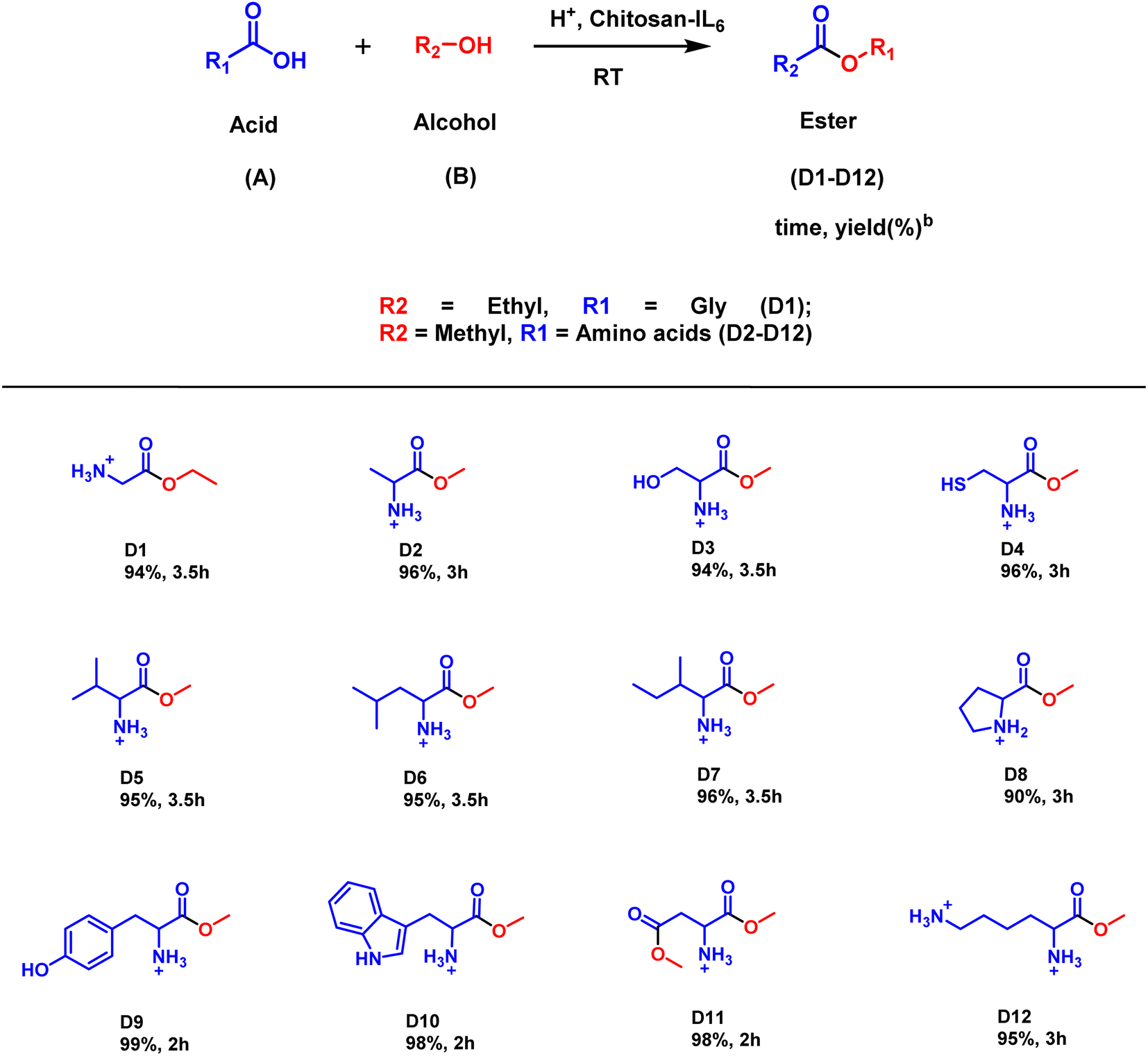 | ||
| Scheme 3 Substrate scope for Chitosan-IL6 catalyzed selective esterification of amino acids to amino acid esters D1–D12.aReaction conditions: amino acid (1 mmol), alcohol (1 mmol), Chitosan-IL6 (10 mg), acetic acid (3 drops) under neat conditions at RT for appropriate time. bIsolated yield. | ||
 | ||
| Scheme 4 Substrate scope for Chitosan-IL6 catalysed selective esterification of carbohydrate to carbohydrate esters E1–E7.aReaction conditions: acetic anhydride (4–5 mmol), carbohydrate (1 mmol), Chitosan-IL6 (10 mg) under neat conditions at RT for appropriate time. bIsolated yield. | ||
Further, the catalytic activity of the ionic liquid catalyst Chitosan-IL6, was applied in the synthesis of esters of amino acids (Scheme 3). In case of amino acid esterification, (Fig. S2†) addition of a few drops of acetic acid as a neutralizing agent leads to the synthesis of an array of amino acid esters having substantially good yield (90–99%). It could be deciphered that amino acids such as tyrosine, tryptophan and aspartic acid (D9–D11) having electron withdrawing groups have better yields (98–99%) than their electron donating equivalents (90–96%).
Additionally, the versatility of the designer catalyst was also explored for the synthesis of carbohydrate esters (Scheme 4). The reaction condition was improvised in this case by using acetic anhydride to facilitate the esterification of carbohydrates in leu of acid. In a solvent less manner a variety of carbohydrate esters, including hexoses, pentoses and sucrose (E1–E7), were synthesized with excellent yields (90–98%).
On the basis of the above mechanistic results and intuitive precedents, a plausible mechanistic pathway for the Chitosan-IL6 catalyzed esterification reaction is being delineated in Scheme 5. In order to gain insight into the reaction pathway, we herein consider the esterification reaction of benzoic acid A1 with ethanol B1 to obtain ethyl benzoate C1. At the onset of the reaction, the oxygen of carbonyl of benzoic acid abstracts a proton from carboxylic group of the Chitosan-IL6 catalyst, which is followed by the attack of ethanolic OH on the carbonyl C of the benzoic acid. This leads to the formation of the intermediate I, which undergoes the loss of a water molecule and final desorption of the benzoate from the catalyst resulting in the formation of the product and the catalyst being ready to be used for the next catalytic cycle. Chitosan provides only surface area for adsorption of the reactants and reagents without themselves participating in the catalytic process.
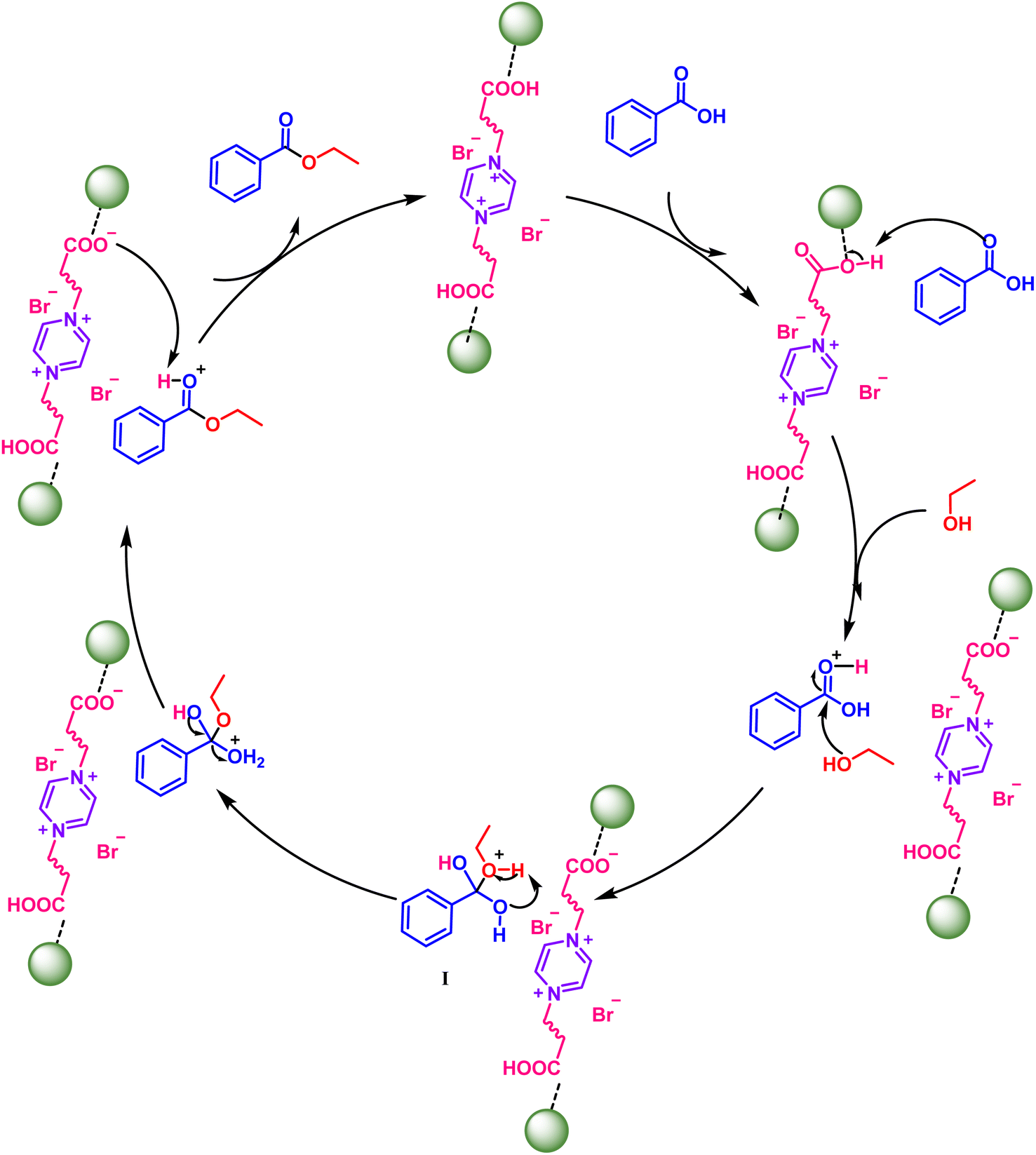 | ||
| Scheme 5 Plausible mechanism for Chitosan-IL6 catalyzed selective esterification of benzoic acids A1 and ethanol B1 to ethyl benzoate C1. | ||
In this report synthesis of Chitosan-IL6 was at first confirmed by FT-IR spectra (Fig. 2a). The spectrum of chitosan shows a peak at 1022 cm−1 which appears due to C–O–C stretching frequency of it. The broad peak around 3328 cm−1 corresponds to –OH and –NH stretching mode of chitosan framework,46 while the –CH stretching mode of chitosan appears at 2886 cm−1. It was observed that the sharp peak of carbonyl of carboxylic acid at 1727 cm−1 of the ionic liquid remains intact on Chitosan-IL6 which corroborates that ionic liquid is incorporated in chitosan.
 | ||
| Fig. 2 (a) FT-IR spectra of IL6, chitosan and Chitosan-IL6. (b) 13C-CPMAS of chitosan and Chitosan-IL6. (c) PXRD of chitosan and Chitosan-IL6. (d) TGA of chitosan and Chitosan-IL6. | ||
The basic chemical structure of Chitosan-IL6 was further established by using 13C-CPMAS NMR (Fig. 2b); peaks ∼20 ppm corresponds to the CH2 of chitosan, addition peaks at ∼30 ppm in Chitosan-IL6 corresponds to aliphatic carbons of the IL moiety incorporated to chitosan. The peaks at ∼55.5 ppm and ∼60.5 ppm corresponds to C–N, peaks at 74 ppm corresponds to C–O. The extra peak at ∼100 ppm appearing at the 13C-CPMAS NMR spectra of Chitosan-IL6 corresponds to pyrazine and peak at 176 ppm corresponds to carbonyl carbon of ionic liquid in Chitosan-IL6. This further establishes the presence of IL6 on chitosan.
To further comprehend the crystallinity of the Chitosan-IL6 we performed the PXRD experiment (Fig. 2c). From the comparative analysis of chitosan and Chitosan-IL6 we observe broad peaks at 2θ = 20° which is in well agreement with the reported literature value.34,46 This also justifies the amorphous nature of chitosan. Further, two additional peaks at 2θ = 10° and 2θ = 15° are observed in Chitosan-IL6 that suggests the incorporation of ionic liquid into the chitosan molecule.
Further to investigate the thermal stability of the ionic liquid TGA was conducted (Fig. 2d). The material was stable up to a considerable temperature after which around 38% weight loss occurred at approximately 300 °C which is attributed to loss of moisture and volatile matter, followed by 60% weight loss at around 600 °C indicating that the material is stable over a wide temperature range. While comparing it with the starting material chitosan, around 10% weight loss was observed at 100 °C followed by approximately 55% weight loss at around 300 °C. From the TGA curves it was deciphered that Chitosan-IL6 is more stable than chitosan.
The N2 adsorption desorption isotherms of chitosan and Chitosan-IL6 and their associated pore distribution curves are displayed in Fig. S5.† Chitosan and Chitosan-IL6 both have BET surface areas of 5.33 m2 g−1 and 1.97 m2 g−1, respectively. The BJH cumulative desorption pore volume and pore size of chitosan were 0.005 cm3 g−1 and 112.5 Å respectively while the pore volume of Chitosan-IL6 is 0.003 cm3 g−1. The pore size obtained indicated towards the mesoporous nature of the solid acid catalyst. The comparative analysis of chitosan and Chitosan-IL6 shows a considerable reduction in surface area and pore volume after the confinement of ionic liquid on chitosan which is attributed to pore blockage by ionic liquid. However, the solid acid catalyst possessed sufficient surface on which the active sites were well dispersed, allowing them easily available to the reactants.
Furthermore, the morphology of the catalyst was determined by SEM analysis (Fig. 3a and b). After careful analysis of the SEM image of Chitosan-IL6 we can observe that particles or roughness appears in an otherwise smooth surface of chitosan molecule,50 which indicates that ionic liquid has been incorporated into the chitosan moiety.46 Moreover, the EDX data (Fig. 3c) of Chitosan-IL6 composite further confirms the existence of bromine as a structural component of the synthesized catalyst.
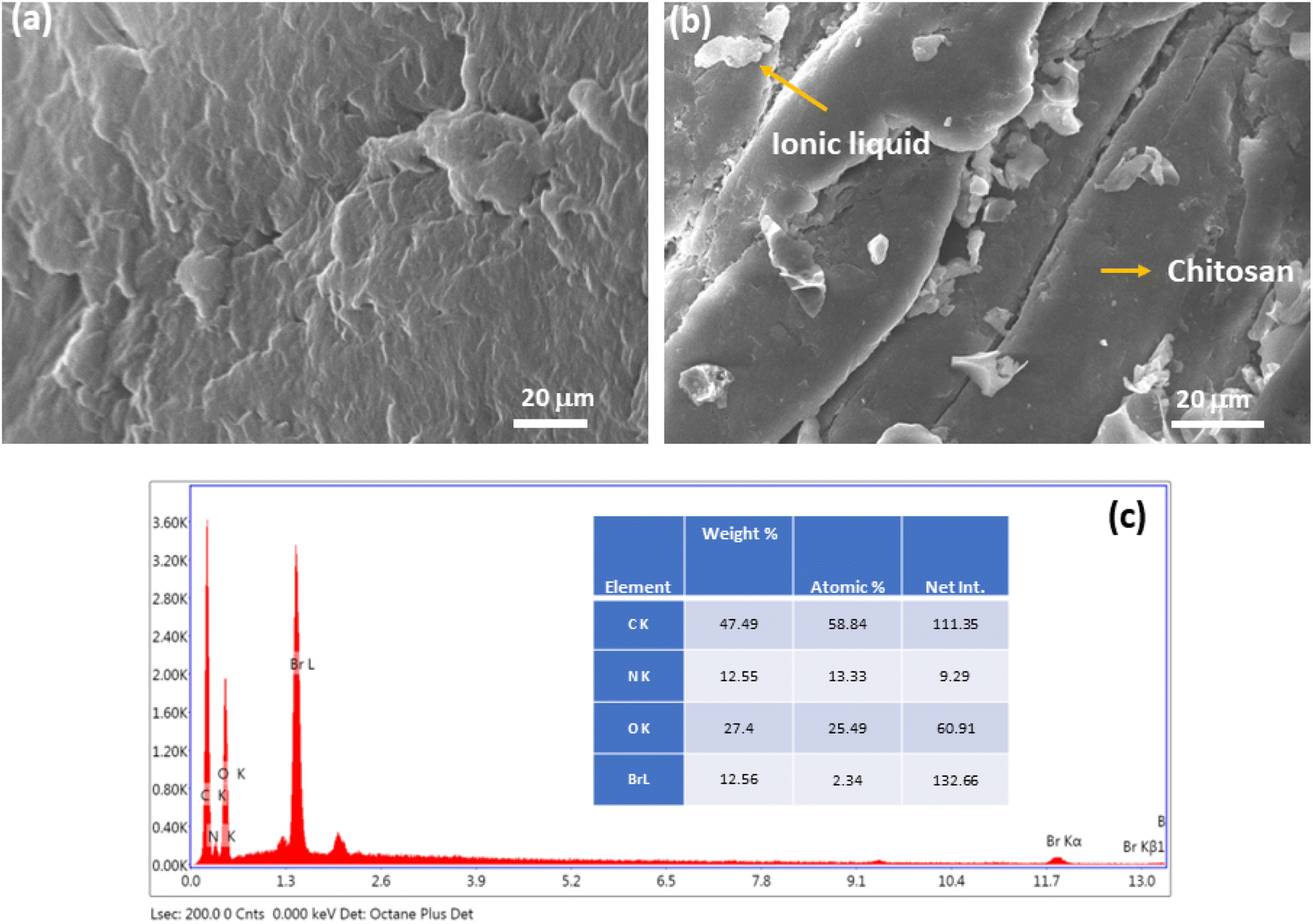 | ||
| Fig. 3 (a) SEM image of chitosan. (b) Sem image of Chitosan-IL6. (c) EDX spectra of Chitosan-IL6. | ||
To order to gain a better understanding of the acidity of this ionic liquid ([BCPPD][Br]) we have studied the UV-Vis absorption spectra of p-nitroaniline which acts as an indicator and molecular probe, I. Hammett acidity functions were utilised to determine the Brønsted acidity.51 This method evaluates the protonation extent of uncharged indicator bases, p-nitroaniline in our case, by measuring the ratio [I]/[IH+]. The Hammett function (H0) is defined as
| H0 = pK(I)aq + log([I]s/[IH+]s) |
The addition of this acidic ionic liquid caused a significant shift in the absorption curve of p-nitroaniline in DMSO in the wavelength area from 350 to 400 nm, as shown in Fig. 4. As the amount of ionic liquid was increased, means as the concertation of acid increases the absorbance of the unprotonated form of the indicator decreases. By taking as the initial reference the total unprotonated form of the indicator, we could determine the [I]/[IH+] ratio from the measured absorbances after each acid concentration, and then the Hammett function is calculated (Table 2). In [BCPPD][Br], the Hammett function is included between 2.37 and 1.32, corresponding to concentrations ranging from 1.33 to 10.66 mmol L−1.
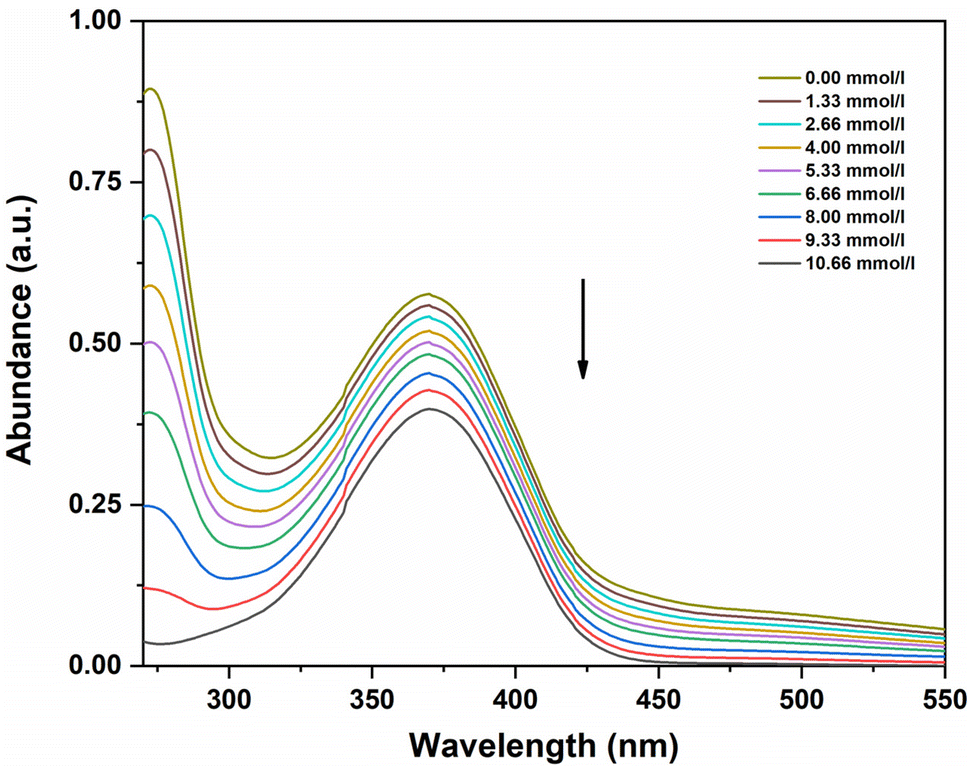 | ||
| Fig. 4 UV-visible spectra of p-nitroaniline for various concentrations of ionic liquid ([BCPPD][Br]) in DMSO. | ||
| Entry | [H+] mmol L−1 | Amax | [I] (%) | [IH+] (%) | H0 |
|---|---|---|---|---|---|
| a p-Nitroaniline (pK(I)aq = 0.99), 10 × 10−6 mol L−1. | |||||
| 1 | 0 | 0.580 | 100 | 0 | |
| 2 | 1.33 | 0.557 | 96 | 4 | 2.37 |
| 3 | 2.66 | 0.542 | 93.4 | 6.6 | 2.14 |
| 4 | 4.00 | 0.519 | 89.5 | 10.5 | 1.92 |
| 5 | 5.33 | 0.501 | 86.4 | 13.6 | 1.79 |
| 6 | 6.66 | 0.481 | 83 | 17 | 1.68 |
| 7 | 8.00 | 0.453 | 78.1 | 21.9 | 1.54 |
| 8 | 9.33 | 0.427 | 73.6 | 26.4 | 1.43 |
| 9 | 10.66 | 0.397 | 68.4 | 31.6 | 1.32 |
To determine the optimal loading amount of ionic liquid on chitosan,46,50 the model reaction between benzoic acid and ethanol was conducted with varying IL loadings, ranging from 5% to 30% w/w (Fig. 5). It was deciphered that the highest product yield was achieved at a 30% w/w IL loading. The ionic liquid loading was calculated to be 0.89 mmol g−1. This was obtained by calculating the amount of ionic liquid that remained unreacted after its reaction with chitosan.
 | ||
| Fig. 5 Loading of IL (w/w%) on chitosan for the reaction of benzoic acid and ethanol catalysed by Chitosan-IL6. | ||
To assess the ionic liquid leaching during the reaction we performed the Shelton test52 for the esterification reaction of benzoic acid A1 and ethanol B1 under the optimized reaction condition (Fig. 6a). During test the catalyst Chitosan-IL6 was filtered off from the reaction mixture after 5 min, and the reaction was continued for an additional 25 min. The reaction mixture was analysed thereafter showed no further product formation, confirming that no ionic liquid had leached into the reaction mixture.
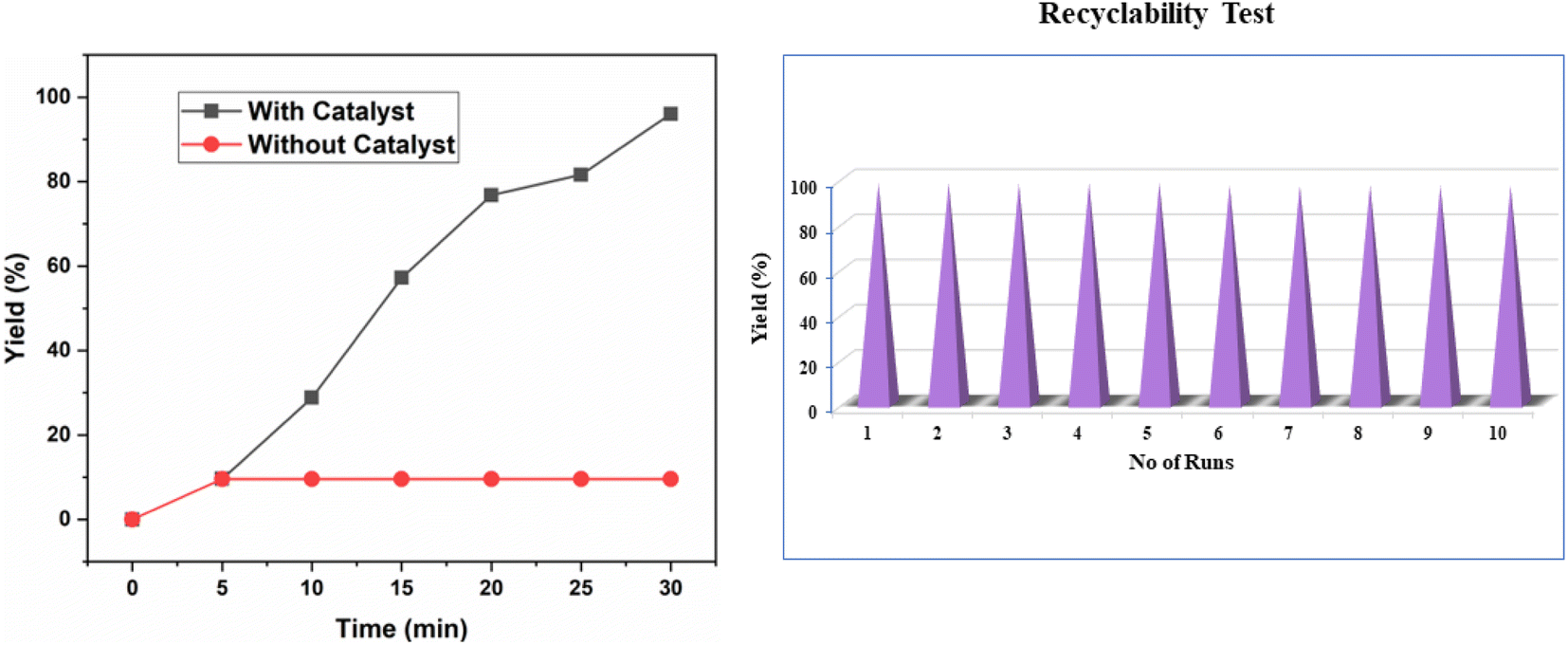 | ||
| Fig. 6 (a) Hot filtration test to determine ionic liquid leaching of Chitosan-IL6. (b) Recyclability test of Chitosan-IL6 catalyzed esterification of benzoic acid and ethanol to ethyl benzoate C1. | ||
Lastly, we evaluated the reusability of the catalyst by taking the model reaction of benzoic acid A1 and ethanol B1 to obtain ethyl benzoate C1 under the optimized reaction conditions. We have observed very inconspicuous loss in the catalytic activity of Chitosan-IL6. It was observed that the catalytic performance of Chitosan-IL6 shows consistent result up to 10 consecutive cycles (Fig. 6b). The recovered catalyst was characterised by FT-IR, XRD and SEM which showed no significant structural or morphological change (Fig. S4†).
Conclusions
In conclusion, we have successfully developed a newer and greener catalyst for the esterification of carboxylic acids and alcohols to their corresponding esters. Chitosan-IL6 showcased optimal efficiency in the esterification reaction out of the synthesized ionic liquids, which were prepared in an affordable approach by embedding the ionic liquid ([BCPPD][Br]) in chitosan. This practice proves superior to the conventional methods of esterification as there is no residual catalyst to shift the equilibrium to the left. The reaction's feasibility, which is determined by the acidity of the catalyst, was further tested by UV visible studies of p-nitroaniline and back titration with HCl giving favourable results. Moreover, the scope of this protocol was extended to include carboxylic acid, amino acid and carbohydrates resulting in good to outstanding yields of their esters in a solvent-free environment. The efficacious use of chitosan-based ionic liquid in the synthesis of esters with a wide range of substrates opens up new avenues for the development of these tailored catalysts and their numerous applications. Furthermore, due to the sustainable nature of ionic liquids, they have received copious attention, and several green syntheses have opened up which are currently being investigated in our laboratory and will be duly reported.Experimental section
Synthesis of ionic liquid (pyrazine based), 1,4-bis(5-carboxypentyl)pyrazine-1,4-diium ([BCPPD][Br])
A mixture of pyrazine (1 mmol) and 6-bromohexanoic acid (1 mmol) in acetonitrile (10 mL) was kept for stirring at RT for 6 h in a round bottomed flask in dark. After completion, the reaction mixture was concentrated under vacuum and the product which was a viscous beige liquid was characterised by 1H NMR.Beige coloured liquid. 1H NMR (400 MHz, DMSO-D6) δ 8.61 (s, 4H), 3.44 (d, J = 11.5 Hz, 4H), 2.16 (t, J = 7.0 Hz, 4H), 1.73 (d, J = 3.4 Hz, 4H), 1.43–1.48 (m, 4H), 1.31–1.34 (m, 4H).
Synthesis of Chitosan-IL6
A mixture of the synthesised ionic liquid 1,4-bis(5-carboxypentyl)pyrazine-1,4-diium (1 mmol) and Chitosan (1 g) was refluxed under acetonitrile medium (20 mL) for 4 h. The solid that was obtained was filtered and Soxhlet with methanol for 8 h which was then finally dried at 72 °C. The obtained catalyst was characterised using FT-IR, 13C-CPMAS, XRD, TGA, SEM and EDX analysis.General procedure for esterification of acid
In a round bottomed flask 1 mmol acid derivative, 1 mmol alcohol and 10 mg of the catalyst was stirred at room temperature for anticipated time (Scheme 2). The progress of the reaction was monitored by a TLC. After completion of the reaction the catalyst was filtered off from the reaction mixture, washed with methanol and dried for further use in subsequent reactions. The solution was then concentrated in vacuum to obtain the desired ester product.General procedure for esterification of amino acid
In a round bottomed flask 1 mmol amino acid derivative, 1 mmol alcohol, 10 mg of the catalyst and 2–3 drops acetic acid was stirred at room temperature for anticipated time (Scheme 3). The progress of the reaction was monitored by a TLC. After completion of the reaction the catalyst was filtered off from the reaction mixture, washed with methanol and dried for further use in subsequent reactions. Ultimately, the solution was concentrated in vacuum to obtain the desired ester product.General procedure for esterification of carbohydrate
In a round bottomed flask at 0 °C 4–5 mmol acetic anhydride, 1 mmol carbohydrate, 10 mg of the catalyst was stirred at room temperature for anticipated time (Scheme 4). The progress of the reaction was monitored by a TLC. After completion of the reaction the catalyst was filtered off from the reaction mixture, washed with methanol and dried for further use in subsequent reactions. Ultimately, the solution was concentrated in vacuum to obtain the desired ester product.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to USIC and the Department of Chemistry, University of Delhi, India for the instrumentation facilities. The authors also acknowledge IISER TVM and IIT ROPAR for their HRMS facilities. PK gratefully acknowledges DST INSPIRE for providing Senior Research Fellowship. SKA acknowledges financial assistance from the Institute of Eminence, University of Delhi (IoE/FRP/ PCMS/2020/27).References
- N. Fattahi, M. Ayubi and A. Ramazani, Amidation and esterification of carboxylic acids with amines and phenols by N,N′-diisopropylcarbodiimide: A new approach for amide and ester bond formation in water, Tetrahedron, 2018, 74(32), 4351–4356 CrossRef CAS.
- J. Li and Y. Sha, A convenient synthesis of amino acid methyl esters, Molecules, 2008, 13(5), 1111–1119 CrossRef CAS PubMed.
- J.-K. Kim, J. Kim, S. Song, O.-S. Jung and H. Suh, Enantiomeric recognition of d-and l-amino acid methyl ester hydrochlorides by new chiral bis-pyridino-18-crown-6 substituted with urea, and diphenyl groups, J. Inclusion Phenom. Macrocyclic Chem., 2007, 58(1), 187–192 CrossRef CAS.
- G. P. Pollini, N. Baricordi, S. Benetti, C. De Risi and V. Zanirato, A simple entry to chiral non-racemic 2-piperazinone derivatives, Tetrahedron Lett., 2005, 46(21), 3699–3701 CrossRef CAS.
- A. Nagai, T. Miyagawa, H. Kudo and T. Endo, Controlled cationic ring-opening polymerization of 1, 3-oxazolidine-2-thione derived from L-serine, Macromolecules, 2003, 36(25), 9335–9339 CrossRef CAS.
- S. W. Chang and J. F. Shaw, Biocatalysis for the production of carbohydrate esters, New Biotechnol., 2009, 26(3–4), 109–116 CrossRef CAS PubMed.
- A. Janssen, C. Klabbers, M. Franssen and K. Van't Riet, Enzymatic synthesis of carbohydrate esters in 2-pyrrolidone, Enzyme Microb. Technol., 1991, 13(7), 565–572 CrossRef CAS.
- R. H. Leonard, Levulinic acid as a basic chemical raw material, Ind. Eng. Chem., 1956, 48(8), 1330–1341 CrossRef.
- H. J. Bart, J. Reidetschlager, K. Schatka and A. Lehmann, Kinetics of esterification of levulinic acid with n-butanol by homogeneous catalysis, Ind. Eng. Chem. Res., 1994, 33(1), 21–25 CrossRef CAS.
- J. Marchetti and A. Errazu, Esterification of free fatty acids using sulfuric acid as catalyst in the presence of triglycerides, Biomass Bioenergy, 2008, 32(9), 892–895 CrossRef CAS.
- J. R. Rachele, The Methyl Esterification of Amino Acids with 2, 2-Dimethoxypropane and Aqueous Hydrogen Chloride1, J. Org. Chem., 1963, 28(10), 2898 CrossRef CAS.
- B. D. Hosangadi and R. H. Dave, An efficient general method for esterification of aromatic carboxylic acids, Tetrahedron Lett., 1996, 37(35), 6375–6378 CrossRef CAS.
- T. Kalita and B. Mandal, One-Pot Synthesis of Amide, Dipeptide, Ester and Hydroxamate Using Oxyma and Thionyl Chloride (SOCl2), ChemistrySelect, 2021, 6(44), 12281–12287 CrossRef CAS.
- I. Rivero, S. Heredia and A. Ochoa, Esterification of amino acids and mono acids using triphosgene, Synth. Commun., 2001, 31(14), 2169–2175 CrossRef CAS.
- N. Shibuya, Phenolic acids and their carbohydrate esters in rice endosperm cell walls, Phytochemistry, 1984, 23(10), 2233–2237 CrossRef CAS.
- H. Seino, T. Uchibori, T. Nishitani and S. Inamasu, Enzymatic synthesis of carbohydrate esters of fatty acid (I) esterification of sucrose, glucose, fructose and sorbitol, J. Am. Oil Chem. Soc., 1984, 61(11), 1761–1765 CrossRef CAS.
- A. Vartolomei, I. Calinescu, M. Vinatoru and A. I. Gavrila, A parameter study of ultrasound assisted enzymatic esterification, Sci. Rep., 2022, 12(1), 1421 CrossRef CAS.
- V. Singhania, M. Cortes-Clerget, J. Dussart-Gautheret, B. Akkachairin, J. Yu, N. Akporji, F. Gallou and B. H. Lipshutz, Lipase-catalyzed esterification in water enabled by nanomicelles. Applications to 1-pot multi-step sequences, Chem. Sci., 2022, 13(5), 1440–1445 RSC.
- L. Gubicza, N. Nemestóthy, T. Fráter and K. Bélafi-Bakó, Enzymatic esterification in ionic liquids integrated with pervaporation for water removal, Green Chem., 2003, 5(2), 236–239 RSC.
- R. Craveiro, L. Meneses, L. Durazzo, A. n. Rocha, J. M. Silva, R. L. Reis, S. Barreiros, A. R. C. Duarte and A. Paiva, Deep eutectic solvents for enzymatic esterification of racemic menthol, ACS Sustainable Chem. Eng., 2019, 7(24), 19943–19950 CrossRef CAS.
- E. Zdybel, T. Zięba, E. Tomaszewska-Ciosk and W. Rymowicz, Effect of the esterification of starch with a mixture of carboxylic acids from Yarrowia lipolitica fermentation broth on its selected properties, Polymers, 2020, 12(6), 1383 CrossRef CAS PubMed.
- A. Janssen, A. Lefferts and K. Van't Riet, Enzymatic synthesis of carbohydrate esters in aqueous media, Biotechnol. Lett., 1990, 12(10), 711–716 CrossRef CAS.
- H. Kitano, H. Ito and K. Itami, Palladium-catalyzed esterification of carboxylic acids with aryl iodides, Org. Lett., 2018, 20(8), 2428–2432 CrossRef CAS PubMed.
- T. Čarný, R. Rocaboy, A. Clemenceau and O. Baudoin, Synthesis of Amides and Esters by Palladium (0)-Catalyzed Carbonylative C (sp3)− H Activation, Angew. Chem., Int. Ed., 2020, 59(43), 18980–18984 CrossRef.
- Q. Zhang, T. Yang, X. Liu, C. Yue, L. Ao, T. Deng and Y. Zhang, Heteropoly acid-encapsulated metal–organic framework as a stable and highly efficient nanocatalyst for esterification reaction, RSC Adv., 2019, 9(29), 16357–16365 RSC.
- B. Pieber, J. A. Malik, C. Cavedon, S. Gisbertz, A. Savateev, D. Cruz, T. Heil, G. Zhang and P. H. Seeberger, Semi-heterogeneous Dual Nickel/Photocatalysis using Carbon Nitrides: Esterification of Carboxylic Acids with Aryl Halides, Angew. Chem., Int. Ed., 2019, 58(28), 9575–9580 CrossRef CAS.
- B. Zhang, M. Gao, J. Geng, Y. Cheng, X. Wang, C. Wu, Q. Wang, S. Liu and S. M. Cheung, Catalytic performance and deactivation mechanism of a one-step sulfonated carbon-based solid-acid catalyst in an esterification reaction, Renewable Energy, 2021, 164, 824–832 CrossRef CAS.
- B. Zhang, M. Gao, W. Tang, X. Wang, C. Wu, Q. Wang, S. M. Cheung and X. Chen, Esterification efficiency improvement of carbon-based solid acid catalysts induced by biomass pretreatments: Intrinsic mechanism, Energy, 2023, 263, 125606 CrossRef CAS.
- A. F. Lee, J. A. Bennett, J. C. Manayil and K. Wilson, Heterogeneous catalysis for sustainable biodiesel production via esterification and transesterification, Chem. Soc. Rev., 2014, 43(22), 7887–7916 RSC.
- W.-J. Liu, K. Tian, H. Jiang and H.-Q. Yu, Facile synthesis of highly efficient and recyclable magnetic solid acid from biomass waste, Sci. Rep., 2013, 3(1), 1–7 Search PubMed.
- S. Chellappan, K. Aparna, C. Chingakham, V. Sajith and V. Nair, Microwave assisted biodiesel production using a novel Brønsted acid catalyst based on nanomagnetic biocomposite, Fuel, 2019, 246, 268–276 CrossRef CAS.
- Z. Yu, X. Chen, Y. Zhang, H. Tu, P. Pan, S. Li, Y. Han, M. Piao, J. Hu and F. Shi, Phosphotungstic acid and propylsulfonic acid bifunctionalized ordered mesoporous silica: A highly efficient and reusable catalysts for esterification of oleic acid, Chem. Eng. J., 2022, 430, 133059 CrossRef CAS.
- I. Ahmad and R. Dhar, Sulfonic acid-functionalized solid acid catalyst in esterification and transesterification reactions, Catal. Surv. Asia, 2017, 21(2), 53–69 CrossRef.
- S. Sadjadi, M. M. Heravi and M. Raja, Composite of ionic liquid decorated cyclodextrin nanosponge, graphene oxide and chitosan: a novel catalyst support, Int. J. Biol. Macromol., 2019, 122, 228–237 CrossRef CAS.
- S. Sadjadi, M. M. Heravi and S. S. Kazemi, Ionic liquid decorated chitosan hybridized with clay: A novel support for immobilizing Pd nanoparticles, Carbohydr. Polym., 2018, 200, 183–190 CrossRef CAS.
- P. Kakati, P. Singh, P. Yadav and S. K. Awasthi, Aiding the versatility of simple ammonium ionic liquids by the synthesis of bioactive 1, 2, 3, 4-tetrahydropyrimidine, 2-aminothiazole and quinazolinone derivatives, New J. Chem., 2021, 45(15), 6724–6738 RSC.
- S. Kumar, S. K. Dixit and S. K. Awasthi, An efficient one pot method for synthesis of carboxylic acids from nitriles using recyclable ionic liquid [bmim] HSO4, Tetrahedron Lett., 2014, 55(28), 3802–3804 CrossRef CAS.
- M. J. Earle, S. P. Katdare and K. R. Seddon, Paradigm confirmed: the first use of ionic liquids to dramatically influence the outcome of chemical reactions, Org. Lett., 2004, 6(5), 707–710 CrossRef CAS PubMed.
- M. J. Earle, P. B. McCormac and K. R. Seddon, Diels–Alder reactions in ionic liquids. A safe recyclable alternative to lithium perchlorate–diethyl ether mixtures, Green Chem., 1999, 1(1), 23–25 RSC.
- Y. Chauvin, L. Mussmann and H. Olivier, A novel class of versatile solvents for two-phase catalysis: hydrogenation, isomerization, and hydroformylation of alkenes catalyzed by rhodium complexes in liquid 1,3-dialkylimidazolium salts, Angew Chem. Int. Ed. Engl., 1996, 34(23–24), 2698–2700 CrossRef.
- M. A. Klingshirn, R. D. Rogers and K. H. Shaughnessy, Palladium-catalyzed hydroesterification of styrene derivatives in the presence of ionic liquids, J. Organomet. Chem., 2005, 690(15), 3620–3626 CrossRef CAS.
- J. Yadav, B. Reddy, G. Baishya, K. Reddy and A. Narsaiah, Conjugate addition of indoles to α,β-unsaturated ketones using Cu (OTf) 2 immobilized in ionic liquids, Tetrahedron, 2005, 61(40), 9541–9544 CrossRef CAS.
- A. Chinnappan and H. Kim, Environmentally benign catalyst: Synthesis, characterization, and properties of pyridinium dicationic molten salts (ionic liquids) and use of application in esterification, Chem. Eng. J., 2012, 187, 283–288 CrossRef CAS.
- S. G. Kalghatgi and B. M. Bhanage, Green syntheses of levulinate esters using ionic liquid 1-Methyl imidazolium hydrogen sulphate [MIM][HSO4] in solvent free system, J. Mol. Liq., 2019, 281, 70–80 CrossRef CAS.
- L. He, S. Qin, T. Chang, Y. Sun and X. Gao, Biodiesel synthesis from the esterification of free fatty acids and alcohol catalyzed by long-chain Brønsted acid ionic liquid, Catal. Sci. Technol., 2013, 3(4), 1102–1107 RSC.
- M. U. Khan, S. Siddiqui and Z. N. Siddiqui, Novel ionic liquid-functionalized chitosan [DSIM][AlCl3] x–@ CS: synthesis, characterization, and catalytic application for preparation of substituted pyrazine derivatives, ACS Omega, 2019, 4(4), 7586–7595 CrossRef CAS PubMed.
- Y. Wang, D. Zhao, G. Chen, S. Liu, N. Ji, H. Ding and J. Fu, Preparation of phosphotungstic acid based poly (ionic liquid) and its application to esterification of palmitic acid, Renewable Energy, 2019, 133, 317–324 CrossRef CAS.
- A. S. Amarasekara, Acidic ionic liquids, Chem. Rev., 2016, 116(10), 6133–6183 CrossRef CAS PubMed.
- Z. Xu, H. Wan, J. Miao, M. Han, C. Yang and G. Guan, Reusable and efficient polystyrene-supported acidic ionic liquid catalyst for esterifications, J. Mol. Catal. A: Chem., 2010, 332(1–2), 152–157 CrossRef CAS.
- J. Sun, J. Wang, W. Cheng, J. Zhang, X. Li, S. Zhang and Y. She, Chitosan functionalized ionic liquid as a recyclable biopolymer-supported catalyst for cycloaddition of CO 2, Green Chem., 2012, 14(3), 654–660 RSC.
- C. Thomazeau, H. Olivier-Bourbigou, L. Magna, S. Luts and B. Gilbert, Determination of an acidic scale in room temperature ionic liquids, J. Am. Chem. Soc., 2003, 125(18), 5264–5265 CrossRef CAS.
- V. Sadhasivam, M. Mathappan, M. Harikrishnan, C. Chithiraikumar, S. Murugesan and A. Siva, Pd (OAc) 2 immobilized on imine-functionalized microporous covalent triazine polymer as efficient heterogeneous catalyst for Mizoroki–Heck cross-coupling reaction, Res. Chem. Intermed., 2018, 44, 2853–2866 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05725b |
| This journal is © The Royal Society of Chemistry 2024 |