
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFuran-rich, biobased transfection agents as potential oligomeric candidates for intracellular plasmid DNA delivery†
Ashique Al Hoque ac,
Prakash Kannaboinab,
Yeabstega Abrahamd,
Masfique Mehedid,
Mukund P. Sibib and
Mohiuddin Quadir*a
ac,
Prakash Kannaboinab,
Yeabstega Abrahamd,
Masfique Mehedid,
Mukund P. Sibib and
Mohiuddin Quadir*a
aDepartment of Coatings and Polymeric Materials, North Dakota State University, Fargo, ND 58108, USA. E-mail: mohiuddin.quadir@ndus.edu; Tel: +1-701-231-6283
bDepartment of Chemistry Biochemistry, North Dakota State University, Fargo, ND 58108, USA
cDepartment of Pharmaceutical Technology, Jadavpur University, Kolkata, India
dDepartment of Biomedical Sciences, University of North Dakota, School of Medicine & Health, Grand Forks, ND 58202, USA
First published on 15th October 2024
Abstract
Biobased, DNA delivery vectors have been synthesized with a core motif composed of 2,5-bishydroxymethylfuran (BHMF) readily available from an important biomass feedstock 5-hydroxymethyl furfural (HMF). To generate the product, BHMF was first converted to 2,5-furan bishydroxymethyl diacrylate (2,5-FDA), which was later conjugated with different types of secondary amines. Rich in tertiary nitrogen, these oligomeric FDA-amino esters demonstrated stable electrostatic interactions with negatively charged plasmid DNA in an aqueous environment. We evaluated synthetic routes toward these plasmid DNA-binding amino esters (pFASTs), identified their nanoscale features, and attempted to establish their structure–property relationship in the context of the DNA delivery. Our preliminary studies show that the pFASTs formed stable complexes with the plasmid DNA. Dynamic light scattering indicated that the DNA polyplexes of pFASTs have hydrodynamic diameters within the size range of 100–150 nm with a surface charge (ζ-potential) ranging from −10 to +33 mV, depending on pFAST type. These oligomeric amino esters rich in furan motif were also found to successfully transfect the GFP-expressing plasmid DNA intracellularly. Collectively, this study establishes a new route to produce DNA transfection agents from sustainable resources that can be used for transferring genetic materials for humans, veterinary, and agrochemical purposes.
1. Introduction
Gene therapy is rapidly emerging as a new therapeutic approach for treating a variety of inherited and acquired diseases that were previously considered incurable. While viral vectors have been widely used for gene delivery in research and some have recently been approved for clinical use, concerns about their safety, including potential mutagenicity and immune reactions, persist. As a result, there is increasing interest in developing safer alternatives, such as polymeric nanomaterials, which offer promising properties for biomedical applications.1 This class of nanomaterials play a crucial role in gene therapy by serving as vectors for delivering genetic material into cells.2 Their versatility of the chemistry of polymers allows for the synthesis of nanoparticles that can be tailored to protect and transport nucleic acids efficiently. Polymers, particularly cationic variants, can form stable complexes with DNA or RNA through electrostatic interactions. Additionally, polymers can be engineered for controlled release of the nucleic acid, biocompatibility, and reduced toxicity, addressing key challenges in gene delivery. However, optimizing polymer vectors for efficient gene release and targeting remains a focus in advancing gene therapy applications.1 To optimize gene delivery properties of polymeric vectors, several key feature of the polymer can be adjusted. For example, excessive levels of cationic charge density can compromise cell membranes and provoke immune responses and inflammation, causing substantial cytotoxicity. Reducing these toxic effects can be achieved by masking these charged groups via covalent- or non-covalent modifications.1,3 Fine-tuning the topology of polymers, introduction of stimuli-sensitive units, and cell-specific targeting ligand further augment the biological efficiency of polymeric gene vectors, chemical stabilization of the genetic cargo, and minimization of cytotoxicity. In addition, variation of polymer topologies was found to improve the efficiency of gene delivery vectors.1,4 In this work, we describe a route to synthesize biobased gene transfection vector. There are no significant reports of biobased gene vectors in the literature. With the increasing emphasis on using sustainable resources to produce a value-added products, pharmaceuticals and biotech industries are pivoting towards the practice of green chemistry principles to improve the techno-environmental impacts of their manufacturing processes. Aligning with the current trend of sustainability in molecular design, we aim to generate a biobased, soft platform for intracellular transport of therapeutically active polynucleic acids, such as DNA, RNA, or other genetic fragments. One of the basic requirements of these biomacromolecules is to provide steric and electrostatic protection to the complexed nucleic acids.5–14 A large fraction of gene transfection agents bear poly-(β-amino ester) as their central molecular motif, owing to their biocompatible complexation with the polynucleotides, biodegradability, and ease of synthesis.15–31 Numerous modifications and structure–activity relationship studies have been done on this class of gene transfection agents, identifying that the efficiency of these amine-rich esters in complexing genetic materials depends on the type, spacing, and charge density of the cationic centers along the macromolecular backbone.28,29,32–37 However, none of these compounds are enriched in monomers derived from sustainable chemical alternatives. We opted out to demonstrate the feasibility of synthesis of oligomeric gene-transfection agents which are composed of monomeric units directly derived from bio-derived starting materials. Our central hypothesis is the fact that, β-amino esters formed via Michael addition between a secondary amine, often time as part of a heterocyclic ring, and an aliphatic biobased acrylate will act as an efficient plasmid DNA vehicle.22,33,38–45 We are particularly interested to investigate how a biobased, furan-based acrylate monomers, that can be connected to bifunctional secondary amines via β-amino ester linkages, and how spacing of the aromatic furan rings within the oligomeric chain affects their DNA complexation capabilities. Our interest stems from two reasons – (1) furans when coupled with an appropriate combination of amines, are supposed to provide interesting steric effects due to molecular planarity and symmetry of the furan conducive to gene transfection,19,46,47 and (2) furan-based acrylates can be obtained in large quantity from biobased materials, and the use of materials obtained with reduced carbon footprint is slowly gaining traction in biotechnology research.48 Therefore, with an aim to expand the molecular repertoire of gene transfection agents with high biobased contents, and to establish a synthetic route to access these macromolecules, we synthesized new β-amino ester-linked macromolecules derived from biobased, 2,5-bishydroxymethyl furan (BHMF). We first prepared bis(acrylate) ester of BHMF, 2,5-furan diacrylate (FDA), which was then reacted with different types of secondary amines via Michael addition to produce plasmid DNA-complexing, furan-amino esters, abbreviated as pFASTs. It is important to note that, the BHMF is a reduction product of 5-hydroxymethylfurfural (HMF), which in turn, is a value-added product of conversion of biomass-derived carbohydrates. As an important biobased platform, HMF can be produced efficiently through the dehydration of carbohydrates, acid-catalyzed hydrolysis and dehydration of cellulose, and even from food waste. This compound features two reactive sites (at the C2 and C5 positions of the furan ring) and an aromatic structure, making it highly versatile for various chemical reactions. As a result, HMF and its derivatives are valuable for creating polymer architectures, as their bi-functional nature enables the formation of diols, diacids, and diamines.49 As such, HMF is listed among the top 14 biobased chemicals by the U.S. Department of Energy.50 To prove our hypothesis that biobased, furan-rich, β-amino ester linked oligomers are able to interact with genetic materials, we evaluated as a proof-of-concept, DNA binding and non-cytotoxic transfection capacities of these compounds inside eukaryotic cells to interact with gene expression machinery. We observed that pFASTs indeed mediate gene transport via the formation of a complex of distinct nanoscale features, i.e., hydrodynamic diameter and zeta (ζ-potential). Collectively, this work demonstrates for the first time the synthetic route of biobased polyamines based on furan acrylates that can potentially be applied for gene delivery applications.2. Materials and methods
2.1. Reagents and instrumentation
Unless otherwise stated, all commercially procured materials were used as received without further purification. 5-Hydroxymethylfurfural was purchased from Avantium and RANEY® Ni catalyst from Sigma-Aldrich. All mention of silica gel refers to Sorbtech standard grade silica gel: 230–400 mesh. N,N′-Dimethylethylenediamine, sodium borohydride, piperazine, and 4,4′-trimethylenedipiperidine were purchased from Sigma-Aldrich (St. Louis, MO, USA). All reagents were used without further purification. Plasmid DNA (pBR322) was purchased from (ThermoFisher Scientific, USA). HFF2, L929, MiaPaca2 cells (ATCC, Manassas, VA, USA) were cultured in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin–streptomycin. Quant-IT PicoGreen dsDNA reagent was purchased from Invitrogen. Melting points were determined on a REACH Devices RDMP digital melting point apparatus and are uncorrected. Nuclear magnetic resonance spectra were recorded on a Bruker Avance 400 MHz instrument and processed with Topspin software. The FTIR figures were prepared using Origin Pro (ESI Fig. S1†). High resolution mass spectra were recorded on Waters Synapt G2-Si high-definition mass spectrometer and were processed using MassLynx (ESI, Fig. S2†). Unless otherwise stated, all reactions were stirred magnetically by polytetrafluoroethylene coated magnetic spin-bars.2.2. Synthesis of 2,5-bis(hydroxymethyl)furan
To a 2000 mL round bottom flask, 5-hydroxymethylfurfural (100 g, 793 mmol) was added in portions to deionized water (200 mL), and then the mixture was chilled on an ice bath. Sodium borohydride (30 g, 793 mmol) was carefully added to the open flask in a well-ventilated fume hood as it was stirred magnetically. The reaction mixture was stirred on ice for 2 h then allowed to react at room temperature overnight. Sodium chloride was added to the reaction mixture, and the mixture was extracted with ethyl acetate at 0 °C several times until no product in the aqueous phase were available. The organic phase was then dried with Na2SO4. The white crystalline solid (99.2 g, 97% yield) was directly used in next step without further purification. The obtained spectroscopic and analytical data for the product corresponded to those reported in the literature.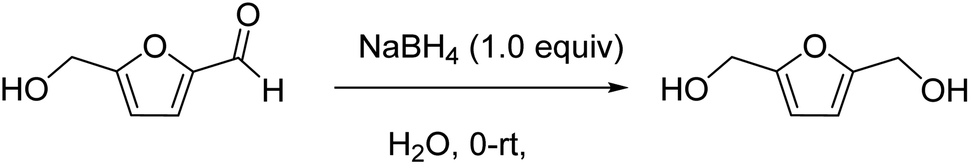
1H (400 MHz, MeOH-d4) δ 6.23 (s, 2H), 4.84 (bs, 2H), 4.48 (s, 4H); 1H (400 MHz, CDCl3) δ 6.10 (s, 2H), 4.42 (s, 4H), 4.35 (bs, 2H); 1H (400 MHz, D2O) δ 6.38 (s, 2H), 4.58 (s, 4H); 13C (101 MHz, MeOH-d4) δ 155.7 (2C), 109.1 (2C), 57.4 (2C); 13C (101 MHz, D2O) δ 153.7 (2C), 109.1 (2C), 55.8 (2C). (NMR spectra is shown in ESI Fig. S2A†).
2.3. Procedure for synthesis of 2,5-bishydroxymethyl furan diacrylate
2,5-Bishydroxymethyl furan diacrylate (2,5-FDA) was prepared through the reaction of hydroxyl groups of BHMF with acryloyl chloride.3 Triethylamine (3.48 mL, 0.025 mol) was added to a solution of BHMF (1.28 g, 0.01 mol) in THF (100 mL) at 0 °C under a nitrogen atmosphere. The mixture was stirred at 0 °C for 15 min. Acryloyl chloride (2.17 mL, 0.025 mol) was slowly added to the solution, and the reaction mixture was stirred for 12 h at room temperature. After the reaction, the solution was filtered to remove triethylamine hydrochloride. The filtrate was concentrated by rotary evaporation. The residue was dissolved in ethyl acetate, and then washed with brine, dried with Na2SO4, and concentrated under vacuum to give a yellow oil. The product was further purified on a silica gel column (0–50% ethyl acetate/hexane) to yield 2,5-bishydroxymethyl furan diacrylate (1.32 g, 56% yield).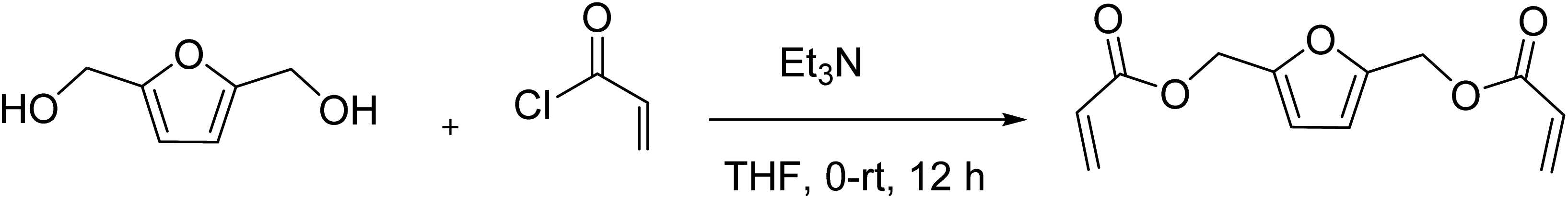
1H NMR (400 MHz, CDCl3) δ 6.45 (d, J = 1.4 Hz, 1H), 6.41 (d, J = 2.6 Hz, 1H), 6.40 (d, J = 1.4 Hz, 2H), 6.15 (dd, J = 10.5, 2.3 Hz, 1H), 6.11 (dd, J = 10.4, 2.3 Hz, 1H), 5.85 (d, J = 1.4 Hz, 1H), 5.83 (d, J = 1.5 Hz, 1H), 5.12 (s, 4H). 13C NMR (100 MHz, CDCl3) δ 165.4, 150.0, 131.3, 127.8, 111.6, 57.9. FTIR (neat) cm−1: 3010, 1725, 1409, 1255, 1172, 1041, 985, 809; HRMS (ESI) calcd for C12H12O5Na+ [M + Na]+ 259.0577, found 259.0583 (NMR spectrum is shown in ESI Fig. S3 and S4†).
2.4. Synthesis of pFASTs
2,5-Furan diacrylate (2,5-FDA) was combined at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratios with three distinct diamines, including N,N′-dimethylethylenediamine, piperazine, and 4,4′-trimethylenedipiperidine, to synthesize three different biobased poly-β-amino esters (pFASTs), such as pFAST 1, 2, and 3. The reaction procedure was modified from that reported earlier (0.5 mM FDCA, 1 mM diamines).5,38,40,51 Individual amines and FDCA were first weighed into two separate glass vials and then dissolved in THF (2 mL). The diamine-containing solution was incorporated into the diacrylate solution via slow addition. The reaction vial was sealed with a Teflon-lined screw cap, and the reaction was heated to 50 °C with constant stirring. The reaction was allowed to run for 72 h after which the mixture was cooled to ambient temperature. The reaction mixture was poured gently into diethyl ether, and the ether suspension was stirred vigorously to initiate precipitation. Finally, the suspension was centrifuged at 10000 rpm to produce pFAST pellets, which were collected via decantation of the ether supernatant. The pellet was dried under a vacuum prior to analysis. The reaction pathways are shown in Scheme 1 in the manuscript. The synthetic pathway is shown in Scheme 1 later.
:2 molar ratios with three distinct diamines, including N,N′-dimethylethylenediamine, piperazine, and 4,4′-trimethylenedipiperidine, to synthesize three different biobased poly-β-amino esters (pFASTs), such as pFAST 1, 2, and 3. The reaction procedure was modified from that reported earlier (0.5 mM FDCA, 1 mM diamines).5,38,40,51 Individual amines and FDCA were first weighed into two separate glass vials and then dissolved in THF (2 mL). The diamine-containing solution was incorporated into the diacrylate solution via slow addition. The reaction vial was sealed with a Teflon-lined screw cap, and the reaction was heated to 50 °C with constant stirring. The reaction was allowed to run for 72 h after which the mixture was cooled to ambient temperature. The reaction mixture was poured gently into diethyl ether, and the ether suspension was stirred vigorously to initiate precipitation. Finally, the suspension was centrifuged at 10000 rpm to produce pFAST pellets, which were collected via decantation of the ether supernatant. The pellet was dried under a vacuum prior to analysis. The reaction pathways are shown in Scheme 1 in the manuscript. The synthetic pathway is shown in Scheme 1 later.
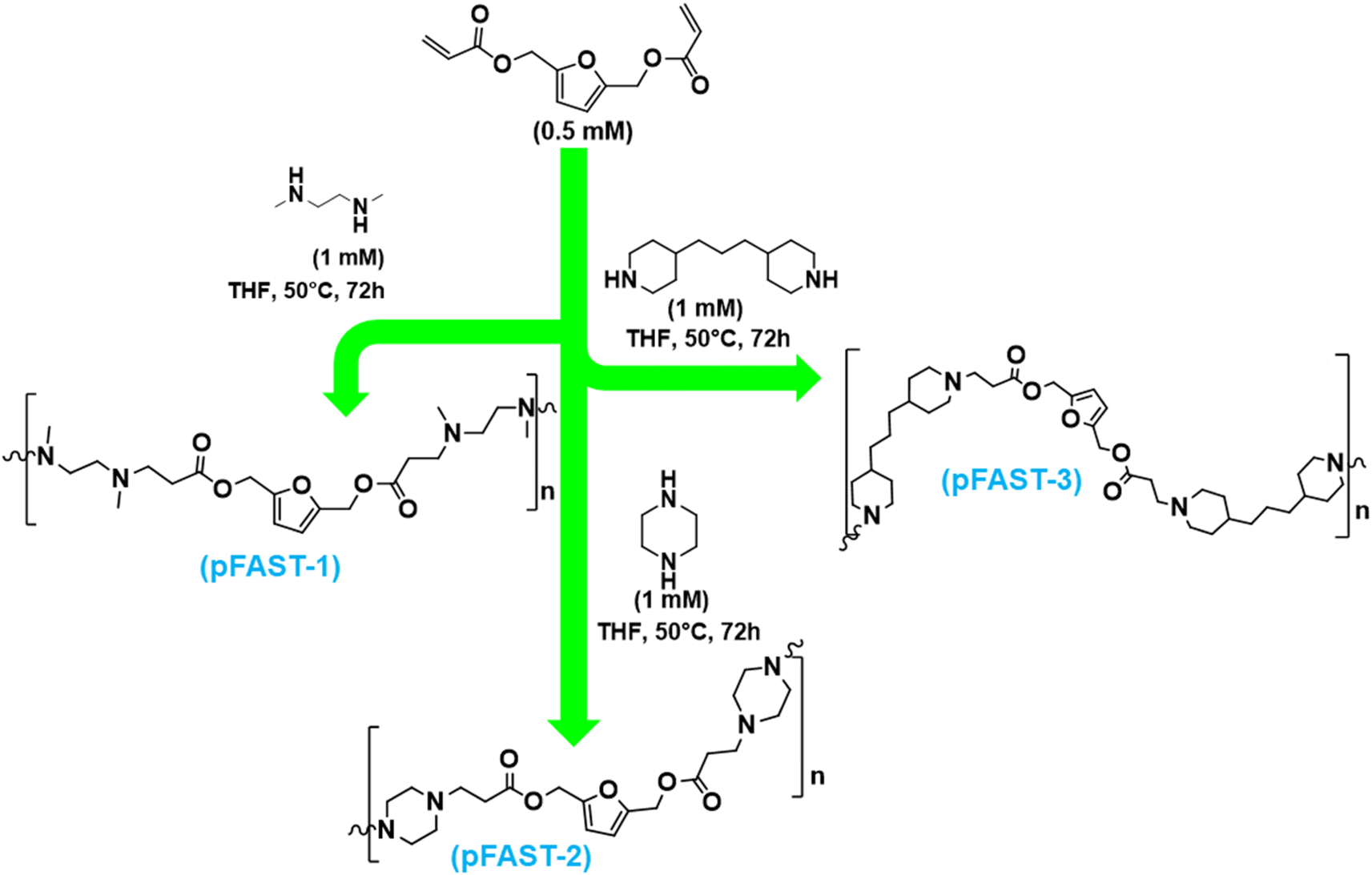 | ||
| Scheme 1 Synthetic pathway to pFAST-1, 2 and 3 via Michael addition of secondary amines to 2,5-FDA. These products were purified by repeated precipitation from diethyl ether. | ||
2.5. Chemical characterization of pFASTs
pFAST-1: 1H NMR (400 MHz, CDCl3) δ 6.39 (s, 2H), 4.92 (s, 4H), 3.30 (dd, J = 10.5, 5.3 Hz, 2H), 2.51–2.41 (m, 5H), 2.34 (t, J = 5.3 Hz, 5H), 2.25 (s, 4H), 2.02 (s, 6H). 13C NMR (100 MHz, DMSO) δ 171.6, 150.0, 111.6, 57.5, 54.7, 52.7, 41.9, 32.0. FTIR (neat) cm−1: 3310, 1730, 1590, 1385, 1172, 1020. pFAST-2: 1H NMR (400 MHz, CDCl3) δ 6.36 (d, J = 1.4 Hz, 2H), 5.04 (d, J = 2.7 Hz, 4H), 3.47 (q, J = 7.0 Hz, 2H), 2.85 (t, J = 4.7 Hz, 3H), 2.68 (t, J = 7.2 Hz, 5H), 2.58–2.46 (m, 7H), 2.46–2.37 (m, 7H). 13NMR (100 MHz, DMSO) δ 171.6, 150.0, 111.6, 57.5, 53.1, 52.4, 31.7. FTIR (neat) cm−1: 3370, 1730, 1590, 1380, 1172, 1020. pFAST-3: 1H NMR (400 MHz, CDCl3) δ 6.36 (s, 2H), 5.03 (s, 4H), 3.47 (q, J = 5.3 Hz, 2H), 2.83 (d, J = 7.8 Hz, 6H), 2.66 (t, J = 5.5 Hz, 6H), 2.53 (t, J = 5.5 Hz, 6H), 1.92 (t, J = 7.9 Hz, 6H), 1.87–1.81 (m, 2H), 1.62 (d, J = 6.3 Hz, 6H), 1.31–1.03 (m, 22). 13CNMR (100 MHz, CDCl3) δ 172.3, 150.2, 111.6, 58.1, 53.8, 36.8, 35.7, 32.4, 32.3, 25.7, 24.0, 15.4. FTIR (neat) cm−1: 3370, 1730, 1590, 1385, 1172, 1020.
2.6. pFAST–DNA polyplex formation and characterization
:10, 1:50, 1:100). The pFAST solutions were prepared in 100 μL in 25 mM sodium acetate buffer, pH-5.5. The concentrations were adjusted to yield desired DNA/pFAST weight ratios. The samples were incubated at room temperature for 30 min for the polyplex formation.:10, 1:50, and 1:100 w/w. The solutions were thoroughly mixed and left undisturbed for 5 minutes to allow for pFAST–DNA complexation to take place. Following this, 100 μL per well of PicoGreen solution was added. This working solution was prepared by diluting 80 μL of stock solution into 15.2 mL sodium acetate buffer. In a black 96-well polystyrene plate, 30 μL per well of the pFAST–DNA, PicoGreen solution was added at 200 μL per well of DMEM medium after 5 minutes. The fluorescence of the plate was then measured using a SpectraMax microplate reader at an excitation wavelength of 485 nm and an emission wavelength of 535 nm. The following equation was used to compute the relative fluorescence (RF):where Fsample is the fluorescence of a pFAST–DNA–PicoGreen sample, Fblank is the fluorescence of a sample that contains neither pFAST nor DNA (only PicoGreen), and FDNA is the fluorescence of a DNA–PicoGreen sample (no pFASTs).
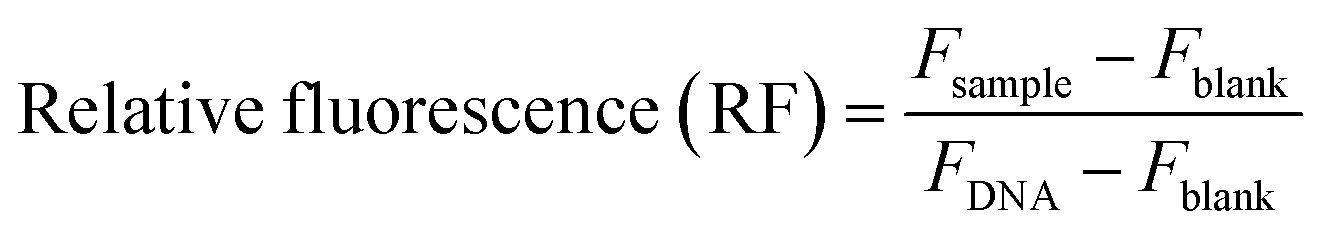 :10, 1:50:1:100 w/w of DNA-to-pFAST ratio. After incubation for 5 min at room temperature, 1.2 mL of 10% serum-containing medium was added to the mixture, which was then immediately subjected to DLS measurements using a zeta sizer (Malvern zeta sizer).000 cells in each well) overnight. On the next day, cells were treated with pFASTs, branched PEI of 25 kDa, and poly(ethylene glycol)-b-poly(L-lysine) of 21 kDa at various concentrations from 10–100 μg mL−1. Here branched PEI and poly(ethylene glycol)-b-poly(L-lysine) were used as controls. AlamarBlue was added to the wells (10% of the media volume) 24, 48, and 72 h of after the initiation of the treatment, and the cells were incubated under 5% CO2 at 37 °C for an additional 3–4 h. Finally, the fluorescence intensities of the samples were checked in a microplate reader (SpectraMax®) at an excitation and an emission wavelength of 560 and 590 nm, respectively.
:10, 1:50:1:100 w/w of DNA-to-pFAST ratio. After incubation for 5 min at room temperature, 1.2 mL of 10% serum-containing medium was added to the mixture, which was then immediately subjected to DLS measurements using a zeta sizer (Malvern zeta sizer).000 cells in each well) overnight. On the next day, cells were treated with pFASTs, branched PEI of 25 kDa, and poly(ethylene glycol)-b-poly(L-lysine) of 21 kDa at various concentrations from 10–100 μg mL−1. Here branched PEI and poly(ethylene glycol)-b-poly(L-lysine) were used as controls. AlamarBlue was added to the wells (10% of the media volume) 24, 48, and 72 h of after the initiation of the treatment, and the cells were incubated under 5% CO2 at 37 °C for an additional 3–4 h. Finally, the fluorescence intensities of the samples were checked in a microplate reader (SpectraMax®) at an excitation and an emission wavelength of 560 and 590 nm, respectively.2.7. Transfection experiments
:500, in 1× DPBS) (Cytoskeleton Inc.) for 30 minutes. Similarly, the cells were stained with Nunc Blue nuclear dye (DAPI) (2 drops in 1000 μL 1× DPBS) (Life Technologies Inc.) for 20 minutes. The coverslip was mounted on a cover glass using Prolong Gold mounting media (Life Technologies Inc.). The slides were Z-stack imaged under a Leica DMI8 fluorescence microscope (Leica Microsystems) or an Olympus FV3000 confocal microscope (Olympus) using a 63× oil objective. The images were processed using IMARIS image processing software (Oxford Instruments).3. Results and discussion
3.1. Chemical synthesis and characterization of biobased DNA transfection agents
To evaluate the feasibility of our strategy, first, we prepared three different pFASTs by using biomass-based 2,5-furan diacrylate (2,5-FDA). This compound, i.e., 2,5-FDA was prepared through the reaction of hydroxyl groups of BHMF with acryloyl chloride, which was purified as a yellow oil.3 In the presence of triethylamine, this reaction was conducted in THF at 0 °C under a nitrogen atmosphere. After silica gel column (0–50% ethyl acetate/hexane) purification, it was possible to synthesize 2,5-FDA at 56% yield. This compound was then reacted with three different secondary amines to produce the PFAST motifs, namely pFAST 1–3 (Scheme 1). The route of acrylate reactions to secondary amines is well-studied in polymer synthesis. For our studies, we reacted 2,5-FDA at 1:2 molar ratios with three distinct diamines, including N,N′-dimethylethylenediamine, piperazine, and 4,4′-trimethylenedipiperidine, to synthesize pFAST 1, 2, and 3, respectively (Scheme 1). A molar ratio of 0.5 mM FDA to 1 mM diamine was maintained. The reactions were carried out in the solution phase, where individual amines and FDA were dissolved in THF (2 mL) separately and then the diamine-containing solutions were added slowly into the diacrylate solution. The reaction vials were not required to purge under inert conditions, however, were tightly sealed with a teflon-lined screw cap. The reaction mixture was heated to 50 °C for 72 h. We employed multiple precipitations from diethyl ether after the stipulated reaction period, followed by centrifugation at 10000 rpm to collect the products. The purified compounds were soluble in THF and in water of acidic pH. The pFAST products were chemically confirmed through FTIR, NMR, and mass spectrometry (ESI, Fig. S1–S6†).
Our initial proof-of-synthesis was derived from FTIR spectra of pFAST products when compared with the amines and furan diacrylate (Fig. S1†). While N,N-dimethylethylenediamine, piperazine, and 4,4-trimethylenedipiperdine showed characteristic peaks around 3230 cm−1 for N–H stretching, 2840 cm−1 for C–H stretching, 1450 cm−1 for C–H bending (methyl group/methylene group), 1135 cm−1 for C–N stretching, and furan diacrylate showed peaks at 1725 cm−1 for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, 1172 cm−1 for C–O stretching (ester), and 985 cm−1 for CC bending (monosubstituted alkene), the synthesized pFASTs showed IR peaks around 3310 cm−1 for N–H stretching, 1730 cm−1 for CO stretching, 1172 cm−1 for C–O stretching (ester), and 1020 cm−1 for C–N stretching. The peak at 985 cm−1 for CC disappeared in all three pFASTs, indicating the chemical reaction that occurred between the CC bond with the amine via the Michael addition route. Since the addition of monomers is a step-growth in nature, we did not expect a very high m.w. macromolecules. We calculated the number average molecular weight of the pFAST products using gel permeation chromatography (GPC) using THF as solvent. The molecular weights were further validated by MALDI-ToF. The polydispersity indices for the pFAST products were found to be within the range of 1.1–1.2. As such, MALDI-ToF (for pFAST-2 and 3) experiment showed that pFAST-2 produced molecules with mass of 1.6 kDa while, with pFAST-3, molecular mass of 1.0 kDa was obtained (ESI, Fig. S2†). Analogous to earlier studies, all pFAST derivatives showed enhanced solubility in acidic environment (pH 5.0–6.0). Finally, 1H and 13C NMR spectroscopy was used to confirm the structures of the starting material, 2,5-diacrylate (Fig. S3 and S4†) and pFASTs (Fig. S5–S10†). We aimed to assign the characteristic signals for individual protons within the segment of the respective pFAST molecule. Analysis of 1H and 13C NMR spectra clearly revealed that furan diacrylates reacted almost quantitatively with secondary amines, where the protons adjacent to the aromatic furan rings resonate at 4.92, 5.04, and 5.03 ppm belonging to (b, 2H, –CH2-) pFAST 1, 2 and 3 respectively (Fig. S5, S7 and S9†). Further, amine proton resonance from the newly formed C–N bond linkages was at 2.34–2.41, 3.30, 2.85, 3.47, and 2.83, 3.47 ppm belongs to (d, 2H, –CH2N-) pFAST 1, 2, and 3, respectively indicating the success of Micheal addition between the diacrylate and the amines (Fig. S5, S7 and S9†). Similarly from 13C NMR, the characteristic carbon resonance signals from the ester linkages of pFAST 1, 2 and 3 were observed at 171.6, 171.6, and 171.3 ppm (d, –CO-) (Fig. S6, S8 and S10†).
O stretching, 1172 cm−1 for C–O stretching (ester), and 985 cm−1 for CC bending (monosubstituted alkene), the synthesized pFASTs showed IR peaks around 3310 cm−1 for N–H stretching, 1730 cm−1 for CO stretching, 1172 cm−1 for C–O stretching (ester), and 1020 cm−1 for C–N stretching. The peak at 985 cm−1 for CC disappeared in all three pFASTs, indicating the chemical reaction that occurred between the CC bond with the amine via the Michael addition route. Since the addition of monomers is a step-growth in nature, we did not expect a very high m.w. macromolecules. We calculated the number average molecular weight of the pFAST products using gel permeation chromatography (GPC) using THF as solvent. The molecular weights were further validated by MALDI-ToF. The polydispersity indices for the pFAST products were found to be within the range of 1.1–1.2. As such, MALDI-ToF (for pFAST-2 and 3) experiment showed that pFAST-2 produced molecules with mass of 1.6 kDa while, with pFAST-3, molecular mass of 1.0 kDa was obtained (ESI, Fig. S2†). Analogous to earlier studies, all pFAST derivatives showed enhanced solubility in acidic environment (pH 5.0–6.0). Finally, 1H and 13C NMR spectroscopy was used to confirm the structures of the starting material, 2,5-diacrylate (Fig. S3 and S4†) and pFASTs (Fig. S5–S10†). We aimed to assign the characteristic signals for individual protons within the segment of the respective pFAST molecule. Analysis of 1H and 13C NMR spectra clearly revealed that furan diacrylates reacted almost quantitatively with secondary amines, where the protons adjacent to the aromatic furan rings resonate at 4.92, 5.04, and 5.03 ppm belonging to (b, 2H, –CH2-) pFAST 1, 2 and 3 respectively (Fig. S5, S7 and S9†). Further, amine proton resonance from the newly formed C–N bond linkages was at 2.34–2.41, 3.30, 2.85, 3.47, and 2.83, 3.47 ppm belongs to (d, 2H, –CH2N-) pFAST 1, 2, and 3, respectively indicating the success of Micheal addition between the diacrylate and the amines (Fig. S5, S7 and S9†). Similarly from 13C NMR, the characteristic carbon resonance signals from the ester linkages of pFAST 1, 2 and 3 were observed at 171.6, 171.6, and 171.3 ppm (d, –CO-) (Fig. S6, S8 and S10†).
3.2. Self-assembly of furan-based pFASTs with DNA
Once the pFASTs were synthesized, we demonstrated their capacity to bind to DNA using fluorescent probes. These probes interact with nucleic acids and form fluorescence complexes that serve as indicators useable in biomedical tests and bioanalytical procedures.52,53 We used PicoGreen (PG) as a fluorescent probe for measuring dye/DNA interactions.54 This molecule binds with dsDNA and creates a highly luminous complex. As such, the fluorescence intensity of PG upon binding to DNA increases dramatically compared to the free dye in the solution. Assuming that the fluorescence intensity is a linear function of bound PG, the change in intensity as a function of the PG in solution may be used to determine the proportion of bound PG during DNA titration with the dye. Our experiment revealed that DNA/pFAST at a 1:10 ratio demonstrated the least binding affinities, while binding increases as the ratio of DNA/pFAST increases from 1:50 to 1:100 ratios. The observation was consistent for all three pFAST products (Fig. 1A). The poly-(β-amino ester), pFAST 1 demonstrated the least extent of DNA binding at any given DNA-to-pFAST ratio from 1:10 to 1:100. The pFAST-1 product was followed by pFAST-3 in terms of DNA binding, while pFAST-2 showed the strongest extent of interactions with the DNA. Relatively weaker binding interactions of DNA with pFAST-1 and pFAST-3 can be attributed to increased steric shielding (for pFAST-1) or non-conducive spacing (for pFAST-3) between the cationic sites of the polymer and the DNA. As a control, we evaluated the DNA binding efficiency of two polymers, namely poly-(ethyleneimine), PEI, and poly(ethylene glycol)-b-poly(L-lysine), abbreviated as PEG-PLK. We used PEI because of their known DNA-binding capacity as well as its role as a well-established positive control (with known cytotoxic character). PEG-PLK on the other hand, is a copolymer, where the cytotoxicity of poly-(L-lysine) residue is masked by the PEG chain. We did not use popular gene transfection agents, such as lipofectamine, This is because, lipofectamine contains a 3:1 mixture of DOSPA (2,3-dioleoyloxy-N-[2(sperminecarboxamido)ethyl]-N,N-dimethyl-1-propaniminium trifluoroacetate) and DOPE (1,2-dioleoyl-sn-glycerophosphoethanolamine) and the mechanism of gene transfection does not match with that of our current product. Interestingly, at a similar concentration range where PG-assay was performed for pFAST products, PEI showed negative fluorescence relative to the control and PEG-PLKC showed reduced DNA binding efficiency at DNA: macromolecule ratio of 1:10 and 1:50 (data not shown). This is most likely the over-compaction of DNA with PEI under the tested concentration. For PEG-conjugated poly(L-lysine), the DNA binding occurs due to the involvement of lysine units with the DNA molecule. It was interesting to observe that at DNA:macromolecule ratio of 1:100, all pFAST products and PEG-PLK showed equivalent DNA binding efficiency. We further validated the capacity of pFASTs to complex plasmid DNA via an agarose gel shift assay. This assay uses both size and charge to separate macromolecules. We used agarose gel electrophoretic assay because this is a straightforward approach to investigate the stoichiometric ratio at which complete DNA charge neutralization is achieved when DNAs are complexing with a polycation present in an increasing concentration in an agarose gel. For this experiment, we prepared DNA/pFAST polyplexes of various ratios (1:10, 1:50, and 1:100) and ran the complexes in an agarose gel plate to check the DNA binding efficiencies of the pFASTs. Fig. 1B shows the results of the electrophoretic mobility assay, which clearly suggests that all three pFASTs showed the most stable binding with a plasmid DNA at a DNA-to-macromolecule ratio of 1:100 irrespective of their interaction affinity obtained via the PG assay.
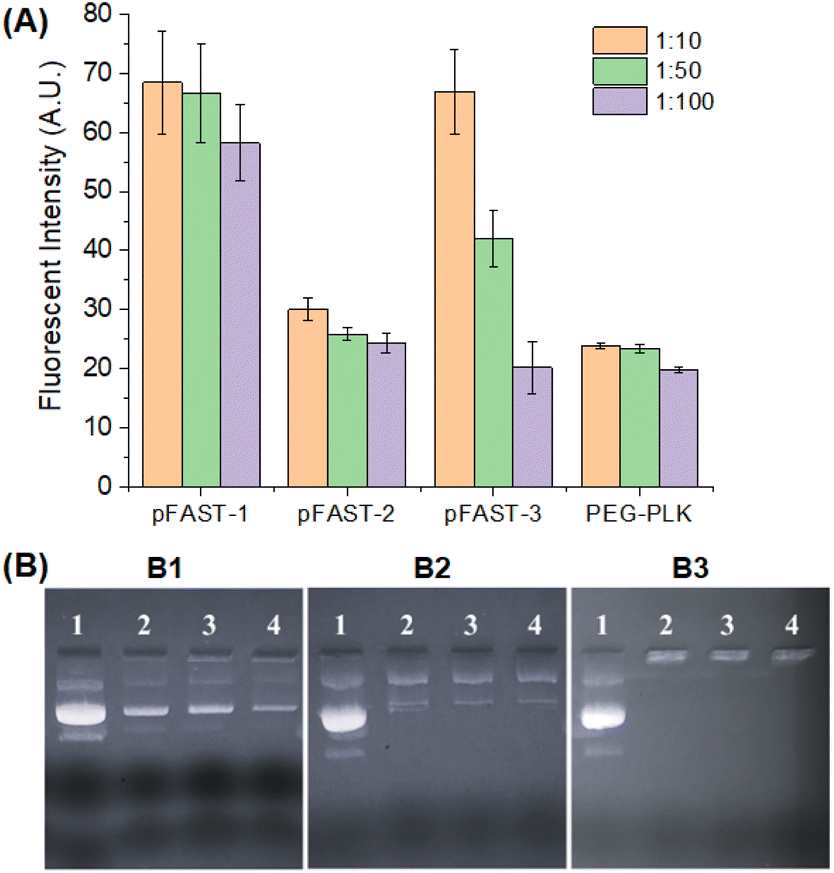 | ||
| Fig. 1 (A) PicoGreen assay indicating the extent of DNA complexation with pFAST systems where PEG-PLK was used as a control (B) DNA binding assay through agarose gel retardation. (B1) DNA–pFASTs at 1:10 ratio where wells 1–4 represent free DNA, and DNA complexed with pFAST-1, pFAST-2, and pFAST-3 respectively, (B2) DNA–pFASTs at 1:50 ratio, and (B3) DNA:pFASTs at 1:100 ratio where wells 1–4 represents free DNA, pFAST-1, pFAST-2, and pFAST-3, respectively. | ||
Fig. 2 shows the nanoscale features of the pFAST polyplexes with plasmid DNA. We anticipated that, interactions of negatively charged polyelectrolytes such as DNA with cationic pFASTs, will result in the formation of stable polyplexes, which are colloidal and stable DNA complexes with distinct nanoscale features. The nanoscale features include hydrodynamic diameters of the complexes that can be detected via dynamic light scattering (DLS) and surface charge directed by the polyelectrolytes present within the media. Since DNA types can also affect the colloidal stability of the polyplexes, we used two types of DNA molecules to perform DLS studies, namely pBR322 and gWiz-GFP plasmid DNA. Analyzing the DLS data for the hydrodynamic diameters of the polyplexes formed at different pBR322 DNA/pFAST ratios (1:10, 1:50, and 1:100) for all three types of pFASTs, were within the range of 65–175 nm. The average effective number average diameters of particles made from pBR322, and pFASTs at various DNA/pFAST ratios are shown in Fig. 2A. The volume average particle size are presented in the ESI Fig. S11.† Polydispersity index for all pFAST/DNA complexes were found to be <0.3. The surface charge (in terms of ζ-potential) of these colloidal polyplexes is important for their stability and transfection capacities. Therefore, we measured the ζ-potential and particle size of polyplexes as a function of DNA and pFAST ratio (Fig. 2B). For this experiment we used the DNA that will be used in transfection experiment later (gWiz-GFP). We observed that at a similar DNA/pFAST ratio, polyplexes were positively charged when pFASTs were complexed with gWiz-GFP DNA. For example, at DNA:pFAST ratio of 10, 50, and 100, the gWiz-GFP DNA/pFAST-3 polyplexes demonstrated zeta potentials of 19, 33, and 27 mV, respectively (Fig. 2B).
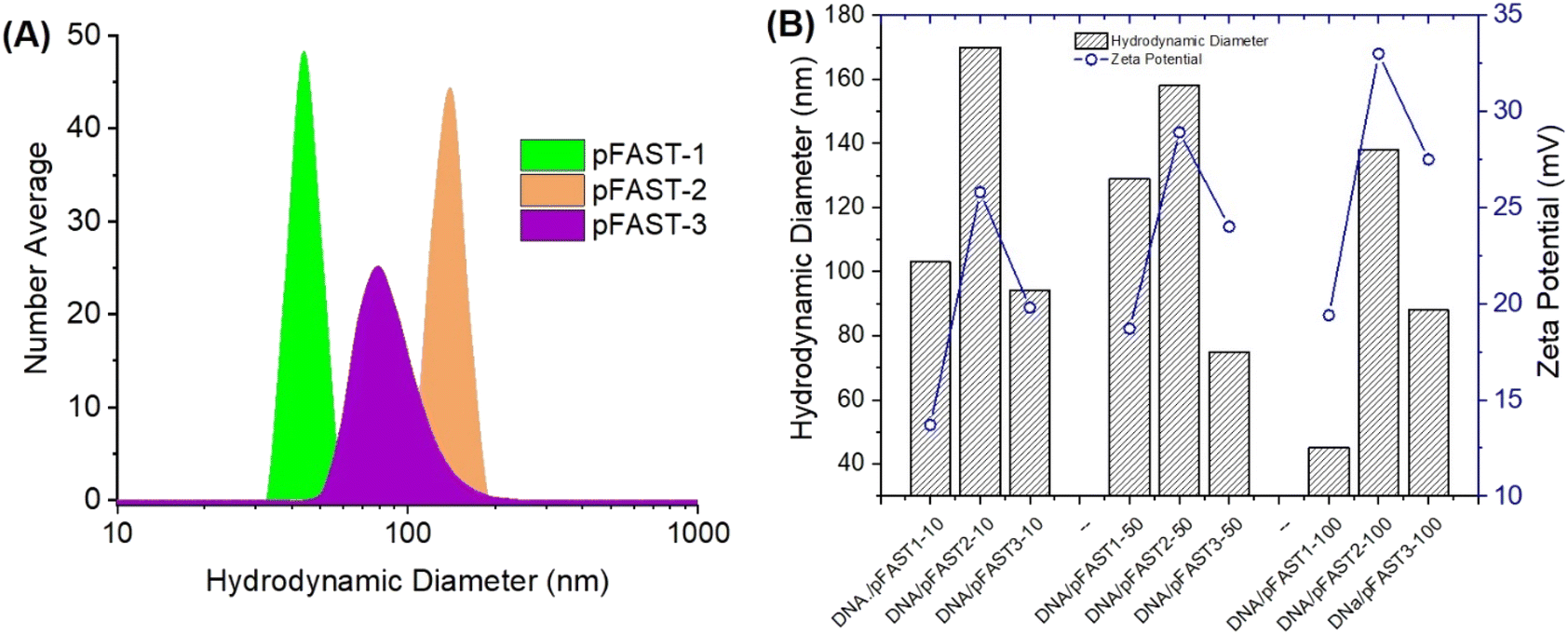 | ||
| Fig. 2 (A) Particle size distribution of pBR322 DNA/pFAST polyplexes prepared with pFAST-1–3 at a DNA:pFAST ratio of 1:50. (B) Effect of complexation ratio of gWiz-GFP DNA/pFAST on the hydrodynamic diameters of polyplexes as a function of pFAST type. | ||
We imaged the complexes of DNA/pFASTs using transmission electron microscopy (TEM). Evaluation of TEM images demonstrated the shape and morphology of stable polyplexes formed via interactions of DNA with pFASTs.46,55–58 As shown in Fig. 3, TEM experiments revealed compact, spherical morphologies of the DNA/pFAST polyplexes with sizes around 60, 150, and 90 nm for the pFAST-1, pFAST-2, and pFAST-3 systems, respectively. Similar to the observation of many preceding work, the positive ζ-potentials of these complexes may play a pivotal role beyond particle size and stability, as net positive charges on particle surfaces may play a role in initiating endocytosis, prompting efficient DNA transfection intracellularly.59,60
 | ||
| Fig. 3 Transmission electron microscopy of DNA/pFAST polyplexes, (A) pFAST-1, (B) pFAST-2 and (C) pFAST-3. | ||
3.3. Evaluation of cytotoxicity and gene transfection capabilities
We evaluated the global biocompatibility of pFAST systems for two types of cells. We used a non-cancerous cell lines, such as HFF2 fibroblasts, and cells with KRAS mutation, such as MIA PaCa 2 (pancreatic ductal adenocarcinoma), and treated the cells for 72 h with the polymeric vectors alone (not complexed with DNA, Fig. 4). We used PEI, and PEG-b-poly(L-lysine) (PEG-PLK) as control for this experiment. We observed that the polymeric vectors studies did not demonstrate significant cytotoxicity against both the cell lines up to 100 μg mL−1 of pFAST (not complexed with DNA) concentration. As such, in terms of biocompatibility, pFAST products showed a comparable cytotoxicity profile as that of PEG-PLK and were far superior to that of the branched PEI systems, which are reported to produce significant cellular toxicity.14,26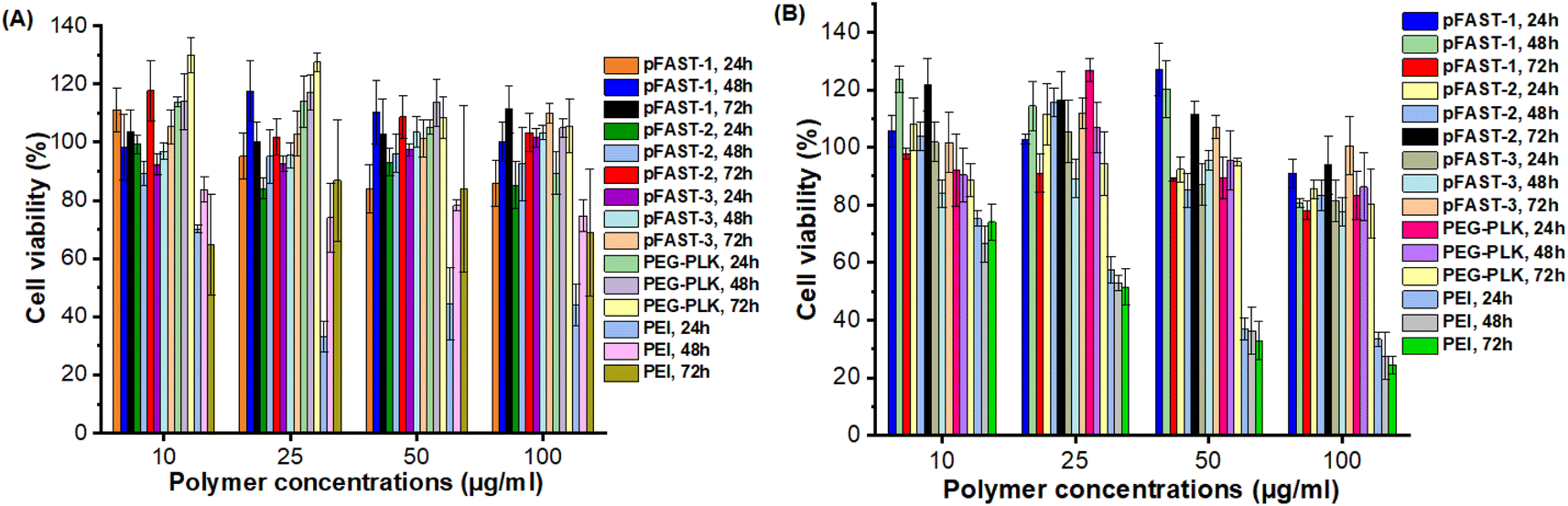 | ||
| Fig. 4 Cytotoxicity profiles of pFAST 1–3 compared to PEI and PEG-PLK in two different cell lines: (A) HFF2 human fibroblasts, and (B) KRAS-mutant MIA PaCa-2 cells. | ||
We used gWiz-GFP plasmid as a model system to determine the successful expression of the GFP gene (green cells) via the polyplex in A549 lung cancer cell lines. We selected A549 cell lines because these cells represents a model for non-small cell lung cancer (NSCLC), specifically adenocarcinoma. We particularly used this cell lines because of their propensity towards expressing multi-drug resistance 1 (MDR1) and multidrug resistance-associated protein (MRP), which is closely related to multidrug resistance (MDR) in lung cancer treatment. Since inhibiting these proteins can reverse MDR, therefore, we selected these cell lines as prospective models for transfection studies. To conduct the experiment, we prepared the complexes by adding 50 μg of plasmid DNA (gWiz-GFP) in 750 μL DNAse free water to either 500 μg, 2.5 mg, or 5 mg pFASTs (i.e., pFAST-1–3) in 750 μL sodium acetate buffer (25 mM, pH-5.5). The three independent mixtures (1:10, 1:50, and 1:100) of DNA and pFASTs were incubated at room temperature for 30 min for nanoparticle formation. We first determined the cytotoxicity of the nanoparticle formulation (DNA/pFAST polyplexes) in human lung airway epithelial A549 cells by using AlamarBlue assay (ESI, Fig. S4†). The cells were incubated with the prepared pFAST/DNA complexes at three intended concentrations (i.e., 1:10, 50, or 100) for 48 hours. At 48 hours post-incubation, the cell viability was determined by using AlamerBlue dye according to the manufacturer's instructions (Life Technologies Inc.), where a 10% volume of AlamarBlue dye was added to the well and incubated for 2 hours before reading the plate by an ELISA reader. From this experiment, we observed that the two nanoparticle formulations, i.e., pFAST-1 and -2 polyplexes showed the least toxic effect to the cells after 48 hours post incubation (ESI, Fig. S4†). Therefore, for we pursued further DNA delivery experiment in the same cell line with these two formulations. Upon identifying the concentration and pFAST:DNA ratio which do not trigger cytotoxic response, the efficiency of the polymeric vectors to deliver DNA intracellularly was then determined. We incubated the A549 cells with the nanoparticle formulation (1:10; DNA:pFAST) either with pFAST-1 or pFAST-2 systems for 48 hours. In the representative images shown in Fig. 5A and B, the identification of GFP positive cells confirmed a successful delivery of gWiz-GFP plasmid DNA in the human lung airway epithelial A549 cells by the both nanoparticle formulations. More detailed study on internalization and release mechanisms of plasmids from these polyplexes are currently undergoing in our laboratory.
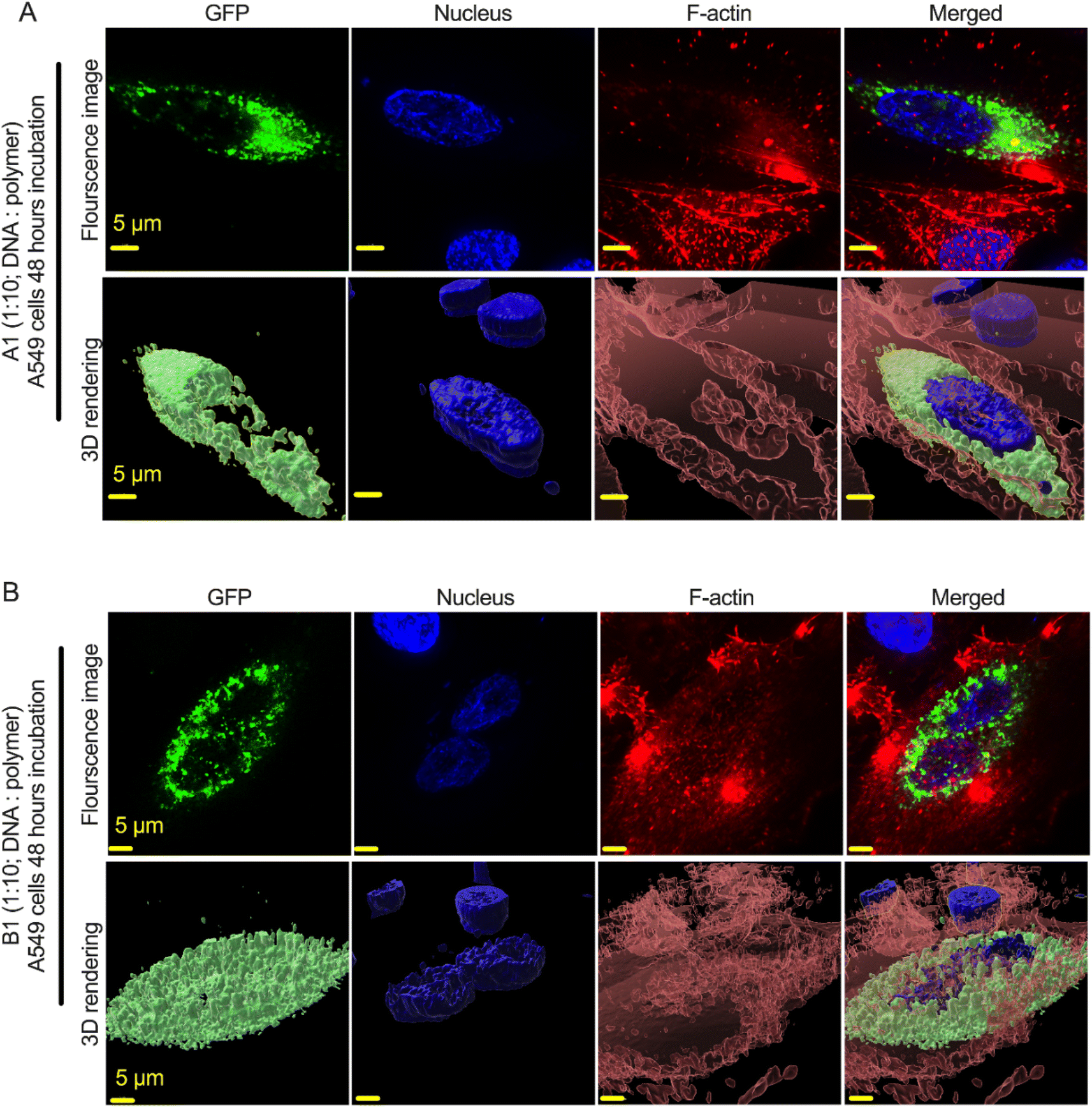 | ||
| Fig. 5 Plasmid DNA delivery in human lung airway epithelial A549 cells using nanoparticle formulations. gWiz-GFP plasmid was delivered to A549 cells using two nanoparticle formulations: (A1) (shown in the top panel) gWiz-GFP DNA/pFAST-1 polyplexes at 1:10 DNA/pFAST ratio and (B1) (shown in the bottom papne) gWiz-GFP DNA/pFAST-2 polyplexes at 1:10 DNA/pFAST ratio. At 48 hours post-incubation, GFP expression (successful intracellular delivery) in the A549 cells was observed under a Leica DMi8 epifluorescence microscope. For convenience, a 3D-rendering in the bottom panel shows the relative position of GFP within the cytosol. | ||
4. Conclusions
In summary, this report reveals that bio-based, BHMF-derived oligomeric amino esters are capable of forming polyplexes with plasmid DNA. Transmission electron microscopic images confirmed the nano size of the biobased polyplexes, while PicoGreen assay and agarose gel electrophoretic study revealed stable binding efficiency between the amino esters and the DNA. We further found out that, the synthesized amino esters were non-toxic to eukaryotic cells in their free- or plasmid-complexed form, stable in physiological pH, and are functionally active in cellular environments. The biomedical field, particularly after the COVID-19 pandemic, has been intensely focusing on the development of polymeric carrier systems capable of delivering therapeutic macromolecules, such as polynucleotides (mRNA and siRNA), antibodies, and enzymes of clinical interest.61–67 Also the drive towards sustainability will push the development of more biobased, greener products and processes. As such, our study collectively showed that furan-derived diacrylates can provide a toolset to generate biobased alternatives to various polyamine-type gene delivery systems. These newer sets of nucleic acid vectors, composed of high biobased contents, can therefore be used to carry therapeutic genes into human cells to treat various diseases by repairing or reconstructing defective genetic materials.Data availability
The data that support the findings of the study are available from the corresponding author [M.Q.] upon reasonable request.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
MAQ acknowledges the support of National Science Foundation (NSF) grant no. CBET 2239629. We are thankful to the University of North Dakota School of Medicine & Health Sciences (UND SMHS) for a pilot grant and national institutes of health (NIH) for P20GM113123 grant funding.References
- Y. Li, R. Tian, J. Xu, Y. Zou, T. Wang and J. Liu, Polym. Chem., 2024, 15, 1908–1931 RSC.
- D. Zhou, L. Cutlar, Y. Gao, W. Wang, J. O’Keeffe-Ahern, S. McMahon, B. Duarte, F. Larcher, B. J. Rodriguez, U. Greiser and W. Wang, Sci. Adv., 2016, 2, e1600102 CrossRef PubMed.
- D.-y. Lee, S. Amirthalingam, C. Lee, A. K. Rajendran, Y.-H. Ahn and N. S. Hwang, Nanoscale Adv., 2023, 5, 3834–3856 RSC.
- M. Assali, N. Kittana, I. Badran and S. Omari, RSC Adv., 2023, 13, 7000–7008 RSC.
- D. M. Lynn and R. Langer, J. Am. Chem. Soc., 2000, 122, 10761–10768 CrossRef CAS.
- M. Keeney, S. G. Ong, A. Padilla, Z. Yao, S. Goodman, J. C. Wu and F. Yang, ACS Nano, 2013, 7, 7241–7250 CrossRef CAS PubMed.
- C. J. Bishop, T. M. Ketola, S. Y. Tzeng, J. C. Sunshine, A. Urtti, H. Lemmetyinen, E. Vuorimaa-Laukkanen, M. Yliperttula and J. J. Green, J. Am. Chem. Soc., 2013, 135, 6951–6957 CrossRef CAS PubMed.
- J. J. Green, G. T. Zugates, R. Langer and D. G. Anderson, Methods Mol. Biol., 2009, 480, 53–63 CrossRef CAS PubMed.
- R. J. Fields, C. J. Cheng, E. Quijano, C. Weller, N. Kristofik, N. Duong, C. Hoimes, M. E. Egan and W. M. Saltzman, J. Controlled Release, 2012, 164, 41–48 CrossRef CAS PubMed.
- P. Mastorakos, E. Song, C. Zhang, S. Berry, H. W. Park, Y. E. Kim, J. S. Park, S. Lee, J. S. Suk and J. Hanes, Small, 2016, 12, 678–685 CrossRef CAS PubMed.
- S. Y. Tzeng and J. J. Green, Adv. Healthcare Mater., 2013, 2, 468–480 CrossRef CAS PubMed.
- Y. Li, Z. He, X. Wang, Z. Li, M. Johnson, R. Foley, A. Sigen, J. Lyu and W. Wang, ACS Macro Lett., 2023, 12, 780–786 CrossRef CAS PubMed.
- Y. Wang and S. M. Grayson, Adv. Drug Delivery Rev., 2012, 64, 852–865 CrossRef CAS PubMed.
- C. Wu, J. Li, W. Wang and P. T. Hammond, ACS Nano, 2018, 12, 6504–6514 CrossRef CAS PubMed.
- M. S. Kim, G. H. Gao, S. W. Kang and D. S. Lee, Macromol. Biosci., 2011, 11, 946–951 CrossRef CAS PubMed.
- T. I. Kim, H. J. Seo, J. S. Choi, J. K. Yoon, J. U. Baek, K. Kim and J. S. Park, Bioconjugate Chem., 2005, 16, 1140–1148 CrossRef CAS PubMed.
- Y. Li, Z. He, J. Lyu, X. Wang, B. Qiu, I. Lara-Saez, J. Zhang, M. Zeng, Q. Xu, A. Sigen, J. F. Curtin and W. Wang, Nanomaterials, 2022, 12, 3892 CrossRef CAS PubMed.
- C. Lin, T. M. Lammens, Z. Y. Zhong, H. Gu, M. C. Lok, X. Jiang, W. E. Hennink, J. Feijen and J. F. J. Engbersen, J. Controlled Release, 2006, 116, E79–E81 CrossRef CAS PubMed.
- S. Liu, Y. Gao, D. Zhou, M. Zeng, F. Alshehri, B. Newland, J. Lyu, J. O’Keeffe-Ahern, U. Greiser, T. Guo, F. Zhang and W. Wang, Nat. Commun., 2019, 10, 3307 CrossRef PubMed.
- Y. Liu, Y. Li, D. Keskin and L. Shi, Adv. Healthcare Mater., 2019, 8, 1801359 CrossRef PubMed.
- T. Lovato, V. Taresco, A. Alazzo, C. Sansone, S. Stolnik, C. Alexander and C. Conte, J. Mater. Chem. B, 2018, 6, 6550–6558 RSC.
- D. Santo, R. A. Cordeiro, A. Sousa, A. Serra, J. F. J. Coelho and H. Faneca, Biomacromolecules, 2017, 18, 3331–3342 CrossRef CAS PubMed.
- N. Segovia, P. Dosta, A. Cascante, V. Ramos and S. Borros, Acta Biomater., 2014, 10, 2147–2158 CrossRef CAS PubMed.
- S. Y. Tzeng, H. Guerrero-Cazares, E. E. Martinez, J. C. Sunshine, A. Quinones-Hinojosa and J. J. Green, Biomaterials, 2011, 32, 5402–5410 CrossRef CAS PubMed.
- R. E. Vandenbroucke, B. G. De Geest, S. Bonne, M. Vinken, T. Van Haecke, H. Heimberg, E. Wagner, V. Rogiers, S. C. De Smedt, J. Demeester and N. N. Sanders, J. Mater. Chem. B, 2008, 10, 783–794 CAS.
- E. Vuorimaa, T.-M. Ketola, J. J. Green, M. Hanzlikova, H. Lemmetyinen, R. Langer, D. G. Anderson, A. Urtti and M. Yliperttula, J. Controlled Release, 2011, 154, 171–176 CrossRef CAS PubMed.
- X. Wang, Z. Zhang and N. Hadjichristidis, Prog. Polym. Sci., 2023, 136, 101634 CrossRef CAS.
- Y. Wang, C.-F. Wang, M. Lie, D.-Z. Zhou, W. Huang and W.-X. Wang, Chin. J. Polym. Sci., 2020, 38, 830–839 CrossRef CAS.
- D. Wu, Y. Liu, X. Jiang, C. He, S. H. Goh and K. W. Leong, Biomacromolecules, 2006, 7, 1879–1883 CrossRef CAS PubMed.
- J. Zhang, X. Cai, R. Dou, C. Guo, J. Tang, Y. Hu, H. Chen and J. Chen, Mol. Ther.--Nucleic Acids, 2023, 32, 568–581 CrossRef CAS PubMed.
- G. T. Zugates, N. C. Tedford, A. Zumbuehl, S. Jhunjhunwala, C. S. Kang, L. G. Griffith, D. A. Lauffenburger, R. Langer and D. G. Anderson, Bioconjugate Chem., 2007, 18, 1887–1896 CrossRef CAS PubMed.
- S. Iqbal, Y. Qu, Z. Dong, J. Zhao, A. Rauf Khan, S. Rehman and Z. Zhao, Eur. Polym. J., 2020, 141, 110097 CrossRef CAS.
- W. Cheng, D. Wu and Y. Liu, Biomacromolecules, 2016, 17, 3115–3126 CrossRef CAS PubMed.
- X. Deng, N. Zheng, Z. Song, L. Yin and J. Cheng, Biomaterials, 2014, 35, 5006–5015 CrossRef CAS PubMed.
- G. Zuo, A. Xie, X. Pan, T. Su, J. Li and W. Dong, ACS Appl. Nano Mater., 2018, 1, 2376–2385 CrossRef CAS.
- H. Devalapally, Z. Duan, M. V. Seiden and M. M. Amiji, Int. J. Cancer, 2007, 121, 1830–1838 CrossRef CAS PubMed.
- L. E. van Vlerken, T. K. Vyas and M. M. Amiji, Pharm. Res., 2007, 24, 1405–1414 CrossRef CAS PubMed.
- A. Akinc, D. G. Anderson, D. M. Lynn and R. Langer, Bioconjugate Chem., 2003, 14, 979–988 CrossRef CAS PubMed.
- J. J. Green, J. Shi, E. Chiu, E. S. Leshchiner, R. Langer and D. G. Anderson, Bioconjugate Chem., 2006, 17, 1162–1169 CrossRef CAS PubMed.
- A. Akinc, D. M. Lynn, D. G. Anderson and R. Langer, J. Am. Chem. Soc., 2003, 125, 5316–5323 CrossRef CAS PubMed.
- Y. Cheng, H. Wei, J.-K. Y. Tan, D. J. Peeler, D. O. Maris, D. L. Sellers, P. J. Horner and S. H. Pun, Small, 2016, 12, 2750–2758 CrossRef CAS PubMed.
- N. L. Forsythe, M. F. Tan, D. Vinciguerra, J. Woodford, A. Z. Stieg and H. D. Maynard, Macromolecules, 2022, 55, 9925–9933 CrossRef CAS PubMed.
- R. Arote, T.-H. Kim, Y.-K. Kim, S.-K. Hwang, H.-L. Jiang, H.-H. Song, J.-W. Nah, M.-H. Cho and C.-S. Cho, Biomaterials, 2007, 28, 735–744 CrossRef CAS PubMed.
- C. Lin, Z. Zhong, M. C. Lok, X. Jiang, W. E. Hennink, J. Feijen and J. F. J. Engbersen, Bioconjugate Chem., 2007, 18, 138–145 CrossRef CAS PubMed.
- Z. Y. Zhong, Y. Song, J. F. J. Engbersen, M. C. Lok, W. E. Hennink and J. Feijen, J. Controlled Release, 2005, 109, 317–329 CrossRef CAS PubMed.
- M. Shi, J. H. Wosnick, K. Ho, A. Keating and M. S. Shoichet, Angew. Chem., Int. Ed., 2007, 46, 6126–6131 CrossRef CAS PubMed.
- J. Barr, G. Colpaert, E. Cadoni and A. Madder, Methods, 2023, 218, 189–197 CrossRef CAS PubMed.
- J. W. A. Langeveld, J. Dixon and J. F. Jaworski, Crop Sci., 2010, 50, 142–151 CrossRef.
- M. Annatelli, J. E. Sánchez-Velandia, G. Mazzi, S. V. Pandeirada, D. Giannakoudakis, S. Rautiainen, A. Esposito, S. Thiyagarajan, A. Richel, K. S. Triantafyllidis, T. Robert, N. Guigo, A. F. Sousa, E. García-Verdugo and F. Aricò, Green Chem., 2024, 26, 8894–8941 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- D. M. Lynn, D. G. Anderson, D. Putnam and R. Langer, J. Am. Chem. Soc., 2001, 123, 8155–8156 CrossRef CAS PubMed.
- A. Biancardi, T. Biver and B. Mennucci, Int. J. Quantum Chem., 2017, 117, e25349 CrossRef.
- C. A. Alabi, K. T. Love, G. Sahay, H. Yin, K. M. Luly, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12881–12886 CrossRef CAS PubMed.
- A. I. Dragan, J. R. Casas-Finet, E. S. Bishop, R. J. Strouse, M. A. Schenerman and C. D. Geddes, Biophys. J., 2010, 99, 3010–3019 CrossRef CAS PubMed.
- D. Ulkoski, A. Bak, J. T. Wilson and V. R. Krishnamurthy, Expert Opin. Drug Delivery, 2019, 16, 1149–1167 CrossRef CAS PubMed.
- G. A. Braun, B. E. Ary, A. J. Dear, M. C. H. Rohn, A. M. Payson, D. S. M. Lee, R. C. Parry, C. Friedman, T. P. J. Knowles, S. Linse and K. S. Akerfeldt, Biomacromolecules, 2020, 21, 4781–4794 CrossRef CAS PubMed.
- S. J. Maassen, J. Huskens and J. Cornelissen, J. Phys. Chem. B, 2019, 123, 9733–9741 CrossRef CAS PubMed.
- B. Zhang, H. Zhang, Y. Li, J. N. Hoskins and S. M. Grayson, ACS Macro Lett., 2013, 2, 845–848 CrossRef CAS PubMed.
- D. Demirci, B. Dayanc, F. A. Mazi and S. Senturk, Cells, 2021, 10, 208 CrossRef CAS PubMed.
- C. Wu, J. Li, Y. Zhu, J. Chen and D. Oupicky, Biomaterials, 2013, 34, 8843–8850 CrossRef CAS PubMed.
- P. Dosta, A. M. Cryer, M. Prado, M. Z. Dion, S. Ferber, S. Kalash and N. Artzi, Adv. NanoBiomed Res., 2021, 1, 2100006 CrossRef CAS.
- N. Segovia, M. Pont, N. Oliva, V. Ramos, S. Borros and N. Artzi, Adv. Healthcare Mater., 2015, 4, 271–280 CrossRef CAS PubMed.
- J. J. Chou, A. G. Berger, S. Jalili-Firoozinezhad and P. T. Hammond, Acta Biomater., 2021, 135, 331–341 CrossRef CAS PubMed.
- J. Hong, N. J. Shah, A. C. Drake, P. C. DeMuth, J. B. Lee, J. Chen and P. T. Hammond, ACS Nano, 2012, 6, 81–88 CrossRef CAS PubMed.
- A. Jaklenec, A. C. Anselmo, J. Hong, A. J. Vegas, M. Kozminsky, R. Langer, P. T. Hammond and D. G. Anderson, ACS Appl. Mater. Interfaces, 2016, 8, 2255–2261 CrossRef CAS PubMed.
- R. C. Smith, A. Leung, B.-S. Kim and P. T. Hammond, Chem. Mater., 2009, 21, 1108–1115 CrossRef CAS PubMed.
- E. C. Gale, G. A. Roth, A. A. A. Smith, M. Alcantara-Hernandez, J. Idoyaga and E. A. Appel, Adv. Ther., 2020, 3, 1900174 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05978f |
| This journal is © The Royal Society of Chemistry 2024 |