
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkoxyhydrosilane-facilitated cross-etherification reaction of secondary benzyl alcohol with aliphatic alcohol: synthesis of unsymmetrical dialkyl ethers†
Toru Hashimoto *ab,
Yakumo Matsunagaa,
Yuki Okamuraa,
Sosuke Takaoa and
Makoto Hojo*a
*ab,
Yakumo Matsunagaa,
Yuki Okamuraa,
Sosuke Takaoa and
Makoto Hojo*a
aDepartment of Applied Chemistry, Faculty of Engineering Sanyo-Onoda City University, Sanyo-Onoda, Yamaguchi 756-0884, Japan. E-mail: hashimototr@stf.teu.ac.jp
bDepartment of Applied Chemistry, Graduate School of Engineering, Tokyo University of Technology, Hachioji, Tokyo 192-0982, Japan
First published on 4th October 2024
Abstract
Alkoxyhydorosilane is found to be an effective mediator for the cross-etherification reaction between two distinct alcohols, namely, a secondary benzyl alcohol and an aliphatic alcohol, providing the unsymmetrical dialkyl ethers in good-to-high yields. The reaction is also successfully applied to the lignin model compound, which is an important renewable non-fossil organic carbon source. Initial mechanistic studies indicated that the carbocation derived from benzyl alcohol was formed under the present reaction conditions.
Alcohol is an inexpensive and environmental benign compound which can be obtained from biomass. Compared to fossil resources, alcohol is a sustainable resource, and its direct use in bulk and fine chemicals is of increasing interest. As environmental issues are growing concern, finding efficient, atom-economical, and step-economical strategies for the directly conversion of distinct alcohols into unsymmetrical dialkyl ethers, which are crucial structural units in many natural products and pharmaceutical,1 is a major challenge in organic chemistry. Many research groups have reported the reaction using various catalysts.2–10
Organosilane compounds have enormous practical advantages due to its non-toxic, environmentally benign nature, and high-abundance in the Earth's crust. Recently, many research groups have focused on the study of the Lewis acidity of neutral tetracoordinated silane compounds.11–14 In 2015, Tilley and Bergman group has reported that bis(perfluorocatecholato)silane, catalyzed hydrosilylation and silylcyanation reactions with aldehydes.12 Greb group and Inoue group also independently reported the electron-deficient tetraalkoxysilane compounds catalyzed the hydrodefluorination of 1-adamantylfluoride.13,14 Recently, our groups reported that Ph3SiH showed the catalytic activity for the synthesis of β-alkoxy alcohols via epoxide ring-opening reaction with aliphatic alcohol (Scheme 1 up).15 As a part of our ongoing study using hydrosilane, we found that alkoxyhydrosilane serves as a Lewis acid in the direct cross-etherification reaction between secondary benzyl alcohols and aliphatic alcohols (Scheme 1 down). In this paper, we disclose a synthesis of the unsymmetrical dialkyl ethers using two distinct alcohols. The reactions proceeded smoothly with the aid of alkoxyhydrosilane mediator to produce the corresponding dialkyl ethers in good-to-high yields.
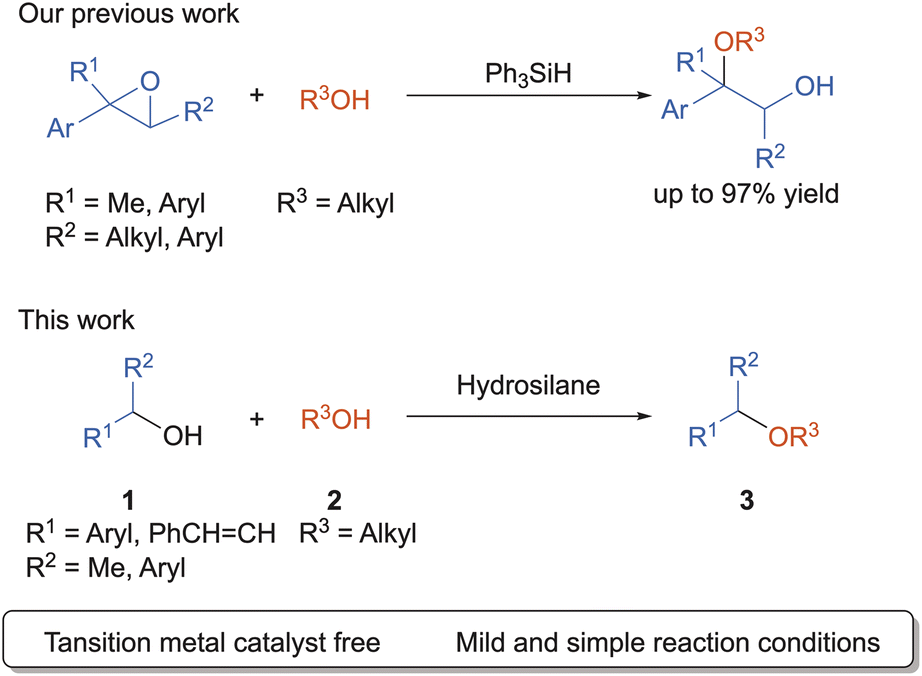 | ||
| Scheme 1 Our recent work and this work. | ||
We started with the optimization of the reaction conditions for the cross-etherification reaction using 1-(p-tolyl)ethanol (1a) with EtOH (2a) as a solvent in the presence of organosilane at 85 °C for 24 h (Table 1). After many trials,16 we identified that the (EtO)2MeSiH (4.0 equiv.) provided the desired ether 3aa in 95% isolated yield (entry 1). The choice of (EtO)2MeSiH is particularly important. When Ph3SiH, which works as the Lewis acid catalyst toward the ring-opening reaction of epoxide to afford the β-alkoxyalcohols,15 was employed, 3aa was not formed, and 1a was recovered (entry 2). (EtO)3SiH, (EtO)4Si, (EtO)2Me2Si and (EtO)2MePhSi were not effective for the reaction (entries 3–6). MePh2SiH afforded 3aa in only 38% yield, while Me2PhSiH and Ph4Si were not effective for this reaction (entries 7–9). When the amount of (EtO)2MeSiH reduced from 4.0 equivs to 3.0 equivs, 3aa was obtained in 67% yield (entry 10). At 60 °C, the reaction proceeded slowly to afford 3aa in only 18% yield (entry 11) When (EtO)2MeSiH was removed from the reaction system, 3aa was not formed and 1a was recovered almost quantitatively (entry 12).17–19
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yield of 3aab (%) |
| a The reaction was carried out using 1a (0.50 mmol) and hydrosilane (4 equiv.) in 2a (2.5 mL) for 24 h in reflux.b The yield of 3aa was determined by NMR analysis using pyrazine as an internal standard.c Isolated yield. | ||
| 1 | None | 95c |
| 2 | Ph3SiH instead of (EtO)2MeSiH | 0 |
| 3 | (EtO)3SiH instead of (EtO)2MeSiH | 0 |
| 4 | (EtO)4Si instead of (EtO)2MeSiH | 0 |
| 5 | (EtO)2Me2Si instead of (EtO)2MeSiH | 7 |
| 6 | (EtO)2MePhSi instead of (EtO)2MeSiH | 0 |
| 7 | MePh2SiH instead of (EtO)2MeSiH | 38 |
| 8 | Me2PhSiH instead of (EtO)2MeSiH | 0 |
| 9 | Ph4Si instead of (EtO)2MeSiH | 0 |
| 10 | 3.0 equiv. of (EtO)2MeSiH | 67c |
| 11 | 60 °C instead of 85 °C | 18c |
| 12 | No (EtO)2MeSiH | 0 |
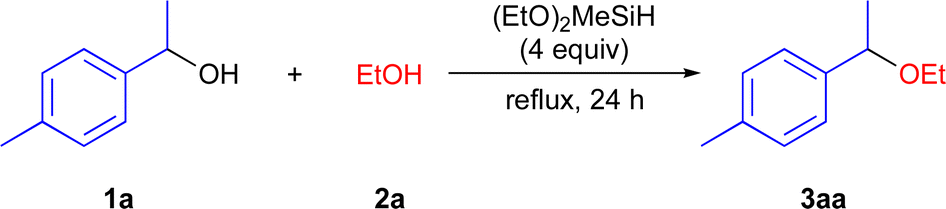
With the optimized reaction conditions in hand, we investigated a variety of secondary benzyl alcohols 1 using EtOH (2a) (Scheme 2). It was found that benzyl alcohols 1b–1d and 1h containing electron-donating substituents in the aryl ring, including MeO, Et2N, and MeS efficiently underwent the present etherification reactions to give the corresponding dialkyl ethers 3ba–3da and 3ha in 84–99% isolated yields, while benzyl alcohols 1e–1g containing electron-withdrawing substituents including Cl, Br, and CF3 in the aryl ring did not give the corresponding ethers 3ea–3ga.20 1-Mesitylethanol (1i) was suitable substrates for the present reaction conditions, affording the ether 3ia in 94% yield. In addition, naphthyl alcohols 1j–1k and cyclic alcohols 1l–1m effectively participated in the direct etherification reactions with 2a to produce the expected products 3ja–3ma in 71–93% yields. 1-(1-Thiophene)ethanol 1n and diphenylmethanol 1o were also applicable in this reaction, affording 3na and 3oa in 65% and 82% yields. Unfortunately, the primary benzylic alcohol 1p was recovered in quantitatively, suggesting the primary carbocation was not formed in the precent reaction conditions. In addition, tertiary benzyl alcohols 1q and aliphatic alcohol 1r were also unreactive under the applied reaction conditions.20
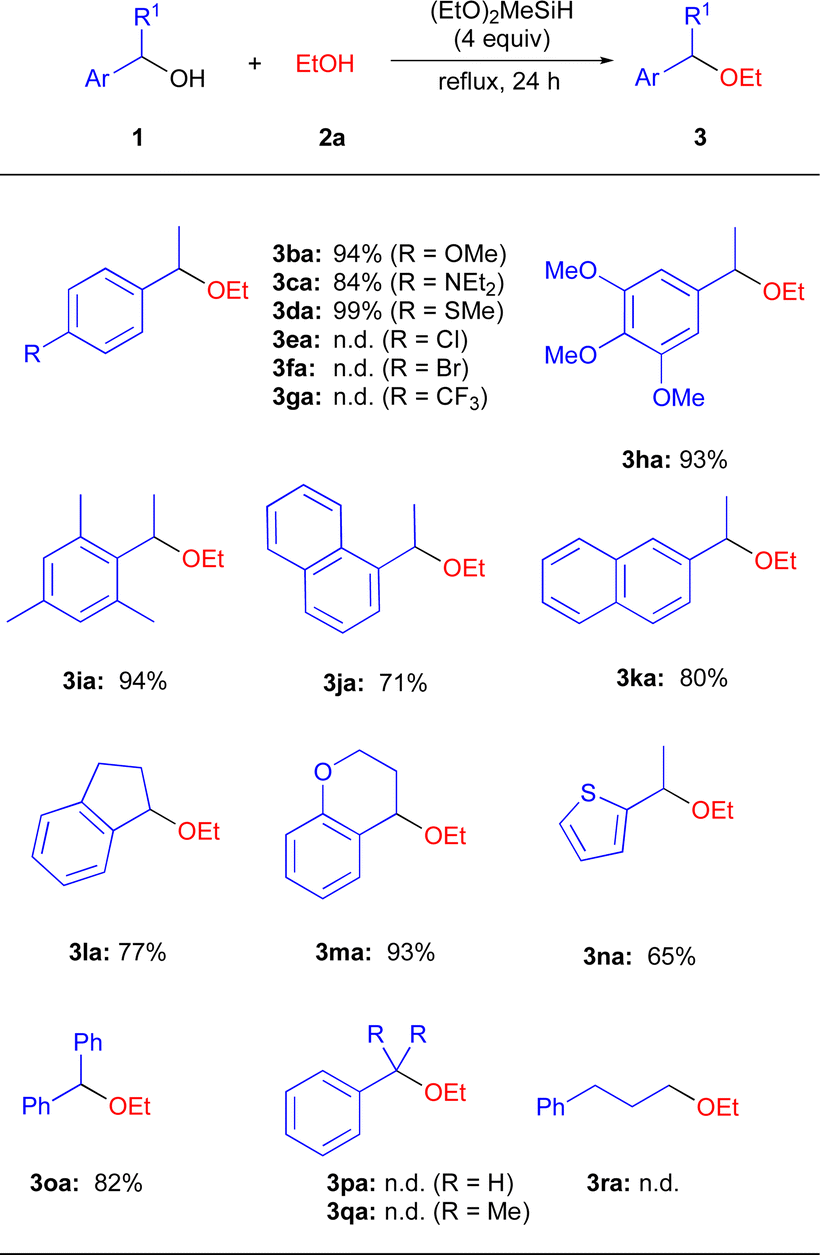 | ||
| Scheme 2 Substrate scope for the etherification reaction of various benzyl alcohols 1 with 2a. | ||
Next, we examined the substrate scope of the etherification reaction between 1a and various aliphatic alcohols 2, as shown in Scheme 3. The reaction using MeOH (2b) afforded 3ab in 78% isolated yield. MeOH-d4 (2b′) can be applied to the present reaction and 3ab′ was obtained in 77% yield. Terminal alkene (2c) and alkyne groups (2d) were tolerated in the present reaction conditions, affording the products 3ac and 3ad in 92% and 97%, respectively. i-PrOH (2e) induced less efficient etherification even at reflux conditions (3ae, 22% isolated yield).21 t-BuOH (2f) did not give the ether 3af.
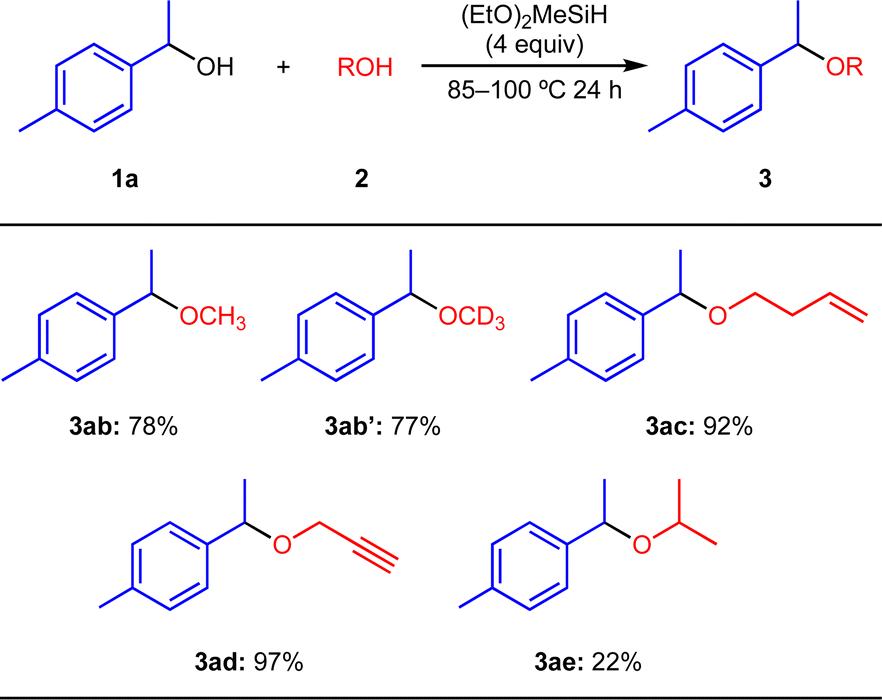 | ||
| Scheme 3 Substrate scope for the etherification reactions of 1a with various primary alcohols 2. | ||
To demonstrate the synthetic utility of the etherification reaction, chemoselective reaction using diol 1s having both primary and secondary hydroxy moieties was performed (Scheme 4). The reaction of 1s with 2a reacted selectively at the secondary hydroxy site to give 3sa in high yield.
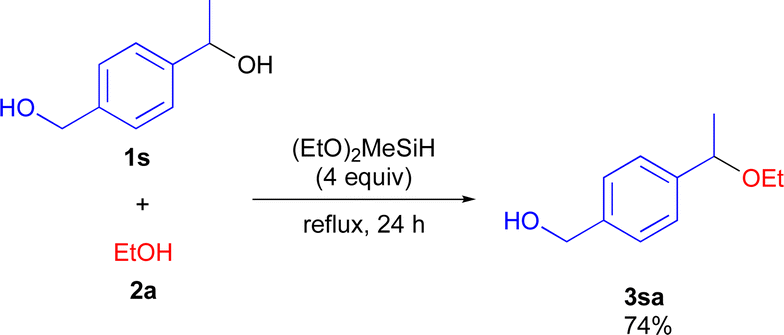 | ||
| Scheme 4 Chemoselective etherification reaction of 1s. | ||
To obtain the mechanistic insight into this reaction, several control experiments were carried out under the optimized reaction conditions (Scheme 5). Initially, we carried out the reaction using cinnamyl alcohol 1t with 2a gave the 3ta in 90% yield, whereas 3ua was not detected (eqn (1)). In addition, when 1u was used, 3ta was obtained in 96% yield as a sole product (eqn (2)). We assumed that the formation of 3ta is more favourable than 3us due to the stability of carbocations (eqn (3)). Next, we examined the reaction using chiral alcohol (S)-1j (97% ee) (Scheme 6). Treatment of (S)-1j with 2a in the presence of (EtO)2MeSiH gave the ether 3ja in 84% isolated yield with 14% ee. In addition, no racemization of the isolated ether 3ja (14% ee) was observed when the enantio-rich ether 3ja was exposed to the present reaction conditions.22 These results indicated the formation of the carbocations derived from secondary benzyl alcohols 1 during the reaction course, although more detailed studies are required for a definitive understanding of the exact mechanism of the present reaction.
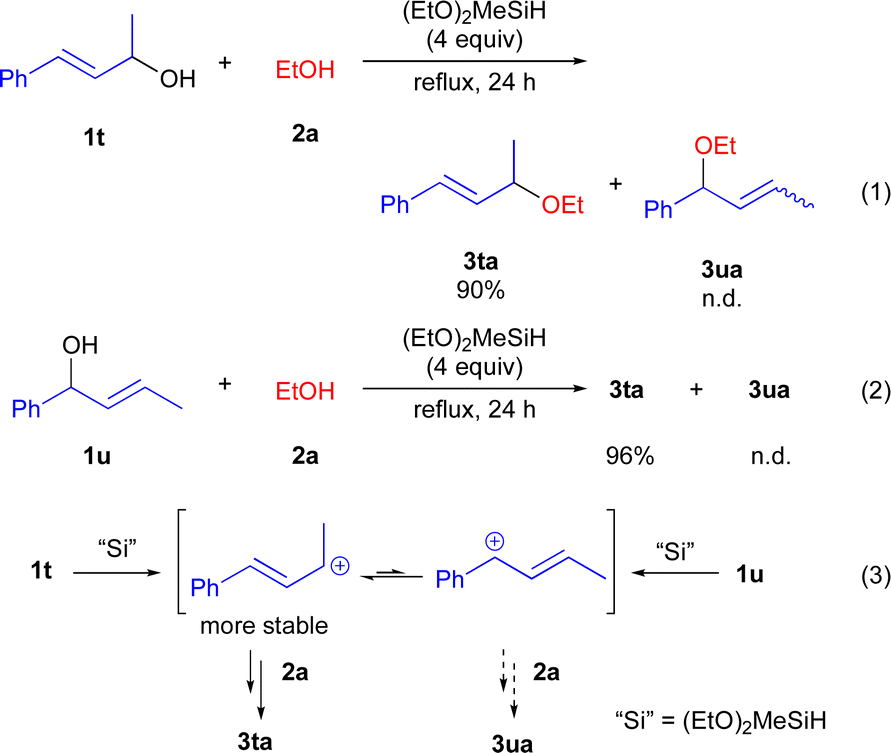 | ||
| Scheme 5 The reaction using alcohols 1t and 1u. | ||
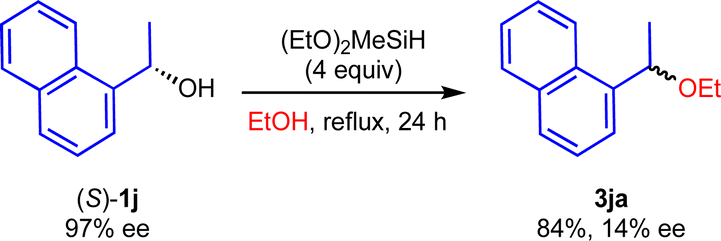 | ||
| Scheme 6 The reaction using chiral alcohol (S)-1j. | ||
Lignin is an important renewable non-fossil organic carbon source.23,24 The selective C(sp3)–O bond functionalization of diol or polyol derivatives could offer a sustainable approach to gain valuable oxygen-containing feedstock chemicals from lignin biomass. To demonstrate the synthetic utility of the (EtO)2MeSiH system, the cross-etherification using lignin model compound was performed (Scheme 7). Compound 1v was converted to the corresponding products 3va in 88% yield.
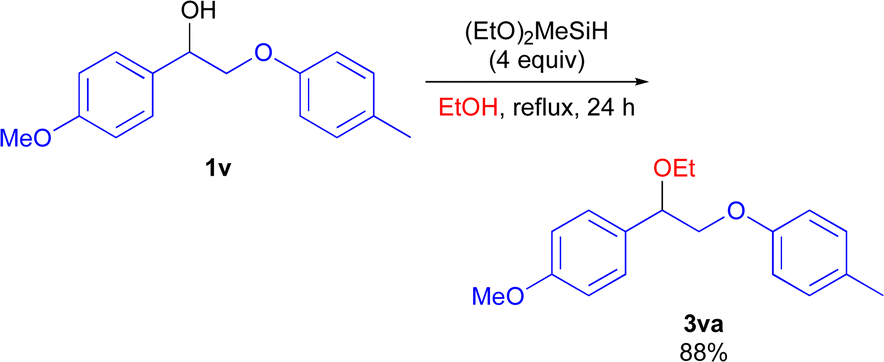 | ||
| Scheme 7 Synthetic application for the etherification reaction of lignin model 1v. | ||
Conclusions
We reported that (EtO)2MeSiH mediated cross-etherification reactions between secondary benzyl alcohol and aliphatic alcohol furnished unsymmetrical dialkyl ethers in good-to-high yields. Initial mechanistic studies suggested that the carbocation derived from benzyl alcohol was formed and subsequently, aliphatic alcohol attacked the in situ formed carbocation to afford the desired product. Further transformations catalyzed/mediated by organosilane and mechanistic studies are currently in progress in our laboratory.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
T. H. and M. H. directed the project. T. H. conceived the idea, designed the experiments and wrote the manuscript. T. H., Y. M., Y. O., and S. T. performed the experiments. All the authors participated in the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported in part by the International Collaborative Research Program of the Institute for Chemical Research, Kyoto University (grant 2022-21 and 2023-11) and the Joint Usage/Research Center for Catalysis (22AY0130, 23AY0204 and 23DS0428). The authors are grateful to Dr Katsuhiro Isozaki and Prof. Masaharu Nakamura (Kyoto Univ.) for their kind help with the HRMS measurements. We also thank Prof. Takashi Nishikata and Mr Tetsuhiro Yamamoto (Yamaguchi Univ.) for their kind help with the HPLC measurements.Notes and references
- A. H. Dempsey and S. R. Kass, J. Org. Chem., 2022, 87, 15466–15482 CrossRef PubMed
.
- T. Weil, M. Kotke, C. M. Kleiner and P. R. Schreiner, Org. Lett., 2008, 10, 1513–1516 CrossRef CAS PubMed
- P. K. Sahoo, S. S. Gawali and C. Gunanathan, ACS Omega, 2018, 3, 124–136 CrossRef CAS PubMed
- J. Kim, D.-H. Lee, N. Kalutharage and C. S. Yi, ACS Catal., 2014, 4, 3881–3885 CrossRef CAS
- B. D. Sherry, A. T. Radosevich and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 6076–6077 CrossRef CAS PubMed
- A. B. Cuenca, G. Mancha, G. Asensio and M. Medio-Simón, Chem.–Eur. J., 2008, 14, 1518–1523 CrossRef CAS PubMed
- G. Hirata, K. Takeuchi, Y. Shimoharai, M. Sumimoto, H. Kaizawa, T. Nokami, M. Abe, E. Shirakawa and T. Nishikata, Angew. Chem., Int. Ed., 2021, 60, 4329–4334 CrossRef CAS PubMed
- L. Zhang, A. Gonzalez-de-Castro, C. Chen, F. Li, S. Xi, L. Xu and J. Xiao, Mol. Catal., 2017, 433, 62–67 CrossRef CAS
- H. Slimi, Z. Litim, T. Ollevier and J. Kraïem, ACS Omega, 2023, 8, 44558–44570 CrossRef CAS PubMed
- Q. Xu, H. Xie, P. Chen, L. Yu, J. Chen and X. Hu, Green Chem., 2015, 17, 2774–2779 RSC
-
(a) S. E. Denmark and T. Wynn, J. Am. Chem. Soc., 2001, 123, 6199–6200 CrossRef CAS PubMed
- A. L. Liberman-Martin, R. G. Bergman and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 5328–5331 CrossRef CAS PubMed
-
(a) R. Maskey, M. Schädler, C. Legler and L. Greb, Angew. Chem., Int. Ed., 2018, 57, 1717–1720 CrossRef CAS PubMed
- F. S. Tschernuth, T. Thorwart, L. Greb, F. Hanusch and S. Inoue, Angew. Chem., Int. Ed., 2021, 60, 25799–25803 CrossRef CAS PubMed
- T. Hashimoto, K. Nishikimura and M. Hojo, ChemistrySelect, 2023, 8, e202303292 CrossRef CAS
- For details, see the ESI†.
- The reaction using mixed solvents (EtOH/DMF and EtOH/acetone) did not give the ether product 3aa and 1a was recovered quantitatively.
- Di(1-phenylethyl)ether did not converted to the 3aa in the present reaction conditions.
- In the reaction mixture, formation of the siloxane derived from (EtO)2MeSiH was confirmed.
- The starting materials 1e, 1f, 1g, 1q, and 1r were recovered in quantitatively.
- In this reaction, the starting material 1a was recovered in 41% and 3aa was not detected at all.
- The reaction using 4-vinylbiphenyl with 2a in the present of (EtO)2MeSiH did not gave the desired ether product at all and the polymerization of styrene derivative was observed.
-
(a) J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed
- C. Margarita, D. D. Francesco, H. Tuñon, I. Kumaniaev, C. J. Rada and H. Lundberg, Green Chem., 2023, 25, 2401–2408 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05997b |
| This journal is © The Royal Society of Chemistry 2024 |