
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, mixed-spin-state structure and Langmuir–Blodgett deposition of amphiphilic Fe(III) quinolylsalicylaldiminate complexes†
Peeranuch Poungsripong b,
Theerapoom Boonpraba,
Phimphaka Hardinga,
Keith S. Murrayc,
Wasinee Phonsric,
Ningjin Zhangd,
Jonathan A. Kitchen*ef and
David J. Harding*a
b,
Theerapoom Boonpraba,
Phimphaka Hardinga,
Keith S. Murrayc,
Wasinee Phonsric,
Ningjin Zhangd,
Jonathan A. Kitchen*ef and
David J. Harding*a
aSchool of Chemistry, Institute of Science, Suranaree University of Technology, Nakhon Ratchasima, 30000, Thailand. E-mail: david@g.sut.ac.th
bFunctional Materials and Nanotechnology Center of Excellence, Walailak University, Thasala, Nakhon Si Thammarat, 80160, Thailand
cSchool of Chemistry, Monash University, Clayton, VIC 3800, Australia
dSchool of Chemistry, University of Southampton, University Road, Southampton, SO17 1BJ, UK
eDepartment of Chemistry, Auckland University of Technology, New Zealand. E-mail: jonathan.kitchen@aut.ac.nz
fThe MacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealand
First published on 10th September 2024
Abstract
Designing and integrating Fe(III)-based spin crossover (SCO) complexes onto substrates remains a challenging goal with only a handful of examples reported. In this work, we successfully synthesized and characterized three [Fe(qsal-OR)2]NO3 (qsal-OR = 5-alkoxy-2-[(8-quinolylimino)methyl]phenolate) complexes, in which R = C12H25 1, C16H33 2, and C22H45 3 to explore the impact of alkyl chain on the modulation of SCO activity and potential for self-assembly on a glass surface. The SCO is found to be gradual and incomplete in all cases, with the LS state more stabilised as the alkyl group shortens. We also demonstrate that all complexes form stable Langmuir films and achieve good transfer ratios to the glass surface, with 2 being the best in terms of stability. This paves the way for the SCO modulation of complexes in this class and the development of SCO devices.
Introduction
The design of switchable molecules is essential to the development of molecular electronic systems.1,2 While a number of different switches are possible, e.g. optical, thermal and mechanical, some of the most promising are magnetic switches. A particularly interesting group of magnetic switches are spin crossover (SCO) compounds. SCO systems exhibit a change in their spin state as the electrons rearrange within the d-manifold, often under thermal stimulus.3–6 An advantage of SCO materials is that they can also be switched under pressure, optically, or by introduction of guest molecules. In the case of Fe(III), the two spin states, high spin (HS, S = 5/2) and low spin (LS, S = 1/2) are both paramagnetic. This has important consequences for the structural changes that accompany SCO with differences in Fe–ligand bond lengths of 0.10–0.13 Å in Fe(III), while ca. 0.20 Å is typical for the more widely studied Fe(II) systems.6,7 Despite this, Fe(III) complexes show the largest magnetic response upon SCO and are often air stable. This latter point is especially useful in the development of devices based on SCO materials.While SCO was first discovered in Fe(III),6 until recently it was largely ignored as the smaller structural changes were thought to limit the chances of abrupt SCO. In 2001, Sato and co-workers reported a large, repeatable hysteresis of 70 K in [Fe(qsal)2]NCSe demonstrating that Fe(III) systems can be just as effective as Fe(II).8,9 Nevertheless, it took a further 10 years before Fe(III) SCO compounds became more actively studied. There are now examples of symmetry-breaking,10–17 stepped,18–20 hysteretic,8,21–23 and even photoswitchable SCO.24–26 Our group has been especially interested in the [Fe(qsal-X)2]+ series of SCO complexes exploring solvent,27,28 substituent29–31 and anion effects.32–35 The success in many of these systems arises from careful control of the supramolecular interactions that link the spin centres. However, control over the SCO temperature, the type of SCO and the presence of hysteresis remains poor. Another problem is the challenge of immobilising SCO molecules onto surfaces, which is vital for tangible applications.
In seeking to address these challenges, we have taken inspiration from the work of Albrecht et al. who explored the impact that long alkyl chains have in [Fe(4-OR-sal2trien)]+ on the abruptness of SCO in solution.36,37 In a series of papers, they demonstrated that longer alkyl chains give rise to increasingly abrupt SCO. Moreover, they were able to show that the complexes do self-assemble to form thin films. However, transfer of the films to a support proved difficult and transfer ratios were generally low,38 limiting the use of these particular compounds. Other groups have been more successful with alkyl chain functionalization of SCO Fe(II) complexes reported with a range of ligand scaffolds.39–46 It was found that the magnetism and some degree of cooperativity were retained in the film, but the detailed studies of the effect of these chains were not widely evaluated. More recent studies on alkylated Fe(II)47–56 and Co(II)57–59 complexes reveal that there can sometimes be a significant correlation between chain length and the SCO temperature, T1/2 (ref. 52) and abruptness.53
We have previously reported that [Fe(qsal-5-OMe)2]+ complexes do undergo SCO while the structurally related [Fe(qsal-4-OMe)2]+ systems are invariably HS.29 Consequently, in this work we sought to answer two questions. Firstly, do [Fe(qsal-4-OR)2]+ compounds (Fig. 1) with long alkyl chains undergo SCO and how does the length of the chain impact the completeness of the transition? Secondly, do these form thin films and how does the change in the head group on the Fe(III) centre influence the stability and transferability of the films relative to [Fe(4-OR-sal2trien)]+.37 Our results show that not only do the long alkyl chains change the packing permitting SCO, but they also permit self-assembly forming stable films that can be readily transferred to glass substrates.
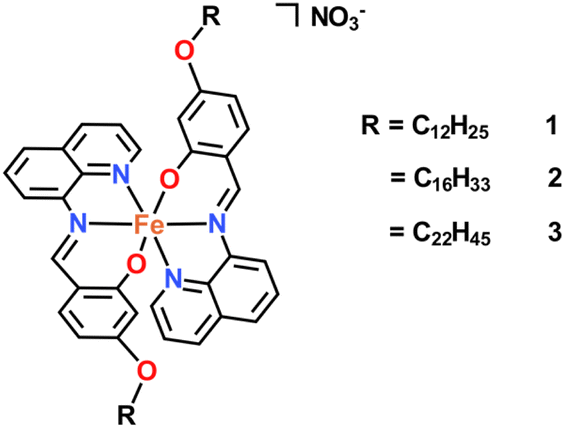 | ||
| Fig. 1 Structure of complexes studied in this work. | ||
Results and discussion
Ligand synthesis and characterisation
Ligands Hqsal-OC12H25, Hqsal-OC16H33 and Hqsal-OC22H45 were synthesised by the same general procedure (Scheme 1) reacting a stoichiometric amount of 8-aminoquinoline with 4-(dodecyloxy)salicylaldehyde, 4-(hexyloxy)salicylaldehyde or 4-(docosyloxy)salicylaldehyde at room temperature in DCM with trifluoroacetic acid as a catalyst. Following the reaction, evaporation to dryness and washing with hexane and diethylether resulted in yellow solids in good yields (83–99%). The ligands were fully characterised using 1H NMR, IR, and UV/vis spectroscopy, mass spectrometry and where possible X-ray crystallography (see ESI†). All spectroscopic data was consistent with the formation of the desired ligands, although trace amounts of the starting aldehyde are present in all cases. Small yellow plate-like single crystals of Hqsal-OC12H25 were obtained by slow evaporation from a DCM solution. Hqsal-OC12H25 crystallized in the triclinic space group, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with two crystallographically independent molecules of Hqsal-OC12H25 and one water molecule (disordered over two sites) in the asymmetric unit (Fig. S14†). Both molecules of Hqsal-OC12H25 crystallize as the keto-amine tautomer [N2⋯O1 = 2.593(2) Å, <(N2–H2⋯O1) = 138.4(2)°; and N102⋯O101 = 2.610(2) Å, <(N102–H102⋯O101) = 136.0(2)°]. One molecule of Hqsal-OC12H25 has the interstitial water molecule hydrogen bonding to the keto oxygen atom [N200⋯O101′ = 2.672(4) Å, <(N200–H20D⋯O101′) = 162.4(2)°]. The overall packing in Hqsal-OC12H25 shows some interdigitation of the alkyl chains (Fig. S14†) which is common for such amphiphilic compounds.60
, with two crystallographically independent molecules of Hqsal-OC12H25 and one water molecule (disordered over two sites) in the asymmetric unit (Fig. S14†). Both molecules of Hqsal-OC12H25 crystallize as the keto-amine tautomer [N2⋯O1 = 2.593(2) Å, <(N2–H2⋯O1) = 138.4(2)°; and N102⋯O101 = 2.610(2) Å, <(N102–H102⋯O101) = 136.0(2)°]. One molecule of Hqsal-OC12H25 has the interstitial water molecule hydrogen bonding to the keto oxygen atom [N200⋯O101′ = 2.672(4) Å, <(N200–H20D⋯O101′) = 162.4(2)°]. The overall packing in Hqsal-OC12H25 shows some interdigitation of the alkyl chains (Fig. S14†) which is common for such amphiphilic compounds.60
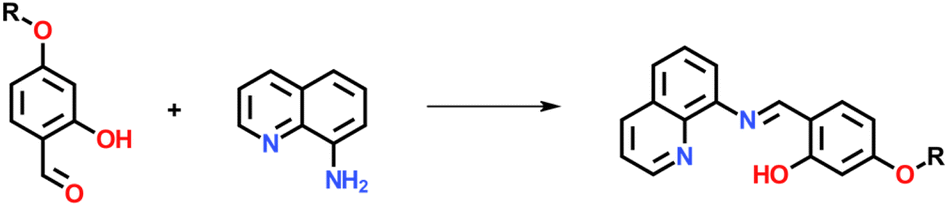 | ||
| Scheme 1 Synthesis of Hqsal-OR. | ||
Complex synthesis and characterisation
Complexation reactions of Hqsal-OC12H25, Hqsal-OC16H33 and Hqsal-OC22H45 were carried out by mixing DCM solutions of the ligand with methanolic solutions of Fe(NO3)3·9H2O and stirring for 10 minutes before adding triethylamine and stirring for a further hour. The resulting dark solution was evaporated and then dissolved in DCM before being eluted through an Al2O3 flashpad. Evaporation of the dark solution gave the complexes as dark purple solids in modest yields (54–74%). Mass Spectrometry (ESI+) data for all complexes showed the expected peaks and isotopic ratios for [Fe(qsal-OC12H25)2]+, [Fe(qsal-OC16H33)2]+ and [Fe(qsal-OC22H45)2]+ with m/z values of 918.47, 1030.60 and 1198.79, respectively (Fig. S9–S11†). Comparison of the infrared spectra of the ligands and complexes shows a decrease in the position of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch, from ∼1620 to ∼1600 cm−1, indicative of complexation (Fig. S4 and S12†). The nitrate counterion stretch is observed around 1380 cm−1. The IR spectra of all complexes are in agreement with those of the related compounds, [Fe(qsal-4-OMe)2]+ confirming that the complexes have successfully formed (Fig. S12†).29 The relative intensity of the aliphatic C–H stretching compared to the CN also increases with alkyl chain elongation, confirming the integration of the chain into the molecule. Absorption spectra of all complexes in methanol showed intense absorption bands at 325 nm and 403 nm (Fig. S13†) most likely arising from π–π* and ligand-to-metal charge transfer (LMCT) transitions respectively.29
N stretch, from ∼1620 to ∼1600 cm−1, indicative of complexation (Fig. S4 and S12†). The nitrate counterion stretch is observed around 1380 cm−1. The IR spectra of all complexes are in agreement with those of the related compounds, [Fe(qsal-4-OMe)2]+ confirming that the complexes have successfully formed (Fig. S12†).29 The relative intensity of the aliphatic C–H stretching compared to the CN also increases with alkyl chain elongation, confirming the integration of the chain into the molecule. Absorption spectra of all complexes in methanol showed intense absorption bands at 325 nm and 403 nm (Fig. S13†) most likely arising from π–π* and ligand-to-metal charge transfer (LMCT) transitions respectively.29
Powder X-ray diffraction studies reveal that the bulk materials are largely amorphous (Fig. S16†). Despite repeated attempts we have been unable to grow crystals of complexes 2 and 3. However, single crystals of [Fe(qsal-OC12H25)2]NO3·CH2Cl2 were grown by evaporation from DCM and the structure determined at 100 K (Fig. 2). The compound crystallized in the triclinic space group P, and contained two molecules of [Fe(qsal-OC12H25)2]NO3 and two interstitial DCM molecules in the asymmetric unit (Fig. S15†). Both complexes show the expected coordination to the Fe(III) centre where two ligands coordinate through the NNO coordination pocket at approximately 90° to each other and give distorted octahedral geometries with N4O2 coordination spheres. Analysis of the Fe–O and Fe–N bond lengths and angles showed that the two Fe(III) complexes adopt different spin states where Fe1–O = 1.883(3) Å, Fe1–Nimine = 1.931(4) and Fe1–Nquinoline = 1.972(4) Å which are typical for LS Fe(III), and Fe2–O = 1.913(3) Å, Fe2–Nimine = 2.115(3) Å and Fe2–Nquinoline = 2.137(4) Å, typical of the HS state (Table 1).19,29 The octahedral distortion parameter (Σ) gave further evidence of the different spin states where ΣFe1 = 49.6° and ΣFe2 = 71.2°. While not unprecedented, [HS-LS] systems remain rare in Fe(III) SCO chemistry, with [Fe(qsal-5-OMe)2]Cl·2MeOH·0.5H2O the most closely related example.19 In contrast, [Fe(qsal-4-OMe)2]NO3·CH2Cl2 is entirely HS29 and confirms that alkyl chains can be used to change the packing and enable SCO (vide infra). One alkyl chain on each complex adopts a trans co-planar arrangement while the other adopts a kink in the chain, this results in the chains being oriented in one direction relative to the Fe(III) centres, which gives an overall bi-layered packing arrangement with interdigitation of alkyl chains – an ideal conformation for Langmuir film formation (Fig. 3). These bilayer configurations and the kinks were also recognized in the SCO complexes for self-assembly application.42,47,49,51,52,54,57,59,61 The ‘head-groups’ are linked to neighboring bi-layer headgroups through non-classical C–H hydrogen bonding interactions between aromatic C–H groups and the NO3 counter anions (see Table S4 in the ESI†) as well as weak C–H⋯π stacking interactions [C106···centroidC32–C37 = 3.538 Å]. By comparison, [Fe(4-OC8H17-sal2trien)]PF6 also contains two independent Fe(III) centres in the asymmetric unit, but both are HS.60 Moreover, unlike 1 the alkyl chains simply face each other with no evidence of interdigitation and the head groups are more loosely associated. Combined, this suggests that 1–3 should form more stable films.
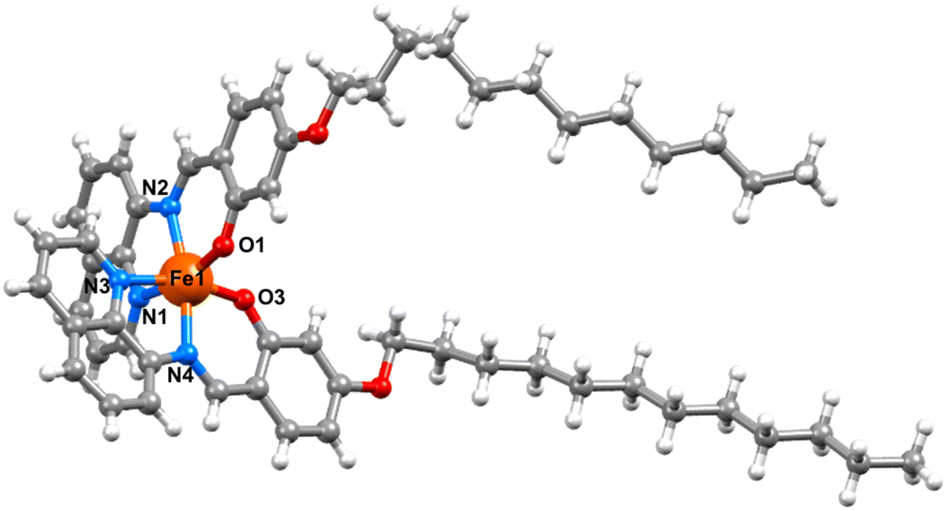 | ||
| Fig. 2 View of the Fe1 molecule in [Fe(qsal-OC12H25)2]NO3·CH2Cl2. | ||
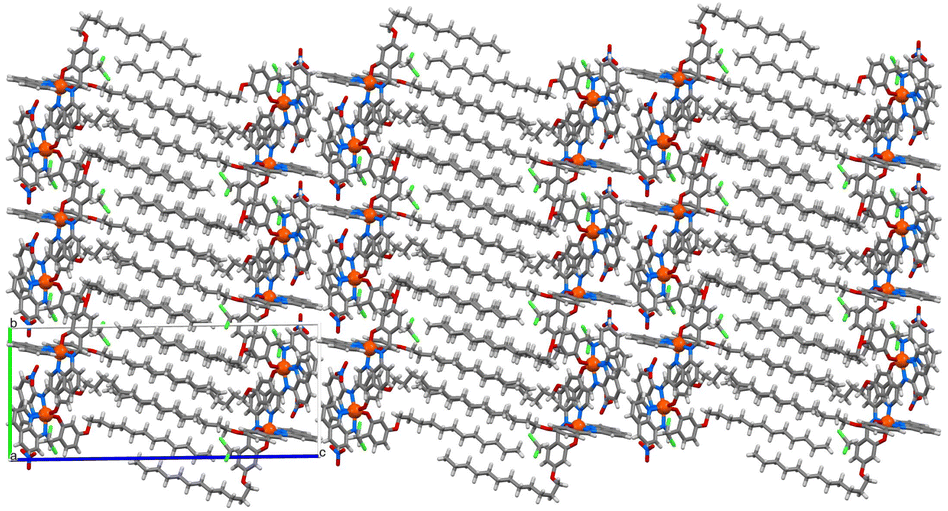 | ||
| Fig. 3 Packing in 1 showing the formation of the bi-layers. | ||
| Fe1 | Fe2 | |
|---|---|---|
| Fe–O | 1.906(3), 1.918(3) | 1.890(3), 1.871(3) |
| Fe–Nquin. | 2.117(3), 2.156(4) | 1.967(4), 1.975(3) |
| Fe–Nimine | 2.115(3), 2.113(3) | 1.928(4), 1.928(4) |
| cis-Bond angle range | 76.5(2)–100.7(2) | 83.1(2)–95.8(2) |
| trans-Bond angle range | 163.9(2)–169.3(2) | 175.9(2)–178.1(2) |
| Σ | 70.9 | 52.7 |
Scanning electron microscopy (SEM)
The morphologies of bulk samples of [Fe(qsal-OR)2]NO3 were evaluated by scanning electron microscopy (Fig. 4). Morphologies were found to heavily depend on the length of the alkyl chains, where [Fe(qsal-OC12H25)2]NO3 existed as micro rods (Fig. 4a and b), [Fe(qsal-OC16H33)2]NO3 appeared to have multiple morphologies and was observed as sheet-like plates (Fig. 4c), and spheres (Fig. 4d). The longest alkyl chain complex [Fe(qsal-OC22H45)2]NO3 adopted a less structured morphology and exists as a network like structure (Fig. 4e and f) and is similar to that observed in [Fe(4-OC22H45-sal2trien)]NO3.37 The alteration in morphologies as the alkyl-chain length changes is clear with more crystalline morphologies observed for the shorter chains. | ||
| Fig. 4 SEM images of [Fe(qsal-OC12H25)2]NO3 (a and b), [Fe(qsal-OC16H33)2]NO3 (c and d) and [Fe(qsal-OC22H45)2]NO3 (e and f). | ||
Magnetic studies
Magnetic susceptibility studies of 1–3 were undertaken using SQUID magnetometry between 30–350 K with the SCO profiles shown in Fig. 5. The SCO is gradual in all cases, being most complete in [Fe(qsal-OC12H25)2]NO3 with χMT increasing from ca. 2.0 cm3 mol−1 K−1 at 30 K to 3.1 cm3 mol−1 K−1 at 350 K. This is consistent with the structural data which show a [HS-LS] state at 150 K (χMT = 2.3 cm3 mol−1 K−1).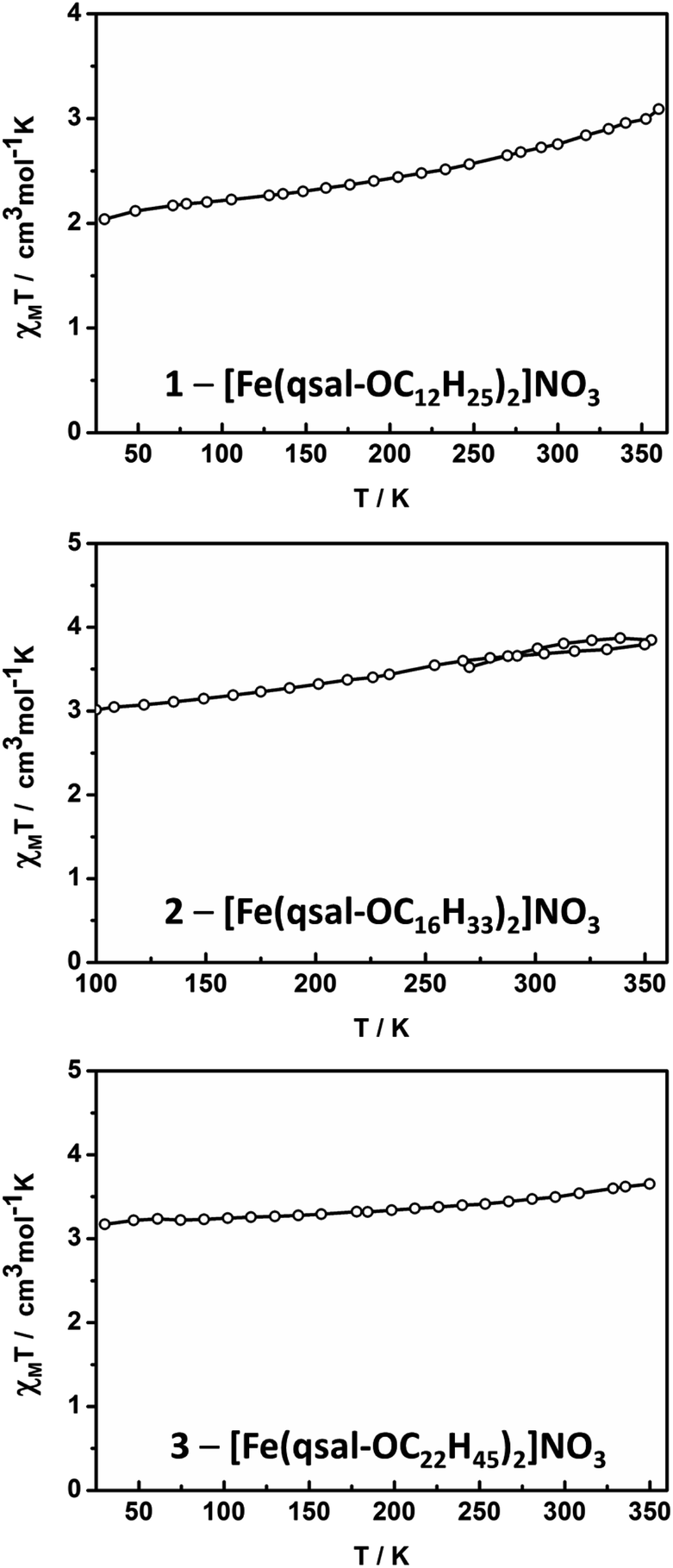 | ||
| Fig. 5 χMT vs. T plots for 1–3. | ||
For the longer chain systems χMT increases by ca. 0.8 and 0.5 cm3 mol−1 K−1, for 2 and 3, respectively. The changes in χMT represent ca. 29%, 21% and 13% SCO in 1, 2 and 3, respectively (assuming χMT = 0.40 cm3 mol−1 K−1 for LS and χMT = 4.2 cm3 mol−1 K−1 for HS). This suggests that as the alkyl chains become longer SCO becomes increasingly difficult, mirroring studies on dimeric Fe(II) SCO complexes.48 It is also probable that the increasingly gradual and incomplete SCO reflects the lower crystallinity noted in the morphological studies. Interestingly, the SCO in 1–3 contrasts with [Fe(4-OR-sal2trien)]NO3 (R = C6H13 to C18H37) which are entirely HS in the solid state,60 indicating that the effect of the alkyl chains on SCO behaviour is dependent on the ligand system. Finally, we note that the presence of SCO in these longer chain systems, compared with the HS behaviour of [Fe(qsal-4-OMe)2]NO3·CH2Cl2,29 confirms that the alkyl chains can be used to tune SCO characteristics.
Langmuir and Langmuir–Blodgett studies
The suitability of these amphiphilic systems for the formation of immobilized mono- and multi-layers was investigated by carrying out a full study of the Langmuir film forming abilities at an air–water interface. Pressure–area isotherms of Hqsal-OC12H25, Hqsal-OC16H33 and Hqsal-OC22H45 showed that, on pure water, the ligands all form Langmuir films with area per molecule values of 35, 25 and 20 Å2 respectively and collapse pressures of 10, 25 and 45 mN m−1 respectively (Fig. S16†). These values are consistent with successful film formation,62 however the low collapse pressure of Hqsal-OC12H25 indicates that the shorter 12-carbon chain system forms a somewhat weaker film. Time stability of the films was determined by holding the monolayers at a fixed molecular area and monitoring the surface pressure over a period of 10–15 minutes. The films showed reasonable stability over this timeframe, with only Hqsal-OC22H45 showing a slight decrease in surface pressure (Fig. S17†). Analogous studies were carried out for the corresponding FeIII complexes.All three complexes showed typical isotherms for the successful formation of ordered monolayers at the air–water interface where [Fe(qsal-OC12H25)2]NO3 formed with an area per molecule value of 70 Å2 and a film collapse pressure of 40 mN m−1; [Fe(qsal-OC16H33)2]NO3 formed with an area per molecule value of 85 Å2 and a film collapse pressure of 45 mN m−1; and [Fe(qsal-OC22H45)2]NO3 formed with an area per molecule value of 85 Å2 and a film collapse pressure of 50 mN m−1 (Fig. 6). Stability studies were again carried out to assess the suitability of these complexes for further deposition studies. The films were compressed to a pressure of 30 mN m−1 and the surface pressure monitored over time as the films were held static. Over a 20 minutes period, [Fe(qsal-OC22H45)2]NO3 was found to form the most stable film with only a small decrease in surface pressure with the least stable film being formed by [Fe(qsal-OC12H25)2]NO3. All films demonstrated sufficient stability for further deposition studies.
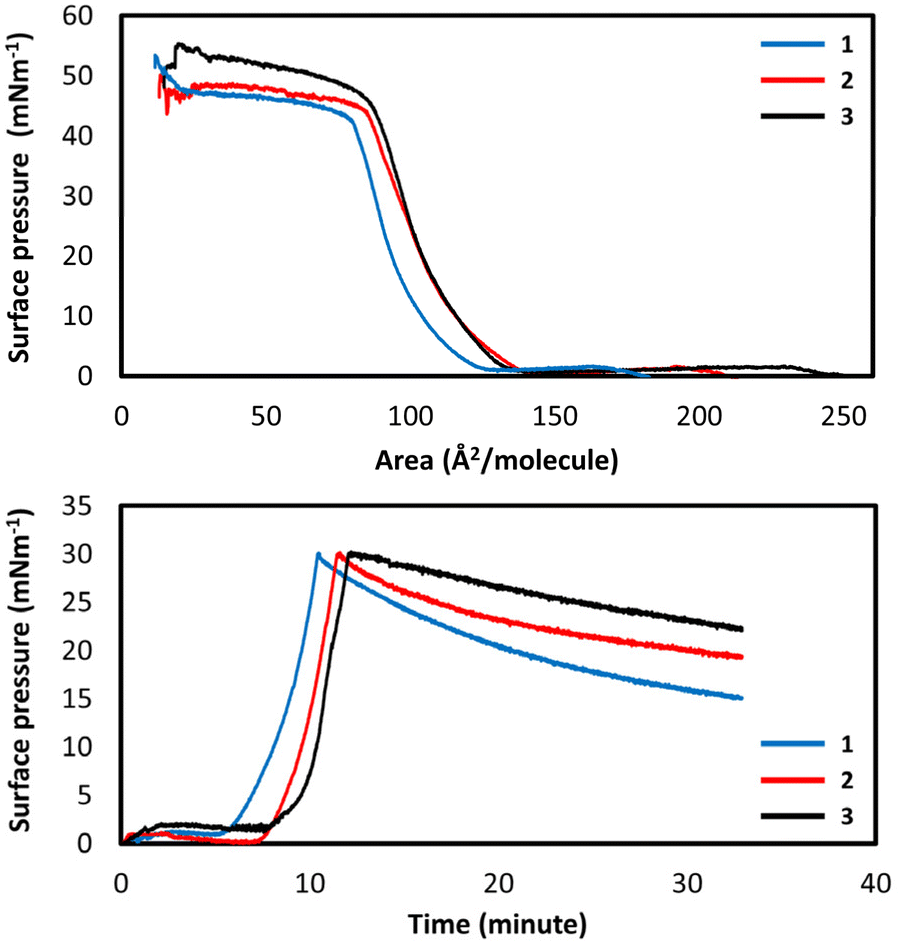 | ||
| Fig. 6 Pressure–area isotherms (top) and stability measurements (bottom) for 1–3. | ||
The ability of the FeIII complexes to form highly ordered mono-layer films on quartz solid supports was investigated. Monolayer film depositions were investigated by vertical deposition of [Fe(qsal-OR)2]NO3 complexes onto quartz at surface pressures of 30 mN m−1. Films were formed for all complexes with good transfer ratios (∼1) on upstroke of the quartz slide – UV/vis measurements confirmed successful deposition of the highly coloured complexes (Fig. 7).
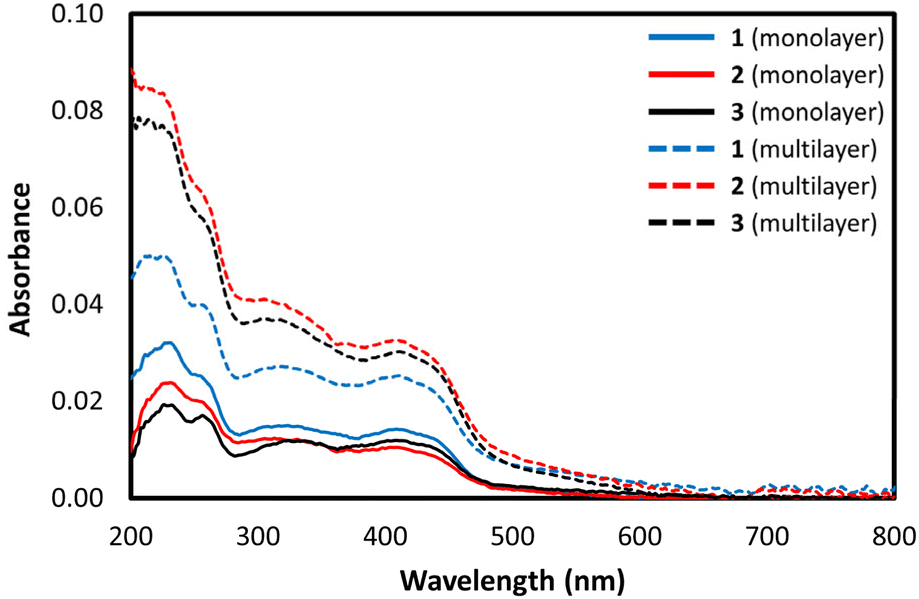 | ||
| Fig. 7 UV-vis spectra of 1–3 as monolayer and multilayer films. | ||
With successful monolayer formation, multi-layering studies were then carried out with emersion (up-stroke) of the slide being the first deposition. [Fe(qsal-OC12H25)2]NO3 showed excellent transfer ratios (∼1) for the first two layers (emersion and immersion), however the third layer (emersion) gave a transfer ratio of only 0.6. The overall result was a system that displayed Y-type film formation with successful deposition of a maximum of two layers. On moving to the longer chain [Fe(qsal-OC16H33)2]NO3 complex the multi-layering ability was improved. This system displayed a maximum of three layers deposited in a Y-type film all with excellent transfer ratios of ∼1 (Fig. 8). The longer chain [Fe(qsal-OC22H45)2]NO3 system was also investigated for Y-type multi-layer film deposition and showed relatively poor deposition characteristics. Initial mono-layer formation on emersion is good (transfer ratio ∼1), however subsequent layers had lower transfer ratios (0.7 and 0.8 respectively) indicating that the optimal chain length from this family is C16 (see Fig. S18 and S19 in ESI†). This may be due to back-folding with this longer alkyl chain, inhibiting the formation of the second layer.
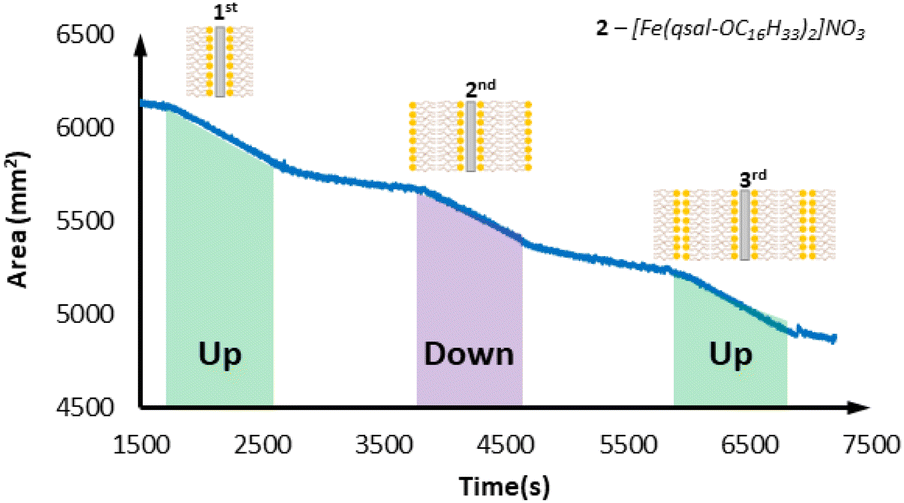 | ||
| Fig. 8 Langmuir film transfer for [Fe(qsal-OC16H33)2]NO3. | ||
UV-vis analysis of the mono- and multi-layered substrates indicated that the complexes remained intact on deposition as they had the same spectral features as was observed for the solution measurements. Multilayer films showed three absorption bands at 222 nm, 326, and 417 nm. The absorption band at 326 nm is assigned to the π–π* transition while the band at 417 nm, is assigned to the charge transfer band.
Conclusions
In this work, we successfully synthesized and characterized a novel class of alkylated [Fe(qsal-OR)2]NO3 complexes 1–3. The solid-state structure of 1 reveals a bi-layer arrangement of the cationic complex, which is ideal for the formation of Langmuir–Blodgett film via self-assembly. The temperature dependent SQUID profile of 1–3 indicates that the integrating the long alkyl chain changes the packing leading to SCO compared with the trapped HS [Fe(qsal-OMe)2]NO3 complex in which R is methyl. Nevertheless, the profile is more gradual with the longer chain, contrasting the effect observed in [Fe(4-OR-sal2trien)]NO3. We can then demonstrate that all complexes can form a stable film and achieve reasonable transfer ratios to the glass surface, with 2 being the best in term of stability. Our results pave the way for the development of SCO devices with self-assembly on more diverse substrates currently being explored.Experimental
General
Infrared spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer with OPUS data collection program in the range of 400–4000 cm−1, either as solid sample or KBr disc media. Electronic spectra were recorded at room temperature on either an Avantes Fiber Optics Spectrometer with Avalight-DHC and Avaspec ULS2048XL-USB2 in the range of 200–1000 nm or a PerkinElmer Lambda35 spectrometer in the range of 200–900 nm. The morphology of [Fe(qsal-OR)2]NO3 compounds were determined by Emission Scanning Electron Microscope (FESEM) at Walailak University, (JEOL JSM-7001 F, Zeiss Supra 55 VP, Zeiss Merlin Compact) operated at 3–5 kV. Samples were placed on a carbon tape and coated with carbon. 1H NMR spectra were recorded on a Bruker DPX400 NMR spectrometer at 300 K in CDCl3. Chemical shifts are reported in parts per million and referenced to the residual solvent peak. Standard conventions indicating multiplicity were used: m = multiplet, t = triplet, d = doublet, s = singlet. Arq = signal originating from the quinoline ring and Ars = signal originating from the salicylic ring. ESI mass spectra were recorded using a MaXis (Bruker Daltonics, Germany) mass spectrometer equipped with a Time of Flight (TOF) analyser. Samples were introduced to the mass spectrometer via a Dionex Ultimate 3000 autosampler and uHPLC pump [Gradient 20% acetonitrile (0.2% formic acid) to 100% acetonitrile (0.2% formic acid) in five minutes at 0.6 mL min−1. Column: Acquity UPLC BEH C18 (Waters) 1.7 micron 50 × 2.1 mm]. Langmuir studies: pressure–area isotherms and time stability were measured at 25 °C on a Kibron MicroTroughXS (MTXS) Langmuir–Blodgett trough. Water for the subphase was purified with a Milli-Q Integral system (Millipore), and its resistivity was measured to be higher than 18 MΩ cm. Chloroform (HPLC grade, Fisher) was used as spreading solvent. Typically, drops (20 μL) of the surfactant solution (∼0.5 mg mL−1) were deposited using a microsyringe onto the subphase. After leaving the solvent to evaporate for ∼10 min, the barriers were compressed at 7 mm min−1 and the surface pressure was monitored using a platinum DyneProbe that had been flamed. The quartz substrate for Langmuir–Blodgett deposition was treated with piranha solution prior to deposition.X-ray crystallography and powder X-ray diffraction
Single-crystal X-ray diffraction data was either collected at 100 K on a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn 724+ detector mounted at the window of an FR-E+ Superbright Mo Kα rotating anode generator (λ = 0.71075 Å) with HF or VHF varimax optics, or a Rigaku 007 HF diffractometer equipped with an enhanced sensitivity Saturn 944+ detector with a Cu-Kα rotating anode generator (λ = 1.5418 Å) with HF varimax optics. Unit cell parameters were refined against all data and an empirical absorption correction applied in either CrystalClear63 or CrysAlisPro.64 All structures were solved by intrinsic phasing using SHELXT65 and refined on F02 by SHELXL using Olex2.66 The CCDC nos. for Hqsal-OC12H25 and 1 are 2357075 and 2357074, respectively and can be downloaded free of charge at https://www.ccdc.cam.ac.uk/. X-ray diffraction patterns (XRD) were collected with a PANalytical Empyrean powder X-ray diffractometer operating at 45 kV and 40 mA. A standard Cu X-ray source was employed with a Kα1 wavelength of 1.5418 Å. Materials were scanned over a two-theta range of 5–80° with a 0.013° step size at a rate of 1.25° per minute. A beam knife was utilised to reduce background scattering at low angle two-theta.Magnetic studies
Data were collected with a Quantum Design MPMS 5 SQUID magnetometer under an applied field of 1 T over the temperature range 30–350 K for 1–3. The powdered or polycrystalline samples were placed in gel capsules and care was taken to allow long thermal equilibration times at each temperature. 1H VT-NMR data were recorded on a Bruker DPX400 NMR spectrometer at 300 K in 0.5 mL THF, with 10 μL t-BuOH added. A reference solution of 10 μL t-BuOH in 50 μL THF was prepared in a small glass capillary.Synthesis and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et2O (1:1, 50 mL) under vacuum to remove organic impurity, followed by ethanol:THF (2:1, 250 mL) to collect the product. The resulting black solution was evaporated to dryness giving the complex as a black solid (0.49 g, 68%). Found (calcd%) for C56H70FeN5O7 (981.03 g mol−1): C 68.27 (68.56), H 7.18 (7.19), N 7.07 (7.14). HRMS (ESI+) m/z 918.47 [M–NO3]+ (Calcd for [Fe(qsal-OC12H25)2]+, 918.47). UV/vis, λmax (MeOH)/nm 325 (ε/dm3 mol−1 cm−1 17175) and 403 (18845). FT-IR, νmax/cm−1 2945, 2920, 2851 (alkyl chain), 1597 (CN), 1379 (NO from anion).:Et2O (1:1, 50 mL) under vacuum to remove organic impurity, followed by ethanol:THF (2:1, 250 mL) to collect the product. The resulting black solution was evaporated to dryness giving the complex as a black solid (0.22 g, 54% for flash chromatography). Found (calcd%) for C64H86FeN5O7 (1093.24 g mol−1): C 70.05 (70.31), H 7.58 (7.93), N 6.50 (6.41). HRMS (ESI+) m/z 1030.60 [M–NO3]+ (Calcd for [Fe(qsal-OC16H33)2]+, 1030.60). UV/vis, λmax (MeOH)/nm 325 (ε/dm3 mol−1 cm−1 20100) and 403 (21135). FT-IR, νmax/cm−1 2945, 2920, 2851 (alkyl chain), 1599 (CN), 1379 (NO from anion).:1, 45 mL) and filtered through Al2O3. A further washing with EtOH–THF (2:1, 30 mL) and evaporation gave the complex as a as a black solid (0.46 g, 74%). Found (calcd%) for C76H110FeN5O7 (1261.56 g mol−1): C 72.09 (72.36) H 7.86 (7.93), N 5.31 (5.55). HRMS (ESI+) m/z 1189.79 [M–NO3]+ (Calcd for [Fe(qsal-OC22H45)2]+, 1189.79). UV/vis, λmax(MeOH)/nm ε/dm3 mol−1 cm−1 325 (21805) and 403 (22360). FT-IR, νmax/cm−1 2941, 2916, 2849 (alkyl chain), 1597 (CN), 1379 (NO from anion).
:Et2O (1:1, 50 mL) under vacuum to remove organic impurity, followed by ethanol:THF (2:1, 250 mL) to collect the product. The resulting black solution was evaporated to dryness giving the complex as a black solid (0.49 g, 68%). Found (calcd%) for C56H70FeN5O7 (981.03 g mol−1): C 68.27 (68.56), H 7.18 (7.19), N 7.07 (7.14). HRMS (ESI+) m/z 918.47 [M–NO3]+ (Calcd for [Fe(qsal-OC12H25)2]+, 918.47). UV/vis, λmax (MeOH)/nm 325 (ε/dm3 mol−1 cm−1 17175) and 403 (18845). FT-IR, νmax/cm−1 2945, 2920, 2851 (alkyl chain), 1597 (CN), 1379 (NO from anion).:Et2O (1:1, 50 mL) under vacuum to remove organic impurity, followed by ethanol:THF (2:1, 250 mL) to collect the product. The resulting black solution was evaporated to dryness giving the complex as a black solid (0.22 g, 54% for flash chromatography). Found (calcd%) for C64H86FeN5O7 (1093.24 g mol−1): C 70.05 (70.31), H 7.58 (7.93), N 6.50 (6.41). HRMS (ESI+) m/z 1030.60 [M–NO3]+ (Calcd for [Fe(qsal-OC16H33)2]+, 1030.60). UV/vis, λmax (MeOH)/nm 325 (ε/dm3 mol−1 cm−1 20100) and 403 (21135). FT-IR, νmax/cm−1 2945, 2920, 2851 (alkyl chain), 1599 (CN), 1379 (NO from anion).:1, 45 mL) and filtered through Al2O3. A further washing with EtOH–THF (2:1, 30 mL) and evaporation gave the complex as a as a black solid (0.46 g, 74%). Found (calcd%) for C76H110FeN5O7 (1261.56 g mol−1): C 72.09 (72.36) H 7.86 (7.93), N 5.31 (5.55). HRMS (ESI+) m/z 1189.79 [M–NO3]+ (Calcd for [Fe(qsal-OC22H45)2]+, 1189.79). UV/vis, λmax(MeOH)/nm ε/dm3 mol−1 cm−1 325 (21805) and 403 (22360). FT-IR, νmax/cm−1 2941, 2916, 2849 (alkyl chain), 1597 (CN), 1379 (NO from anion).Data availability
The data supporting this article have been included as part of the ESI.† The CCDC no. for Hqsal-OC12H25 and 1 are 2357075 and 2357074, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has received funding support from the NSRF via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation (Grant Number: B39G670018). The Development and Promotion of Science and Technology Talents project is thanked for a scholarship to P. P. The University of Auckland Shared Research Equipment Platform (ShaRe) and Dr Timothy Christopher for collecting powder X-ray diffraction data.Notes and references
- M. A. Halcrow, Spin-Crossover Materials: Properties and Applications, 2013 Search PubMed.
- A. Enriquez-Cabrera, A. Rapakousiou, M. Piedrahita Bello, G. Molnár, L. Salmon and A. Bousseksou, Coord. Chem. Rev., 2020, 419, 213396 CrossRef CAS.
- P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC.
- M. Nihei, T. Shiga, Y. Maeda and H. Oshio, Coord. Chem. Rev., 2007, 251, 2606–2621 CrossRef CAS.
- A. B. Gaspar, M. Seredyuk and P. Gütlich, J. Mol. Struct., 2009, 924–926, 9–19 CrossRef CAS.
- D. J. Harding, P. Harding and W. Phonsri, Coord. Chem. Rev., 2016, 313, 38–61 CrossRef CAS.
- S. Hayami, K. Hiki, T. Kawahara, Y. Maeda, D. Urakami, K. Inoue, M. Ohama, S. Kawata and O. Sato, Chem.–Eur. J., 2009, 15, 3497–3508 CrossRef CAS.
- S. Hayami, Z. Z. Gu, H. Yoshiki, A. Fujishima and O. Sato, J. Am. Chem. Soc., 2001, 123, 11644–11650 CrossRef CAS.
- S. Hayami, T. Kawahara, G. Juhász, K. Kawamura, K. Uehashi, O. Sato and Y. Maeda, J. Radioanal. Nucl. Chem., 2003, 255, 443–447 CrossRef CAS.
- M. Shatruk, H. Phan, B. A. Chrisostomo and A. Suleimenova, Coord. Chem. Rev., 2015, 289–290, 62–73 CrossRef CAS.
- E. Trzop, D. Zhang, L. Piñeiro-Lopez, F. J. Valverde-Muñoz, M. Carmen Muñoz, L. Palatinus, L. Guerin, H. Cailleau, J. A. Real and E. Collet, Angew. Chem., Int. Ed., 2016, 55, 8675–8679 CrossRef CAS PubMed.
- Z. Y. Li, H. Ohtsu, T. Kojima, J. W. Dai, T. Yoshida, B. K. Breedlove, W. X. Zhang, H. Iguchi, O. Sato, M. Kawano and M. Yamashita, Angew. Chem., Int. Ed., 2016, 55, 5184–5189 CrossRef CAS.
- Y. Meng, Q. Q. Sheng, M. N. Hoque, Y. C. Chen, S. G. Wu, J. Tucek, R. Zboril, T. Liu, Z. P. Ni and M. L. Tong, Chem.–Eur. J., 2017, 23, 10034–10037 CrossRef CAS.
- C. Zheng, S. Jia, Y. Dong, J. Xu, H. Sui, F. Wang and D. Li, Inorg. Chem., 2019, 58, 14316–14324 CrossRef CAS PubMed.
- F. F. Martins, A. Joseph, H. P. Diogo, M. E. Minas da Piedade, L. P. Ferreira, M. D. Carvalho, S. Barroso, M. J. Romão, M. J. Calhorda and P. N. Martinho, Eur. J. Inorg. Chem., 2018, 2018, 2976–2983 CrossRef CAS.
- Z. Y. Li, J. W. Dai, Y. Shiota, K. Yoshizawa, S. Kanegawa and O. Sato, Chem.–Eur. J., 2013, 19, 12948–12952 CrossRef CAS PubMed.
- M. Griffin, S. Shakespeare, H. J. Shepherd, C. J. Harding, J. F. Létard, C. Desplanches, A. E. Goeta, J. A. K. Howard, A. K. Powell, V. Mereacre, Y. Garcia, A. D. Naik, H. Müller-Bunz and G. G. Morgan, Angew. Chem., Int. Ed., 2011, 50, 896–900 CrossRef CAS PubMed.
- T. Boonprab, S. J. Lee, S. G. Telfer, K. S. Murray, W. Phonsri, G. Chastanet, E. Collet, E. Trzop, G. N. L. Jameson, P. Harding and D. J. Harding, Angew. Chem., Int. Ed., 2019, 58, 11811–11815 CrossRef CAS.
- D. J. Harding, D. Sertphon, P. Harding, K. S. Murray, B. Moubaraki, J. D. Cashion and H. Adams, Chem.–Eur. J., 2013, 19, 1082–1090 CrossRef CAS.
- D. J. Harding, W. Phonsri, P. Harding, K. S. Murray, B. Moubaraki and G. N. L. Jameson, Dalton Trans., 2014, 44, 15079–15082 RSC.
- N. Phukkaphan, D. L. Cruickshank, K. S. Murray, W. Phonsri, P. Harding and D. J. Harding, Chem. Commun., 2017, 53, 9801–9804 RSC.
- B. J. C. Vieira, J. C. Dias, I. C. Santos, L. C. J. Pereira, V. Da Gama and J. C. Waerenborgh, Inorg. Chem., 2015, 54, 1354–1362 CrossRef CAS PubMed.
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC.
- K. Fukuroi, K. Takahashi, T. Mochida, T. Sakurai, H. Ohta, T. Yamamoto, Y. Einaga and H. Mori, Angew. Chem., Int. Ed., 2014, 53, 1983–1986 CrossRef CAS.
- M. Nakaya, R. Ohtani, L. F. Lindoy and S. Hayami, Inorg. Chem. Front., 2021, 8, 484–498 RSC.
- R. Díaz-Torres, G. Chastanet, E. Collet, E. Trzop, P. Harding and D. J. Harding, Chem. Sci., 2023, 14, 7185–7191 RSC.
- R. Díaz-Torres, T. Boonprab, S. Gómez-Coca, E. Ruiz, G. Chastanet, P. Harding and D. J. Harding, Inorg. Chem. Front., 2022, 22–24 Search PubMed.
- W. Phonsri, P. Harding, L. Liu, S. G. Telfer, K. S. Murray, B. Moubaraki, T. M. Ross, G. N. L. Jameson and D. J. Harding, Chem. Sci., 2017, 8, 3949–3959 RSC.
- D. Sertphon, D. J. Harding, P. Harding, K. S. Murray, B. Moubaraki, H. Adams, A. Alkaş and S. G. Telfer, Eur. J. Inorg. Chem., 2016, 2016, 432–438 CrossRef CAS.
- R. Díaz-Torres, W. Phonsri, K. S. Murray, P. Harding and D. J. Harding, Cryst. Growth Des., 2022, 22, 1543–1547 CrossRef.
- B. J. C. Vieira, L. C. J. Pereira, V. da Gama and J. C. Waerenborgh, CrystEngComm, 2023, 25, 6472–6477 RSC.
- R. Diáz-Torres, W. Phonsri, K. S. Murray, L. Liu, M. Ahmed, S. M. Neville, P. Harding and D. J. Harding, Inorg. Chem., 2020, 59, 13784–13791 CrossRef.
- S. Zhao, H. Zhou, C.-Y. Qin, H.-Z. Zhang, Y.-H. Li, M. Yamashita and S. Wang, Chem.–Eur. J., 2023, 29, e202300554 CrossRef CAS.
- I. R. Jeon, O. Jeannin, R. Clérac, M. Rouzières and M. Fourmigué, Chem. Commun., 2017, 53, 4989–4992 RSC.
- B. J. C. Vieira, L. C. J. Pereira, V. da Gama, I. C. Santos, A. C. Cerdeira and J. C. Waerenborgh, Magnetochemistry, 2022, 8, 1–19 CAS.
- P. N. Martinho, C. J. Harding, H. Müller-Bunz, M. Albrecht and G. G. Morgan, Eur. J. Inorg. Chem., 2010, 675–679 CAS.
- C. Gandolfi, G. G. Morgan and M. Albrecht, Dalton Trans., 2012, 41, 3726–3730 RSC.
- C. Gandolfi, N. Miyashita, D. G. Kurth, P. N. Martinho, G. G. Morgan and M. Albrecht, Dalton Trans., 2010, 39, 4508–4516 RSC.
- P. Coronel, A. Barraud, R. Claude, O. Kahn, A. Ruaudel-Teixier and J. Zarembowitch, J. Chem. Soc., Chem. Commun., 1989, 193–194 RSC.
- H. Soyer, E. Dupart, C. Mingotaud, C. J. Gomez-Garcia and P. Delhaès, Colloids Surf., A, 2000, 171, 275–282 CrossRef CAS.
- A. Ruaudel-Teixier, A. Barraud, P. Coronel and O. Kahn, Thin Solid Films, 1988, 160, 107–115 CrossRef CAS.
- N. G. White, H. L. C. Feltham, C. Gandolfi, M. Albrecht and S. Brooker, Dalton Trans., 2010, 39, 3751–3758 RSC.
- Y. Bodenthin, U. Pietsch, H. Möhwald and D. G. Kurth, J. Am. Chem. Soc., 2005, 127, 3110–3114 CrossRef CAS.
- H. Soyer, C. Mingotaud, M. L. Boillot and P. Delhaes, Langmuir, 1998, 14, 5890–5894 CrossRef CAS.
- O. Roubeau, B. Agricole, R. Clérac and S. Ravaine, J. Phys. Chem. B, 2004, 108, 15110–15116 CrossRef CAS.
- J. T. Culp, J. H. Park, D. Stratakis, M. W. Meisel and D. R. Talham, J. Am. Chem. Soc., 2002, 124, 10083–10090 CrossRef CAS PubMed.
- J. Weihermüller, S. Schlamp, W. Milius, F. Puchtler, J. Breu, P. Ramming, S. Hüttner, S. Agarwal, C. Göbel, M. Hund, G. Papastavrou and B. Weber, J. Mater. Chem. C, 2019, 7, 1151–1163 RSC.
- S. Sundaresan, J. A. Kitchen and S. Brooker, Inorg. Chem. Front., 2020, 7, 2050–2059 RSC.
- J. A. Kitchen, N. G. White, C. Gandolfi, M. Albrecht, G. N. L. Jameson, J. L. Tallon and S. Brooker, Chem. Commun., 2010, 46, 6464–6466 RSC.
- R. Akiyoshi, Y. Hirota, D. Kosumi, M. Tsutsumi, M. Nakamura, L. F. Lindoy and S. Hayami, Chem. Sci., 2019, 10, 5843–5848 RSC.
- I. Galadzhun, R. Kulmaczewski, N. Shahid, O. Cespedes, M. J. Howard and M. A. Halcrow, Chem. Commun., 2021, 57, 4039–4042 RSC.
- H. L. C. Feltham, C. Johnson, A. B. S. Elliott, K. C. Gordon, M. Albrecht and S. Brooker, Inorg. Chem., 2015, 54, 2902–2909 CrossRef CAS.
- Y. Guo, A. Rotaru, H. Müller-Bunz, G. G. Morgan, S. Zhang, S. Xue and Y. Garcia, Dalton Trans., 2021, 50, 12835–12842 RSC.
- D. Rosario-Amorin, P. Dechambenoit, A. Bentaleb, M. Rouzières, C. Mathonière and R. Clérac, J. Am. Chem. Soc., 2018, 140, 98–101 CrossRef CAS PubMed.
- M. Seredyuk, M. C. Muñoz, V. Ksenofontov, P. Gütlich, Y. Galyametdinov and J. A. Real, Inorg. Chem., 2014, 53, 8442–8454 CrossRef CAS.
- Q. Zhao, J. P. Xue, Z. K. Liu, Z. S. Yao and J. Tao, Dalton Trans., 2021, 50, 11106–11112 RSC.
- R. Akiyoshi, R. Ohtani, L. F. Lindoy and S. Hayami, Dalton Trans., 2021, 50, 5065–5079 RSC.
- R. Akiyoshi, K. Kuroiwa, S. Alao Amolegbe, M. Nakaya, R. Ohtani, M. Nakamura, L. F. Lindoy and S. Hayami, Chem. Commun., 2017, 53, 4685–4687 RSC.
- Y. Komatsu, K. Kato, Y. Yamamoto, H. Kamihata, Y. H. Lee, A. Fuyuhiro, S. Kawata and S. Hayami, Eur. J. Inorg. Chem., 2012, 2769–2775 CrossRef CAS.
- C. Gandolfi, C. Moitzi, P. Schurtenberger, G. G. Morgan and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 14434–14435 CrossRef CAS PubMed.
- H. L. C. Feltham, A. S. Barltrop and S. Brooker, Coord. Chem. Rev., 2017, 344, 26–53 CrossRef CAS.
- M. Bodik, M. Jergel, E. Majkova and P. Siffalovic, Adv. Colloid Interface Sci., 2020, 283, 102239 CrossRef CAS.
- Rigaku Oxford Diffraction CrystalClear-SM Expert 3.1. 2012.
- Rigaku Oxford Diffraction, CrysAlisPro v1.171.39.34 2017.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2357075 and 2357074. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra06111j |
| This journal is © The Royal Society of Chemistry 2024 |