
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceP-Sulfonatocalix[4]arene turns peptide aggregates into an efficient cell-penetrating peptide†
Mahsima Heydaria,
Najmeh Salehib,
Reza Zadmard c,
Werner M. Naud,
Khosro Khajehe,
Zahra Azizif and
Amir Norouzy*a
c,
Werner M. Naud,
Khosro Khajehe,
Zahra Azizif and
Amir Norouzy*a
aBioprocess Engineering Department, National Institute of Genetic Engineering and Biotechnology (NIGEB), Tehran, Iran. E-mail: a.norouzy@nigeb.ac.ir
bSchool of Biology, College of Science, University of Tehran, Tehran, Iran
cDepartment of Organic Chemistry, Chemistry and Chemical Engineering Research Center of Iran, Tehran, Iran
dSchool of Science, Constructor University, Bremen, Germany
eDepartment of Biochemistry, Faculty of Biological Sciences, Tarbiat Modares University, Tehran, Iran
fDepartment of Molecular Medicine, School of Advanced Technologies in Medicine, Tehran University of Medical Sciences, Tehran, Iran
First published on 15th October 2024
Abstract
A novel cell-penetrating peptide (CPP) called FAM-Y4R4, with FAM as a fluorescent probe, was developed. Initially, we aimed to use Y4 as a supramolecular host for water-insoluble drugs, with R4 driving the complex into cells. However, an unexpected hurdle was discovered; the peptide self-assembled into amorphous aggregates, rendering it ineffective for our intended purpose. Molecular dynamics simulations revealed that intermolecular cation–π interactions between arginine and tyrosine caused this aggregation. By decorating the R4 sidechains with p-sulfonatocalix[4]arene (CX4), we successfully dissolved most of the aggregates, significantly improved the peptide's solubility and enhanced the cell uptake with MCF7 and A549 cells via both direct penetration and endocytosis. The binding strength between CX4 and R4, as well as the interaction between curcumin and tyrosines was quantified. Encouragingly, our results showed that FAM-Y4R4, with CX4, effectively delivered curcumin – as a model for poorly water-soluble drugs – into cells which exhibited potent anticancer activity. Using R4/CX4 instead of the conventional R7–9 oligoarginine-based CPP simplifies peptide synthesis and offers higher yields. CX4 shows promise for addressing aggregation issues in other peptides that undergo a similar aggregation mechanism.
Introduction
The poor solubility and cell membrane impermeability often limit the effectiveness of many therapeutic agents.1 Although significant progress has been made in drug delivery, intracellular delivery of many drugs and therapeutic agents still remains a challenge.2 Among the various drug carriers, cell-penetrating peptides (CPPs) are capable and effective carriers for the intracellular delivery of a variety of cargoes, such as small molecules, nucleic acids, viruses, imaging contrast agents, and nanoparticles.3 CPPs are easily synthesized on a large scale and because of their low molecular weight do not trigger an immune response. Numerous CPP-conjugated drugs are making their way to the bedside for reducing inflammation and pain, cancer therapy, curing heart diseases, anti-aging medications and even for diagnostic purposes.4Based on their physicochemical properties, CPPs are classified into three groups: cationic, hydrophobic and amphipathic. Amphipathic CPPs are composed of a hydrophobic part at one end and a hydrophilic part at the other end. Poorly water-soluble drugs (PWSDs) are hydrophobic molecules that often exhibit limited bioavailability. Using CPPs for increasing their solubility and bioavailability is therefore the goal of many research studies. Cationic CPPs frequently have the least affinity for the PWSDS. Hydrophobic CPPs on the other hand can bind to PWSDs but do not increase their solubility, whereas amphipathic CPPs can bind to PWSDs from their hydrophobic part and increase drug solubility with their hydrophilic part. The hydrophilic part is usually composed of cationic amino acids.
In this study, we designed FAM-Y4R4 (Fig. 1) as an amphipathic CPP to deliver curcumin to cells. Beside its therapeutic property curcumine served as a model for PWSD. FAM is the commercial name for 5-carboxyfluorescein –a fluorescent probe– that is extensively used for labeling peptides. FAM-Y4 is the hydrophobic part designed for biding to curcumin. R4 is both the hydrophilic and the cell-penetrating part. Arginine is the most effective amino acid for enhancing the cellular uptake of cationic or amphipathic CPPs.5,6 The permeability of arginine residues can be attributed to the ability of their guanidine group to form hydrogen bonds with the negatively charged groups on the cell membrane, such as carboxylic acid, sulfate, and phosphate.7
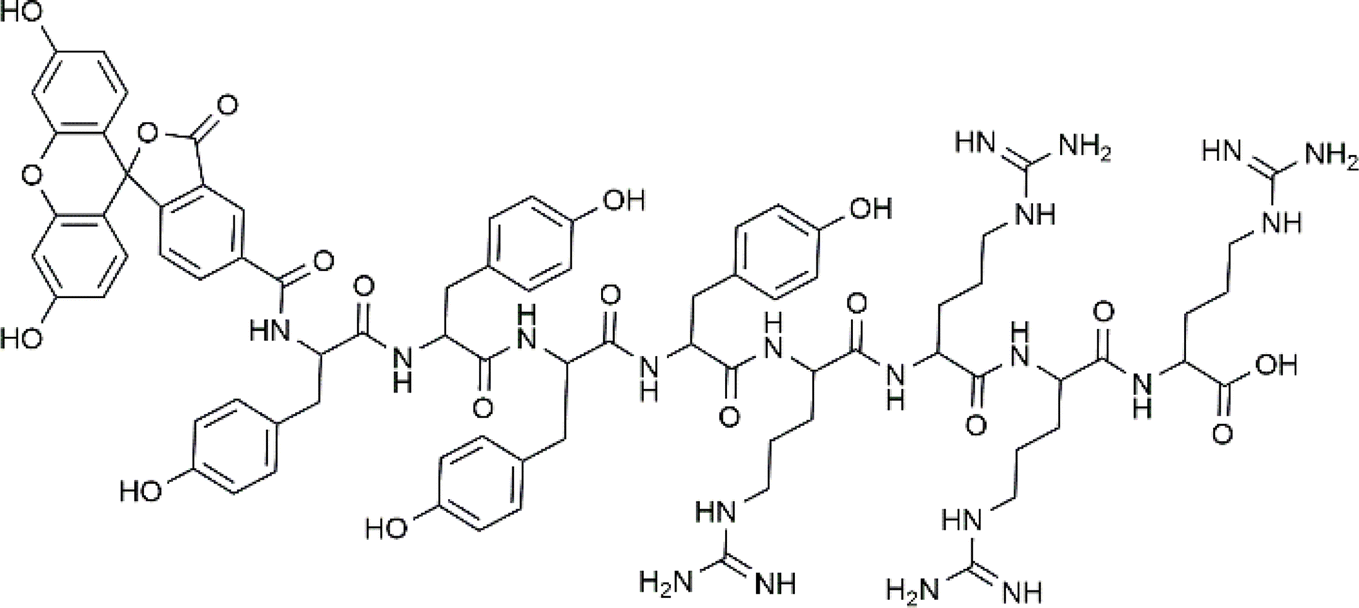 | ||
| Fig. 1 Chemical structure of FAM-Y4R4 peptide, with FAM as a 5-carboxyfluorescein fluorescent dye attached to the N-terminus. | ||
Unfortunately, extensive intermolecular cation–π interactions between the sidechains of arginine and tyrosine residues led to aggregation. These aggregates were rendered useless as they precipitated and did not enter the cells. Amphipathic peptides, in general, have a tendency to aggregate or to form micelles. Aggregation reduces the effective hydrophobicity of amphiphilic peptides by concealing their nonpolar parts from the surrounding aqueous environment.8 Sometimes amphipathic peptides aggregated on the surface of the cells abolish their uptake.9 Developing the strategies to inhibit amphipathic peptides from aggregation is therefore crucially important.10 We successfully solubilized the majority of peptide aggregates by decorating the arginine side chains with p-sulfonatocalix[4]arenes (CX4). CX4 is a member of calixarene family. Calixarens are efficient molecular host for variety of cargos including peptides.6,11,12 Decoration of the arginine sidechain with CX4 liberated the arginines from the tyrosines and drastically improved the solubility of the peptides. The solubilized peptide demonstrated promising cell uptake and cargo-carrying capabilities. This approach serves as a proof-of-principle for addressing similarly-aggregated peptides resulting from intramolecular cation–π interactions involving lysine or arginine side chains (as cation donors) and aromatic side chains of tryptophan, tyrosine, or phenylalanine (as π donors). It is worth noting that CX4 also binds to lysine side chains,13 similar to arginine,14 and is able to prevent its binding to the aromatic side chains.
Curcumin has been identified as the most important active ingredient in turmeric with a wide range of therapeutic benefits such as anti-cancer, anti-oxidant, anti-inflammatory, anti-thrombotic, and anti-microbial effects.15 However, its usage is limited due to its poor aqueous solubility, low bioavailability, and rapid metabolism. The aim of using a CPP is to increase the solubility of curcumin and translocate it across the cell membrane. Due to its pharmaceutical importance and its aromatic structure, to bind to Y4, we chose curcumin as a PWSD model to be carried with our peptide into the cells.
Materials and methods
Peptide preparation
FAM-Y4R4 was synthesized with the solid-phase peptide synthesis (SPPS) method using the Fmoc strategy.16–18 We used 2-chlorotrityl chloride (CTC) resin as the solid phase. The synthesized peptide was cleaved from the resin using a cleavage cocktail containing 90% trifluoroacetic acid, 5% phenol, 2.5% water, and 2.5% triisopropylsilane as a scavenger. The crude peptide was precipitated using cold diethyl ether and subsequently dissolved in a solution consisting of 50% acetonitrile in water. The crude peptide was purified with a reversed-phased semi-preparative HPLC instrument equipped with a C18 column (21.2 × 150 mm, 5 μm). The purity of the peptide was assayed by an analytical HPLC instrument. The accuracy of the synthesis was confirmed by mass spectrometry. See the ESI† for details of the synthesis, purification, and mass spectroscopy.Dynamic light scattering (DLS)
FAM-Y4R4 solutions were prepared at a concentration of 8 μM in 10 mM phosphate buffer with a pH of 7. Both solutions with and without 4 millimolar CX4 were prepared. The particle diameter was measured using a NanoBrook Omni instrument from Malvern, which was equipped with a 4 mW He/Ne laser at a wavelength of 633 nm. The measurements were taken at a fixed detector angle of 173°, utilizing an avalanche photodiode detector.Field emission scanning electron microscopy (FE-SEM)
Peptide solution by the concentration of 50 μM were prepared in the phosphate buffer. For dissolving the peptide aggregates, CX4 was added to the peptide and the mixture was sonicated for 15 min followed by stirring at room temperature for an hour. The FE-SEM images were obtained using a MIRA3 TESCAN-XMU electron microscope at a voltage of 15 kV. The mean particle diameters of the images were calculated using ImageJ.Molecular dynamics (MD) simulation of the peptide aggregation
The 3D structure of the FAM-Y4R4 peptide was modeled. Three of 50 ns MD simulations were performed for 150 ns to sample different conformations of FAM-Y4R4. The four most stable monomer conformations of FAM-Y4R4 were extracted using clustering analysis on the MD simulations trajectory of the FAM-Y4R4 monomer. These four peptide conformations were randomly placed in the two different simulation boxes with at least a 6 Å distance between them. In each system, after equilibration, the production run was carried out for 100 ns. All MD simulation parameters and structural analysis were explained in the ESI.†Cellular uptake
MCF-7 and A549 cells were purchased from the Iranian Biological Resource Center. The cells were grown in DMEM/F12 culture medium containing 10% heat-inactivated fetal bovine serum (FBS) and 1% of penicillin/streptomycin in a humidified incubator with 5% CO2 and temperature fixed to 37 °C. Cellular uptake of the peptide and peptide/CX4 complex was confirmed using flow cytometry, fluorescence microscopy, and confocal laser scanning microscopy (CLSM).Measuring the binding constants
The binding constants (Ka) of the arginine sidechains to CX4 and the Ka of curcumin to tyrosine and phenol (as representatives of the binding domain, Y4) were determined using the following methods.In vitro cytotoxicity study
The viability of MCF-7 and A549 cells were assessed in the presence of peptide, curcumin, peptide/curcumin, peptide/CX4, peptide/CX4/curcumin or CX4 using the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The cells were seeded at a density of 1 × 104 cells per well in a 96-well plate and treated with different concentrations of the aforementioned samples in triplicates for 18 h. The cell's viability was assayed with the MTT stablished protocol. The absorption of the treated and untreated cells at 570 nm was measured using the BioTek Epoch2 microplate spectrophotometer. The results are presented as mean ± standard error. The Tukey test was employed to determine the statistical significance with P-values < 0.05.Results and discussion
Peptide design, synthesis and purification
We have designed FAM-Y4R4 which is composed of three parts: tetraarginine as the cell-penetrating part, tetratyrosine as the binding site and FAM as the fluorescence label (Fig. 1). As a result, this peptide is capable of: (i) entering the cells by its tetraarginine part (ii) binding to hydrophobic cargos via its tetratyrosine part and (iii) is fluorescently traceable inside live cells. The FAM molecule, being hydrophobic, is intentionally attached to the Y4 part to ensure that all hydrophobic groups are located on one side, away from the R4. This arrangement makes FAM-Y4R4 amphipathic. The amphipathicity allows the peptide to bind to hydrophobic cargos and simultaneously increase the solubility of the cargo.In the pursuit of finding shorter peptides capable of cell penetration, researchers have explored truncated versions of Tat37–72. Vivès et al. identified a positively charged, 13 amino acid domain (Tat48–60) responsible for cellular uptake.20 Subsequent research by Park et al. further truncated it to Tat49–57 without significant loss of penetration capability.21 In our study, we opted for a shorter oligoarginine sequence instead of the traditional R7–R15 due to synthetic concerns, such as low yield associated with longer homorepeat of arginine. Adding even a single arginine to any peptide sequence proved challenging during peptide synthesis. On the other hand, CPPs shorter than eight amino acids, particularly oligoarginine peptides with fewer than eight arginine residues, exhibit poor cellular uptake.22 To address this issue, we compensated for the poor cellular uptake of R4 by attaching it to CX4. The preparation of R4/nCX4 is easier and more cost-effective compared to synthesizing R8. As a result, FAM-Y4R4 was synthesized with a high yield, and the peptide purity is approximately 99% while it exhibited promising cell uptake. The accuracy of the synthesis was confirmed by the mass spectrum (Fig. S1†).
Peptide self assembly
The peptide underwent self-assembly, forming nanostructural aggregates as observed through DLS and FE-SEM imaging (Fig. 2).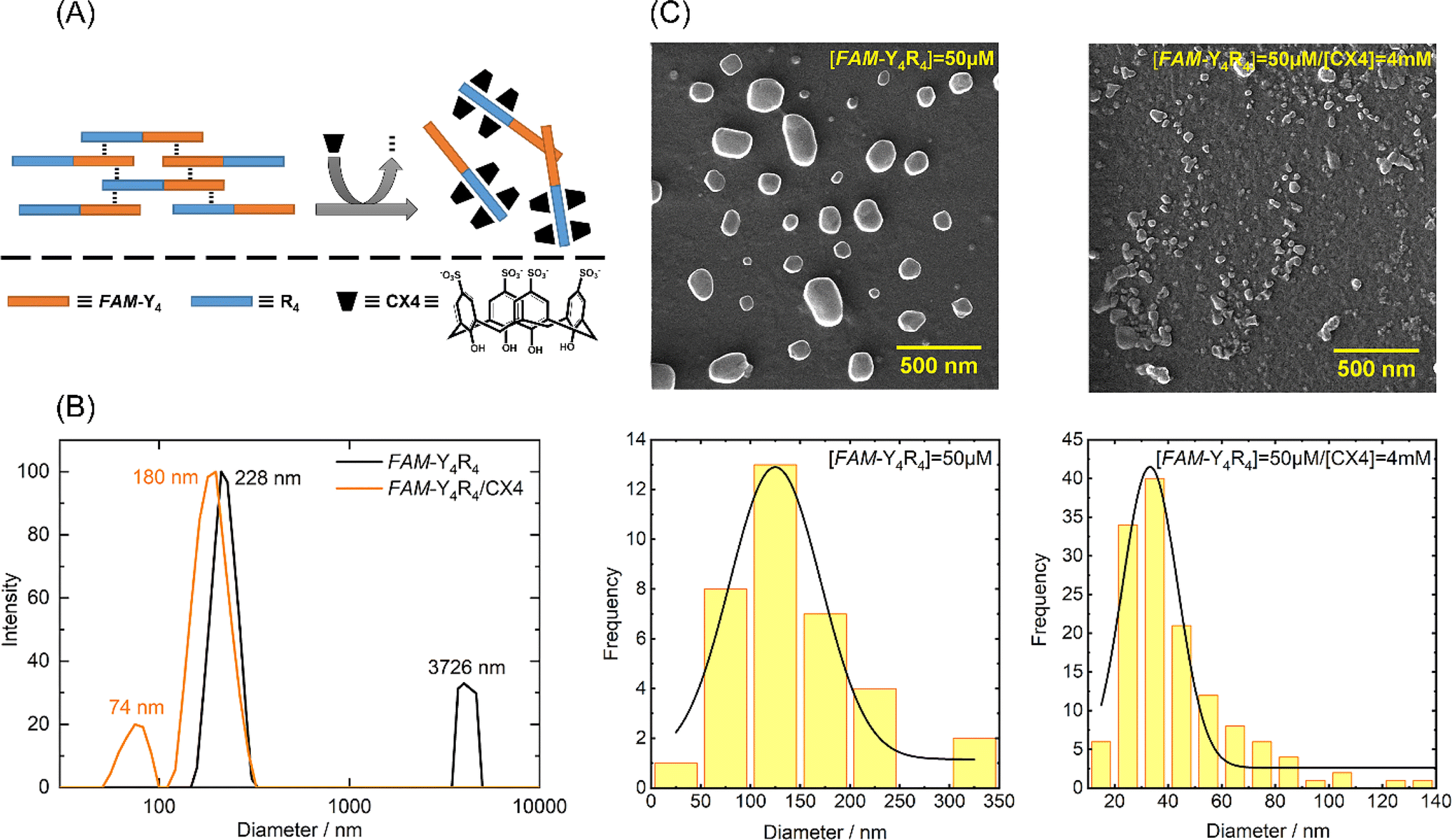 | ||
| Fig. 2 (A) Schematic illustration of the cation–π and π–π interactions that drive peptide aggregation. Subsequently, the addition of CX4 triggers the breakdown of these aggregates. (B) Size distribution of particles in a solution containing either the peptide or peptide/CX4 measured by dynamic light scattering (DLS). (C) FE-SEM images depicting the morphology of the peptide and the peptide/CX4 aggregates, along with their respective size distributions. | ||
The predominant particle sizes, as determined by DLS, were approximately 3726 nm and 228 nm. We conducted circular dichroism (CD) spectroscopy on FAM-Y4R4 in order to identify α-helix or β-sheet structures. However, the CD spectrum did not exhibit any characteristics of these secondary structures (Fig. S2†).23 Upon adding CX4 to the peptide solution, some of the aggregates completely dissolved, while the remaining aggregates shrank into nanoparticles with a mean size ranging from 74 to 180 nm. FE-SEM images were taken before and after the addition of CX4 to visualize the peptide aggregates. The average diameter of the aggregates in the images was measured to be 142 ± 19.1 nm. Following the addition of CX4, the mean size of the aggregates in the images decreased to 42 ± 8.7 nm. To explain this observation, we hypothesized that there was a cation–π interaction between the sidechains of arginines and tyrosines. Because when the guanidino group of the arginine sidechains entered the CX4 cavity it became inaccessible for interaction with the tyrosine sidechains, resulted in dissolving the aggregates (Fig. 2A).19 To illustrate the cation–π interaction, we performed a spectral titration of phenol, representing the tyrosine side chain, with guanidine HCl, as the side chain of arginine. The spectral titration yielded a binding isotherm, from which a Ka value of 9.7 × 105 M−1 and the binding energy of 8.13 kcal mol−1 was calculated (Fig. S3†). In general, smaller cations exhibit stronger interactions with π systems. For instance, the binding energies of Li+ and NH4+ to benzene are 38 and 19 kcal mol−1 respectively.24 Since guanidino is larger than these ions, its binding energy is comparatively lower. Numerous studies have extensively reported the presence of cation–π interactions within protein structures, wherein the side chains of lysine or arginine interact with the side chains of phenylalanine, tyrosine, or tryptophan residues governing important biological processes.24
To gain further insights into the aggregation mechanism, we conducted MD simulations. The clustering of peptide MD simulations revealed four stable conformations of FAM-Y4R4 (Fig. S4†), randomly distributed in two separate boxes. Two independent MD simulations, named MD I and MD II, were conducted for a duration of 100 ns each. The radius of gyration (Rg) value, which represents the average root mean square distance of all atoms from the center of mass of the peptide, was used to assess structural compactness. In both MD I and II trajectories, the Rg values decreased from approximately 20 to 13 Å (Fig. S5A†), indicating a more compact structure. The solvent-accessible surface area (SASA) value, which quantifies the surface area of the peptide interacting with solvent molecules, also decreased throughout MD I and II, further supporting the compactness and aggregation of the peptides (Fig. S5B†). Additionally, the aggregation process was associated with an increase in hydrogen bonding. The number of hydrogen bonds formed between and across all peptides increased during the MD I and II simulations (Fig. S5C†). To visualize the aggregation process during the production MD simulations, representative systems at 25, 50, 75, and 100 ns were depicted (Fig. S5D†). These snapshots provide insight into the kinetics of aggregation for the four FAM-Y4R4 peptides over the course of the 100 ns simulations. Notably, MD II exhibited faster aggregation kinetics, as evidenced by a more rapid decrease in Rg and SASA values, as well as a quicker increase in the number of hydrogen bonds.
The clustering of MD I and II trajectories depicted two more stable clusters for the aggregated form of FAM-Y4R4. The aggregated structure at 100 ns of MD I and II which are belonged to the most stable clusters and the details of their interactions are shown in Fig. 3 and S6.†
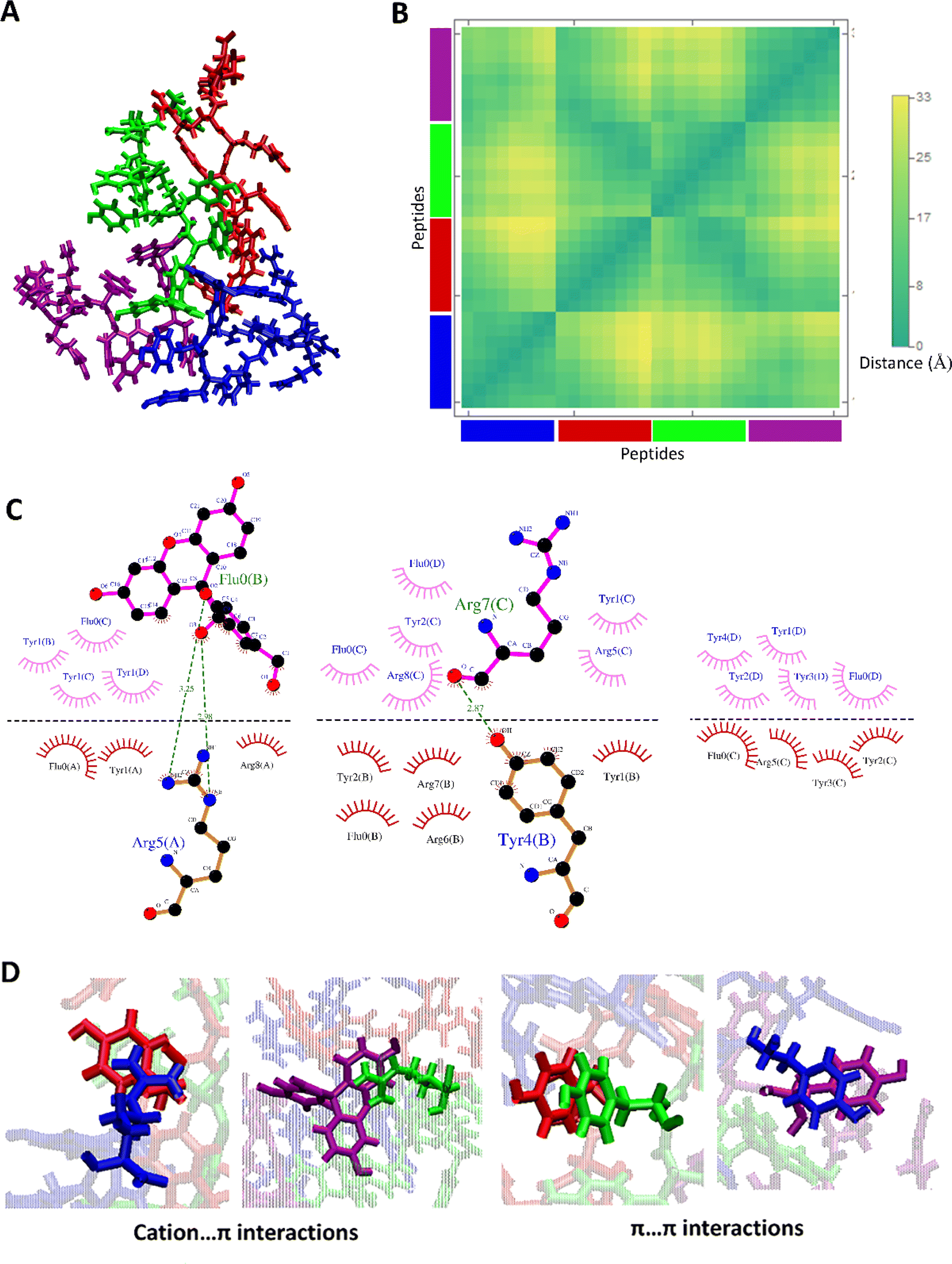 | ||
| Fig. 3 (A) The representation of the MD II system at 100 ns. (B) The distance map of the four FAM-Y4R4 peptides in the structure of panel A. (C) The hydrogen bonds, and hydrophobic contacts between four FAM-Y4R4 peptides in the structure of panel A, and (D) some examples of other non-covalent interactions such as cation–π and π–π. | ||
The distance map represents the distance between all possible amino acid residue pairs of a biomolecule in the 3D structure. The distance map of these aggregated peptides showed short distances between some residues of these peptides (Fig. 3B and S6B†). The hydrogen bonds and hydrophobic contacts (carbon atoms in contact with other carbon atoms) as important non-covalent interactions and aggregation driving forces were detected between all pairs of amino acids (Fig. 3C and S6C†). More importantly, some other non-covalent interactions such as cation–π and π–π are detected in this aggregated form of FAM-Y4R4 (Fig. 3D).
A cation–π interaction is defined between a cationic side chain of either lysine or arginine and an aromatic side chain with a distance of less than 6 Å.25 The cation–π interactions were detected between R–Y and R–FAM in these aggregated structures which was confirmed by our experimental results (Fig. 2 and S3†). The π–π interactions occur between two aromatic rings with a distance and angle of less than 6.0 Å and 30°, respectively.26 The π–π interactions were detected between Y–Y, Y–FAM, and FAM–FAM in these aggregated structures. All of these interactions stabilized the aggregates.
Peptides lack well-defined secondary structures, which necessitates a structural ensemble to describe their structure and function. During the aggregation process, the peptides undergo conformational motions and come close to each other, which causes the formation and breaking of some non-covalent interactions to reach a stable aggregated form. Aggregation is a complex phenomenon with different influencing factors, of which amino acid sequence is one of the most important.27,28
Tyrosine is a hydrophobic amino acid with an aromatic side chain. On the other hand, arginine is a positively charged amino acid with a guanidino group, resulting in a cation. FAM contains both aromatic and hydrophilic parts. Here, the most two stable clusters were detected for aggregated structures. The peptide aggregated structures are stabilized with different non-covalent interactions explained above. Conventional hydrogen bonds are the dominant interactions for stabilizing aggregated structures.29 But in these aggregated structures more stabilization and ordering were offered with packing interactions between two aromatic rings (π–π) and the side chain of arginine and aromatic rings (cation–π). A wide range of roles in proteins has been identified for interactions involving the π electron cloud of aromatic rings. Cation–π and π–π interactions were highlighted in the stability of peptide and protein structures,25,30,31 and supramolecular assemblies.32–34 The cation–π interaction is essential in the formation of the aggregate because in the absence of the cation–π interactions, the π–π interactions would lead to the formation of micelles or liposomes.
Cellular uptake
The uptake of the peptide with MCF-7 and A549 cells was investigated using flowcytometry and fluorescence imaging. Flowcytometry was utilized to quantify the amount of the peptide inside the live cells in terms of MFI values, as a function of the peptide and CX4 concentration. The recorded MFI values in Fig. 4 demonstrate that both FAM-Y4R4 and FAM-Y4R4/CX4 were uptake by the cells after 18 hours of incubation. The maximum cellular uptake of FAM-Y4R4 occurs at a concentration of 100 nM. However, as the concentration increases, the cellular uptake decreases due to peptide aggregation. It is important to note that peptide aggregation is a concentration-dependent phenomenon,27,35 and very often the cellular uptake of peptide aggregates is lower compared to individual peptide molecules. The solubilization of FAM-Y4R4 peptides with CX4 has been observed to enhance the internalization. Notably, FAM-Y4R4/CX4 exhibits the most efficient cell uptake at concentrations equal to or greater than 900 nM of the peptide in the presence of 4 mM of CX4. Previous studies have also shown that CX4 can enhance the cell uptake of LCG, acting as its host fluorescent dye.19 A549 cells demonstrate higher uptake compared to MCF7 cells. Repotente Jr et al. have also reported higher cellular uptake of gold nanoparticles with A549 cells compared to MCF7 cells.36 This higher uptake in A549 cells may be attributed to their alveolar origin tissue.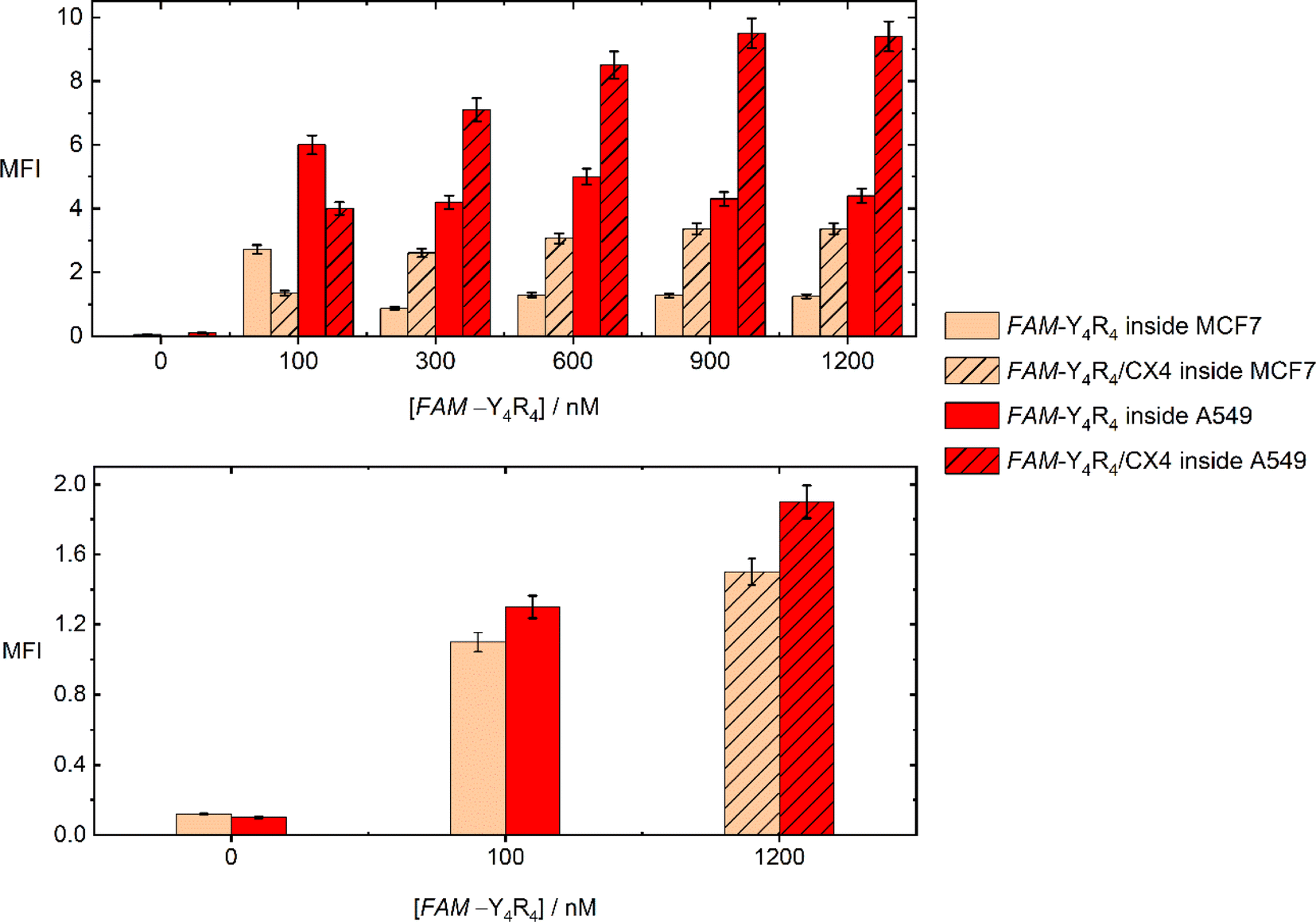 | ||
| Fig. 4 Mean fluorescence intensity (MFI) values of MCF7 and A549 cells incubated with varying concentrations of FAM-Y4R4, with and without 4 mM of CX4, were measured after 18 hours (top) and 1 hour (bottom) of incubation. | ||
Endocytosis and diffusion (direct penetration) are the main cellular uptake mechanisms. The diffusion across the cell membrane is faster than endocytosis. Eighteen hours was a plentiful time for the cells to endocytose the peptide. To favor diffusion, the peptide incubation time was shortened to one hour, and the MFI was remeasured (Fig. 4). The most effective concentration of FAM-Y4R4 (100 nM) and FAM-Y4R4/CX4 (1200 nM/4 mM) – in which the maximum MFI values were obtained after 18 hours – was applied. The result showed that the peptide in the presence and absence of CX4 diffused to the cells; however, compared to the 18 hours experiment, a lower amount of peptide molecules was found inside the live cells. The more internalized peptide molecules after 18 hours of incubation indicates that endocytosis is more efficient than diffusion in peptide uptake. After 1 h incubation, the MFI of peptide in the presence of CX4 is fairly higher than the peptide alone which means that CX4 has slightly facilitated the diffusion.37 Decorating the hydroxyl group of CX4 with alkyl chains and incubating with octaarginine has been shown to increase the octaarginine cell entry via diffusion.6 Our results indicates that CX4 alone – even in the absence of CX4 alkylation – paves the way for diffusion. Nevertheless, endocytosis remains the main uptake pathway, as the MFI values of FAM-Y4R4/CX4 are almost five times higher after 18 hours of incubation.
In order to further validate the uptake and intracellular distributions of the peptide, the cells were treated with FAM-Y4R4 or FAM-Y4R4/CX4 and subjected to fluorescence microscopy (Fig. S8†) and CLSM (Fig. 5). FAM-Y4R4, with and without CX4, was abundantly observed in the cytoplasm of the cells. FAM-Y4R4 is observable in the nucleus of some MCF7 and A549 cells (distinguished with the white arrows).
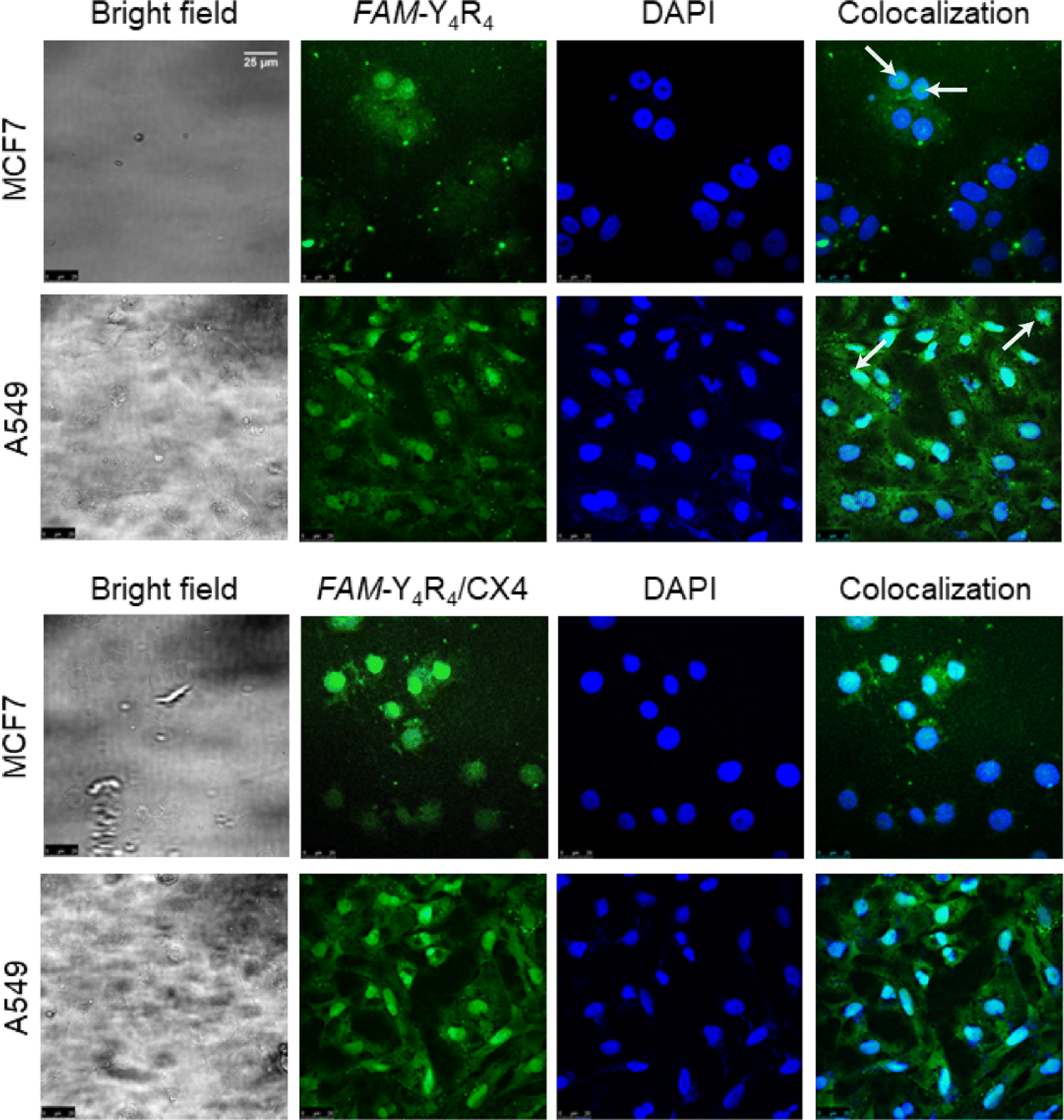 | ||
| Fig. 5 Confocal microscopy images of MCF7 and A549 cells. The cells were incubated with FAM-Y4R4 (100 nM) or FAM-Y4R4/CX4 (50 μM/1 mM) at 37 °C for 18 hours. Cell nuclei stained with DAPI dye are represented in blue, while the peptides are represented in green. The white arrows distinguish the peptide inside the nuclei. | ||
A similar observation has been reported for some amphipathic CPPs such as Pep-1 (KETWWETWWTEWSQPKKKRKV). Its cationic domain (KKKRKV) can facilitate the transportation of the peptide into the cell nucleus through nuclear pores. This cationic domain is referred to as the nuclear localization signal (NLS).38 Pep-1 consists of a tryptophan-rich, hydrophobic domain fused to an NLS domain. Despite their differences in length, both FAM-Y4R4 and Pep-1 are amphipathic in nature, composed of an aromatic-rich, hydrophobic domain fused to a cationic domain that functions as an NLS. Detailed explanation of the mechanism behind the direct penetration of guanidinium-rich molecules has been given in the literature.7,39 The guanidino group of the arginine side chains drives the internalization of arginine-rich CPPs, including FAM-Y4R4, by creating pores in the cell membrane and inducing membrane multilamellarity.
Supramolecular bindings of curcumin and CX4 to FAM-Y4R4
CPPs bind to their cargo either covalently or non-covalently (supramolecularly). Covalently conjugating the CPP with its cargo carries the risk of potentially altering the biological activity of the cargo. Conversely, non-covalent binding allows for reversible association between the cargo and CPP, enabling the cargo to dissociate from the CPP intact. The CPP/cargo supramolecular complex is able to traverse the cell membrane, and upon reaching the cytosolic side, the CPP dissociates from its cargo and undergoes proteolysis by cytosolic proteases; therefore, re-association with its cargo is not possible. An additional advantage of this method is that the CPP provides protection to its cargo, thereby increasing its serum half-life.40Curcumin contains aromatic chemical groups that can interact with the sidechains of Y4 through a process known as π-stacking (Fig. 6A). This interaction is characterized by a decrease in the fluorescence intensity of tyrosine and an increase in the molar absorption of curcumin. We utilized these spectral properties to outline the binding isotherms. To exclude the nonspecific binding between the side chains of Y4 and R4 we used tyrosine in the form of free amino acid instead of FAM-Y4R4 for fluorescence titration with curcumin (Fig. 6B).
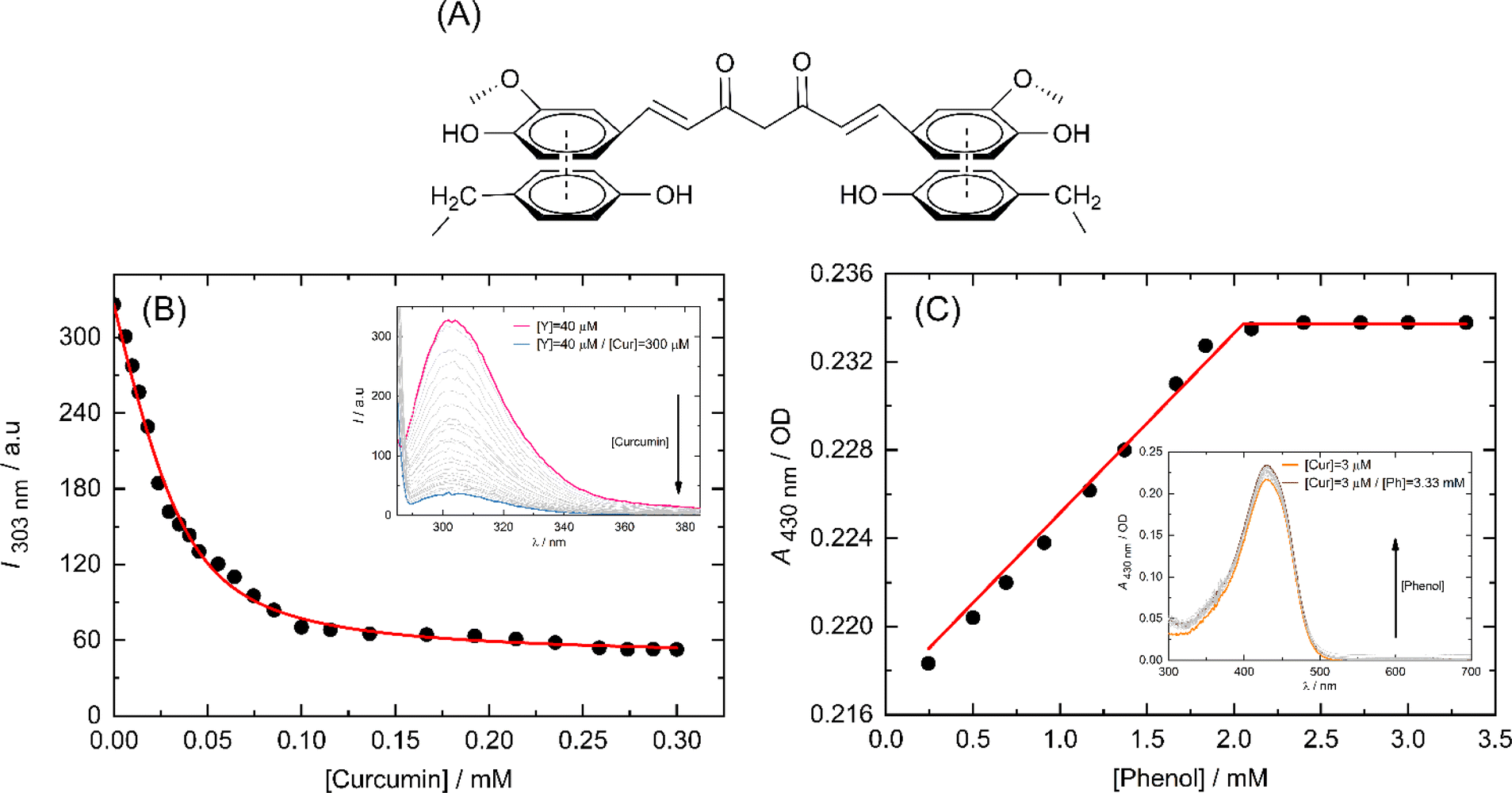 | ||
| Fig. 6 Schematic representation of π-stacking interactions between two tyrosine side chains and a curcumin molecule (A). The binding isotherms of curcumin (Cur) to tyrosine (Y) and phenol (Ph) were depicted in B and C respectively which were obtained through fluorescence and UV-vis spectral titration, respectively. The insets show the spectral titrations. | ||
We double demonstrate the binding of curcumin to the tyrosine side chain using a UV-vis spectrometer (Fig. 6C). We opted to use phenol instead of FAM-Y4R4 as a representative of the tyrosine sidechain during the spectral titration. Because our primary focus was to investigate the π-stacking interaction between the aromatic portion of Y4 and curcumin. The presence of FAM and R4 in the spectral titration could potentially complicate the results by influencing the molar absorption of curcumin.
We employed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 model (one host to one guest) in a nonlinear regression algorithm28 to calculate the binding constants. The Ka values obtained from fluorescence and UV-vis titration were found to be 1.6 × 105 M−1 and 4.6 × 106 M−1 respectively. Taking the average of these two values, the overall binding constant was determined to be Ka = 2.38 × 106 M−1. The ability of Y4 to bind strongly to curcumin is a promising indication for considering our peptide for transporting other PWSDs into live cells.
:1 model (one host to one guest) in a nonlinear regression algorithm28 to calculate the binding constants. The Ka values obtained from fluorescence and UV-vis titration were found to be 1.6 × 105 M−1 and 4.6 × 106 M−1 respectively. Taking the average of these two values, the overall binding constant was determined to be Ka = 2.38 × 106 M−1. The ability of Y4 to bind strongly to curcumin is a promising indication for considering our peptide for transporting other PWSDs into live cells.
Curcumin is widely recognized as an insoluble molecule, with a solubility of 30 nM (11 ng mL−1) in water.41 However, when bound to the tyrosines of FAM-Y4R4, the solubility of curcumin enormously increased to 400 nM. To assess the solubility of curcumin in the presence and absence of the peptide, we measured its absorption peak at 425 nm and extrapolated the concentrations from the curcumin standard curve.
Arginine side chain enters the cavity of CX4. The average binding strength of CX4 molecules to arginine side chains of FAM-Y4R4 was measured with IDA method. The obtained binding strength is Ka (CX4/R4) = 4.5 × 105 M−1 (Fig. S7†). CX4 can weakly bind to FAM with Ka (CX4/FAM) = 3.9 × 104 M−1.42
Cell viability and toxicity assays
To evaluate the toxicity of various samples including the peptide, curcumin, the peptide/curcumin, the peptide/CX4, the (peptide/curcumin)/CX4 and CX4 on the cells, MTT viability test was conducted (Fig. 7). The cells were treated with different concentrations of the aforementioned samples for 18 hours. The results indicated that at its highest concentration, FAM-Y4R4 caused a maximum decrease in MCF-7 cell viability of 20%. However, when combined with CX4 (FAM-Y4R4/CX4), the cell viability decreased by 40%. This finding suggests that CX4 drives more peptides into the cells. In the mixture of FAM-Y4R4 and CX4, the concentration of CX4 was fixed at 4 mM, which was significantly higher than the concentration of the peptide in the micromolar range. This ensured an excess of CX4 molecules (by 80 times) to ensure accessibility to every arginine's side chain. CX4, even up to 200 μM, did not significantly reduce the toxicity of MCF7 and A549 cells beyond 22% (results not shown in the graph). In vivo studies have demonstrated that CX4 exhibits no hemolytic toxicity at concentrations up to 5 mM and does not induce non-specific immunological reactions.43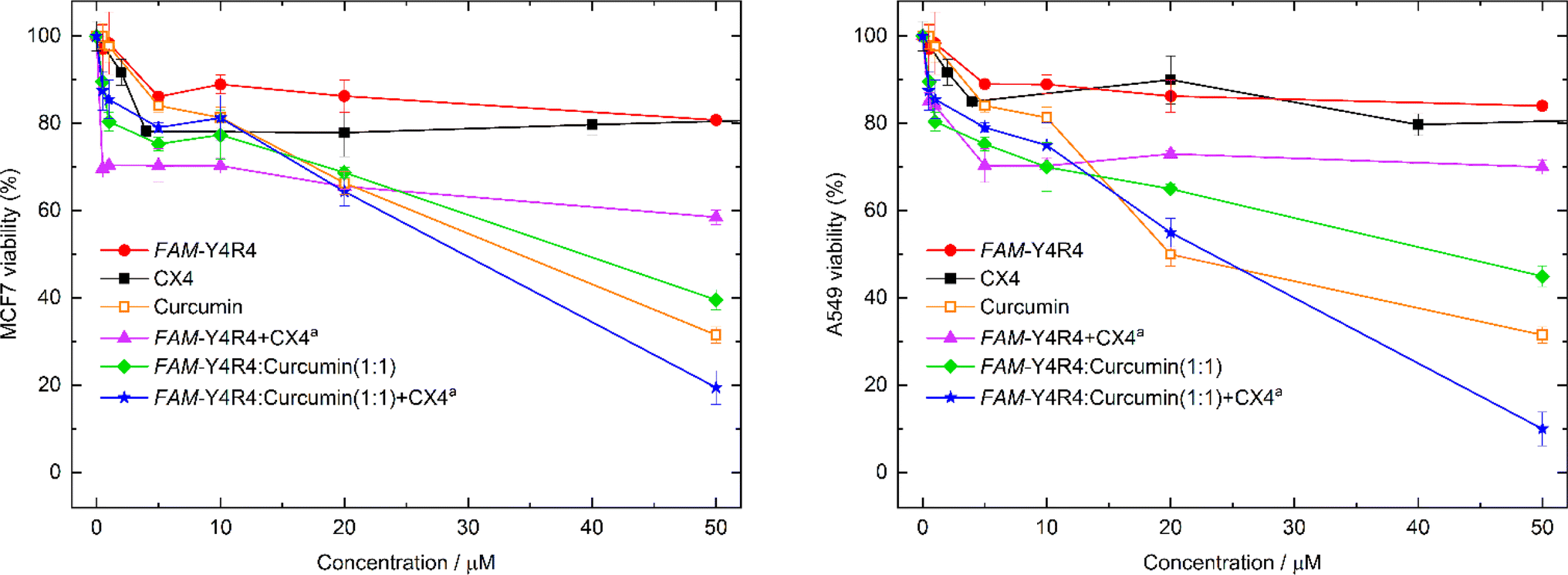 | ||
| Fig. 7 Viability of MCF-7 cells (left) and A549 cells (right) after incubation with various concentrations of FAM-Y4R4, curcumin, CX4, FAM-Y4R4/curcumin, FAM-Y4R4/CX4, or (FAM-Y4R4/curcumin)/CX4a. The concentration of CX4 was maintained at a maximum of 4 mM in FAM-Y4R4/CX4 and (FAM-Y4R4/curcumin)/CX4 complexes. | ||
Curcumin exhibits cytotoxicity in a manner that is dependent on the dosage administered. Notably, the FAM-Y4R4/curcumin and the (FAM-Y4R4/curcumin)/CX4 complexes demonstrated a more pronounced dose-dependent suppression of cell growth compared to curcumin alone. To validate these findings, we conducted the toxicity experiment on A549 cells. The results obtained for FAM-Y4R4 and CX4 were consistent with those observed in the MCF7 cells. However, the peptide/CX4 complex exhibited higher toxicity in MCF7 cells. Conversely, curcumin and peptide/curcumin showed equal toxicity in both cell lines, while the (FAM-Y4R4/curcumin)/CX4 complex displayed greater toxicity in A549 cells compared to MCF7 cells. This discrepancy can be attributed to the increased uptake of the peptide/CX4 complex by A549 cells discussed earlier (Fig. 4), resulting in a higher uptake of the toxic cargo (curcumin) when complexed with the cell-penetrating peptide (CPP).
Various carriers for curcumin, including nanoparticles, hydrogels, nanostructures, liposomes, polymeric micelles, quantum dots, and polymeric blend films have been documented.44 In a recent study, a cyclic peptide was synthesized and utilized as a carrier for curcumin and doxorubicin. Both covalent and supramolecular modifications were employed to bind curcumin with the peptide. The covalent modification of curcumin resulted in reduced toxicity towards LLCPK, SKOV-3, and CCRF-CEM cell lines. On the other hand, in consistency to our toxicity results; the supramolecular binding of curcumin/doxorubicin to the peptide exhibited enhanced efficacy in killing cancer cells.45
Ratrey et al. used an octaarginine CPP (R8) to bind to curcumin via cation–π interactions. The resulting complex of R8/curcumin exhibited enhanced antibacterial and anticancer activity compared to curcumin alone.41 However, directly engaging the cargo with the sidechain of R8 may hinder its direct penetration across the cell membrane.7 In contrast, our peptide utilizes Y4, which exhibits a stronger affinity for curcumin or any other PWSD (peptide-wrapped small drug), enabling the R4 portion to bind to CX4 and fulfill its role in cellular penetration.
Conclusion
FAM-Y4R4/CX4 complex represents a novel amphipathic CPP with promising potential. This complex consists of a binding component (Y4) that hosts PWSDs, and a cell-penetrating component (R4/CX4) that facilitates the entry of the peptide into live cells. The shorter length of R4 in this peptide offers an advantage, as it allows for more efficient synthesis compared to conventional oligoarginine-based CPPs, which typically contain 7 to 9 arginine residues. Additionally, CPP aggregation often leads to reduced effectiveness, but the presence of CX4 in our complex prevents the side chains of R4 from forming cation–π interaction with the Y4 sidechain that avoids aggregation and enhances cell uptake. Importantly, our CPP complex significantly improves the solubility of curcumin and effectively delivers it into live cells, thereby enhancing curcumin's anticancer properties.Data availability
All data generated and/or analyzed during this study are included in this published article or its ESI.†Author contributions
ZA conducted the toxicity assays on A549 cells. NS performed the molecular dynamics simulations. MH carried out the remaining experiments. RZ, WN, and KK provided scientific guidance as advisors. AN supervised the project and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Iran National Science Foundation for financing this work with Grant No. 96009524. We also thank the National Institute of Genetic Engineering and Biotechnology (NIGEB) for its support.References
- A. Komin, L. Russell, K. Hristova and P. Searson, Adv. Drug Delivery Rev., 2017, 110, 52–64 CrossRef PubMed.
- A. F. Schneider, M. Kithil, M. C. Cardoso, M. Lehmann and C. P. Hackenberger, Nat. Chem., 2021, 13, 530–539 CrossRef PubMed.
- J. D. Ramsey and N. H. Flynn, Pharmacol. Ther., 2015, 154, 78–86 CrossRef PubMed.
- J. Xie, Y. Bi, H. Zhang, S. Dong, L. Teng, R. J. Lee and Z. Yang, Front. Pharmacol, 2020, 11, 697 CrossRef PubMed.
- J. B. Rothbard, E. Kreider, C. L. VanDeusen, L. Wright, B. L. Wylie and P. A. Wender, J. Med. Chem., 2002, 45, 3612–3618 CrossRef PubMed.
- C. Huang, Y. C. Liu, H. Oh, D. S. Guo, W. M. Nau and A. Hennig, Chem.–Eur. J., 2024, 30, e202400174 CrossRef PubMed.
- H. D. Herce, A. E. Garcia and M. C. Cardoso, J. Am. Chem. Soc., 2014, 136, 17459–17467 CrossRef.
- Z. Vaezi, A. Bortolotti, V. Luca, G. Perilli, M. L. Mangoni, R. Khosravi-Far, S. Bobone and L. Stella, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183107 CrossRef PubMed.
- W. P. R. Verdurmen, R. Wallbrecher, S. Schmidt, J. Eilander, P. Bovee-Geurts, S. Fanghänel, J. Bürck, P. Wadhwani, A. S. Ulrich and R. Brock, J. Controlled Release, 2013, 170, 83–91 Search PubMed.
- L. Vasconcelos, T. Lehto, F. Madani, V. Radoi, M. Hällbrink, V. Vukojević and Ü. Langel, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 491–504 CrossRef PubMed.
- L. Baldini, A. Casnati, F. Sansone and R. Ungaro, Chem. Soc. Rev., 2007, 36, 254–266 RSC.
- A. D'Urso, G. Brancatelli, N. Hickey, E. Farnetti, R. De Zorzi, C. Bonaccorso, R. Purrello and S. Geremia, Supramol. Chem., 2016, 28, 499–505 CrossRef.
- M. Selkti, A. W. Coleman, I. Nicolis, N. Douteau-Guével, F. Villain, A. Tomas and C. de Rango, Chem. Commun., 2000, 161–162 Search PubMed.
- A. Lazar, E. Da Silva, A. Navaza, C. Barbey and A. W. Coleman, Chem. Commun., 2004, 2162–2163 Search PubMed.
- M. R. Islam, A. Rauf, S. Akash, S. I. Trisha, A. H. Nasim, M. Akter, P. S. Dhar, H. A. Ogaly, H. A. Hemeg, P. Wilairatana and M. Thiruvengadam, Biomed. Pharmacother., 2024, 170, 116034 Search PubMed.
- M. Shahabi, R. Hajihosseini, W. M. Nau, K. A. Noghabi and A. Norouzy, Int. J. Pept. Res. Ther., 2020, 26, 2633–2640 Search PubMed.
- A. Norouzy, A. I. Lazar, M. H. Karimi-Jafari, R. Firouzi and W. M. Nau, Amino Acids, 2022, 1–11 Search PubMed.
- P. Fattahi, N. Salehi, J. Mohammadi, A. Norouzy and S. M. Moazzeni, J. Pept. Sci., 2023, 29, e3480 CrossRef PubMed.
- A. Norouzy, Z. Azizi and W. M. Nau, Angew. Chem., Int. Ed., 2015, 54, 792–795 CrossRef PubMed.
- E. Vivès, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef PubMed.
- J. Park, J. Ryu, K.-A. Kim, H. J. Lee, J. H. Bahn, K. Han, E. Y. Choi, K. S. Lee, H. Y. Kwon and S. Y. Choi, J. Gen. Virol., 2002, 83, 1173–1181 CrossRef PubMed.
- G. Tünnemann, G. Ter-Avetisyan, R. M. Martin, M. Stöckl, A. Herrmann and M. C. Cardoso, J. Pept. Sci., 2008, 14, 469–476 CrossRef PubMed.
- A. Rodger, in Encyclopedia of Biophysics, ed. G. Roberts and A. Watts, Springer Berlin Heidelberg, Berlin, Heidelberg, 2018, pp. 1–6, DOI:10.1007/978-3-642-35943-9_634-1.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef PubMed.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef PubMed.
- A. Jain and R. Sankararamakrishnan, J. Chem. Inf. Model., 2011, 51, 3208–3216 CrossRef.
- K. L. Zapadka, F. J. Becher, A. Gomes dos Santos and S. E. Jackson, Interface Focus, 2017, 7, 20170030 CrossRef PubMed.
- A. Norouzy, A. I. Lazar, M. H. Karimi-Jafari, R. Firouzi and W. M. Nau, Amino Acids, 2022, 54, 277–287 CrossRef PubMed.
- M. Meli, G. Morra and G. Colombo, Biophys. J., 2008, 94, 4414–4426 CrossRef.
- N. Queralt-Rosinach and J. Mestres, Eur. Biophys. J., 2010, 39, 1471–1475 CrossRef PubMed.
- M. L. Waters, Biopolymers, 2004, 76, 435–445 CrossRef.
- S. A. Vuai, M. G. Sahini, I. Onoka, L. W. Kiruri and D. M. Shadrack, RSC Adv., 2021, 11, 33136–33147 RSC.
- A. Norouzy, M. Habibi-Rezaei, D. Qujeq, M. Vatani and A. Badiei, Bull. Korean Chem. Soc., 2010, 31, 157 CrossRef.
- A. Norouzy, D. Qujeq and M. Habibi-Rezaei, React. Kinet. Catal. Lett., 2009, 98, 391–401 CrossRef.
- S.-j. Choi, W.-j. Jeong, S.-K. Kang, M. Lee, E. Kim, D. Y. Ryu and Y.-b. Lim, Biomacromolecules, 2012, 13, 1991–1995 CrossRef PubMed.
- E. C. Repotente Jr, A. J. Carreon, M. K. Devanadera, M. S. Esmalla and M. Santiago-Bautista, Front. Mater., 2022, 9, 933749 CrossRef.
- R. Lalor, H. Baillie-Johnson, C. Redshaw, S. E. Matthews and A. Mueller, J. Am. Chem. Soc., 2008, 130, 2892–2893 CrossRef PubMed.
- G. Guidotti, L. Brambilla and D. Rossi, Trends Pharmacol. Sci., 2017, 38, 406–424 CrossRef.
- C. Allolio, A. Magarkar, P. Jurkiewicz, K. Baxová, M. Javanainen, P. E. Mason, R. Šachl, M. Cebecauer, M. Hof and D. Horinek, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11923–11928 CrossRef.
- J. Regberg, A. Srimanee and Ü. Langel, Pharmaceuticals, 2012, 5, 991–1007 CrossRef PubMed.
- P. Ratrey, S. V. Dalvi and A. Mishra, ACS Omega, 2020, 5, 19004–19013 CrossRef.
- S. Gawhale, Y. Thakare, D. Malkhede and G. J. O. Chaudhari, Opt. Photonics J., 2014, 4, 237 CrossRef.
- D.-S. Guo and Y. Liu, Acc. Chem. Res., 2014, 47, 1925–1934 CrossRef PubMed.
- S. Sharifi, N. Fathi, M. Y. Memar, S. M. Hosseiniyan Khatibi, R. Khalilov, R. Negahdari, S. Zununi Vahed and S. Maleki Dizaj, Phytother. Res., 2020, 34, 1926–1946 CrossRef PubMed.
- S. Darwish, S. Mozaffari, K. Parang and R. Tiwari, Tetrahedron Lett., 2017, 58, 4617–4622 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06124a |
| This journal is © The Royal Society of Chemistry 2024 |