
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and in vitro anti-proliferative evaluation of new pyridine-2,3-dihydrothiazole/thiazolidin-4-one hybrids as dual CDK2/GSK3β kinase inhibitors†
Asmaa F. Kassema,
Ashraf A. Sediekb,
Mervat M. Omranc,
Doaa S. Fodad and
Aisha A. K. Al-Ashmawy *d
*d
aChemistry of Natural and Microbial Products Department, National Research Centre, Dokki, 12622, Cairo, Egypt
bChemical Industries Institute, National Research Centre, Dokki, 12622, Cairo, Egypt
cPharmacology Unit, Cancer Biology Department, National Cancer Institute, Cairo University, Cairo, Egypt
dTherapeutic Chemistry Department, Pharmaceutical and Drug Industries Research Institute, National Research Centre, Dokki, 12622, Cairo, Egypt. E-mail: aisha_pharmacy@yahoo.com
First published on 7th October 2024
Abstract
Herein, the molecular hybridization drug discovery approach was used in the design and synthesis of twelve novel pyridine-2,3-dihydrothiazole hybrids (2a,b–5a,b and 13a,b–14a,b) and fourteen pyridine-thiazolidin-4-one hybrids (6a,b–12a,b) as anti-proliferative analogues targeting CDK2 and GSK3β kinase inhibition. Almost all of the newly synthesized hybrids, including their precursors (1a,b), were evaluated for their anti-proliferative activity against three human cancer cell lines—MCF-7, HepG2 and HEp-2—as well as normal Vero cell lines. Both compounds 1a (pyridine-thiourea precursor) and 8a (pyridine-5-acetyl-thiazolidin-4-one hybrid) exhibited excellent anti-proliferative activity against HEp-2 (IC50 = 7.5 μg mL−1, 5.9 μg mL−1, respectively). Additionally, 13a (pyridine-5-(p-tolyldiazenyl-2,3-dihydrothiazole)) hybrid demonstrated excellent anti-proliferative activity against HepG2 (IC50 = 9.5 μg mL−1), with an acceptable safety profile against Vero (<45% inhibition at 100 μg mL−1) in the cases of 8a and 13a alone. The three promising anti-proliferative hybrids (1a, 8a, 13a) were selected for the assessment of their in vitro inhibitory kinase activity against CDK2/GSK3β using roscovitine (IC50 = 0.88 μg mL−1) and CHIR-99021 (IC50 = 0.07 μg mL−1) as references, respectively. Compound 13a was the most potent dual CDK2/GSK3β inhibitor (IC50 = 0.396 μg mL−1, 0.118 μg mL−1, respectively) followed by 8a (IC50 = 0.675 μg mL−1, 0.134 μg mL−1, respectively), and the weakest was 1a. To elucidate the mechanism of the most potent anti-proliferative 13a hybrid, further cell cycle analysis was performed revealing that it caused G1 cell cycle arrest and induced apoptosis. Moreover, it resulted in an increase in Bax and caspase-3 with a decrease in Bcl-2 levels in HepG2 cells compared with untreated cells. Finally, in silico drug likeness/ADME prediction for the three potent compounds as well as a molecular docking simulation study were conducted in order to explore the binding affinity and interactions in the binding site of each enzyme, which inspired their usage as anti-proliferative leads for further modification.
1 Introduction
Cancer is considered as the second largest cause of death and has been redefined as a broad category of various diseases.1 According to a recent report of the International Agency for Research on Cancer (IARC), there were 10 million deaths and 19.5 million new cancer cases in 2020, and within 4 years, cancer is projected to become the first cause of death.2 In conventional cancer treatments, including chemotherapy, radiotherapy and surgery, normal cells are observed to be badly affected, which is a major concern. Additionally, the development of multi-drug resistance to common chemotherapy presents a major challenge. All of these challenges prompted healthcare providers to use targeted therapies as a safer choice.3 Protein kinase inhibitors (PKIs) provide the most attractive targeted oncotherapy in clinical use and the development of new cancer therapeutics, as they target the tumor cell microenvironment and signalling cascades with a minimal negative effect on normal cells.4,5 Protein kinases, with 538 proteins, are classified into tyrosine, serine/threonine and dual-specificity kinases, as they catalyse the transfer of the terminal phosphate in ATP to the target protein.6 This regulates and propagates the growth cell signal followed by differentiation, proliferation and apoptosis.6 Any disturbance in PK regulation will result in many diseases such as autoimmune, cardiovascular, and neurological diseases as well as cancers. Consequently, the development of PKIs may serve as a treatment method for related diseases.7The complicated nature of cancer implies the use of multi-targeted kinase inhibitors, where a single compound targets more than one kinase that were previously reported to have cross-linked pathways (this is due to a certain percentage of the ATP binding sites being conserved), in an attempt to obtain a synergistic effect, with fewer side effects and to overcome drug resistance.4,8,9
An example of the serine/threonine kinase family is the cyclin-dependent kinases (CDKs), which are responsible for cell cycle progression and control cellular division, proliferation, gene transcription, protein synthesis and finally cell death.10 One of the CDK isoforms is CDK2, whose activation requires its binding with one of its regulatory subunits cyclin A or E to be able to share in the essential cellular processes.10 Furthermore, various types of cancers, such as hepatic, pancreatic, ovarian, melanoma, cervical and breast cancers, are linked to dysregulation of CDK2 or its cyclins.11 Based on the promising outcomes of marketed CDK2 inhibitors on tumour cells, CDK2 can be considered a valuable target in the drug discovery of cancer-targeting therapeutics.12–14
Another example of the constitutive serine/threonine kinase family is glycogen synthase kinase (GSK3), a multi-functioning kinase, which controls glycogen metabolism as well as involves in many cellular processes, including the cell cycle, gene transcription, proliferation, metabolism and cell death (including apoptosis and autophagy). It presents in two isoforms: GSK3α (alpha) and GSK3β (beta).15 Particularly, the dysregulation in GSK3β activity was reported in neurological, metabolic and neoplastic diseases.16 A literature survey revealed that GSK3β is a valuable therapeutic target in more than twenty types of malignancies.17 Moreover, there is growing evidence that emphasizes the protective role of GSK3β inhibitors on normal cells from the adverse side effects of conventional tumour therapies and their important role in managing radioresistance and chemoresistance.18 Therefore, the design of novel GSK3β inhibitors is considered a valuable approach in tumour-targeting therapies.19
The molecular hybridization drug design approach is widely used in medicinal chemistry research in which two or more therapeutically active pharmacophores are combined to obtain a novel single compound with an expected greater therapeutic effect.20
Pyridine, as a core structure, has been found in a wide range of pharmacologically active synthetic or natural compounds, which has anti-proliferative,21 antimicrobial, and anti-hepatitis B virus22 actions and is a promising candidate in the management of cardiovascular disorders.23 When pyridine carbonitrile was hybridized with methyl benzenesulfonamide in compound A (Fig. 1), it afforded a CDK2 inhibitor with IC50 = 1.79 μM and with anti-proliferative activity against MCF-7 with IC50 = 18.3 μM.24 In compound B, pyridine was hybridized with thiazolidin-4-one-triazole-glycoside through an ethylidenehydrazono spacer to afford a CDK2 inhibitor with IC50 = 0.18 μM and good anti-proliferative activity against HepG2 and MCF-7 with IC50 = 2.09 and 0.15 μM, respectively.25 Moreover, there are a lot of promising pyridines incorporating GSK3β inhibitors in different clinical phases, such as CHIR99021 (ref. 26) (pan GSK3), AZD1080,27 and AZD2858,28 in addition to the newly reported potent bi-pyridine single-digit GSK3β inhibitor in compound C with IC50 = 3.4 nM.29
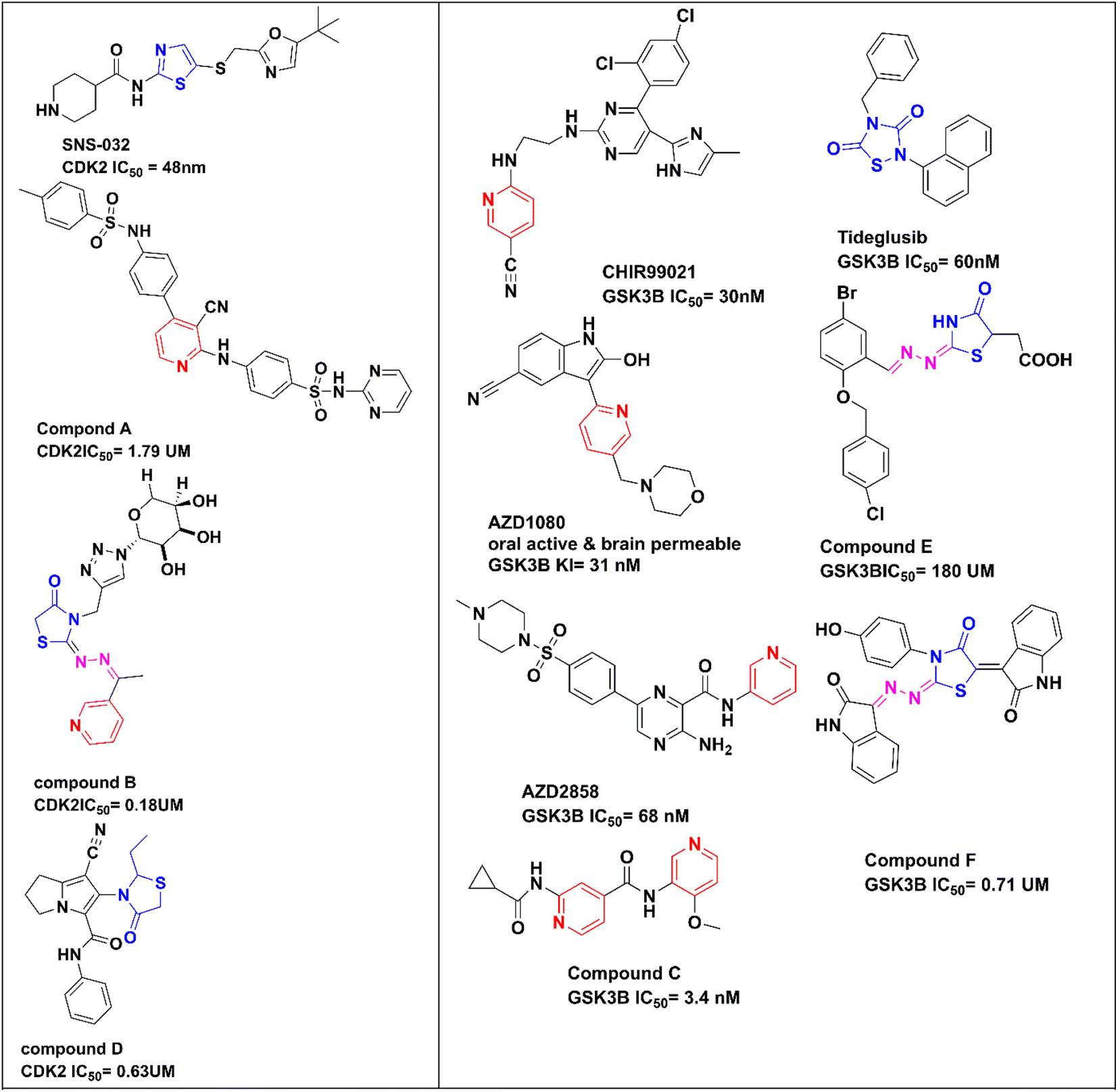 | ||
| Fig. 1 Promising CDK2 inhibitors and GSK3β inhibitors. | ||
Compounds containing thiazole, dihydrothiazol or thiazolidinone core have a wide range of therapeutic activities, such as anticancer,30 antibacterial,31 antifungal,32 anticonvulsant33 and anti-inflammatory34 actions. SNS-032, with thiazole as a central core structure, is a potent and selective CDK2 inhibitor with IC50 = 48 nM.35 The 4-thiazolidinone compound D showed CDK2 inhibitory activity with IC50 = 0.63 μM and induced apoptosis in the MCF-7 cell line.36 A potent selective GSK3β inhibitor incorporating a thiazolidinedione core structure is tideglusib with IC50 = 60 nM.37 New compounds E and F with a thiazolidinedione core structure and hydrazono linker were reported as GSK3β inhibitors with IC50 = 180 and 0.71 μM, respectively29 (Fig. 1).
Based upon the aforementioned findings, we designed a series of twenty-six novel pyridine-2,3-dihydrothiazole and pyridine-thiazolidin-4-one hybrids through an ethylidenehydrazono spacer, as presented in Fig. 2. The newly synthesized derivatives were evaluated for their anti-proliferative activities against hepatocellular, breast and laryngeal carcinoma cell lines, which were previously reported to overexpress CDK2 (ref. 38–40) and GSK3β41–43 as well as were evaluated against the normal African green monkey kidney cell line in order to detect their safety limits. The promising compounds were investigated for their in vitro inhibition of CDK2/cyclin A and GSK3β, as well as their effect on the cell cycle, apoptosis, caspase-3, Bax and Bcl-2 proteins. An in silico study of the promising compounds was performed, including ADME and molecular docking in the ATP-binding pocket of CDK2 and GSK3β, to predict the binding affinity and binding interactions with the amino acid residues in the active sites.
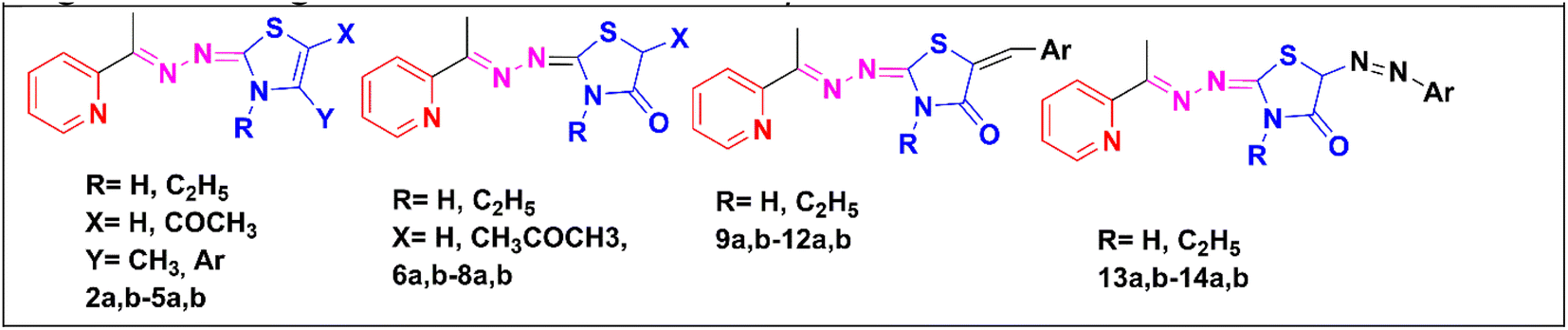 | ||
| Fig. 2 The newly designed pyridine-2,3-dihydrothiazole and pyridine-thiazolidin-4-one hybrids using a molecular hybridization approach. | ||
2 Results and discussion
2.1. Chemistry
The current work synthesized two kinds of substituted 1,3-thiazoles, 1 and 2, connected to the pyridine moiety (Scheme 1).
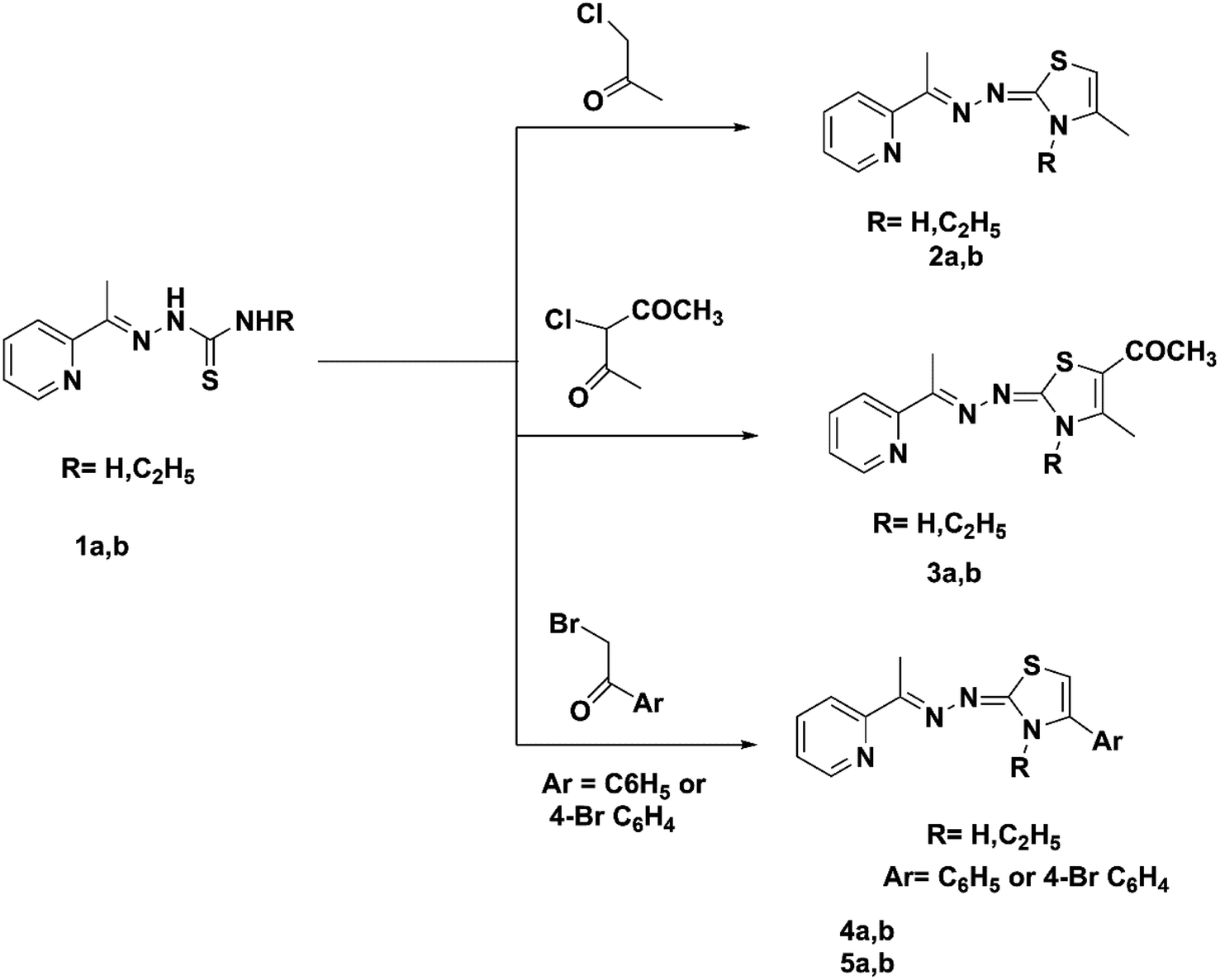 | ||
| Scheme 1 Synthesis of pyridin-2-yl-2,3-dihydrothiazole hybrids (2a,b–5a,b). | ||
A facile synthesis of the key (E)-2-(1-(pyridin-2-yl)ethylidene)hydrazine-1-carbothioamide 1a was prepared via the reaction of 2 acetyl pyridine with thiosemicarbazide in the presence of conc. HCl following a previously described method.44 The alkyl thiosemicarbazide derivative 1b was synthesized with high yields through the reaction of 2 acetyl pyridine with ethyl thiosemicarbazide in the presence of a catalytic amount of HCl following a previously described method.45 The Hantzsch reaction of thiosemicarbazones derivatives 1a,b with chloroacetone, chloroacetyl acetone, phenacyl bromide, or 4-bromo phenacyl bromide in absolute ethanol and fused sodium acetate under reflux led to the new pyridine-2,3-dihydrothiazole derivatives 2a,b–5a,b, respectively (Scheme 1). Compounds 4a and 5a were prepared using a previously described method.46,47 1H NMR spectra of compounds 2b, 3b, 4b, and 5b contained no signals of the NH groups recorded in the ethyl-thiosemicarbazide derivatives of their precursors. Compounds 2a, 2b, 4b and 5b demonstrated the singlet attributed to the CH of the thiazole ring at 6.04–6.44 ppm. The IR spectra for the previous derivatives 3a and 3b showed the presence of a carbonyl group as evidence for the formation of the new derivatives at 1632 and 1625 cm−1. Moreover, the 13C NMR spectra for derivatives 3a and 3b showed characteristic signals for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 190.95 ppm.
O at 190.95 ppm.
The reaction of thiosemicarbazone derivatives 1a and 1b with ethyl bromoacetate, ethyl-2-bromopropionate or ethyl-2-chloroacetoacetate prepared pyridin-2-yl-thiazolidin-4-one derivatives 6a,b–8a,b, respectively (Scheme 2). Compound 6a was prepared using a previously described procedure.25 The IR spectrum of compound 6b exhibited bands at 1715 cm−1 (CO). The 1H NMR spectra of compounds 7a and 7b exhibited the quartet of CH of the thiazole ring at 4.13 and 4.22 ppm. The 1H NMR spectra of 8a and 8b exhibited the singlet signal of CH of the thiazole ring at 4.17 and 4.17 ppm. The 13C NMR spectra of hydrazonothiazolidin-4-one derivatives 6b–8a,b showed characteristic signals for CO at 170.64–176.23 ppm.
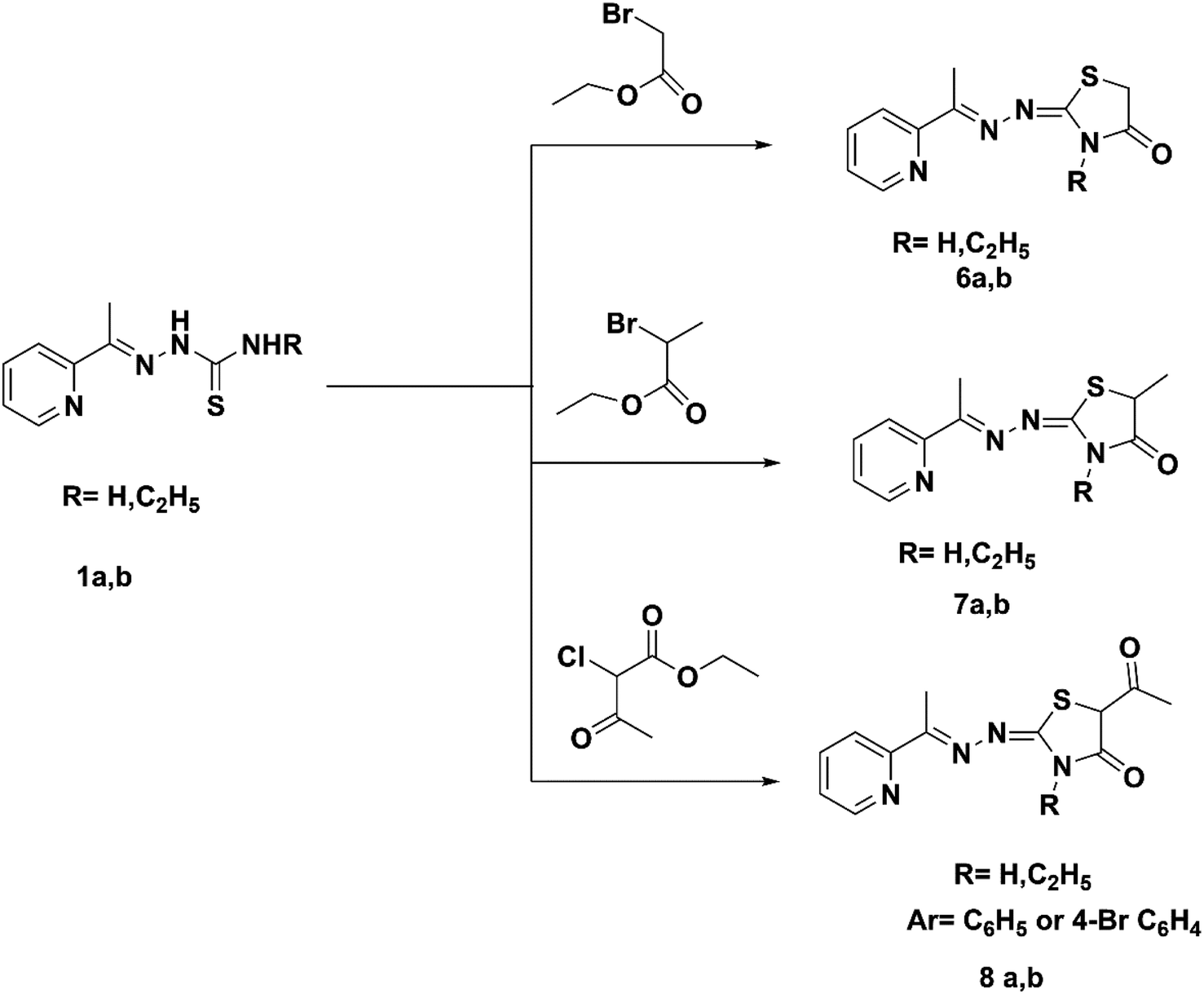 | ||
| Scheme 2 Synthesis of pyridin-2-yl-thiazolidin-4-one hybrids (6a,b–8a,b). | ||
Thiosemicarbazones 1a and 1b were allowed to react with ethyl bromoacetate and an equimolar amount of an appropriate aromatic aldehyde, 4-fluorobenzaldehyde, 4-chlorobenzaldehyde, 4-methoxybenzaldehyde, or 3,4-dimethoxybenzaldehyde, in the presence of a mixture of glacial acetic acid and anhydrous sodium acetate. The pyridin-2-yl-5-substituted benzylidene-thiazolidin-4-one derivatives 9a,b–12a,b were obtained in 70–80% yields. Another pathway for the formation of compounds 9a,b–12a,b was based on the reaction of compounds 6a and 6b with aromatic aldehydes in the presence of a mixture of glacial acetic acid and anhydrous sodium acetate (Scheme 3). The 13C NMR spectra of compounds 9a,b–12a,b revealed the presence of a characteristic CH benzylic peak, recorded from their precursor thiosemicarbazide derivatives 1a and 1b at 142.91–143.72 ppm.
 | ||
| Scheme 3 Synthesis of pyridin-2-yl-5-substituted benzylidene-thiazolidin-4-one hybrids (9a,b–12a,b) and synthesis of pyridin-2-yl-5-diazenyl-2,3 dihydrothiazole hybrids (13a,b–14a,b). | ||
Pyridin-2-yl-5-diazenyl-2,3-dihydrothiazole derivatives 13a,b–14a,b were formed by the reaction of compounds 1a and 1b and an appropriate hydrazonoyl chloride in absolute ethanol catalyzed by TEA (Scheme 3). Compound 14a was prepared by a procedure described earlier.48 Mass spectra of compounds 13a, 13b and 14b displayed molecular ion peaks at m/z 350, 378 and 398, respectively.
2.2. Biological evaluation
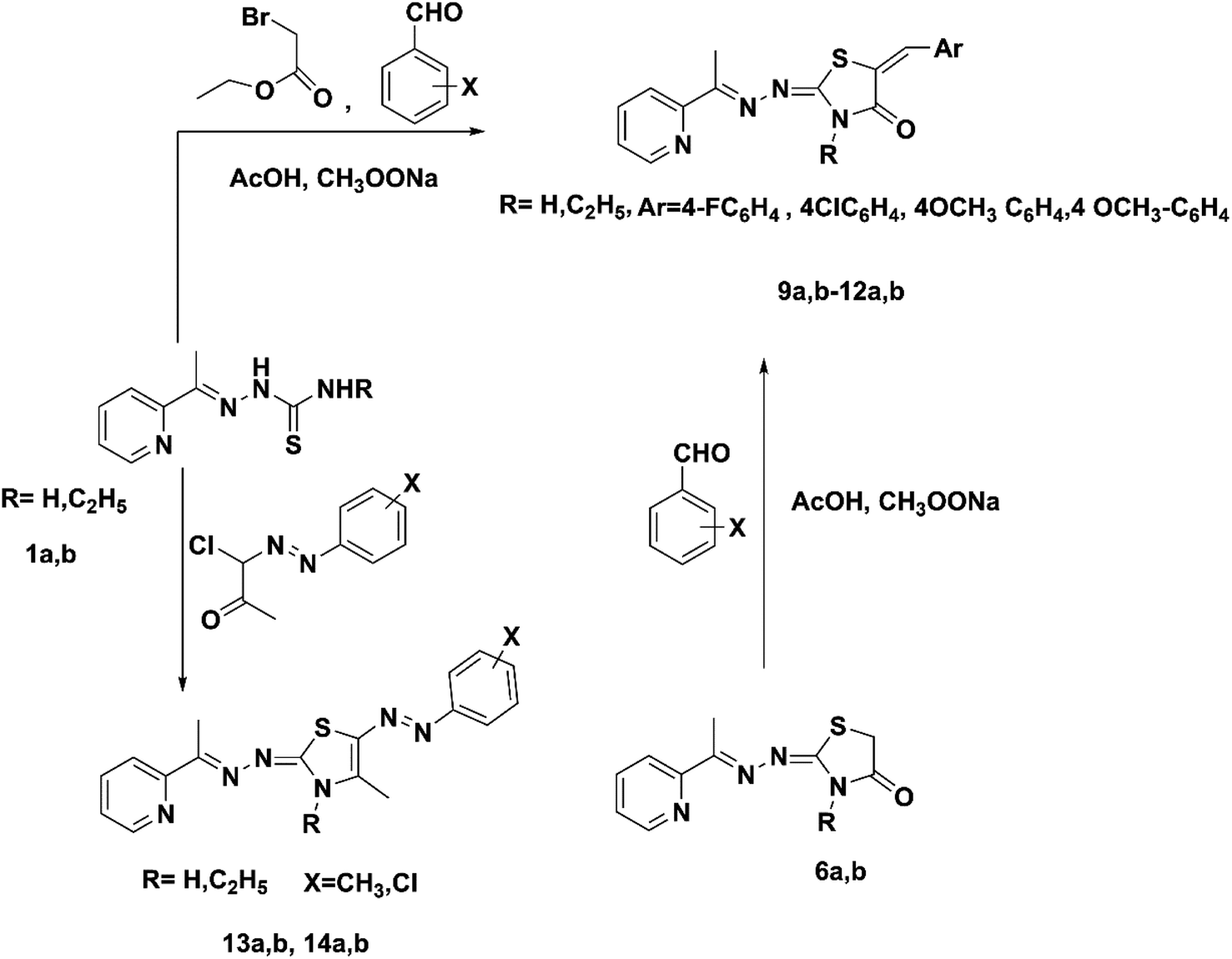
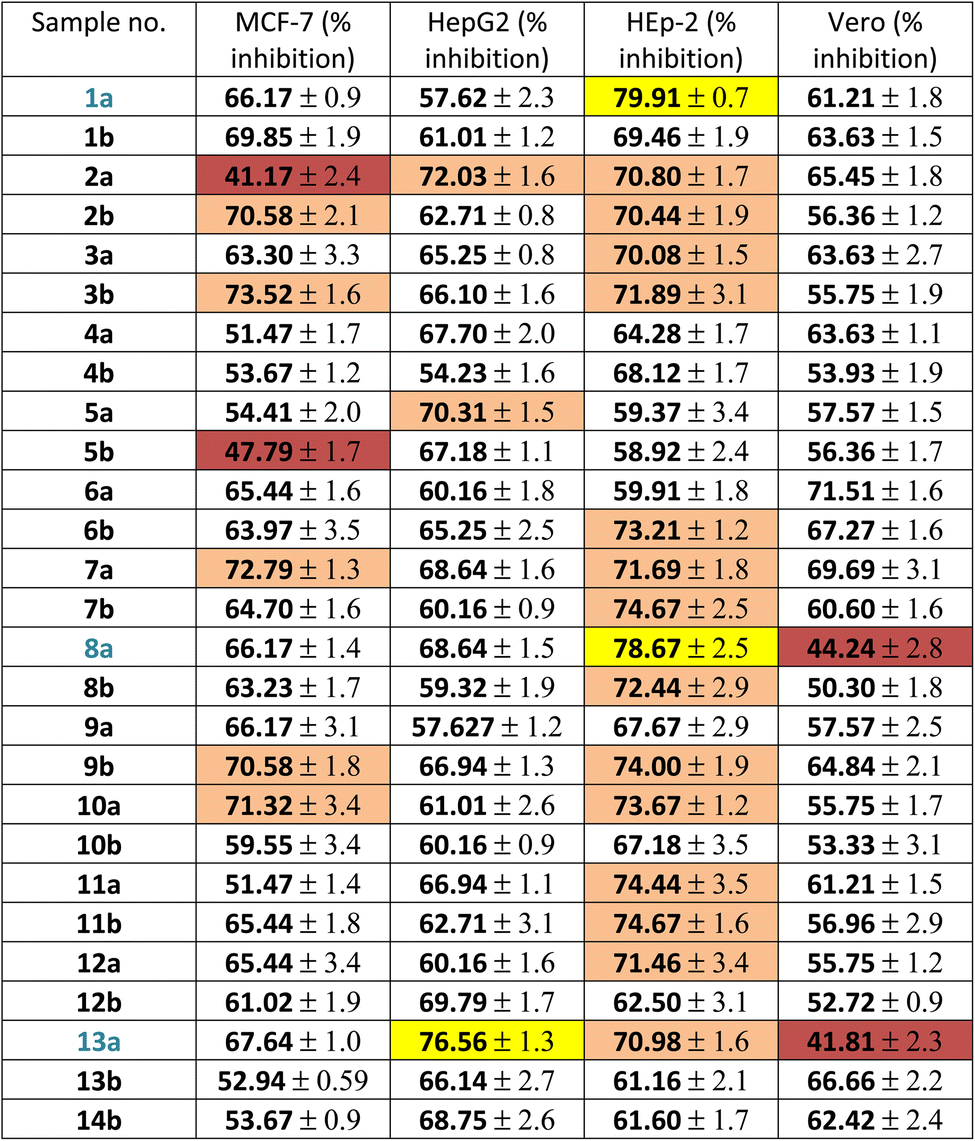
Firstly, the % inhibition at a single concentration (100 μg mL−1) of all the tested compounds was assessed. Only the promising compounds that exhibited % inhibition > 75% at 100 μg mL−1 in the cancer cell lines and % inhibition < 45% against normal Vero cells were evaluated for IC50 (the concentration that produces 50% inhibition of cell growth). The cells were treated at different concentrations (0, 5, 12.5, 25 and 50 μg mL−1) of compounds 1a, 8a and 13a together with doxorubicin as a reference, as shown in Table 1 and Fig. 3. For each compound, its IC50 was calculated using dose–response curve-fitting models (GraphPad Prism software, version 5).
| a Vero: normal African green monkey kidney cell line; HepG2: human liver carcinoma cell line; HEp-2: human laryngeal carcinoma cell line; MCF-7: human breast carcinoma cell line. Values are the means ± SD of three independent experiments performed in triplicate. Yellow: excellent effect (>75% inhibition), orange: good activity (>70% inhibition), not highlighted moderate effect (50–70% inhibition) and brown: weak activity. |
|---|
 |
 | ||
| Fig. 3 Cytotoxicity of the promising compounds: (A) 1a, 8a against the HEp-2 cell line; (B) 13a against the HepG2 cell line; (C) 1a, 8a, 13a against the Vero cell line after 48 h. The values represents the means ± SD of three independent experiments performed in triplicate. | ||
By inspecting the tabulated results, it was found that three compounds possessed excellent anti-proliferative activity (>75% inhibition): 1a and 8a against HEp-2 in addition to 13a against HepG2. Fortunately, compounds 8a (5-acetyl-thiazolidin-4-one derivative) and 13a (5-(p-tolyldiazenyl-2,3-dihydrothiazole derivative) showed a good safety profile against the normal Vero cells (<45% inhibition) with higher selectivity towards HEp-2 and HepG2 cancer cell lines, respectively, which was not the case in compound 1a, the pyridine-thiourea precursor (Fig. 3C). Moreover, compound 13a exhibited good anti-proliferative activity against HEp-2 (70.98% inhibition). However, the presence of an N-ethyl moiety in the 2,3-dihydrothiazole derivative 13b resulted in a decrease in anti-proliferative activity against HepG2 and HEp-2, along with higher toxicity against the normal Vero cell line. The two pyridine-2,3-dihydrothiazole derivatives 2a and 5b showed weak anti-proliferative activities against MCF-7 with 41.17% and 47.79%, respectively, in the whole series. However, 2a showed good anti-proliferative activity against HepG2 (72.03%) and HEp-2 (70.80%). The presence of N-ethyl-2,3-dihydrothiazole derivative 2b showed good % inhibition against MCF-7 (70.58%) and retained a good % inhibition against HEp-2 (70.44%) as 2a. In addition to compound 2b, in the whole series, compounds 3b (5-acetyl-2,3-dihydrothiazole derivative), 7a (5-methyl-thiazolidin-4-one derivative), 9b and 10a (5-substituted-benzylidene-thiazolidin-4-one derivatives) showed good anti-proliferative activity against MCF-7 (>70% inhibition). The rest of the compounds (excluding 2a and 5b) showed moderate anti-proliferative activity (50–70% inhibition). Regarding the HepG2 cell lines, all of the tested compounds showed moderate cytotoxic activity, except that 13a was excellent and 2a and 5a showed good activity. In the case of HEp-2, for the anti-proliferative activity of the tested compounds, two compounds 1a and 8a showed excellent inhibitory activity, and 14 compounds showed good inhibitory activity, namely 2a, 2b, 3a, 3b (2,3-dihydrothiazole derivatives) 6b, 7a, 7b, 8b (thiazolidin-4-one derivatives), 9b, 10a, 11a, 11b, 12a (5-substituted-benzylidene-thiazolidi-4-one), and 13a (5-(p-tolyldiazenyl-2,3-dihydrothiazole derivative). The rest of the compounds showed moderate cytotoxic activity.
As presented in the dose–response curves (Fig. 3A–C), compounds 8a and 1a showed anticancer activity against the HEp-2 cell line with IC50 of 5.9 and 7.5 μg mL−1, respectively. Conversely, compound 13a showed anticancer activity against HepG2 with IC50 = 9.5 μg mL−1.
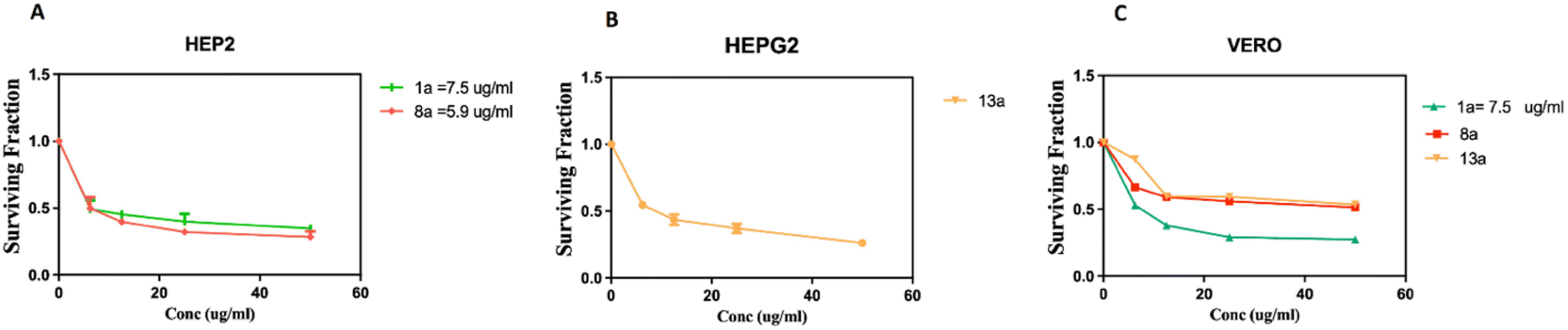
As depicted in Fig. 4A, compounds 8a and 13a significantly increased the NOx level compared to untreated cells by 19% and 25%, respectively. Moreover, there was a slight reduction in the GSH content by 14% in HEp-2 and a significant reduction in the GSH level in HepG2 cells by 33% compared to untreated cells (Fig. 4B). It was noticed that the production of reactive oxygen species (ROS) was elevated after treatment with 8a and 13a, resulting in the induction of nuclear damage, which might also lead to an increase in apoptotic protein expression. Therefore, it can be concluded that 8a and 13a possess strong chemo-preventive potential.
 | ||
| Fig. 4 Effect of treatment with 8a and 13a on oxidative stress and antioxidants in HEp-2 and HepG2 cell lines, respectively: NOx (A), GSH level (B). Data are expressed as means ± SD of two independent experiments. *Compounds are significantly different from the control group at a P value < 0.05. | ||
| Cpd | IC50 (mean ± SEM) (μg mL−1) | |
|---|---|---|
| CDK2/cyclin A | GSK3β | |
| a IC50: compound concentration necessary to inhibit the enzyme activity by 50%; SEM: standard error of the mean; each value is the mean of three values; (—) not detected. | ||
| 1a | 3.25 | 0.438 |
| 8a | 0.675 | 0.134 |
| 13a | 0.396 | 0.118 |
| Roscovitine | 0.88 | — |
| CHIR-99021 | — | 0.07 |
According to the results in Table 2, compounds 13a and 8a showed stronger inhibitory activity against CDK2/cyclin A by 2.2 fold and 1.3 fold compared with roscovitine. Unfortunately, 1a (the pyridine-thiourea) precursor exhibited the weakest activity with roscovitine demonstrating more than 3.5 times its inhibitory activity. In the case of GSK3β inhibitory activity, the three tested compounds showed moderate inhibitory activity, with 13a and 8a showing approximately twice the CHIR-99021 inhibitory activity. However, 1a showed very weak inhibitory activity with the reference drug showing nearly six times its inhibitory activity. Thus, it can be concluded that 13a exhibited anti-proliferative activity against HepG2 by acting as a dual CDK2 (strong)/GSK3β (moderate) inhibitor. Moreover, 8a showed anti-proliferative activity against HEp-2 by acting as a dual CDK2/GSK3β inhibitor but was slightly weaker than 13a. The pyridine-thiourea precursor 1a showed excellent anti-proliferative activity against HEp-2 but was the weakest dual CDK2/GSK3β inhibitor.
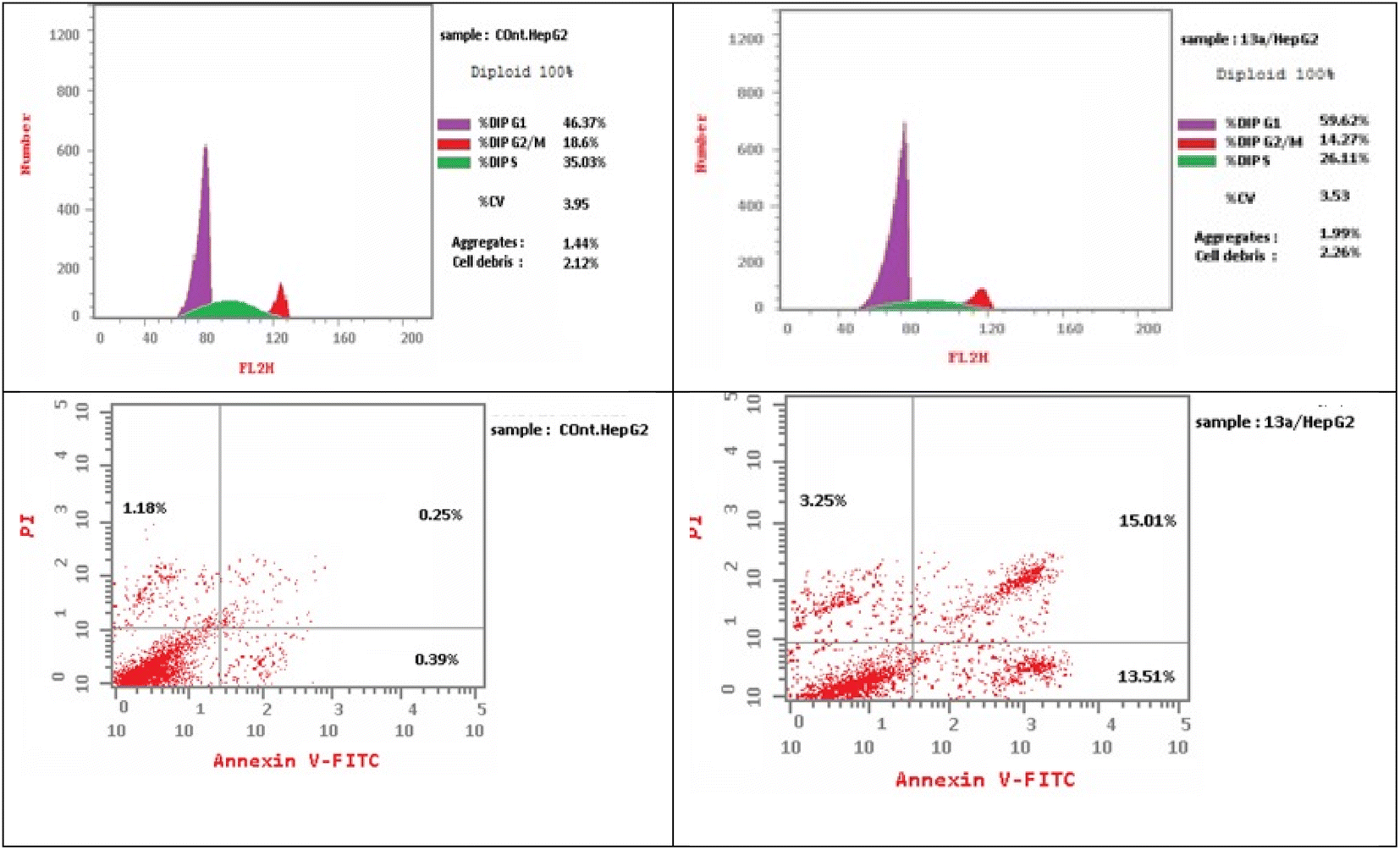 | ||
| Fig. 5 Cell cycle analysis and the effect of the pyridine-5-(p-tolyldiazenyl-2,3-dihydrothiazole) hybrid 13a on the percentage of V-FITC-positive annexin staining in HepG2 cells compared with the control. | ||
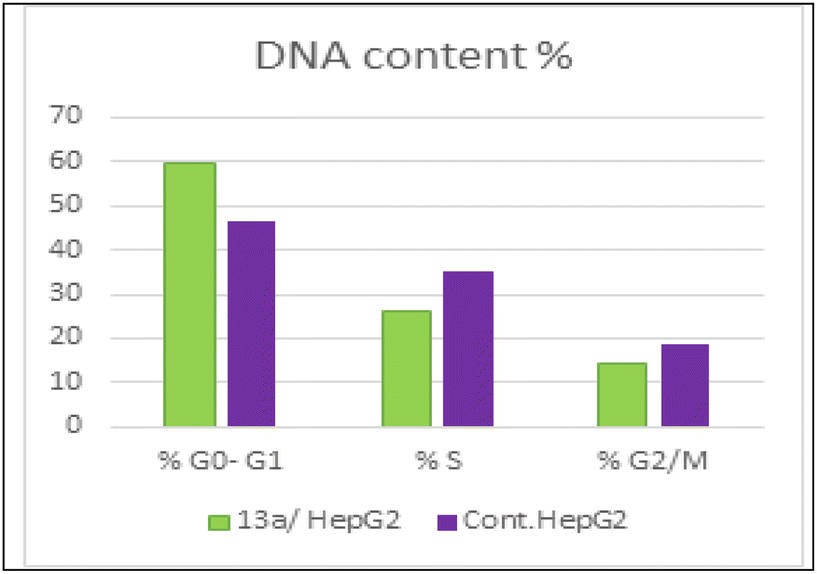 | ||
| Fig. 6 Cell cycle analysis of pyridine-5-(p-tolyldiazenyl-2,3-dihydrothiazole) hybrid compound 13a. | ||
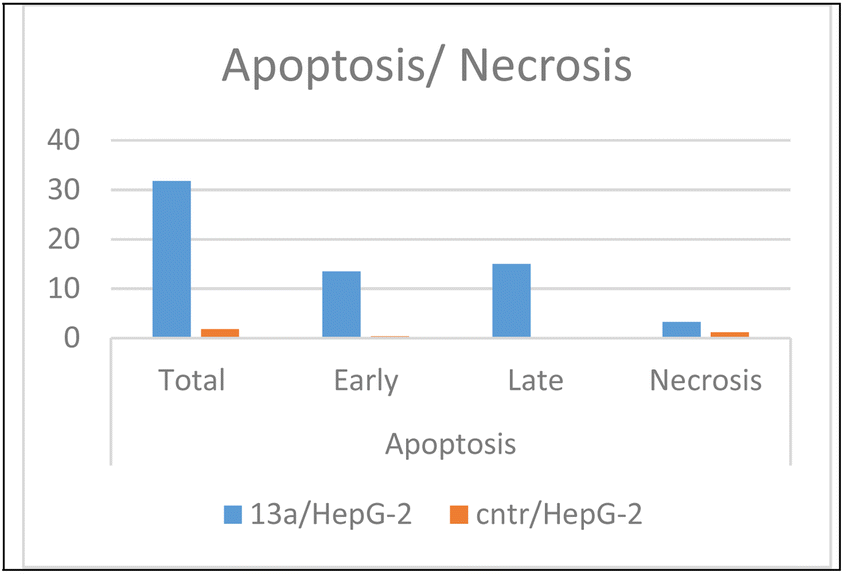 | ||
| Fig. 7 Apoptotic activity of pyridine-5-(p-tolyldiazenyl-2,3-dihydrothiazole) hybrid compound 13a. | ||
As presented in Fig. 6, compound 13a caused cell accumulation of 59.62% in the G0–G1 phase compared with untreated HepG2 cells. Therefore, compound 13a caused G1 cell cycle arrest. Moreover, there was an elevation in the total number of apoptotic cells by 31.77% compared with 1.82% for the DMSO control, as shown in detail in Fig. 6 (% of early and late apoptosis). Additionally, it is noticeable that the % of necrotic cells was 3.25% in 13a-treated HepG2 cells compared with 1.18% in the DMSO control. All of these results pointed to the ability of compound 13a to induce apoptosis.
In the present study, major apoptotic parameters were evaluated to trace the anti-proliferative (or apoptosis induction) effect of compound 13a on HepG2 cancer cells. The gene expression of the pro-apoptotic protein Bax58 and the anti-apoptotic protein Bcl-2 (ref. 59) in addition to the death protein caspase-3 (ref. 60) were evaluated using the RT-PCR technique.
As shown in Table 3, after the treatment of HepG2 cells with 9.5 μg mL−1 (IC50) for 24 h, there was a 4.33-fold increase in Bax protein and a decrease in Bcl-2 protein by a value equal to 0.3 fold in 13a/HepG2 treated cells compared to the control untreated HepG2 cancer cells. Additionally, an increase in the caspase-3 gene expression by a value equal to 4.9 fold was observed in 13a/HepG2 compared to its corresponding control. These results pointed to the impact of compound 13a in inducing apoptosis in the HepG2 cancer cell.
| Compound | Cells | RT-PCR fold change | ||
|---|---|---|---|---|
| Bax | Bcl-2 | Caspase 3 | ||
| 13a/HepG2 | HepG2 | 4.337 | 0.303 | 4.916 |
| Control HepG2 | HepG2 | 1 | 1 | 1 |
2.3. In silico study of promising compounds 1a, 8a and 13a
Based on the promising in vitro biochemical screening results, three compounds were selected for further in silico study comprising ADME prediction and a docking study in the ATP binding pocket of CDK2 and GS3Kβ.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logS) for compounds 1a, 8a and 13aa
logS) for compounds 1a, 8a and 13aa
| Compound | Lipinski rules | Violation | Water solubility | ||||||
|---|---|---|---|---|---|---|---|---|---|
| MW ≤ 500 | HB acceptor ≤ 10 | HB donor ≤ 5 | TPSA | MLOGP ≤ 4.15 | Rotatable bonds ≤ 9 | Yes or 0 | AlilogS |
Ali class | |
| a MW: molecular weight ≤ 500; logP: lipophilicity < 4.15; HBA: hydrogen bond acceptor ≤ 10; HBD: hydrogen bond donor ≤ 5; TPSA (topological polar surface area) 20–130 Å2. |
|||||||||
| 1a | 194.26 | 2 | 2 | 95.39 | −0.14 | 3 | 0 | −2.31 | Very soluble |
| 8a | 276.31 | 5 | 1 | 109.08 | −0.14 | 3 | 0 | −2.31 | Very soluble |
| 13a | 350.44 | 5 | 1 | 106.36 | 2.19 | 4 | 0 | −6.33 | Moderate |
Favourably, the three tested compounds were predicted to fulfil the Pfizer Lipinski rule of five without any violation.62 Additionally, the water solubility was predicted as having AlilogS scale values ranging from ≤10 insoluble to ≤2 soluble, where 1a and 8a were very soluble and 13a showed moderate solubility.63 According to Fig. 8, the boiled-egg plot64 showed that the three compounds were expected to be highly absorbed from the gastrointestinal tract and would not cross the blood–brain barrier, and they were not expected to be a substrate for the efflux transporter P-glycoprotein.
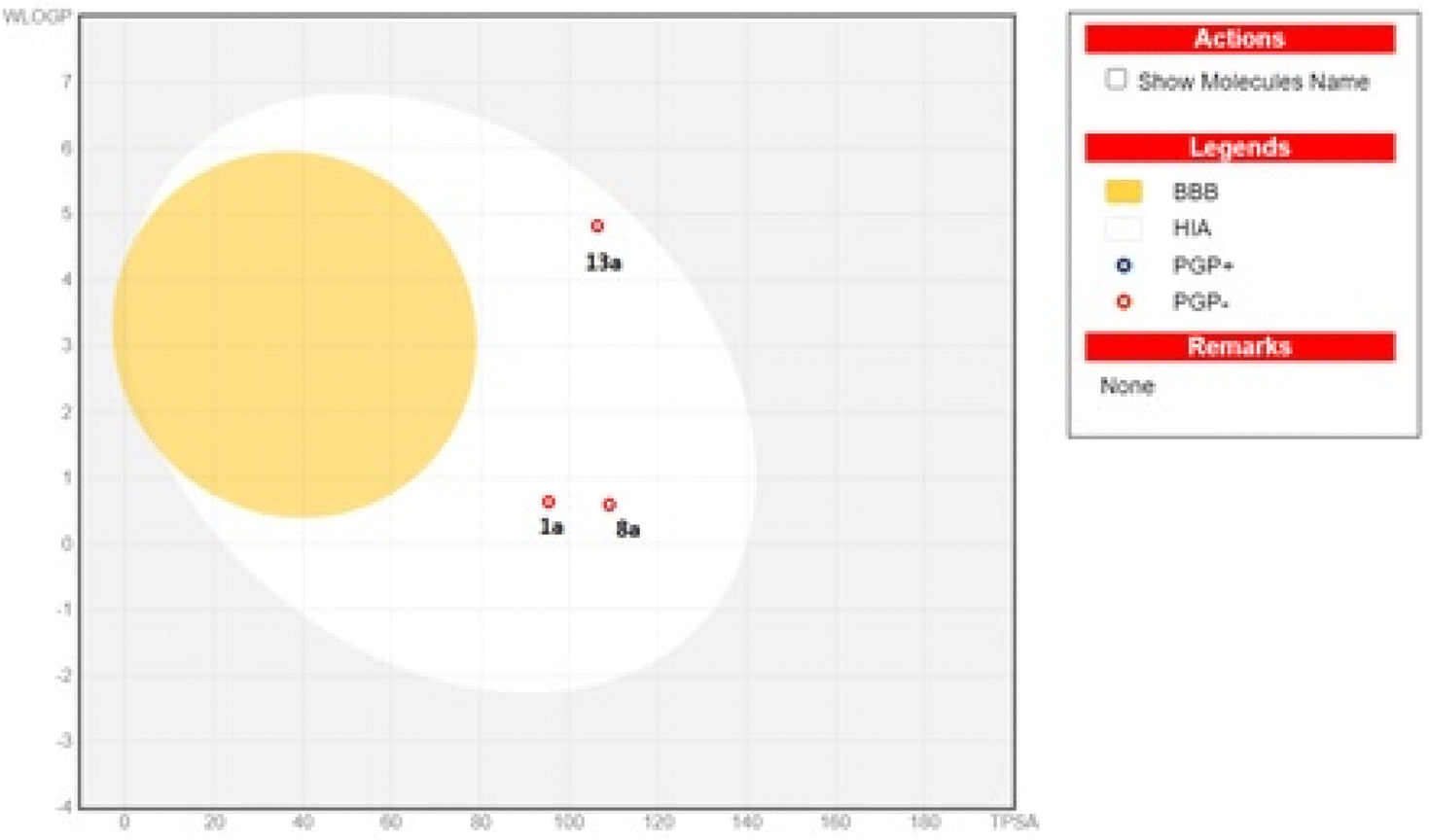 | ||
| Fig. 8 Boiled-egg plot from the SwissADME website for compounds 1a, 8a and 13a. | ||
| Compound | Binding affinity (kcal mol−1) CDK2 | Binding affinity (kcal mol−1) GSK3β |
|---|---|---|
| 1a | −6.4 | −5.5 |
| 8a | −8.2 | −7.8 |
| 13a | −8.6 | −8.0 |
| AZD5438 | −8.6 | — |
| 2WF | — | −8.6 |
In the CDK2 ATP binding site, it was predicted that 13a had the highest binding affinity (−8.6 kcal mol−1), which was similar to that of the native ligand AZD5438 (−8.6 kcal mol−1), followed by 8a (−8.2 kcal mol−1). Both were superior to 1a (−8.2 kcal mol−1), the pyridine thiourea precursor (Table 5). Regarding the binding interactions (Fig. 9A and B), compound 13a formed two hydrogen bonds with Leu83 (2.06 Å) and His84 (2.10 Å) considering that the interaction with Leu83 in the hinge region is essential, as previously reported. Additionally, there were several hydrophobic interactions, including pi–pi interaction with Phe80 (5.61 Å), pi–sigma interaction with Ile10 (3.5 Å), several pi–alkyl interactions and van der Waals interactions (Fig. 9A). In Fig. 9B, compound 13a was typically superimposed over AZD5438 with the pyridine ring extension in the binding site forming more hydrophobic interactions, which may increase the stability of the compound in the pocket. In Fig. 10, compound 8a formed two hydrogen bonds one with the essential Leu83 (2.08 Å) and the other with Lys20 (2.02 Å), in addition to hydrophobic alkyl and van der Waals interactions. Compound 1a formed three hydrogen bonds with Ile10 (2.74 Å), Asp86 (2.40 Å) and Gln131 (2.21 Å) with the NH of the thiourea moiety, highlighting the previously reported essential Asp86 interaction in the hinge region, but this interaction was not sufficient to potently inhibit CDK2 (Fig. 11).
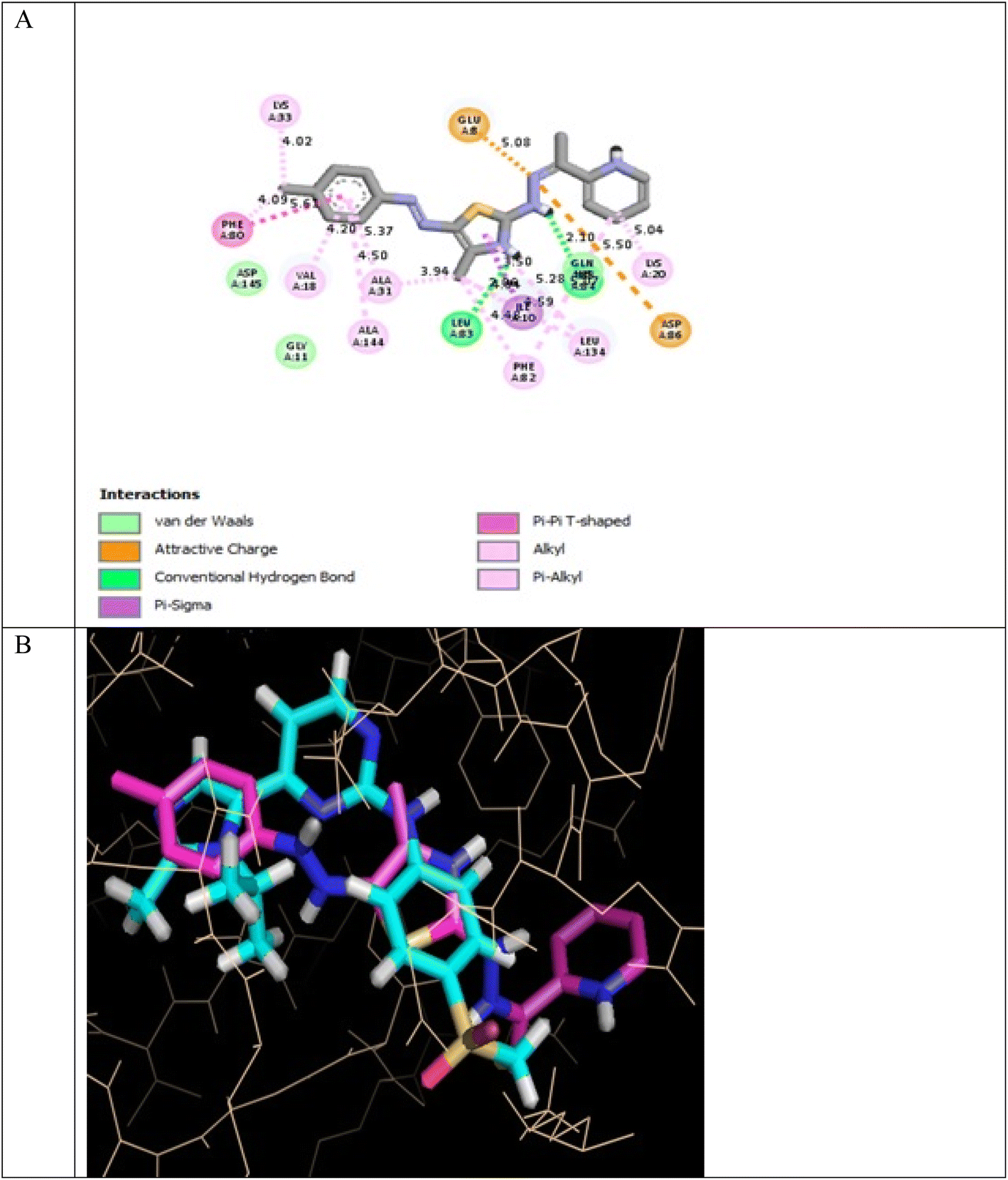 | ||
| Fig. 9 (A) 2D binding interactions of compound 13a in CDK2 (PDB ID 6GUH), (B) 3D structure of 13a (pink) superimposed with AZD5438 (pink) in CDK2 (PDB ID 6GUH) in tint sticks. | ||
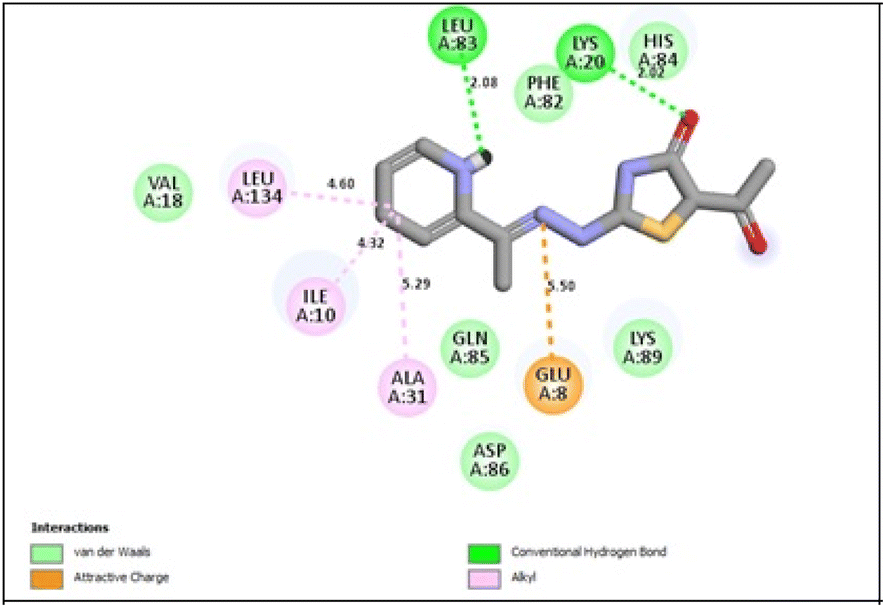 | ||
| Fig. 10 2D binding interactions of compound 8a in CDK2. | ||
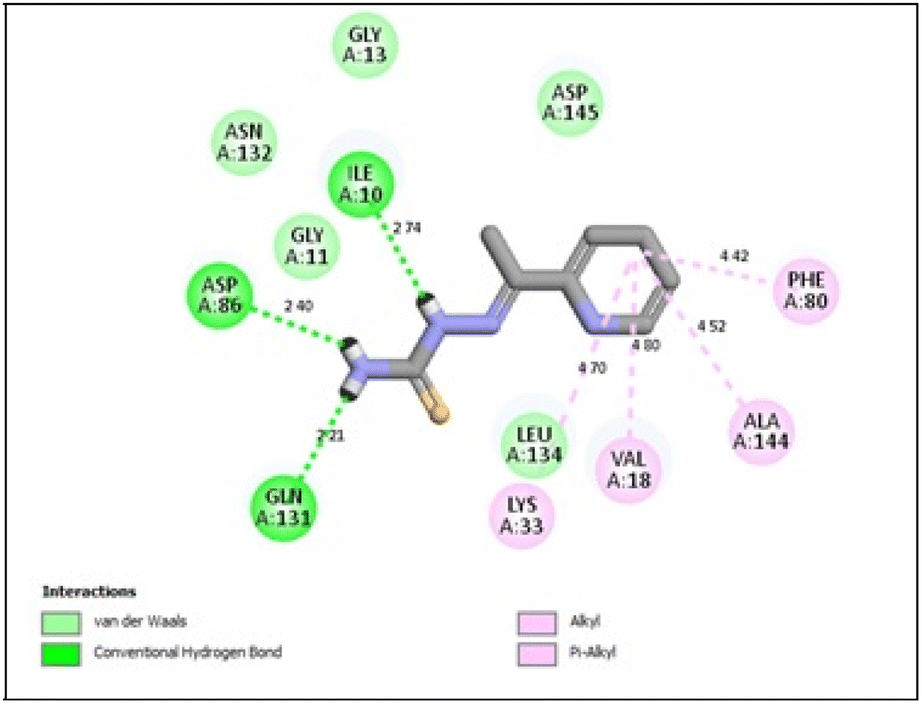 | ||
| Fig. 11 2D binding interactions of compound 1a in CDK2. | ||
In conclusion, the docking simulation study coincides with the in vitro enzymatic assay, which pointed out that 13a was the most potent CDK2 inhibitor among the tested compounds followed by 8a and the pyridine thiourea precursor 1a was the weakest. Therefore, hybridization of the pyridine core structure with 2,3-dihydrothiazole (13a) or thiazolidin-4-one (8a) through an ethylidenehydrazono spacer was significant for CDK2 inhibitory activity.
In the GSK3β ATP binding site, it was predicted that 13a had the highest binding affinity (−8.0 kcal mol−1) but less than that of the native ligand 2WF (−8.6 kcal mol−1), followed by 8a (−7.8 kcal mol−1). Both were superior to 1a (−5.5 kcal mol−1), the pyridine thiourea precursor (Table 5).
It was reported that 2WF formed hydrogen bonds with Lys85 and Val135. Unlikely compound 13a formed hydrophobic interactions with Lys85 and Val135 and instead formed two hydrogen bonds with Asn64 (2.28 Å and 2.66 Å). There were several hydrophobic interactions exemplified in pi–sigma interaction with Leu188 (3.93 Å), pi–anion interaction with Asp200 (4.62 Å), many pi–alkyl and van der Waals interactions, as presented in Fig. 12A. Compound 13a was slightly superimposed, mainly the 5(p-tolyldiazenyl)-2,3-dihydrothiazole moiety, over the native ligand 2WF, which might explain the slight decrease in its binding affinity (Fig. 12B). It was predicted that compound 8a would keep the essential hydrogen bond between thiazolidin-4-one carbonyl and Lys85 (3.06 Å) with alkyl and van der Waals hydrophobic interactions (Fig. 13). Conversely, although 1a possessed three hydrogen bonds two of which were with the essential Val135, it was the weakest GSK3β inhibitor in the tested set (Fig. 14). Depending on the agreement of the docking study results with the in vitro GSK3β inhibitory screening results, the importance of pyridine hybridization with 2,3-dihydrothiazole (13a) and with thiazolidin-4-one (8a) through an ethylidenehydrazono spacer was highlighted for affording a GSK3β inhibitor.
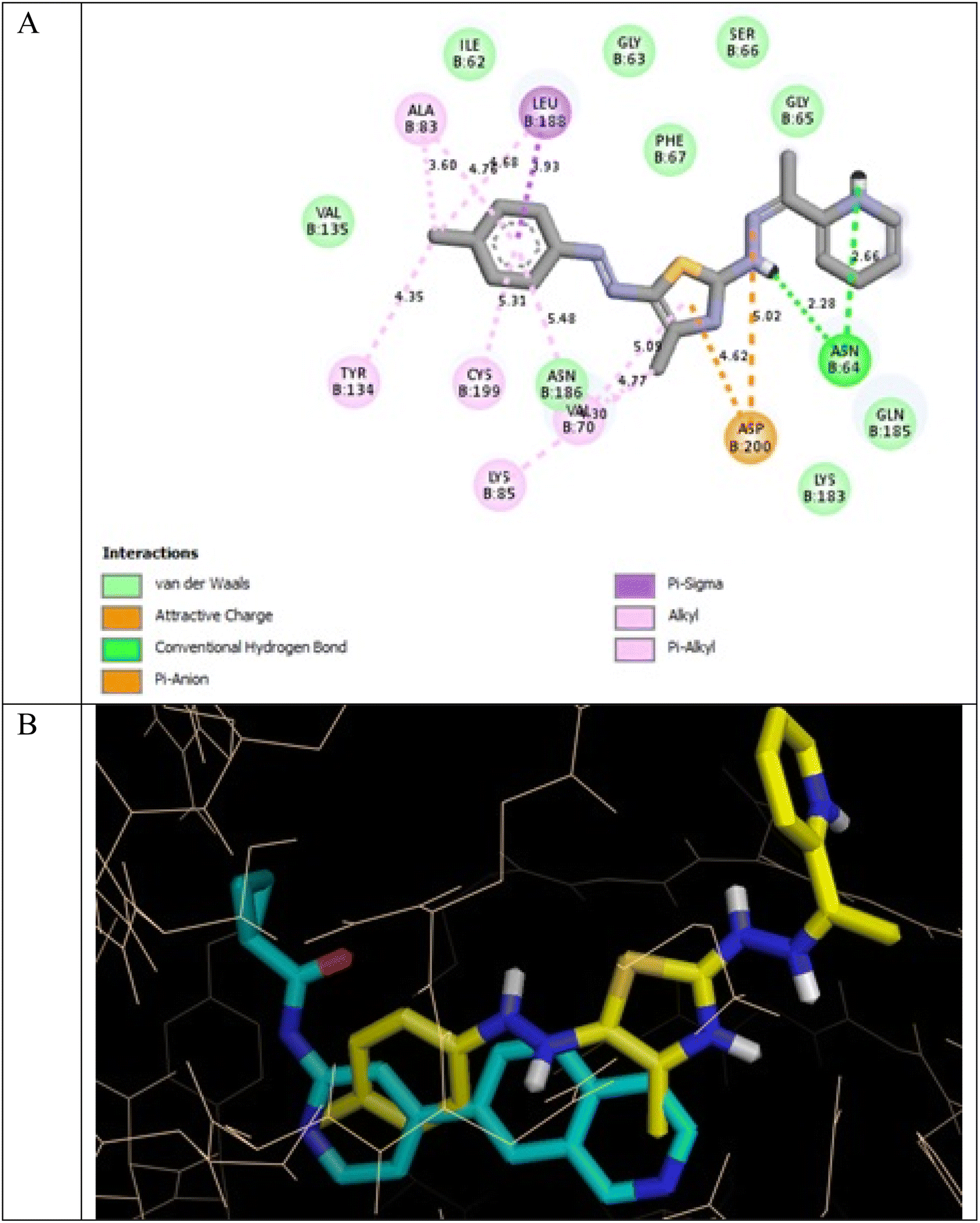 | ||
| Fig. 12 (A) 2D binding interactions of compound 13a in GSK3β (PDB ID 4PTE); (B) 3D structure of 13a (yellow) superimposed with 2WF (cyan) in GSK3β (PDB ID 4PTE) in tint sticks. | ||
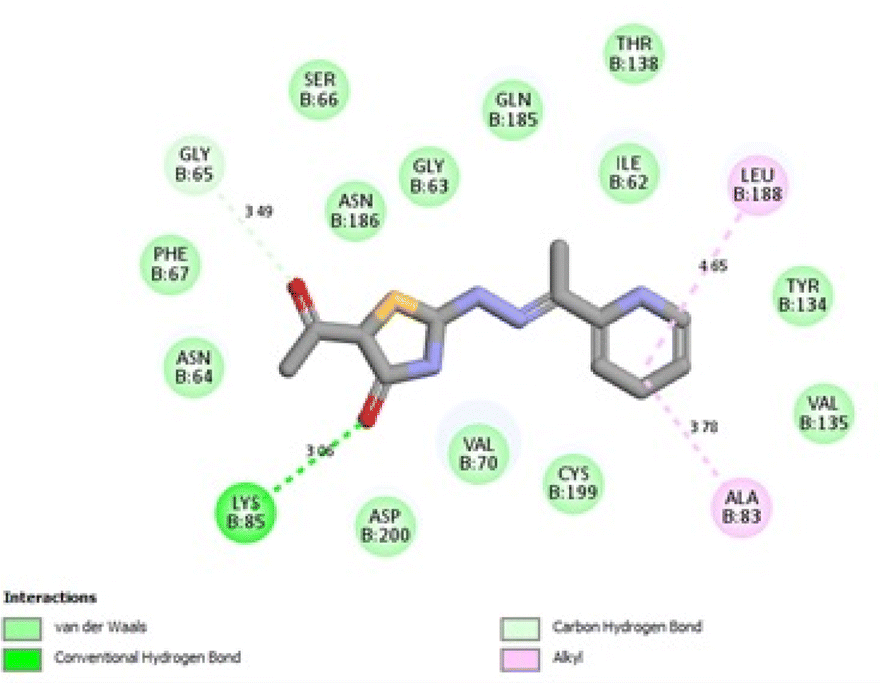 | ||
| Fig. 13 2D binding interactions of compound 8a in GSK3β (PDB ID 4PTE). | ||
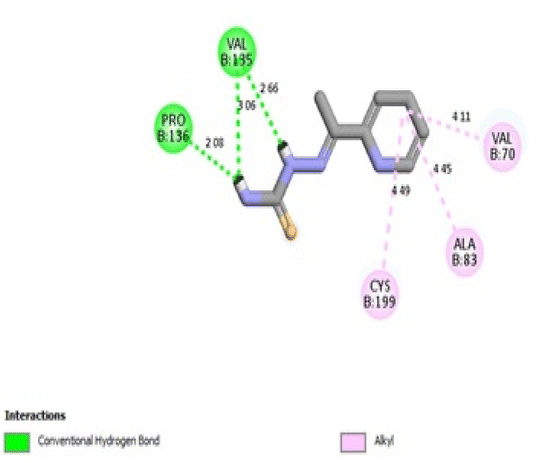 | ||
| Fig. 14 2D binding interactions of compound 1a in GSK3β (PDB ID 4PTE). | ||
3 Conclusions
In this research, twenty-six novel hybrids were designed and synthesized as follows: pyridin-2-yl-2,3-dihydrothiazoles (2a,b–5a,b), pyridin-2-yl-thiazolidin-4-ones (6a,b–8a,b), pyridin-2-yl-5-substituted benzylidene-thiazolidin-4-ones (9a,b–12a,b) and pyridin-2-yl-5-diazenyl-2,3-dihydrothiazoles (13a,b–14a,b) as dual CDK2/GSK3β inhibitors. Almost all of the synthesized compounds including their precursors 1a and 1b were in vitro screened for anti-proliferative activity against MCF-7, HepG2, and HEp-2 cancer cell lines and the Vero normal cell lines. Compound 8a (pyridine-5-acetyl-thiazolidin-4-one) hybrid exhibited excellent anti-proliferative activity against HEp-2, and 13a (pyridine-5-(p-tolyldiazenyl-2,3-dihydrothiazole)) hybrid possessed excellent anti-proliferative activity against HepG2 with an acceptable safety profile against normal cells. Additionally, 1a (pyridine thiourea) precursor showed excellent anti-proliferative activity against HEp-2, but it negatively affected normal cells. The three promising anti-proliferative compounds 1a, 8a and 13a were evaluated for inhibitory activity against CDK2 and GSK3β using roscovitine and CHIR-99021 as references, respectively. Compound 13a was the most potent dual CDK2/GSK3β inhibitor followed by 8a, and the weakest was 1a. Further study of compound 13a revealed that it caused G1 cell cycle arrest and induced apoptosis. Moreover, it caused an increase in Bax and caspase-3 levels with a decrease in Bcl-2 levels in HepG2. Finally, an in silico study of the three promising compounds predicted an acceptable drug likeness and ADME profiles. The molecular modelling simulation results were in agreement with the in vitro kinase inhibitory results and highlighted the importance of pyridine hybridization with 2,3-dihydrothiazole (13a) and with thiazolidin-4-one (8a) through an ethylidenehydrazono spacer for dual CDK2/GSK3β inhibitory activity. These might act as promising leads which may be subjected to further modification for better anti-proliferative activity in future work.4 Experimental
4.1. Chemistry
(E)-4-Methyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)-2,3-dihydrothiazole (2a). Yield 88%; mp (110–112) °C. Analysis for C11H12N4S (232.31) calcd: % C, 59.87; H, 5.21; N, 24.12; S, 13.80. Found: % C, 59.85; H, 5.26; N, 24.21; S, 13.84. IR (cm−1, KBr): 3344 (NH), 1551 (C
N). 1H NMR (DMSO-d6, δ, ppm): 2.15 (s, 3H, CH3), 2.34 (s, 3H, CH3), 6.36 (s, 1H, CH thiazole), 7.31 (1H, t, Ar–H), 7.77 (1H, t, Ar–H), 7.96 (1H, d, Ar–H), 8.52 (1H, m, Ar–H), 11.24 (1H, s, NH). 13C NMR (DMSO-d6, δ, ppm): 15.16 (CH3), 19.64 (CH3), 99.63 (CH2 thiazole), 122.54, 126.30, 135.99, 147.99, 153.48, 153.92, 158.85 (Ar–C + C1, C4 thiazole), 164.57 (CN). MS m/z (RA%): 232 (M+) (34%).
(E)-3-Ethyl-4-methyl-2-(((E)-1-(pyridin-2-yl)ethylidene) hydrazono)-2,3-dihydrothiazole (2b). Yield 80%; mp (87–89) °C. Analysis for C13H16N4S (260.36) calcd: % C, 59.97; H, 6.19; N, 21.52; S, 12.31. Found: % C, 59.89; H, 6.21; N, 21.43; S, 12.45. IR (cm−1, KBr): 1554 (C
N). 1H NMR (DMSO-d6, δ, ppm): 1.21 (t, 3H, CH3), 2.15 (s, 3H, CH3), 2.37 (s, 3H, CH3), 3.88 (q, 2H, CH2), 6.04 (s, 1H, CH thiazole), 7.28 (H, t, Ar–H), 7.74 (H, t, Ar–H), 8.02 (H, d, Ar–H), 8.52 (H, d, Ar–H). 13C NMR (DMSO-d6, δ, ppm): 13.46 (CH2CH3), 13.63 (CH3), 13.99 (CH3), 40.59 (CH2CH3), 97.44 (CH2 thiazole), 120.15, 123.54, 136.55, 149.06, 154.96, 156.85 (Ar–C + C1, C4 thiazole), 169.87 (CN). MS m/z (RA%): 260 (M+) (3.55%).
1-((E)-4-Methyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)-2,3-dihydrothiazol-5-yl)ethan-1-one (3a). Yield 75%; mp (115–117) °C. Analysis for C13H14N4OS (274.34) calcd: % C, 56.92; H, 5.14; N, 20.42; S, 11.69. Found: % C, 56.89; H, 5.11; N, 20.39; S, 11.59. IR (cm−1, KBr): 3378 (NH), 1623 (C
O), 1551 (CN). 1H NMR (DMSO-d6, δ, ppm): 2.36 (s, 6H, CH3, COCH3), 2.39 (s, 3H, CH3), 7.34 (1H, t, Ar–H), 7.80 (1H, t, Ar–H), 7.82 (1H, d, Ar–H), 8.02 (1H, d, Ar–H), 11.82 (1H, s, NH). 13C NMR (DMSO-d6, δ, ppm): 14.11 (CH3), 19.09 (CH3), 25.46 (CH3CO), 105.77, 122.05, 127.09, 136.69, 149.03, 155.16, 158.34, 158.38 (Ar–C), 164.74 (CN), 190.95 (CO). MS m/z (RA%): 274 (M+) (32%).
1-((E)-3-Ethyl-4-methyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)-2,3-dihydrothiazol-5-yl)ethan-1-one (3b). Yield 75%; mp (166–168) °C. Analysis for C15H18N4OS (302.40) calcd: % C, 59.58; H, 600; N, 18.53; S, 10.60. Found: % C, 59.54; H, 603; N, 18.56; S, 10.57. IR (cm−1, KBr): 1625 (C
O), 1556 (CN). 1H NMR (DMSO-d6, δ, ppm): 1.18 (t, 3H, CH3), 2.20, 2.22 (s, 6H, CH3, COCH3, CH3), 2.41 (s, 3H, CH3), 3.94 (q, 2H, CH2), 7.38 (1H, t, Ar–H), 7.84 (1H, t, Ar–H), 7.38 (1H, d, Ar–H), 8.06 (1H, d, Ar–H), 8.37 (1H, s, NH). 13C NMR (DMSO-d6, δ, ppm): 7.38 (CH3), 13.21 (CH2–CH3), 20.45 (CH3), 25.56 (CH3CO), 42.26 (CH2–CH3), 106.10, 122.03, 127.09, 136.69, 149.12, 155.16, 158.34, 158.38 (Ar–C), 164.91 (CN), 190.95 (CO). MS m/z (RA%): 302 (M+) (100%).
(E)-3-Ethyl-4-phenyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)-2,3-dihydrothiazole (4b). Yield 65%; mp (97–99) °C. Analysis for C18H18N4S (322.43) calcd: % C, 67.05; H, 5.63; N, 17.38; S, 9.94. Found: % C, 67.15; H, 5.66; N, 17.40; S, 9.96. IR (cm−1, KBr): 1555 (C
N). 1H NMR (DMSO-d6, δ, ppm): 1.14 (t, 3H, CH3), 2.43 (s, 3H, CH3), 3.81 (q, 2H, CH2), 6.40 (s, 1H, CH thiazole), 7.43–8.56 (8H, m, Ar–H). 13C NMR (DMSO-d6, δ, ppm): 13.45 (CH2–CH3), 14.11 (CH3), 39.59 (CH2–CH3), 109.34, 121.08, 125.23, 129.14, 129.18, 132.06, 136.07, 144.70, 149.43, 153.96, 157.79 (ArC), 164.91 (CN), MS m/z (RA%): 322 (M+) (34%).
(E)-4-(4-Bromophenyl)-3-ethyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)-2,3-dihydrothiazole (5b). Yield 65%; mp (106–108) °C. Analysis for C18H17BrN4S (401.33) calcd: % C, 53.87; H, 4.27; N, 13.96; S, 7.99. Found: % C, 53.84; H, 4.27; N, 13.91; S, 7.93. IR (cm−1, KBr): 1557 (C
N). 1H NMR (DMSO-d6, δ, ppm): 1.14 (t, 3H, CH3), 2.43 (s, 3H, CH3), 3.81 (q, 2H, CH2), 6.44 (s, 1H, CH thiazole), 7.43–8.61 (8H, m, Ar–H). 13C NMR (DMSO-d6, δ, ppm): 13.43 (CH2–CH3), 14.12 (CH3), 39.59 (CH2–CH3), 109.54, 121.28, 125.34, 129.45, 129.20, 131.56, 136.23, 144.34, 149.40, 154.06, 158.09 (Ar–C), 164.54 (CN), MS m/z (RA%): 402 (M+ + 1) (52%).
(E)-3-Ethyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (6b). Yield 90%; mp (133–135) °C. Analysis for C12H14N4OS (262.089) calcd: % C, 54.94; H, 5.38; N, 21.36; S, 12.22. Found: % C, 54.85; H, 5.36; N, 21.32; S, 12.26. IR (cm−1, KBr): 1715 (C
O), 1554 (CN). 1H NMR (DMSO-d6, δ, ppm): 1.19 (t, 3H, CH3), 2.44 (s, 3H, CH3), 3.77 (q, 2H, CH2), 3.94 (S, 2H, CH2 thiazole), 7.43 (H, t, Ar–H), 7.84 (H, t, Ar–H), 8.04 (H, d, Ar–H), 8.61 (H, d, Ar–H), 13C NMR (DMSO-d6, δ, ppm): 12.85 (CH2–CH3), 14.13 (CH3), 32.39 (CH2 thiazole), 38.13 (CH2–CH3), 121.99, 136.57, 149.05, 155.95 (Ar–C), 158.23 (C4 thiazole), 164.46 (CN), 170.64 (CO), MS m/z (RA%): 262 (M+) (13%).
(E)-5-Methyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (7a). Yield 85%; mp (176–178) °C. Analysis for C11H12N4OS (248.30) calcd: % C, 53.21; H, 4.87; N, 22.56; S, 12.91. Found: % C, 53.24; H, 4.83; N, 22.51; S, 12.97. IR (cm−1, KBr): 3375 (NH), 1712 (C
O), 1554 (CN), 1H NMR (DMSO-d6, δ, ppm): 1.47 (d, 3H, CH3), 2.39 (s, 3H, CH3), 4.13 (q, 2H, CH2), 7.39 (H, t, Ar–H), 7.83 (H, t, Ar–H), 8.02 (H, d, Ar–H), 8.59 (H, d, Ar–H), 12.0 (1H, s, NH). 13C NMR (DMSO-d6, δ, ppm): 14.07 (CH3), 19.33 (CH3), 42.67 (CH2 thiazole), 120.91, 124.91, 137.02, 149.33, 154.61 (Ar–C), 155.76 (C1 thiazole), 161.71 (CN), 177.56 (CO), MS m/z (RA%): 248 (M+) (10%).
(E)-3-Ethyl-5-methyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (7b). Yield 80%; mp (95–97) °C. Analysis for C13H16N4OS (276.36) calcd: % C, 56.50; H, 5.84; N, 20.27; S, 11.60. Found: % C, 56.55; H, 5.80; N, 20.29; S, 11.65. IR (cm−1, KBr): 1708 (C
O), 1561 (CN), 1H NMR (DMSO-d6, δ, ppm): 1.18 (t, 3H, CH3), 1.49 (d, 3H, CH3), 2.43 (s, 3H, CH3), 3.76 (q, 2H, CH2–CH3), 4.22 (q, 2H, S–CH2), 7.41 (H, t, Ar–H), 7.83 (H, t, Ar–H), 8.04 (H, d, Ar–H) 8.60 (H, d, Ar–H), 13C NMR (DMSO-d6, δ, ppm): 12.78 (CH2–CH3), 14.07 (CH3), 19.32 (CH3), 38.59 (CH2–CH3), 41.90 (CH2 thiazole), 120.97, 125.14, 137.13, 149.42, 155.57 (Ar–C), 157.13 (C1 thiazole), 162.58 (CN), 176.23 (CO) MS m/z (RA%): 276 (M+) (12%).
(E)-5-Acetyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (8a). Yield 75%; mp (118–120) °C. Analysis for C12H12N4O2S (276.31) calcd: % C, 56.16; H, 4.38; N, 20.28; S, 11.60. Found: % C, 56.18; H, 4.34; N, 20.18; S, 11.65. IR (cm−1, KBr): 3330 (NH), 1542 (C
N), 1995, 1703 (2CO), 1H NMR (DMSO-d6, δ, ppm): 2.40 (s, 3H, COCH3), 2.47 (s, 3H, CH3), 4.17 (s, 1H, CH thiazole), 7.43 (1H, t, Ar–H), 7.79 (1H, t, Ar–H), 8.06 (1H, d, Ar–H), 8.55 (1H, d, Ar–H), 11.92 (1H, s, NH). 13C NMR (DMSO-d6, δ, ppm): 14.82 (CH3), 22.52 (COCH3), 61.06 (CH thiazole), 122.18, 126.77, 136.54, 149.27, 154.12, 158.80 (C1 thiazole), 164.53 (CN), 170.95 (CO), 197.98 (COCH3), MS m/z (RA%): 276 (M+) (0.75%).
(E)-5-Acetyl-3-ethyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (8b). Yield 60%; mp (94–96) °C. Analysis for C14H16N4O2S (304.37) calcd: % C, 55.25; H, 5.30; N, 18.41; S, 10.53. Found: % C, 55.28; H, 5.35; N, 18.46; S, 10.48. IR (cm−1, KBr): 1543 (C
N), 1703 (2CO), 1H NMR (DMSO-d6, δ, ppm): 1.24 (3H, t, CH3), 2.40 (s, 3H, COCH3), 2.43 (s, 3H, CH3), 3.98 (q, 2H, CH2), 4.19 (s, 1H, CH thiazole), 7.34 (1H, t, Ar–H), 7.79 (1H, t, Ar–H), 8.08 (1H, d, Ar–H), 8.55 (1H, d, Ar–H). 13C NMR (DMSO-d6, δ, ppm): 13.49 (CH2–CH3), 14.83 (CH3), 22.52 (COCH3), 35.72 (CH2–CH3), 61.09 (CH2 thiazole), 122.18, 126.72, 136.54, 149.27, 154.12 (Ar–C), 158.80 (C1 thiazole), 164.53 (CN), 170.80 (CO), 197.38 (COCH3). MS m/z (RA%): 304 (M+) (0.98%).
(b) In glacial acetic acid (10 mL) and anhydrous sodium acetate (25 mmol), a mixture of compounds 6a and 6b (10 mmol) with an equimolar amount of an appropriate aromatic aldehyde, namely, 4-fluoro-benzaldehyde, 4-chloro-benzaldehyde, 4-methoxy benzaldehyde and/or 3,4-dimethoxybenzaldehyde (10 mmol), was refluxed for 6–8 hours, cooled, and then placed into cold water. The resulting chemicals were obtained by collecting and crystallizing the solid that was generated from absolute ethanol (9a,b–12a,b).
(E)-5-((E)-4-Fluorobenzylidene)-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (9a). Yield 75%; mp (204–206) °C. Analysis for C17H13FN4OS (340.38) calcd: % C, 59.99; H, 4.85; N, 16.46; S, 9.42. Found: % C, 59.94; H, 4.75; N, 16.41; S, 9.46. IR (cm−1, KBr): 3325 (NH), 1692 (C
O), 1550 (2CN). 1H NMR (DMSO-d6, δ, ppm): 2.46 (s, 3H, CH3), 7.18–8.63 (9H, m, Ar–H + CH benzylic), 12.16 (1H, s, NH). 13C NMR (DMSO-d6, δ, ppm): 14.50 (CH3), 115.52, 115.56, 124.10, 125.45, 131.10, 136.42, 142.91, 149.19, 154.13, 159.62, 162.14 (Ar–C + CH benzylic + C2, C5 thiazole), 164.53 (CN), 164.34 (CO), MS m/z (RA%): 340 (M+) (5.78%).
(E)-3-Ethyl-5-((E)-4-fluorobenzylidene)-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (9b). Yield 70%; mp (125–127) °C. Analysis for C19H17FN4OS (368.43) calcd: % C, 61.94; H, 4.65; N, 15.21; S, 8.70. Found: % C, 61.99; H, 4.60; N, 15.26; S, 8.67. IR (cm−1, KBr): 1695 (C
O), 1554 (CN). 1H NMR (DMSO-d6, δ, ppm): 1.13 (t, 3H, CH3), 2.47 (s, 3H, CH3), 3.78 (q, 3H, CH2), 7.72–8.51 (9H, m, Ar–H + CH benzylic). 13C NMR (DMSO-d6, δ, ppm): 12.85 (CH2–CH3), 14.59 (CH3), 35.58 (CH2–CH3), 115.52, 115.56, 124.10, 125.45, 131.10, 136.42, 142.91, 149.19, 154.13, 159.62, 162.14 (Ar–C + CH benzylic + C1, C5 thiazole), 164.53 (CN), 164.34 (CO), MS m/z (RA%): 368 (M+) (0.78%).
(E)-5-((E)-4-Chlorobenzylidene)-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (10a). Yield 65%; mp (119–121) °C. Analysis for C17H13ClN4OS (356.83) calcd: % C, 57.22; H, 3.67; N, 15.70; S, 8.98. Found: % C, 57.28; H, 3.69; N, 15.10; S, 8.94. IR (cm−1, KBr): 3326 (NH), 1696 (C
O), 1552 (CN). 1H NMR (DMSO-d6, δ, ppm): 2.47 (s, 3H, CH3), 7.46–8.51 (9H, m, Ar–H + CH benzylic), 11.67 (1H, s, NH). 13C NMR (DMSO-d6, δ, ppm): 14.55 (CH3), 116.33, 120.90, 126.20, 127.55, 129.99, 133.10, 135.74, 143.72, 149.72, 154.13, 158.07 (Ar–C + CH benzylic + C1, C5 thiazole), 164.40 (CN), 172.02 (CO), MS m/z (RA%): 356 (M+) (2.89%).
(E)-5-((E)-4-Chlorobenzylidene)-3-ethyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (10b). Yield 80%; mp (146–148) °C. Analysis for C19H17ClN4OS (384.88) calcd: % C, 59.29; H, 4.45; N, 14.56; S, 8.33. Found: % C, 59.19; H, 4.49; N, 14.52; S, 8.38. IR (cm−1, KBr): 1698 (C
O), 1556 (CN). 1H NMR (DMSO-d6, δ, ppm): 1.13 (t, 3H, CH3), 2.47 (s, 3H, CH3), 3.70 (q, 3H, CH2), 7.47–8.51 (8H, m, Ar–H + CH benzylic). 13C NMR (DMSO-d6, δ, ppm): 12.42 (CH2–CH3), 14.66 (CH3), 35.63 (CH2–CH3), 116.62, 120.90, 126.26, 127.99, 129.99, 133.83, 135.74, 143.79, 149.62, 154.18, 158.12 (Ar–C + CH benzylic + C2, C5 thiazole), 164.77 (CN), 166.32 (CO), MS m/z (RA%): 384 (M+) (20%).
(E)-5-((E)-4-Methoxybenzylidene)-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (11a). Yield 50%; mp (155–157) °C. Analysis for C18H16N4O2S (352.41) calcd: % C, 61.35; H, 4.58; N, 15.90; S, 9.10. Found: % C, 61.29; H, 4.52; N, 15.98; S, 9.14. IR (cm−1, KBr): 3328 (NH), 1694 (C
O), 1552 (CN). 1H NMR (DMSO-d6, δ, ppm): 2.47 (s, 3H, CH3), 3.73 (s, 3H, OCH3), 6.99–8.44 (9H, m, Ar–H + CH benzylic), 11.62 (1H, s, NH). 13C NMR (DMSO-d6, δ, ppm): 14.54 (CH3), 56.65 (OCH3), 114.54, 116.62, 122.76, 126.26, 127.64, 130.64, 137.36, 143.72, 148.70, 154.13, 156.80 (Ar–C + CH benzylic + C2, C5 thiazole), 164.40 (CN), 174.53 (CO), MS m/z (RA%): 352 (M+) (24.14%).
(E)-3-Ethyl-5-((E)-4-methoxybenzylidene)-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (11b). Yield 75%; mp (150–152) °C. Analysis for C20H20N4O2S (380.47) calcd: % C, 63.14; H, 5.30; N, 14.73; S, 8.43. Found: % C, 63.19; H, 5.37; N, 14.70; S, 8.47. IR (cm−1, KBr): 1699 (C
O), 1554 (CN). 1H NMR (DMSO-d6, δ, ppm): 1.14 (t, 3H, CH3), 2.47 (s, 3H, CH3), 3.69 (s, 3H, OCH3), 3.71 (q, 2H, CH2), 6.99–8.44 (9H, m, Ar–H + CH benzylic), 13C NMR (DMSO-d6, δ, ppm): 12.96 (CH2–CH3), 14.66 (CH3), 35.63 (CH2–CH3), 56.61 (OCH3), 114.54, 116.75, 122.92, 126.26, 127.99, 130.55, 137.29, 143.27, 148.65, 154.18, 156.80 (Ar–C + CH benzylic + C2, C5 thiazole), 164.36 (CN), 166.55 (CO), MS m/z (RA%): 380 (M+) (1.31%).
(E)-5-((E)-3,4-Dimethoxybenzylidene)-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (12a). Yield 70%; mp (143–145) °C. Analysis for C19H18N4O3S (382.44) calcd: % C, 59.67; H, 4.74; N, 14.65; S, 8.38. Found: % C, 59.71; H, 4.64; N, 14.60; S, 8.32. IR (cm−1, KBr): 3329 (NH), 1696 (C
O), 1554 (CN). 1H NMR (DMSO-d6, δ, ppm): 2.47 (s, 3H, CH3), 3.78, 3.81 (s, 6H, 2OCH3), 7.01–8.44 (9H, m, Ar–H + CH benzylic), 11.90 (s, 1H, NH), 13C NMR (DMSO-d6, δ, ppm): 14.59 (CH3), 56.69 (OCH3), 110.97, 111.01, 116.62, 120.14, 122.93, 122.97, 126.20, 136.27, 143.68, 149.40, 149.24, 154.24, 158.07 (Ar–C + CH benzylic + C2, C5 thiazole), 164.62 (CN), 174.46 (CO), MS m/z (RA%): 382 (M+) (5.14%).
(E)-5-((E)-3,4-Dimethoxybenzylidene)-3-ethyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)thiazolidin-4-one (12b). Yield 65%; mp (173–175) °C. Analysis for C21H22N4O3S (410.141) calcd: % C, 61.45; H, 5.40; N, 13.65; S, 7.81. Found: % C, 61.49; H, 5.30; N, 13.62; S, 7.87. IR (cm−1, KBr): 1698 (C
O), 1559 (CN). 1H NMR (DMSO-d6, δ, ppm): 1.16 (t, 3H, CH3), 2.47 (s, 3H, CH3), 3.71, 3.72 (2s, 6H, 2OCH3), 3.74 (q, 2H, CH2), 6.99–8.44 (9H, m, Ar–H + CH benzylic), 13C NMR (DMSO-d6, δ, ppm): 12.85 (CH2–CH3), 14.55 (CH3), 35.63 (CH2–CH3), 56.68 (OCH3), 115.47, 116.54, 122.93, 127.99, 129.99, 138.57, 143.27, 148.70, 154.84, 156.12, (Ar–C + CH benzylic + C2, C5 thiazole), 164.62 (CN), 166.06 (CO), MS m/z (RA%): 410 (M+) (0.34%).
(E)-4-Methyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)-5-((E)-p-tolyldiazenyl)-2,3-dihydrothiazole (13a). Yield 60%; mp (150–152) °C. Analysis for C18H18N6S (350.44) calcd: % C, 61.69; H, 5.18; N, 23.98; S, 9.15. Found: % C, 61.60; H, 5.28; N, 23.93; S, 9.18. IR (cm−1, KBr): 3310 (NH); 1550 (C
N). 1H NMR (DMSO-d6, δ, ppm): 2.44 (s, 3H, CH3), 2.45 (s, 3H, CH3), 2.47 (s, 3H, CH3), 7.37–8.59 (8H, m, Ar–H), 10.65 (1H, s, NH). 13C NMR (DMSO-d6, δ, ppm): 12.17 (CH3), 14.99 (CH3), 21.76 (CH3), 94.02 (C5 thiazole), 122.93, 126.08, 129.01, 129.38, 136.46, 137.80, 144.41, 149.66, 153.73, 154.18, 158.67 (Ar–C + C2, C4 thiazole), 165.28 (CN), MS m/z (RA%): 350 (M+) (37%).
(E)-3-Ethyl-4-methyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)-5-((E)-p-tolyldiazenyl)-2,3-dihydrothiazole (13b). Yield 65%; mp (148–150) °C. Analysis for C20H22N6S (378.50) calcd: % C, 63.47; H, 5.86; N, 22.20; S, 8.47. Found: % C, 63.41; H, 5.80; N, 22.27; S, 8.37, IR (cm−1, KBr): 1552 (C
N). 1H NMR (DMSO-d6, δ, ppm): 1.34 (t, 3H, CH3), 2.43 (s, 3H, CH3), 2.45 (s, 3H, CH3), 2.47 (s, 3H, CH3), 4.12 (q, 2H, CH2), 7.46–8.60 (8H, m, Ar–H). 13C NMR (DMSO-d6, δ, ppm): 10.15 (CH3), 12.58 (CH3), 14.59 (CH3), 21.62 (CH3), 42.35 (CH2–CH3), 93.69 (C5 thiazole), 122.88, 126.08, 129.06, 129.89, 136.51, 138.42, 144.41, 153.73, 155.07, 158.71 (Ar–C + C2, C4 thiazole), 164.41 (CN). MS m/z (RA%): 378 (M+) (49%).
(E)-5-((E)-(4-Chlorophenyl)diazenyl)-3-ethyl-4-methyl-2-(((E)-1-(pyridin-2-yl)ethylidene)hydrazono)-2,3-dihydrothiazole (14b). Yield 60%; mp (150–152) °C. Analysis for C19H19ClN6S (398.91) calcd: % C, 57.21; H, 4.80; N, 21.07; S, 8.04. Found: % C, 57.28; H, 4.85; N, 21.12; S, 8.08, IR (cm−1, KBr): 1553 (C
N). 1H NMR (DMSO-d6, δ, ppm): 1.34 (t, 3H, CH3), 2.45 (s, 3H, CH3), 2.47 (s, 3H, CH3), 4.13 (q, 2H, CH2), 7.37–8.63 (8H, m, Ar–H). 13C NMR (DMSO-d6, δ, ppm): 10.03 (CH3), 13.30 (CH3), 14.56 (CH3), 42.89 (CH2–CH3), 93.71 (C5 thiazole), 122.94, 126.26, 129.43, 129.99, 136.22, 138.30, 145.76, 149.54, 153.73, 154.18, 158.16 (Ar–C + C2, C4 thiazole), 164.32 (CN), MS m/z (RA%): 398 (M+) (47%).
4.2. Biological evaluation
Data availability
The data supporting this article have been included as part of the ESI† associated with this article and can be found in the online version.Author contributions
Asmaa F. Kassem: conceptualization, methodology, validation, formal analysis, investigation, resources, data curation, writing – original draft, writing – review & editing. Ashraf A. Sediek: conceptualization, methodology, validation, formal analysis, investigation, resources, data curation. Mervat M. Omran: software, investigation, data curation, writing – original draft. Doaa S. Foda: software, investigation, data curation, writing – review & editing. Aisha A. K. Al-Ashmawy: conceptualization, methodology, validation, formal analysis, investigation, resources, software, data curation, writing – original draft, writing – review & editing. All persons who meet authorship criteria are listed as authors, and all authors certify that they have participated sufficiently in the work to take public responsibility for the content, including participation in the concept, design, analysis, writing, or revision of the manuscript. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There is no conflict to declare.Acknowledgements
The authors thank the National Cancer Institute and National Research Centre for the equipment and facilities.References
- Y. Chen, X. F. Zhang and L. Ou-Yang, Comput. Struct. Biotechnol. J., 2023, 21, 974–990 CrossRef CAS PubMed.
- World Health Organization International Agency for Research on Cancer (IARC), World Cancer Report: Cancer Research for Cancer Prevention, 2020, vol. 199 Search PubMed.
- R. V. J. Chari, Acc. Chem. Res., 2008, 41, 98–107 CrossRef CAS.
- M. Brindisi, S. M. Kessler, V. Kumar and C. Zwergel, Front. Oncol., 2022, 12, 980141 CrossRef.
- K. S. Bhullar, N. O. Lagarón, E. M. McGowan, I. Parmar, A. Jha, B. P. Hubbard and H. P. V. Rupasinghe, Mol. Cancer, 2018, 17, 48 CrossRef.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- A. A. K. Al-Ashmawy, K. M. Elokely, O. Perez-Leal, M. Rico, J. Gordon, G. Mateo, A. M. Omar, M. Abou-Gharbia and W. E. Childers, ACS Med. Chem. Lett., 2020, 11, 2156–2164 CrossRef CAS PubMed.
- A. A. K. Al-Ashmawy, F. A. Ragab, K. M. Elokely, M. M. Anwar, O. Perez-Leal, M. C. Rico, J. Gordon, E. Bichenkov, G. Mateo, E. M. M. Kassem, G. H. Hegazy, M. Abou-Gharbia and W. Childers, Bioorg. Med. Chem. Lett., 2017, 27, 3117–3122 CrossRef CAS.
- M. Malumbres, Physiol. Rev., 2011, 91, 973–1007 CrossRef.
- S. Tadesse, A. T. Anshabo, N. Portman, E. Lim, W. Tilley, C. E. Caldon and S. Wang, Drug Discovery Today, 2020, 25, 406–413 CrossRef.
- T. Chohan, H. Qian, Y. Pan and J.-Z. Chen, Curr. Med. Chem., 2014, 22, 237–263 CrossRef.
- T. A. Chohan, A. Qayyum, K. Rehman, M. Tariq and M. S. H. Akash, Biomed. Pharmacother., 2018, 107, 1326–1341 CrossRef PubMed.
- O. A. E. F. M. Fathalla, M. A. H. Ismail, M. M. Anwar, K. A. M. Abouzid and A. A. K. Ramadan, Med. Chem. Res., 2013, 22, 659–673 CrossRef.
- S. M. A. Ruiz and H. Eldar-Finkelman, Front. Mol. Neurosci., 2022, 14, 22 Search PubMed.
- B. W. Doble and J. R. Woodgett, J. Cell Sci., 2003, 116, 1175–1186 CrossRef CAS.
- K. D. Miller, M. Fidler-Benaoudia, T. H. Keegan, H. S. Hipp, A. Jemal and R. L. Siegel, Cancer statistics for adolescents and young adults, Wiley Online Libr., 2020, vol. 70, pp. 443–459 Search PubMed.
- T. Domoto, M. Uehara, D. Bolidong and T. Minamoto, Cells, 2020, 9, 1–31 CrossRef PubMed.
- J. A. Mccubrey, L. S. Steelman, F. E. Bertrand, N. M. Davis, M. Sokolosky, S. L. Abrams, G. Montalto, A. B. D'Assoro, M. Libra, F. Nicoletti, R. Maestro, J. Basecke, D. Rakus, A. Gizak, Z. Demidenko, L. Cocco, A. M. Martelli and M. Cervello, Oncotarget, 2014, 5, 2881–2911 CrossRef PubMed.
- M. Brindisi, S. M. Kessler, V. Kumar and C. Zwergel, Front. Oncol., 2022, 12, 985363 CrossRef.
- A. M. Alqahtani and A. A. Bayazeed, Arabian J. Chem., 2021, 14(1), 102914 CrossRef CAS.
- Z. Lv, C. Sheng, T. Wang, Y. Zhang, J. Liu, J. Feng, H. Sun, H. Zhong, C. Niu and K. Li, J. Med. Chem., 2010, 53, 660–668 CrossRef CAS PubMed.
- M. Ravinder, B. Mahendar, S. Mattapally, K. V. Hamsini, T. N. Reddy, C. Rohit, K. Srinivas, S. K. Banerjee and V. J. Rao, Bioorg. Med. Chem. Lett., 2012, 22, 6010–6015 CrossRef CAS PubMed.
- M. M. Ghorab, F. A. Ragab, H. I. Heiba, M. S. A. Elsayed and W. M. Ghorab, Bioorg. Chem., 2018, 80, 276–287 CrossRef CAS PubMed.
- A. F. Kassem, M. A. Omar, E. S. Nossier, H. M. Awad and W. A. El-Sayed, J. Mol. Struct., 2023, 1294, 136358 CrossRef CAS.
- C. Li, S. Zhang, Y. Lu, Y. Zhang, E. Wang and Z. Cui, PLoS One, 2013, 8(12), e84659 CrossRef.
- A. Kazi, S. Xiang, H. Yang, D. Delitto, J. Trevino, R. H. Y. Jiang, M. Ayaz, H. R. Lawrence, P. Kennedy and S. M. Sebti, Nat. Commun., 2018, 9(1), 5154 CrossRef.
- Y. Xu, D. Cheng, L. Hu, X. Dong, L. Lv, C. Zhang and J. Zhou, Biocell, 2023, 47, 825–836 CAS.
- B. S. Choudhary, Sukanya, P. Mehta, S. Bach, S. Ruchaud, T. Robert, B. Josselin, S. Filipek and R. Malik, Bioorg. Med. Chem. Lett., 2021, 52, 128375 CrossRef CAS.
- A. F. Kassem, M. A. Omar, E. S. Nossier, H. M. Awad and W. A. El-Sayed, J. Mol. Struct., 2023, 1294, 136358 CrossRef CAS.
- P. Makam, R. Kankanala, A. Prakash and T. Kannan, Eur. J. Med. Chem., 2013, 69, 564–576 CrossRef CAS PubMed.
- S. Carradori, B. Bizzarri, M. D'Ascenzio, C. De Monte, R. Grande, D. Rivanera, A. Zicari, E. Mari, M. Sabatino, A. Patsilinakos, R. Ragno and D. Secci, Eur. J. Med. Chem., 2017, 140, 274–292 CrossRef PubMed.
- H. A. Ghabbour, A. A. Kadi, K. E. H. Eltahir, R. F. Angawi and H. I. El-Subbagh, Med. Chem. Res., 2015, 24, 3194–3211 CrossRef CAS.
- C. Tratrat, M. Haroun, E. Tsolaki, A. Petrou, A. Gavalas and A. Geronikaki, Curr. Top. Med. Chem., 2020, 21, 257–268 CrossRef PubMed.
- A. Conroy, D. E. Stockett, D. Walker, M. R. Arkin, U. Hoch, J. A. Fox and R. E. Hawtin, Cancer Chemother. Pharmacol., 2009, 64, 723–732 CrossRef CAS PubMed.
- A. M. Shawky, M. A. S. Abourehab, A. N. Abdalla and A. M. Gouda, Eur. J. Med. Chem., 2020, 185(1), 111780 CrossRef CAS PubMed.
- J. M. Domínguez, A. Fuertes, L. Orozco, M. Del Monte-Millán, E. Delgado and M. Medina, J. Biol. Chem., 2012, 287, 893–904 CrossRef.
- Y. Dong, L. Sui, Y. Tai, K. Sugimoto and M. Tokuda, Anticancer Res., 2001, 21, 103–108 Search PubMed.
- S. Shen, D. C. Dean, Z. Yu and Z. Duan, Hepatol. Res., 2019, 49, 1097–1108 CrossRef.
- L. Ding, J. Cao, W. Lin, H. Chen, X. Xiong, H. Ao, M. Yu, J. Lin and Q. Cui, Int. J. Mol. Sci., 2020, 21, 1960 CrossRef.
- C. Desbois-Mouthon, M.-J. Blivet-Van Eggelpoë, E. Beurel, M. Boissan, R. Delélo, A. Cadoret and J. Capeau, Hepatology, 2002, 36, 1528–1536 CrossRef PubMed.
- H. Goto, K. Kawano, I. Kobayashi, H. Sakai and S. Yanagisawa, Oral Oncol., 2002, 38, 549–556 CrossRef PubMed.
- C. Xu, Z. Du, S. Ren and Y. Pian, Ann. Clin. Lab. Sci., 2021, 51, 285–294 Search PubMed.
- J. Qi, Y. Zheng, K. Qian, L. Tian, G. X. Zhang, Z. Cheng and Y. Wang, J. Inorg. Biochem., 2017, 177, 110–117 CrossRef.
- N. S. M. Mokhtaruddin, E. N. M. Yusof, T. B. S. A. Ravoof, E. R. T. Tiekink, A. Veerakumarasivam and M. I. M. Tahir, J. Mol. Struct., 2017, 1139, 1–9 CrossRef.
- F. Ali, K. M. Khan, U. Salar, M. Taha, N. H. Ismail, A. Wadood, M. Riaz and S. Perveen, Eur. J. Med. Chem., 2017, 138, 255–272 CrossRef.
- M. D'Ascenzio, P. Chimenti, M. C. Gidaro, C. De Monte, D. De Vita, A. Granese, L. Scipione, R. Di Santo, G. Costa, S. Alcaro, M. Yáñez and S. Carradori, J. Enzyme Inhib. Med. Chem., 2015, 30, 908–919 CrossRef.
- R. E. Khidre, S. R. El-Gogary and M. S. Mostafa, J. Heterocycl. Chem., 2017, 54, 2511–2519 CrossRef.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. Mcmahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef.
- D. Baskić, I. Jovanović, P. Ristić, V. Jakovljević, D. Delibašić and N. Arsenijević, in Nitric Oxide, 2005, vol. 6, pp. 49–52 Search PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef PubMed.
- U. Asghar, A. K. Witkiewicz, N. C. Turner and E. S. Knudsen, Nat. Rev. Drug Discovery, 2015, 14, 130–146 CrossRef.
- J. A. Mccubrey, L. S. Steelman, F. E. Bertrand, N. M. Davis, M. Sokolosky, S. L. Abrams, G. Montalto, A. B. D'Assoro, M. Libra, F. Nicoletti, R. Maestro, J. Basecke, D. Rakus, A. Gizak, Z. Demidenko, L. Cocco, A. M. Martelli and M. Cervello, Oncotarget, 2014, 5, 2881–2911 CrossRef PubMed.
- M. B. Labib, J. N. Philoppes, P. F. Lamie and E. R. Ahmed, Bioorg. Chem., 2018, 76, 67–80 CrossRef PubMed.
- R. Jan and G. e. S. Chaudhry, Adv. Pharm. Bull., 2019, 9, 205–218 CrossRef.
- P. Hussar, Encyclopedia, 2022, 2, 1624–1636 CrossRef.
- H. Kalkavan and D. R. Green, Cell Death Differ., 2018, 25, 46–55 CrossRef PubMed.
- S. Qian, Z. Wei, W. Yang, J. Huang, Y. Yang and J. Wang, Front. Oncol., 2022, 12, 985363 CrossRef PubMed.
- E. Eskandari and C. J. Eaves, J. Cell Biol., 2022, 221, e202201159 CrossRef.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- J. Ali, P. Camilleri, M. B. Brown, A. J. Hutt and S. B. Kirton, J. Chem. Inf. Model., 2012, 52, 420–428 CrossRef.
- A. Daina and V. Zoete, ChemMedChem, 2016, 1117–1121 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef PubMed.
- N. M. Abdelazeem, W. M. Aboulthana, Z. A. Elshahid, M. El-Hussieny and A. A. K. Al-Ashmawy, J. Mol. Struct., 2024, 1310, 138224 CrossRef.
- D. J. Wood, S. Korolchuk, N. J. Tatum, L. Z. Wang, J. A. Endicott, M. E. M. Noble and M. P. Martin, Cell Chem. Biol., 2019, 26, 121–130 CrossRef.
- P. Sivaprakasam, X. Han, R. L. Civiello, S. Jacutin-Porte, K. Kish, M. Pokross, H. A. Lewis, N. Ahmed, N. Szapiel, J. A. Newitt, E. T. Baldwin, H. Xiao, C. M. Krause, H. Park, M. Nophsker, J. S. Lippy, C. R. Burton, D. R. Langley, J. E. Macor and G. M. Dubowchik, Bioorg. Med. Chem. Lett., 2015, 25, 1856–1863 CrossRef PubMed.
- E. Krieger, K. Joo, J. Lee, J. Lee, S. Raman, J. Thompson, M. Tyka, D. Baker and K. Karplus, Proteins, 2009, 77, 114–122 CrossRef.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06146b |
| This journal is © The Royal Society of Chemistry 2024 |