
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfonated porous organic polymer supported Ziegler–Natta catalysts for the synthesis of ultra-high molecular weight polyethylene
Wenqian Kang,
Xiong Wang *,
Yue Ren,
Pingsheng Zhang,
Anping Huang and
Guangquan Li
*,
Yue Ren,
Pingsheng Zhang,
Anping Huang and
Guangquan Li
Lanzhou Petrochemical Research Center, Petrochemical Research Institute, PetroChina, Lanzhou 730060, China. E-mail: wangxiong1@petrochina.com.cn; suewang001@163.com
First published on 29th October 2024
Abstract
Porous organic polymers (POPs) are attracting attention for their easy functionalization and potential as catalyst supports in olefin polymerization. In this study, sulfonated POP (s-POP) supported Ziegler–Natta catalysts were used for ethylene polymerization, producing ultra-high molecular weight polyethylene, with Mν reaching up to 6.83 × 106 g mol−1. The maximum Mν of polyethylene was achieved by Cat-3 with DIBP as the internal donor, albeit with a partial loss of catalytic activity. Polymerization conditions also play a pivotal role in determining the molecular weight of polyethylene. Hydrogen, being the most efficient chain transfer agent, can decrease the molecular weight to 9.68 × 104 g mol−1 at higher hydrogen concentrations ([H2]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[C2H4] = 0.83), and the s-POP-supported ethylene polymerization catalysts were observed to exhibit high sensitivity to hydrogen response. The effects of polymerization temperature, [Al]:[Ti] molar ratio, and ethylene pressure on ethylene polymerization were thoroughly investigated.
:[C2H4] = 0.83), and the s-POP-supported ethylene polymerization catalysts were observed to exhibit high sensitivity to hydrogen response. The effects of polymerization temperature, [Al]:[Ti] molar ratio, and ethylene pressure on ethylene polymerization were thoroughly investigated.
1. Introduction
Ultra-high molecular weight polyethylene (UHMWPE) is a specialized type of semi-crystalline thermoplastic engineering plastic known for its good chemical resistance, high impact resistance, high wear resistance, durability and biocompatibility. It plays an important role in fields such as national defense, military, marine engineering, ropes and textiles, and sports equipment. Additionally, UHMWPE has a Young's modulus of approximately 0.5–0.8 GPa, similar to human bones, with low friction coefficient and good biocompatibility, making it suitable for the production of artificial knee joints, artificial hip joints, artificial heart valves, and more.1–6The performance of UHMWPE is closely related to its molecular weight, and place high demands on the catalyst system. Single-site catalysts, including metallocene catalysts, Fhenoxy imine-based catalysts, and post-transition metal catalysts, among others, can be utilized for the production of UHMWPE.7–15 Through the design and synthesis of ligands, precise control over polymer structure can be achieved, resulting in high molecular weight narrow distribution UHMWPE, disentangled UHMWPE, branched UHMWPE, and other UHMWPE variants with specific structural properties. However, the industrial application of single-site catalyst systems still faces challenges such as reaction conditions, catalyst cost, and activity. Currently, most commercial UHMWPE is manufactured using conventional Ziegler–Natta (Z–N) catalytic systems employing TiCl4/MgCl2.16 In the industry, Z–N catalysts continue to be the predominant catalyst system utilized for olefin polymerization. Ongoing research on Z–N catalyst systems primarily focuses on innovation and refinement catalyst supports. In comparison with traditional inorganic supports like MgCl2 and SiO2, polymer supports can be synthesized using versatile strategies, allowing for easy modification with substituents and controlled pore structures.17,18 And polystyrene or polysiloxane based supports have been proved to effectively prepare Z–N catalysts and catalyze ethylene/propylene polymerization.19–24
Porous organic polymers (POPs) composed mainly of C, H and O atoms are garnering increasing attention due to their easier functionalization to accommodate active cites, while maintaining minimal impact on the produced polymer properties.25–28 Based on our prior research, POPs based metallocene catalysts have demonstrated outstanding capabilities in ethylene homo-polymerization and ethylene/α-olefin copolymerization. Moreover, we found that the 4-hydroxyethylmethacrylate (HEMA) functionalized and sulfonated POPs could be modified using a Grignard reagent to synthesize Z–N catalysts. Despite the slightly lower activity of the resulting Z–N catalysts compared to the MgCl2-based catalysts, the prepared polypropylene displayed notable characteristics with broad molecular weight distribution (MWD > 11) and high stereoselectivity.29,30 The functional comonomer not only influences the properties of POPs, but also affects the microchemical environment of immobilized active centers. Thus, polymers with specific structure and property can be prepared through the integrated design and preparation of functional POPs.
In the present study, 4-vinylbenzenesulfonic acid sodium salt and sodium allylsulfonate were selected as functional groups for synthesizing s-POPs, which reacted with methylmagnesium chloride to prepare Z–N catalysts. The findings from ethylene polymerization experiments revealed that the s-POP-based Z–N catalysts exhibited strong ethylene polymerization capability, yielding a polymer with a viscosity average molecular weight (Mν) as high as 6.83 × 106 g mol−1, indicating their considerable potential for UHMWPE production.
2. Materials and methods
2.1 Materials
Divinylbenzene (80% mixtures of isomers, DVB), 2,2′-azo-bis-isobutyronitrile (≥98%, AIBN), 4-vinylbenzenesulfonic acid sodium salt (≥90%), sodium allylsulfonate (≥90%), diisobutyl phthalate (DIBP, ID-1), decahydronaphthalene and methylmagnesium chloride (3 M in tetrahydrofuran solution) were all purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). DVB was pretreated before use according to our previous work.26,28 Poloxamer 407 (F127, BASF), titanium tetrachloride (Tianjing Yongda Chemical Reagent Co. Ltd), 3-methyl-5-tert-butyl-1,2-phenylene dibenzoate (Tianjin Scaxchem Limited Company, ID-2) and ethanol (≥99.5%, Sinopharm Chemical Reagent Co., Ltd.) were used as received. Deionized water, ethylene, nitrogen, toluene, hexane and triethylaluminium (TEAL, 10% in hexane solution) were kindly donated by PetroChina Lanzhou Petrochemical Company.2.2 Sulfonated porous organic polymer and catalysts preparation
Sulfonated porous organic polymers were synthesized through dispersion polymerization as previously reported, utilizing 4-vinylbenzenesulfonic acid sodium salt (A) and sodium allylsulfonate (B) as functional comonomers.29 Some of the resulting s-POPs underwent further acidification, while all s-POPs were subjected to vacuum treatment at 80 °C for 8 hours to prepare the catalysts. Under a nitrogen atmosphere, 20 mL of CH3MgCl (3 mol L−1 in THF) was added into a toluene solution containing s-POPs (5 g), stirred at 35 °C for 3 hours. The reaction mixture was then filtered, washed with toluene, and 50 mL of fresh toluene was added. Subsequently, 50 mL of TiCl4 was added dropwise, and the slurry was heated to 80 °C. Upon reaching the desired temperature, 0.75 g of internal donor (Cat-3 and Cat-4) was added and stirring for 3 hours. Finally, the slurry was filtered, washed sequentially with three portions of toluene and hexane, and dried under vacuum at 80 °C for 1 hour.2.3 Ethylene polymerization in slurry process
Ethylene polymerization was conducted in a nitrogen-replaced 2 L stainless steel reactor. Initially, 300 g of hexane and 5–20 mL of aluminum triethyl (TEAL, 10% in hexane) were introduced into the reactor, stirred for approximately 10 minutes at room temperature to remove the impurities. Following this, a moderate number of catalysts was added, along with an additional 400 g of hexane. Ethylene was continuously supplied under pressure of 0.4–1.1 MPa, while maintaining a temperature of 60–80 °C. Subsequently, the reactor was cooled to room temperature, excess gas vented, and the polyethylene collected through filtration and drying.2.4 Characterization
The N2 adsorption/desorption isotherms of s-POPs were examined using a Quantachrome Nova 2000e N2 sorption instrument (Boynton Beach, FL, USA) at 77.3 K. Prior to analysis, the particles were degassed at 120 °C overnight. X-ray diffraction (XRD) measurements of the catalysts were performed on a Bruker D8 ADVANCE using Cu Kα radiation (λ = 0.154 nm) with 2θ scanning angle from 10° to 65°, and samples were protected from contact with air by a polyethylene film. X-ray photoelectron spectroscopy (XPS) analysis utilized an ESCA Lab250 spectrometer from Thermo Fisher Scientific (USA) with Al Kα radiation at 1486.6 eV (500 μm spot-size). The FT-IR of s-POPs and s-POP supported Z–N catalysts were examined on the NEXUS 670 FTIR. Ti and Mg content analysis of the catalysts were carried out on a VISTAICP-MPX (VARIAN, Palo Alto, CA, USA). The viscosity molecular weight (Mν) of the polymer was measured in decahydronaphthalene at 135 °C following ASTM D-4020. The Mν was calculated according to the formula (1).| Mν = 5.37 × 104[η]1.37 | (1) |
3. Results and discussion
3.1 Preparation of sulfonated porous organic polymers
In this study, s-POPs were synthesized via a dispersion polymerization strategy with two functional comonomers. POP-SO3Na-A and POP-SO3H-A used 4-vinylbenzenesulfonic acid sodium salt (A) as functional comonomer, POP-SO3Na-B and POP-SO3H-B used sodium allylsulfonate (B) as functional comonomer, respectively. POP-SO3H-A and POP-SO3H-B were acidified to convert –SO3Na groups to –SO3H. Conversely, non-acidified s-POPs (POP-SO3Na-A and POP-SO3Na-B) retained their original composition. The specific surface area (SSA) and total pore volume (TPV) of obtained s-POPs were assessed by nitrogen sorption analysis. As presented in Table 1, s-POPs exhibited good porosity with the SSA exceeding 300 m2 g−1. In comparison, POP-SO3H-B, employing functional comonomer B, obtained a higher SSA of 471 m2 g−1 with a TPV value of 0.31 cm3 g−1 compared to POP-SO3H-A. This difference could be attributed to the different thermodynamic compatibility among comonomers, solvent systems and the prepared s-POPs.27| SSA (m2 g−1) | TPV (cm3 g−1) | |
|---|---|---|
| POP-SO3H-A | 385 | 0.22 |
| POP-SO3H-B | 471 | 0.31 |
3.2 Catalysts preparation
In this work, Mg–Cl moiety was anchored onto the s-POPs via CH3MgCl mediated reaction, allowing the modified s-POPs to immobilize TiCl4 and form Ti/Mg@s-POP Z–N catalysts. For Cat-2 and Cat-6, TiCl4 was immobilized onto the Mg–Cl moiety generated from the –SO3Na on the surface of non-acidified POP-SO3Na-A and POP-SO3Na-B. To investigate the effect of internal donor on the catalyst's polymerization performance and the prepared polymers, DIBP (ID-1) and 3-methyl-5-tert-butyl-1,2-phenylene dibenzoate (ID-2) were employed as internal donor in Cat-3 and Cat-4, respectively.The contents of magnesium and titanium loaded in s-POPs were determined by ICP analysis after calcination and acidolysis. As detailed in Table 2, the magnesium contents in the Ti/Mg@s-POP catalysts were controlled in a relatively narrow range of 3.4–4.2%, which was significantly lower than that of MgCl2 supported catalysts.31 Moreover, the titanium contents were all controlled below 4%. Catalysts (Cat-2 and Cat-6) derived from unacidified POP-SO3Na-A and POP-SO3Na-B exhibited lower magnesium contents compared to catalysts prepared from acidified supports. This indicates that the unacidified s-POPs have a weaker interaction with the Grignard reagent, possibly resulting in the physical adsorption of the Grignard reagent onto the s-POP structure. However, the titanium exhibited opposite trend, catalysts derived from unacidified supports showed higher titanium loading content. TiCl4 probably adsorbed onto the s-POPs via polarized adsorption of –SO3Na groups.
| Catalyst | Support | Internal donor(ID) | Mg (wt%) | Ti (wt%) | Activity (g PE per g cat per h) | Mν × 10−4 g mol−1 |
|---|---|---|---|---|---|---|
| a Polymerization conditions: Cat = 50–60 mg; TEAL = 10 mL; temperature = 70 °C; pressure = 0.6 MPa; time = 60 min. | ||||||
| Cat-1 | POP-SO3H-A | No ID | 4.1 | 2.8 | 4966 | 235 |
| Cat-2 | POP-SO3Na-A | No ID | 3.4 | 3.2 | 2677 | 302 |
| Cat-3 | POP-SO3H-A | ID-1 | 3.8 | 2.7 | 2397 | 683 |
| Cat-4 | POP-SO3H-A | ID-2 | 4.0 | 2.0 | 2208 | 265 |
| Cat-5 | POP-SO3H-B | No ID | 4.2 | 2.8 | 7356 | 428 |
| Cat-6 | POP-SO3Na-B | No ID | 3.7 | 3.6 | 3347 | 459 |
3.3 Ethylene polymerization results
The ethylene polymerization was conducted using the six prepared heterogeneous catalysts in conjunction with TEAL, and the polymerization results are presented in Table 2. The Ti/Mg@s-POP catalytic system was observed to produce polyethylene with Mν exceeding 2 million g mol−1, making it suitable for producing UHMWPE. Specifically, Cat-1, originating from acidified POP-SO3H-A, exhibited high activity of 4966 g PE per g cat per h, resulting in polyethylene with a Mν of 2.35 × 106 g mol−1. In contrast, Cat-2, prepared from unacidified s-POP, showed lower activity of 2677 g PE per g cat per h. Similarly, Cat-5, derived from acidified POP-SO3H-B without an internal donor, yielded polyethylene with a Mν of 4.28 × 106 g mol−1, demonstrating the highest polymerization activity of 7356 g PE per g cat per h. However, the unacidified POP-SO3Na-B supported catalyst Cat-6 exhibited lower activity but yield polyethylene with a higher molecular weight. This disparity could be attributed to the weaker reaction of CH3MgCl with the unacidified –SO3Na groups, leading to most of the CH3MgCl and TiCl4 being physically adsorbed onto the s-POP. Consequently, only a small portions of effective Mg–Ti active centers were loaded onto the s-POP and took effect during the ethylene polymerization. Moreover, Cat-5, prepared from POP-SO3H-B, exhibited higher polymerization activity and produced polyethylene with a higher molecular weight, suggesting that the choice of functional comonomer impacted the active sites of the prepared catalysts. Compared to traditional MgCl2 or MgCl2 (ethoxide type) supported Z–N catalysts, the decrease in activity observed in s-POP based Z–N catalysts for ethylene polymerization is not as pronounced as the decrease observed for propylene polymerization.32Regarding the results of Cat-3 and Cat-4, catalysts prepared with internal donors exhibited lower activity but higher molecular weight. Cat-3, employing DIBP as the internal donor, produced polyethylene with the highest Mν of 6.83 × 106 g mol−1, almost three times that of Cat-1 without internal donor. Cat-4 with ID-2 as the internal donor produced polyethylene with a similar Mν to Cat-1 but showed lower catalytic activity. Internal donors can bind to the Mg–Ti active centers formed during the preparation process and affect the molecular weight. This may be cause the disruption of active centers responsible for producing low molecular weight polymer by the internal donor, or the reduction in chain transfer to TEAL.16,33
3.4 FTIR, XPS and XRD analysis of catalysts
The FTIR spectra of Ti/Mg@s-POP catalysts are depicted in the Fig. 1. A prominent, wide absorption band near 3200–3700 cm−1 indicates the presence of –OH stretching vibrations, likely due to water adsorbed in Ti/Mg@s-POP catalysts. Peaks around 2920 cm−1 correspond to C–H stretching vibrations, aligning with the chemical structure of the supports used. Notably, a sharp absorption peak at approximately 1600 cm−1 can be ascribed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations in aromatic rings, particularly associated with divinylbenzene. Additionally, bands near 1170 cm−1 and 1030 cm−1 are indicative of asymmetric and symmetric SO stretching, respectively. These spectral features exhibit variations across different catalyst samples, suggesting alterations in the sulfonic groups' environment, potentially due to interactions with metal centers or other structural changes during the catalyst synthesis on s-POP.34,35 For samples Cat-3 and Cat-4, additional absorption bands are observed near 1700 cm−1 and 1300 cm−1, corresponding to CO and C–O stretching vibrations, respectively. These are attributed to the inclusion of ID-1 and ID-2 during the catalyst preparation. Furthermore, a band centered at approximately 580 cm−1 is identified as C–S stretching vibration.
C stretching vibrations in aromatic rings, particularly associated with divinylbenzene. Additionally, bands near 1170 cm−1 and 1030 cm−1 are indicative of asymmetric and symmetric SO stretching, respectively. These spectral features exhibit variations across different catalyst samples, suggesting alterations in the sulfonic groups' environment, potentially due to interactions with metal centers or other structural changes during the catalyst synthesis on s-POP.34,35 For samples Cat-3 and Cat-4, additional absorption bands are observed near 1700 cm−1 and 1300 cm−1, corresponding to CO and C–O stretching vibrations, respectively. These are attributed to the inclusion of ID-1 and ID-2 during the catalyst preparation. Furthermore, a band centered at approximately 580 cm−1 is identified as C–S stretching vibration.
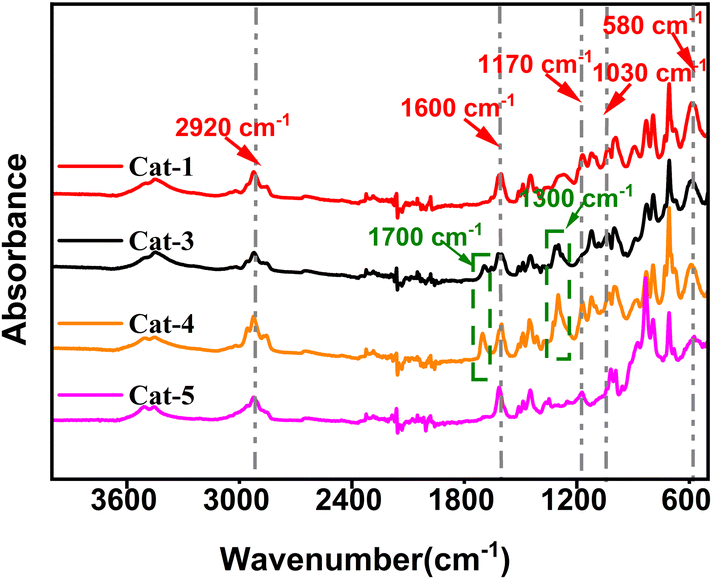 | ||
| Fig. 1 FTIR spectra of Ti/Mg@s-POP catalysts. | ||
To investigate the influence of s-POP on the localized microchemical environments of magnesium and titanium sites, X-ray photoelectron spectroscopy (XPS) analysis was conducted. The obtained spectra, shown in Fig. 2, provide detailed insights into the binding energies (BE) of S, O, Ti, Mg and Cl atoms. As shown in the Fig. 2(a), compared to the binding energy (BE) of the S 2p orbital in pure s-POP (POP-SO3H-A), the Ti/Mg@s-POP catalysts display a slight increase in the BE of the S 2p, indicating an electron density decrease around the sulfur atoms in the s-POP. Additionally, as illustrated in the Fig. 2(b), the broadened shape of the O 1s spectra in the s-POP supported catalysts indicates the presence of oxygen in at least three distinct states, contrasting with the narrow peak observed in standalone s-POP at a binding energy around 531.5 eV. The varied peaks within the O 1s range can be attributed to different functional groups: SO/CO bonds appearing in the 533.8–532.5 eV range, S–O/C–O bonds around 532.5–531.0 eV, and Ti–O or Mg–O bonds manifesting in the 531.0–529.5 eV range.33 This diversity in oxygen states reflects the complex interactions and bonding environments in the s-POP supported catalysts. Cat-3 and Cat-4, contain similar BE curves around the range of 533.8–532.5 eV, it could be ascribed to the addition of internal donor, which contribute CO bonds to the prepared catalysts. In contrast, Cat-1 and Cat-5 exhibit distinct metal–oxygen peak profile, reflecting variations in their chemical composition and structure.
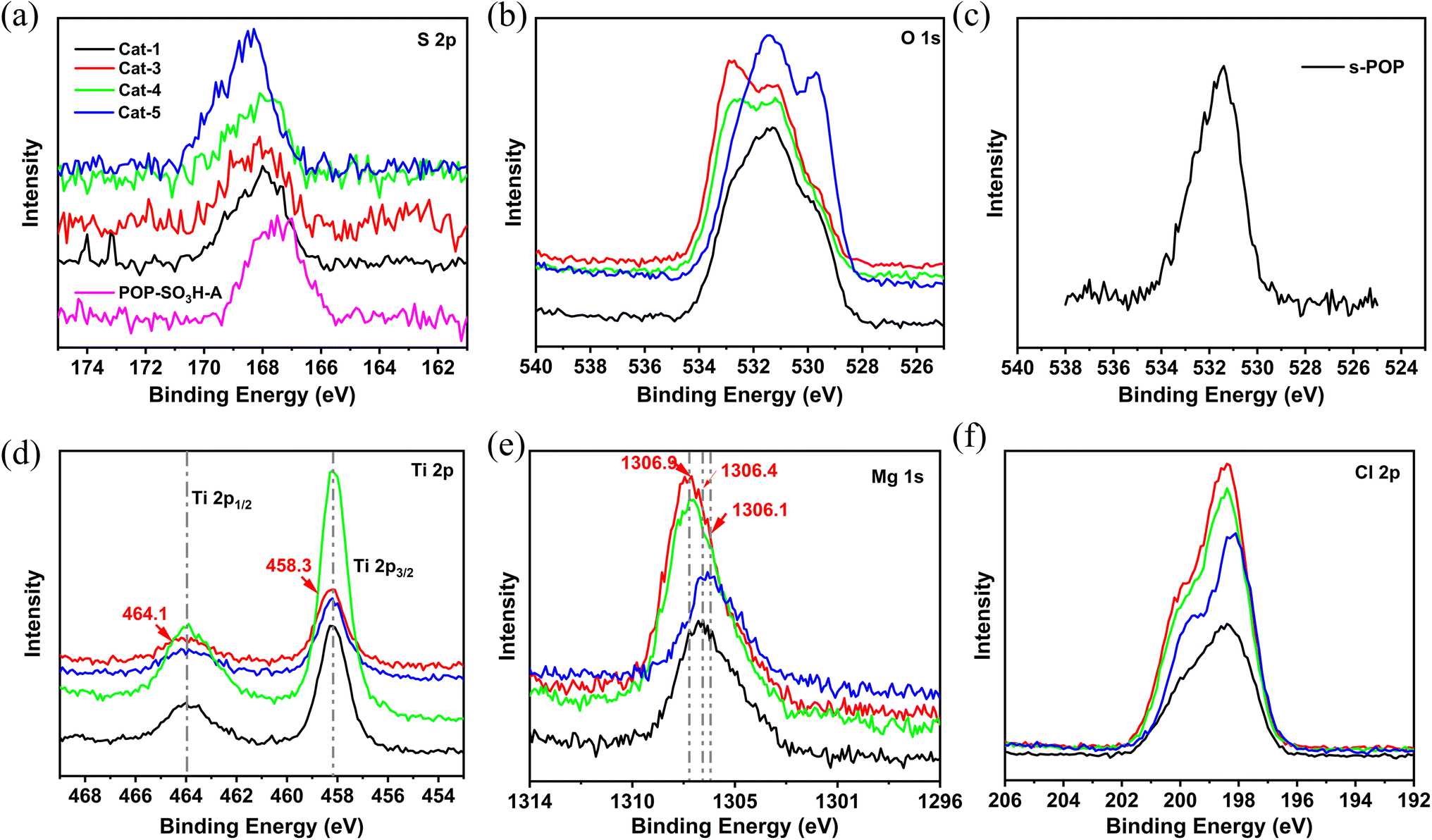 | ||
| Fig. 2 (a) S 2p XPS spectra of s-POP and s-POP supported catalysts; (b) O 1s XPS spectra of s-POP supported catalysts; (c) O 1s XPS spectra of s-POP; (d) Ti 2p XPS spectra of s-POP supported catalysts; (e) Mg 1s XPS spectra of s-POP supported catalysts; (f) Cl 2p XPS spectra of s-POP supported catalysts. | ||
Fig. 2(d) illustrates the Ti 2p spectra of the catalysts, characterized by a doublet resulting from spin–orbit coupling of the 2p electrons, corresponding to Ti 2p3/2 and Ti 2p1/2 photoelectrons.36 The BE values for all supported catalysts are nearly identical. As shown in Table 3, Cat-3 with DIBP as internal donor shows a marginally higher BE of 458.3 eV and 464.1 eV for Ti 2p3/2 and Ti 2p1/2, respectively, potentially due to the enhanced electron-withdrawing effect by the combination DIBP and s-POP. However, the BE for Ti 2p3/2 of Cat-3 was lower than that reported in the literature in the case of Ti/DIBP/MgCl2.37 BE shifts are influenced by both the oxidation state and the charge on atoms adjacent to the excited atom.38 In comparison with Cat-1, Cat-5 displays a slightly lower BE for Ti 2p3/2 (458.1 eV) and Ti 2p1/2 (463.9 eV). This suggests that POP-SO3H-B has weaker electron-withdrawing capability than the s-POP employed sodium p-styrene sulfonate as comonomer. In turn, Cat-3 and Cat-4 obtained increased BE for Mg 1s, attributed to the formed Mg–O by the interaction with internal donor. Additionally, POP-SO3H-B exhibits the weakest electron withdrawal from the Mg atom, reflected in the smallest Mg 1s BE at 1306.1 eV. Furthermore, Fig. 2(f) reveals a slight increase in the BE of Cl 2p3/2 for Cat-3 and Cat-4 to 200.0 eV. This increase is likely due to the electron-withdrawing effect from oxygen atoms in the internal donors, indicating a complex interaction between these components within the catalyst structure.
| Entry | Ti 2p1/2 | Ti 2p3/2 | Mg 1s | Cl 2p1/2 | Cl 2p3/2 |
|---|---|---|---|---|---|
| Cat-1 | 464.0 | 458.2 | 1306.4 | 198.2 | 199.7 |
| Cat-3 | 464.1 | 458.3 | 1306.9 | 198.3 | 200.0 |
| Cat-4 | 464.0 | 458.2 | 1306.9 | 198.3 | 200.0 |
| Cat-5 | 463.9 | 458.1 | 1306.1 | 198.1 | 199.7 |
Fig. 3 illustrate the powder X-ray diffraction (XRD) patterns for Ti/Mg@s-POP catalysts alongside anhydrous MgCl2. The anhydrous MgCl2 exhibits distinct and sharp diffraction peaks at 2θ values of 15°, 30°, 35° and 50°, corresponding to the (003), (012), (104), and (110) crystallographic planes of the α-MgCl2 phase, respectively. The XRD patterns of the Ti/Mg@s-POP catalysts mirror the MgCl2 pattern to some extent, suggesting the presence of MgCl2 crystals on the s-POP support. However, the specific peak for the (003) plane is conspicuously absent, implying that the Cl–Mg–Cl layers within the s-POP based catalysts structure are limited and possibly mainly monolayered. Moreover, the (012) and (104) peaks appear merged into a single broadened peak (2θ ranges at 27–37°), combining with the broad reflection at 48–54°, indicating the formation of stacking disorder δ-MgCl2 within the s-POP based Z–N catalysts.39 The diminished intensity of the (110) peak, compared to that of MgCl2, likely signifies a reduction in the size of the MgCl2 crystallites within the catalyst.40–42
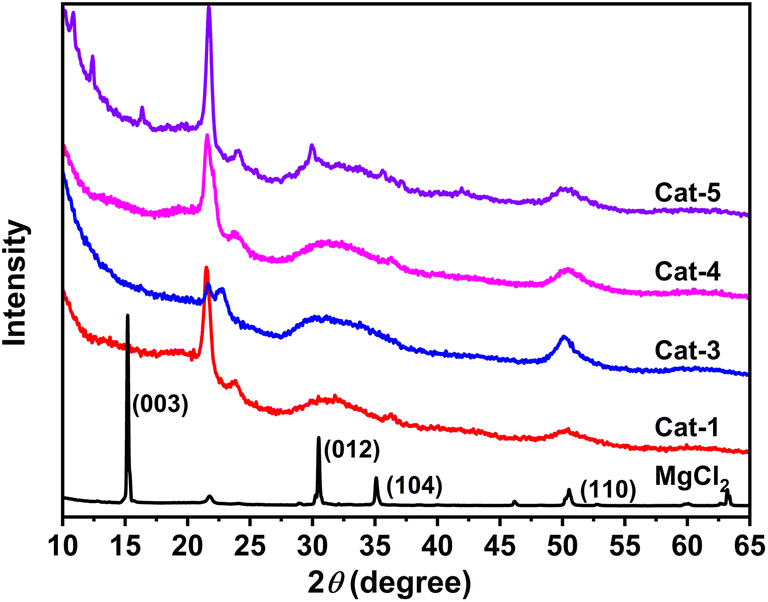 | ||
| Fig. 3 Powder X-ray diffraction patterns of s-POP based catalysts and MgCl2. The peak around 2θ = 21.7° is mainly due to the crystal of the polyethylene protection film. | ||
3.5 Effect of polymerization conditions on the polyethylene
Multiple factors could affect the polymerization activity and the molecular weight of the obtained UHMWPE. The effect of temperature, [Al]:[Ti] molar ratio, hydrogen concentration and ethylene pressure on catalyst activity and the Mν of the polyethylene was investigated. For example, while polymerization temperature positively affects activity, it adversely affects polymer molecular weight. As shown in Table 4 and Fig. 4(a), Cat-1 exhibited activity below 3000 g PE per g cat per h at 55 °C, but increased to 6800 g PE per g cat per h at 80 °C, nearly 2.5 times higher than at 55 °C. Within the temperature range of 60–70 °C, polymerization activity remained relatively stable. However, the Mν of the resulting polyethylene decreased significantly from 4.83 × 106 to 1.70 × 106 g mol−1, due to an increased chain transfer rate. Alkylaluminum plays a crucial role in olefin polymerization, influencing catalyst behavior and polymer properties as transfer agent.16,43 In this study, the [Al]:[Ti] molar ratio was consistently maintained between 40 and 294 while keeping the catalyst content constant. An increase in [Al]:[Ti] molar ratio corresponded to higher catalytic activity, ranging from 2964 to 4084 g PE per g cat per h. Moreover, no maximum activity was observed within the studied [Al]:[Ti] range, possibly because both the Ti3+ and Ti2+ are active species for ethylene polymerization.44 However, due to chain transfer to TEAL, the viscosity average molecular weight decreased to 2.05 × 106 g mol−1 at a [Al]:[Ti] ratio of 294 (Table 5).
| Entry | PE no. | Temp (°C) | Activity (g PE per g cat per h) | Mν × 10−4 (g mol−1) |
|---|---|---|---|---|
| a Polymerization conditions: Cat-1 = 50–60 mg; pressure = 0.6 MPa, TEAL = 10 mL, time = 60 min. | ||||
| 1 | PE-1 | 55 | 2570 | 483 |
| 2 | PE-2 | 60 | 4500 | 342 |
| 3 | PE-3 | 65 | 4740 | 317 |
| 4 | PE-4 | 70 | 4966 | 235 |
| 5 | PE-5 | 80 | 6800 | 170 |
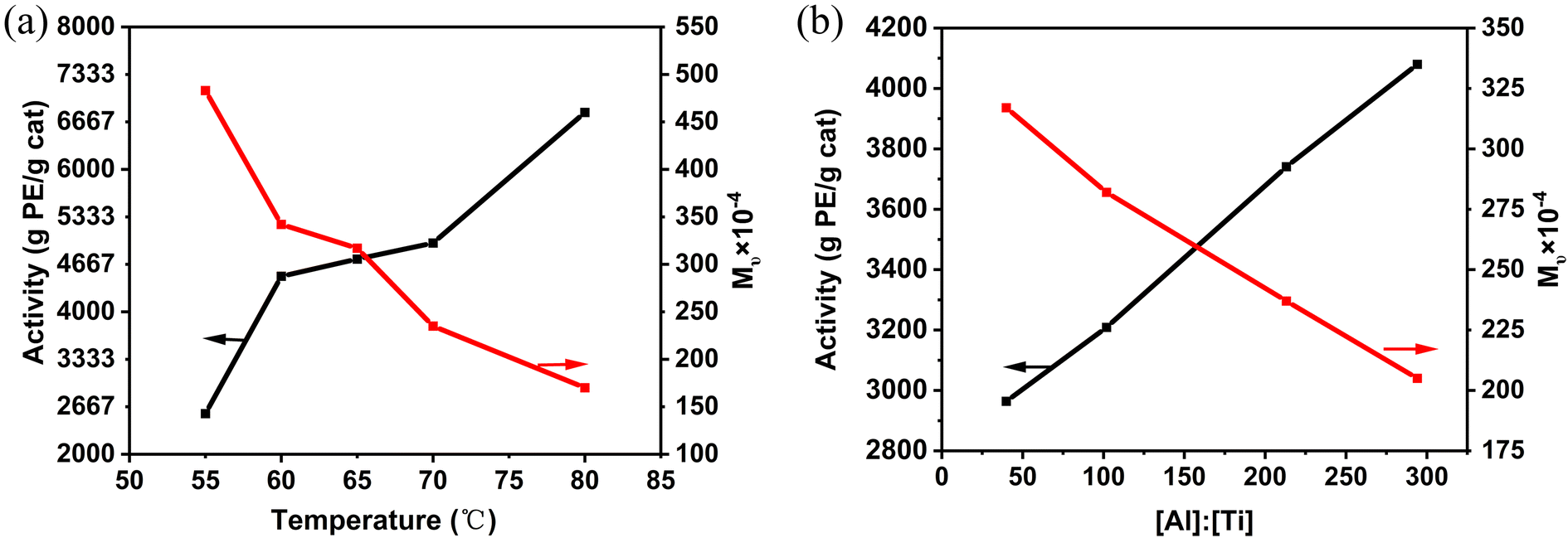 | ||
| Fig. 4 (a) Effect of temperature on polymerization results; (b) effect of [Al]:[Ti] molar ratio on polymerization results. | ||
:[Ti] molar ratio on polymerization activity of Cat-1 and Mν of the UHMWPE obtaineda
| Entry | PE no. | [Al]:[Ti] |
Activity (g PE per g cat per h) | Mν × 10−4 (g mol−1) |
|---|---|---|---|---|
| a Polymerization conditions: Cat-1 = 50–60 mg; temperature = 70 °C; pressure = 0.6 MPa; time = 60 min. | ||||
| 6 | PE-6 | 40 | 2964 | 317 |
| 7 | PE-7 | 102 | 3209 | 282 |
| 8 | PE-8 | 213 | 3741 | 237 |
| 9 | PE-9 | 294 | 4080 | 205 |
Hydrogen serves as the principal chain transfer agent in the polymerization of all olefins with heterogeneous Ti-based catalysts.45 As shown in Table 6, the catalyst activity decreased with increasing hydrogen concentration, completely opposite to propylene polymerization.46,47 Compared to polymerization without hydrogen, the catalyst activity decreased by 2.5 fold when [H2]:[C2H4] = 0.50. Numerous hypotheses have been proposed to explain the reduction in ethylene polymerization activity in the presence of hydrogen, but the mechanism still requires further elucidation.48,49 Sukulova et al.50 suggested that this reduction is mainly caused by the decrease in the calculated propagation rate constant. Due to the chain transfer reaction of hydrogen, the molecular weight of the prepared polyethylene sharply decreases. In entry 10, a small amount of hydrogen ([H2]:[C2H4] = 0.17) reduced the Mν to 44.5 × 104 g mol−1, nearly 20% of the polyethylene (2.35 × 106 g mol−1) obtained without hydrogen, and the polyethylene's Mν was only 9.68 × 104 g mol−1 at higher hydrogen concentration ([H2]:[C2H4] = 0.83). It can be concluded that the s-POP-supported ethylene polymerization catalyst exhibit high sensitivity to hydrogen response.
| Entry | PE no. | [C2H4] pressure (MPa) | [H2]:[C2H4] molar ratio |
Activity (g PE per g cat per h) | Mν × 10−4 (g mol−1) |
|---|---|---|---|---|---|
| a Polymerization conditions: Cat-1 = 50–60 mg; temperature = 70 °C; TEAL = 10 mL; time = 60 min. | |||||
| 10 | PE-10 | 0.6 | 0.17 | 3672 | 44.5 |
| 11 | PE-11 | 0.6 | 0.50 | 2054 | 13.3 |
| 12 | PE-12 | 0.6 | 0.83 | 1068 | 9.68 |
| 13 | PE-13 | 1.0 | 0 | 15146 |
313 |
Ethylene polymerization without hydrogen at high pressure was conducted in entry 13. As shown in Table 6, the catalytic activity was two times higher than that of Cat-1 under the pressure of 0.6 MPa. However, in contrast, the molecular weight did not change significantly.
4. Conclusion
In conclusion, this study demonstrated the effectiveness of using s-POP supported Ziegler–Natta catalysts for ethylene polymerization. These s-POP-based catalysts successfully produced UHMWPE, with Mν reaching up to 6.83 × 106 g mol−1, particularly with Cat-3 using DIBP as the internal donor. The effects of polymerization temperature, [Al]:[Ti] molar ratio, and ethylene pressure on the polymerization process were thoroughly explored, providing valuable insights for optimizing catalyst performance and polymer characteristics. The s-POP-supported Ziegler–Natta catalysts with the collaborative influence of internal electron donors and supports on the active centers. These functionalized POPs offer a new strategy and a straightforward approach for producing UHMWPE with tailored molecular weights.
Data availability
All data supporting the findings of this study are included within the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author acknowledges the financial support of PetroChina Company Limited through Projects 2020B-2621 and 22-LH-03-01.References
- M. C. Sobieraj and C. M. Rimnac, J. Mech. Behav. Biomed. Mater., 2009, 2, 433–443, DOI:10.1016/j.jmbbm.2008.12.006.
- P. Bracco, A. Bellare, A. Bistolfi and S. Affatato, Materials, 2007, 10, 791, DOI:10.3390/ma10070791.
- J. M. Kelly, J. Macromol. Sci., Part C: Polym. Rev., 2002, 42, 355–371, DOI:10.1081/MC-120006452.
- Z. X. Liu and H. C. Zhang, J. Phys.: Conf. Ser., 2022, 2229, 012006, DOI:10.1088/1742-6596/2229/1/012006.
- D. L. P. Macuvele, J. Nones, J. V. Matsinhe, M. M. Lima, C. Soares, M. A. Fiori and H. G. Riella, Mater. Sci. Eng., C, 2017, 76, 1248–1262, DOI:10.1016/j.msec.2017.02.070.
- K. Patel, S. H. Chikkali and S. Sivaram, Prog. Polym. Sci., 2020, 109, 101290, DOI:10.1016/j.progpolymsci.2020.101290.
- F. Zhang, Y. Mu, J. H. Wang, Z. Shi, W. M. Bu, S. R. Hu, Y. T. Zhang and S. H. Feng, Polyhedron, 2000, 19, 1941–1947, DOI:10.1016/S0277-5387(00)00484-8.
- K. A. O. Starzewski, B. S. Xin, N. Steinhauser, J. Schweer and J. Benet-Buchholz, Angew. Chem., Int. Ed., 2006, 45, 1799–1803, DOI:10.1002/anie.200504173.
- B. Choi, J. Lee, S. Lee, J.-H. Ko, K.-S. Lee, J. Oh, J. Han, Y.-H. Kim, I. S. Choi and S. Park, Macromol. Rapid Commun., 2013, 34, 533–538, DOI:10.1002/marc.201200768.
- S. Matsui, M. Mitani, J. Saito, Y. Tohi, H. Makio, N. Matsukawa, Y. Takagi, K. Tsuru, M. Nitabaru, T. Nakano, H. Tanaka, N. Kashiwa and T. Fujita, J. Am. Chem. Soc., 2001, 123, 6847–6856, DOI:10.1021/ja0032780.
- M. Mitani, J. Mohri, Y. Yoshida, J. Saito, S. Ishii, K. Tsuru, S. Matsui, R. Furuyama, T. Nakano, H. Tanaka, S. Kojoh, T. Matsugi, N. Kashiwa and T. Fujita, J. Am. Chem. Soc., 2002, 124, 3327–3336, DOI:10.1021/ja0117581.
- Y. Bando, P. Chammingkwan, M. Terano and T. Taniike, Macromol. Chem. Phys., 2018, 219, 1800011, DOI:10.1002/macp.201800011.
- H.-X. Zhang, Y.-J. Shin, D.-H. Lee and K.-B. Yoon, Polym. Bull., 2011, 66, 627–635, DOI:10.1007/s00289-010-0299-5.
- Q. H. Tran, M. Brookhart and O. Daugulis, J. Am. Chem. Soc., 2020, 142, 7198–7206, DOI:10.1021/jacs.0c02045.
- Z. Chen, M. Mesgar, P. S. White, O. Daugulis and M. Brookhart, ACS Catal., 2015, 5, 631–636, DOI:10.1021/cs501948d.
- S. Padmanabhan, K. R. Sarma and S. Sharma, Ind. Eng. Chem. Res., 2009, 48, 4866–4871, DOI:10.1021/ie802000n.
- Z. G. Zeng, Y. Chen, X. M. Zhu and L. Yu, Chin. Chem. Lett., 2023, 34, 107728, DOI:10.1016/j.cclet.2022.08.008.
- W. Li, F. Wang, Y. C. Shi and L. Yu, Chin. Chem. Lett., 2023, 34, 107505, DOI:10.1016/j.cclet.2022.05.019.
- F. H. Meng, G. Q. Yu and B. T. Huang, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 37–46, DOI:10.1002/(SICI)1099-0518(19990101)37:13.0.CO;2-O.
- J. M. Zhou, N. H. Li, N. Y. Bu, D. T. Lynch and S. E. Wanke, J. Appl. Polym. Sci., 2003, 90, 1319–1330, DOI:10.1002/app.12773.
- A. G. M. Barrett and Y. R. de Miguel, Tetrahedron, 2002, 58, 3785–3792, DOI:10.1016/S0040-4020(02)00325-3.
- A. S. Shearer, Y. R. de Miguel, E. A. Minich, D. Pochan and C. Jenny, Inorg. Chem. Commun., 2007, 10, 262–264, DOI:10.1016/j.inoche.2006.10.020.
- S. S. Liu, F. H. Meng, G. Q. Yu and B. T. Huang, J. Appl. Polym. Sci., 1999, 71, 2253–2258, DOI:10.1002/(SICI)1097-4628(19990328)71:13<2253::AID-APP16>3.0.CO;2-E.
- B. Heurtefeu, C. Bouilhac, É. Cloutet, D. Taton, A. Deffieux and H. Cramail, Prog. Polym. Sci., 2011, 36, 89–126, DOI:10.1016/j.progpolymsci.2010.09.002.
- X. Wang, G. Q. Li, P. L. He, D. W. Chen and W. Q. Kang, Ann. Chem. Sci. Res., 2023, 3, 000572, DOI:10.31031/ACSR.2023.03.000572.
- X. Wang, W. Kang, L. Gao, G. Li, X. Chen and Y. Guo, Nanomaterials, 2020, 11, 60, DOI:10.3390/nano11010060.
- W. Q. Kang, S. Chen, X. Wang, G. Q. Li, X. Y. Han and M. F. Da, Catalysts, 2022, 12, 270, DOI:10.3390/catal12030270.
- X. Wang, W. Q. Kang, G. Q. Li, P. S. Zhang, H. Q. Jia and D. J. Gao, J. Mater. Sci., 2021, 56, 19253–19266, DOI:10.1007/s10853-021-06518-5.
- X. Wang, W. Q. Kang, C. L. Zhang, G. Q. Li, P. S. Zhang and Y. Q. Li, Microporous Mesoporous Mater., 2022, 343, 112151, DOI:10.1016/j.micromeso.2022.112151.
- X. Wang, D. Wu, X. M. Mu, W. Q. Kang, G. Q. Li, A. P. Huang and Y. Xie, Polymers, 2023, 15, 555, DOI:10.3390/polym15030555.
- M. M. De Camargo Forte, F. V. Da Cunha and J. H. Z. Dos Santos, J. Mol. Catal. A: Chem., 2001, 175, 91–103, DOI:10.1016/S1381-1169(01)00172-8.
- R. Jamjah, G. H. Zohuri, M. Javaheri, M. Nekoomanesh, S. Ahmadjo and A. Farhadi, Macromol. Symp., 2008, 274, 148–153, DOI:10.1002/masy.200851420.
- B. Zhang, L. T. Zhang, Z. S. Fu and Z. Q. Fan, Catal. Commun., 2015, 69, 147–149, DOI:10.1016/j.catcom.2015.06.011.
- Z. M. Sun, F. Y. Liu, X. Y. Yang, X. P. Huang, M. M. Zhang, G. M. Bian, Y. L. Qi, X. L. Yang and W. Q. Zhang, New J. Chem., 2020, 44, 9546–9556, 10.1039/D0NJ01357A.
- F. J. Liu, W. P. Kong, C. Z. Qi, L. F. Zhu and F. S. Xiao, ACS Catal., 2012, 2, 565–572, DOI:10.1021/cs200613p.
- F. Huang, M. Humayun, G. Li, T. T. Fan, W. L. Wang, Y. L. Cao, A. Nikiforov, C. D. Wang and J. Wang, Rare Met., 2024, 43, 3161–3172, DOI:10.1007/s12598-024-02688-8.
- A. A. Da Silva Filho, M. D. C. Martins Alves and J. H. Z. Dos Santos, J. Appl. Polym. Sci., 2008, 109, 1675–1683, DOI:10.1002/app.28310.
- H. Mori, K. Hasebe and M. Terano, J. Mol. Catal. A: Chem., 1999, 140, 165–172, DOI:10.1016/S1381-1169(98)00225-8.
- S. Pirinen and T. T. Pakkanen, J. Mol. Catal. A: Chem., 2015, 398, 177–183, DOI:10.1016/j.molcata.2014.12.005.
- A. Shams, M. Mehdizadeh, H. Teimoury, M. Emami, S. A. Mirmohammadi, S. Sadjadi, E. Bardají, A. Poater and N. Bahri-Laleh, J. Ind. Eng. Chem., 2022, 116, 359–370, DOI:10.1016/j.jiec.2022.09.026.
- T. Wada, G. Takasao, A. Piovano, M. D'Amore, A. Thakur, P. Chammingkwan, P. C. Bruzzese, M. Terano, B. Civalleri, S. Bordiga, E. Groppo and T. Taniike, J. Catal., 2020, 385, 76–86, DOI:10.1016/j.jcat.2020.03.002.
- T. Wada, A. Thakur, P. Chammingkwan, M. Terano, T. Taniike, A. Piovano and E. Groppo, Catalysts, 2020, 10, 1089, DOI:10.3390/catal10091089.
- M. Masoori, M. Nekoomanesh, S. Posada-Pérez, R. Rashedi and N. Bahri-Laleh, J. Polym. Res., 2022, 29, 197, DOI:10.1007/s10965-022-03050-1.
- J. J. A. Dusseault and C. C. Hsu, J. Appl. Polym. Sci., 1993, 50, 431–447, DOI:10.1002/app.1993.070500307.
- Y. V. Kissin, R. I. Mink and T. E. Nowlin, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4255–4272, DOI:10.1002/(SICI)1099-0518(19991201)37:23<4255::AID-POLA2>3.0.CO;2-H.
- Y. V. Kissin, L. A. Rishina and E. I. Vizen, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1899–1911, DOI:10.1002/pola.10273.
- S. Ahmadjo, R. Jamjah, G. H. Zohuri, S. Damavandi, M. N. Haghighi and M. Javaheri, Iran, Polym. J., 2007, 16, 31–37 CAS.
- B. M. Grieveson, Makromol. Chem., 1965, 84, 93–107, DOI:10.1002/macp.1965.020840108.
- G. A. Mortimer, J. Polym. Sci., Part A: Polym. Chem., 1966, 4, 881–900, DOI:10.1002/pol.1966.150040414.
- V. V. Sukulova, A. A. Barabanov, T. B. Mikenas, M. A. Matsko and V. A. Zakharov, Mol. Catal., 2018, 445, 299–306, DOI:10.1016/j.mcat.2017.11.017.
| This journal is © The Royal Society of Chemistry 2024 |