
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a highly stable Mo–Fe bimetallic metal–organic framework and its catalytic activity for the selective oxidation of sulfides under mild conditions†
Xin Li *a,
Mohan Zua,
Linzhuo Yana,
Qiang Zhao*a and
Liu Liu*b
*a,
Mohan Zua,
Linzhuo Yana,
Qiang Zhao*a and
Liu Liu*b
aCollege of Chemistry and Pharmacy Engineering, Nanyang Normal University, Nanyang, 473061, P. R. China
bNanyang Branch, Henan Provincial Tobacco Company, Nanyang, 473061, P. R. China
First published on 28th November 2024
Abstract
A novel Mo–Fe bimetallic metal–organic framework [Fe2(4,4′-bipy)Mo2O8]·2H2O (1) was synthesized and characterized by X-ray single crystal diffraction, infrared spectroscopy (IR), thermogravimetric analysis (TGA), ultraviolet-visible spectroscopy (UV-vis), and its magnetic properties were studied. The compound demonstrates insolubility in most solvents and exhibits significant chemical stability across a wide pH range. Typically, exceptional selectivity and efficient conversion of sulfides to sulfoxides using 1 with H2O2 have been verified under ambient conditions.
1. Introduction
Metal–organic frameworks (MOFs) have emerged as a prominent class of materials, attracting increasing interest in recent years due to their inherent advantages such as diverse inorganic metal ions and organic linkers, tunable porosity, multi-channel design, and versatile functionality.1,2 Over the past few decades, these materials have found extensive applications in adsorptive separation, biomedicine, catalysis, and various other fields.3,4 In the recent exploration, it has been revealed that MOFs containing two different metals (named as bimetallic MOFs) offer a wider range of applications than those containing only one metal, thus garnering extensive attention from chemists.5–7 However, most of these are prepared by replacing the original metal moieties or oxometalates/metal clusters encased in the MOF pores, such as ZIF-67(Zn/Co), ZIF-8(Cu/Zn), CoZn(1,2,4-triazole)3Cl, Zn/Cu-BTC, MIL-53(Fe), Fe/M-MOF-91, Fe/M-MOF-71, Fe/M-MOF-51, and Fe/M-MOF-31 (M = Mn, Co, Ni).8–14 This often results in challenges with matching the metal/metal cluster sizes. Only a limited number of bimetallic MOFs have been synthesized, in which secondary metals or metal clusters act as linkers, enabling the formation of distinctive architectures, such as V–M-MOF (M = Ni, Co), [Ni(bix)2]{V4O11}, [Co(bib)]{V2O6} and [Ni(en)(bib)]{V2O6}·2H2O, where bix = 1,4-bis(imidazol-1-ylmethyl)benzene, bib = 1,4-bis(1H-imidazoly-1-yl) benzene, en = ethylenediamine,15,16 and {[In2Tm2(BDCP)2(μ2-OH)2(H2O)2]·4DMF·3H2O}n (NUC-56)17. Hence, the self-assembly of bimetallic MOFs incorporating secondary metals or metal clusters as linkers presents a particularly formidable challenge.Organic sulfoxides, which are crucial active intermediates, have found extensive applications in biology, medicine, and the polymer industry.18,19 Sulfoxidation serves as a common and direct approach for their synthesis. Key considerations in this process encompass the reaction kinetics, cost-effective oxidants, and catalysts.20 However, the use of metal and iodine-containing oxidants in industrial settings presents a challenge to sustainable development as companies strive for profit maximization.21 Therefore, it is imperative to explore environmentally friendly oxidants such as molecular oxygen, hydrogen peroxide, or organic hydrogen peroxide along with efficient recyclable catalysts to address this issue. Hydrogen peroxide has been extensively studied due to its ability to activate various transition metal catalysts.22 While various types of catalysts, including metal oxides, organic–inorganic hybrid compounds or composites, and some organic molecules, have been employed for this purpose, their practical application is hindered by their harsh reaction condition requirements, low specific surface area, limited availability of active sites and poor selectivity.23,24 Hence, there is strong interest in developing a catalyst with a strictly controlled ecological footprint during reactions while providing more accessible catalytic sites and decent performance under mild conditions.
Transition metal-based catalytic systems have emerged as potential catalysts in the oxidation of sulfides to corresponding sulfoxides due to their high reactivity and selectivity. Among these, Mo- and Fe-based catalysts have shown promising results in promoting the desired oxidation reactions.25–29 Additionally, combining the advantages of the MOFs mentioned above and those included in our previous reports,30–32 a novel bimetallic MOF [Fe2(4,4′-bipy)Mo2O8]·2H2O (1) has been prepared. The compound features unusual {Fe4} short chains connected by MoO42− and 4,4′-bipy. As anticipated, it exhibits outstanding heterogeneous catalytic performance in sulfoxidation, utilizing H2O2 at ambient temperature with exceptional selectivity and favorable conversions.
2. Experimental
2.1 Reagent and apparatus
The raw materials (NH4)6Mo7O24·4H2O and 4,4′-bpy for preparing compound 1 and sulfoether derivatives were purchased from Aladdin and J&K Scientific Ltd., and used without further purification. The raw material FeCl3·6H2O was purchased from Tianjin Wind Boat Chemical Reagent Technology Co. Ltd., and used without further purification. The recording of the FTIR spectra data within the range of 4500–400 cm−1 was accomplished by using a FT-IR spectrophotometer with KBr pellets. Thermogravimetric analysis for 1 was measured on a Shimadzu DTG-60H instrument under a nitrogen atmosphere and temperature range of 25–1000 °C. X-ray photoelectron spectroscopy (XPS) measurements were recorded using a Thermo Scientific Escalab 250 spectrometer with an Al Kα X-ray source (1486.6 eV). X-ray powder diffraction (XRPD) patterns were obtained by a Philips X'Pert Pro diffractometer with λ = 1.54178 Å and data collected within 5–60° (2-theta). Magnetic measurements were recorded by a Quantum Design MPMS XL-5 SQUID magnetometer with the temperature range from 1.8 to 300 K. Solid UV-vis absorption spectra were monitored by a Shimadzu UV-540 spectrophotometer from 200 to 2000 nm. The electrochemical behavior of 1-CPE (carbon paste modified electrode), consisting of compound 1 and graphite in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, was investigated in detail at 0.5 mol per L Na2SO4 to 0.1 mol per L H2SO4 solution (pH value 2.2) by cyclic voltammogram (CV) technique with a three-electrode system, in which 1-CPE was used as the working electrode, Ag/AgCl functioned as a reference electrode and Pt wire acted as the counter electrode. The bond valence sum (BVS) calculation was based on
:2 ratio, was investigated in detail at 0.5 mol per L Na2SO4 to 0.1 mol per L H2SO4 solution (pH value 2.2) by cyclic voltammogram (CV) technique with a three-electrode system, in which 1-CPE was used as the working electrode, Ag/AgCl functioned as a reference electrode and Pt wire acted as the counter electrode. The bond valence sum (BVS) calculation was based on 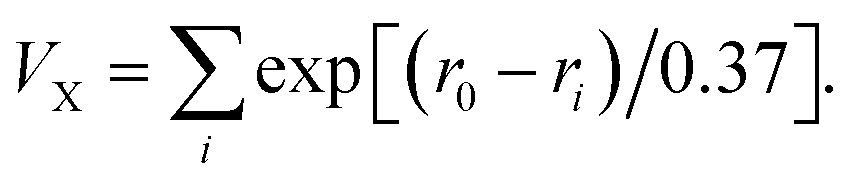 33
33
2.2 Preparation of the compound
A mixture of (NH4)6Mo7O24·4H2O (0.185 g, 0.15 mmol), FeCl3·6H2O (0.162 g, 0.6 mmol), NH4VO3 (0.116 g, 1 mmol), 4,4′-bipy (0.036 g, 0.25 mmol) and 2 mL N2H4·HCl (0.1 mol L−1) in 20 mL deionized water was stirred at room temperature for 20 min. The pH value was changed to ∼1.8 by adding HCl (2 mol L−1) and subsequently stirred for another 20 min. The mixture was then transferred to a stainless steel reactor and heated at 150 °C. Numerous dark brown rod-like-shaped crystals were isolated 4 days later, and washed thoroughly with distilled water. Yield, 65%, based on 4,4′-bipy. Elemental analysis (%) calcd: C 18.65; H 2.50; N 4.35; found: C 18.96; H 2.62; N 4.28. IR (KBr, cm−1): 3466(s), 3233(m), 3101(m), 2061(w), 1639(m), 1609(s), 1537(w), 1493(m), 1414(m), 1226(m), 1069(m), 936(s), 881(s), 862(s), 812(s), 744(s), 636(s), 458(w).2.3 X-ray structure determination
A perfect single crystal of the compound was prepared by choosing from the reactor, and subsequently installed in a capillary glass tube. Suitable diffraction data were recorded on a Bruker D8 VENTURE detector with graphite monochromatized Mo–Kα radiation (λ = 0.71073 Å) under room temperature. The direct method and difference Fourier synthesis method were used to analyze the crystal structure by SHELXTL-2017 programs and Olex2 software.34,35 Crystal data and structure refinement for 1 are listed in Table 1. Selected bond lengths and angles for 1 are listed in Table S1.† Crystallographic data for the structure have been deposited to the Cambridge Crystallographic Data Centre with CCDC 2377471.| Compound 1 | |||
|---|---|---|---|
| Empirical formula | C20N2H24Mo4Fe4O20 | γ (°) | 103.745(2) |
| Formula weight | 1247.59 | V (Å3) | 870.3(2) |
| Crystal system | Triclinic | Z | 1 |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Dc (g cm−3) | 2.380 |
| a (Å) | 7.0109(12) | GOF on F2 | 1.060 |
| b (Å) | 9.4488(15) | R1, wR2 [I > 2σ(I)] | 0.0444, 0.1471 |
| c (Å) | 14.183(2) | R1, wR2 [all data] | 0.0483, 0.1549 |
| α (°) | 106.746(2) | Rint | 0.0273 |
| β (°) | 90.909(2) | ||
2.4 General procedure for the oxidation of sulfides
The reaction mixture of sulfides, hydrogen peroxide, compound 1 and solution (CH3OH, CH3CN, etc.) was put into a glass reaction tube with a 10 mL capacity, and stirred under a water bath at room temperature (25 °C). After the reaction was completed, the product yield was detected with gas chromatography (GC) (Agilent 7890B equipped with a DB-FFAP capillary column, 30 m × 0.320 mm × 0.25 m) and nuclear magnetic resonance spectroscopy (1H-NMR) (Bruker AVANCE 400, 0.5 mL deuterochloroform (CDCl3) was used to fully dissolve the sample). The recyclability experiment was carried out under the optimum conditions. The catalyst was retrieved by centrifugation or filtration, washed with water and ethanol, and dried under vacuum for 10 h. Subsequently, the dry sample was used for the next run.3. Results and discussion
3.1 Crystal structures
The X-ray crystallographic analysis reveals that 1 crystallizes in the triclinic space group P, and features uncommon MOFs with MoO42− and 4,4′-bipy as linkers. The unsymmetric unit has two crystallographically independent Fe(II) ions, two MoO42− units, one 4,4′-bipy ligand and two free H2O molecules (Fig. 1a and S1†). As clearly shown in Fig. 1b, four Fe ions from two groups Fe1 and Fe2 are connected by corner or edge share, and resulted in a tetra-nucleate iron {Fe4} short chain. Such shaped {Fe4} short chains and four Mo1 tetrahedron are oriented in the alternating corner-sharing fashion to generate a 1D ladder type chain running parallel along the a-axis (Fig. 1c). Then, in the ab plane, the 1D ladder type chain links to adjacent two 1D chains by the Mo2 tetrahedron, resulting in a 2D layer (Fig. 1d). An analogous layer of Mo–Fe bimetallic oxides was reported in α-FeMoO4, β-FeMoO4, FeMoO4 II, and some organic/inorganic hybrid Mo–Fe bimetallic oxides,36–38 but such {Fe4} short chains linked by MoO42− have never been seen before. Each 2D layer covalently links through 4,4′-bipy ligands into three-dimensional MOFs (Fig. 1e), in which this special structure is similar to [Cu(4,4′-bipy)0.5MoO4]·1.5H2O prepared by Zubieta.39 The MOF contains two kinds of channels viewed along the a- and b-axes (Fig. S2†). The dimensions of the window along axis a measure approximately 10.8269–14.3983 Å, as depicted in the figure, while the dimensions along axis b also range from 10.8269–14.3983 Å. Both of them are straight and filled with the lattice water molecules.
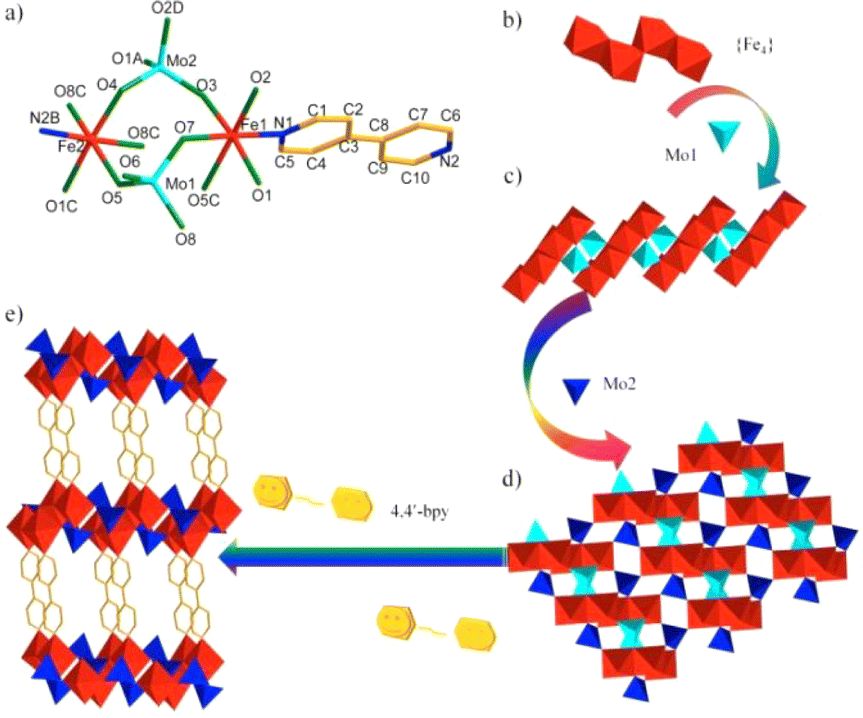 | ||
| Fig. 1 (a) Coordination environment diagram of 1. (All the H atoms and dissociative H2O are omitted). (b) {Fe4} short chain; (c) 1D ladder type chain along the a-axis; (d) 2D layer of 1 viewed along the ab plane; (e) the 3D framework in 1. Polyhedron in red octahedron for [FeNO5], shallow blue tetrahedron for Mo1 and dark blue for Mo2. | ||
In the framework, Fe1 and Fe2 adopted a six-coordinated environment showing a distorted octahedron geometry with a NO5 set (O1, O2, O3, O5C, O7, N1 for Fe1 and O1C, O4, O5, O8A, O8C N2B for Fe2), of which those surrounded atoms are from the MoO42− units and 4,4′-bipy ligand. The Fe(II)–O bond lengths vary from 1.984(6) Å to 2.117(7) Å and the Fe(II)–N bond distances are 2.170(7) Å and 2.123(7) Å, respectively. Two MoO42− units adopt different linking fashions, μ3-O5, μ3-O8 and μ2-O7 from Mo1, and μ3-O1, μ2-O2, μ2-O3 and μ2-O4 from Mo2, as shown in Fig. S3.† The distances of Mo–Ob (Ob, bridging oxygen) are in the range of 1.732(6)–1.872(7) Å and O–Mo–O angles of 106.9(9) to 114.0(9). Moreover, one Mo1Ot (Ot, terminal oxygen) distance is 1.699 Å. These M–O (M = Mo, Fe) bond distances in compound 1 are similar with the other corresponding bond distances; for example, the range from 1.729 Å to 1.813 Å for Mo–Ob, and 2.093(3) Å to 2.205(2) Å for Fe–O in KH3FeII2MoVI2O10.37 Based on BVS, the values 2.2 and 2.3 of Fe indicate the +2 oxidation state, while the values 5.9 and 6.2 of Mo indicate the +6 oxidation state, as estimated. Furthermore, supplementary XPS measurements confirm the presence of Fe in its +2 oxidation state. As shown in Fig. S4,† the deconvolution of the Fe 2p peak revealed three signals at around 710.7 eV, 724.1 eV and 719.3 eV, corresponding to Fe 2p3/2, Fe 2p3/1 and Fe satellite, respectively, indicating that the oxidation state of Fe is +2.40,41
3.2 IR, and TGA
To further demonstrate the structure, IR and TGA techniques were utilized. As shown in Fig. S5,† the characteristic absorption peaks observed in the IR spectrum of compound 1 closely resemble those of the organic ligands 4,4′-bipy, iron, and molybdenum. The broad absorption peak near 3466 cm−1 is attributed to the H–O–H stretching vibration of solvent water molecules, while the v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and m(CN) stretching vibrations of the ligand pyridine occur between 1639 and 1069 cm−1. The multiple strong absorptions between 936 and 744 cm−1 may be attributed to metal–oxygen stretching vibrations, thus providing comprehensive evidence for the diverse bridging modes existing between molybdenum and oxygen within the structural framework.42,43 Furthermore, thermal analysis results also support the structural analysis. As depicted in Fig. S6,† an initial weight loss of 5.2% occurs at 80–180 °C, closely aligning with the water molecule content within the structure (5.4%); subsequently, a second weight loss is observed at 370–440 °C, accounting for a reduction of 25.7%, which mirrors closely with the organic ligand 4,4′-bipy content within the structure (25.04%). A small amount of error may be due to systematic errors in testing or variations in the sample water content.
C) and m(CN) stretching vibrations of the ligand pyridine occur between 1639 and 1069 cm−1. The multiple strong absorptions between 936 and 744 cm−1 may be attributed to metal–oxygen stretching vibrations, thus providing comprehensive evidence for the diverse bridging modes existing between molybdenum and oxygen within the structural framework.42,43 Furthermore, thermal analysis results also support the structural analysis. As depicted in Fig. S6,† an initial weight loss of 5.2% occurs at 80–180 °C, closely aligning with the water molecule content within the structure (5.4%); subsequently, a second weight loss is observed at 370–440 °C, accounting for a reduction of 25.7%, which mirrors closely with the organic ligand 4,4′-bipy content within the structure (25.04%). A small amount of error may be due to systematic errors in testing or variations in the sample water content.
3.3 UV-vis-NIR spectra
As is known, the diffuse reflectance spectroscopy not only was used to evaluate the spectral performance of the compound but also used to determine the symmetry and coordination environment.44 In the UV-vis absorption spectrum, there are three strong bands at 254, 390, and 500 nm and a wide absorption region from 900 to 1700 nm (Fig. 2a). According to previous reports, the absorption band of the Mo(VI) ion with d0 electronic configuration only occurs in the UV-visible range of 200–400 nm. This is because of the ligand metal charge transfer process (LMCT): O2− → Mo. Much more have concluded that the absorption bands around 260 nm are characteristic of tetrahedral molybdate [MoO4], and that the characteristic absorption peaks of octahedral species [MoO6] are higher at 300 nm or above. So, the 254 nm absorption peak should be attributed to tetrahedral molybdate [MoO4].42 The absorption bands at 390 nm, 500 nm and 900–1700 nm agree with the charge-transfer transitions between the iron(II) ion and ligand, and the d–d transition of Fe(II) ions by 1A1g → 1T1g and 5T2g → 5Eg modality.45–47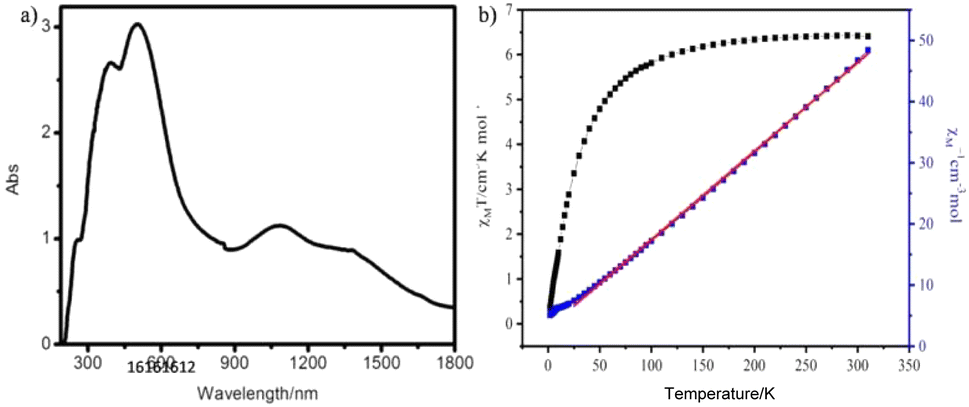 | ||
| Fig. 2 (a) The UV-vis absorption spectrum of compound 1 in the solid state at room temperature. (b) The χmT and χm−1 vs. T plots for 1. The red line is generated by the Curie–Weiss law. | ||
3.4 Magnetic properties
Iron-containing compounds have attracted increasing attention owing to their charming magnetism exchange interactions. Thus, the magnetic property of 1 has been measured through varying temperature from 2 to 300 K. The data of χmT and χm−1 vs. T for 1 have been plotted in Fig. 2b. In the χmT vs. T plot of 1, the initial χmT value decreased gradually from 6.42 cm3 K mol−1 at 300 K to 5.81 cm3 K mol−1 at 100 K, then rapidly reaches the minimum of 0.39 cm3 K mol−1 at 2 K. The χmT value of 6.40 cm3 K mol−1 at 298 K is slightly higher than that of the two isolated spin-only Fe(II) cations.48–50 The χm−1 vs. T plot was fitted based on the Curie–Weiss law [χ = C/(T − θ)] from 35 to 300 K, and resulted in a Curie constant of C = 6.88 cm3 K mol−1 and Weiss constant of θ = −2.83 K. The above dates and fitting result have shown antiferromagnetic coupling for 1. On the other hand, the χmT value of 1 is close to that of two isolated spin-only Fe(II) at room temperature, which is in accordance with the BVS calculation on Fe.3.5 PXRD and stability study
The phase purity and stability of the sample were verified by comparing the XRPD pattern with the single crystal simulation data. Firstly, the peak positions of the experimental pattern in fresh 1 (untreated by other solvents) are in good agreement with the simulation, indicating the good phase purity of the products (Fig. S7†). Next, the compound was immersed in various solutions, such as CH3OH, CH3CN, DMF, and n-hexane, for two days, and vacuum-dried after repeated washing with distilled water. All collected samples were characterized by XRPD, showing nearly unchanged characteristic peaks. This indicated that 1 shows a remarkable stability in various solutions (Fig. S8†). Finally, the stability of 1 in H2O with different pH values has also been studied, and the results show that the compound maintains a stable structure in a wide range of pH values from 3.6 to 13.0 (Fig. S9†). The slight difference in intensity for all data would be due to the preferred orientation of the samples. From the above, the compound shows good stability in various solutions and a wide pH range from 3.6 to 13.0.3.6 Electrochemical properties
The electrochemical behavior of 1-CPE was investigated in H2SO4–Na2SO4 aqueous solution by cyclic-voltammetry (CV) method in the potential range from 0.16 to 0.43 V. As shown in Fig. 3, the CV result shows that 1-CPE undergoes three couples of reversible redox peaks with half-wave potentials of 0.29, 0.15 and 0.03 V (vs. Ag/AgCl). These reversible redox potentials should be attributed to the reversible transfer processes of MoV/MoVI, which is similar to other molybdenum-containing compounds; for example, 0.27, 0.15 and −0.03 V for [SiMo12O40]4−; and 0.31, 0.16 and −0.07 V for [PMo12O40],51 respectively. With the increased scan rate, the peak currents of both oxidation and reduction processes increase gradually with increasing scan rates, and their cathodic (ipc) and anodic (ipa) peak current intensities are proportional to the scan rates (Fig. S10†), indicating that the redox process of 1-CPE is a surface-controlled process.52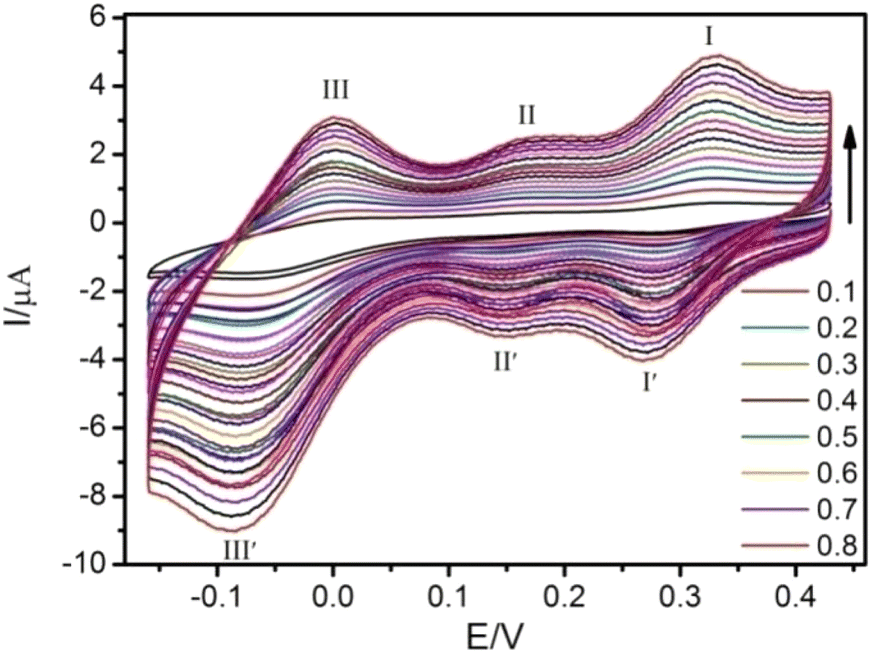 | ||
| Fig. 3 Cyclic voltammogram of 1-CPE in Na2SO4–H2SO4 aqueous solution with different scan rates from 0.1 to 0.8 V s−1. | ||
3.7 Chemoselective oxidation of sulfides to sulfoxides
The performance of sulfides oxidized to their corresponding sulfoxides for 1 was explored in a model reaction with methyl phenyl sulfide as the substrate under room temperature for 1 h. Varying reaction parameters have been taken into account to find the optimized conditions, like the catalyst loading, the effect of solvent, the effect of the oxygen source and concentration, reaction temperature and time, as summarized in Table 2. Firstly, the model reaction was carried out in different solvents such as acetonitrile (CH3CN), methanol (CH3OH), dichloromethane (CH2Cl2), n-hexane (C6H14), and p-dioxane (C4H8O2). From Table 2, entries 1–5, the nonpolar solvents of n-hexane and p-dioxane showed very low conversions; in contrast, polar solvents distinctly improved the conversions, which is in line with the relevant literature.53 Among the polar solvents, methanol would be a suitable candidate with a high conversion (96%) and selectivity (98%) of sulfoxide. This result is comparable with most Mo-based heterogeneous catalysts (Table S2†). To further identify the reaction products, the 1H-NMR of the reaction mixtures has been analyzed (Fig. S11†). It can be seen that the predominant product observed is sulfoxide, with only trace amounts of weak sulfone detected, wherein the sulfoxide content reaches an impressive 98%, aligning perfectly with the GC results. Thus, we spontaneously examined the effect of the catalyst concentration by changing the volume of solvent (CH3OH) from 1 to 3 mL, and the result shows that the conversion and selectivity are almost unchanged (Table 2, entries 1, 11 and 12). The amount of catalyst was also studied, and the result shows that the conversion was improved with the dosage increasing from 2.5 to 10 mg. Furthermore, the conversion speed of methyl phenyl sulfide was increased at dosages above 10 mg (Table 2, entries 1 and 6–10). Moreover, when we replaced 30% H2O2 with 70% tert-butyl hydroperoxide (TBHP), the conversion and selectivity were decreased to 29% and 99% under the same conditions (Table 2, entry 13), respectively. Based on the above results, the optimum catalytic condition of 1 for this model reaction would be summarized as follows: 1 mmol sulfides, 1 mmol H2O2, 10 mg (1.5 mol%) compound 1 dispersed in CH3OH solution for 1 h under room temperature. The reaction kinetics for this model reaction was carried out under this optimum condition. As shown in Fig. S12,† the reaction for oxidizing thioanisole to the corresponding sulfoxide exhibits pseudo-first-order kinetics with rate constant k = 0.054 min−1 (R2 = 0.986).54
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (mg) | Vol. of solv. (mL) | Solv. | Conv.b (%) | Sel.c/% |
| a Reaction conditions: 1 mmol of methyl phenyl sulfide, 1 mmol of H2O2 under stirring for 1 h at room temperature.b Conversion of sulfides determined by GC with naphthalene as an internal standard.c Selectivity towards methyl phenyl sulfoxide.d 1 mmol of methyl phenyl sulfide, 1 mmol of TBHP under stirring for 1 h. | |||||
| 1 | 10 | 2 | CH3OH | 97 | 98 |
| 2 | 10 | 2 | CH3CN | 79 | 87 |
| 3 | 10 | 2 | C6H14 | 12 | 86 |
| 4 | 10 | 2 | CH2Cl2 | 7 | 95 |
| 5 | 10 | 2 | C4H8O2 | 4 | 24 |
| 6 | 2.5 | 2 | CH3OH | 87 | 92 |
| 7 | 5 | 2 | CH3OH | 96 | 98 |
| 8 | 7.5 | 2 | CH3OH | 96 | 98 |
| 9 | 12.5 | 2 | CH3OH | 98 | 99 |
| 10 | 15 | 2 | CH3OH | 98 | 97 |
| 11 | 10 | 1 | CH3OH | 96 | 95 |
| 12 | 10 | 3 | CH3OH | 94 | 97 |
| 13d | 10 | 2 | CH3OH | 29 | 99 |
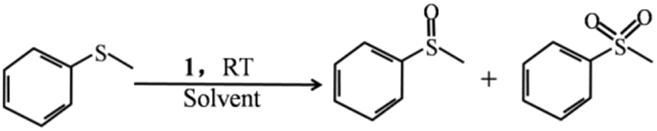
To confirm the heterogeneous process of this catalytic reaction, a hot filtration experiment was carried out. As shown in Fig. 4a, the conversion was nearly stopped when the catalyst was moved out from the reaction system. In addition, no color change was observed when the filtrate joined to the KSCN solution, showing that no Fe3+ ion was detected. Both of them are strong evidence that the catalytic reaction is an authentic heterogeneous process. Because of its heterogeneous nature, the catalyst can be reused easily by simple centrifugal separation. In Fig. 4b, no significant decrease in the activity of 1 was observed after 4 runs of continuous use, showing that the catalyst has a good recyclable capacity. Beyond that, the structural stability of the compound was checked by IR spectra and PXRD patterns. Compared with the fresh samples that did not take part in the catalytic experiment, those parameters are almost unchanged after 4 cycles (Fig. S13 and S14†) showing the excellent structural stability for the compound.
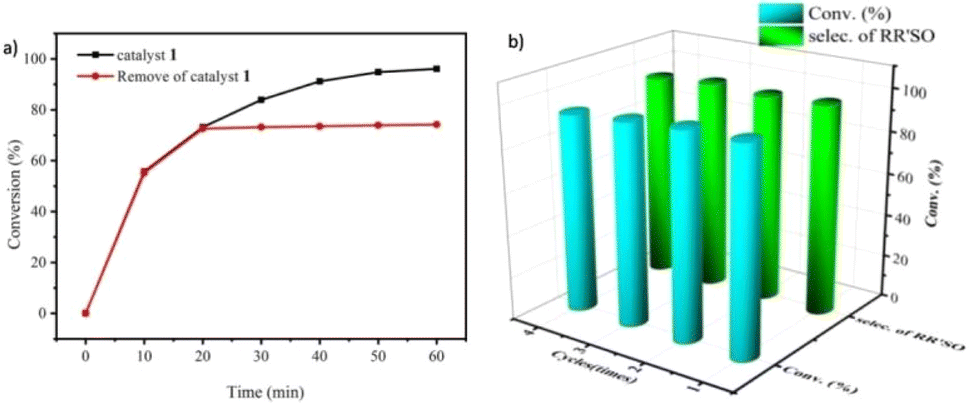 | ||
| Fig. 4 (a) Hot filtration experiment using catalyst 1. (b) Recycling of catalyst 1 for the catalytic oxidation reactions. | ||
Except for the heterogeneous nature, good activity, recyclable capacity and excellent stability, general applicability is another important indicator to estimate the performance of catalysts. Thus, a variety of sulfides acting as substrates were tested under the optimum catalytic condition, and the results are given in Table 3. Similar to thioanisole, the catalytic substrate of some dialkyl sulfides and differently substituted phenyl methyl sulfides were rapidly oxidized in considerably good to high yield and chemoselectivity to the corresponding sulfoxides in one hour (Table 3, entries 1–6). It is worth mentioning that the catalytic activity and selectivity were less affected by the electronic character and the position of the phenyl-ring substituted thioanisoles, even when an ortho-position substituent derivate (o-bromoanisole) was used as the substrate (Table 3, entries 7–12).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Conv. (%) | Selec. (%) | Entry | Substrate | Conv. (%) | Sel./% R1R2SO |
| 1 | 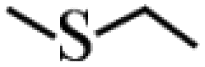 |
≥99 | 98 | 2 | 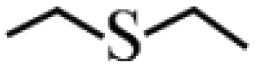 |
99 | 97 |
| 3 | 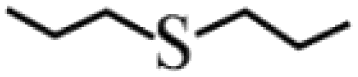 |
≥99 | 98 | 4 | 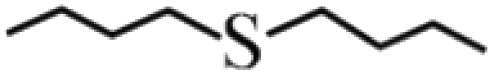 |
99 | 98 |
| 5 | 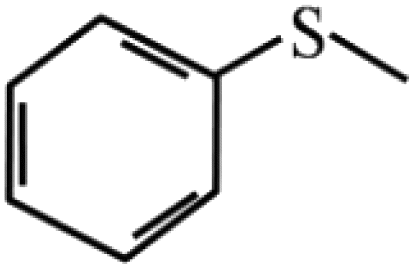 |
96 | 98 | 6 | 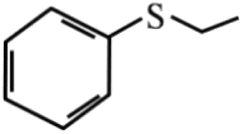 |
98 | 99 |
| 7 | 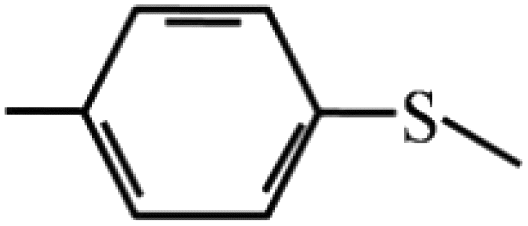 |
98 | 97 | 8 | 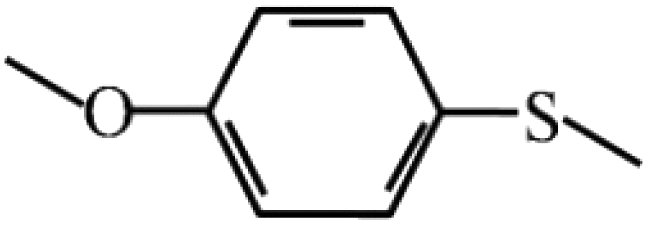 |
89 | ≥99 |
| 9 | 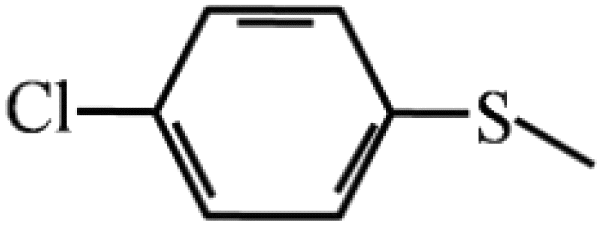 |
95 | 95 | 10 | 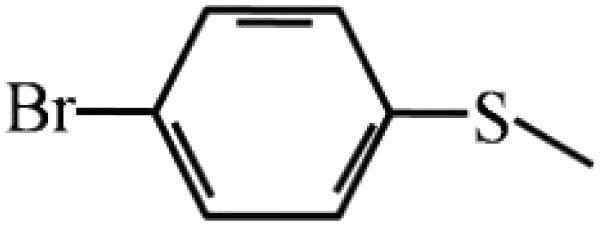 |
93 | 98 |
| 11 | 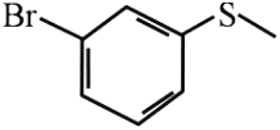 |
87 | 95 | 12 | 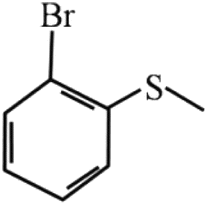 |
79 | 99 |
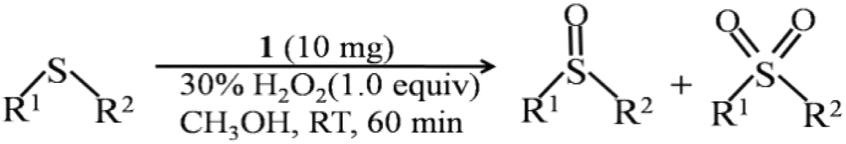
Based on the characteristics of the 3D porous structure of the catalyst and previous research,55–57 we hypothesized that the mechanism of the catalytic reaction is as follows: first, the oxidizing agent oxidizes molybdenum into the peroxido molybdenum(VI) unit; secondly, thioether is attracted to the pore through the hydrogen bonding of ligand units in the pore channel and attacks the peroxido molybdenum(VI) unit through nucleophilic action to achieve oxygen transfer, generating sulfoxide and regenerating the catalyst. The catalyst then enters the next catalytic oxidation cycle (Fig. S15†).
4. Conclusions
In conclusion, a rare Mo–Fe bimetallic MOF [Fe2(4,4′-bipy)Mo2O8]·2H2O (1) was prepared with simple hydrothermal method and characterized in detail. It has been successfully used in the selective oxidation of organic sulfides to sulfoxides under mild conditions with hydrogen peroxide. Importantly, the excellent stability and significant catalytic activity for the compound not only indicates that incorporating MoO42− into MOF as a linker is an effective strategy to prepare the Mo-based heterogeneous catalyst, but also would open a new road for the performance research of bimetallic MOFs. In future work, major efforts will be devoted to optimizing the architectures and excellent catalytic properties by adjusting the pore size, specific surfaces and components.Data availability
The data that are used to support the findings of this study are available from the corresponding author upon request.Author contributions
Xin Li: writing – original draft, investigation, funding acquisition; Mohan Zu: formal analysis, data curation; Linzhuo Yan: investigation; Qiang Zhao: writing – review & editing, funding acquisition, conceptualization, resources, project administration; Liu Liu: investigation, data curation.Conflicts of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.Acknowledgements
This work is financially supported by the Natural Science Foundation of Henan Province (242300420567), the Henan Provincial Class Postdoctoral Research Grant (19030054), the Program of Nanyang Normal University (2019ZX005, 2022PY017 and 2023PY001) and the Nanyang Normal University Laboratory Open Project (SYKF2023016).References
- H. Y. Li, X. J. Kong, S. D. Han, J. D. Pang, T. He, G. M. Wang and X. H. Bu, Chem. Soc. Rev., 2024, 53, 5626 RSC.
- G. R. Cai, P. Yan, L. L. Zhang, H. C. Zhou and H. L. Jiang, Chem. Rev., 2021, 121, 12278 CrossRef CAS PubMed.
- R. E. Sikma, K. S. Butler, D. J. Vogel, J. A. Harvey and D. F. S. Gallis, J. Am. Chem. Soc., 2024, 146, 5715 CrossRef CAS PubMed.
- F. Xing, J. W. Xu, Y. X. Zhou, P. Y. Yu, M. Zhe, Z. Xiang, X. Duan and U. Ritz, Nanoscale, 2024, 16, 4434 RSC.
- X. Y. Lu, K. Zhang, X. K. Niu, D. D. Ren, Z. Zhou, L. L. Dang, H. R. Fu, C. L. Tan, L. F. Ma and S. Q. Zang, Chem. Soc. Rev., 2024, 53, 6694 RSC.
- M. Ahmed, Inorg. Chem. Front., 2022, 9, 3003 RSC.
- S. S. Li, Y. Q. Gao, N. Li, L. Ge, X. H. Bu and P. Y. Feng, Energy Environ. Sci., 2021, 14, 1897 RSC.
- S. Daliran, A. R. Oveisi, C. W. Kung, U. Sen, A. Dhakshinamoorthy, C. H. Chuang, M. Khajeh, M. Erkartal and J. T. Hupp, Chem. Soc. Rev., 2024, 53, 6244 RSC.
- L. Y. Chen, H. F. Wang, C. X. Li and Q. Xu, Chem. Sci., 2020, 11, 5369 RSC.
- K. Zhou, B. Mousavi, Z. X. Luo, S. Phatanasri, S. Chaemchuen and F. Verpoort, J. Mater. Chem. A, 2017, 5, 952 RSC.
- X. Y. Li, S. J. Li, J. H. Liu, J. Zhang, Y. P. Ren and J. G. Zhao, RSC Adv., 2024, 14, 20780 RSC.
- Q. Sun, M. Liu, K. Y. Li, Y. T. Han, Y. Zuo, F. F. Chai, C. S. Song, G. L. Zhang and X. W. Guo, Inorg. Chem. Front., 2017, 4, 144 RSC.
- C. S. Song, G. L. Zhang and X. W. Guo, Inorg. Chem. Front., 2017, 4, 144 RSC.
- C. Shuai, C. Kong, Y. Y. Li, L. Zhang, C. J. Qi and Z. L. Mo, RSC Adv., 2024, 14, 18367 RSC.
- T. Y. Dang, R. H. Li, H. R. Tian, Q. Wang, Y. Lu and S. X. Liu, Inorg. Chem. Front., 2021, 8, 4367 RSC.
- H. R. Tian, Z. Zhang, S. M. Liu, T. Y. Dang, X. H. Li, Y. Lu and S. X. Liu, J. Mater. Chem. A, 2020, 8, 12398 RSC.
- H. X. Lv, H. T. Chen, T. P. Hu and X. T. Zhang, Inorg. Chem. Front., 2022, 9, 5788 RSC.
- J. L. Han, V. A. Soloshonok, K. D. Klika, J. Drabowicz and A. Wzorek, Chem. Soc. Rev., 2018, 47, 1307 RSC.
- S. G. Collins, O. C. M. O'Sullivan, P. G. Kelleher and A. R. Maguire, Org. Biomol. Chem., 2013, 11, 1706 RSC.
- J. Y. Wu, S. Anselmi, A. T. P. Carvalho, J. Caswell, D. J. Quinn, T. S. Moody and D. Castagnolo, Green Chem., 2024, 26, 8685 RSC.
- R. D. Chakravarthy, V. Ramkumar and D. K. Chand, Green Chem., 2014, 16, 2190 RSC.
- J. J. Boruah, S. P. Das, S. R. Ankireddy, S. R. Gogoi and N. S. Islam, Green Chem., 2013, 15, 2944 RSC.
- H. Vardhan, G. Verma, S. Ramani, A. Nafady, A. M. Al-Enizi, Y. X. Pan, Z. Y. Yang, H. Yang and S. Q. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 3070 CrossRef CAS PubMed.
- L. Fang, Q. H. Xu, Y. Y. Qi, X. X. Wu, Y. H. Fu, Q. Xiao, F. M. Zhang and W. D. Zhu, Mol. Catal., 2020, 486, 110863 CrossRef CAS.
- L. L. Zong, C. Wang, A. M. P. Moeljadi, X. Y. Ye, R. Ganguly, Y. X. Li, H. Hirao and C. H. Tan, Nat. Commun., 2016, 7, 13455 CrossRef PubMed.
- A. Lazar, W. R. Thiel and A. P. Singh, RSC Adv., 2014, 4, 14063 RSC.
- H. Keypour, M. Balali, M. M. Haghdoostb and M. Bagherzadeh, RSC Adv., 2015, 5, 53349 RSC.
- Y. M. Zhu, X. Liu, S. G. Jin, H. J. Chen, W. Lee, M. L. Liu and Y. Chen, J. Mater. Chem. A, 2019, 7, 5875 RSC.
- S. D. M. Jacques, O. Leynaud, D. Strusevich, A. M. Beale, G. Sankar, C. M. Martin and P. Barnes, Angew. Chem., Int. Ed., 2006, 118, 459 CrossRef.
- Q. Zhao, X. M. Liu, H. R. Li, Y. H. Zhang and X. H. Bu, Dalton Trans., 2016, 45, 1083 Search PubMed.
- Z. Shen, Q. Y. Li, S. K. Shi, G. Zang, W. J. Lv and Q. Zhao, J. Mol. Struct., 2024, 1295, 136661 CrossRef CAS.
- Q. Zhao, Q. Y. Li and J. Li, CrystEngComm, 2022, 24, 6751 RSC.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B, 1954, 41, 244 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- W. Maalej, S. Vilminot, G. Andr, Z. Elaoud, T. Mhiri and M. Kurmoo, Inorg. Chem., 2011, 50, 9191 CrossRef CAS PubMed.
- L. j. Zhang, Y. S. Zhou, J. L. Zuo, Z. Yu, H. K. Fun, I. Abdul and X. Z. You, Inorg. Chem. Commun., 2000, 3, 697 CrossRef CAS.
- T. Tanakaa, Y. Sunatsukia and T. Suzuki, Inorg. Chim. Acta, 2020, 502, 119373 CrossRef.
- R. S. Rarig, R. Lam, P. Y. Zavalij, J. K. Ngala, R. L. LaDuca, J. E. Greedan and J. Zubieta, Inorg. Chem., 2002, 41, 2124 CrossRef CAS PubMed.
- F. Hayati, S. Moradi, S. F. Saei, Z. Madani and S. Giannakis, J. Environ. Manage., 2022, 321, 115851 CrossRef CAS PubMed.
- Y. Oh, J. O. Hwang, E.-S. Lee, M. Yoon, V.-D. Le, Y.-H. Kim, D. H. Kim and S. O. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 25438 CrossRef CAS PubMed.
- H. I. Buvailo, V. G. Makhankova, V. N. Kokozay, I. V. Omelchenko, O. V. Shishkin, D. Matoga and J. Jeziersk, Inorg. Chim. Acta, 2016, 443, 36 CrossRef CAS.
- W. S. El-Yazeed, Y. G. El-Reash, L. A. Elatwy and A. I. Ahmed, RSC Adv., 2020, 10, 9693 RSC.
- M. Fournier, C. Louis, M. Che, P. Chquin and D. Masure, J. Catal., 1989, 119, 400 CrossRef CAS.
- L. Li, S. Y. Niu, D. Li, J. Jin, Y. X. Chi and Y. H. Xing, Inorg. Chem. Commun., 2011, 14, 993 CrossRef CAS.
- Y. M. Chiou and L. Que Jr, J. Am. Chem. Soc., 1995, 117, 3999 CrossRef CAS.
- H. Zheng, Y. Zang, Y. H. Dong, V. G. Young and L. Que, J. Am. Chem. Soc., 1999, 121, 2226 CrossRef CAS.
- S. D. M. Jacques, O. Leynaud, D. Strusevich, A. M. Beale, G. Sankar, C. M. Martin and P. Barnes, Angew. Chem., Int. Ed., 2006, 118, 459 CrossRef.
- R. J. Wei, Q. Huo, J. Tao, R. B. Huang and L. S. Zheng, Angew. Chem., Int. Ed., 2011, 50, 8940 CrossRef CAS PubMed.
- T. Tanakaa, Y. Sunatsukia and T. Suzuki, Inorg. Chim. Acta, 2020, 502, 119373 CrossRef.
- Y. T. Chang, K. C. Lin and S. M. Chen, Electrochim. Acta, 2005, 51, 450 CrossRef CAS.
- J. Q. Sha, J. Peng, Y. Q. Lan, Z. M. Su, H. J. Pang, A. X. Tian, P. P. Zhang and M. Zhu, Inorg. Chem., 2008, 47, 12 CrossRef PubMed.
- C. B. Yang, Q. Q. Jin, H. Zhang, J. Liao, J. Zhu, B. Yu and J. E. Deng, Green Chem., 2009, 11, 1401 RSC.
- R. Wan, P. P. He, Z. Liu, X. Y. Ma, P. T. Ma, V. Singh, C. Zhang, J. Y. Niu and J. P. Wang, Chem.–Eur. J., 2020, 26, 1 CrossRef.
- M. Amini, M. M. Haghdoost and M. Bagherzadeh, Coord. Chem. Rev., 2013, 257, 1093 CrossRef CAS.
- H. Keypour, M. Balali, M. M. Haghdoost and M. Bagherzadeh, RSC Adv., 2015, 5, 53349 RSC.
- P. Patel, B. Parmar, R. I. Kureshy, N. H. Khan and E. Suresh, Dalton Trans., 2018, 47, 8041 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2377471. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra06324d |
| This journal is © The Royal Society of Chemistry 2024 |