
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of 4′-C-α-aminoethoxy-2′-O-methyl-5-propynyl-uridine for antisense therapeutics†
Yujun Zhoua,
Hitotaka Satoa,
Miwa Kawadeb,
Kenji Yamagishie and
Yoshihito Ueno*abcd
aThe United Graduate School of Agriculture Science (UGSAS), Gifu University, Japan. E-mail: ueno.yoshihito.e7@f.gifu-u.ac.jp; Fax: +81-58-293-2919; Tel: +81-58-293-2919
bFaculty of Applied Biological Sciences, Gifu University, Japan
cGraduate School of Natural Sciences and Technology, Gifu University, Japan
dCenter for One Medicine Innovative Translational Research (COMIT), Tokai National Higher Education and Research System, Gifu University, 1-1 Yanagido, Gifu, 501-1193, Japan
eDepartment of Chemical Biology and Applied Chemistry, College of Engineering, Nihon University, 1 Nakagawara, Tokusada, Tamuramachi, Koriyama, Fukushima 963-8642, Japan
First published on 11th December 2024
Abstract
Owing to the increased public interest and advances in chemical modifications, the approval of antisense therapeutics, a class of mRNA-targeting DNA-based oligonucleotide therapeutics, has accelerated in recent years. It was previously reported that siRNAs with several 4′-C-α-aminoethoxy-2′-O-methyl-uridine (4AEoU) analogs could maintain moderate thermal stability similar to the native ones while showing robust nuclease stability. In this study, we further expanded the application of 4AEo modification to antisense therapeutics and achieved superior thermal stability by adding the uracil 5-propynyl modification. Antisense oligonucleotides containing 4′-C-α-aminoethoxy-2′-O-methyl-5-propynyl-uridine (4AEopU) could efficiently activate RNase H-mediated antisense in vitro in the presence of native DNA gaps. These results encourage future studies of 4AEopU-containing antisense therapeutics.
Introduction
The COVID-19 pandemic has brought public attention to mRNA vaccines that use long-stranded RNAs encoding antigens as drug molecules. As a novel and attractive modality, nucleotide-based therapeutics are considered useful in treating diseases related to viral infections and genetic mutations. Compared to long mRNA vaccines, antisense oligonucleotides (ASOs) composed of short single-stranded DNA-like oligonucleotides are representative and constitute the majority of approved oligonucleotide therapeutics.1–3 Since they were first approved by the Food and Drug Administration (FDA) in 1998, 12 types of antisense therapeutics have been approved till 2024.4–15 ASOs hybridize with their target mRNA strands in the cytoplasm or nucleus and modulate the gene expression by the RNase H-mediated mRNA-selective cleavage or steric hindrance during pre-mRNA splicing or protein translation.16–18 Because native oligonucleotides are too unstable to survive nuclease-mediated degradation and exhibit poor cellular uptake due to their negatively charged phosphate linkages, various chemically modified ASOs have been developed and utilized instead of native oligonucleotides.3,19–21 There is a special platform for RNase H-dependent ASOs called a “gapmer,” which has been employed in several approved antisense therapies.5,8,9,13,14 The antisense gapmer consists of a central DNA gap region to maintain the ability to activate the RNase H-mediated antisense mechanism and two chemically modified wing regions on either side to improve thermal and enzymatic stabilities.22 Generally, phosphorothioate (PS) linkages are utilized instead of native phosphate (PO) linkages to enhance cellular uptake and protect ASOs from enzymatic degradation.23–27In our previous studies, siRNAs, another typical oligonucleotide therapeutic consisting of double-strand RNAs, containing 4′-C-α-aminoalkyl-2′-O-methyl-modified uridine analogs showed superior nuclease resistance and tended to be more stable against nucleases with relatively longer side chains.28 Nevertheless, incorporation of the aminoalkyl side chain into C4′ led to a C2′-endo preference and the significant thermal destabilization of siRNAs. Through inserting an oxygen atom to form a 4′-C-α-aminoalkyl-2′-O-methyl (4AEo) modification, the modified siRNAs showed moderate thermal stability as the native ones and were more stable than those with a 4′-C-α-aminopropyl-2′-O-methyl (4AP) modification.29 This improvement prompted further investigations into the application of 4AEo modification in antisense therapeutics. Furthermore, C2′-O-modifications such as 2′-O-methyl (2′-OMe) and 2′-O-methoxyethyl (2′-MOE) are well known to inhibit RNase H recruitment, resulting in poor RNase H-mediated cleavage.30,31 Another study showed that gapmers containing 4AEo-modified deoxythymidine without any 2′-OMe modification in the gap region reduced the antisense activity, indicating an unfavorable effect of the aminoethoxy side chain on the RNase H mechanism.32 Therefore, we hypothesized that it would be better for the 4AEo-modified analogs to be located in the wing regions of antisense gapmers. Based on this hypothesis, the thermal stability of the 4AEo-modified analogs must be further improved to obtain sufficient target RNA binding affinity for the antisense gapmer.
In this study, we designed two novel 4AEo-modified uridine analogs: 4′-C-α-aminoethoxy-2′-O-methyl-5-methyl-uridine (4AEomU) and 4′-C-α-aminoethoxy-2′-O-methyl-5-propynyl-uridine (4AEopU) (Fig. 1). Both 5-methyl and 5-propynyl modifications have been used to stabilize the siRNAs formation.33 The same thermal stabilization was also confirmed in ASOs, and no inhibition towards RNase H was observed, as the 5-propynyl moiety was located in the major groove of the DNA/RNA duplexes.34 The synthesis and physical and biochemical evaluations of the 16-mer thymidine–cytidine(TC)-repeat DNA oligomers containing these novel uridine analogs are described. A series of 4AEo-modified antisense gapmers based on a previously well-studied 16-mer KRAS-targeting sequence was prepared and used for cellular evaluation.
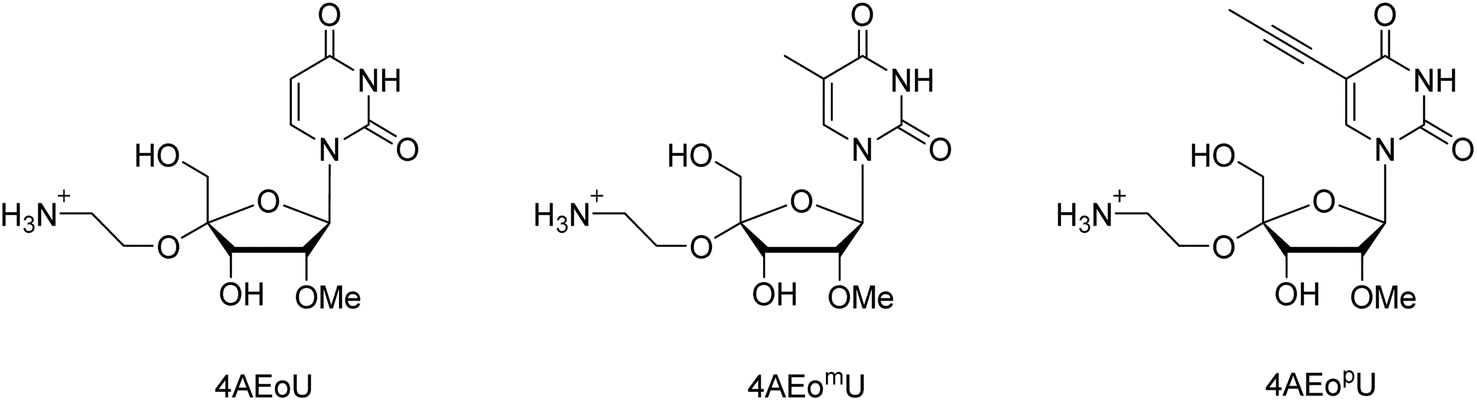 | ||
| Fig. 1 The 4AEo-modified uridine analogs. | ||
Synthesis of novel nucleoside analogs
The novel synthesis of the phosphoramidite corresponding to 4′-C-α-aminoethoxy-2′-O-methyl-5-methyl-uridine (4AEomU) is shown in Scheme 1. Commercially available 2′-O-methyl-5-methyl-uridine (1) was subjected to iodination via the Appel reaction, followed immediately by dehalogenation using 1,8-diazabicyclo[5.4.0]-7-undecene (DBU) to obtain the olefin derivative. Subsequently, 3′-O-silyl protection was performed to obtain compound 4. After epoxidation with Oxone® in acetone, the aminoethoxy side chain was incorporated into the C4′ position using N-trifluoroacetylaminoethanol in the presence of a Lewis acid. Two kinds of products were obtained from this reaction with a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. According to an earlier study in 1993, epoxidation of the 4′,5′-double bonds could yield diastereomeric epoxide derivatives, which would be attacked by the alcohol reactant and transform into the corresponding diastereomeric 4′-C-substituted ones.35 Furthermore, the 4′-C-α-aminoethoxy derivatives would be the major products in the presence of the 3′-O-silyl protecting group.29,32,36,37 Therefore, we hypothesized that the desired compound 6 was also predominantly obtained in this study. The configurations of the C4′ position in the major product were determined via nuclear Overhauser effect spectroscopy (NOESY) to confirm our hypothesis. As shown in ESI,† consistent with the previous studies, the aminoethoxy side chain was proven to be inserted at the 4′-C-α-site by the correlations observed between H1′ and H6′, H3′ and H5′, and H3′ and OH5′.29,32,38 Thus, compound 6 was the major product, as expected, and was used for the following process.
:1. According to an earlier study in 1993, epoxidation of the 4′,5′-double bonds could yield diastereomeric epoxide derivatives, which would be attacked by the alcohol reactant and transform into the corresponding diastereomeric 4′-C-substituted ones.35 Furthermore, the 4′-C-α-aminoethoxy derivatives would be the major products in the presence of the 3′-O-silyl protecting group.29,32,36,37 Therefore, we hypothesized that the desired compound 6 was also predominantly obtained in this study. The configurations of the C4′ position in the major product were determined via nuclear Overhauser effect spectroscopy (NOESY) to confirm our hypothesis. As shown in ESI,† consistent with the previous studies, the aminoethoxy side chain was proven to be inserted at the 4′-C-α-site by the correlations observed between H1′ and H6′, H3′ and H5′, and H3′ and OH5′.29,32,38 Thus, compound 6 was the major product, as expected, and was used for the following process.
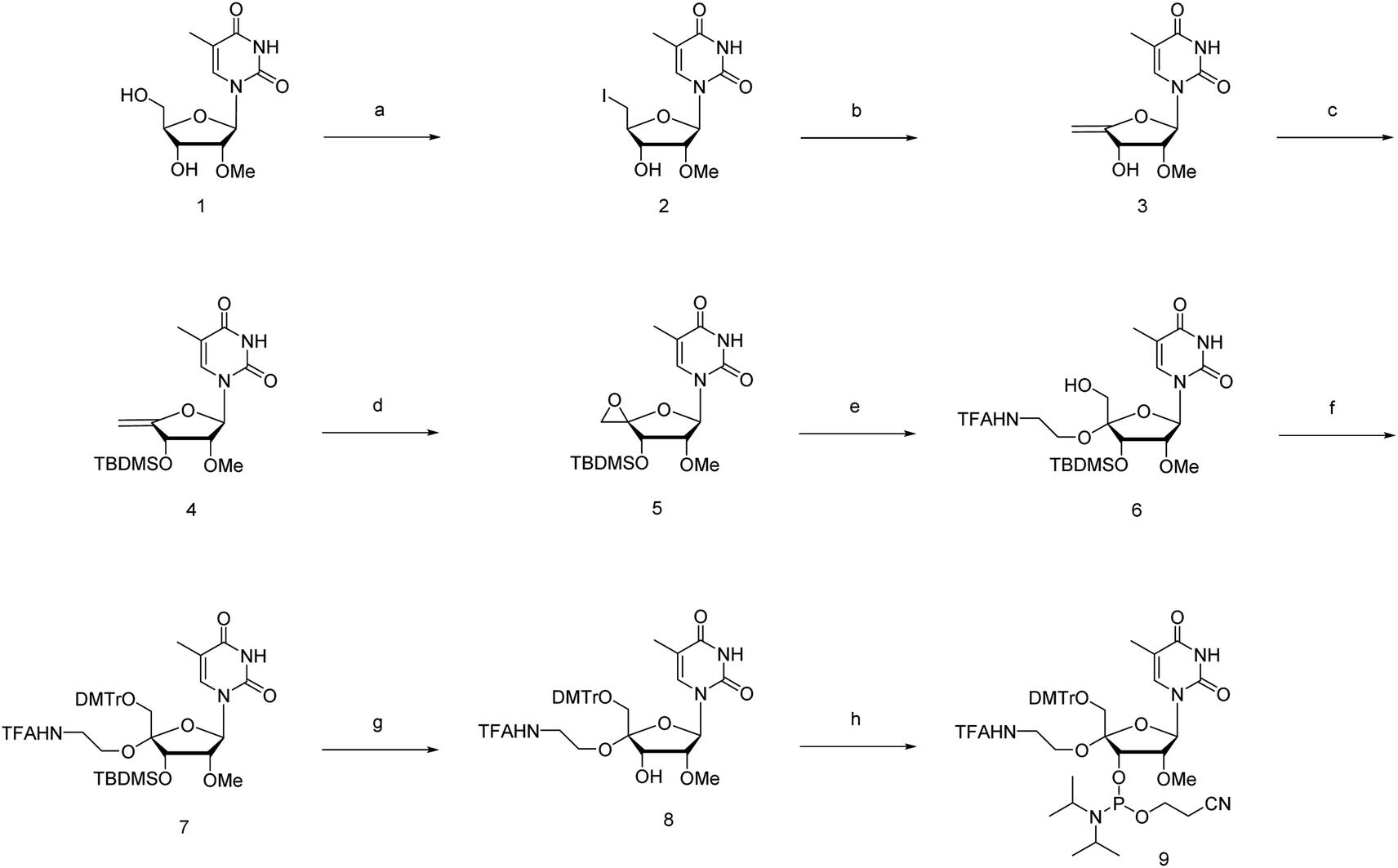 | ||
| Scheme 1 Synthesis of 4′-C-α-aminoethoxy-2′-O-methyl-5-methyl-uridine phosphoramidite 9.a aReagents and conditions: (a) PPh3, imidazole, I2, THF, r. t., 4 h; (b) DBU, 24 h; (c) TBDMSCl, imidazole, DCM, 3 h; 4: 74% in 3 steps; (d) Oxone®, sat. NaHCO3 aq., DCM, acetone, r. t., 2 h; (e) N-trifluoroacetylaminoethanol, ZnCl2, THF, −60 °C; then, r. t., 24 h; 6: 29% in 2 steps; (f) DMTrCl, DIPEA, pyridine, r. t., overnight; 7: 79%; (g) TBAF, THF, r. t., 1 h; 8: 86%; (h) CEPCl, DIPEA, THF, r. t., 2 h; 9: 55%. | ||
Next, the 5′-hydroxy moiety was protected using a 4,4′-dimethoxytrityl (DMTr) group, followed by deprotection of the 3′-O-silyl group with 0.1 M tetrabutylammonium fluoride (TBAF) solution in tetrahydrofuran (THF). Phosphoramidite 9 was synthesized using a standard phosphitylation procedure with N,N-diisopropylethylamine (DIPEA) and 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (CEPCl) as reagents.
The phosphoramidite corresponding to 4′-C-α-aminoethoxy-2′-O-methyl-5-propynyl-uridine (4AEopU) was prepared from a previously reported compound 10 (Scheme 2).29 Initially, the 3′-O-silyl protecting group was removed due to its low stability in the subsequent steps. Selective 5-iodination was accomplished using diammonium cerium(IV) nitrate (CeAN) as the catalyst, and the propynyl group was introduced by Sonogashira–Hagihara cross-coupling. Finally, after 5′-O-DMTr protection, the corresponding phosphoramidite 15 was prepared using the standard phosphitylation procedure.
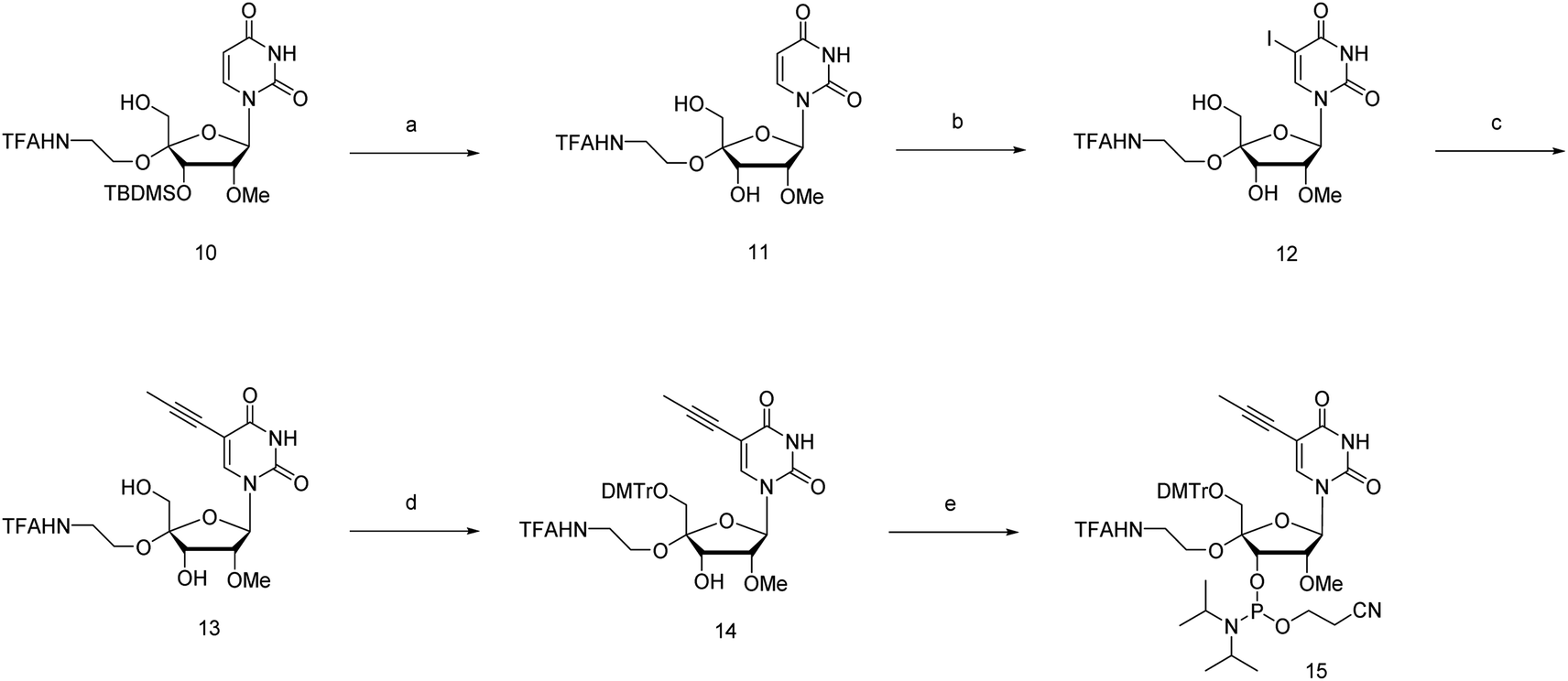 | ||
| Scheme 2 Synthesis of 4′-C-α-aminoethoxy-2′-O-methyl-5-propynyl-uridine phosphoramidite 15.a aReagents and conditions: (a) TBAF, THF, r. t., 1 h, 11: 95%; (b) CeAN, I2, 80 °C (reflux), 1 h; (c) Pd(PPh3)2Cl2, CuI, TEA, propyne, DMF, 35 °C, overnight; 13: 79% in 2 steps; (d) DMTrCl, DIPEA, pyridine, 50 °C, 24 h, 14: 73%; (e) CEPCl, DIPEA, THF, r. t., 2 h, 15: 72%. | ||
Synthesis of oligonucleotides
Novel nucleoside analogs were incorporated into the DNA-based oligonucleotides utilizing the solid-phase phosphoramidite method with a DNA/RNA synthesizer. After oligo-synthesis, to prevent the reaction of acrylonitrile with the amino groups at the end of the 4′-C-α-aminoethoxy side chain, controlled-pore glass (CPG) beads were treated with 10% dimethylamine in acetonitrile (MeCN), followed by rinsing with MeCN to selectively remove the cyanoethyl groups. The oligonucleotides were then treated with an aqueous ammonia solution for cleavage from the CPG beads and deprotection and finally purified by 20% denaturing polyacrylamide gel electrophoresis (PAGE) containing 7 M urea. The sequences of the oligonucleotides used in this study are shown in Tables 1 and S1.†| Oligo | Sequencea | Tmb (°C) | ΔTmc (°C) | ΔH°d (kcal mol−1) |
ΔS°d (cal mol−1 K−1) |
ΔG°d (kcal mol−1) |
|
|---|---|---|---|---|---|---|---|
| a T and C denote native thymidine and deoxycytidine, respectively. m and p in red denote the 4AEomU and 4AEopU analogs, respectively.b The Tm values were measured in 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl. The sequence of the complementary RNA strand was 5′-r(a agg gaa aga gaa aga)-3′, where a and g denote native adenosine and guanosine, respectively. The concentrations of the duplexes were 3 μM. All measurements were performed in triplicate, and the data are presented as average values.c ΔTm represents [Tm (modified) – Tm (unmodified)].d Thermodynamic parameters were calculated based on the slope of the 1/Tm vs. ln(CT/4) plot. All measurements were performed in triplicate, and the data are presented as average values. | |||||||
| TC-Nat. | 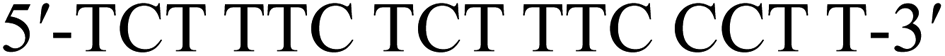 |
59.3 | — | −148.4 | −417.7 | −23.9 | (25 °C) |
| −18.9 | (37 °C) | ||||||
| TC-m | 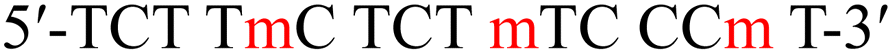 |
58.1 | −1.2 | −129.9 | −364.1 | −21.4 | (25 °C) |
| −17.1 | (37 °C) | ||||||
| TC-p | 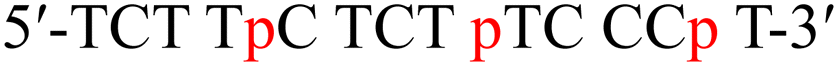 |
63.5 | +4.2 | −156.5 | −436.2 | −26.5 | (25 °C) |
| −21.3 | (37 °C) | ||||||
Thermal stability
Because it is important for RNase H-dependent antisense oligonucleotides to hybridize to their target mRNAs with excellent affinity, the 50% melting temperature (Tm) values of DNA/RNA duplexes containing modified oligonucleotides with their complementary RNAs strand should be evaluated. Here, we employed a TC-repeat modeling sequence tested in our previous studies.32,39–42 For the Tm measurement, each unmodified or 25% modified 16-mer DNA was hybridized with a 16-mer complementary RNA strand, and 3 μM of these DNA/RNA duplexes were utilized to obtain the Tm values. The results in Table 1 show that, compared with the unmodified TC-Nat., the DNA/RNA duplex containing four 4AEomU analogs resulted in a slight thermal destabilization (ΔTm −1.2 °C; −0.3 °C per modification). Conversely, incorporating 4AEopU led to a significant increase in the Tm value (ΔTm +4.2 °C; +1.4 °C per modification), suggesting thermal stabilization by the 5-propynyl moiety. Furthermore, thermodynamic parameters were calculated using 1, 3, 6, 12, 15, 30, and 60 μM of these DNA/RNA duplexes, providing more insight into the differences in Tm values. As shown in Table 1, the free energy of duplex formation (expressed as ΔG°) of the unmodified DNA/RNA duplexes was −23.9 kcal mol−1 at 25 °C and −18.9 kcal mol−1 at 37 °C. In contrast, the ΔG° values of the 4AEomU-containing duplex were slightly lower (−21.4 kcal mol−1 at 25 °C and −17.1 kcal mol−1 at 37 °C). This result was consistent with the Tm measurements. Similarly, the decrease in ΔG° for duplexes containing 4AEopU (−26.5 kcal mol−1 at 25 °C and −21.3 kcal mol−1 at 37 °C) indicated that it could more readily bind with the target RNA strand.Circular dichroism (CD) spectra showed that the 4AEo-modified DNA/RNA duplexes formed the A-type-like helix, similar to those of the unmodified ones (Fig. S3†), which is consistent with a previous study on the secondary structure of duplexes consisting of homopyrimidine DNA strands and homopurine RNA strands.43 No significant change was observed in the negative maximum around 210 nm, but either the wavelength or the intensity of the positive maximum shifted slightly due to the incorporation of the 4AEomU analogs and significantly shifted in the case of the 4AEopU analogs.
Since all nucleotides adopt the C3′-endo sugar puckering when forming a typical A-type helix, a more favorable pre-orientation and a resulting helix more akin to the A-type could be predicted if nucleoside analogs in the DNA strand prefer a C3′-endo sugar puckering. In a previous study, we investigated whether RNA/RNA duplexes containing several 4AEoU analogs could achieve thermal stability equivalent to the unmodified uridines.29 It was possible that a favorable C3′-endo sugar puckering resulted from the 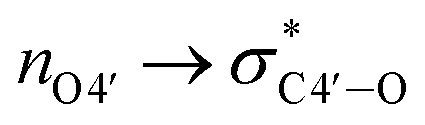 anomeric and
anomeric and 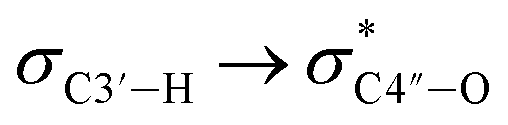 gauche effects of the 4′-C-α-aminoethoxy side chain, as well as the 2′-O-methyl modification.44 It could also explain the favorable entropy observed in the 4AEomU-containing DNA/RNA duplex (ΔS° −364.1 cal mol−1 K−1) compared with the unmodified one (ΔS° −417.7 cal mol−1 K−1). However, regardless of the favorable entropy, the 4AEomU-containing duplex exhibited slightly lower thermal stability, according to the Tm measurements. Several factors affect the thermal stability of the nucleotide duplexes, including hydrogen bonding, stacking interactions, conformational entropy, counterion condensation, and hydration.45 Since there was favorable counterion condensation due to the aminoethoxy moiety and no change in thymine, its thermal destabilization might be due to unfavorable hydration resulting from steric repulsion between the 4′-C-α-aminoethoxy side chain and the 2′-O-methyl group. Conversely, the ΔS° value for the 4AEopU-modified duplex (−436.2 kcal mol−1) was unfavorable, while the ΔH° value (−156.5 kcal mol−1) was more favorable than the other duplexes. It has been previously reported that incorporating a propynyl group at the C5 position of thymine can result in an enhanced stacking effect.46 Therefore, we hypothesized that 4AEopU analogs also prefer a C3′-endo sugar puckering similar to 4AEomU analogs, and the improved stacking interaction seems to play a major role in this case, showing more favorable enthalpy.
gauche effects of the 4′-C-α-aminoethoxy side chain, as well as the 2′-O-methyl modification.44 It could also explain the favorable entropy observed in the 4AEomU-containing DNA/RNA duplex (ΔS° −364.1 cal mol−1 K−1) compared with the unmodified one (ΔS° −417.7 cal mol−1 K−1). However, regardless of the favorable entropy, the 4AEomU-containing duplex exhibited slightly lower thermal stability, according to the Tm measurements. Several factors affect the thermal stability of the nucleotide duplexes, including hydrogen bonding, stacking interactions, conformational entropy, counterion condensation, and hydration.45 Since there was favorable counterion condensation due to the aminoethoxy moiety and no change in thymine, its thermal destabilization might be due to unfavorable hydration resulting from steric repulsion between the 4′-C-α-aminoethoxy side chain and the 2′-O-methyl group. Conversely, the ΔS° value for the 4AEopU-modified duplex (−436.2 kcal mol−1) was unfavorable, while the ΔH° value (−156.5 kcal mol−1) was more favorable than the other duplexes. It has been previously reported that incorporating a propynyl group at the C5 position of thymine can result in an enhanced stacking effect.46 Therefore, we hypothesized that 4AEopU analogs also prefer a C3′-endo sugar puckering similar to 4AEomU analogs, and the improved stacking interaction seems to play a major role in this case, showing more favorable enthalpy.
Molecular dynamics simulation
As described above, the incorporation of the 4′-C-α-aminoethoxy side chain was expected to further induce the sugar puckering into C3′-endo because of the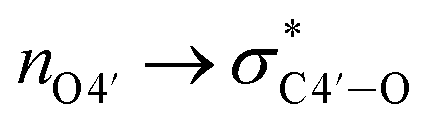 anomeric and
anomeric and 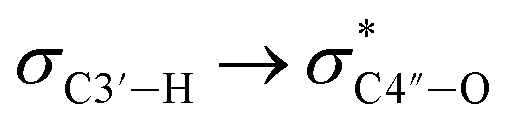 gauche effects. Based on molecular dynamics (MD) simulations, we performed in silico calculations of these 4AEo-modified nucleoside analogs to investigate the changes in nucleoside sugar puckering and molecular fluctuations caused by chemical modifications. The results of the MD simulations are presented in Table 2, Fig. S4 and S5.† Native thymidine prefers the C2′-endo sugar puckering due to the absence of the C2′-substituent. Consistent with that, the percentage of C3′-endo sugar puckering in the case of thymidine was only 5% from the MD simulation. In contrast, the incorporation of the 4AEo modification resulted in a significant shift towards the C3′-endo sugar puckering, as expected, with 76% for the 4AEomU analog and 67% for the 4AEopU analog. The 4AEopU analog was slightly less favorable for C3′-endo sugar puckering than the 4AEomU analog, suggesting steric hindrance of the 5-propynyl moiety. The root mean square deviations (RMSDs) were calculated relative to the reference structure obtained from the energy minimization calculation. The distribution range of the RMSD, shown as the standard deviation (SD), is considered an indicator of fluctuations in nucleoside molecules. As shown in Table 2, the SD of the RMSDs of thymidine, 4AEomU analog, and 4AEopU analog were ±0.15, ±0.23 and ±0.33, respectively. Therefore, the fluctuation of the 4AEomU analog was more pronounced than that of native thymidine because of the flexibility of the 4′-C-α-aminoethoxy side chain. Furthermore, the nucleoside molecule vibrated more violently when the 5-methyl group was changed to a propynyl group, indicating a negative effect of the 5-propynyl modification on the nucleoside structural stability.
gauche effects. Based on molecular dynamics (MD) simulations, we performed in silico calculations of these 4AEo-modified nucleoside analogs to investigate the changes in nucleoside sugar puckering and molecular fluctuations caused by chemical modifications. The results of the MD simulations are presented in Table 2, Fig. S4 and S5.† Native thymidine prefers the C2′-endo sugar puckering due to the absence of the C2′-substituent. Consistent with that, the percentage of C3′-endo sugar puckering in the case of thymidine was only 5% from the MD simulation. In contrast, the incorporation of the 4AEo modification resulted in a significant shift towards the C3′-endo sugar puckering, as expected, with 76% for the 4AEomU analog and 67% for the 4AEopU analog. The 4AEopU analog was slightly less favorable for C3′-endo sugar puckering than the 4AEomU analog, suggesting steric hindrance of the 5-propynyl moiety. The root mean square deviations (RMSDs) were calculated relative to the reference structure obtained from the energy minimization calculation. The distribution range of the RMSD, shown as the standard deviation (SD), is considered an indicator of fluctuations in nucleoside molecules. As shown in Table 2, the SD of the RMSDs of thymidine, 4AEomU analog, and 4AEopU analog were ±0.15, ±0.23 and ±0.33, respectively. Therefore, the fluctuation of the 4AEomU analog was more pronounced than that of native thymidine because of the flexibility of the 4′-C-α-aminoethoxy side chain. Furthermore, the nucleoside molecule vibrated more violently when the 5-methyl group was changed to a propynyl group, indicating a negative effect of the 5-propynyl modification on the nucleoside structural stability.
| Thymidine | 4AEomU | 4AEopU | |
|---|---|---|---|
| a The percentages of C3′-endo sugar puckering were calculated based on a criterion that considers the sugar puckering as C3′-endo if 0° < P < 90° and 270° < P < 360°.b The mean values and standard deviations of the RMSD (Å). The values are shown as average (±standard deviation). | |||
| C3′-endoa (%) | 5 | 76 | 67 |
| RMSDb (Å) | 0.52 (±0.15) | 1.18 (±0.23) | 1.04 (±0.33) |
Consequently, our hypothesis proved that either the 4AEomU or 4AEopU analog that prefers C3′-endo sugar puckering would result in favorable entropy for the DNA/RNA duplex formation. Notably, the 4AEopU analog was more structurally unstable than the 4AEomU analog, resulting in unfavorable entropy due to the extra energy required to strain the structure for duplex formation. However, the enhanced stacking effect of the 5-propynyl modification was strong enough to recover the structural energy loss, resulting in favorable enthalpy and higher thermal stability than the 4AEomU analog.
Base-discrimination ability
To investigate the base-discrimination ability of the 4AEo-modified analogs, the binding affinities of these modified DNAs towards one-base mismatched RNA strands were evaluated according to their Tm values. As the results show in Table 3 and Fig. S1,† the Tm values of the unmodified duplexes with a mismatched base pair with uridine (u), guanosine (g), and cytidine (c) were 49.1, 55.9, and 49.2 °C, respectively. Compared with the full-matched duplex (Tm(T:a) 59.3 °C), pyrimidine:pyrimidine mismatching decreased the Tm value significantly by approximately 10 °C, whereas a mismatched base pair consisting of thymidine and guanosine led to a decrease of approximately 3 °C. The incorporation of one 4AEomU analog yielded results similar to those of the unmodified thymidine. The Tm values were 59.3 °C for the full-matched duplex and 49.3 and 49.2 °C for the pyrimidine:pyrimidine mismatched ones. However, a highly beneficial decrease of 5 °C was observed for 4AEomU:guanosine mismatching. The same trend was observed for the 4AEopU analog. One thymidine substituted with a 4AEopU analog increased the Tm value of the full-matched duplex by 2 °C (Tm(p:a) 61.5 °C). The Tm values of the p:u and p:c mismatching were 51.0 and 51.2 °C, respectively. These Tm values were 2 °C higher than those of the unmodified and 4AEomU-containing ones, while the ΔTm values were the same as the others, approximately −10 °C. Conversely, the Tm(p:g) was 54.2 °C, which was not significantly different from Tm(m:g) (54.0 °C), but 7 °C lower than the full-matched Tm(p:a).| Tmc (°C)(ΔTmd (°C)) | ![[X with combining low line]](https://www.rsc.org/images/entities/b_char_0058_0332.gif) b b |
||||
|---|---|---|---|---|---|
| a | u | g | c | ||
a The sequence of the DNA strand: 5′-TCT TTC ![[T with combining low line]](https://www.rsc.org/images/entities/b_char_0054_0332.gif) CT TTC CCT T-3′. T and C denote native thymidine and deoxycytidine, respectively. denotes native thymidine, 4AEomU, or 4AEopU analog, as shown in the table.b The sequence of the RNA strand: 3′-aga aag ga aag gga a-5′. a, u, g, and c denote native adenosine, uridine, guanosine and cytidine, respectively. denotes a mismatched site.c The Tm values were measured in 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl. The concentrations of the duplexes were 3 μM. All measurements were performed in triplicate and the data are presented as average values.d ΔTm represents [Tm (mismatched) – Tm (full-matched)]. CT TTC CCT T-3′. T and C denote native thymidine and deoxycytidine, respectively. denotes native thymidine, 4AEomU, or 4AEopU analog, as shown in the table.b The sequence of the RNA strand: 3′-aga aag ga aag gga a-5′. a, u, g, and c denote native adenosine, uridine, guanosine and cytidine, respectively. denotes a mismatched site.c The Tm values were measured in 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl. The concentrations of the duplexes were 3 μM. All measurements were performed in triplicate and the data are presented as average values.d ΔTm represents [Tm (mismatched) – Tm (full-matched)]. |
|||||
| a |
Thymidine (T) | 59.3(−) | 49.1(−10.2) | 55.9(−3.4) | 49.2(−10.1) |
| 4AEomU (m) | 59.3(−) | 49.3(−10.0) | 54.0(−5.3) | 49.2(−10.1) | |
| 4AEopU (p) | 61.5(−) | 51.0(−10.5) | 54.2(−7.3) | 51.2(−10.3) | |
Thymidine not only hybridizes with adenosine to form the Watson–Crick base pairs but can also hybridize with other nucleobases to form the so-called “wobble” base pairs, which have been studied to be as stable as the Watson–Crick base pairs.47 While the formation of wobble base pairs is important for some biological mechanisms, such as translation, it is unsuitable for antisense therapeutics because of the associated risk of off-target effects. In this study, the thymidine:pyrimidine mismatched duplexes were severely destabilized, suggesting that no wobble base pairs were formed. However, a 2 °C increase was still observed in the case of Tm(p:u) and Tm(p:c). This indicates that thermal stabilization from improved stacking interactions by the 5-propynyl modification was still present without base pairing. Conversely, because the ΔTm value of thymidine: guanosine mismatching was −3 °C, there is no doubt that thymidine formed a wobble base pair with guanosine. The ΔTm values were also moderate in the cases of 4AEomU:guanosine and 4AEopU:guanosine mismatching, suggesting the formation of wobble base pairs. However, their Tm values were comparable (Tm(m:g) 54.0 °C vs. Tm(p:g) 54.2 °C), indicating that the formation of wobble base pairs altered the torsion of nucleobases and changed the relative position of the 5-propynyl moiety, resulting in no enhanced stacking effect. Overall, the 4′-C-α-aminoethoxy-2′-O-methyl-5-propynyl modification exhibited higher thermal stability and base discrimination ability.
Nuclease resistance
The enzymatic stability of the modified DNAs was investigated in OPTI-MEM supplemented with 3% (v/v) bovine serum (BS) containing various exonucleases and endonucleases. In this study, the 5′-fluorescein-labeled TC-Nat.-F, TC-m-F, and TC-p-F, corresponding to TC-Nat., TC-m, and TC-p, respectively, were used. Their sequences are shown in Table S1.† Samples aliquoted at each timepoint were analyzed using 20% denaturing PAGE. The resulting images were acquired using LAS-4000 (Fujifilm) and quantified using ImageJ software, as shown in Fig. 2. Unmodified TC-Nat.-F was degraded to half its amount within 15 min, with no complete strand remaining after 180 min. In contrast, more than 90% of the full-length strands of 4AEo-modified TC-m-F and TC-p-F remained even after 360 min of the BS treatment. We further tested the nuclease resistances of TC-m-F and TC-p-F under 10%, 25% and 50% BS conditions (Fig. S6†). No significant differences were observed between the 4AEo-modified DNAs. With 360 min of incubation, approximately 20% of the full-length strands of the 4AEo-modified DNAs survived in a 50% BS-containing environment, indicating robust nuclease resistance due to the 4AEo modification. Substitution of the 2′-hydroxy group with a methoxy group might avoid nuclease-mediated hydrolysis of the 3′-side phosphate diester linkage. Additionally, the aminopropyl modification at the C2′ site was shown to be highly nuclease-resistant and contributed to an electrostatic effect of the positively charged amino group occupying the metal ion binding site of exonucleases instead of a magnesium ion, as well as the steric effect of removing the phosphate group from a zinc ion.48 Altering the aminoalkyl side chain into the C4′ or C5′ site could also significantly increase stability against nucleases. Furthermore, because the variety of cleavage fragments from 4AEo-modified DNAs was reduced to 3 or 4, the 4AEo modification appeared to prevent not only the 3′-phosphate but also at least two phosphate diester bonds on the 5′-side.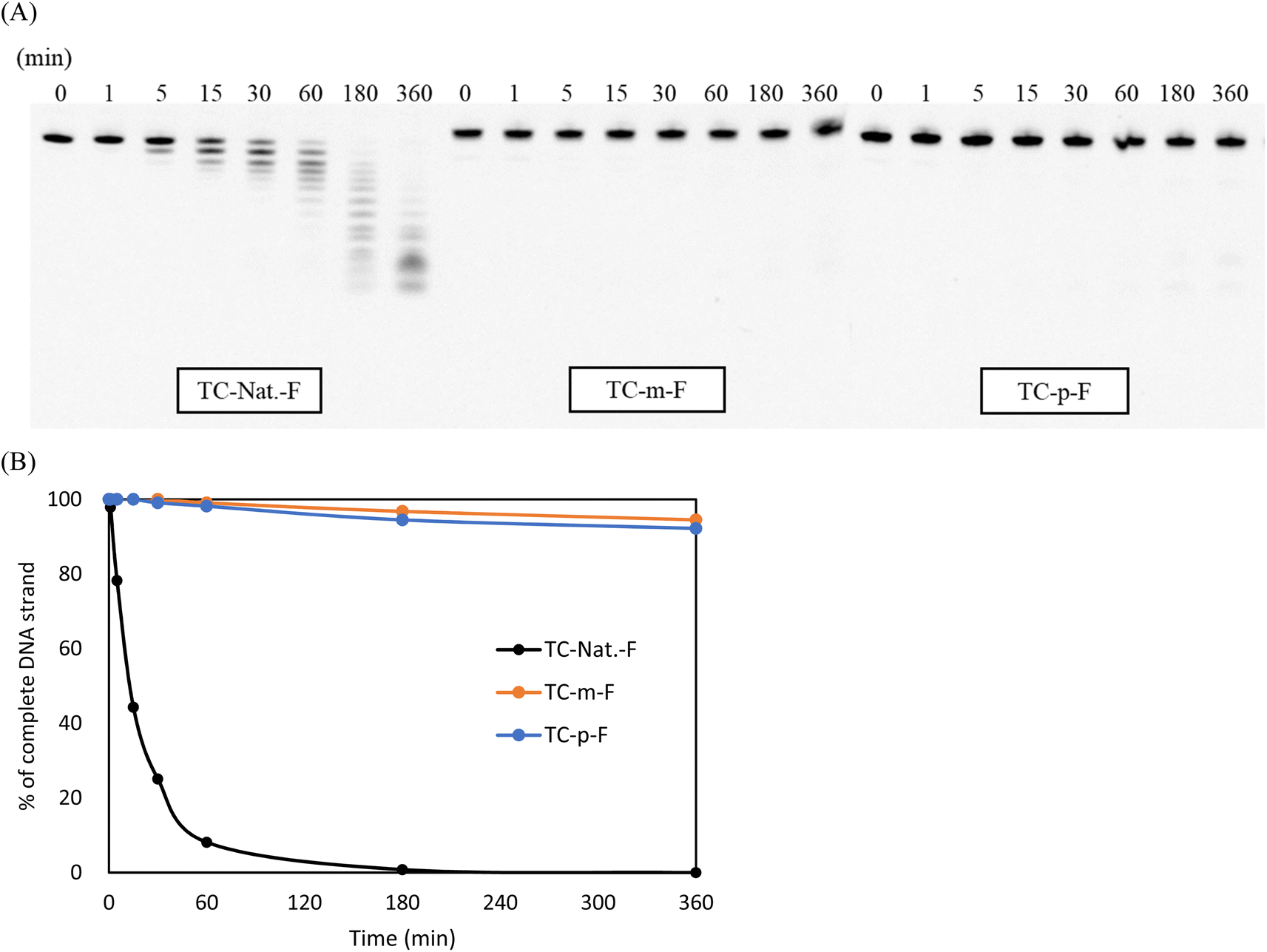 | ||
| Fig. 2 PAGE analysis of single-stranded oligomers treated with 3% bovine seruma. aThe 5′-fluorescein-labeled TC-Nat.-F, TC-m-F, and TC-p-F, corresponding to TC-Nat., TC-m, and TC-p, respectively, were incubated in OPTI-MEM containing 3% bovine serum, and samples from the reaction mixtures at various incubation time points (0, 1, 5, 15, 30, 60, 180, and 360 min) were analyzed via 20% PAGE with 7 M urea. (A) PAGE images obtained from luminescent analysis; (B) plot of the percentage of complete DNA strands remaining at each time point analyzed via ImageJ. | ||
E. coli RNase H1 activation
To confirm the ability of the modified DNAs to activate the RNase H-mediated antisense mechanism, we utilized RNase H1 purified from Escherichia coli (E. coli) and the TC-repeat modeling sequence containing several native 4-mer DNA gaps that are sufficient for recognition and cleavage by E. coli RNase H1.49 To evaluate the turnover, unmodified TC-Nat., modified TC-m, and TC-p were annealed to their complementary 5′-fluorescein-labeled RNA strand in a ratio of 1:5. Then, these duplexes treated with 3.5 U of E. coli RNase H1 at 37 °C and samples aliquoted at each time point were analyzed by 20% denaturing PAGE (Fig. 3). Consequently, the E. coli RNase H1-mediated cleavage was efficiently activated by the unmodified DNA/RNA duplex, leaving no full-length RNA strands within 5 min. However, when comparing each result at the 5 min timepoint, the cleavage rate of duplexes containing the 4AEomU analogs was slower than that of the unmodified ones and even slower in the case of 4AEopU analogs. It was reported that chemical modifications inside the minor groove of DNA/RNA duplexes, such as 2′-O-methyl modification, influence RNase H1 recognition, which was not cleaved when fully modified with the 2′-O-methyl group. In addition, metal ions are required for the E. coli RNase H1-mediated cleavage.49–51 There was concern that the 4′-C-aminoethoxy side chain would electronically and sterically interfere with RNase H1 recognition. Conversely, 5-propynyl modification, a chemical modification located in the major groove, efficiently knockdowned the target mRNA in cells.34 However, it has also been reported that the 5-propynyl moiety incorporated into siRNAs can interfere with interactions at the major groove of the RISC complex, adversely affecting RNAi activity.33 Similar disruptions might also appear in DNA/RNA duplexes and RNase H1. Therefore, considering the change in the variety of cleavage products, it seems that the binding, recognition, and migration of E. coli RNase H1 were affected by the 4AEo modification, resulting in a shift in the initial and final cleavage sites and a decrease in the cleavage rate.
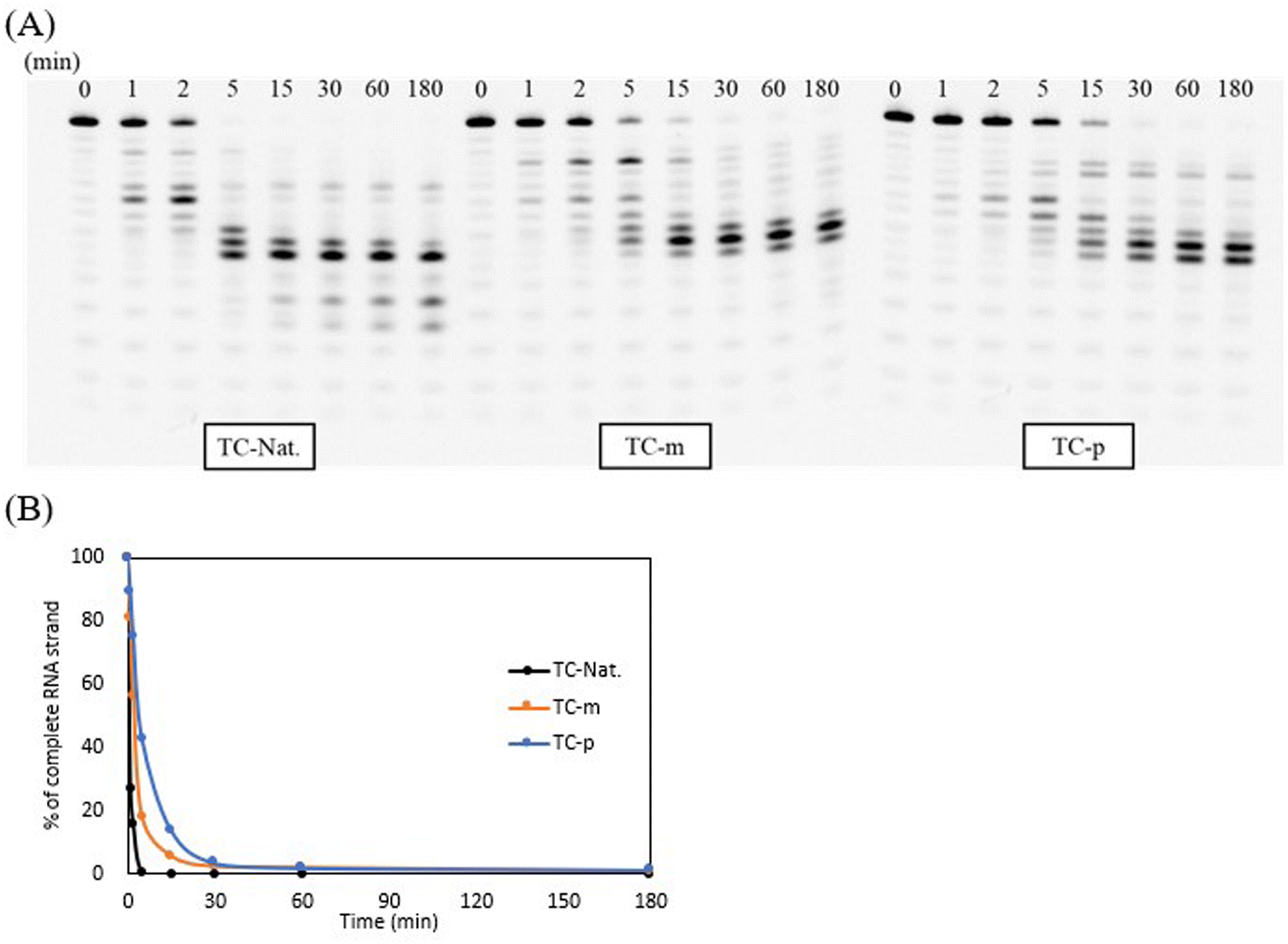 | ||
| Fig. 3 PAGE analysis of native and modified duplexes treated with E. coli RNase Ha. aThe duplexes containing TC-Nat., TC-m, and TC-p within the 5′-fluorescein-labeled complementary RNA strand were incubated in a reaction buffer with a diluted RNase H1 solution (50 units per L in water). Samples from the reaction mixtures at various incubation time points (0, 1, 2, 5, 15, 30, 60, and 180 min) were analyzed via 20% PAGE with 7 M urea. (A) PAGE images obtained from luminescent analysis; (B) plot of the percentage of complete RNA strands remaining at each time point analyzed via ImageJ. | ||
Nevertheless, the 4AEo-modified duplexes also activated E. coli RNase H1 for RNA-selective cleavage if native DNA sections were long enough for activation, indicating their potential for application as antisense gapmers.
Antisense activity in mammal cells
As aforementioned, we demonstrated that the 4′-C-α-aminoethoxy-2′-O-methyl-5-propynyl modification exhibited superior thermal and biochemical stability in the TC-repeat modeling sequence and was able to activate the RNase H-mediated RNA-selective cleavage with several DNA gaps. We further investigated its effect on antisense activity when incorporated into antisense gapmers. Here, a cancer-related gene, KRAS, was chosen as the endogenous target in NCI-H460 cells, and a 3-10-3 LNA-wing antisense gapmer served as a positive control. Based on this platform, a series of KRAS-targeting antisense gapmers (“KRASs” in short) were prepared by replacing an LNA-T in the wing region as well as a thymidine in the gap region with a 4AEomU or 4AEopU analog, leaving at least an 8-mer DNA-gap for Human RNase H activation.52Before operating the cell assays, duplexes composed of the KRASs and their complementary RNA strands were checked to determine if they were sufficiently stable under biological conditions (37 °C). As shown in Table 4, substituting an LNA-T with a 4AEomU analog caused a significant decrease in Tm values, −4.6 °C for KRAS-m-1 and −3.6 °C for KRAS-m-4, whereas substituting a native thymidine showed comparable or even higher Tm values, −0.2 °C for KRAS-m-2 and +1.3 °C for KRAS-m-3. Notably, when thymidine was replaced, the 4AEomU analog caused a slight decrease in the TC-repeat modeling sequence but an increase in KRAS-m-3, indicating the possibility of being affected by the position inside the oligonucleotide and the variety of surrounding nucleobases. It was considered that it would be better to add the 4AEo modification into the 3′-side of the gapmer rather than the 5′-side in terms of thermal stability. The same trend was also confirmed in the case of the 4AEopU analog since the Tm (ΔTm) values of KRAS-p-1, 2, 3 and 4 were 59.4 (−3.0), 63.8 (+1.4), 66.4 (+4.0) and 62.3 (−0.2) °C, respectively. Meanwhile, despite the dispersion of Tm values, each gapmer exhibited a Tm value much higher than 37 °C, suggesting that it could form stable duplexes with the target mRNA.
| No. of gapmer | Sequencea | Tmb (°C) | ΔTmc (°C) |
|---|---|---|---|
| a Underlined letters denote LNAs. m and p in red denote the 4AEomU and 4AEopU analogs, respectively. Other letters denote native DNAs. The dots denote PS linkages.b The Tm values were measured in 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl. The sequence of the complementary RNA strand was 5′-r(aaa gac ucc uaa uag c)-3′. The concentrations of the duplexes were 3 μM. All measurements were performed in triplicate, and the data are shown as average values.c ΔTm represents [Tm (KRAS-m/p-1-4) − Tm (KRAS-pos)]. | |||
| KRAS-pos | 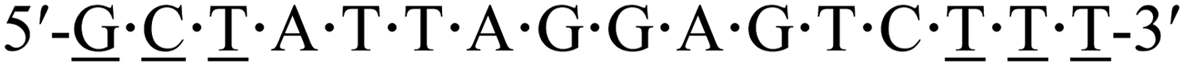 |
62.5 | — |
| KRAS-m-1 | 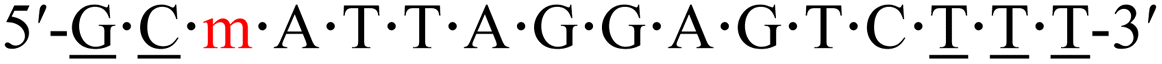 |
57.9 | −4.6 |
| KRAS-m-2 | 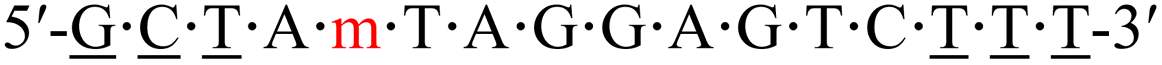 |
62.2 | −0.2 |
| KRAS-m-3 | 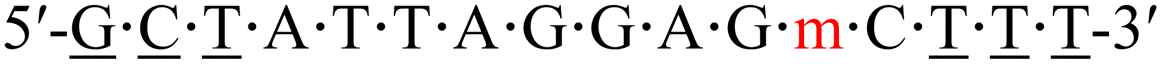 |
63.8 | +1.3 |
| KRAS-m-4 | 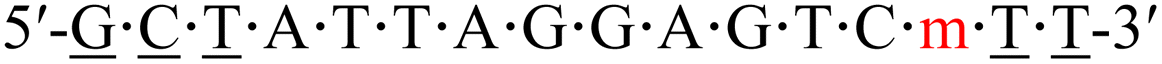 |
58.9 | −3.6 |
| KRAS-p-1 | 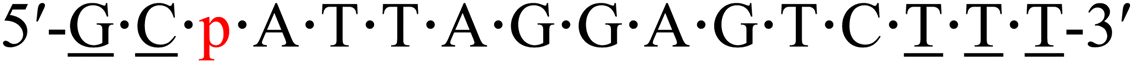 |
59.4 | −3.0 |
| KRAS-p-2 | 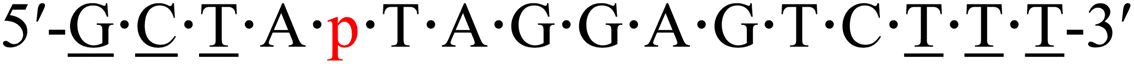 |
63.8 | +1.4 |
| KRAS-p-3 | 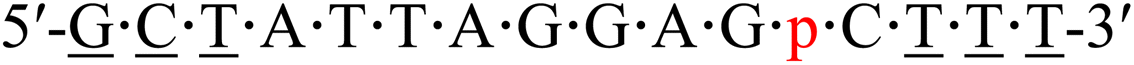 |
66.4 | +4.0 |
| KRAS-p-4 | 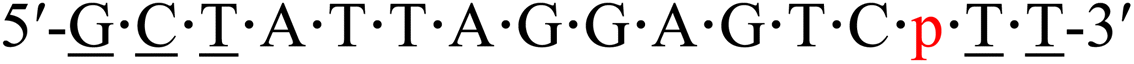 |
62.3 | −0.2 |
The intracellular antisense activity of KRASs was then evaluated in NCI-H460 cells. The cells were treated with KRASs at final concentrations of 0.5 and 5.0 μM without lipofection for 48 h at 37 °C with 5% carbon dioxide. The relative KRAS mRNA levels corresponding to each KRAS-treated group were calculated by qRT-PCR using the ΔΔCT method with beta-actin (ACTB) as a reference. A group treated only with PBS buffer was designated as the negative control, “par.” As shown in Fig. 4, the 4AEomU-containing gapmers showed significantly lower antisense activity than KRAS-pos, except for KRAS-m-1 and KRAS-m-4 at a concentration of 5.0 μM. Conversely, KRASs containing one 4AEopU analog, except for KRAS-p-2, efficiently knocked down the target mRNA without any significant difference from the corresponding positive control. Incorporating these 4AEo-modified analogs at the gap-2 position negatively influenced the intracellular antisense activity, suggesting a disadvantage to Human RNase H recruitment, which is consistent with the results for E. coli RNase H1. Nevertheless, compared with 4AEomU and 4AEopU, the combination with 5-propynyl modification obviously improved the antisense performance. As the thermal stability and antisense activity of KRAS-p-4 and its LNA counterpart were comparable, LNA might be replaced with 4AEopU to mitigate LNA-associated hepatoxicity.26,53
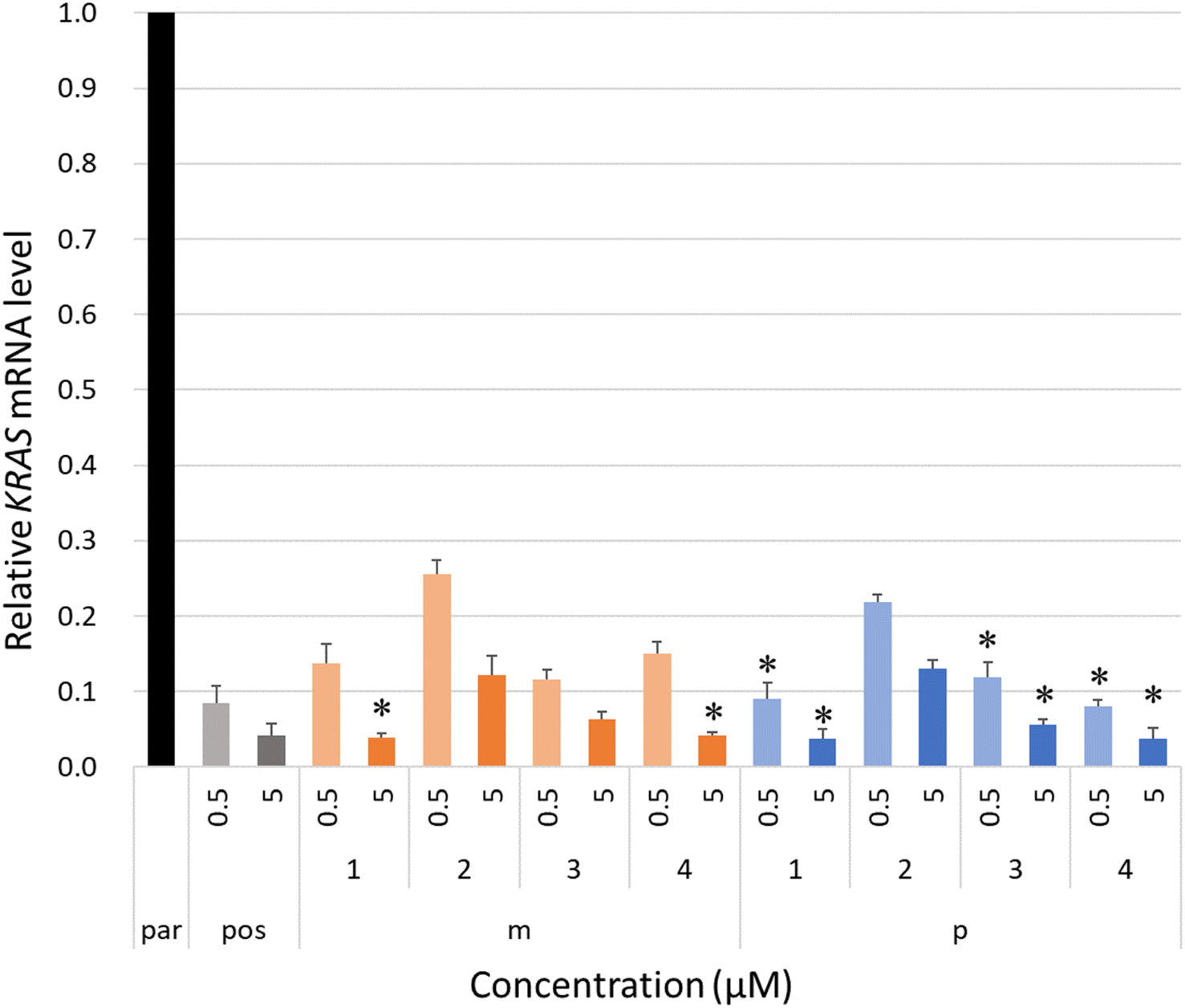 | ||
| Fig. 4 Relative KRAS mRNA levels of each KRAS gapmer.a aNCI-H460 cells were treated with the KRAS gapmers at various final concentrations (0.5 or 5.0 μM). Subsequently, the cells were incubated with the gapmers at 37 °C for 24 h and further incubated for 24 h after replacing the medium. Treatment only with PBS was used as a control (“par”). Total cellular mRNA was extracted, and the target KRAS mRNA was reverse transcribed. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed in triplicate, and relative KRAS mRNA levels were calculated using the ΔΔCT method. Results are expressed as the mean ± SE (n = 3 or 4). Asterisks indicate no significant difference compared with the KRAS-pos-treated group at the corresponding concentration (*P ≥ 0.05). | ||
Overall, the 4′-C-α-aminoethoxy-2′-O-methyl-5-propynyl modification exhibited excellent thermal and biochemical stability and could be tolerated while maintaining sufficient antisense activity in the 3′-wing region of the LNA gapmer, indicating its potential clinical use as an alternative to LNA.
Conclusion
To summarize, two novel uridine analogs, 4′-C-α-aminoethoxy-2′-O-methyl-5-methyl-uridine (4AEomU) and 4′-C-α-aminoethoxy-2′-O-methyl-5-propynyl-uridine (4AEopU), were designed and synthesized to expand the application of the 4AEo modification in antisense therapeutics. In the case of 4AEopU, the 5-propynyl moiety was introduced after the addition of the 4′-aminoethoxy side chain, providing an opportunity to combine the 4AEo modification with other base modifications in future studies. These uridine analogs could be smoothly incorporated into the DNA-based oligonucleotides using an automated DNA/RNA synthesizer in a manner similar to native uridine. In the TC-repeat modeling sequence, the presence of three 4AEomU analogs in the complementary DNA/RNA duplex resulted in a slight thermal destabilization, whereas the incorporation of the 4AEopU analogs thermally stabilized the duplex significantly. According to the results obtained from calculations of the thermodynamic parameters, CD spectra and molecular dynamics simulations, both 4AEomU and 4AEopU analogs preferred the C3′-endo sugar puckering, which is entropically favorable for the thermal stability of the DNA/RNA duplexes. Furthermore, these analog molecules exhibited slight structural fluctuations in in silico calculations. However, the enhanced stacking effect of the 5-propynyl moiety in the 4AEopU analog could overcome this structural disadvantage, resulting in favorable enthalpy and much higher thermal stability. The difference between these novel uridine analogs in terms of thermal stability was attributed to the 5-propynyl modification. Combination with the 5-propynyl modification also improved the base discrimination ability by reducing the affinity with the complementary RNA strand containing a single guanosine mismatch. From a biochemical perspective, the novel uridine analogs used in this study showed either robust nuclease resistance or efficient E. coli RNase H activation. These results encouraged us to prepare LNA-wing antisense gapmers containing one 4AEomU or 4AEopU analog and to confirm their effects on cellular antisense activity. Incorporating one 4AEomU analog in the wing region resulted in a significant decrease in thermal stability and antisense activity, whereas in the gap region, the changes were modest. In contrast, the 4AEopU analog was as thermally stable as its LNA counterpart in the 3′-wing region, and no negative effects were observed in antisense activity except for KRAS-p-2.Owing to its excellent thermal stability, the novel 4AEopU analog could be an alternative to LNA, providing insight into the design of antisense gapmers containing the 4AEopU wings for further evaluation. This analog was also expected to reduce the number of phosphorothioate linkages surrounding the 4AEopU analog to avoid additional interactions with undesirable proteins while maintaining robust nuclease resistance. This approach might be suitable for splicing-switching ASOs. Therefore, the 4′-C-α-aminoethoxy-2′-O-methyl-5-propynyl-modified nucleoside analogs are potential candidates as antisense therapeutics.
Experimental section
General remark
All chemicals and dry solvents (DCM, DMF, MeCN, THF, and pyridine) were obtained from commercial sources and used without any further purification. Thin layer chromatography (TLC) was performed on silica gel plates pre-coated with a fluorescent indicator and visualization by UV light or by dipping into a solution of 5% (v/v) concentrated H2SO4 in a mixture of p-anisaldehyde and methanol, followed by heating. Silica gel (63–210 mesh) was used for column chromatography. 1H NMR (400 or 500 MHz), 13C {1H} NMR (101 MHz), and 31P NMR (162 MHz) were recorded on 400 or 500 M Hz NMR equipment. CDCl3 or DMSO-d6 was used as a solvent to obtain the NMR spectra. Chemical shifts (δ) are given in parts per million (ppm) from CDCl3 (7.26 ppm) and DMSO-d6 (2.50 ppm) for 1H NMR spectra, and CDCl3 (77.2 ppm) and DMSO-d6 (39.52 ppm) for 13C NMR spectra. The abbreviations s, d, t, q, and m signify singlet, doublet, triplet, quadruplet, and multiplet, respectively. High-resolution mass spectra (HRMS) were obtained in the positive ion electrospray ionization (ESI-TOF) mode. The purity of each oligonucleotide synthesized in this study was confirmed by MALDI-TOF/MS.3′-O-(tert-Butyldimethysilyl)-4′,5′-didehydro-5′-deoxy-2′-O-methyl-5-methyl-uridine (4)
Under an argon atmosphere, triphenylphosphine (2.31 g, 8.81 mmol), imidazole (21.00 g, 14.69 mmol) and iodine (2.23 g, 8.81 mmol) were added to the solution of compound 1 (2.01 g, 7.34 mmol) in THF (38.20 mL). After stirring at room temperature for 4 hours, DBU (4.40 mL, 29.38 mmol) was added dropwise to the reaction solution and stirred at room temperature for 24 hours. The solution was then evaporated, and the resulting residue was dried overnight in vacuo. After that, the crude residue was dissolved in DCM (20.00 mL) under the argon atmosphere. Imidazole (1.51 g, 22.02 mmol) and tert-butyldimethylsilyl chloride (3.33 g, 22.09 mmol) were added to the reaction solution and stirred at room temperature for 3 hours. The reactant was then quenched by saturated NaHCO3 aq. and extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and then concentrated in vacuo. The resulting residue was purified by column chromatography (33% ethyl acetate in hexane) to afford the desired product 4 (2.01 g, 5.44 mmol, 74%). 1H NMR (400 M Hz, CDCl3) δ: 9.02 (s, 1H), 6.98 (d, J = 1.4 Hz, 1H), 6.07 (d, J = 3.7 Hz, 1H), 4.60–4.59 (m, 2H), 4.33 (q, J = 1.2 Hz, 1H), 3.83 (t, J = 4.4 Hz, 1H), 3.49 (s, 3H), 1.94 (d, J = 1.4 Hz, 3H), 0.93 (s, 9H), 0.14 (d, J = 1.8 Hz, 6H); 13C{1H} NMR (101 M Hz, CDCl3) δ: 163.7, 160.7, 150.1, 134.9, 111.8, 88.8, 86.7, 82.2, 69.9, 58.4, 25.8, 18.4, 12.8, −4.5, −4.6; HRMS (ESI-TOF) m/z calcd for C17H28N2NaO5Si [M + Na]+, 391.1665, found 391.1682.3′-O-(tert-Butyldimethysilyl)-2′-O-methyl-5-methyl-4′-C-α-trifluoroacetylaminoethoxy-uridine (6)
Compound 4 (2.01 g, 5.44 mmol) was dissolved in a mixture of DCM (30.00 mL), acetone (30.00 mL) and saturated NaHCO3 aq. (30.00 mL), followed by cooling in an ice bath. Oxone® (2.48 g, 16.32 mmol) in water (20.00 mL) was added to the reaction solution. After stirring at room temperature for 2 hours, the reactant was extracted with chloroform and water. The organic layer was washed with brine, dried by Na2SO4, filtered, and then concentrated in vacuo. The crude residue was dissolved in THF (20.00 mL) under the argon atmosphere. N-Trifluoroacetylaminoethanol (1.83 mL, 16.32 mmol) was then added and immediately stirred at −60 °C for at least 10 minutes. After adding zinc chloride (1.0 M solution in THF; 5.40 mL, 5.40 mmol), the reaction solution was slowly brought to room temperature and stirred for 24 hours. The reactant was then quenched by saturated NaHCO3 aq. and filtered by Celite®. The resulting filtrate was extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and then concentrated in vacuo. The resulting residue was purified by column chromatography (50% ethyl acetate in hexane) to afford the desired product 6 (0.86 g, 1.60 mmol, 29%). 1H NMR (400 M Hz, DMSO-D6) δ: 11.41 (s, 1H), 9.22 (bs, 1H), 7.67 (s, 1H), 6.01 (d, J = 5.5 Hz, 1H), 5.40 (t, J = 5.5 Hz, 1H), 4.44 (d, J = 6.0 Hz, 1H), 3.93 (t, J = 5.7 Hz, 1H), 3.76–3.66 (m, 2H), 3.49 (dq, J = 31.1, 5.8 Hz, 2H), 3.30 (s, 5H), 1.78 (s, 3H), 0.88 (s, 9H), 0.08 (d, J = 2.3 Hz, 6H); 13C{1H} NMR (101 M Hz, DMSO-D6) δ: 163.7, 150.5, 136.0, 109.9, 106.7, 86.3, 81.5, 71.1, 62.0, 60.2, 58.2, 40.1, 40.0, 25.7, 18.1, 12.3, −4.7, −5.0; HRMS (ESI-TOF) m/z calcd for C21H34F3N3NaO8Si [M + Na]+, 564.1965, found 564.1963.3′-O-(tert-Butyldimethysilyl)-5′-O-(4,4′-dimethoxytrityl)-2′-O-methyl-5-methyl-4′-C-α-trifluoroacetylaminoethoxy-uridine (7)
Under an argon atmosphere, N,N-diisopropylethylamine (0.70 mL, 4.00 mmol) and 4,4′-dimethoxytrityl chloride (0.67 g, 1.98 mmol) were added to the solution of compound 6 (0.87 g, 1.60 mmol) in pyridine (8.70 mL). After stirring overnight at room temperature, the reactant was quenched by saturated NaHCO3 aq. and extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and then concentrated in vacuo. The resulting residue was purified by column chromatography (50% ethyl acetate in hexane) to afford the desired product 7 (1.06 g, 1.26 mmol, 79%). 1H NMR (500 M Hz, CDCl3) δ: 8.19 (s, 1H), 7.64 (s, 1H), 7.43 (s, 1H), 7.35 (d, J = 6.9 Hz, 2H), 7.31–7.22 (m, 7H), 6.83 (q, J = 4.6 Hz, 4H), 6.00 (s, 1H), 4.60 (d, J = 6.3 Hz, 1H), 3.79 (d, J = 1.1 Hz, 6H), 3.75–3.71 (m, 2H), 3.63–3.57 (m, 2H), 3.55 (s, 3H), 3.54–3.50 (m, 1H), 3.42–3.36 (m, 1H), 3.23 (d, J = 9.7 Hz, 1H), 1.26 (s, 3H), 0.80 (s, 9H), 0.05 (s, 3H), −0.04 (s, 3H); 13C{1H} NMR (101 M Hz, CDCl3) δ: 163.8, 159.1, 150.0, 143.8, 135.2, 134.8, 130.4, 130.3, 128.4, 128.2, 127.6, 113.4, 113.4, 111.6, 106.7, 90.1, 87.5, 84.1, 71.4, 61.7, 60.6, 60.0, 55.4, 39.9, 25.7, 18.2, 11.6, −4.4, −4.8; HRMS (ESI-TOF) m/z calcd for C42H52F3N3NaO10Si [M + Na]+, 866.3272, found 866.3275.5′-O-(4,4′-Dimethoxytrityl)-2′-O-methyl-5-methyl-4′-C-α-trifluoroacetylaminoethoxy-uridine (8)
Under an argon atmosphere, tetrabutylammonium fluoride (1.0 M solution in THF, 0.48 mL, 0.48 mmol) was added to a solution of compound 7 (0.20 g, 0.24 mmol) in THF (4.30 mL). After stirring at room temperature for 1 hour, the reactant was quenched with saturated NaHCO3 aq. and extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The resulting residue was purified by column chromatography (67% ethyl acetate in hexane) to afford the desired product 8 (0.15 g, 0.21 mmol, 86%). 1H NMR (400 M Hz, CDCl3) δ: 8.78 (s, 1H), 7.52 (d, J = 0.9 Hz, 1H), 7.39–7.35 (m, 2H), 7.32–7.9 (m, 10H), 6.84 (d, J = 8.7, 4H), 5.89 (s, 1H), 4.70 (dd, J = 11.2, 6.6 Hz, 1H), 3.83–3.79 (m, 7H), 3.75–3.70 (m, 2H), 3.65 (s, 3H), 3.50–3.45 (m, 2H), 3.37 (d, J = 10.1 Hz, 1H), 3.26 (dd, J = 14.2, 10.5 Hz, 1H), 2.94 (d, J = 11.4 Hz, 1H), 1.34 (d, J = 0.9 Hz, 3H); 13C{1H} NMR (101 M Hz, CDCl3) δ: 163.7, 159.0, 149.9, 144.0, 135.0, 134.9, 130.2, 128.3, 128.2, 127.5, 113.5, 111.6, 106.2, 89.9, 87.5, 84.2, 70.4, 61.4, 61.2, 60.3, 55.4, 39.4, 11.8, 1.2; HRMS (ESI-TOF) m/z calcd for C36H38F3N3NaO10 [M + Na]+, 752.2407, found 752.2430.3′-O-[2-Cyanoethoxy(diisopropylamino)phosphino]-5′-O-(4,4′-dimethoxytrityl)-2′-O-methyl-5-methyl-4′-C-α-trifluoroacetylaminoethoxy-uridine (9)
Under an argon atmosphere, N,N-diisopropylethylamine (0.88 mL, 5.04 mmol) and 2-cyanoethyl-N,N-diisopropylchlorophosphoroamidite (0.45 mL, 2.00 mmol) were added to a solution of compound 8 (0.73 g, 1.00 mmol) in THF (7.30 mL). After stirring at room temperature for 2 hours, the reactant was quenched by saturated NaHCO3 aq. and extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and then concentrated in vacuo. The resulting residue was purified by column chromatography (50% ethyl acetate in hexane) to afford the desired product 9 (0.51 g, 0.55 mmol, 55%). 31P NMR (162 M Hz, CDCl3) δ: 151.8, 151.3; HRMS (ESI-TOF) m/z calcd for C45H55F3N5NaO11P [M + Na]+, 952.3486, found 952.3458.2′-O-Methyl-4′-C-α-trifluoroacetylaminoethoxy-uridine (11)
Under an argon atmosphere, tetrabutylammonium fluoride (1.0 M solution in THF, 9.80 mL, 9.80 mmol) was added to a solution of compound 10 (2.57 g, 4.87 mmol) in THF (88.00 mL). After stirring at room temperature for 1 hour, the reactant was quenched with saturated NaHCO3 aq. and extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The resulting residue was purified by column chromatography (9% methanol in chloroform) to afford the desired product 11 (1.92 g, 4.64 mmol, 95%). 1H NMR (400 MHz, DMSO-D6) δ: 11.39 (s, 1H), 9.25 (s, 1H), 7.79 (d, J = 8.2 Hz, 1H), 5.96 (d, J = 4.1 Hz, 1H), 5.67 (d, J = 8.2 Hz, 1H), 5.34 (t, J = 5.5 Hz, 1H), 4.67 (d, J = 9.2 Hz, 1H), 4.29–4.25 (m, 1H), 3.83–3.80 (m, 1H), 3.70–3.54 (m, 3H), 3.43 (q, J = 5.8 Hz, 1H), 3.35 (s, 4H); 13C{1H} NMR (101 M Hz, CDCl3) δ: 163.0, 150.3, 140.4, 106.3, 102.2, 87.0, 81.9, 79.2, 69.6, 60.8, 60.1, 58.1; HRMS (ESI-TOF) m/z calcd for C14H18F3N3NaO8 [M + Na]+, 436.0944, found 436.0956.2′-O-Methyl-5-(1-propynyl)-4′-C-α-trifluoroacetylaminoethoxy-uridine (13)
Under an argon atmosphere, diammonium cerium(IV) nitrate (1.14 g, 2.08 mmol) and iodine (0.64 g, 2.52 mmol) were added to a solution of compound 11 (2.18 g, 5.28 mmol) in MeCN (40.00 mL). After stirring at reflux at 80 °C for 1 hour, the reactant was quenched with methanol, and the solvent was evaporated. The resulting residue was then dried in vacuo for 2 hours. After that, the crude residue was dissolved in DMF (10 mL) under argon atmosphere. Bis(triphenylphosphine) palladium(II) dichloride (0.34 g, 0.49 mmol), copper(I) iodide (0.19 g, 0.98 mmol), triethylamine (1.40 mL, 9.80 mmol) and propyne (1.0 M solution in DMF; 15.00 mL, 15.00 mmol) were added to the reaction solution. After stirring overnight at 35 °C, the reactant was quenched with methanol, and the solvent was evaporated. The resulting residue was purified by column chromatography (9% methanol in chloroform) to afford the desired product 13 (1.89 g, 4.18 mmol, 79%).1H NMR (400 MHz, DMSO-D6) δ: 11.65 (s, 1H), 9.24 (s, 1H), 8.08 (s, 1H), 5.92 (d, J = 3.7 Hz, 1H), 5.45 (t, J = 5.5 Hz, 1H), 4.65 (d, J = 9.2 Hz, 1H), 4.28–4.24 (m, 1H), 3.87–3.84 (m, 1H), 3.68 (d, J = 10.1 Hz, 1H), 3.59 (q, J = 5.2 Hz, 2H), 3.44 (q, J = 5.5 Hz, 1H), 3.35 (s, 4H), 1.98 (s, 3H); 13C{1H} NMR (101 M Hz, DMSO-D6) δ: 161.7, 149.3, 142.4, 117.4, 106.5, 99.5, 89.4, 87.4, 82.0, 71.9, 69.4, 60.6, 60.1, 58.2, 4.1; HRMS (ESI-TOF) m/z calcd for C17H20F3N3NaO8 [M + Na]+, 474.1100, found 474.1103.
5′-O-(4,4′-Dimethoxytrityl)-2′-O-methyl-5-(1-propynyl)-4′-C-α-trifluoroacetylaminoethoxy-uridine (14)
Under an argon atmosphere, N,N-diisopropylethylamine (1.00 mL, 5.73 mmol) and 4,4′-dimethoxytrityl chloride (1.36 g, 4.01 mmol) were added to a solution of compound 13 (0.80 g, 2.00 mmol) in pyridine (9.00 mL). After stirring at 50 °C for 24 hours, the reactant was quenched by saturated NaHCO3 aq. and extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and then concentrated in vacuo. The resulting residue was purified by column chromatography (60% ethyl acetate in hexane) to afford the desired product 14 (1.10 g, 1.46 mmol, 73%). 1H NMR (400 MHz, CDCl3) δ: 9.23 (s, 1H), 7.87 (s, 1H), 7.42 (d, J = 6.9 Hz, 2H), 7.34–7.20 (m, 8H), 6.84 (d, J = 8.2 Hz, 4H), 5.81 (s, 1H), 4.70 (q, J = 6.0 Hz, 1H), 3.86 (d, J = 6.4 Hz, 1H), 3.79 (s, 6H), 3.75–3.67 (m, 2H), 3.65 (s, 3H), 3.47–3.43 (m, 1H), 3.42 (s, 2H), 3.29–3.22 (m, 1H), 3.02 (d, J = 11.4 Hz, 1H), 1.81 (s, 1H), 1.60 (s, 3H); 13C{1H} NMR (101 M Hz, CDCl3) δ: 162.0, 158.8, 149.1, 144.3, 140.9, 135.3, 135.1, 130.1, 130.0, 128.2, 127.9, 127.1, 113.5, 106.4, 101.4, 91.7, 90.1, 87.4, 84.0, 70.2, 69.7, 61.4, 61.2, 60.3, 55.4, 39.3, 4.5; HRMS (ESI-TOF) m/z calcd for C38H38F3N3NaO10 [M + Na]+, 776.2407, found 776.2398.3′-O-[2-Cyanoethoxy(diisopropylamino)phosphino]-5′-O-(4,4′-dimethoxytrityl)-2′-O-methyl-5-(1-propynyl)-4′-C-α-trifluoroacetylaminoethoxy-uridine (15)
Under an argon atmosphere, N,N-diisopropylethylamine (2.00 mL, 11.45 mmol) and 2-cyanoethyl-N,N-diisopropylchlorophosphoroamidite (1.00 mL, 4.48 mmol) were added to a solution of compound 14 (1.88 g, 2.50 mmol) in THF (19.00 mL). After stirring at room temperature for 2 hours, the reactant was quenched by saturated NaHCO3 aq. and extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and then concentrated in vacuo. The resulting residue was purified by column chromatography (60% ethyl acetate in hexane) to afford the desired product 115 (1.72 g, 1.80 mmol, 72%). 31P NMR (162 M Hz, CDCl3) δ: 151.2, 151.1; HRMS (ESI-TOF) m/z calcd for C47H55F3N5NaO11P [M + Na]+, 976.3486, found 976.3514.Solid-phase oligonucleotide synthesis
The synthesis was carried out with an automated DNA/RNA synthesizer using the solid-phase phosphoramidite method. After the synthesis, oligonucleotides containing 4AEo modification were treated with a mixture of dimethylamine/MeCN (1:9, v/v) for 5 minutes, followed by rinsing with MeCN to selectively remove the cyanoethyl groups. After that, the DNA oligonucleotides were treated with 28% aqueous ammonium solution for 12–14 hours at 55 °C. In contrast, the RNA oligonucleotides were cleaved from CPG beads and deprotected by treatment with 28% aqueous ammonium solution/40% methylamine (1:1, v/v) for 10 minutes at 65 °C. Then, 2′-O-TBDMS groups in RNA oligonucleotides were removed by Et3N·3HF (125 μL) in DMSO (100 μL) for 1.5 hours at 65 °C. The reaction was quenched with 0.1 M TEAA buffer (pH 7.0), and the mixture was desalted using a Sep-Pak C18 cartridge. All oligonucleotides were purified by 20% PAGE containing 7 M urea to give highly purified ones. The analysis of each synthesized oligonucleotide in this study was carried out with MALDI-TOF/MS, and the results are shown in Table S1.†
Tm measurement
The solution containing 3.0 μM DNA/RNA duplexes was prepared by mixing each DNA with an equal amount of cRNA1 in a buffer of 10 mM sodium phosphate (pH 7.0) containing 100 mM NaCl. The solution was heated at 90–100 °C and allowed to stand for at least 1.5 hours at room temperature to cool down gradually. Thermally induced transitions were monitored at 260 nm with a UV/vis spectrometer fitted with a temperature controller in quartz cuvettes with a path length of 1.0 cm. The sample temperature was increased by 0.5 °C min−1. The thermodynamic parameters of the duplexes on duplex formation were determined by calculations based on the slope of a 1/Tm vs. ln(CT/4) plot, where CT (1, 3, 6, 12, 15, 21, 30 and 60 μM) is the total concentration of the single strands.CD spectroscopy
All CD spectra were recorded at 25 °C. The following instrument settings were used: resolution, 1 nm; response, 1.0 s; speed, 50 nm min−1; accumulation, 10.Molecular dynamics simulation
The initial geometries of the native thymidine, 4′-C-α-aminoethoxy-2′-O-methyl-5-methyl-uridine and 4′-C-α-aminoethoxy-2′-O-methyl-5-propynyl-uridine were modeled by a molecular modeling software, GaussView. The geometry of each nucleoside was optimized, and the electrostatic potential (ESP) of each nucleoside was calculated using Gaussain16 at the HF/6-31G* level of theory.54 The restrained ESP (RESP) charges were derived using the Antechamber RESP fitting procedure.55 The generalized AMBER force field (gaff) was used for the native thymidine and modified nucleosides.56 Each of the nucleosides was solvated in a cubic box with the TIP3P water model, and the size of the cubic box was determined to be 42.0 Å (in length).57 Chloride ions were added to the solution to neutralize the charge of the systems. The energy minimization calculation was performed using the SANDER module of AMBER18. The solute molecule was optimized by 2500 steps of the steepest descent method, followed by 2500 steps of the conjugate gradient method by applying a restraint force constant of 2.0 kcal mol−1 Å−2 to the nucleosides. Then, the calculation of the entire system was carried out using 5000 steps of the conjugate gradient method without any restraints. The minimized structure assigned to the atom type and calculated RESP charges for each nucleoside are shown in Fig. S7.† After that, the temperature of the system was heated gradually from 0 K to 300 K over a period of 100 ps of NVT dynamics with a restraint force constant of 2.0 kcal mol−1 Å−2 to the nucleoside, followed by 100 ps of NPT equilibration at 300 K and 1 bar pressure with the same restraint force. After the equilibration phase, 500 ns MD simulations were performed in an NPT ensemble at 300 K and 1 bar pressure without any restraints. During the MD simulations, a time step of 2 fs was used. The cut-off distance towards the van der Waals force was 10 Å, the SHAKE algorithm was used to constrain all bonds involving hydrogen atoms, and the Particle Mesh Ewald (PME) method was used for long-range electrostatic interaction.58,59 The temperature was regulated by Langevin dynamics with a collision frequency of 2.0 ps−1.60 All the MD simulations were performed using the PMEMD module of AMBER18. Trajectory analysis was done for 50000 snapshots, every 10 ps, sampled during the full 500 ns simulation.
Nuclease resistance of single-strand DNAs
Fluorescein-labeled DNAs (300 pmol) were dissolved in OPTI-MEM (37 μL) and used for the serum stability test. 1.0 μL of the oligomer solution was diluted in 10 μL stop solution (10% formamide in 10 mM EDTA) as the control sample (0 min). Then, bovine serum was added to achieve various final concentrations (3%, 10%, 25% or 50%; v/v), and subsequently, the mixture was incubated at 37 °C for the required time. Aliquots of 2.0 μL were diluted with a 10 μL stop solution. Samples were subjected to electrophoresis in 20% PAGE containing 7 M urea and quantified by Luminescent Image analyzer LAS-4000 (Fujifilm).E. coli RNase H activation
The solution of DNA/RNA duplexes used for the RNase H assay was prepared by mixing the native or modified DNAs (600 pmol) with fluorescein-labeled cRNA-2 (3000 pmol) in 75 μL of 50 mM Tris–HCl (pH 8.0) containing 75 mM KCl, 3 mM MgCl2 and 10 mM dithiothreitol, followed by heating at 90–100 °C for 5 min and cooling gradually to room temperature. Then, 70 μL diluted RNase H solution (60 units per L in H2O) was added, and subsequently, the mixture was incubated at 37 °C for the required time. Aliquots of 5 μL were diluted with 100% formamide (10 μL). Samples were subjected to electrophoresis in 20% PAGE containing 7 M urea and quantified by a Luminescent Image analyzer LAS-4000 (Fujifilm).In vitro antisense activity of KRAS-gapmers
NCI-H460 cells, transfected with KRAS and ACTB genes, were used for the gene expression suppression assays, and the KRAS & ACTB primer probes for qPCR were provided by Professor Tokuhirro Chano (Shiga University of Medical Science). Cells were cultured in D-MEM containing 10% fetal bovine serum (FBS), penicillin (50 units per mL) and streptomycin (50 mg mL−1). Cells were incubated at 37 °C in a humidified chamber supplemented with 5% carbon dioxide. NCI-H460 cells were plated into 96-well plates at 400 cells per well in 50 μL D-MEM containing 10% FBS. Then, the KRAS-targeting antisense gapmers were added into specified wells at the final concentration of 0.5 or 5.0 μM. After a 24 hours incubation at 37 °C with 5% carbon dioxide, an additional 50 μL medium was added, followed by a further 24 hours incubation. Reverse transcription was directly performed in each of the 96-well plates using a TaqMan™ Fast Advanced Cells to CT™ Kit (Thermo Fisher Scientific). Treatment without gapmers was used as a control, named “par”. Quantitative real-time polymerase chain reaction (qRT-PCR) of the targeted KRAS mRNA was performed using a TaqMan® Fast Advanced master mix and StepOnePlus™ Real-Time PCR system (Thermo). Each PCR reaction was performed in duplicate, and the relative KRAS mRNA levels were calculated by the ΔΔCT method using beta-actin (ACTB) as a reference.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and/or its ESI.†Author contributions
Yujun Zhou: investigation, data curation, writing – original draft. Hitotaka Sato: data curation. Miwa Kawade: data curation. Kenji Yamagishi: data curation. Yoshihito Ueno: writing – review & editing, funding acquisition, conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Agency for Medical Research and Development (AMED) through its funding program for the research project number 23ae0121029h0003 and Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number JP23KJ1044. The first author, Yujun ZHOU, was supported by a fellowship of the Nagoya University CIBoG WISE program from MEXT. The authors would like to thank Professor Tokuhirro Chano (Shiga University of Medical Science Hospital) for supplying the cells and providing technical advice.References
- M. Gagliardi and A. T. Ashizawa, The challenges and strategies of antisense oligonucleotide drug delivery, Biomedicines, 2021, 9(4), 433 CrossRef CAS.
- A. M. Quemener, M. L. Centomo, S. L. Sax and R. Panella, Small drugs, huge impact: the extraordinary impact of antisense oligonucleotides in research and drug development, Molecules, 2022, 27(2), 536 CrossRef CAS.
- M. Egli and M. Manoharan, Chemistry, structure and function of approved oligonucleotide therapeutics, Nucleic Acids Res., 2023, 51(6), 2529–2573 CrossRef CAS PubMed.
- C. M. Perry and J. A. B. Balfour, Fomivirsen, Drugs, 1999, 57, 375–380 CrossRef CAS PubMed.
- P. Hair, F. Cameron and K. McKeage, Mipomersen sodium: first global approval, Drugs, 2013, 73, 487–493 CrossRef CAS.
- Y. Y. Syed, Eteplirsen: first global approval, Drugs, 2016, 76, 1699–1704 CrossRef CAS.
- S. M. Hoy, Nusinersen: first global approval, Drugs, 2017, 77, 473–479 CrossRef CAS.
- S. J. Keam, Inotersen: first global approval, Drugs, 2018, 78, 1371–1376 CrossRef CAS.
- J. Paik and S. Duggan, Volanesorsen: first global approval, Drugs, 2019, 79(12), 1349–1354 CrossRef CAS PubMed.
- Y. A. Heo, Golodirsen: first approval, Drugs, 2020, 80(3), 329–333 CrossRef.
- S. Dhillon, Viltolarsen: first approval, Drugs, 2020, 80(10), 1027–1031 CrossRef CAS.
- M. Shirley, Casimersen: first approval, Drugs, 2021, 81, 875–879 CrossRef CAS PubMed.
- H. A. Blair, Tofersen: first approval, Drugs, 2023, 83(11), 1039–1043 CrossRef CAS PubMed.
- T. Nie, Eplontersen: First Approval, Drugs, 2024, 1–6 CAS.
- X. Wang, C. S. Hu, B. Petersen, J. Qiu, F. Ye, J. Houldsworth and R. Hoffman, Imetelstat, a telomerase inhibitor, is capable of depleting myelofibrosis stem and progenitor cells, Blood Adv., 2018, 2(18), 2378–2388 Search PubMed.
- M. Manoharan, Oligonucleotide conjugates as potential antisense drugs with improved uptake, biodistribution, targeted delivery, and mechanism of action, Antisense Nucleic Acid Drug Dev., 2002, 12(2), 103–128 CrossRef CAS PubMed.
- N. Dias and C. A. Stein, Antisense oligonucleotides: basic concepts and mechanisms, Mol. Cancer Ther., 2002, 1(5), 347–355 CAS.
- S. T. Crooke, Molecular mechanisms of antisense oligonucleotides, Nucleic Acid Ther., 2017, 27(2), 70–77 CrossRef CAS.
- J. Kurreck, Antisense technologies: improvement through novel chemical modifications, Eur. J. Biochem., 2003, 270(8), 1628–1644 CrossRef CAS PubMed.
- V. K. Sharma, R. K. Sharma and S. K. Singh, Antisense oligonucleotides: modifications and clinical trials, MedChemComm, 2014, 5(10), 1454–1471 RSC.
- H. Kim, S. Kim, D. Lee, D. Lee, J. Yoon and H. Lee, Oligonucleotide therapeutics and their chemical modification strategies for clinical applications, J. Pharm. Invest., 2024, 1–19 Search PubMed.
- W. F. Lima, J. B. Rose, J. G. Nichols, H. Wu, M. T. Migawa, T. K. Wyrzykiewicz and S. T. Crooke, The positional influence of the helical geometry of the heteroduplex substrate on human RNase H1 catalysis, Mol. Pharmacol., 2007, 71(1), 73–82 CrossRef CAS PubMed.
- E. F. Phosphorothioates, essential components of therapeutic oligonucleotides, Nucleic Acid Ther., 2014, 24, 374–387 CrossRef PubMed.
- S. T. Crooke, S. Wang, T. A. Vickers, W. Shen and X. H. Liang, Cellular uptake and trafficking of antisense oligonucleotides, Nat. Biotechnol., 2017, 35(3), 230–237 CrossRef CAS.
- W. Shen, C. L. De Hoyos, H. Sun, T. A. Vickers, X. H. Liang and S. T. Crooke, Acute hepatotoxicity of 2′ fluoro-modified 5–10–5 gapmer phosphorothioate oligonucleotides in mice correlates with intracellular protein binding and the loss of DBHS proteins, Nucleic Acids Res., 2018, 46(5), 2204–2217 CrossRef.
- W. Shen, C. L. De Hoyos, M. T. Migawa, T. A. Vickers, H. Sun, A. Low and S. T. Crooke, Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index, Nat. Biotechnol., 2019, 37(6), 640–650 CrossRef CAS.
- Q. Laurent, R. Martinent, D. Moreau and S. Matile, et al., Oligonucleotide Phosphorothioates Enter Cells by Thiol-Mediated Uptake, Angew. Chem., Int. Ed., 2021, 60, 19102 CrossRef CAS PubMed.
- K. Koizumi, Y. Maeda, T. Kano, H. Yoshida, T. Sakamoto, K. Yamagishi and Y. Ueno, Synthesis of 4′-C-aminoalkyl-2′-O-methyl modified RNA and their biological properties, Bioorg. Med. Chem., 2018, 26(12), 3521–3534 CrossRef CAS PubMed.
- R. Tsukimura, R. Kajino, Y. Zhou, A. Chandela and Y. Ueno, 4′-C-Aminoethoxy modification enhanced nuclease resistance of RNAs and improved thermal stability of RNA duplexes, Results Chem., 2021, 3, 100231 CrossRef CAS.
- H. Inoue, Y. Hayase, S. Iwai and E. Ohtsuka, Sequence-dependent hydrolysis of RNA using modified oligonucleotide splints and RNase H, FEBS Lett., 1987, 215(2), 327–330 CrossRef CAS PubMed.
- A. T. Daniher, J. Xie, S. Mathur and J. K. Bashkin, Modulation of RNase H activity by modified DNA probes: Major groove vs. minor groove effects, Bioorg. Med. Chem., 1997, 5(6), 1037–1042 CrossRef CAS PubMed.
- Y. Katsuzaki, R. Tsukimura, A. Chandela, T. Chano and Y. Ueno, 4′-C-Aminoethoxy-Modified DNAs Exhibit Increased Nuclease Resistance, Sustained RNase H Activity, and Inhibition of KRAS Gene Expression, Chem. Biodiversity, 2022, 19(8), e202200125 CrossRef CAS.
- M. Terrazas and E. T. Kool, RNA major groove modifications improve siRNA stability and biological activity, Nucleic Acids Res., 2009, 37(2), 346–353 CrossRef CAS PubMed.
- R. W. Wagner, M. D. Matteucci, J. G. Lewis, A. J. Gutierrez, C. Moulds and B. C. Froehler, Antisense gene inhibition by oligonucleotides containing C-5 propyne pyrimidines, Science, 1993, 260(5113), 1510–1513 CrossRef CAS PubMed.
- W. Tong, P. Agback and J. Chattopadhyaya, Synthesis of Diastereomerically Pure 4'-Alkoxy-alpha-(L)-and-beta (D)-nucleosides and their Conformational Analysis by 500 MHz 1H NMR Spectroscopy, Acta Chem. Scand., 1993, 47(2), 145–156 CrossRef CAS.
- J. M. Harp, D. C. Guenther, A. Bisbe, L. Perkins, S. Matsuda, G. R. Bommineni and M. Egli, Structural basis for the synergy of 4′- and 2′-modifications on siRNA nuclease resistance, thermal stability and RNAi activity, Nucleic Acids Res., 2018, 46(16), 8090–8104 CrossRef CAS PubMed.
- E. Malek-Adamian, D. C. Guenther, S. Matsuda, S. Martínez-Montero, I. Zlatev, J. Harp and M. J. Damha, 4′-C-Methoxy-2′-deoxy-2′-fluoro modified ribonucleotides improve metabolic stability and elicit efficient RNAi-mediated gene silencing, J. Am. Chem. Soc., 2017, 139(41), 14542–14555 CrossRef CAS PubMed.
- M. Petrová, O. Páv, M. Budesinsky, E. Zborníková, P. Novák, S. Rosenbergová and I. Rosenberg, Straightforward synthesis of purine 4′-alkoxy-2′-deoxynucleosides: first report of mixed purine–pyrimidine 4′-alkoxyoligodeoxynucleotides as new RNA mimics, Org. Lett., 2015, 17(14), 3426–3429 CrossRef.
- T. Tsuchihira, R. Kajino, Y. Maeda and Y. Ueno, 4′-C-Aminomethyl-2′-deoxy-2′-fluoroarabinonucleoside increases the nuclease resistance of DNA without inhibiting the ability of a DNA/RNA duplex to activate RNase H, Bioorg. Med. Chem., 2020, 28(16), 115611 CrossRef CAS.
- Y. Zhou, R. Kajino, S. Ishii, K. Yamagishi and Y. Ueno, Synthesis and evaluation of (S)-5′-C-aminopropyl and (S)-5′-C-aminopropyl-2′-arabinofluoro modified DNA oligomers for novel RNase H-dependent antisense oligonucleotides, RSC Adv., 2020, 10(68), 41901–41914 RSC.
- H. Ueda and Y. Ueno, Synthesis of 4′-C-(aminoethyl) thymidine and 4′-C-[(N-methyl) aminoethyl] thymidine by a new synthetic route and evaluation of the properties of the DNAs containing the nucleoside analogs, Bioorg. Med. Chem., 2022, 60, 116690 CrossRef CAS PubMed.
- S. Onishi, A. Chandela and Y. Ueno, Synthesis and Evaluation of 4′-C-(S)- and 4′-C-(R)-2-Aminopropoxy-Thymidine-Modified DNAs for Thermal Stability, RNase H Digestion, and Nuclease Resistance, ChemistrySelect, 2024, 9(23), e202305090 CrossRef CAS.
- S. H. Hung, Q. Yu, D. M. Gray and R. L. Ratliff, Evidence from CD spectra that d(purine)-r(pyrimidine) and r(purine)-d(pyrimidine) hybrids are in different structural classes, Nucleic Acids Res., 1994, 22(20), 4326–4334 CrossRef CAS.
- H. Inoue, Y. Hayase, A. Imura, S. Iwai, K. Miura and E. Ohtsuka, Synthesis and hybridization studies on two complementary nona (2'-O-methyl) ribonucleotides, Nucleic Acids Res., 1987, 15(15), 6131–6148 CrossRef CAS.
- S. Takahashi and N. Sugimoto, Stability prediction of canonical and non-canonical structures of nucleic acids in various molecular environments and cells, Chem. Soc. Rev., 2020, 49(23), 8439–8468 RSC.
- J. Parkinson, DNA duplexes stabilized by modified monomer residues: synthesis and stability, J. Chem. Soc., Perkin Trans. 1, 1998,(6), 1131–1138 Search PubMed.
- G. Varani and W. H. McClain, The G· U wobble base pair, EMBO Rep., 2000, 1(1), 18–23 CrossRef CAS PubMed.
- M. Teplova, S. T. Wallace, V. Tereshko, G. Minasov, A. M. Symons, P. D. Cook and M. Egli, Structural origins of the exonuclease resistance of a zwitterionic RNA, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(25), 14240–14245 CrossRef CAS PubMed.
- H. Nakamura, Y. Oda, S. Iwai, H. Inoue, E. Ohtsuka, S. Kanaya and K. Morikawa, How does RNase H recognize a DNA/RNA hybrid?, Proc. Natl. Acad. Sci. U. S. A., 1991, 88(24), 11535–11539 CrossRef CAS.
- R. Y. Walder and J. A. Walder, Role of RNase H in hybrid-arrested translation by antisense oligonucleotides, Proc. Natl. Acad. Sci. U. S. A., 1988, 85(14), 5011–5015 CrossRef CAS PubMed.
- M. Nowotny, S. A. Gaidamakov, R. J. Crouch and W. Yang, Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis, Cell, 2005, 121(7), 1005–1016 CrossRef CAS.
- M. Nowotny, S. A. Gaidamakov, R. Ghirlando, S. M. Cerritelli, R. J. Crouch and W. Yang, Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription, Mol. Cell, 2007, 28(2), 264–276 CrossRef CAS PubMed.
- A. Dieckmann, P. H. Hagedorn, Y. Burki, C. Brügmann, M. Berrera, M. Ebeling and F. Schuler, A sensitive in vitro approach to assess the hybridization-dependent toxic potential of high affinity gapmer oligonucleotides, Mol. Ther.–Nucleic Acids, 2018, 10, 45–54 CrossRef CAS PubMed.
- J. F. Michael, W. T. Gary and H. S. Bernhard, Gaussian, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model, J. Phys. Chem., 1993, 97(40), 10269–10280 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79(2), 926–935 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. Berendsen, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, J. Comput. Phys., 1977, 23(3), 327–341 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An Nlog (N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98(12), 10089–10092 CrossRef CAS.
- R. W. Pastor, B. R. Brooks and A. Szabo, An analysis of the accuracy of Langevin and molecular dynamics algorithms, Mol. Phys., 1988, 65(6), 1409–1419 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06376g |
| This journal is © The Royal Society of Chemistry 2024 |