
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceπ–π stacking assisted regioselectivity regulation in palladium-catalyzed cyclization reactions: a theoretical study†
Song Liu *ab,
Dianmin Zhang*a,
Yue Gonga,
Lianli Maa,
Li Lia and
Wei Chena
*ab,
Dianmin Zhang*a,
Yue Gonga,
Lianli Maa,
Li Lia and
Wei Chena
aChongqing Key Laboratory for Resource Utilization of Heavy Metal Wastewater, College of Chemistry and Environmental Engineering, Chongqing University of Arts and Sciences, Yongchuan, 402160, PR China. E-mail: sliu@cqwu.edu.cn
bSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing, 400030, China
First published on 4th December 2024
Abstract
The regulation of regioselectivity is an objective often pursued by organic chemists, and the comprehension of its mechanisms is crucial for devising efficient synthetic pathways. In this report, we conducted theoretical calculations to explore the regioselectivity regulatory mechanisms of two palladium-catalyzed cyclization reactions. In these cyclization reactions, manipulating the structural differences in the reaction substrates leads to the formation of distinct products. A detailed reaction mechanism and reactivity profile for this reaction were revealed. Furthermore, a π–π stacking assisted regioselectivity regulatory mechanism was proven by distortion–interaction energy analysis and noncovalent interaction calculations. The calculated results presented herein provide a theoretical guide for further experimental investigations of regioselectivity regulation.
Introduction
Regioselectivity is an important field in organic synthetic chemistry that mainly focuses on the differences in reactivity at various positions on a substrate.1 Several reactive sites may exhibit similar activity, leading to the formation of a mixture of isomers in the final product. Catalytic methods are ideal synthetic approaches in organic chemistry, as they allow for the prediction and switching of regioselectivity to access site-diverse regioisomers by altering the fewest reaction parameters.2Various regulatory strategies in organic chemistry, such as steric effects,3 electronic effects,4 chelation control,5 and ligand control,6 have been used to achieve regioselectivity7 (Scheme 1a). Chemists can use these strategies to manipulate the outcome of organic reactions to achieve certain modifications at specific sites within complex molecules, but the regulatory mechanism of regioselectivity is often ambiguous. In organic synthesis, understanding the regulatory mechanisms of these strategies is essential for designing efficient synthetic routes to obtain the targeted products.8
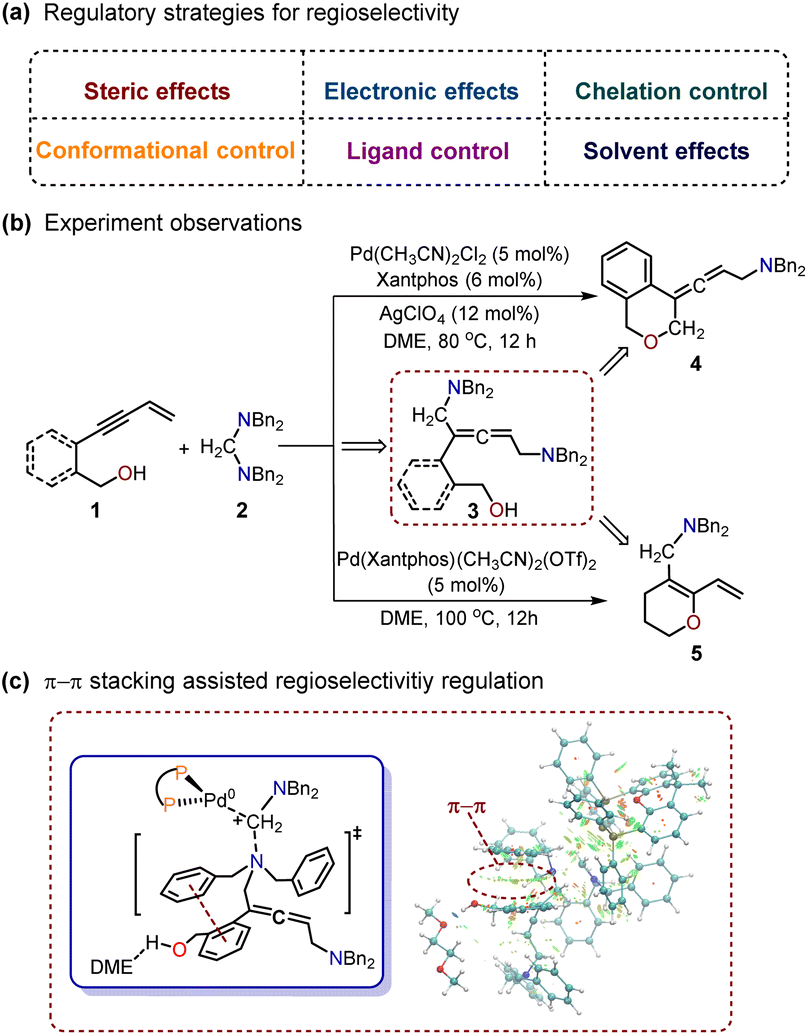 | ||
| Scheme 1 Palladium-catalyzed cyclization of enynols with aminals. | ||
Recently, Huang et al. described two palladium (Pd)-catalyzed cyclization reactions of enynols with aminals.9 In their reports, different products were obtained by manipulating the skeletal variances of substrates. As shown in Scheme 1b, the use of (2-(but-3-en-1-yn-1-yl)phenyl)methanol as the substrate led to the predominant formation of the O-heterocycle-containing allenic amine product 4. Furthermore, the use of hept-6-en-4-yn-1-ol as the substrate resulted in the formation of polysubstituted 1,3-diene embedded in O-heterocycle 5 as the main product (Scheme 1b). Control experiments confirmed the non-cyclic allenic 1,5-diamine 3 as the key intermediate in both cyclization reactions. However, the detailed regioselectivity regulatory mechanism for these Pd-catalyzed cyclizations is still not clear. Herein, we used density functional theory (DFT) calculations to explore these Pd-catalyzed cyclization reactions and proposed a π–π stacking assisted regioselectivity regulatory mechanism (Scheme 1c). A detailed reaction mechanism and reactivity profile were also provided.
Computational methods
All DFT calculations were carried out with Gaussian 16.10 The B3LYP functional11 with a standard 6-31G(d) basis set (the SDD basis set for Pd) was used for geometry optimizations. Harmonic frequency calculations were performed for all stationary points to confirm them as local minima or transition structures and to derive the thermochemical corrections of the enthalpies and free energies. All minima had zero imaginary frequency and all transition states had only one imaginary frequency. The solvent effects were considered by single-point calculations on the gas-phase stationary points with the SMD continuum solvation model.12 The M06 (ref. 13) functional with the 6-311+g(d) basis set (the SDD basis set for Pd) was used to calculate the single-point energies with dimethyl ether as the solvent. For the 1,2-dimethoxyethane (DME) solvent data not included in Gaussian 16, the key parameters were set in the input file manually (for 1,2-dimethoxyethane, eps = 7.2, while the other parameters were the same as those for diethyl ether). The energies given in this report were the M06 calculated Gibbs free energies in 1,2-dimethoxyethane solvent. The optimized structures were displayed using CYLview.14 The distortion and interaction energies were calculated B3LYP functional with the 6-31G(d) basis set. Additionally, a basis set superposition error (BSSE)15 correction was applied when calculating the interaction energies. The noncovalent interactions (NCIs)16 were calculated using B3LYP-D3 functional with the 6-31G(d) basis set.The global reactivity index values were also calculated using the M06 functional and the 6-311+g(d) basis set for all atoms. The N (global nucleophilicity index)17 values were calculated with eqn (1), where εHOMO refers to the orbital energy of the highest occupied molecular orbital (HOMO) and εLOMO refers to the orbital energy of the lowest unoccupied molecular orbital (LUMO). In eqn (1), tetracyanoethylene (TCE) with the lowest HOMO energy among the organic molecules was used as a ref. 18.
| N = εHOMO (Nu) − εHOMO (TCE) | (1) |
The Nk (local nucleophilicity index) values were calculated according to eqn (2), where the fk− (condensed Fukui function) values can be obtained by fk− = qk(N − 1) − qk(N).19
| Nk = N × fk− | (2) |
Results and discussion
The proposed catalytic cycle for the Pd-catalyzed cyclization of enynols with aminals is illustrated in Schemes 2 and 3. Scheme 2 depicts the catalytic cycle for the allenic 1,5-diamine intermediate 3. The aminomethyl cyclopalladated complex A was the active catalyst in both cyclizations. The coordination of the terminal alkene in the enynol substrate 1 with the Pd(II) center generated intermediate B. The subsequent migratory insertion of the enyne triple bond into the Pd(II)–C bond provided the π-allylpalladium intermediate C, which was intercepted by an aminal to form intermediate D via an SN2-type reductive elimination process. Finally, the SN2-type oxidative addition of the amino cation to the Pd(0) center afforded the non-cyclic allenic 1,5-diamine intermediate 3 together and regenerated the active Pd complex A to complete the catalytic cycle.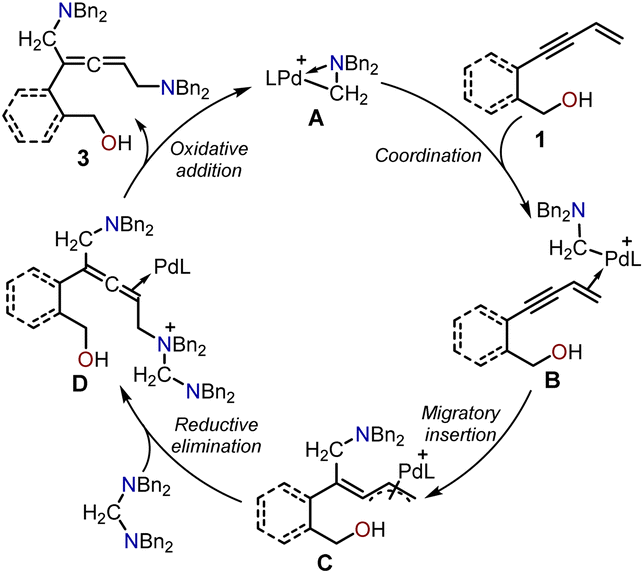 | ||
| Scheme 2 The proposed catalytic cycle for the production of intermediate 3. | ||
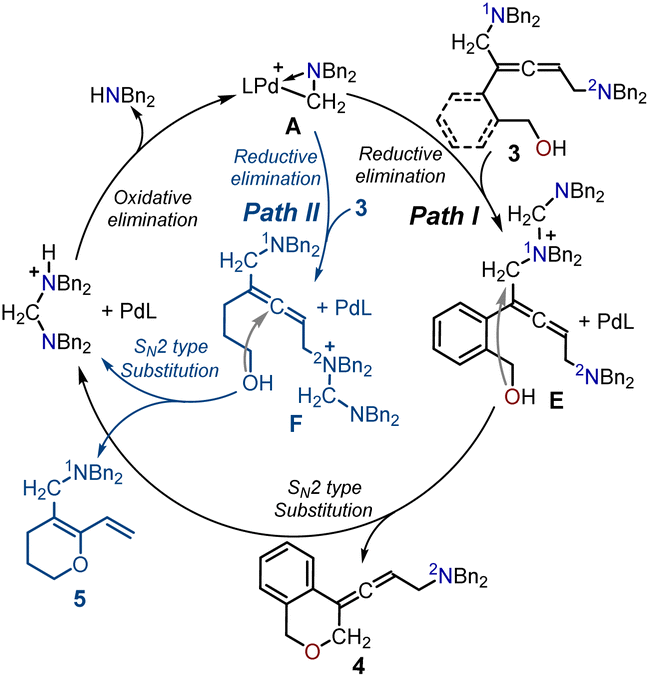 | ||
| Scheme 3 Proposed catalytic cycle for the generation of the allenic amine product 4 and 1,3-diene O-heterocycle 5. | ||
The proposed catalytic cycles in the production of the allenic amine 4 and the 1,3-diene O-heterocycle product 5 are depicted in Scheme 3. By employing (2-(but-3-en-1-yn-1-yl)phenyl)methanol 1 as the substrate in Path I, the 1N atom in intermediate 3 can serve as a nucleophilic site to interact with the cyclopalladated complex A through SN2-type reductive elimination, resulting in the formation of the quaternary ammonium intermediate E and Pd(0). The intermediate E is intramolecularly intercepted by the pendent alcohol through SN2-type substitution to yield the desired product 4. Finally, the SN2-type oxidative addition of the generated amino cation to the Pd(0) center regenerated the active palladium-complex A to complete the catalytic cycle. Moreover, the use of hept-6-en-4-yn-1-ol 1′ as the substrate led to the 2N atom in intermediate 3 acting as a nucleophilic site to attack the Pd complex A, forming the quaternary ammonium intermediate F. The allene in intermediate F was intercepted by the pendent alcohol to give the 1,3-diene O-heterocycle product 5.
The calculated free energy profiles for the generation of intermediate 3 using (2-(but-3-en-1-yn-1-yl)phenyl)methanol 1 as the substrate are presented in Fig. 1, with the aminomethyl cyclopalladated complex INT1 serving as the reference zero point. The binding of the terminal alkene in the enynol substrate 1 to the Pd(II) center led to the endergonic generation of intermediate INT2 (12.9 kcal mol−1). The subsequent migratory insertion of the enyne triple bond into the Pd(II)–C bond took place through the transition state TS1, resulting in the formation of the π-allylpalladium intermediate INT3, with a free energy barrier of 12.2 kcal mol−1. In transition state TS1, the length of the formed C–C bond is 2.19 Å. The intermediate INT3 would then be intercepted by an aminal, leading to the formation of allene-coordinated Pd(0) intermediate INT4 through an SN2-type reductive elimination transition state TS2 (activation free energy = 27.0 kcal mol−1). In the transition state TS2, the length of the formed C–N bond is 1.97 Å. The following SN2-type oxidative addition occurred between Pd(0) and the amino cation via transition state TS3, with an energy barrier of 4.4 kcal mol−1. The release of the allenic 1,5-diamine intermediate 3 led to the exothermic formation of the aminomethyl cyclopalladated complex INT1 (4.3 kcal mol−1) in comparison with substrates 1 and 2. Moreover, the calculated free energy profiles for the generation of intermediate 3' using hept-6-en-4-yn-1-ol 1' as the substrate are presented in Fig. S1.†
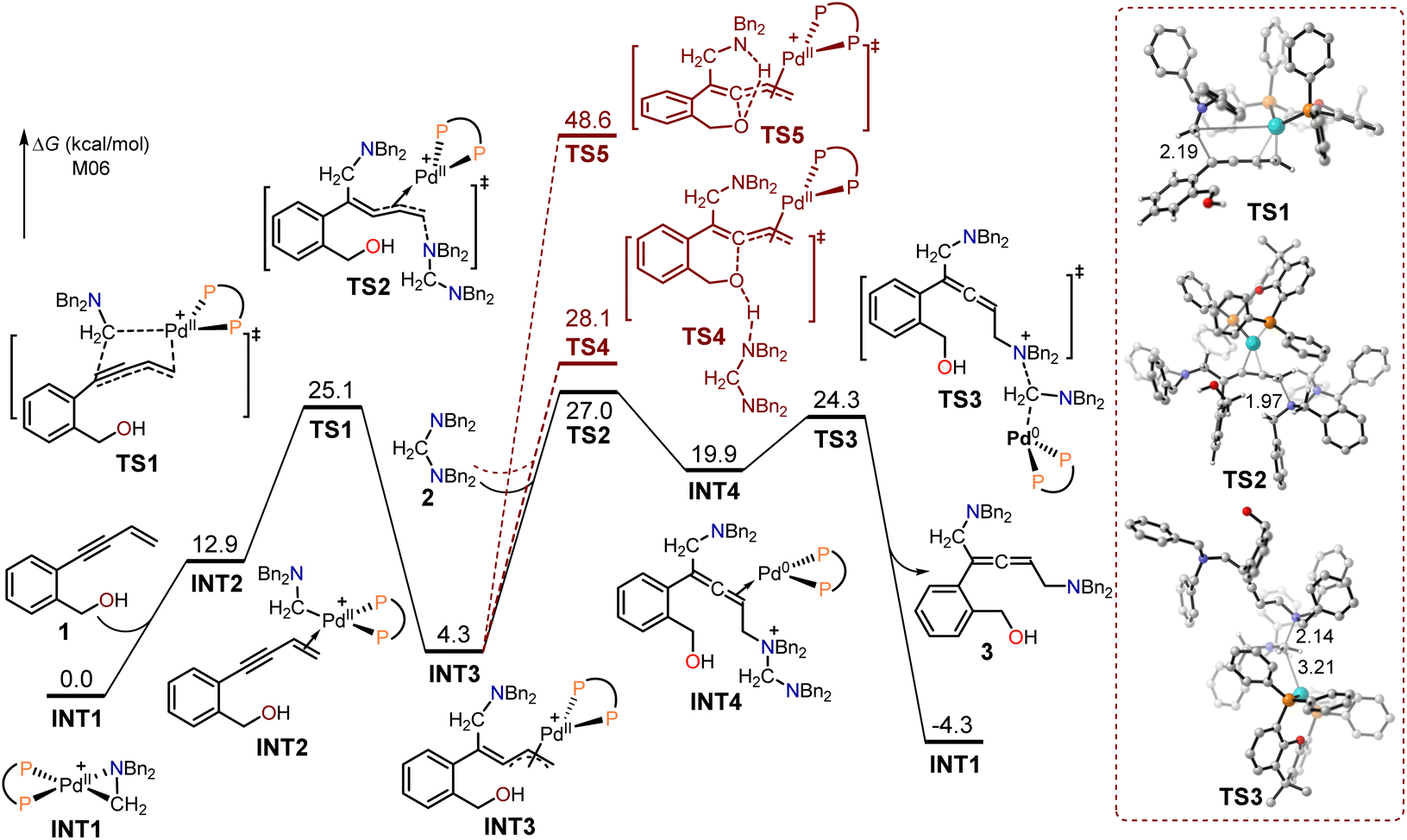 | ||
| Fig. 1 Calculated free energy profiles for the production of intermediate 3 using (2-(but-3-en-1-yn-1-yl)phenyl)methanol 1 as the substrate. | ||
The π-allylpalladium in INT3 can be intercepted by the tethered alcohol to form the 1,3-diene O-heterocycle product 5 through the SN2-type reductive elimination transition states TS4 and TS5. In TS4, the aminal substrate functioned as a base to facilitate reductive elimination, while the intramolecular amido group in transition state TS5 assisted in reductive elimination. The energies of TS4 and TS5 were respectively 1.1 and 21.6 kcal mol−1 higher than that of TS2, indicating that these pathways were unfavorable.
The free energy profiles for the generation of the allenic amine product 4, using (2-(but-3-en-1-yn-1-yl)phenyl)methanol 1 as the substrate are presented in Fig. 2. To investigate the effect of solvation on the system's structure and energy, an explicit DME molecule was included with substrate 3 to allow for the possibility of intermolecular hydrogen bonding between the hydroxyl group of 3 and a solvent oxygen atom. In the absence of the solvent molecule, an artefactual and misleading intramolecular O–H⋯π hydrogen bond would occur. The 1N atom in intermediate 3-DME served as a nucleophilic site to interact with the cyclopalladated complex INT1 through SN2-type reductive elimination to provide the quaternary ammonium intermediate INT5 and Pd(0). The free energy barrier for this SN2-type reductive elimination transition state TS6 is 26.3 kcal mol−1; the formed C–N bond and broken C–Pd bond had lengths of 2.05 and 3.41 Å, respectively. The resulting Pd(0) was captured by the amino cation, produced through the protonation of the aminal with HClO4, to form the cyclopalladated complex, INT1. The generated intermediate INT5 was then intramolecularly intercepted by the pendent alcohol through the SN2-type substitution transition state TS7 with a free energy barrier of 24.2 kcal mol−1, to yield the desired allenic amine product 4.
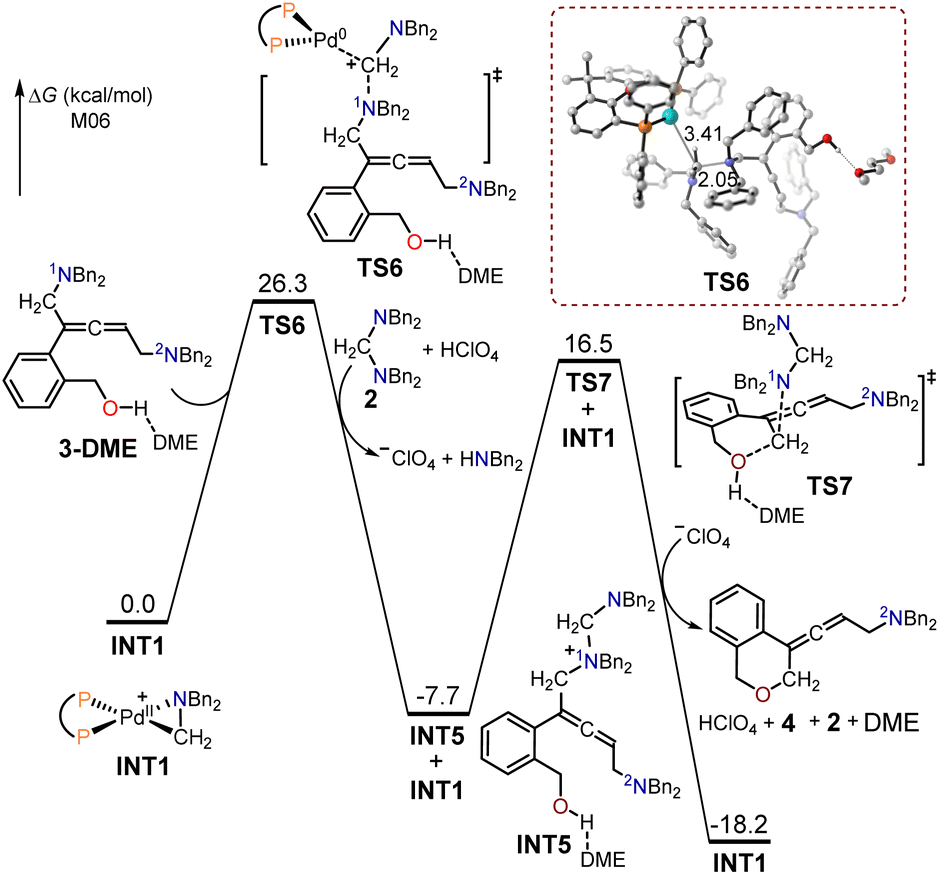 | ||
| Fig. 2 Calculated free energy profiles for the production of the allenic amine product 4 using (2-(but-3-en-1-yn-1-yl)phenyl)methanol 1 as the substrate. | ||
The free energy profiles for the formation of the 1,3-diene O-heterocycle product 5′ using (2-(but-3-en-1-yn-1-yl)phenyl)methanol 1 as the substrate are depicted in Fig. 3. The 2N atom in intermediate 3-DME acted as a nucleophilic site to undergo SN2-type reductive elimination with the cyclopalladated complex INT1 to generate the quaternary ammonium intermediate INT6. The free energy barrier for the SN2-type reductive elimination transition state TS8 (28.8 kcal mol−1) was 2.5 kcal mol−1 higher than that of TS6, as the use of (2-(but-3-en-1-yn-1-yl)phenyl)methanol as the substrate primary resulted in the formation of the allenic amine product 4. In TS8, the formed C–N bond had a length of 2.13 Å, while the broken C–Pd bond was 3.26 Å. The intermediate INT6 is expected to undergo an intramolecular interception by the pendant alcohol through an SN2-type substitution transition state TS9, resulting in the formation of the 1,3-diene O-heterocycle product 5′.
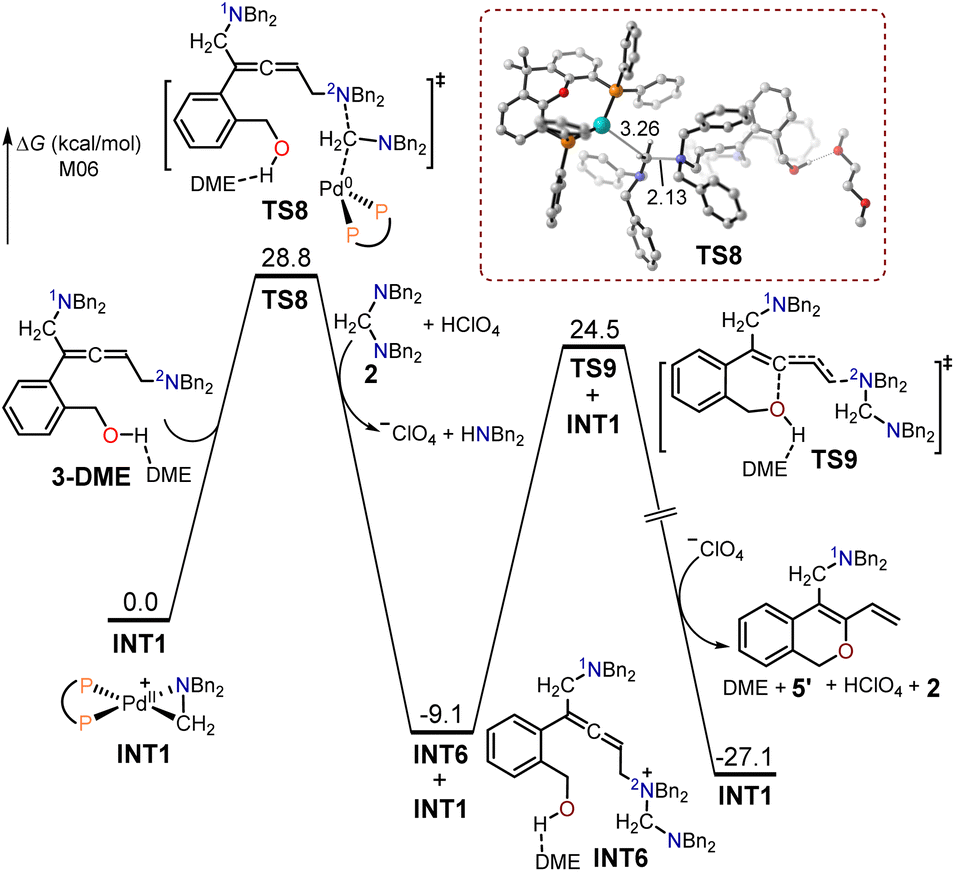 | ||
| Fig. 3 Calculated free energy profiles for the production of the 1,3-diene O-heterocycle product 5′ using (2-(but-3-en-1-yn-1-yl)phenyl)methanol 1 as the substrate. | ||
The free energy profiles for the formation of the allenic amine product 4′ using hept-6-en-4-yn-1-ol 1′ as the substrate are depicted in Fig. 4. The 1N atom in intermediate 3′-DME functioned as a nucleophilic site that interacted with the cyclopalladated complex INT1 via SN2-type reductive elimination to create the quaternary ammonium intermediate INT5′ and Pd(0). The energy barrier for this SN2-type reductive elimination transition state TS6′ was 33.1 kcal mol−1. The resulting intermediate INT5′ was further reacted intramolecularly with the adjacent alcohol via an SN2-type substitution transition state TS7′, with a free energy barrier of 27.7 kcal mol−1, to yield the allenic amine product 4′.
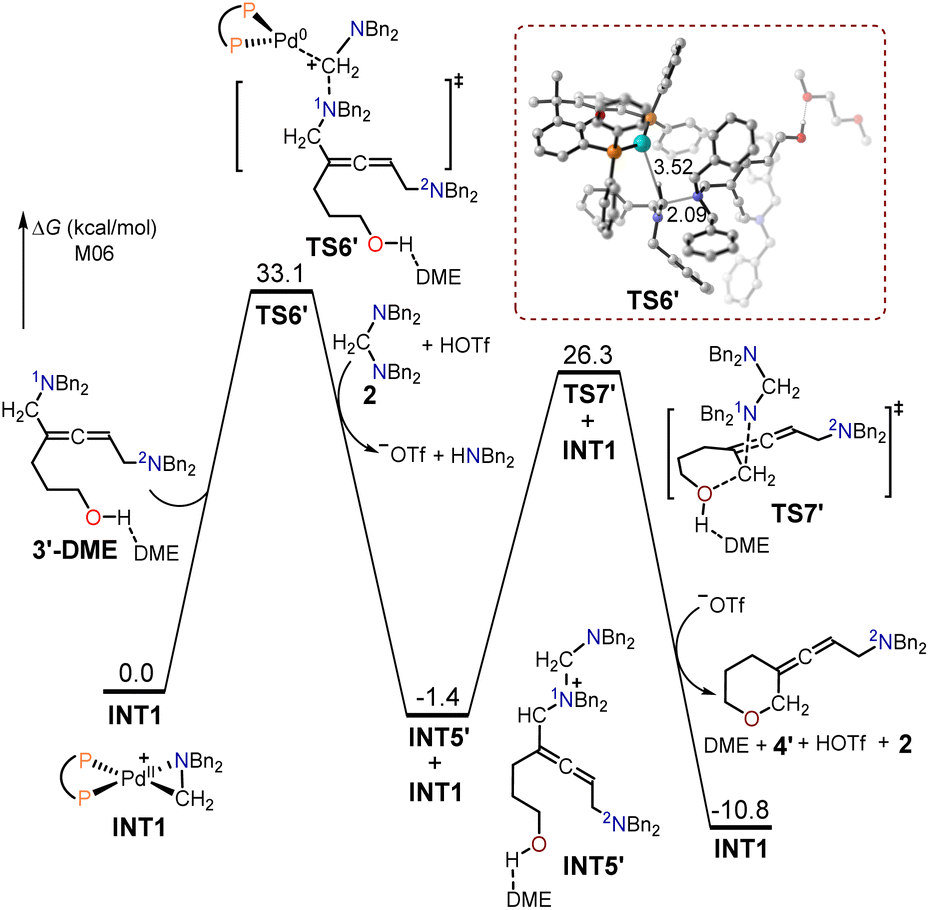 | ||
| Fig. 4 Calculated free energy profiles for the production of the allenic amine product 4′ using hept-6-en-4-yn-1-ol 1′ as the substrate. | ||
The free energy profiles for the synthesis of the 1,3-diene O-heterocycle product 5 from the hept-6-en-4-yn-1-ol 1′ substrate are displayed in Fig. 5. The nitrogen atom (2N) in intermediate 3′-DME functioned as a nucleophilic site to undergo SN2-type reductive elimination with the cyclopalladated complex INT1, producing the quaternary ammonium intermediate INT6′. The energy barrier for the SN2-type reductive elimination transition state TS8′ was 31.8 kcal mol−1. The intermediate INT6′ underwent intramolecular interception by the adjacent alcohol through an SN2-type substitution transition state TS9′, with a free energy barrier of 29.9 kcal mol−1, to yield the 1,3-diene O-heterocycle product 5. TS8′ had a free energy 1.3 kcal mol−1 lower than that of TS6′, and the free energy of the 1,3-diene O-heterocycle product 5 was 12.4 kcal mol−1 lower than that of the allenic amine product 4′. These findings elucidate that the primary product formed was the 1,3-diene O-heterocycle product 5 when using hept-6-en-4-yn-1-ol as the substrate.
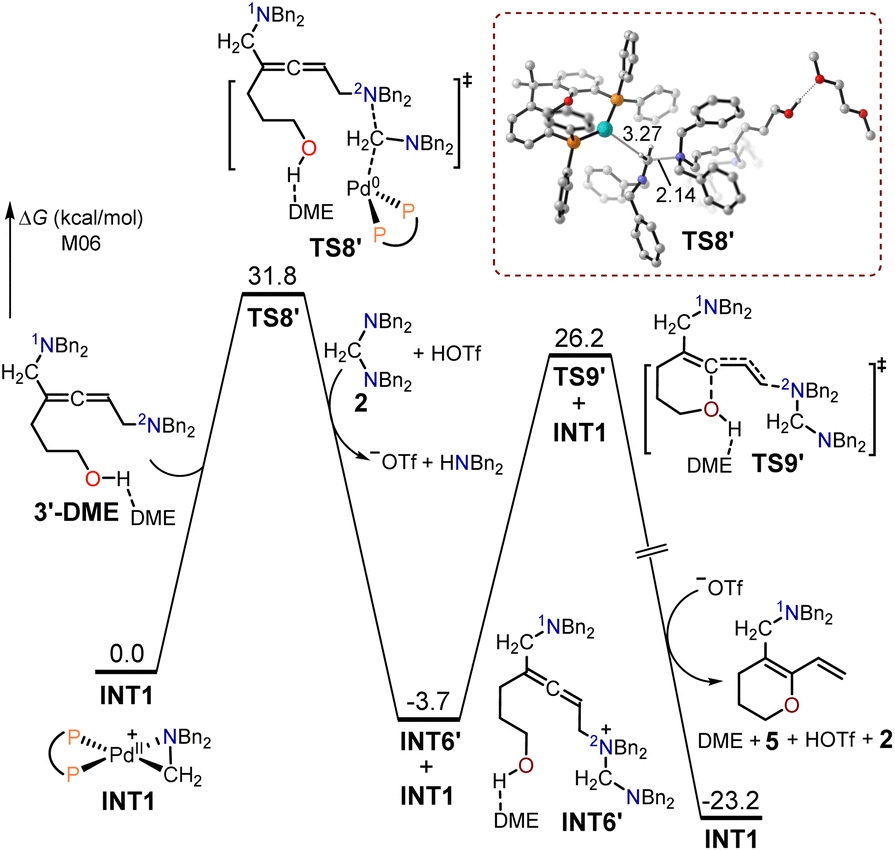 | ||
| Fig. 5 Calculated free energy profiles for the production of the 1,3-diene O-heterocycle product 5 using hept-6-en-4-yn-1-ol 1′ as the substrate. | ||
The free energy barrier for TS6 was 6.8 kcal mol−1 lower than that of TS6′, whereas the free energy barriers for TS8 was 3.0 kcal mol−1 lower than that of TS8′. The reactivity of the 1N atom decreased when the substrate was changed from (2-(but-3-en-1-yn-1-yl)phenyl)methanol 1 to hept-6-en-4-yn-1-ol 1′, whereas the reactivity of the 2N atom remained unchanged. This difference in the reactivities of 1N and 2N contributed to the varying selectivity observed with different substrates.
The local nucleophilicity index Nk values were calculated to analyze the nucleophilicity of the nitrogen atoms (1N and 2N) in intermediates 3 and 3′. The calculated Nk values for 1N and 2N in intermediate 3 were respectively 1.294 and 0.122 eV, while the values were 1.464 and 0.003 eV for intermediate 3′ (Fig. 6). The nucleophilicity of 1N was stronger than that of 2N in both intermediates. Notably, the nucleophilicity of 1N and 2N in non-cyclic allenic 1,5-diamine intermediates 3 and 3′ remained unaffected when using (2-(but-3-en-1-yn-1-yl)phenyl)methanol 1 or hept-6-en-4-yn-1-ol 1′ as substrates.
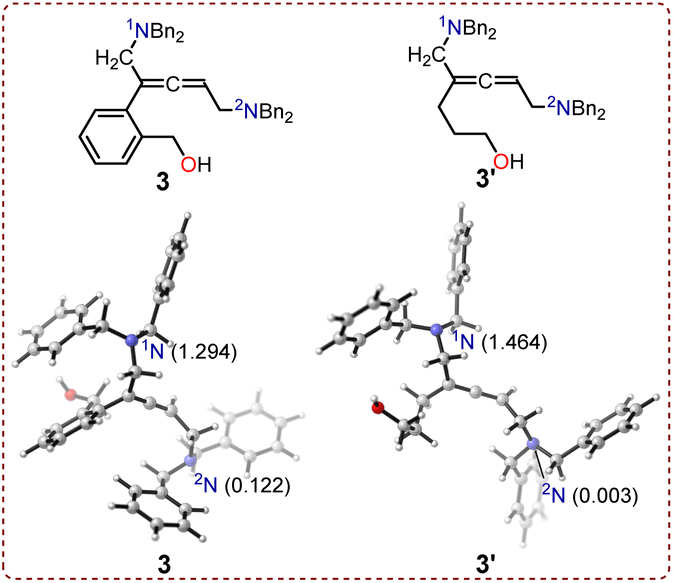 | ||
| Fig. 6 Calculated local nucleophilicity index Nk values. | ||
Distortion–interaction energy analysis was used to elucidate the disparity in the free energy barrier between transition states TS6 and TS6′. The distortion energy of TS6′ was 3.0 kcal mol−1 higher than that of TS6, while their interaction energies were similar (Table 1). The distortion energies of intermediates 3-DME and 3′-DME in TS6 and TS6′ were 9.5 and 11.2 kcal mol−1, respectively. The distortion energy of INT1 in both TS6 and TS6′ were similar. Analysis of the distortion energy revealed that the difference in energy barriers of TS6 and TS6′ was controlled by the distortion energies of intermediates 3-DME and 3′-DME.
| ΔE‡dist(3-DME or 3′-DME) | ΔE‡dist(INT1) | ΔE‡dist | ΔE‡int | ΔE‡ | |
|---|---|---|---|---|---|
| TS6 | 9.5 | 35.9 | 45.4 | −15.9 | 29.5 |
| TS6′ | 11.2 | 37.2 | 48.4 | −16.5 | 32.0 |
The structural information for intermediate 3-DME and transition state TS6 are shown in Fig. 7. In the optimized structure of intermediate 3-DME, the dihedral angle of 1C–1N–2C–3C is 95.7°, whereas this dihedral angle changes to 54.7° in TS6. In the optimized structure of intermediate 3′-DME, the dihedral angle of 1C–1N–2C–3C is −64.8°, which changes to 55.2° in TS6′ (Fig. 8). These calculated results indicated that the difference in free energy barrier between TS6 and TS6′ was controlled by the distortion of the benzyl group associated with 1N in intermediates 3-DME and 3′-DME.
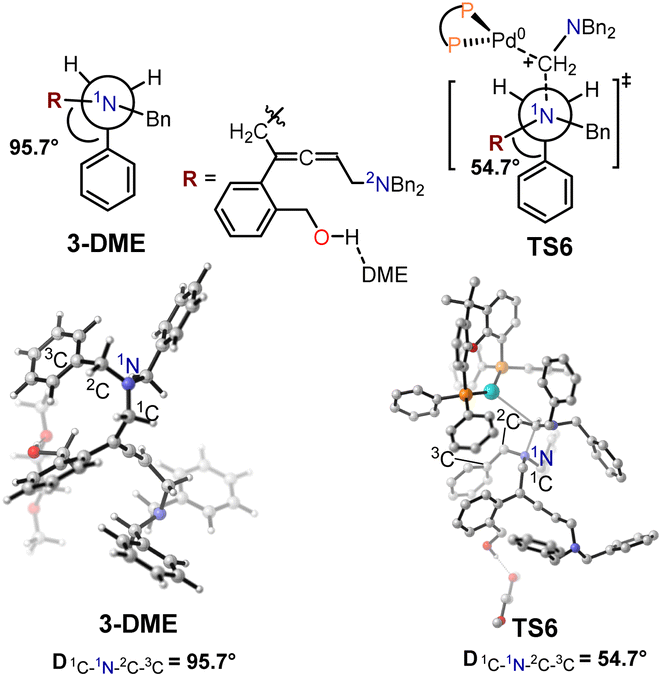 | ||
| Fig. 7 Structural information for intermediate 3-DME and transition state TS6. | ||
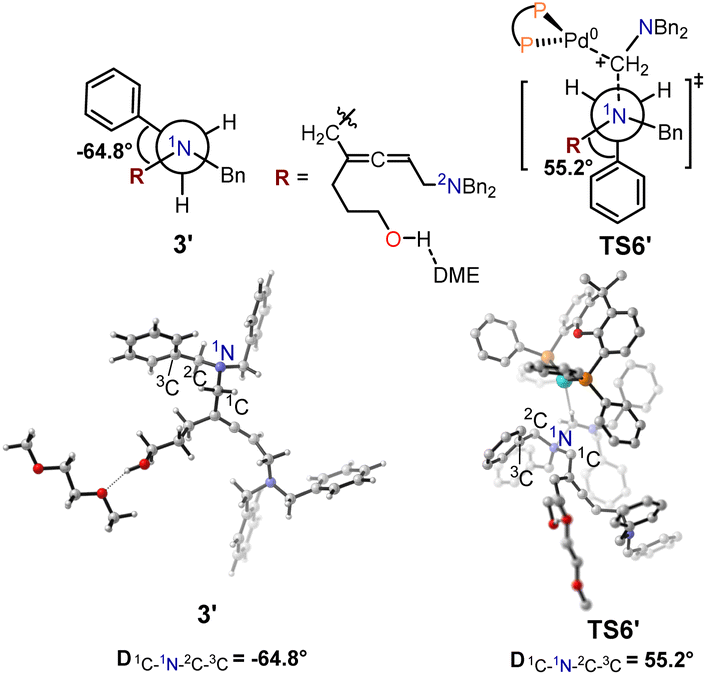 | ||
| Fig. 8 Structural information for intermediate 3'-DME and transition state TS6'. | ||
To further investigate the regioselectivity regulatory mechanism behind these Pd-catalyzed cyclization reactions, NCI analysis was conducted for transition states TS6 and TS6′. The π–π stacking of two phenyl rings in transition state TS6 occurred when (2-(but-3-en-1-yn-1-yl)phenyl)methanol 1 was used as the substrate (Fig. 9). By contrast, in transition state TS6′, there is solely a C–H⋯π interaction between the alkyl chain and phenyl group. NCI calculations revealed that the π–π stacking in TS6 are stronger than the C–H⋯π interactions in TS6′. This difference results in a lower distortion energy for TS6 compared to TS6′, which consequently reduces the reactivity of the 1N atom when the substrate is changed from (2-(but-3-en-1-yn-1-yl)phenyl)methanol 1 to hept-6-en-4-yn-1-ol 1′.
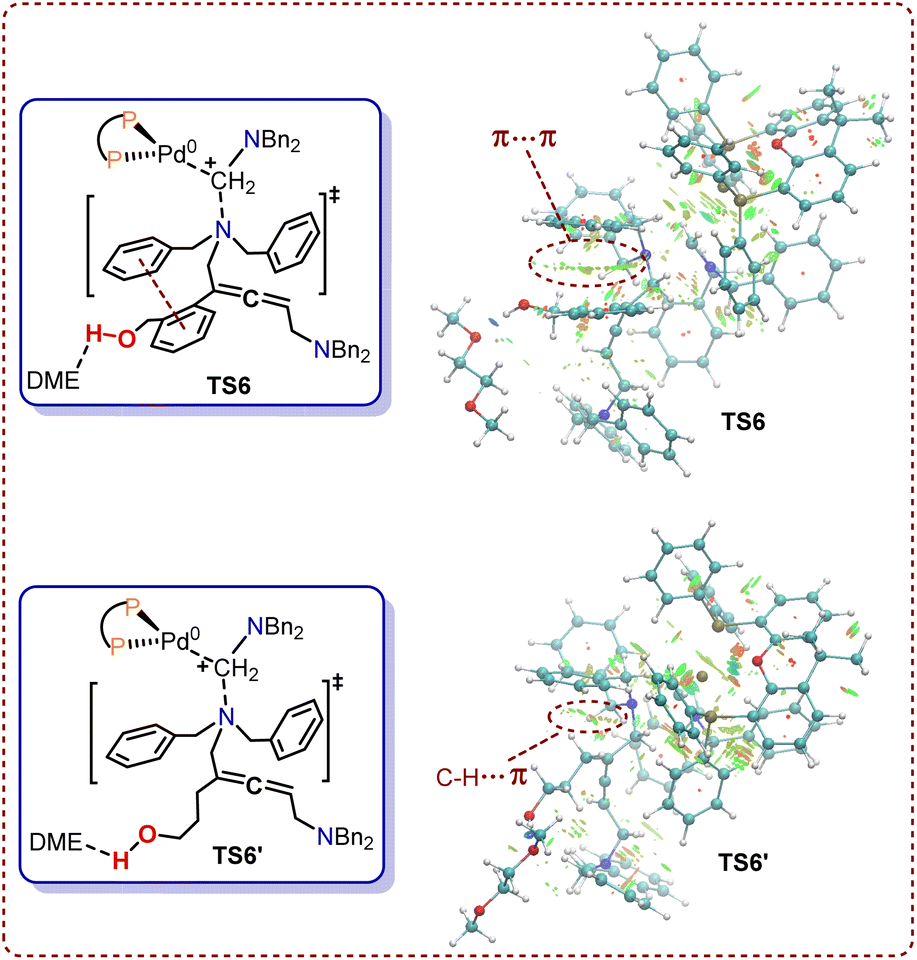 | ||
| Fig. 9 Noncovalent interactions of TS6 and TS6′ (blue, attraction; green, weak interaction; red, steric effect). | ||
Conclusions
In this work, DFT calculations were used to explore the mechanism of Huang's Pd-catalyzed cyclization reactions with an emphasis on the regioselectivity regulation mechanism. Theoretical calculations revealed that these reactions proceeded via migratory insertion, SN2-type reductive elimination, and oxidative addition to give the non-cyclic allenic 1,5-diamine intermediate. The allenic amine and 1,3-diene O-heterocycle products were then obtained through subsequent reductive elimination, SN2-type substitution, and oxidative addition.The reactivity of the 1N atom decreased when (2-(but-3-en-1-yn-1-yl)phenyl)methanol instead of hept-6-en-4-yn-1-ol was used as the substrate, while the reactivity of the 2N atom exhibited minimal change. This difference in reactivities of 1N and 2N contributed to the varying selectivity observed with different substrates. Distortion–interaction energy analysis and NCI calculations indicated that the presence of π–π stacking in intermediate 3-DME led to the lower distortion energy of TS6 than that of TS6′, consequently reducing the reactivity of the 1N atom upon changing the substrate from (2-(but-3-en-1-yn-1-yl)phenyl)methanol to hept-6-en-4-yn-1-ol for regioselective control.
Data availability
The data that support the findings of this study have been included in the main text and ESI† and are available from the corresponding author upon reasonable request.Author contributions
S. Liu conceived and designed the project. S. Liu designed the computational studies. D. Zhang, Y. Gong, L. Ma and L. Li performed the DFT calculations. S. Liu and W. Chen prepared the manuscript, D. Zhang, Y. Gong, L. Ma and L. Li prepared the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22303010 and 22271034). We are thankful to the Scientific Research Fund of Chongqing University of Arts and Sciences (no. R2020SHH01) for financial support.References
- (a) S. Kaltenberger and M. V. Gemmeren, Controlling Reactivity and Selectivity in the Nondirected C–H Activation of Arenes with Palladium, Acc. Chem. Res., 2023, 56(18), 2459–2472 CrossRef CAS PubMed; (b) K. Yamatsugu and M. Kanai, Catalytic Approaches to Chemo- and Site-Selective Transformation of Carbohydrates, Chem. Rev., 2023, 123(10), 6793–6838 CrossRef CAS PubMed; (c) F. Juliá, Q. Shao, M. Duan, M. B. Plutschack, F. Berger, J. Mateos, C. Lu, X.-S. Xue, K. N. Houk and T. Ritter, High Site Selectivity in Electrophilic Aromatic Substitutions: Mechanism of C–H Thianthrenation, J. Am. Chem. Soc., 2021, 143(39), 16041–16054 CrossRef PubMed; (d) S. Bhadra and H. Yamamoto, Substrate Directed Asymmetric Reactions, Chem. Rev., 2018, 118, 3391–3446 CrossRef CAS PubMed.
- (a) L. Veth, H. A. Grab and P. Dydio, Recent Trends in Group 9 Catalyzed C–H Borylation Reactions: Different Strategies To Control Site-, Regio-, and Stereoselectivity, Synthesis, 2022, 54, 3482–3498 CrossRef CAS; (b) C. Nájera, I. P. Beletskaya and M. Yus, Metal-catalyzed regiodivergent organic reactions, Chem. Soc. Rev., 2019, 48, 4515–4618 RSC.
- (a) J. F. Hartwig, Regioselectivity of the borylation of alkanes and arenes, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC; (b) B. E. Haines, Y. Saito, Y. Segawa, K. Itami and D. G. Musaev, Flexible Reaction Pocket on Bulky Diphosphine–Ir Complex Controls Regioselectivity in para-Selective C–H Borylation of Arenes, ACS Catal., 2016, 6, 7536–7546 CrossRef CAS; (c) K. Liao, T. C. Pickel, V. Boyarskikh, J. Bacsa, D. G. Musaev and H. M. L. Davies, Nature, 2017, 551, 609–613 CrossRef CAS PubMed; (d) M. Duan, C. D. Díaz-Oviedo, Y. Zhou, X. Chen, P. Yu, B. List, K. N. Houk and Y. Lan, Chiral Phosphoric Acid Catalyzed Conversion of Epoxides into Thiiranes: Mechanism, Stereochemical Model, and New Catalyst Design, Angew. Chem., Int. Ed., 2022, 61, e202113204 CrossRef CAS PubMed.
- (a) H. Chen, M. Farizyan and M. van Gemmeren, Regioselective Olefination of 3-Substituted Five-Membered Heteroarenes, Eur. J. Org Chem., 2020, 40, 6318–6327 CrossRef; (b) S. Negretti, A. R. H. Narayan, K. C. Chiou, P. M. Kells, J. L. Stachowski, D. A. Hansen, L. M. Podust, J. Montgomery and D. H. Sherman, Directing Group-Controlled Regioselectivity in an Enzymatic C–H Bond Oxygenation, J. Am. Chem. Soc., 2014, 136, 4901–4904 CrossRef CAS PubMed; (c) J. M. Medina, J. L. Mackey, N. K. Garg and K. N. Houk, The Role of Aryne Distortions, Steric Effects, and Charges in Regioselectivities of Aryne Reactions, J. Am. Chem. Soc., 2014, 136, 15798–15805 CrossRef CAS PubMed; (d) A. K. Hubbell and G. W. Coates, Nucleophilic Transformations of Lewis Acid-Activated Disubstituted Epoxides with Catalyst-Controlled Regioselectivity, J. Org. Chem., 2020, 85, 13391–13414 CrossRef CAS PubMed.
- (a) B. Zhou, T.-D. Tan, X.-Q. Zhu, M. Shang and L.-W. Ye, Reversal of Regioselectivity in Ynamide Chemistry, ACS Catal., 2019, 9, 6393–6406 CrossRef CAS; (b) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (c) Y. Xia and G. Dong, Temporary or removable directing groups enable activation of unstrained C–C bonds, Nat. Rev. Chem, 2020, 4, 600–614 CrossRef CAS PubMed; (d) G. Xia, J. Weng, L. Liu, P. Verma, Z. Li and J.-Q. Yu, Reversing conventional site-selectivity in C(sp3)–H bond activation, Nat. Chem., 2019, 11, 571–577 CrossRef CAS PubMed; (e) T. Zhang, K. Zhong, Z.-K. Lin, L. Niu, Z.-Q. Li, R. Bai, K. M. Engle and Y. Lan, Revised Mechanism of C(sp3)−C(sp3) Reductive Elimination from Ni(II) with the Assistance of a Z-Type Metalloligand, J. Am. Chem. Soc., 2023, 145, 2207–2218 CrossRef CAS PubMed.
- (a) W.-T. Zhao, H. Meng, J.-N. Lin and W. Shu, Ligand-Controlled Nickel-Catalyzed Regiodivergent Cross-Electrophile Alkyl-Alkyl Couplings of Alkyl Halides, Angew. Chem., Int. Ed., 2023, 62, e202215779 CrossRef CAS PubMed; (b) L. Zhao, Y. Zhu, M. Liu, L. Xie, J. Liang, H. Shi, X. Meng, Z. Chen, J. Han and C. Wang, Ligand-Controlled NiH-Catalyzed Regiodivergent Chain-Walking Hydroalkylation of Alkenes, Angew. Chem., Int. Ed., 2022, 61, e202204716 CrossRef CAS PubMed; (c) Q. Pan, Y. Ping, Y. Wang, Y. Guo and W. Kong, Ni-Catalyzed Ligand-Controlled Regiodivergent Reductive Dicarbofunctionalization of Alkenes, J. Am. Chem. Soc., 2021, 143, 10282–10291 CrossRef CAS PubMed; (d) T. Xu, F. Sha and H. Alper, Highly Ligand-Controlled Regioselective Pd-Catalyzed Aminocarbonylation of Styrenes with Aminophenols, J. Am. Chem. Soc., 2016, 138, 6629–6635 CrossRef CAS PubMed; (e) F. Schoenebeck and K. N. Houk, Ligand-Controlled Regioselectivity in Palladium-Catalyzed Cross Coupling Reactions, J. Am. Chem. Soc., 2010, 132, 2496–2497 CrossRef CAS PubMed.
- (a) H. J. Davis and R. J. Phipps, Harnessing non-covalent interactions to exert control over regioselectivity and site-selectivity in catalytic reactions, Chem. Sci., 2017, 8, 864–877 RSC; (b) F. O'Hara, D. G. Blackmond and P. S. Baran, Radical-Based Regioselective C–H Functionalization of Electron-Deficient Heteroarenes: Scope, Tunability, and Predictability, J. Am. Chem. Soc., 2013, 135, 12122–12134 CrossRef PubMed; (c) C. Schnepel and N. Sewald, Enzymatic Halogenation: A Timely Strategy for Regioselective C−H Activation, Chem.–Eur. J., 2017, 23, 12064–12086 CrossRef CAS PubMed; (d) C. Wang, L. Luo and H. Yamamoto, Metal-Catalyzed Directed Regio- and Enantioselective Ring-Opening of Epoxides, Acc. Chem. Res., 2016, 49, 193–204 CrossRef CAS PubMed.
- (a) P.-S. Wang and L.-Z. Gong, Palladium-Catalyzed Asymmetric Allylic C–H Functionalization: Mechanism, Stereo- and Regioselectivities, and Synthetic Applications, Acc. Chem. Res., 2020, 53, 2841–2854 CrossRef CAS PubMed; (b) H. Yue, C. Zhu, R. Kancherla, F. Liu and M. Rueping, Regioselective Hydroalkylation and Arylalkylation of Alkynes by Photoredox/Nickel Dual Catalysis: Application and Mechanism, Angew. Chem., Int. Ed., 2020, 132, 5787–5795 CrossRef; (c) S. Deng, C. Qu, Y. Jiao, W. Liu and F. Shi, Insights into 2-Indolylmethanol-Involved Cycloadditions: Origins of Regioselectivity and Enantioselectivity, J. Org. Chem., 2020, 85, 11641–11653 CrossRef CAS PubMed; (d) B. Zhang, X. Xu, L. Tao, Z. Lin and W. Zhao, Rhodium-Catalyzed Regiodivergent Synthesis of Alkylboronates via Deoxygenative Hydroboration of Aryl Ketones: Mechanism and Origin of Selectivities, ACS Catal., 2021, 11, 9495–9505 CrossRef CAS; (e) Q. Zhang, D. Dong and W. Zi, Palladium-Catalyzed Regio- and Enantioselective Hydrosulfonylation of 1,3-Dienes with Sulfinic Acids: Scope, Mechanism, and Origin of Selectivity, J. Am. Chem. Soc., 2020, 142, 15860–15869 CrossRef CAS PubMed.
- (a) H. Yu, B. Yu, H. Zhang and H. Huang, Palladium-Catalyzed Aminomethylation and Cyclization of Enynol to O-Heterocycle Confined 1,3-Dienes, Org. Lett., 2021, 23, 3891–3896 CrossRef CAS PubMed; (b) B. Yu, M. Yu and H. Huang, Palladium-catalyzed ring-closing reaction of enynols with aminals via methylene transfer and C–N bond activation, Tetrahedron Chem, 2023, 7, 100042 CrossRef CAS.
- M. J. Frisch, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016. The full author list is given in the ESI Search PubMed.
- (a) A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetticorrelation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (c) H. L. Schmider and A. D. Becke, Optimized density functionals from the extended G2 test set, J. Chem. Phys., 1998, 108, 9624–9631 CrossRef CAS; (d) K. Zhong, S. Liu, X. He, H. Ni, W. Lai, W. Gong, C. Shan, Z. Zhao, Y. Lan and R. Bai, Oxidative cyclopalladation triggers the hydroalkylation of alkynes, Chin. Chem. Lett., 2023, 34, 108339 CrossRef CAS.
- (a) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Ab initio study of solvated molecules: a new implementation of the polarizable continuum model, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS; (b) T. Liu, R. Duan, Y. Wang, S. Li, L. Qu, J. Song, Q. Liu and Y. Lan, Mechanistic investigation of zwitterionic MOF-catalyzed enyne annulation using UNLPF-14-MnIII as catalyst, Chin. Chem. Lett., 2022, 33, 4281–4286 CrossRef CAS.
- (a) Y. Zhao and D. G. Truhlar, A New Local Density Functional for Main-Group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed; (b) Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry,mThermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (c) Y. Zhao and D. G. Truhlar, Density Functionals with Broad Applicability in Chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- C. Y. Legault, CYLView, 1.0b, Université de Sherbrooke, Quebec, Canada, 2009, https://www.cylview.org, accessed 1 October 2017 Search PubMed.
- (a) F. Jensen, The magnitude of intramolecular basis set superposition error, Chem. Phys. Lett., 1996, 261, 633–636 CrossRef CAS; (b) M. L. Senent and S. Wilson, Intramolecular basis set superposition errors, Int. J. Quantum Chem., 2001, 82, 282–292 CrossRef CAS; (c) F. Jensen, Using valence bond methods to estimate intramolecular basis set superposition errors, J. Chem. Phys., 2017, 146, 184109 CrossRef; (d) F. Jensen, Basis Set Superposition Errors Are Partly Basis Set Imbalances, J. Chem. Theory Comput., 2024, 20, 767–774 CrossRef CAS PubMed.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing Noncovalent Interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed; (b) T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- (a) L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs Diels-Alder reactions, Tetrahedron, 2002, 58, 4417–4423 CrossRef CAS; (b) L. R. Domingo, E. Chamorro and P. Pérez, Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study, J. Org. Chem., 2008, 73, 4615–4624 CrossRef CAS PubMed.
- (a) L. R. Domingo and P. Pérez, The nucleophilicity N index in organic chemistry, Org. Biomol. Chem., 2011, 9, 7168–7175 RSC; (b) L. R. Domingo and P. Pérez, Global and local reactivity indices for electrophilic/nucleophilic free radicals, Org. Biomol. Chem., 2013, 11, 4350–4358 RSC.
- P. K. Chattaraj, S. Duleya and L. R. Domingo, Understanding local electrophilicity/nucleophilicity activation through a single reactivity difference index, Org. Biomol. Chem., 2012, 10, 2855–2861 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06552b |
| This journal is © The Royal Society of Chemistry 2024 |