
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the photophysical properties of rhodamines using a spectroscopic single-molecule fluorescence method†
Shangyuan Deng‡
ab,
Deqi Yi‡ab,
Thitima Rujiralaic,
Qinghua Ren§
 d,
Chuang Tan§ab and
Jie Ma§ab
d,
Chuang Tan§ab and
Jie Ma§ab
aSchool of Physics, Sun Yat-sen University, Guangzhou 510275, China. E-mail: tanch26@mail.sysu.edu.cn; majie6@mail.sysu.edu.cn
bState Key Laboratory of Optoelectronic Materials and Technologies, Sun Yat-sen University, Guangzhou 510006, China
cDivision of Physical Science, Faculty of Science, Prince of Songkla University, Songkhla, 90110, Thailand
dDepartment of Chemistry, Shanghai University, Shanghai 200444, China. E-mail: qinghua.ren@shu.edu.cn
First published on 6th December 2024
Abstract
The photophysical properties of rhodamine molecules play a critical role in their performance across various applications. The spectroscopic single-molecule fluorescence (sSMF) technique overcomes the limitations of conventional SMF by distinguishing individual fluorophores based on their emission spectra. This enables precise measurement and direct comparison of photophysical properties among distinct molecules under identical conditions, without requiring separation of molecules. In this study, using a custom sSMF instrument, we successfully identified individual rhodamine B molecules and their various N-dealkylated intermediates, allowing for simultaneous investigation of their photophysical properties. Notably, we observed that rhodamine B undergoing a single dealkylation step exhibited a striking enhancement in photostability compared to its fully intact counterparts and those undergoing two dealkylation steps. This enhancement persisted across various buffer conditions, including different pH levels and the presence or absence of an oxygen scavenger system (OSS). Despite these differences in photostability, time-dependent density functional theory (TD-DFT) calculations revealed that all these rhodamine molecules examined shared a similar energy gap (∼0.6 eV) between their first excited singlet and triplet states.
Introduction
Over the past several decades, single-molecule fluorescence (SMF) techniques have become an essential tool for addressing various biological questions regarding RNA1 or protein folding,2 enzyme kinetics,3 and protein-DNA interactions.4 Interestingly, although SMF was originally developed as a spectroscopic technique,5 most early applications primarily focused on measuring fluorescence intensity, often overlooking the spectral information. Recently, however, there has been growing interest in single-molecule spectrum measurement, which leverages the unique spectral “fingerprint” within each molecule's fluorescence spectrum for a wide range of applications. These include multi-color single-molecule detection in DNA sequencing,6 multiplexed super-resolution imaging,7 chemical reaction mechanism probing,8 etc. Generally, in these and other studies, spectrally resolved single-molecule imaging was achieved using either gratings or dispersive prisms, followed by various calibration methods.6–12 Advances in these instruments have made the spectroscopic single-molecule fluorescence (sSMF) technique a more accessible tool for use in diverse research contexts.In addition, fluorophores, such as organic dyes and fluorescent proteins, are indispensable for SMF experiments. Their photophysical properties, particularly brightness and photostability, play a critical role in determining their suitability for various applications.13–15 For instance, single-molecule tracking experiments often require prolonged observation periods,13,16 which is challenging due to the limited photobleaching lifetime of fluorophores. Photobleaching commonly occurs through reactions of fluorophores with reactive oxygen species (ROS), such as singlet oxygen (1O2).13,17 Although the use of an oxygen scavenging system (OSS) can prolong the lifespan of fluorophores to some extent,14,18 the improvement remains limited. Therefore, there is a pressing need to deepen our understanding of the photobleaching mechanism and explore structural modifications to fluorophores to enhance their photostability.
The conventional SMF method, which mainly relies on fluorescence intensity detection, has been widely employed to characterize the brightness and photobleaching lifetime of fluorophores.19 However, this approach would face challenges under certain conditions. For example, synthesized fluorophores may contain fluorescent impurities or contaminants. Even highly pure fluorophores can degrade or be oxidized over time, resulting in unwanted reaction byproducts in the samples. Additionally, fluorophores like fluorescent proteins could exhibit significant molecular heterogeneity.20 In such case, the conventional SMF method struggles to distinguish the target molecules from others, potentially introducing bias in the measurements of average brightness and photobleaching lifetimes. In contrast, sSMF method effectively addresses these limitations by identifying each fluorophore molecule based on its unique emission spectrum. This capability allows for precise measurement and comparison of photophysical properties of different fluorophore molecules under identical conditions, thereby overcoming the limitations of the conventional method.
In this study, we employed a custom-built sSMF instrument to investigate the photophysical properties of rhodamine molecules. Rhodamine was chosen as the model system for three key reasons. First, rhodamine represents an important class of dye molecules that have been extensively utilized in chemical and biological research.21,22 For instance, rhodamine and its derivatives are among the most commonly used fluorophores in stimulated emission depletion (STED) microscopy,15 owing to their high brightness and photostability. There is also ongoing interest in enhancing their characteristics, including brightness,15,23 photostability,8,15 cell permeability,24 fluorogenicity25 and Stokes Shift,15,26 to meet the demands of diverse fluorescence imaging applications. Second, rhodamine-like fluorophores with N-alkyl substituents are well-known for their propensity to undergo N-dealkylation, a process that typically causes a hypsochromic shift of approximately 10–15 nm in the emission spectra of the dealkylated products.17 This offers a unique opportunity to monitor this photochemical transformation in real-time using sSMF and potentially identify each intermediate species. Third, the emission spectra of various rhodamine molecules involved in the N-dealkylation process, including both reagents and products, can be distinctively identified. This capability enables us to examine their photophysical properties under identical conditions and make direct comparisons. It is noteworthy that recently Zhang et al.27 have employed the sSMF technique to explore the origins of large spectral heterogeneity in rhodamine B molecules. Additionally, Butkevich et al.8 have utilized high-performance liquid chromatography (HPLC) and nuclear magnetic resonance spectroscopy (NMR) to separate and characterize various rhodamine B derivatives generated during its N-dealkylation process. However, the photophysical properties of these rhodamine B derivatives, such as photostability and brightness, have not been directly assessed and compared at the single-molecule level across various buffer conditions. Consequently, this work aims to harness a custom sSMF system (which differs from previous sSMF instruments using a dual-optical path to split the light into two channels7,11 and can make a full utilization of the field of view for spectrally resolved single-molecule imaging) and an improved spectral peak analysis method (i.e., skewed Gaussian fitting method28 which can enhance the precision in locating the peak wavelength in a single-molecule spectrum) to investigate the disparities in photophysical properties among rhodamine B and its derivatives within the same solution, as well as the changes in these properties under different solution conditions. We also aim to elucidate the underlying causes for these observations. These investigations are anticipated to provide valuable insights for the future design and optimization of fluorophores with enhanced photophysical properties.
Results and discussion
The custom sSMF system and measured single-molecule fluorescence spectra of Cy3 and TAMRA
In our experiments, the sSMF system was built on a commercial total internal reflective fluorescence (TIRF) microscope (Ti-E 2000, Nikon Instruments Inc.). Fluorophores were excited using a 532 nm laser (MW-SL-532, Changchun Laser Optoelectronics) under TIRF illumination. The fluorescence signal was collected with a 100× objective (CFI Apochromat TIRF 100XC Oil, Nikon Instruments Inc.) and an intermediate image was formed at the front focal plane of relay lens L3, as shown in Fig. 1a. This design allowed full utilization of the field of view (FOV) in both modes. Specifically, two reflective mirrors, M3 and M4, mounted on a translation stage, guided the light either directly into the imaging lens, L4 (for imaging mode), or through a prism to form spectral images (for spectral mode) on a highly sensitive COMS camera (Prime 95B Scientific CMOS camera, Teledyne Photometrics). The typical images of fluorescent molecules acquired in both imaging and spectral modes are presented in Fig. 1b and c. After calibrating the wavelength–pixel relationship, our sSMF system demonstrated precise spectral measurement capabilities spanning from 517 to 670 nm. As an example, Fig. 1d shows typical single-molecule emission spectra of cyanine 3 (Cy3) and carboxytetramethylrhodamine (TAMRA), non-specifically adhered to the coverslip surface under solution conditions. To precisely determine the peak position of a single-molecule emission spectrum, we applied a skewed Gaussian function to fit the data around the peak.28 By analysing 80 Cy3 and 40 TAMRA single-molecule spectra, we determined average peak positions of 567.1 (±1.9) nm for Cy3 and 584.3 (±8.5) nm for TAMRA, closely matching their respective expected values (570 nm for Cy3 and 580 nm for TAMRA). Furthermore, both Cy3 and TAMRA peak positions exhibited single-peak distributions (Fig. 1e and f), confirming the presence of a uniform molecular species for each of them in solution.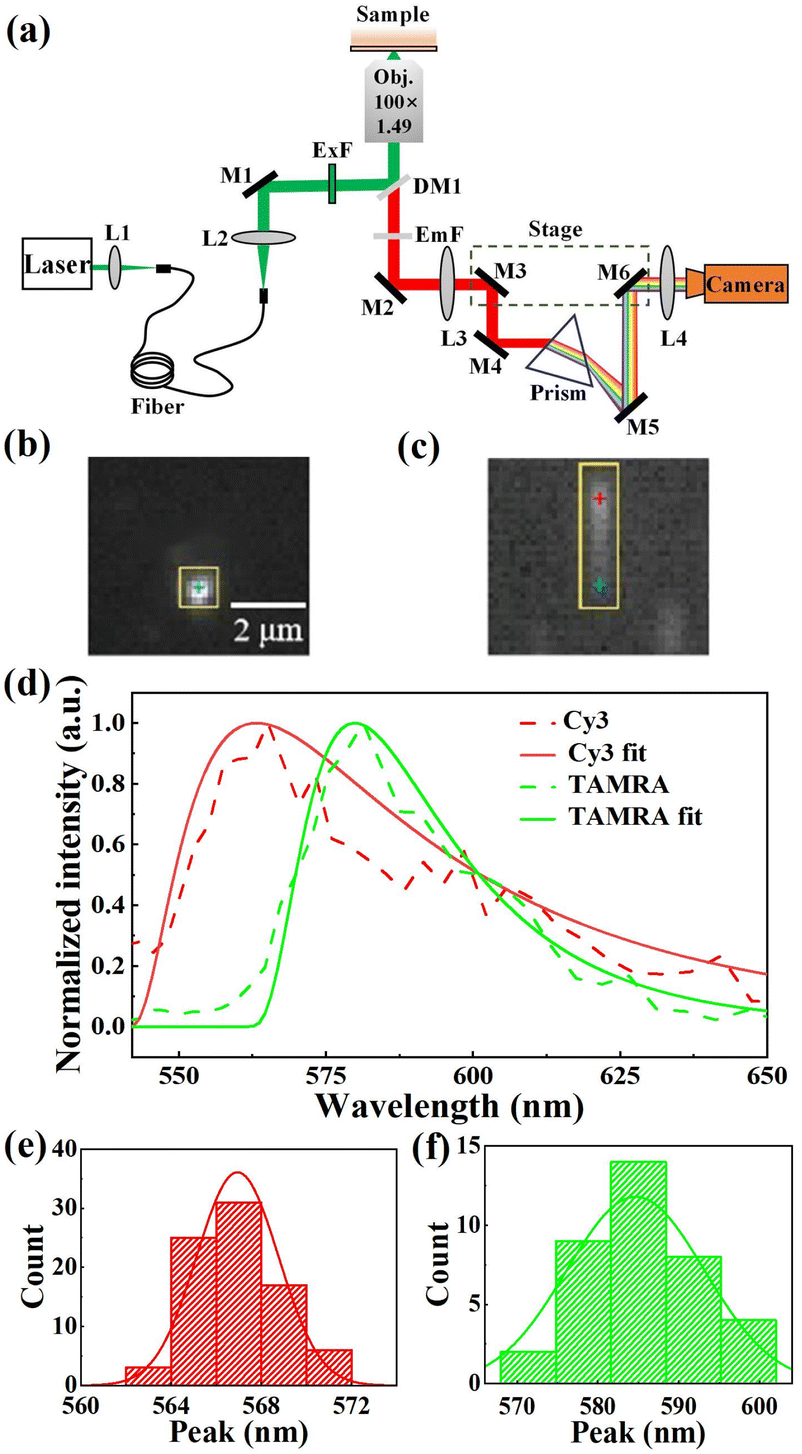 | ||
| Fig. 1 Spectroscopic single-molecule fluorescence (sSMF) system and single-molecule fluorescence spectra of Cy3 and TAMRA. (a) Schematic of the sSMF setup (L: lens; M: mirror; DM: dichroic mirror; ExF: excitation filter; EmF: emission filter). (b and c) Images of fluorescent molecules in imaging mode (b) and spectral mode (c). The yellow rectangles indicate the same molecule in imaging mode and spectral mode, and the blue crosshairs mark the corresponding points in the two modes, while the red crosshair depicted in (c) marks the spectral peak position. (d) Single-molecule emission spectra of Cy3 (peak at 566.8 nm) and TAMRA (peak at 581.9 nm). A skewed Gaussian fit is applied to improve peak determination accuracy. (e and f) Spectral peak distribution of Cy3 (e) and TAMRA (f). | ||
Identification of different kinds of rhodamine molecules based on sSMF
We then applied sSMF technique to investigate rhodamine molecules. The sample was prepared from a commercial stock of rhodamine B (Macklin) and diluted to a concentration of 1 pM using T50 buffer (10 mM Tris–HCl, 50 mM NaCl, pH 8.0) prior to the sSMF experiments. The typical single-molecule emission spectra of rhodamine molecules and their peak position distributions are shown in Fig. 2. Notably, the peak position distribution of rhodamine exhibits three distinct peaks, suggesting the presence of three different rhodamine species in the sample, which is in a striking contrast to the results observed for Cy3 and TAMRA. A recent study on the photo-bluing effect of rhodamine B has identified several photoproducts resulting from photo-oxidative dealkylation.8 High-performance liquid chromatography (HPLC) and NMR analysis revealed six major rhodamine species in the solution, including RhB-1 (rhodamine B), RhB-2, RhB-3a, RhB-3b, RhB-4 and RhB-5 (rhodamine 110), as shown in Fig. S1.† Interestingly, our measured mean peak wavelengths (573, 561 and 551 nm) for the three rhodamine species (Fig. 2d) closely match the reported maximum emission wavelengths of RhB-1 (578 nm), RhB-2 (564 nm), RhB-3a/3b (556/554 nm).8 The slight differences here are probably due to the different buffer conditions or the measurement uncertainties. Therefore, we attributed these three observed species to RhB-1, RhB-2 and RhB-3, representing rhodamine B and its photoproducts with one or two steps of dealkylation (Fig. 2e). Among these, RhB-1 was the most abundant (∼43%), followed by RhB-2 (∼38%), and RhB-3 (∼19%). Note here, given the small difference in emission peaks between RhB-3a and RhB-3b, we grouped them as RhB-3. Additionally, due to the optical filters used in the experiments, RhB-4 and RhB-5 were not detected. Unexpectedly, we rarely observed the transition of a rhodamine molecule to its dealkylated products before photobleaching occurred (see Fig. S2†). Possibly, the photobleaching process could be much faster than the photo-dealkylation process for rhodamine molecules and hence prevented real-time observation of this transition.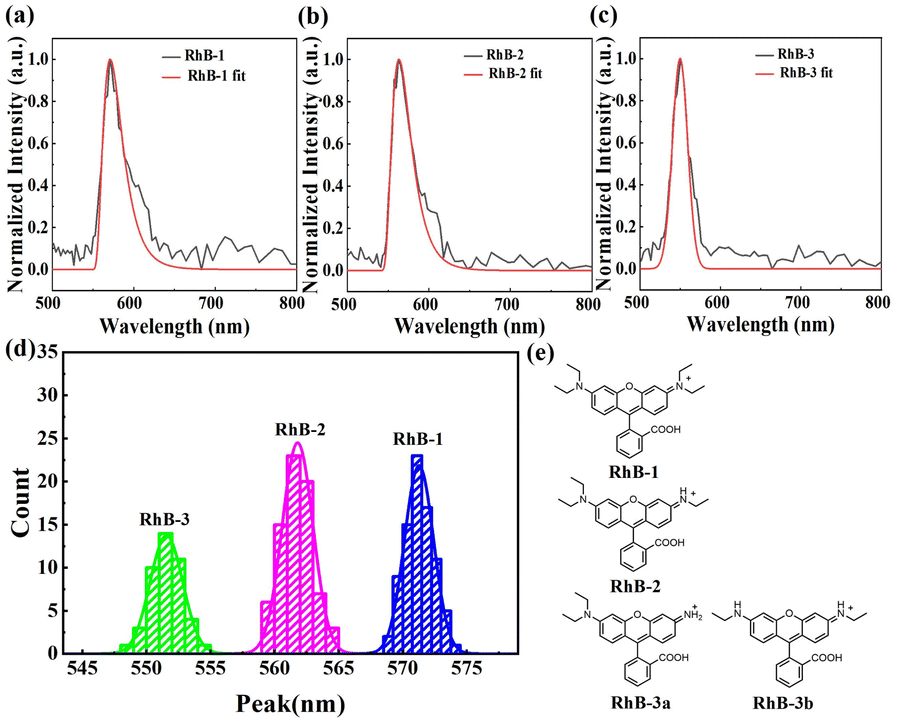 | ||
| Fig. 2 Identification of different types of rhodamine molecules. (a)–(c) Single-molecule fluorescence spectra of RhB-1 (a), RhB-2 (b), and RhB-3 (c) molecules in T50 buffer (pH 8.0), with skewed Gaussian fitting curves to determining peak positions. (d) Statistical distribution of RhB-1, RhB-2, and RhB-3 within the same sample in T50 buffer (pH 8.0), where RhB-1 accounts for 85 molecules (∼43%), RhB-2 for 76 molecules (∼38%), and RhB-3 for 39 molecules (∼19%). (e) The molecular structures of RhB-1, RhB-2, RhB-3a/3b in their “open” forms. | ||
Investigation of photophysical properties of different kinds of rhodamine molecules using sSMF
Since sSMF can distinguish between various rhodamine molecules based on their emission spectra, it allows for the direct comparison of the photophysical properties of different rhodamine molecules under identical conditions without requiring HPLC separation, which is a significant advantage of sSMF. In our experiments, we quantitatively assessed the brightness and photobleaching lifetimes of RhB-1, RhB-2, and RhB-3 within the same chamber under buffer conditions at pH 8.0. Fig. 3a–c show representative fluorescence intensity traces over time recorded for these three molecules. The observed single-step photobleaching in the figures confirms that the measured fluorescence originates from individual molecules.29 The photobleaching time distribution for each molecule, illustrated in Fig. 3d–f, are well-characterized by a single exponential decay function, allowing for accurate determination of their characteristic photobleaching lifetimes. Notably, despite their similar molecular brightness, RhB-2 demonstrated a significantly longer photobleaching lifetime (73.6 s) compared to RhB-1 (37.8 s) and RhB-3 (27.4 s), highlighting the superior photostability of RhB-2.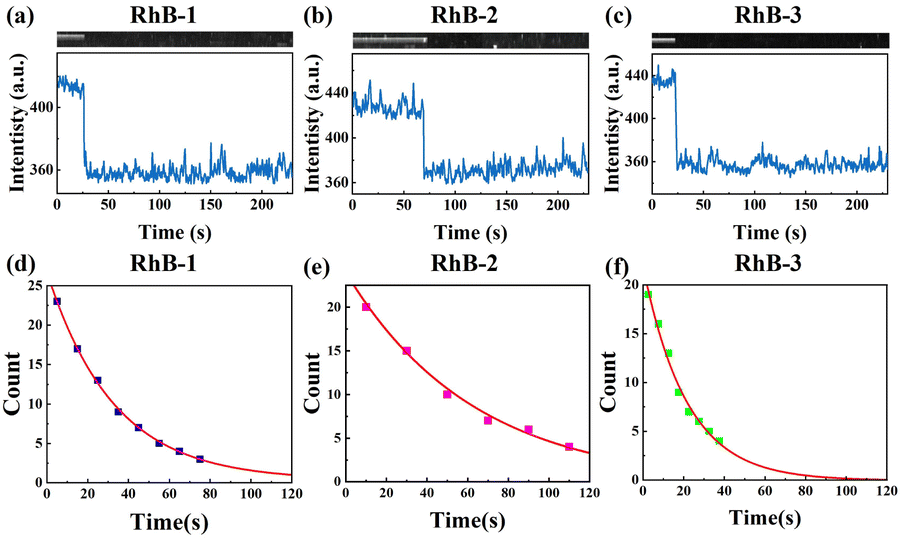 | ||
| Fig. 3 Photobleaching lifetimes of different types of rhodamine molecules. (a)–(c) Representative single-molecule fluorescence trajectories recorded for RhB-1 (a), RhB-2 (b), and RhB-3 (c) molecules in T50 buffer (pH 8.0). (d)–(f) Photobleaching time distributions of RhB-1 (d), RhB-2 (e), and RhB-3 (f) molecules in T50 buffer (pH 8.0), fitted with single exponential decay functions to determine the characteristic photobleaching lifetimes. The photobleaching lifetimes are 37.8 ± 2.4 s for RhB-1, 73.6 ± 2.8 s for RhB-2, and 27.4 ± 1.6 s for RhB-3. | ||
We further investigated the effect of varying buffer conditions, including the changes in pH and addition of protocatechuate-3,4-dioxygenase (PCA)–protocatechuic acid (PCD) oxygen scavenging system (OSS), on the photophysical properties of the three types of rhodamine molecules. As shown in Fig. 4a, increasing the pH of the buffer resulted in a blue shift in the emission spectra and a slight reduction in brightness for all three rhodamine species. It is well-known that rhodamine molecules can adopt different forms in solution (Fig. 4d), and the equilibrium between these forms depends on conditions such as pH and solvent polarity.30,31 For instance, in acidic solutions, rhodamine tends to exist in its cationic (“open”) form due to the protonation of the carboxyl group. In contrast, under basic conditions, rhodamine may convert into a zwitterion or even a non-fluorescent spirolactam (“closed”) form. When in its zwitterion form, the negative charge on the oxygen atom has been proposed to have an inductive effect on the central carbon atom of the xanthene chromophore, leading to a hypsochromic shift in both absorption and fluorescence maxima,30 consistent with our experimental results for all three RhB species. Interestingly, changes in pH had minimal impact on the relative abundance of the three rhodamine species (see Table S2†), suggesting that the dealkylation process remains largely unaffected within the observation window of our experiments.
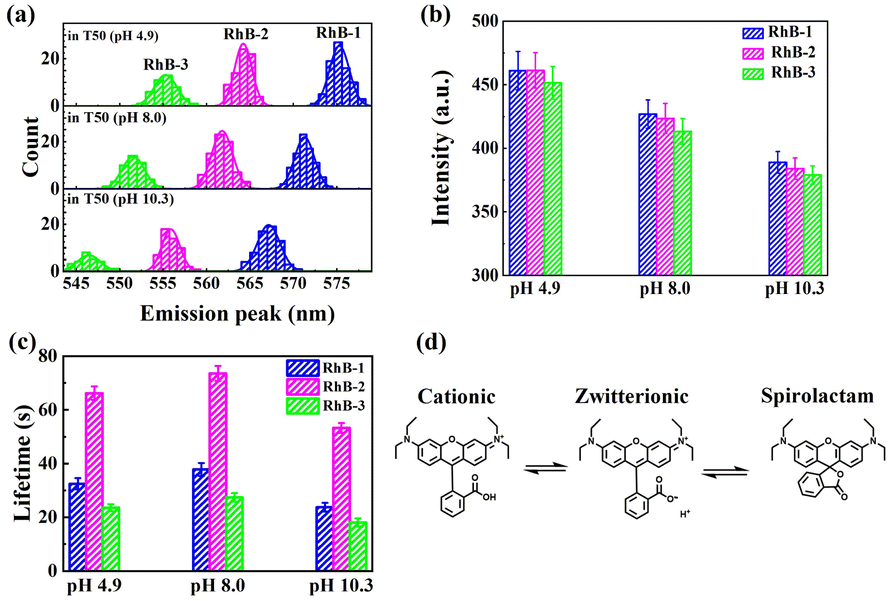 | ||
| Fig. 4 Photophysical properties of different types of rhodamine molecules under various buffer conditions. (a)–(c) Population (a), fluorescence intensities (b), and photobleaching lifetimes (c) of rhodamine molecules in T50 buffer of different pH (pH 4.9, pH 8.0, and pH 10.3) within the same sample. (d) The equilibrium between different forms of rhodamine B (RhB-1). Similar equilibria are also proposed for RhB-2 and RhB-3. | ||
In Fig. 4b, the fluorescence intensities of all three rhodamine species exhibited a significant decrease as the pH was raised from 4.9 to 10.3, consistent with prior reported ensemble measurements of rhodamine B and its derivatives.31,32 As the pH increased, the equilibrium shifted towards the zwitterionic and spirolactam forms of rhodamine molecules (see Fig. 4d). Since the spirolactam state is non-fluorescent, the single-molecule fluorescence signal measured at high pH shall be attributed to the zwitterionic state. Therefore, the decrease in single-molecule fluorescence intensity suggests that the zwitterionic forms of rhodamine molecules possess lower quantum yields, rendering them less luminous in comparison to their “open” form counterparts.
Fig. 4c demonstrates that pH variations also influence the photostability of these three rhodamine molecules. Specifically, both acidic or basic conditions led to a reduction in photobleaching lifetimes, with the effect being more pronounced under basic conditions. A possible explanation is that, in alkaline environments, rhodamine molecules, predominantly in their zwitterionic forms (see Fig. 4d), may interact more readily with oxygen compared to those in the cationic form, whereas in acidic solutions, ROS tend to be more reactive,33,34 thereby accelerating the photobleaching process.
Table 1 presents the impact of incorporating PCA–PCD OSS into the buffer. The data indicated that the addition of OSS significantly extended the photobleaching lifetimes for all three types of RhB molecules, underscoring the critical role of oxygen in the degradation of rhodamine molecules. Additionally, the introduction of OSS had a negligible effect on the fluorescence intensity of the RhB molecules, in contrast to the notable effects observed for pH variations.
| T50 (pH 8.0) | T50 (pH 8.0 with OSS) | ||
|---|---|---|---|
| Lifetime (s) | RhB-1 | 37.8 | 78.1 |
| RhB-2 | 73.6 | 113.3 | |
| RhB-3 | 27.4 | 62.9 | |
| Intensity (a.u.) | RhB-1 | 426.8 | 435.8 |
| RhB-2 | 423.5 | 421.1 | |
| RhB-3 | 413.3 | 410.5 | |
Interestingly, the three types of rhodamine molecules (RhB-1, RhB-2 and RhB-3) responded similarly to those changes in buffer conditions, such as OSS addition and pH adjustments, suggesting a shared mechanism in their photobleaching processes. Regardless, RhB-2 consistently exhibited the longest photobleaching lifetime among the three.
Computation of the first excited singlet and triplet state energies of different kinds of rhodamine molecules
To better understand why RhB-2 exhibits greater stability under excitation compared to RhB-1 and RhB-3, theoretical calculations were performed for four rhodamine molecular structures (RhB-1, RhB-2, RhB-3a and RhB-3b). Detailed information on the calculations is provided in the ESI.† The computed emission peak wavelengths of these rhodamine molecules are summarized in Table 2, successfully predicting the blue-shift in emission peaks from RhB-1 to RhB-3a or RhB-3b. The discrepancy between the theoretical and experimental peak wavelengths is approximately 30 nm, which falls within the typical accuracy (∼0.2 eV or ∼50 nm at 580 nm) of TD-DFT calculations.35 Additionally, the computations reveal that the energies of the first singlet excited states are generally higher than those of the triplet states for all four types of rhodamine molecules, with a similar energy gap of ∼0.6 eV, as indicated by the calculated fluorescence and phosphorescence emission energies in Table 3. However, this does not necessarily imply that the transition rates from singlet to triplet excited states are similar. Since the triplet state has been proposed to play a critical role in fluorophore photobleaching processes,17,36 further calculations of the intersystem crossing (ISC) transition rates are necessary to clarify the difference in triplet accessibility among these rhodamine molecules. Additionally, rhodamine molecules in their excited states may exhibit different reactivities with oxygen, which could further explain the observed disparities in photostability among them. However, theoretically assessing the reactivity of molecules in excited states is much more complex than determining their inherent properties such as the energies of ground and excited states, and thus falls outside the scope of this study. These results thus underscore the complexity and challenges in understanding the photophysical behavior of fluorophores in their excited states.| RhB-1_open | RhB-2_open | RhB-3a_open | RhB-3b_open | |
|---|---|---|---|---|
| Experimental values (nm) | 578 | 564 | 556 | 554 |
| Computational values (nm) | 549 | 535 | 525 | 525 |
| RhB-1_open | RhB-2_open | RhB-3a_open | RhB-3b_open | |
|---|---|---|---|---|
| Vertical excited energy (eV) | 2.5888 | 2.6578 | 2.7236 | 2.7092 |
| Fluorescence emission energy (eV) | 2.2580 | 2.3178 | 2.3620 | 2.3599 |
| Phosphorescence emission energy (eV) | 1.6723 | 1.7181 | 1.7612 | 1.7608 |
Conclusions
In this study, we employed a custom-built single-molecule fluorescence spectrometer to successfully distinguish three different species of rhodamine within a sample, based on their unique fluorescence spectra. These species, designated RhB-1, RhB-2, and RhB-3, represent different stages of rhodamine B dealkylation. By leveraging this technique, we could simultaneously assess the photophysical properties of these molecules under identical conditions, eliminating the need for chromatographic separation.8Notably, the analysis of photobleaching lifetimes among the rhodamine species revealed that RhB-2 exhibits superior photostability. This enhanced stability may be linked to its molecular structure, the energy levels of its excited states, or its lower reactivity with oxygen. Furthermore, theoretical calculations using TD-DFT demonstrated that all rhodamine species (including RhB-1, RhB-2, RhB-3a, and RhB-3b) share a nearly identical energy gap of approximately 0.6 eV between their first singlet and triplet excited states. However, the specific molecular mechanisms responsible for these differences remain unclear and warrant further investigation.
We also systematically explored the influence of buffer conditions on the photophysical properties of these rhodamine molecules. Our results indicated that variations in pH and the presence of oxygen scavengers significantly affected the emission spectra, brightness, and photostability of the molecules. These observations can be rationalized by shifts in equilibrium between the different forms of rhodamine molecules and their interactions with oxygen during the photobleaching process.
In summary, our research demonstrates the potential of single-molecule spectroscopy in elucidating the photophysical characteristics of fluorophores, while also emphasizing the complexity and challenges involved in understanding the fluorophore behavior in excited states. The insights gained from this study are expected to contribute to the development and optimization of more stable fluorescent probes for various applications, including those for pH sensing.
Methods
Buffer
All reagents were purchased from Sigma (Shanghai, China). T50 buffer consisted of 20 mM Tris–HCl (pH 8.0) and 50 mM NaCl, unless otherwise stated. To prepare T50 buffers with pH 4.9 and 10.3, pH adjustments were made with NaOH and HCl, respectively.PCA stock was diluted with deionized water to 500 mM and the pH was adjusted to 8 using NaOH. Likewise, 50 mM Trolox and 500 mM glucose solutions were also prepared by the same method.
For T50 buffer containing PCA–PCD OSS, protocatechuate 3,4-dioxygenase (PCD) was used at a concentration of 5 U mL−1, with 2 mM Trolox and 10 mM PCA, all mixed in T50 buffer.
Reagent
The rhodamine B sample stock were prepared by dissolving the rhodamine B powder (Macklin) in ultrapure water with a concentration of 1 M. This stock solution was subsequently diluted to 1 fM using T50 buffer for various sSMF experiments.Experimental setup
The single-molecule fluorescence experiments were performed using a commercial inverted microscope (Ti-E 2000, Nikon Instruments). The samples were illuminated in objective-type total internal reflection fluorescence (TIRF) mode with a 532 nm green solid-state continuous laser (MW-SL-532, Changchun Laser Optoelectronics). During the SMF experiments, the laser power density was approximately 25 kW cm−2. A filter cube (LF405/488/532/635-4X-B-NTE, Semrock) was used to direct the laser to objective and select the emission light from the sample. Fluorescence images were captured with an oil-immersion objective (CFI Apochromat TIRF 100XC Oil, Nikon Instruments Inc.) and recorded by a highly sensitive CMOS camera (Prime 95B Scientific CMOS Camera, Teledyne Photometrics). The camera exposure time was set to 300 ms for all experiments, and images were acquired using the Micro-manager software. All experiments were conducted at room temperature (23 ± 1 °C).Experimental methods
To prepare the experimental reaction chamber, a solution of 1 fM rhodamine B was injected into the chamber and incubated for 5 minutes to facilitate nonspecific attachment of rhodamine B molecules to the coverslip surface. The chamber was then rinsed with different T50 buffers (i.e., pH 4.9, pH 8.0, pH 8.0 with PCA–PCD OSS or pH 10.3) to remove any unbound rhodamine B molecules. Subsequently, the sample chamber was fixed on the microscope stage, and TIRF mode was activated. The excitation laser was turned on using the OXXIUS LASER software.For single-molecule fluorescence spectroscopy, the sSMF setup was initially configured for imaging mode (i.e., M1 and M2 mirrors, shown in Fig. 1, were removed from the optical path using the precise translation stage) to acquire a short video of fluorescence points for approximately 5 s. The setup was then switched to spectral mode (i.e., M3 and M6 mirrors were returned to their previously marked positions using the precise translation stage) to record a long video of spectral lines for several minutes. Afterward, the microscope stage was manipulated to examine a different region of the sample and collect additional data.
The videos captured in both imaging and spectral modes were combined into a single file. A custom-written MATLAB script was then utilized to split this file into separate video files for each mode.
The script further correlated fluorescence spots from the imaging mode with spectral lines from the spectral mode and plotted spectral profiles over time for each fluorescence spot using pre-calibrated parameters, followed by fitting the profiles with skewed Gaussian functions to determine peak positions.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. Deng: methodology, software, investigation, formal analysis, validation; D. Yi: methodology, investigation, formal analysis, visualization, validation; T. Rujiralai: writing – review & editing; Q. Ren: software, methodology, writing – original draft; C. Tan: supervision, writing – review & editing; J. Ma: supervision, funding acquisition, writing – original draft, writing – review & editing.Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this article. The authors confirmed that the paper is free of plagiarism.Acknowledgements
This work was supported by the National Natural Science Foundation of China (12074445 and 32371284) and the Open Fund of the State Key Laboratory of Optoelectronic Materials and Technologies, Sun Yat-sen University (OEMT-2024-ZTS-04). We also wish to thank for the support from the Physical Research Platform in the School of Physics, Sun Yat-sen University (PRPSP, SYSU), the High-Performance Computing Center of Shanghai University and the Shanghai Technical Service Center of Science and Engineering Computing, Shanghai University.References
- X. Zhuang, L. E. Bartley and H. P. Babcock, et al., Science, 2000, 288, 2048–2051 CrossRef CAS PubMed.
- X. Michalet, S. Weiss and M. Jäger, Chem. Rev., 2006, 106, 1785–1813 CrossRef CAS.
- A. Yildiz and P. R. Selvin, Acc. Chem. Res., 2005, 38, 574–582 CrossRef CAS PubMed.
- S.-J. Lee, S. Syed and E. J. Enemark, et al., Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E827–E835 CAS.
- M. Orrit and J. Bernard, Phys. Rev. Lett., 1990, 65, 2716–2719 CrossRef CAS PubMed.
- J. Eid, A. Fehr and J. Gray, et al., Science, 2009, 323, 133–138 CrossRef CAS.
- Z. Zhang, S. J. Kenny and M. Hauser, et al., Nat. Methods, 2015, 12, 935–938 CrossRef CAS PubMed.
- A. N. Butkevich, M. L. Bossi and G. Lukinavičius, et al., J. Am. Chem. Soc., 2019, 141, 981–989 CrossRef CAS PubMed.
- B. Dong, L. Almassalha and B. E. Urban, et al., Nat. Commun., 2016, 7, 12290 CrossRef CAS PubMed.
- M. J. Phillips and G. K. Voeltz, Nat. Rev. Mol. Cell Biol., 2016, 17, 69–82 CrossRef CAS PubMed.
- R. Yan, S. Moon and S. J. Kenny, et al., Acc. Chem. Res., 2018, 51, 697–705 CrossRef CAS PubMed.
- J. Jeffet, A. Ionescu and Y. Michaeli, et al., Biophys. Rep., 2021, 1, 100013 CAS.
- R. Roy, S. Hohng and T. Ha, Nat. Methods, 2008, 5, 507–516 CrossRef CAS PubMed.
- C. E. Aitken, R. A. Marshall and J. D. Puglisi, Biophys. J., 2008, 94, 1826–1835 CrossRef CAS.
- G. Jiang, T.-B. Ren and E. D’Este, et al., Nat. Commun., 2022, 13, 2264 CrossRef CAS.
- M. Kümmerlin, A. Mazumder and A. N. Kapanidis, ChemPhysChem, 2023, 24, e202300175 CrossRef.
- K. Kikuchi, L. D. Adair and J. Lin, et al., Angew. Chem., Int. Ed., 2023, 62, e202204745 CrossRef CAS.
- M. Swoboda, J. Henig and H.-M. Cheng, et al., ACS Nano, 2012, 6, 6364–6369 CrossRef CAS PubMed.
- Y. Lin, J. Hu and W. Zhang, et al., Nanoscale, 2022, 14, 17550–17560 RSC.
- A. Maity, J. Wulffelé and I. Ayala, et al., Adv. Sci., 2024, 11, 2306272 CrossRef CAS.
- S. Zeng, X. Liu and Y. S. Kafuti, et al., Chem. Soc. Rev., 2023, 52, 5607–5651 RSC.
- Y. Wang, X. Wang and W. Ma, et al., Chemosensors, 2022, 10, 399 CrossRef CAS.
- X. Liu, Q. Qiao and W. Tian, et al., J. Am. Chem. Soc., 2016, 138, 6960–6963 CrossRef CAS.
- L. Wang, M. Tran and E. D'Este, et al., Nat. Chem., 2020, 12, 165–172 CrossRef CAS.
- N. Lardon, L. Wang and A. Tschanz, et al., J. Am. Chem. Soc., 2021, 143, 14592–14600 CrossRef CAS.
- T.-B. Ren, W. Xu and W. Zhang, et al., J. Am. Chem. Soc., 2018, 140, 7716–7722 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhang and K. H. Song, et al., J. Phys. Chem. Lett., 2021, 12, 3914–3921 CrossRef CAS.
- T. P. J. Krüger, V. I. Novoderezhkin and C. Ilioaia, et al., Biophys. J., 2010, 98, 3093–3101 CrossRef.
- J. C. Gebhardt, D. M. Suter and R. Roy, et al., Nat. Methods, 2013, 10, 421–426 CrossRef CAS PubMed.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC.
- E. Birtalan, B. Rudat and D. K. Kölmel, et al., Pept. Sci., 2011, 96, 694–701 CrossRef CAS.
- R. Tachibana, M. Kamiya and S. Suzuki, et al., Commun. Chem., 2020, 3, 82 CrossRef CAS PubMed.
- J. Wei, T. Fang and M. Shiraiwa, ACS Environ. Au, 2022, 2, 336–345 CrossRef CAS PubMed.
- J. Vogelsang, T. Cordes and P. Tinnefeld, Photochem. Photobiol. Sci., 2009, 8, 486–496 CrossRef CAS.
- J. Ren, S. Meng and C. E. Lekka, et al., J. Phys. Chem. B, 2008, 112, 1845–1850 CrossRef CAS.
- C. Eggeling, J. Widengren and R. Rigler, et al., Anal. Chem., 1998, 70, 2651–2659 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06577h |
| ‡ These authors contributed equally to this work. |
| § Regarded as co-corresponding authors. |
| This journal is © The Royal Society of Chemistry 2024 |