
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of corticiolic acid—a bioactive pharmacophore from natural sources†
Sunil Kumar Deevi a,
Bhadra Anilkumara,
Priyanka Gladys Pintoc,
Prasanna Ramaniad,
Chethala N. Vishnuprasad*c,
Sankarasekaran Shanmugaraju*e and
Nanjan Pandurangan*ab
a,
Bhadra Anilkumara,
Priyanka Gladys Pintoc,
Prasanna Ramaniad,
Chethala N. Vishnuprasad*c,
Sankarasekaran Shanmugaraju*e and
Nanjan Pandurangan*ab
aDhanvanthri Lab, Department of Chemistry, Amrita School of Physical Sciences, Amrita Vishwa Vidyapeetham, Coimbatore-641112, Tamil Nadu, India. E-mail: n_pandurangan@cb.amrita.edu
bFunctional Materials Lab, Amrita School of Engineering, Amrita Vishwa Vidyapeetham, Coimbatore-641112, Tamil Nadu, India
cAyurveda Biology and Holistic Nutrition, The University of Trans-Disciplinary Health Sciences and Technology, Bangalore-560064, India, drcnvp@tdu.edu.in
dCenter of Excellence in Advanced Materials & Green Technologies (CoE-AMGT), Amrita School of Engineering, Amrita Vishwa Vidyapeetham, Coimbatore-641112, India
eDepartment of Chemistry, Indian Institute of Technology Palakkad, Palakkad 678557, Kerala, India. E-mail: shanmugam@iitpkd.ac.in
First published on 22nd November 2024
Abstract
Fungal strains have inspired us to find the untapped sources of secondary metabolites. Corticiolic acid (CA, 2,4-dihydroxy-6-pentadecylbenzoic acid; from fungus, Hapalopilus mutans) is one of the core active scaffolds in natural compounds such as Aquastatin-A, B, & C. CA can also be isolated from the plant Lysimachia japonica. CA is a selective inhibitor of PTB1B, a crucial biomarker for anti-diabetic activity. Herein, we report the total synthesis of corticiolic acid achieved via the 9-BBN-based reductive Suzuki–Miyaura coupling of aryl bromide and pentadecane, a key reaction in this strategy. Further, this approach has been explored for the protection-free synthesis of corticiolic acid. The improved synthesis is short, requires mild reaction conditions, and avoids the use of hydrogenation and pyrophoric reagents. Further, the reaction is scalable and does not require protection–deprotection steps. Preliminary studies on cancer cells indicated that corticiolic acid and cordol significantly inhibited the proliferation of HepG2, N2A, and CaCo-2 cancer cells.
Introduction
Natural products (NP) have long been considered privileged scaffolds for drug discovery programs.1,2 Further, NPs occupy a larger chemical space, with more than 50% of drugs being derived from natural products or related compounds.3 Over the past two decades, in addition to plants, microorganisms have contributed significantly as an untapped source of secondary metabolites with infinite structural diversity.4,5 This has led to biological activities targeting a range of diseases, from metabolic diseases to infectious diseases.6 One such active molecule derived from fungi is depside,7 including CRM646-A, CRM646-B, Aquastatin-A, B, and C (see Fig. 1 for structures).8–10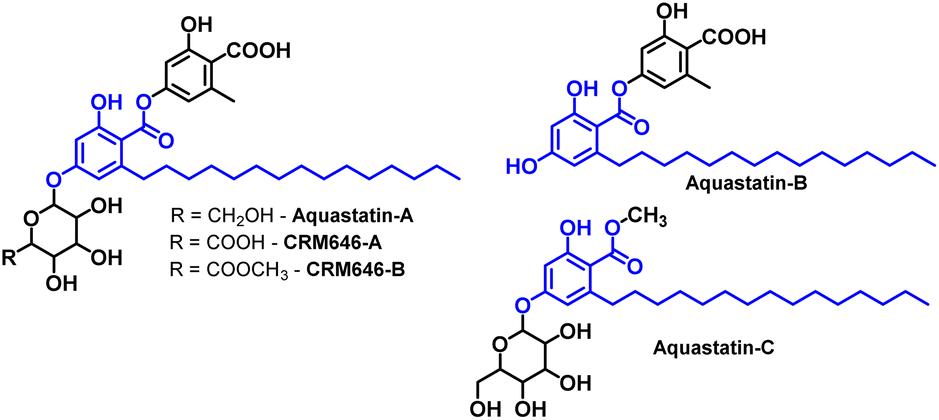 | ||
| Fig. 1 Bioactive metabolites derived from fungus. | ||
A detailed survey suggests that most of the bioactivities are attributed to the active entity, corticiolic acid (CA).7 CA is a core scaffold in the molecules mentioned (see Fig. 1). It has also been found in another fungus, Corticium caeruleum (Aphyllophorales),11 and in the plant Lysimachia japonica in Japan.12 CA is an important compound with various applications, such as marked inhibition of Na+, K+, and ATPase activity.12 CA also shows selective inhibitory activity against protein tyrosine phosphatase 1B (PTP1B), an important enzyme for diabetic activity.7 Similarly, it exhibits effective antibacterial activity against S. aureus (MRSA) strain.9 Despite its promising biological activities, fewer studies have been conducted on this molecule in the last 10 to 15 years. One reason for this is that these derivatives may not be available in sufficient quantities, either through isolation or bio-production from fungal extracts. The next option would be the synthesis of these compounds using reliable methods. A literature review suggests two primary methods for synthesizing non-isoprenoid phenolic acids: (i) nucleophilic addition of phenolic acids by alkynyl lithium, and (ii) conversion of pre-synthesized 1-haloalkyl(en)yl groups into an unsaturated side chain by an activated methyl group.13
Based on these observations, Yu et al. reported the total synthesis of fungal metabolites CRM646-A and CRM646-B, where corticiolic acid, one of the constituents, was synthesized through activated methyl addition (from orcinol) by an LDA-based reaction involving orcinol ester and tetradecyl bromide.14 Further, corticiolic acid was prepared by Massimo et al. from the triflate derivative through Sonogashira coupling with various terminal alkynes. The strategy provides good overall yield of corticiolic acid.15 Similarly, another non-isoprenoid derivative, olivetolic acid, which is a biosynthetic intermediate of cannabinoids, has also been synthesized via the same route.16 From the above studies, it was observed that all the routes required the hydrogenation reaction after the attachment of side chains (because of either double or triple bonds). Further, they involved a tedious isolation procedure, use of pyrophoric reagents,14 and the starting materials used are costly, which are challenges to handle on a larger scale. These difficulties slow down the biological studies on these reported compounds even though they show selective biological functions. Specific inhibitions on biological targets using pyrophoric reagents in existing methods, extra steps in hydrogenation, and scalability issues made us look into a practical approach for synthesizing corticiolic acid. From the literature search, it has been identified that the Suzuki–Miyaura Coupling (SMC)17 reaction would be the choice of synthesis method for making such naturally-derived molecules.
SMC is one of the vital tools for the synthesis of carbon–carbon bonds.17–19 Due to the versatile conditions, SMC is considered a gold-standard protocol for developing many commercial products and pharmaceuticals.20 Similarly, hydroboration in combination with Suzuki–Miyaura cross-coupling reactions has also gained a special interest in making complex products. In this, 9-BBN coupling occupies a major role due to its various advantages. In this communication, we report a novel method for synthesizing corticiolic acid through the 9-BBN–pentadecene complex and aryl bromide via C(sp3)–C(sp2) coupling. The described method is rapid and efficient and the reaction conditions are mild and scalable. Further, the hydroborated–pentacene linker (avoids hydrogenation reaction), followed by coupling in a single step, enables us to achieve corticiolic acid. Further, it has been extended for the anticancer activities in HepG2, CaCo-2, and N2A cell lines. The results showed that corticiolic acid (7) and cordol (12) significantly inhibited the proliferation of HepG2, N2A, and CaCo-2 cells (Fig. 2).
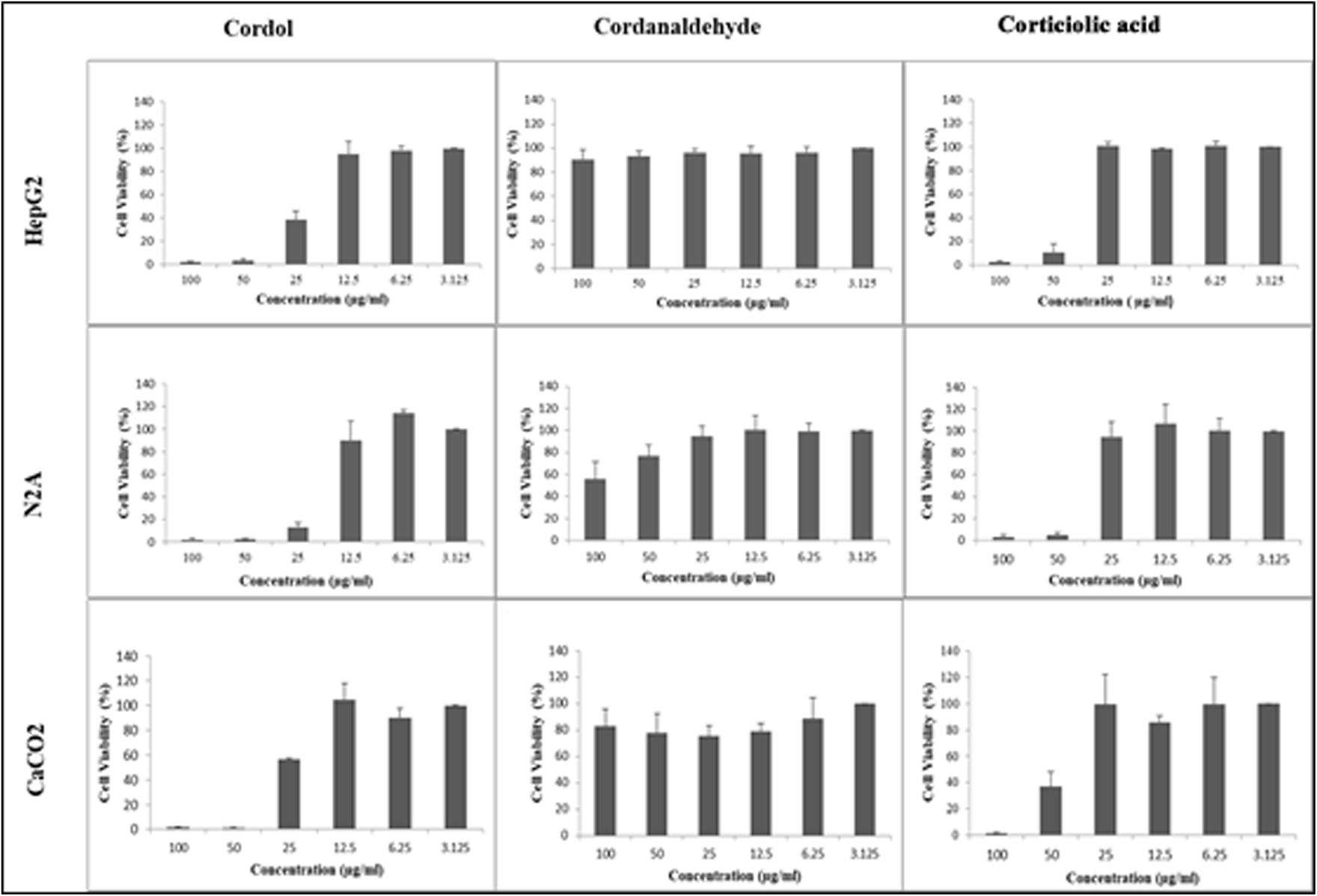 | ||
| Fig. 2 Cytotoxicity studies of corticiolic acid and its derivatives. | ||
Results and discussion
As shown in Scheme 1, corticiolic acid synthesis was started from a commercially available reagent, 3,5-dimethoxy-bromobenzene (1). The synthesis begins with a formylation reaction of 1 using DMF/POCl3, which gave dimethoxy bromobenzaldehyde 2 in good yield (91%). Compound 4 was obtained by the oxidation of 2 (69%), followed by the esterification reaction, which leads to 3 in a good yield (82%). The SMC of 4 with the BBN complex of petadec-1-ene (5) afforded 6 with a moderate yield (44%). The demethylation followed, by the ester hydrolysis of 6, resulted in corticiolic acid (7) with a maximum isolated yield of 34%. The coupling reaction was attempted initially with 2 and 5. From the analysis, it is identified that the reaction ended up with a very low conversion (<10% product formation). The reason would be due to the stability issues of aldehyde under aqueous Suzuki conditions wherein we used base and water at higher temperature. Further developments were not done in this regard due to the poor conversion rate. It is important to know that the use of alkylborane (B-alkyl-9-BBN) in sp3 SMCs suffered from isolation difficulties, affected by various factors such as atom economy and sensitivity to air functional group intolerance. Hence, extending this approach to make C(sp3)–C(sp2) coupling partners has been challenging.17 Hence, the optimization of the reaction conditions of the 9-BBN complex are important. Thus, the SMC was optimized based on the observations indicated in Table 1 (see ESI).† Initially, 9-BBN complex formation with 1-pentadecene was optimized with various reaction conditions. The careful formylation reaction was carried out under the specified conditions. Similarly, the 9-BBN complex reactions followed by the Suzuki reaction were optimized. The palladium catalyst [Pd(dppf)Cl2·DCM] was used throughout the optimization process. The analysis identified that the 9-BBN complex was unsuccessful at room temperature under specific reagent conditions (ESI; entry 1–3 in Table 1†), indicated by the SMC (the coupled product) yield, which was about 5–21%. The 9-BBN complex yield was almost quantitative when the temperature was raised to 50 °C (monitored by TLC), where the yield of the coupled product was increased from 21% to 44%. The reactions at 80 °C yielded only 45% of the product, but when the temperature increased to 100 °C, maximum yield (72%) of the coupling product (8) was obtained. The deprotection of methyl and ester would lead to corticiolic acid in good to moderate yields (7; 65% yield). This route provides an optimum condition of 9 by oxidation, giving a maximum yield of 86%. The final corticiolic acid (7) was obtained in 75% yield by the deprotection of the methyl group. After the optimization reaction, it was identified that the overall yield increased from 7% to 30%.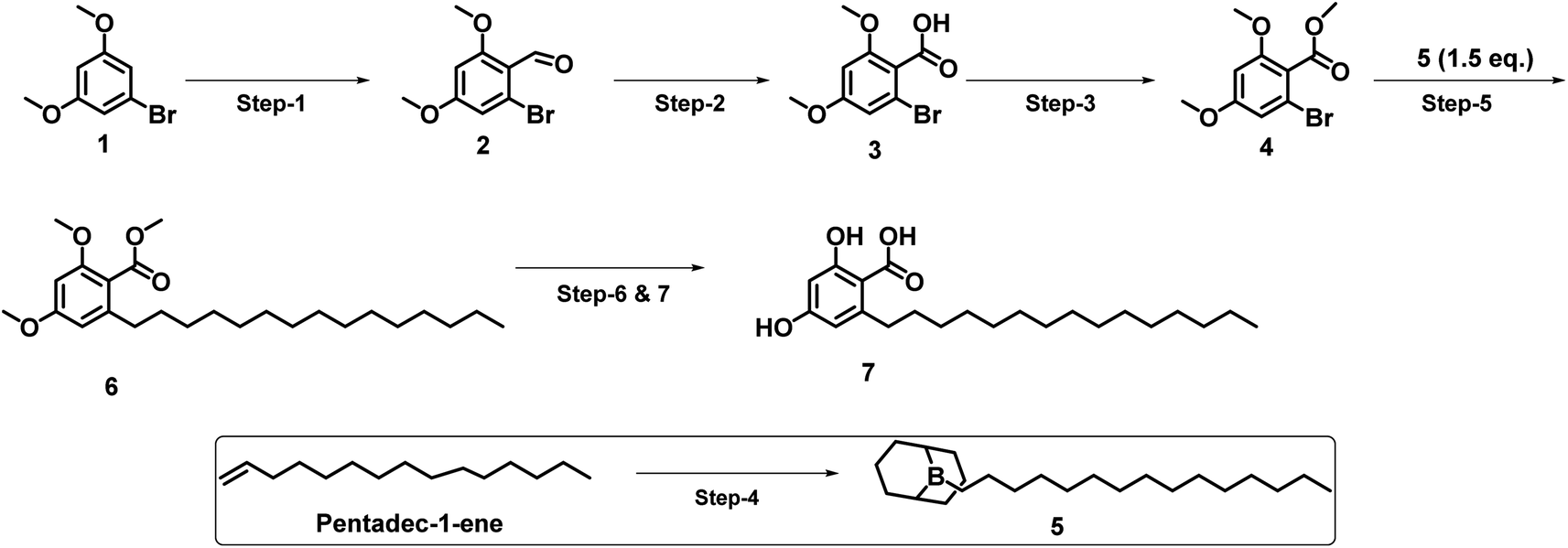 | ||
| Scheme 1 Synthesis of corticiolic acid (inset, step-4, 9-BBN-pentadecene preparation). Reaction conditions: step-1: POCl3, DMF, 0–90 °C, 91% yield, step-2: NaClO2, NaH2PO4, DMSO/H2O, 0 °C-RT, 69% yield, step-3: K2CO3, CH3I, DMF, 50 °C, 82% yield, step-4: 9-BBN, THF, 50 °C (crude), step-5: K2CO3, [Pd(dppf)Cl2·DCM], DMF/H2O, 100 °C, 44% yield, step-6: BBr3, DCM, −78 °C-RT, (crude), step-7: 1 N NaOH, THF/H2O, RT, 34% yield. | ||
After understanding the advantages and disadvantages of the present method (as shown in Scheme 1), we devised a new strategy (see Scheme 2) to improve the yield of corticiolic acid synthesis. According to this novel strategy, the 9-BBN-based SMC reaction was carried out in the first step, where the coupling product was obtained in a maximum yield of 72%. To show the scalability of the strategy, the reaction was carried at a 5 g scale level. It was identified that the yield of the reaction does not change when it is performed at gram scale, which indicates that the process can be scaled without much difficulty. Further, the reaction optimization was extended for the preparation of hydroxylated derivatives. Most biological studies were attributed to the active functional groups (pharmacophores) in their structural scaffold. Based on the prerequisites, the same SMC was extended to develop hydroxylated derivatives. Hence, 1,3-dihydroxy bromoderivative (11) and 9-BBN were taken for the SMC under the conditions specified in Scheme 3 to synthesize cordol (12). The formylation of 12 under the same condition (from Schemes 1 and 2) yielded product 13. Finally, corticiolic acid (7) was obtained by the oxidation of compound 13. From this strategy, the overall yield of corticiolic acid (7) was increased from 30% to 41% with hydroxylated derivatives directly. Hence, these compounds are directly taken for biological studies without the deprotection steps, which is a major disadvantage of the reported method.14
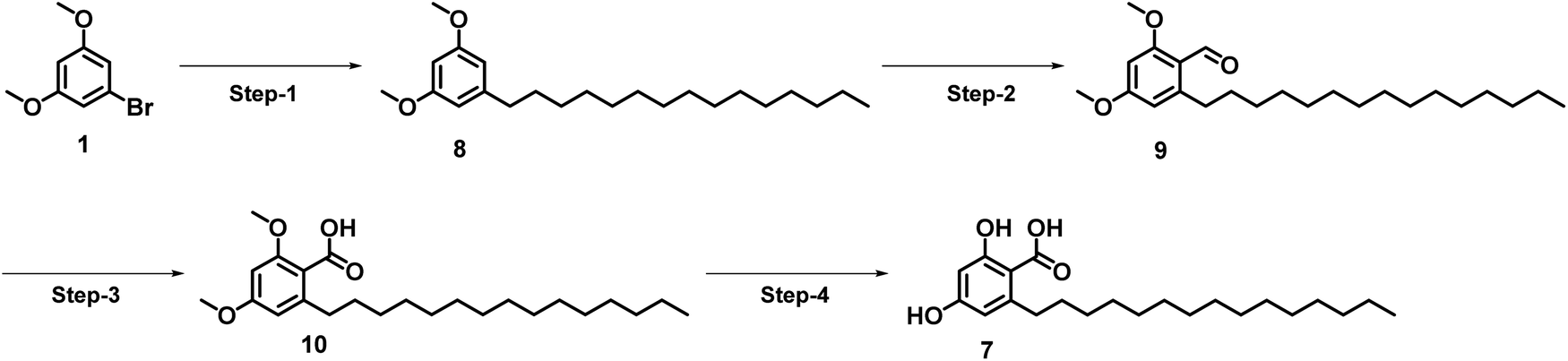 | ||
| Scheme 2 An improved method for the synthesis of corticiolic acid. Reaction conditions: step-1: pentadec-1-ene, 9-BBN, THF, 50 °C, K2CO3, [Pd(dppf)Cl2DCM], DMF/water, 100 °C; 72% yield, step-2: POCl3, DMF, 0–90 °C, 91% yield, step-3: NaClO2, NaH2PO4, DMSO/H2O, 0 °C-RT; 86% yield, step-4: BBr3, DCM, −78 °C-RT; 75% yield. | ||
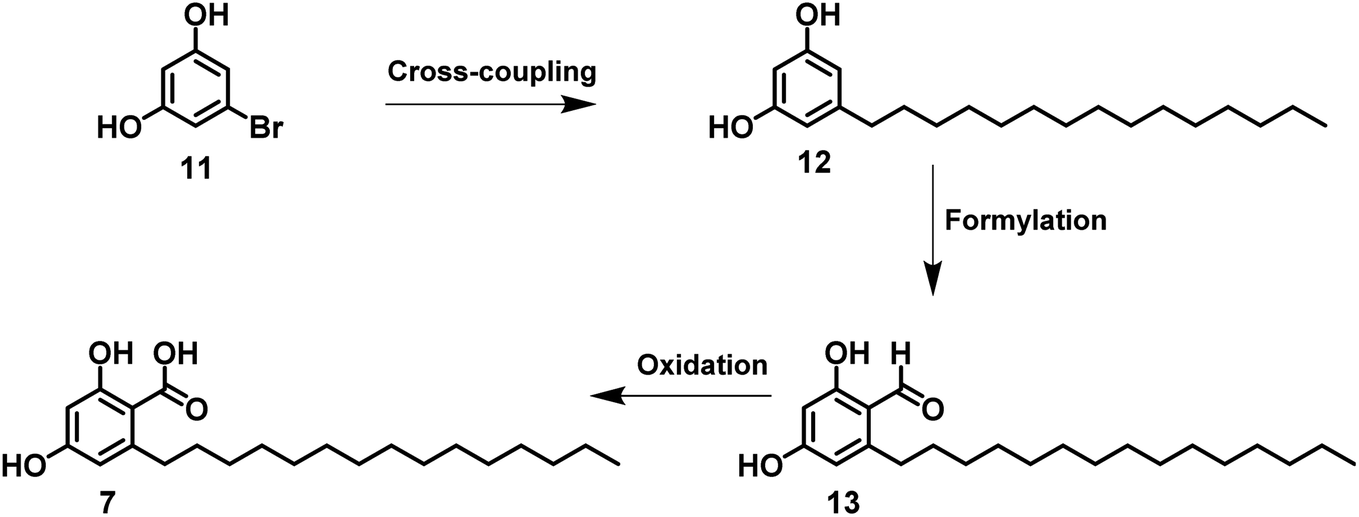 | ||
| Scheme 3 Protection–deprotection-free synthesis of corticiolic acid. Reaction conditions: step-1: pentadec-1-ene, 9-BBN, THF, 50 °C, K2CO3, [Pd(dppf)Cl2·DCM], DMF/water, 100 °C; 78% yield, step-2: POCl3, DMF, 0–90 °C, 91% yield, step-3: NaClO2 (4.0 eq.), NaClO2, NaH2PO4, DMSO/H2O, 0 °C-RT; 80% yield. | ||
Our phytochemistry lab is constantly looking for bioactive entities from natural sources. During the search on molecules, we identified that many naturally occurring derivatives were are active for various biological key biomarkers, which are crucial for metabolic and infectious diseases.21–26 In general, not many reports are available on the biological properties of corticiolic acid (7), in particular for its anticancer studies. With this view, the synthesized compounds, i.e., corticiolic acid (7), cardol (12), and crotonaldehyde (13), were taken for the preliminary studies to determine their inhibition potential of cell proliferation using three cancer cell models, viz., HepG2 (human hepato-carcinoma cells), N2A (derived from peripheral nervous system carcinoma of mice), and CaCo-2 (human colon carcinoma-derived epithelial cells) (Fig. 2). The results showed that cordol (12) and corticiolic acid (7) significantly inhibited the proliferation of HepG2, N2A, and CaCo-2, whereas inhibition by cordanaldehyde (13) was minimal even at the highest concentration of 100 μg mL−1 tested in the assay. From this study, corticiolic acid was identified as an active molecule, and other selective biological studies are in progress.
Conclusions
In summary, the total synthesis of corticiolic acid (CA) has been achieved. The 9-BBN-based Suzuki–Miyaura cross-coupling reaction was used as a key reaction for preparing CA and its derivatives. Further, CA synthesis has been improved based on practical and commercial applicability to an efficient and short synthesis (three-step) using easily accessible starting materials and mild reaction conditions. Three different reaction strategies were developed to improve the yield (overall yield; 50–55%) of the products, whereas other reported methods showed only 10–20% overall yield. Further, SMC was extended for the protection-free strategy and it was found that the reported coupling reaction was efficient even without the protection of hydroxyl functionalities. The reported synthesis avoids hydrogenation and pyrophoric reagents. The strategy was found to be scalable. The described strategy does not warrant protection–deprotection steps. Further, it was extended for studying the anticancer activities on HepG2, CaCo2, and N2A cell lines. The results showed that cordol and corticiolic acid significantly inhibited the proliferation of HepG2, N2A, and CaCo-2.Experimental details
All reagents and solvents were purchased from commercial suppliers and used as received. NMR spectra were obtained with a Bruker NMR spectrometer (1H NMR, 400 MHz and 13C NMR – 101, 151 MHz). Chemical shift (δ) values are reported in ppm using CDCl3 solvent as the reference relative to TMS. Coupling constant (J) values are reported in Hz, and HRMS spectra were collected using a Xevo G2-XS QTOF. The single-crystal X-ray diffraction data of the compound were collected using a Bruker Apex II CCD diffractometer with a Cu and Mo source at room temperature with the monochrome beam method. The structure was generated using the full-matrix least squares method using the SHELKS program.Preparation of 2-bromo-4,6-dimethoxybenzaldehyde (2)
To a stirred solution of 3,5-dimethoxybromobenzene (2.0 g, 9.21 mmol, 1.0 eq.) in DMF (4.3 mL) at 0 °C, POCl3 (2.58 mL, 27.63 mmol, 3.0 equivalents) was added dropwise under an inert atmosphere. The prepared orange-colored reaction mixture was heated to 90 °C and stirred for 6 hours. After completion of the reaction, the reaction mixture was poured into ice water (100 mL) and the reaction mixture was quenched by slowly adding solid KOH until a pH of 14 was reached. The mixture was stirred for 12 h at room temperature. The crude material was extracted with EtOAc (3 × 100 mL). The combined organic extracts were then washed with water (3 × 100 mL) and brine (100 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to afford the desired aldehyde 2 (2.05 g, 90.8% yield) as a white solid. 1H-NMR: (400 MHz, CDCl3); δ 10.31 (s, 1H), 6.78 (d, J = 2.4, Hz, 1H), 6.44 (d, J = 2.0 Hz, 1H), 3.89 (s, 3H), 3.87 (s, 3H). LCMS: 96.21% purity at RT = 1.68; m/z = 245.08 [M + H]+, 247.09 [M + H + 2]+ (observed); m/z = 244.97 [M]+.Preparation of 2-bromo-4,6-dimethoxybenzoic acid (3)
To a stirred solution of 2-bromo-4,6-dimethoxybenzaldehyde (1.5 g, 6.12 mmol, 1.0 equivalent) in DMSO (10 mL) and water (5 mL) at 0 °C, NaClO2 (2.2 g, 24.48 mmol, 4.0 equivalents) was added, followed by NaH2PO4 (4.4 g, 36.72 mmol, 6.0 equivalents). The resulting reaction mixture was stirred at ambient temperature for 24 h. After the completion of the reaction, the reaction mixture was quenched by adding 2 N HCl (50 mL) and extracted with EtOAc (2 × 50 mL). The combined organic extract was washed with water (3 × 50 mL), followed by brine solution (50 mL). The organic layer was separated, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to afford the desired acid 3 (1.1 g, 69% yield) as an off-white solid. 1H-NMR (400 MHz, DMSO-d6): δ 13.11 (s, 1H), 6.77 (d, J = 2.0 Hz, 1H), 6.64 (d, J = 2.0 Hz, 1H), 3.79 (s, 3H), 3.78 (s, 3H). LCMS: 96.33% purity at RT = 1.44; m/z = 260.95 [M + H]+ (observed).Preparation of methyl-2-bromo-4,6-dimethoxybenzoate (4)
To a stirred solution of 2-bromo-4,6-dimethoxybenzoic acid (0.5 g, 1.92 mmol, 1.0 equivalent) in DMF (5 mL) at 0 °C, K2CO3 (663.4 mg, 4.8 mmol, 2.5 equivalents) was added, followed by CH3I (0.24 mL, 3.84 mmol, 2.0 equivalents). The resulting reaction mixture was stirred at 50 °C for 16 h. After completion, the reaction mixture was diluted with EtOAc (20 mL) and washed with water (3 × 50 mL), followed by brine solution (50 mL). The organic extract was separated, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to afford the desired ester 4 (0.43 g, 81% yield) as an off-white solid. LCMS: 93.86% purity at RT = 1.81; m/z = 275.19 [M + H]+; m/z = 277.17 [M + H + 2]+ (observed); m/z = 274.98 [M + H]+.Preparation of 9-BBN-pentadec-1-ene complex (5)
To a stirred solution of pentadec-1-ene (0.25 g, 1.19 mmol, 1.0 equivalent) in THF (5 mL), 9-BBN [0.5 M in THF] (11.9 mL, 5.95 mmol, 5 equivalents) was added at RT under inert atmosphere. The resulting reaction mixture was allowed to stir at 50 °C for 16 h under sealed tube conditions. Progress of the reaction was monitored by TLC [eluent: 5% EtOAc in petroleum ether; Rf = 0.5]. The reaction mixture was evaporated under reduced pressure in an inert atmosphere to half the volume. The crude solution of the BBN-complex was taken forward to the next coupling reaction without further delay.Preparation of methyl-2,4-dimethoxy-6-pentadecylbenzoate (6)
To a stirred solution of methyl-2-bromo-4,6-dimethoxybenzoate (0.2 g, 0.727 mmol, 1.0 equivalent) in DMF (3.5 mL) and water (0.1 mL), K2CO3 (251.5 mg, 1.82 mmol, 2.5 equivalents) was added and degassed by argon purging for 10 min. To the resulting reaction mixture, the above prepared 9-BBN complex of pentadec-1-ene (solution in THF) was added, followed by [Pd(dppf)Cl2.DCM] (59.4 mg, 0.0727 mmol, 0.1 eq.) and further degassed for 5 min. The reaction mixture was allowed to stir at 100 °C for 16 h under an inert atmosphere. After completion, the reaction mixture was diluted with EtOAc (20 mL) and washed with water (3 × 20 mL), followed by brine solution (20 mL). The organic extract was separated, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to obtain the crude product. The crude product was purified by flash column chromatography [eluent: 5–8% EtOAc in pet. ether] to afford the desired product, 5 (0.13 g, 44% yield) as a low melting off-white solid. 1H-NMR: (400 MHz, CDCl3): δ 6.32 (d, J = 2.4 Hz, 1H), 6.31 (d, J = 2.0 Hz, 1H), 3.87 (s, 3H), 3.81 (s, 3H), 3.79 (s, 3H), 2.55–2.51 (m, 2H), 1.28–1.25 (m, 24H), 0.88 (t, J = 6.8 Hz, 3H). LCMS: 96.75% purity at RT = 3.38; m/z = 407.55 [M + H]+ (observed).Preparation of corticiolic acid (7)
To a stirred solution of methyl-2,4-dimethoxy-6-pentadecylbenzoate (0.13 g, 0.32 mmol, 1.0 equivalent) in DCM (2 mL), BBr3 [1 M in DCM] (0.96 mL, 0.96 mmol, 3.0 equivalents) was added and stirred at RT for 16 h under inert atmosphere. The resulting reaction mixture was evaporated under reduced pressure. The crude product was dissolved in THF (3 mL) and 2 N NaOH solution (1.6 mL, 3.2 mmol, 10.0 equivalent) was added and stirred for 6 h at ambient temperature. After the completion of the reaction, as monitored by TLC, the reaction mixture was evaporated under reduced pressure. The residue was diluted with water (10 mL) and washed with EtOAc (2 × 10 mL). The aqueous layer was acidified with conc. HCl to acidic pH and extracted with EtOAc (2 × 10 mL). The organic layer was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to afford 2,4-dihydroxy-6-pentadecylbenzoic acid (corticiolic acid, 7) (40 mg, 34% yield) as an off-white solid.In Scheme 3, compound 7 was synthesized from compound 13 using the general procedure for the synthesis of compound 3 in Scheme 1. Off-white solid 1.16 g (52% yield).1H-NMR: (400 MHz, DMSO-d6): δ 13.31 (br s, 1H), 11.83 (br s, 1H), 10.01 (br s, 1H), 6.14 (s, 1H), 6.11 (s, 1H), 2.75–2.71 (m, 2H), 1.47–1.45 (m, 2H), 1.23 (br s, 24H), 0.85 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 172.6, 163.5, 161.5, 147.1, 109.9, 105.1, 100.5, 35.3, 31.2, 29.3, 29.0, 28.8, 28.6, 22.06, 14.05. LCMS: 98.15% purity at RT = 2.86; m/z = 363.44 [M–H]− (observed).
Preparation of 1,3-dimethoxy-5-pentadecylbenzene (8)
The general method used for the synthesis of 6 was used here for the synthesis of compound 8. Low melting off-white solid 0.58 g (72% yield). 1H-NMR: (400 MHz, CDCl3): δ 6.34 (d, J = 2.0 Hz, 1H), 6.29 (d, J = 2.0 Hz, 1H), 3.78 (s, 6H), 2.54 (t, J = 7.6 Hz, 2H), 1.59–1.53 (m, 2H), 1.30–1.25 (m, 24H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 160.8, 145.6, 106.7, 97.7, 55.4, 36.5, 32.1, 31.5, 29.9, 29.8, 29.7, 29.5, 22.9, 14.3. LCMS: 91.27% purity at RT = 1.09; m/z = 349.31 [M + H]+ (observed).Preparation of 2,4-dimethoxy-6-pentadecylbenzaldehyde (9)
The general method used for the synthesis of 2 was used here for the synthesis of compound 9. Off-white solid 0.41 g (65% yield). 1H-NMR: (400 MHz, CDCl3): δ 10.46 (br s, 1H), 6.33 (d, J = 2.4 Hz, 1H), 6.31 (d, J = 2.0 Hz, 1H), 3.87 (s, 3H), 3.86 (s, 3H), 2.94 (t, J = 7.6 Hz, 2H), 1.62–1.53 (m, 2H), 1.37–1.25 (m, 24H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 193.0, 166.6, 163.4, 150.5, 112.9, 109.8, 101.4, 32.9, 32.2, 32.1, 29.9, 29.8, 29.7, 29.6, 22.9, 14.3. LCMS: 81.30% purity at RT = 3.30; m/z = 377.32 [M + H]+ (observed).Preparation of 2,4-dimethoxy-6-pentadecylbenzoic acid (10)
The general method used for the synthesis of 3 was used here for the synthesis of compound 10. Off-white solid 0.37 g (89% yield).1H-NMR: (400 MHz, CDCl3): δ 6.43 (d, J = 1.6 Hz, 1H), 6.38 (d, J = 2.0 Hz, 1H), 3.93 (s, 3H), 3.84 (s, 3H), 2.88 (t, J = 8.4 Hz, 2H), 1.60–1.56 (m, 2H), 1.35–1.25 (m, 24H), 0.88 (t, J = 7.2 Hz, 3H). LCMS: 80.17% purity at RT = 3.02; m/z = 393.52 [M + H]+ (observed).Preparation of 5-pentadecylbenzene-1,3-diol (12)
The general method used for the synthesis of 6 was used here for the synthesis of compound 12. Low melting off-white solid 3.31 g (65% yield).1H-NMR: (400 MHz, CDCl3): δ 6.24 (s, 2H), 6.17 (s, 1H), 2.48 (t, J = 7.6 Hz, 2H), 1.56–1.54 (m, 2H), 1.25 (br s, 24H), 0.88 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 156.7, 146.4, 108.2, 100.3, 36.04, 32.1, 31.2, 29.9, 29.8, 29.7, 29.5, 22.9, 14.3. LCMS: 85.12% purity at RT = 1.43; m/z = 321.48 [M + H]+ (observed).Preparation of 2,4-dihydroxy-6-pentadecylbenzaldehyde(13)
The general method used for the synthesis of 2 was used here for the synthesis of compound 13. Off-white solid 2.29 g (70% yield).1H-NMR: (400 MHz, DMSO-d6): δ 12.09 (br s, 1H), 10.68 (br s, 1H), 10.05 (br s, 1H), 6.21 (d, J = 2.0 Hz, 1H), 6.13 (d, J = 2.0 Hz, 1H), 2.80 (t, J = 7.6 Hz, 2H), 1.58–1.51 (m, 2H), 1.26–1.22 (m, 24H), 0.88 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 192.7, 165.4, 165.1, 149.5, 111.7, 109.9, 100.2, 31.8, 31.3, 31.2, 28.9, 28.7, 28.6, 22.07, 13.9. LCMS: m/z = 349.53 [M + H]+ (observed).Cell culture
Cell lines HepG2 (Human Hepatocarcinoma cell line), N2A (Murine Neural cell line), and CaCo-2 (Human Colon adenocarcinoma cell line) were purchased from the National Centre for Cell Sciences, NCCS, Pune. Cells were cultured in MEM (Minimal Essential media) medium with 10% Fetal Bovine Serum (FBS) and incubated at 37 °C with 5% CO2.Cytotoxicity evaluation
In vitro cytotoxicity evaluation of corticiolic acid (7), cordol (12), and cordanaldehyde (13) was performed using MTT assay, as described by Mossman.27 Approximately 1 × 104 mL−1 cells (HepG2, CaCo-2, and N2A) in their exponential growth phase were seeded in a flat-bottomed 96-well polystyrene-coated plate and incubated for 24 h at 37 °C in a 5% CO2 incubator. A series of dilutions (100, 50, 25, 12.5, and 6.25 μg mL−1) of the synthesized compounds in the medium was done in the plate in triplicates. After 24 hours of incubation, 100 μL of MTT reagent (0.5 mg mL−1) was added to each well and further incubated for 4 hours. Formazan crystals formed after 4 hours in each well were dissolved in 100 μL of DMSO and the plates were read immediately using a microplate reader (BIO-RAD microplate reader) at 570 nm and 630 nm. Untreated cells were used as a negative control for the experiment. Cordol and corticiolic acid reduced the viability in all three cell lines at higher concentrations, as shown in Fig. 2.Data availability
Data files can be shared on reasonable request.Author contributions
SKD-synthesis and analysis; BA-analysis; PGP-carryout biological studies; VCN-design, discussion, and paper writing; design of bioassays; PR-writing and analysis; SS and NP-conceptualization, synthesis, characterization, analysis, and writing the manuscript.Conflicts of interest
There are no conflicts to declare.Notes and references
- F. I. Saldívar-González, V. D. Aldas-Bulos, J. L. Medina-Franco and F. Plisson, Chem. Sci., 2022, 13, 1526–1546 RSC
.
- N. E. Thomford, D. A. Senthebane, A. Rowe, D. Munro, P. Seele, A. Maroyi and K. Dzobo, Int. J. Mol. Sci., 2018, 19, 1578 CrossRef
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS
- A. S. Abdel-Razek, M. E. El-Naggar, A. Allam, O. M. Morsy and S. I. Othman, Processes, 2020, 8, 470 CrossRef CAS
- P. J. Rutledge and G. L. Challis, Nat. Rev. Microbiol., 2015, 13, 509–523 CrossRef CAS PubMed
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, I. E. Orhan, M. Banach, J. M. Rollinger, D. Barreca, W. Weckwerth, R. Bauer, E. A. Bayer, M. Majeed, A. Bishayee, V. Bochkov, G. K. Bonn, N. Braidy, F. Bucar, A. Cifuentes, G. D'Onofrio, M. Bodkin, M. Diederich, A. T. Dinkova-Kostova, T. Efferth, K. El Bairi, N. Arkells, T. P. Fan, B. L. Fiebich, M. Freissmuth, M. I. Georgiev, S. Gibbons, K. M. Godfrey, C. W. Gruber, J. Heer, L. A. Huber, E. Ibanez, A. Kijjoa, A. K. Kiss, A. Lu, F. A. Macias, M. J. S. Miller, A. Mocan, R. Müller, F. Nicoletti, G. Perry, V. Pittalà, L. Rastrelli, M. Ristow, G. L. Russo, A. S. Silva, D. Schuster, H. Sheridan, K. Skalicka-Woźniak, L. Skaltsounis, E. Sobarzo-Sánchez, D. S. Bredt, H. Stuppner, A. Sureda, N. T. Tzvetkov, R. A. Vacca, B. B. Aggarwal, M. Battino, F. Giampieri, M. Wink, J. L. Wolfender, J. Xiao, A. W. K. Yeung, G. Lizard, M. A. Popp, M. Heinrich, I. Berindan-Neagoe, M. Stadler, M. Daglia, R. Verpoorte and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed
- S. R. M. Ibrahim, A. Sirwi, B. G. Eid, S. G. A. Mohamed and G. A. Mohamed, Metabolites, 2021, 11, 943 CrossRef
- K. Hamanof, M. Kinoshita-Okami, K. Minagawa, H. Haruyama, T. Kinoshita, T. Hosoyac, K. Furuyac, K. Kinoshita, K. Tabata, A. Hemmiand and K. Tanzawa, J. Antibiot., 1993, 46, 1648–1657 CrossRef
- Y.-J. Kwon, Y. Fang, G. H. Xu and W.-G. Kim, Biol. Pharm. Bull., 2009, 32, 2061–2064 CrossRef CAS
- C. Seo, J. H. Sohn, H. Oh, B. Y. Kim and J. S. Ahn, Bioorg. Med. Chem. Lett., 2009, 19, 6095–6097 CrossRef CAS PubMed
- B. Sontag, J. Dasenbrock, N. Arnold and W. Steglich, Eur. J. Org Chem., 1999, 1051–1055 CrossRef CAS
- N. Shoji, A. Umeyama, T. Takemoto, M. Kobayashi and Y. Ohizumi, J. Nat. Prod., 1984, 47, 530–532 CrossRef CAS
- A. Kozubek and J. H. P. Tyman, Stud. Nat. Prod. Chem., 2005, 36, 111–190 Search PubMed
- P. Wang, Z. Zhang and B. Yu, J. Org. Chem., 2005, 70, 8884–8889 CrossRef CAS PubMed
- M. Ghizzoni, A. Boltjes, C. D. Graaf, H. J. Haisma and F. J. Dekker, Bioorg. Med. Chem., 2010, 18, 5826 CrossRef CAS PubMed
- M. Ghizzoni, J. Wu, T. Gao, H. J. Haisma, F. J. Dekker and Y. G. Zheng, Eur. J. Med. Chem., 2012, 47, 337 CrossRef CAS PubMed
- J. L. Alterman and G. A. Kraus, Nat. Prod. Commun., 2019, 14, 6 CrossRef
- J. El-Maiss, T. M. El Dine, C. S. Lu, I. Karamé, A. Kanj, K. Polychronopoulou and J. Shaya, Catalysts, 2020, 10, 296 CrossRef CAS
- G. A. Molander and B. Canturk, Angew. Chem. Int. Ed., 2009, 48, 9240–9261 CrossRef CAS PubMed
- M. Farhang, A. R. Akbarzadeh, M. Rabbani and A. M. Ghadiri, Polyhedron, 2022, 227, 116124 CrossRef CAS
- S. K. Shaji, G. Drishya, D. Sunilkumar, N. Pandurangan, G. B. Kumar and B. G. Nair, Eur. J. Pharmacol., 2021, 893, 173808 CrossRef CAS PubMed
- D. Nedungadi, A. Binoy, N. Pandurangan, B. G. Nair and N. Mishra, Cell Biol. Int., 2021, 45, 164–176 CrossRef CAS
- A. Omanakuttan, C. Bose, N. Pandurangan, G. B. Kumar, A. Banerji and B. G. Nair, Exp. Cell Res., 2016, 346, 167–175 CrossRef CAS PubMed
- J. Nambiar, G. Vijayakumar, G. Drishya, S. K. Shaji, N. Pandurangan, G. B. Kumar and B. G. Nair, Mol. Cell. Biochem., 2019, 451, 79–90 CrossRef CAS
- P. Nanjan, J. Nambiar, B. G. Nair and A. Banerji, Bioorg. Med. Chem., 2015, 23, 3781–3787 CrossRef CAS
- K. Amrutha, P. Nanjan, S. K. Shaji, D. Sunilkumar, K. Subhalakshmi, L. Rajakrishna and A. Banerji, Bioorg. Med. Chem. Lett., 2014, 24, 4735–4742 CrossRef CAS PubMed
- T. Mosmann, J. Immun. Meth, 1983, 65, 55–63 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and NMR (1H and 13C) spectral data. See DOI: https://doi.org/10.1039/d4ra06585a |
| This journal is © The Royal Society of Chemistry 2024 |