
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhenylsulfonate as a photolabile group for intramolecular carbon–carbon cross-coupling reactions†
Simon Plaize and
Jean-François Morin *
*
Département de Chimie and Centre de Recherche sur les Matériaux Avancés (CERMA), Université Laval, 1045 Ave de La Médecine, Québec, G1V 0A6, Canada. E-mail: jean-francois.morin@chm.ulaval.ca
First published on 5th November 2024
Abstract
Efficient cyclization reactions play a pivotal role in the synthesis of extended polycyclic aromatic hydrocarbons (PAHs) and graphene nanoribbons. Although efficient reactions have been developed, a simple yet versatile method that is compatible with most functional groups is still lacking. Herein, we report the use of phenylsulfonates as a photolabile group to generate aryl radicals that undergo a radical cyclization reaction to produce triphenylene derivatives. The phenylsulfonate group proves to be a highly adaptable and robust photolabile group, and compatible with Suzuki cross-coupling conditions. Kinetic and optimization experiments have been conducted, shedding light on the potential of this reaction as a versatile tool for the synthesis of PAHs.
The synthetic methodologies employed for the preparation of polycyclic aromatic hydrocarbons (PAHs) and graphene nanoribbons (GNRs) involve the synthesis of a phenyl-rich precursor, followed by multiple intramolecular C–C cross-coupling reactions.1–4 The oxidative dehydrogenation reaction, commonly known as the Scholl reaction, is the primary method used for the latter.5 This reaction requires an excess of oxidant such as iron(III) chloride, aluminum(III) chloride and DDQ,5–10 which can be incompatible with several chemical functions.5,11,12 Moreover, the Scholl reaction often leads to rearrangement, oligomerization and halogenation, making it unsuitable for the synthesis of several structures.13–18
Recently, our research group revisited and optimized the photochemical cyclodehydrochlorination (CDHC) for the synthesis of PAHs and GNRs.18–20 While this reaction offers numerous advantages that we exploited for producing a variety of PAHs and GNRs,1,18–20 it exhibits significant drawbacks, some of which are intrinsic and related to the reaction mechanism under irradiation.21 Among others, the need for anchimeric assistance by a freely rotating neighboring aryl and the relatively high energy required for the homolysis of the C–Cl bond cause significant limitations for the use of this reaction.
To overcome these issues, the substitution of the chlorine atom with a photocleavable group capable of cleanly generating either a phenyl radical or a cation intermediate, which would subsequently undergo an intramolecular SRN1 or SN1 reaction, has been explored. In this context, photochemical methods utilizing triazene22–24 and arylazo sulfone25 have been developed. While elegant, the preparation of both triazene and azosulfone moieties necessitates the use of sensitive diazonium intermediates, which is a significant disadvantage for introducing multiple functionalities on a single aryl, as often required for the synthesis of large PAHs.
Another photocleavable group that has been used to generate aryl radicals is the phenylsulfonate.26 The photolysis mechanism proposed by Scaiano and coworkers involves the generation of the phenoxy radical, leading to the extrusion of SO2 and the subsequent generation of the aryl radical (Fig. 1, left pathway). More recently, Fagnoni and coworkers explored the use of aryl sulfonates to generate triplet aryl cations, through the photoheterolysis of the Ar–OS bond, that can undergo a coupling reaction with different π nucleophiles (Fig. 1, right pathway).27 Reactions with monosubstituted alkenes28 and various substituted phenyl moieties29 have been reported with relative success. In most cases, the yields are limited by a competing mechanism that involved the homolytic cleavage of the ArO–S bond. Fagnoni and coworkers hypothesized that the later mechanism is largely preferred as the ArO–S bond cleavage proceeds from the singlet excited state (fast process) while the mechanism yielding to the cleavage of the Ar–OS bond requires intersystem crossing (ISC) to the triplet state (slow process). Despite the predominance of the former mechanism, the photochemical homolytic cleavage of the ArO–S has been underexploited for the generation of aryl radicals, which is an increasingly demanded intermediate for carbon–carbon cross-coupling reactions.30
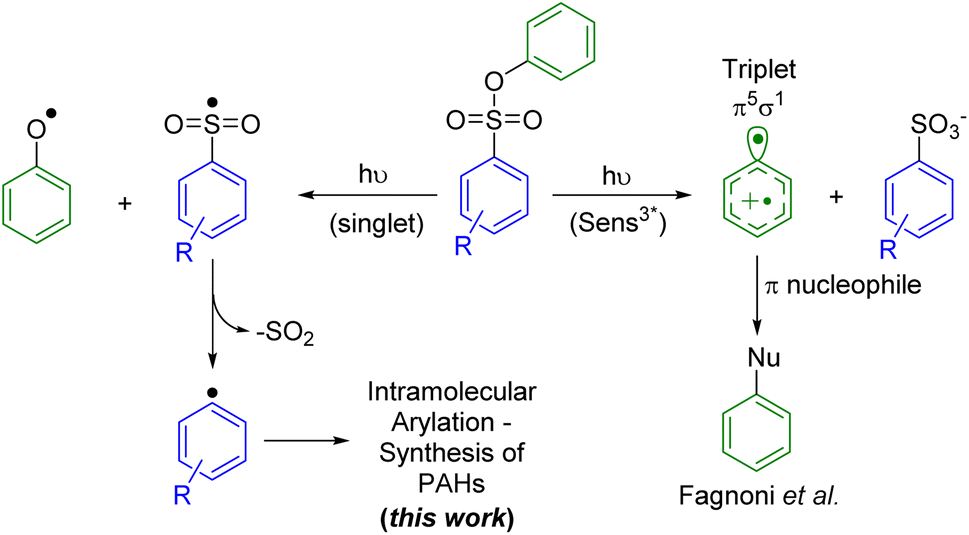 | ||
| Fig. 1 Mechanistic pathways for the photocleavage of the phenylsulfonate moiety. | ||
Herein, we report the use of phenylsulfonate as a photocleavable group to generate aryl radicals that further engaged in an intramolecular radical cyclization to afford various triphenylene derivatives. Reaction parameters, including the concentration, the solvent and the nature of the functional group added on the phenolic part of the phenylsulfonate have been studied. Experiments conducted using triplet quenchers suggested that this reaction proceed through the triplet state.
Results and discussion
To study the efficiency of the phenylsulfonate moiety as a photocleavable group for the synthesis of PAHs, a simple o-terphenyl derivative (compound 3, strategy 1, Scheme 1) was prepared. Starting from the commercially available compound 1, the phenylsulfonate group was formed using a standard condensation reaction with phenol is the presence of a base to provide compound 2 in excellent yield. This compound was engaged in a Suzuki cross-coupling reaction with 2-(biphenyl-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane to produce compound 3 in 70% yield. The later reaction demonstrates the compatibility of the phenylsulfonate group with Suzuki–Miyaura reaction conditions, which is crucial for the use of this functional group in the synthesis of larger, more complex PAHs and GNRs.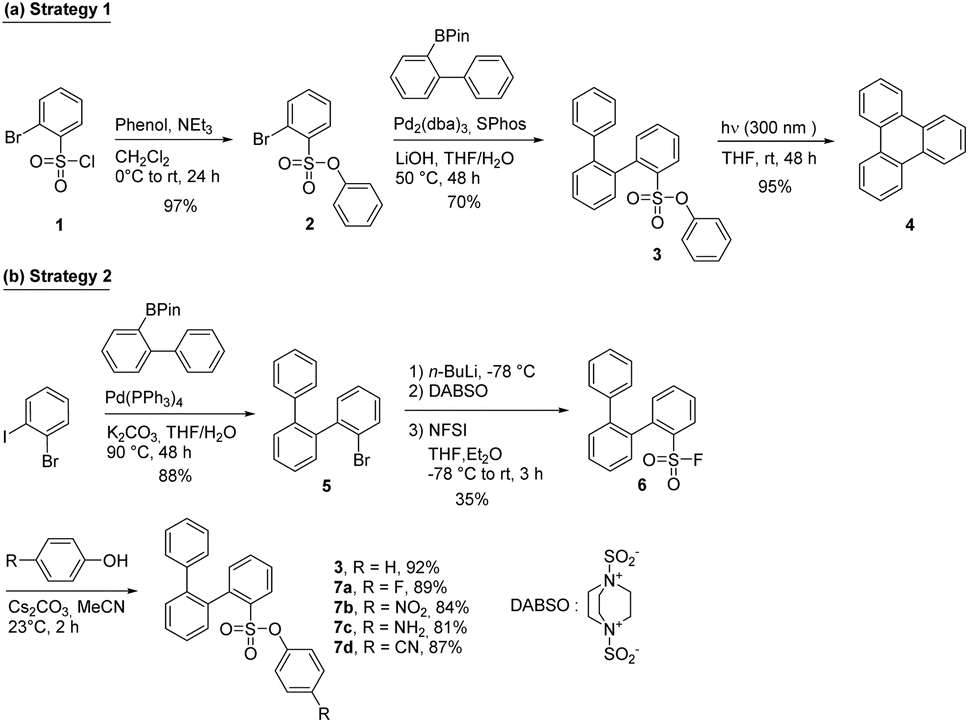 | ||
| Scheme 1 (a) Synthetic pathway for the preparation of triphenylene from chlorosulfonyl derivatives and (b) an alternative strategy involving the preparation of fluorosulfonyl derivatives as precursors. | ||
Despite the success of this synthetic pathway, we decided to explore a different strategy in which the phenylsulfonate group is introduced after the Suzuki–Miyaura coupling. This approach allows to eventually overcome the steric hindrance caused by the phenylsulfonate group (strategy 2, Scheme 1). The o-terphenyl was first prepared using a selective Suzuki–Miyaura coupling between 2-iodobromobenzene and 2-(biphenyl-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane to give compound 4 in 88% yield. Then, a fluorosulfonyl group was introduced in 35% yield using the modified method developed by Willis and coworkers.31 Finally, condensations with different substituted phenol derivatives were achieved in good to excellent yield. Phenol substitution in para position with either electron-donating (NH2) or electron-withdrawing (F, NO2 and CN) group allows for a modification of the electronic properties of the phenylsulfonate group, which can influence its reactivity towards irradiation with UV light (vide infra).
With different o-terphenyl derivatives in hands, attempts at preparing triphenylene 4 through UV irradiation have been made. As the reference set of conditions, the unsubstituted compound 3 was irradiated at 300 nm in degassed THF. Surprisingly, compound 3 was totally consumed after 48 hours, leading to the formation of the cyclized product (triphenylene 4) in 95% yield (Scheme 1). Then, an investigation was undertaken to identify the optimal solvent and concentration for the cyclization reaction. The concentration of the reagent is a critical factor in photochemistry as low concentrations reduce the solution absorption. As depicted in Fig. 2a, a direct correlation was observed between concentration and reaction rate, with lower concentrations resulting in faster conversions. In fact, fast conversion was observed at 10−3 M and further dilution did not improve the reaction rate. Increasing the concentration to 5 × 10−3 M resulted in only a marginal decrease in the reaction rate, whereas doubling that concentration to 1.0 × 10−2 M led to a substantial decrease. For practical reasons, further reactions were carried out at a concentration of 5 × 10−3 M.
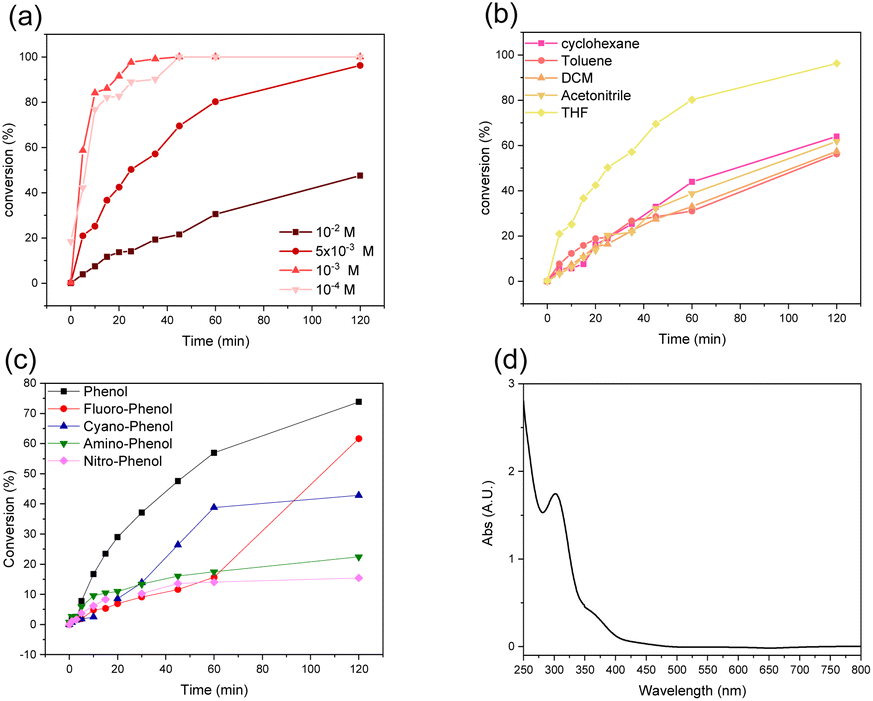 | ||
| Fig. 2 (a) Photolysis of 3 in different concentrations in THF. (b) Photolysis of 3 in different solvents at a constant concentration (5 × 10−3M). (c) Photolysis of different substrates (Scheme 1) in THF. (d) UV-vis spectrum of 3 in THF. All these experiments have been conducted at 300 nm. | ||
The effect of the solvent was studied, and the results are reported in Fig. 2b. Interestingly, acetonitrile, dichloromethane, toluene and cyclohexane yielded almost the same reaction rate. Moreover, none of these solvents allowed for a total conversion of compound 3, even after 48 hours of irradiation. Fortunately, THF prove to be way more efficient, both in terms of reaction rate and conversion, the latter being almost complete after 120 minutes.
Next, the reaction rates for the formation of triphenylene from substrates with various functional groups attached to the phenolic part were examined in THF under irradiation at 300 nm for 2 hours. The results are presented in Fig. 2c. Unsubstituted phenol derivative 3 underwent the fastest conversion to form compound 4. Unexpectedly, both the electron-donating (NH2, 7c) and electron-withdrawing (F, NO2, CN, 7a, 7b and 7d, respectively) groups led to a significant decrease of the reaction rate, the –NH2 and –CN group giving the worst result with a negligible amount of compound 4 produced after 2 hours. It is noteworthy that other parameters such as the molar extinction coefficient and the intersystem crossing (ISC) rate might be influenced by the functionalization of the phenylsulfonate group, changing its photochemical behaviour.
To broaden the scope of the reaction, the formation of a 5-membered ring to form fluoranthene 9 was attempted (Scheme 2). Following a 96 hours photolysis at 300 nm in THF, two distinct reaction products were observed: the desired fluoranthene 9 and 1-phenylnaphthalene 10, resulting from the desulfonylated product, in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (GC result, 10% yield). This result is consistent with reported radical ring closure experiments using halogens (Br and I) as photolabile groups, suggesting that the phenylsulfonate group indeed generates aryl radical upon irradiation with UV light as shown in Fig. 1 (singlet pathway).32
:4 (GC result, 10% yield). This result is consistent with reported radical ring closure experiments using halogens (Br and I) as photolabile groups, suggesting that the phenylsulfonate group indeed generates aryl radical upon irradiation with UV light as shown in Fig. 1 (singlet pathway).32
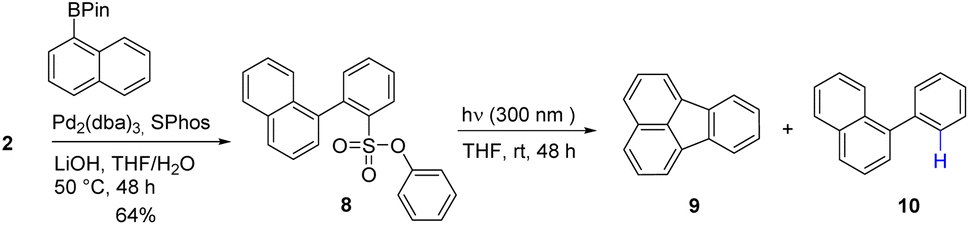 | ||
| Scheme 2 Synthetic pathway giving fluoranthene after photolysis. | ||
To gain better insight into the reaction mechanism, a triplet sensitization experiment was conducted using thioxanthone. Two solutions of compound 3 in THF at 5 × 10−3 M, one without a triplet sensitizer and the other with 20% molar of thioxanthone, were irradiated at 365 nm in THF for 48 hours. Interestingly, the reaction mixture containing the thioxanthone yielded complete conversion of compound 3 within 48 hours while the one without produced only traces amount of compound 4. This result indicates that the photochemical decomposition of phenylsulfonyl to form the aryl radical proceeds through the triplet state, contradicting the initial hypothesis stating that the generation of aryl radicals through the homolysis of the phenylsulfonate group goes through a singlet state as shown in Fig. 1. To confirm this result, a triplet quenching experiment was performed using triethylamine and DABCO as quenchers to validate this result.33 Interestingly, these experiments showed that the photocyclization is ca. four times slower for both quenchers compared to the same reaction conducted without a quencher (see ESI† section for the experimental details). These results support the hypothesis of a triplet-mediated pathway for the fragmentation of phenylsulfonate to generate an aryl radical.
In earlier work, Scaiano26 and Protti34 independently showed that the photolysis of phenylsulfonate produced three distinct radicals. Thus, attempts at trapping these radicals using TEMPO were made in various conditions. Interestingly, none of these radicals were either trapped by TEMPO (20 eq.) or led to the formation of a photo-Fries product, commonly observed for similar substrates.27 In addition, the mechanism proposed by Scaiano26 involved the homolysis of the ArS–O bond first to form a phenoxy radical, followed by the extrusion of SO2. Despite all our attempts, phenol was never isolated or detected by HRMS or NMR, but unidentified peaks can be observed in the NMR spectrum of the crude mixture (see the ESI† section). This result suggests that phenol might be engaged in side reactions that do not involve the desired cyclized product.
Conclusion
In conclusion, we have successfully developed a novel intramolecular photochemical cyclization reaction utilizing the phenylsulfonate group as an aryl radical precursor to prepare PAH derivatives. In contrast to other reports, this study suggests that both direct and triplet-sensitized irradiation of aryl bearing a phenylsulfonate group lead to the formation of an aryl radical. The possibility of using a triplet sensitizer to generate an aryl radical is a major advantage since low energy visible light can be used instead of high energy UV irradiation. The phenylsulfonate group demonstrated robustness, modularity and versatility, making it a good alternative to known photocleavable groups for aryl radical generation. Future work will focus on incorporating the phenylsulfonate group into more complex structures to explore the potential of multicyclization reactions in collaboration with photocatalysts.Data availability
The data that support the findings of this study are available from the corresponding author, Prof. Jean-François Morin (E-mail: jean-francois.morin@chm.ulaval.ca), upon reasonable request.Author contributions
S. P.: conceptualization (equal), investigation (lead), methodology (lead), data curation (lead), validation (lead), formal analysis (lead), writing original draft, review, and editing (lead). J.-F. M.: conceptualization (equal), investigation, methodology, project administration (lead), resources (lead), validation, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Natural Sciences and Engineering Research Council of Canada (NSERC) through the Discovery Grant Program for financial support. We are also grateful for professional support from CQMF and CERMA.Notes and references
- A. Jolly, D. Miao, M. Daigle and J.-F. Morin, Emerging Bottom-Up Strategies for the Synthesis of Graphene Nanoribbons and Related Structures, Angew. Chem., Int. Ed., 2020, 59, 4624–4633 CrossRef CAS.
- L. Chen, Y. Hernandez, X. Feng and K. Müllen, From Nanographene and Graphene Nanoribbons to Graphene Sheets: Chemical Synthesis, Angew. Chem., Int. Ed., 2012, 51, 7640–7654 CrossRef CAS PubMed.
- A. Narita, Z. Chen, Q. Chen and K. Müllen, Solution and on-surface synthesis of structurally defined graphene nanoribbons as a new family of semiconductors, Chem. Sci., 2019, 10, 964–975 RSC.
- K. Y. Yoon and G. Dong, Liquid-phase bottom-up synthesis of graphene nanoribbons, Mater. Chem. Front., 2020, 4, 29–45 RSC.
- P. Rempala, J. Kroulik and B. T. King, Investigation of the Mechanism of the Intramolecular Scholl Reaction of Contiguous Phenylbenzenes, J. Org. Chem., 2006, 71, 5067–5081 CrossRef CAS.
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Comparison of Oxidative Aromatic Coupling and the Scholl Reaction, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed.
- R. S. Jassas, E. U. Mughal, A. Sadiq, R. I. Alsantali, M. M. Al-Rooqi, N. Naeem, Z. Moussa and S. A. Ahmed, Scholl reaction as a powerful tool for the synthesis of nanographenes: a systematic review, RSC Adv., 2021, 11, 32158–32202 RSC.
- M. Grzybowski, B. Sadowski, H. Butenschön and D. T. Gryko, Synthetic Applications of Oxidative Aromatic Coupling—From Biphenols to Nanographenes, Angew. Chem., Int. Ed., 2020, 59, 2998–3027 CrossRef CAS.
- L. Zhai, R. Shukla and R. Rathore, Oxidative C−C Bond Formation (Scholl Reaction) with DDQ as an Efficient and Easily Recyclable Oxidant, Org. Lett., 2009, 11, 3474–3477 CrossRef CAS PubMed.
- R. Scholl and J. Mansfeld, meso-Benzdianthron (Helianthron), meso-Naphthodianthron, und ein neuer Weg zum Flavanthren, Ber. Dtsch. Chem. Ges., 1910, 43, 1734–1746 CrossRef CAS.
- B. T. King, J. Kroulík, C. R. Robertson, P. Rempala, C. L. Hilton, J. D. Korinek and L. M. Gortari, Controlling the Scholl reaction, J. Org. Chem., 2007, 72, 2279–2288 CrossRef CAS PubMed.
- X. Dou, X. Yang, G. J. Bodwell, M. Wagner, V. Enkelmann and K. Mullen, Unexpected Phenyl Group Rearrangement during an Intramolecular Scholl Reaction Leading to an Alkoxy-Substituted Hexa-peri-hexabenzocoronene, Org. Lett., 2007, 9, 2485–2488 CrossRef CAS PubMed.
- M. Danz, R. Tonner and G. Hilt, Understanding the regioselectivity in Scholl reactions for the synthesis of oligoarenes, Chem. Commun., 2012, 48, 377–379 RSC.
- D. Lorbach, M. Wagne, M. Baumgarten and M. Klaus, The right way to self-fuse bi- and terpyrenyls to afford graphenic cutouts, Chem. Commun., 2013, 49, 10578–10580 RSC.
- M. Müller, V. S. Iyer, C. Kübel, V. Enkelmann and K. Müllen, Polycyclic Aromatic Hydrocarbons by Cyclodehydrogenation and Skeletal Rearrangement of Oligophenylenes, Angew. Chem., Int. Ed. Engl., 1997, 36, 1607–1610 CrossRef.
- N. Ponugoti and V. Parthasarathy, Rearrangements in Scholl Reaction, Chem.–Eur. J., 2022, 28, e202103530 CrossRef CAS.
- A. Pradhan, P. Dechambenoit, H. Bock and F. Durola, Highly twisted arenes by scholl cyclizations with unexpected regioselectivity, Angew. Chem., Int. Ed., 2011,(50), 12582–12585 CrossRef CAS.
- M. Daigle, A. Picard-Lafond, E. Soligo and J. F. Morin, Regioselective Synthesis of Nanographenes by Photochemical Cyclodehydrochlorination, Angew. Chem., Int. Ed., 2016, 55, 2042–2047 CrossRef CAS PubMed.
- M. Daigle, D. Miao, A. Lucotti, M. Tommasini and J. F. Morin, Helically Coiled Graphene Nanoribbons, Angew. Chem., Int. Ed., 2017, 56, 6213–6217 CrossRef CAS PubMed.
- D. Miao, M. Daigle, A. Lucotti, J. Boismenu-Lavoie, M. Tommasini and J. F. Morin, Toward Thiophene-Annulated Graphene Nanoribbon, Angew. Chem., Int. Ed., 2018, 57, 3588–3592 CrossRef CAS.
- A. Jolly, C. É. Fecteau, P. A. Johnson and J. F. Morin, Parameters Influencing the Photochemical Cyclodehydrochlorination (CDHC) Reaction, ChemPlusChem, 2024, 89, e202300677 CrossRef CAS.
- T. B. Patrick, R. P. Willaredt and D. J. DeGonia, Synthesis of biaryls from aryltriazenes, J. Org. Chem., 1985, 50, 2232–2235 CrossRef CAS.
- J. Zhou, W. Yang, B. Wang and H. Ren, Friedel–Crafts Arylation for the Formation of Csp2–Csp2 Bonds: A Routeto Unsymmetrical and Functionalized Polycyclic AromaticHydrocarbons from Aryl Triazenes, Angew. Chem., Int. Ed., 2012, 51, 12293–12297 CrossRef CAS PubMed.
- E. Barragan, A. Noonikara Poyil, C. H. Yang, H. Wang and A. Bugarin, Metal-free cross-coupling of π-conjugated triazenes with unactivated arenes via photoactivation, Org. Chem. Front., 2019, 6, 152–161 RSC.
- S. Crespi, S. Protti and J. Fagnoni, Wavelength Selective Generation of Aryl Radicals and Aryl Cations for Metal-Free Photoarylations, J. Org. Chem., 2016, 81(20), 9612–9619 CrossRef CAS PubMed.
- J. Andraos, G. G. Barclay, D. R. Medeiros, M. V. Baldovi, J. C. Scaiano and R. Sinta, Model Studies on the Photochemistry of Phenolic Sulfonate Photoacid Generators, Chem. Mater., 1998, 10(6), 1694–1699 CrossRef CAS.
- (a) M. Terpolilli, D. Merli, S. Protti, V. Dichiarante, M. Fagnoni and A. Albini, Cationic and radical intermediates in the acid photorelease from aryl sulfonates and phosphates, Photochem. Photobiol. Sci., 2011, 10, 123–127 CrossRef CAS PubMed; (b) S. Lazzaroni, D. Ravelli, S. Protti, M. Fagnoni and A. Albini, Photochemical synthesis: Using light to build C–C bonds under mild conditions, Compt. Rendus Chem., 2017, 20, 261–271 CAS.
- M. De Carolis, S. Protti, M. Fagnoni and A. Albini, Metal-Free Cross-Coupling Reactions of Aryl Sulfonates and Phosphates through Photoheterolysis of Aryl–Oxygen Bonds, Angew. Chem., Int. Ed., 2005, 44, 1232–1236 CrossRef CAS PubMed.
- C. Raviola, V. Canevari, S. Protti, A. Albini and M. Fagnoni, Metal-free arylations via photochemical activation of the Ar–OSO2R bond in aryl nonaflates, Green Chem., 2013, 15, 2704–2708 RSC.
- (a) N. Kvasovs and V. Gevorgyan, Contemporary methods for generation of aryl radicals, Chem. Soc. Rev., 2021, 50, 2244–2259 RSC; (b) A. Das and K. R. Justin Thomas, Generation and Application of Aryl Radicals Under Photoinduced Conditions, Chem.–Eur. J., 2024, 30, e202400193 CrossRef CAS PubMed.
- A. T. Davies, J. M. Curto, S. W. Bagley and M. C. Willis, One-pot palladium-catalyzed synthesis of sulfonyl fluorides from aryl bromides, Chem. Sci., 2017, 8, 1233–1237 RSC.
- W. A. Henderson and A. Zweig, Photolytic rearrangement and halogen-dependent photocyclization of halophenylnaphthalenes, J. Am. Chem. Soc., 1967, 89(25), 6778–6779 CrossRef CAS.
- J. F. Rodrigues, F. De Assis Da Silva and C. P. Netto-Ferreira, Laser Flash Photolysis Study of the Photochemistry of Thioxanthone in Organic Solvents, J. Braz. Chem. Soc., 2010, 21, 960–965 CrossRef CAS.
- E. Torti, G. Della Giustina, S. Protti, D. Merli, G. Brusatin and M. Fagnoni, Aryl tosylates as non-ionic photoacid generators (PAGs): photochemistry and applications in cationic photopolymerizations, RSC Adv., 2015, 5, 33239–33248 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06592a |
| This journal is © The Royal Society of Chemistry 2024 |