
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIonic resorcinarenes as drug solubilization agents in water†
Frank Boateng Osei a,
Kwaku Twuma,
Barbara Manfredib,
Mariana Fatohia,
Yvonne Bessem Ojonga,
Valance Washingtonb and
Ngong Kodiah Beyeh*a
a,
Kwaku Twuma,
Barbara Manfredib,
Mariana Fatohia,
Yvonne Bessem Ojonga,
Valance Washingtonb and
Ngong Kodiah Beyeh*a
aOakland University, Department of Chemistry, 146 Library Drive, Rochester, MI 48309-4479, USA. E-mail: beyeh@oakland.edu
bOakland University, Department of Biological Sciences, 146 Library Drive, Rochester, MI 48309-4479, USA
First published on 28th October 2024
Abstract
Resorcinarenes are capable of host–guest complexation with small molecules, however, they are less studied as pharmaceutical drug delivery aids. This article reports on the aqueous-solubility enhancing effect of an octa-sulfonated resorcinarene and a C-hydroxybenzyl ammonium resorcinarene chloride on three hydrophobic drugs: isoniazid, caffeine, and griseofulvin. The findings are backed by dynamic light scattering, isothermal calorimetric titration, and nuclear magnetic resonance experiments in water. Aqueous mixtures of equal volumes of drug compounds and resorcinarene solutions produced a more soluble and clearer unit than solutions of pure drug compounds in water. Light scattering experiments revealed shifts in particle sizes of pure drug compounds to the range of resorcinarene hosts. 1H NMR measurements of resorcinarene-drug mixtures confirmed interactions with shift changes ranging from −0.20 to 0.81 ppm. Binding affinities quantified through ITC experiments ranged between 0.54 and 211 mM, signifying interactions between resorcinarenes and drug compounds necessary for the solubility of the drugs. Cytotoxicity studies suggest that resorcinarenes alone, or complexed with any of the drug compounds, do not exert cytotoxicity in mammalian cells HEK-293 up to 200 μM. We herein propose a set of hydrophilic resorcinarene macrocycles as potential drug solubilizers.
Introduction
Resorcinarenes are widely investigated organic macrocycles for their host properties towards many guest systems.1–3 They interact with other molecular guests through multiple non-covalent interactions within their well-defined internal cavities.4–7 These host–guest interactions confer various properties on the guest molecules, which include solubility modifications, guest-release modifications, enhanced targeting, enhanced stability, and milder side effects in the case of drug agents.8–12 Drug solubility in aqueous environments, including that of the human body, is a major challenge in the pharmaceutical industry.13,14 The biopharmaceutical classification system (BCS) defines groups II and IV drugs as having lower solubility, which inhibits their gastrointestinal absorption, permeability, and efficacy.15,16 Enhancing the solubility of hydrophobic drugs plays a key role in formulation development as it enhances the bioavailability and therapeutic action of drugs at their target sites.17–19 Methods for enhancing drug solubility include particle size reduction through micronization20 and nanosuspension,21 modification of the crystal nature of the active pharmaceutical ingredient through crystal engineering, polymorphism, and use of hydrated or solvated forms of the drugs,22 drug dispersion in carriers,23 complexation techniques, and chemical modifications.24Griseofulvin is an antifungal agent mainly used to treat Tinea capitis infections, especially in children.25 It belongs to BCS group II drugs26 and is known to have low aqueous solubility, leading to low bioavailability.27 Methods that have been studied in the solubility enhancement of griseofulvin include nanoformulation techniques and melt granulation.28,29 Other BCS groups I and III drugs are known to have varying degrees of solubility that affect their permeability and bioavailability. Isoniazid, an anti-tubercular agent, is reported to be on the borderline of groups I and III because lactose and other deoxidizing saccharides can form condensation products with isoniazid, which limits its permeability.30 This calls for solid dosage forms of isoniazid that are very rapidly dissolving.30 Methods, such as cocrystal formation, have been employed to enhance the aqueous solubility and subsequent drug release.31 Caffeine, a psychomotor stimulant, is known to be hydrophobic in nature due to a planar purine structure and the presence of 3-methyl groups and, therefore, weakly polar in water undergoing oligomerization and aggregation in aqueous solutions, which limits its bioavailability and efficacy.32,33 Methods such as the use of sugars and deep eutectic solvents have been employed in enhancing the aqueous solubility of caffeine.33,34
Water-soluble resorcinarenes are not cytotoxic, especially at lower concentrations, and have been shown to exhibit proper biodegradability and biocompatibility, making them attractive in improving drug solubility.35 In this work, we explore the interactions between two synthetic ionic water-soluble resorcinarene macrocycles (R1 and R2, (Fig. 1)) and three hydrophobic drugs, isoniazid, caffeine, and griseofulvin (ISO, CAF, and GRI, (Fig. 1)). Herein, we investigate two water-soluble resorcinarenes that successfully interact and solubilize three drugs through a combination of NMR, isothermal titration calorimetry, and cytotoxicity analyses.
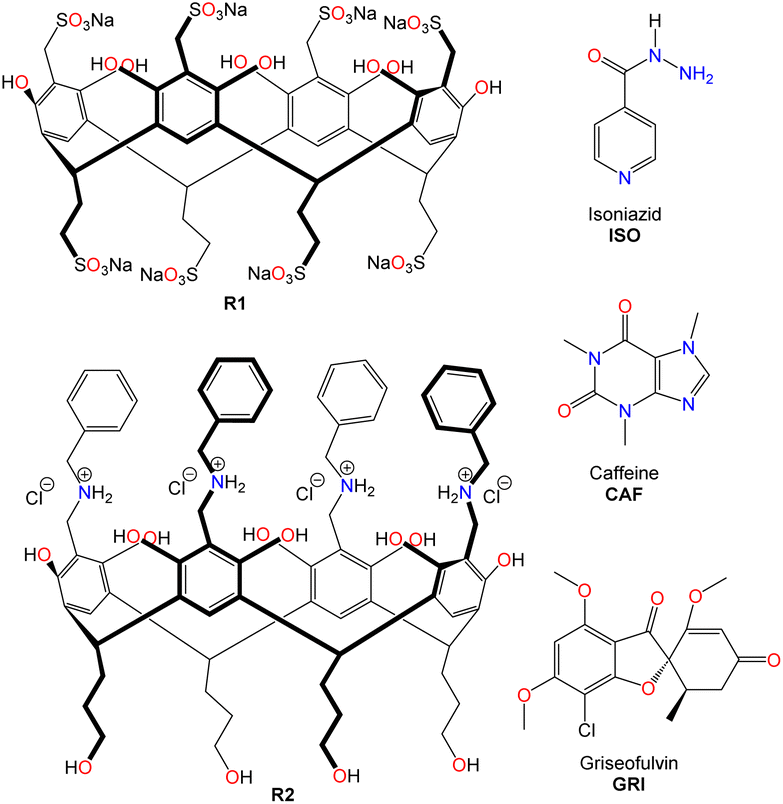 | ||
| Fig. 1 Structures of octa-sulfonated resorcinarene (R1) and C-hydroxybenzylammonium resorcinarene chloride (R2) as hosts, and drug compounds: isoniazid (ISO), caffeine (CAF) and griseofulvin (GRI) as guests. | ||
Materials and methods
Synthesis of resorcinarene receptors and guests
In this study, we selected two water-soluble resorcinarenes receptors. The first receptor is an octa-anionic poly-sulfonated resorcinarene with both the upper and lower rims decorated with sulfonate groups. A two-phase mixture of 2-(2-bromoethyl)-1,3-dioxane, 1 (20 mmol) and an aqueous solution (20 mL) of Na2SO3, 2 (40 mmol) was stirred at 100 °C for 24 hours. Water (20 mL) was added to the resulting homogeneous solution, and the mixture was washed with ether (40 mL × 2) to eliminate unreacted 1. To this mixture was added ethanol (40 mL), resorcinol, 3 (36 mmol), and concentrated HCI (6 mL). The mixture was stirred under nitrogen at 100 °C on a VWR® hotplate/stirrer for 24 h. After reflux, the solvent was evaporated, and the residue was taken in water (60 mL) and dialyzed three times against water (2 L) using a dialysis membrane with a transport critical molecular weight of 1000 (Spectra/Por membrane MWCO 1000) to remove inorganic salts. Most of the water was removed under vacuum, and the residue was triturated from cold methanol to give the lower rim sulfonated resorcinarene, 4 (Fig. 2). Resorcinarene 4 (0.01 mol), a solution of 37% formaldehyde (0.01 mol) and sodium sulfite (0.01 M) in H2O (30 mL) was stirred and heated at 90–95 °C for 4 h. Dilute hydrochloric acid was added after cooling until pH 7, then methanol (50 mL or more) was added to precipitate the product R1. The solid was filtered and dried (Fig. 2 and S1–S4†).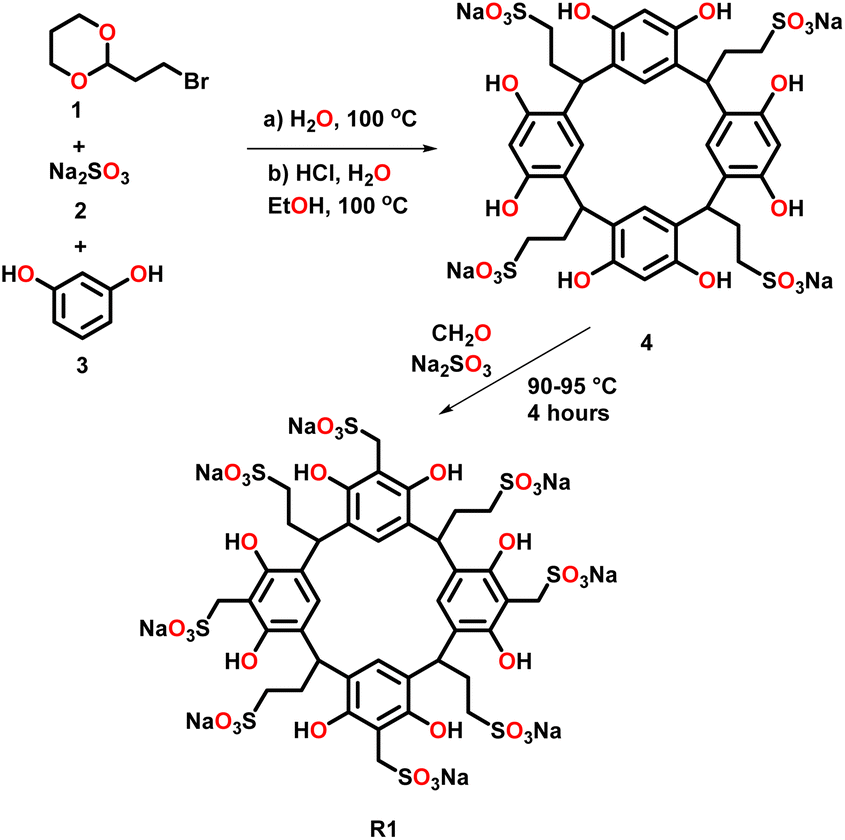 | ||
| Fig. 2 Schematic representation of the synthesis of R1 with 4 as an intermediate. | ||
The second receptor is a tetra-cationic resorcinarene with the cationic groups on the upper rim and four hydroxyl groups on the lower rim for enhanced solubility. This cationic receptor also possesses four flexible benzyl units with the potential to interact with the aromatic fragments of the drugs. The synthesis of this macrocycle is reported elsewhere (Fig. S5 and S6†).36–38 Both resorcinarene macrocycles exist in the C4v conformation with a persistent hydrophobic cavity. For this pilot study, three drugs, isoniazid, ISO, caffeine, CAF, and griseofulvin, GRI, were selected. These drugs were selected due to their hydrophobic nature and very limited solubility in water. In addition, they all possess pyridinium, imidazolium, and methyl phenolic aromatic groups, respectively, which can interact with different components of the two resorcinarenes receptors. All the drugs were purchased from Sigma Aldrich.
Dynamic light scattering (DLS) experiments
Dynamic light scattering measurements were performed to determine the particle size distributions within associations between host and guest molecules, which can be a determinant of the solubility of drugs. This technique has been employed to study the solubility of drugs such as doxorubicin, hydrochlorothiazide, lamotrigine, erythromycin, ibuprofen, and aspirin.39–42 DLS was conducted using a Malvern® zetasizer Nano from Malvern Panalytical to measure the particle size distribution of drug compounds alone and in combination with macrocycles. The Malvern Instruments DLS device (Zetasizer Nano ZS Series) has a 4 mW He–Ne ion laser at a wavelength of 633 nm and an avalanche photodiode detector at an angle of 173°. Experiments were carried out at 25 °C. Plastibrand semi-micro PMMA cuvettes were used to measure the size. Zetasizer software (Malvern Instruments) was used to obtain the particle size distributions. Equimolar concentration samples were prepared in deionized water. DLS experiments involved 1 mL of each pure sample. Mixtures were prepared by pipetting 1 mL of drug compound solution and 1 mL of resorcinarene into a clean cuvette.Nuclear magnetic resonance (NMR)
NMR is a suitable method to study the interaction between organic species in solution by monitoring changes between the isolated components and the physical mixtures. This technique also provides information on the orientation of the drug in the macrocycle. 1H and 13C NMR are useful as the chemical and electronic environments of the protons and/or carbons are affected in macrocycle-drug complexations.42 This method has been used to assess the solubility of drugs such as ibuprofen and carbamazepine.43–45 The qualitative binding properties of drugs as guests towards hosts R1-2 were probed in solution through 1H NMR experiments. The interaction between the host and guest in deuterated water was observed from equimolar mixtures of both species. Spectra were recorded on a Bruker Avance DRX 400 spectrometer (400 MHz for 1H). All signals are given as δ values in ppm using residual solvent signals as the internal standard. For sample preparation, stock solutions of the macrocycles R1-2 (5 mg mL−1) and drug compounds (10 mg mL−1) were prepared in D2O. For pure sample measurements, 250 μL of the 5 mg mL−1 stock solution was measured in NMR tube and diluted with 250 μL of D2O. To measure possible binding interactions for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest complex, 250 μL of 5 mg mL−1 of a sample drug and 250 μL of 5 mg mL−1 of a host were measured into an NMR sample tube to give a resulting mixture of 2.5 mg mL−1 of both samples. Due to the very low aqueous solubility of GRI, NMR measurements in deuterated water were not possible.
:1 host–guest complex, 250 μL of 5 mg mL−1 of a sample drug and 250 μL of 5 mg mL−1 of a host were measured into an NMR sample tube to give a resulting mixture of 2.5 mg mL−1 of both samples. Due to the very low aqueous solubility of GRI, NMR measurements in deuterated water were not possible.
Isothermal calorimetric (ITC) titrations
ITC titrations are useful in obtaining the quantitative parameters related to interactions between the drug compounds and macrocyclic hosts R1-2. This approach has been used in studying the effects of cyclodextrin and surfactants on the solubility of drug compounds such as simvastatin and sertaconazole.42,46,47 The complexation of the drug guests by the host R1-2 was quantified through a series of ITC experiments in deionized water. This was carried out by filling the sample cell with a receptor and the drug sample as titrants in the syringe in a 1:10 molar ratios. Titrations were done via a computer-automated injector at 298 K. The thermodynamic parameters of host–guest binding (Ka, ΔH, ΔS, and ΔG) were determined by fitting the ITC data to one and two-site binding models (Table 1).
| Ka × 103 (M−1) | ΔH (kcal mol−1) | TΔS (kcal mol−1) | ΔG (kcal mol−1) | |
|---|---|---|---|---|
| a ITC was done in H2O at 298 K and fitted to two binding sites.b Fit to one binding site. | ||||
| CAF@R1 | K1 = 5.36 ± 0.30 | −0.86 ± 0.77 | 4.23 | −5.10 ± 0.77 |
| K2 = 0.54 ± 0.16 | 2.24 ± 0.81 | 5.96 | −3.72 ± 0.81 | |
| CAF@R2 | K1 = 58.40 ± 5.48 | −0.11 ± 0.01 | 6.41 | −6.51 ± 0.01 |
| K2 = 1.97 ± 0.15 | 1.21 ± 0.04 | 5.69 | −4.48 ± 0.04 | |
| GRI@R1 | K1 = 10.94 ± 6.01 | 0.18 ± 0.23 | 5.69 | −5.51 ± 0.20 |
| K2 = 0.68 ± 0.25 | 2.69 ± 0.83 | 6.56 | −3.86 ± 0.83 | |
| bGRI@R2 |
K = 24.50 ± 2.60 | 3.54 ± 0.10 | 9.51 | −5.97 ± 0.10 |
| ISO@R1 | K1 = 211.25 ± 60.01 | −0.66 ± 0.03 | 6.58 | −7.25 ± 0.03 |
| K2 = 1.99 ± 0.16 | 0.31 ± 0.01 | 4.83 | −4.51 ± 0.01 | |
| bISO@R2 |
K = 2.14 ± 0.90 | 1.72 ± 1.47 | 6.26 | −4.53 ± 1.47 |
Cell culture
HEK-293 cells (ATCC CRL-11268) were maintained in complete media as a datasheet. All cultures were incubated at 37 °C and 5% CO2. All cells were tested for mycoplasma (data not shown) as a kit datasheet (rep-mys-10 by InvivoGen, San Diego, CA). Cells had at least 95% viability by Trypan Blue Exclusion Assay (15250-061, Gibco, Billings, MT), and not exceeded passage 30 (data not shown).MTT assay
HEK-293 were seeded in cell-culture-treated 96 multiwell plates (CytoOne CC7682-7596, USA Scientific Inc., Ocala, FL, USA), in complete media at a concentration of 40000 cells per well resuspended in 100 μL per well and incubated at 37 °C overnight. The media was replaced with complete media containing the compounds of interest at increasing concentration up to 2000 μM. Plates were incubated at 37 °C up to 20 or 68 hours, after which cells were assayed with 10 μL of 5 mg mL−1 MTT reagent (Calbiochem, San Diego, CA, USA) in ultra-purified water for 4 hours. Formazan crystals were dissolved in the incubator overnight with 100 μL of a stop solution (80% 2-propanol, 10% 1 N HCl, and 10% Triton X-100). A Tecan plate-reader model Spark (Tecan Austria GmbH, Grödig, Austria) and Magellan software or Epoch microplate (BioTek Instruments, Winooski, VT, USA) and Microsoft Excel software were used to detect colorimetric changes measured at A570 (test) and A690 (reference) wavelengths. Absorbance was normalized to untreated controls using Microsoft Excel software. Statistical analyses (significance set at p < 0.05) were performed using GraphPad Prism version 10.0. Assays were performed at least as two independent experiments, repeated in at least three biological replicates, and in technical replicates, when possible, unless otherwise stated. Data were presented as mean values ± standard deviation (SD).
Crystal violet assay
Cells were seeded in 6 multiwell plates (CC7682-7506, CytoOne, USA) at a concentration of 0.5 × 106 per well in a final volume of 2 mL per well and let adhere overnight. Cells were treated for 24 or 72 hours with a low (6 μM) or high (200 μM) concentration of compounds of interest or controls. DMSO vehicle serves as negative control, and nocodazole (in DMSO) 500 μM or hydrogen peroxide (H2O2) 200 μM serve as positive controls. Bright field microscope (Nikon) pictures of the cells were taken after fixing them with paraformaldehyde (PFA) 4% 20 minutes at room temperature and gently washing the wells to remove unattached cells. Cells were stained with 0.05% (w/v) crystal violet solution for 10 minutes (Sigma-Aldrich, Milwaukee, WI, USA) and distained with 10% acetic acid solution using a benchtop shaker for 30 minutes (Thermo Fisher Scientific, Waltham, MA, USA). A plate reader model Epoch (BioTek Instruments, Winooski, VT, USA) was used to measure the absorbance at 570 nm in three biological replicates in technical triplicate. Relative viability (%), calculated from the raw data of background signal subtracted from the optical density (OD), was plotted in GraphPad Prism software.Results and discussion
Synthesis of receptor R1 starts with resorcinarene 4, which was synthesized using established procedures.48 Resorcinarene 4, in the presence of sodium sulfite and 37% formaldehyde, undergoes electrophilic aromatic substitution, leading to sulfonation of the upper rim (Fig. 2). Full characterization through HRMS, 1H, and 13C NMR confirmed the purity of the compound (Fig. S2 and S4†).Light scattering experiments revealed that in isolation, molecules of the drugs ISO, CAF, and GRI formed smaller-sized assemblies as compared to the hydrophilic host R1 and R2. A mixture of ISO and R1 led to larger assemblies, which can be attributed to the complexation of ISO to R1, enhancing the former's solubility in water. This association can be evident in the differences in the clarity of solutions of ISO in water and ISO-R1 mixture in water (Fig. 3). A similar observation was made in the molecular size distribution of GRI and GRI-R1 mixture. However, the effect of host R1 on the solubility of ISO is greater than that of GRI (Fig. 3). This may be attributed to stronger host–guest interaction between ISO and R1 due to enhanced complementarity of weak interactions. Molecules of ISO and GRI formed respective assemblies with host R2, the sizes of which are skewed towards the sizes of the molecules of the host R2, indicative of host–guest interactions between ISO, GRI, and R2. A similar trend in particle size distribution has been reported by Patel et al.49 by using an amphiphilic cyclodextrin to enhance the solubility and release of the hydrophobic drug tamoxifen citrate. The association of ISO to R2 produced a clearer solution than GRI, which may be due to better complementarity of weak interactions, therefore enhancing the solubility of ISO in a hydrophilic environment in the presence of R2 (Fig. 3). For the same guest, the solubility is markedly enhanced with R1 than R2, which can be explained in terms of better host–guest complementarity of R1 for host–guest interactions than R2. A similar trend in particle size distribution was observed with both hosts, R1 and R2 when mixed with CAF. The particle sizes of CAF in the presence of either host were skewed towards that of the host, thereby imparting the water-soluble effects of the hosts on CAF (Fig. S14 and S15†).
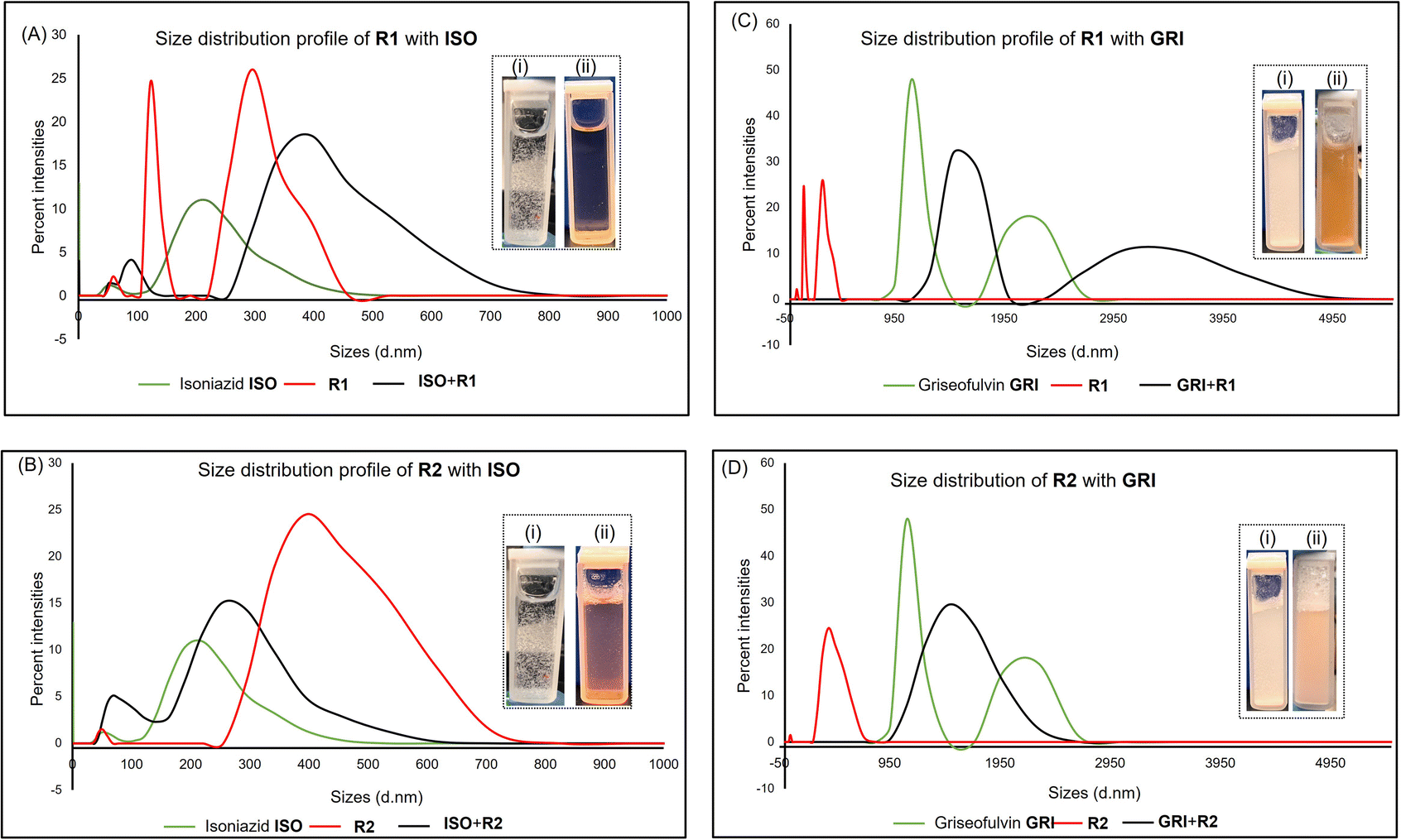 | ||
| Fig. 3 Illustration of the dynamic light scattering (DLS) experiment showing the size distribution profile of the pure drug, pure receptor, and equimolar mixtures of the receptor and the drugs (A) R1 + ISO, (B) R1 + GRI, (C) R2 + ISO, (D) R2 + GRI. Inset: picture showing (i) the pure drug and (ii) the equimolar receptor-drug mixtures, respectively. | ||
Results from NMR measurements indicate that in solution, the complexes are in rapid equilibrium with the free components. Therefore, only one set of signals is observed in the NMR spectra of the mixtures. Despite the dynamic system and fast exchange process, endo-cavity binding of the guests can be determined by monitoring the shielding effects of the guest signals. Lower ppm values (shielding) of a guest's proton signals signify a guest predominantly in the cavity of the receptor. In addition, the orientation of the guest within the cavity can be inferred by comparing the degree of shielding of the guest protons with the effect greater for those deeper in the cavity of the receptor. The binding of CAF to either R1 or R2 led to the shielding of the CAF protons, which is indicative of the binding of CAF within the cavities of the macrocycles, R1 and R2 most likely through hydrophobic effects. At lower equivalents of the guest, more significant shielding of the signals is observed since most of the guests are bound by the host in a fast exchange process. Larger shielding of CAF protons was observed with R2 compared to R1. This can be attributed to the size match and aromaticity of R2, which complement CAF and promote more π–π stacking than R1. In contrast, R1 has fewer flexible groups on its upper rim (Fig. 4 and S7–S10†).
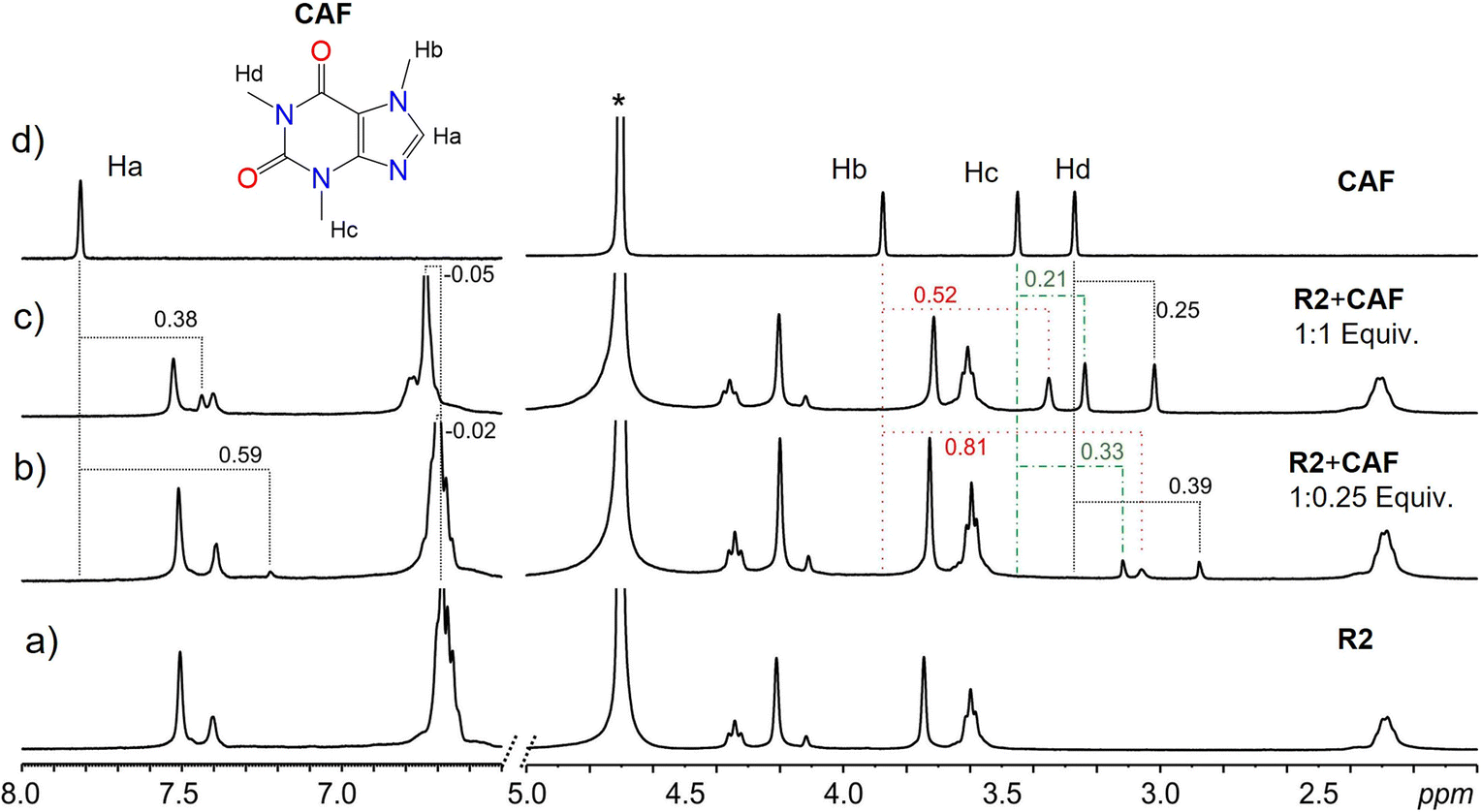 | ||
| Fig. 4 Sections of the 1H NMR spectra in D2O at 298 K of pure (a) receptor R2 and (d) drug CAF, and mixtures of R2 and CAF (b) 1:0.25 eq and (c) 1:1 eq. The broken lines and colors indicate the signal changes in ppm. The star represents the residual D2O solvent. | ||
Deshielding of ISO guest protons was observed in macrocycle R2, indicating binding interactions on the surface of R2 via π–π interactions (Fig. 5). Comparatively, less shielding of ISO aromatic protons was observed with R1, indicating binding inside the hydrophobic cavity of R1 via π–π stacking. The difference in binding dynamics can be attributed to the flexibility of the upper rim substituents in R2 compared to R1. Poor aqueous solubility of GRI at concentrations suitable for NMR did not favor NMR measurements in water. Dissolution in DMSO, however, barred the detection of signal changes due to the more hydrophilic nature of DMSO, which prevents possible hydrophobic interactions between GRI and either R1 or R2.
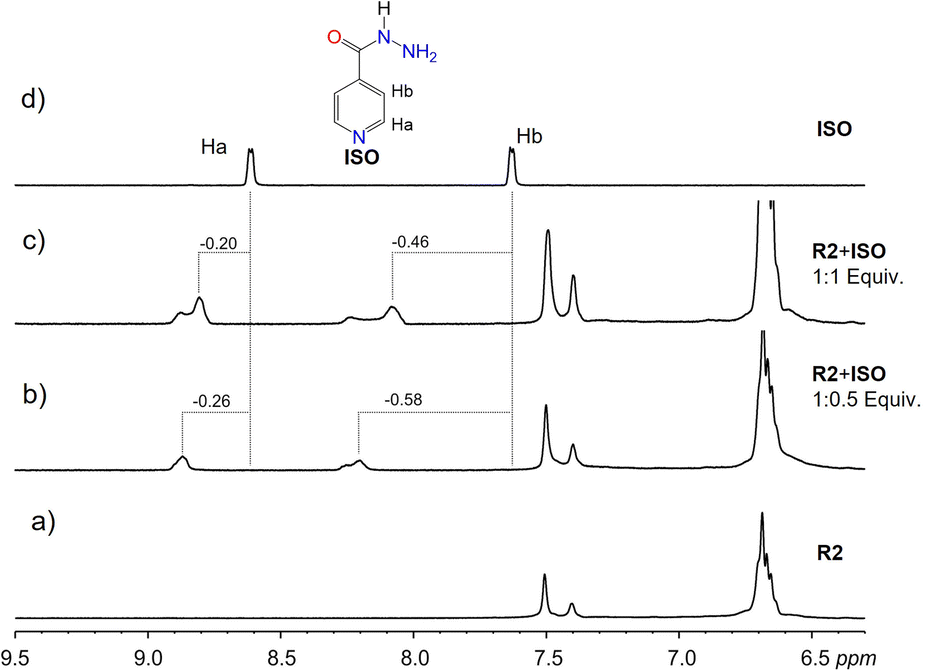 | ||
| Fig. 5 Sections of the 1H NMR spectra in D2O at 298 K of pure (a) receptor R2 and (d) drug ISO, and mixtures of R2 and ISO (b) 1:0.5 eq and (c) 1:1 eq. The dashed lines indicate the signal changes in ppm. | ||
Quantification and binding thermodynamics were obtained from a series of ITC experiments. Complex formation between any combination of the hosts' Rn and the drug guests is spontaneous (ΔG < 0) at the experimental temperature (298 K) (Table 1 and Fig. S11–S13†). Spontaneous associations between resorcinarenes and guest molecules, such as quinoline, naphthalene, pyrophosphates, N-oxides, and heparin in water, have been reported.36–38,50,51 The positive ΔH and TΔS values indicate that the complexation of the guests by R1 and R2 is driven mainly by entropy. Desolvation of guest species because of their poor solubility in water may account for the entropy-driven complexations. Only associations between CAF and the hosts are both enthalpy and entropy-driven. The highest binding affinity among all the guests was observed with ISO on the first binding sites with R1 (Ka = 2.11 × 105 M−1) (Table 1) which may be due to its ability to fit well into the cavity of the host.
Once it was established that the cell culture was mycoplasma-free, MTT and crystal violet assays were performed to test the compounds' possible cytotoxic effect using the human cell line HEK-293, a well-established model for drug discovery.52,53
The MTT assay is usually used to infer cell cytotoxicity and cell proliferation, but also measures cell metabolic activity. More specifically, it measures mainly but not exclusively, the activity of the mitochondrial succinic dehydrogenases.54,55 Usually, in drug discovery, the hit compounds are tested up to 10 μM concentration.56,57 However, it is preferable to reach higher concentrations to better understand the effects of the compounds under investigation. This approach guides the researchers in selecting the optimal concentration range for further studies. Data showed that none of the compounds exerted a significant cytotoxic activity at lower concentrations compared to the negative control (Fig. 6). More in detail, isoniazid, the gold standard treatment for tuberculosis, is associated with a well-known hepatotoxicity side effect that may be severe in rare cases, leading to liver failure.58,59 Cytotoxicity studies in Chinese hamster V97 cell lines showed decreased viability up to about 40% at 50 μM and between 60 and 80% at 200 μM isoniazid concentration at 24 hours.60 Our data confirmed that the inferred viability is between 60 and 80% at a concentration of 200 μM at 24 hours of treatment, as reported in the literature.60 Our study showed that isoniazid alone decreases MTT activity, remaining slightly above 50% at a concentration of 2000 μM, both at 24 and 72 hour treatment (Fig. 6A and C). The co-treatment with R1 or R2 and isoniazid together (R1 + ISO or R2 + ISO) decreases the MTT activity at concentrations higher than 600 μM, at 24 (Fig. 6A and B) and 72 hours (Fig. 6C and D).
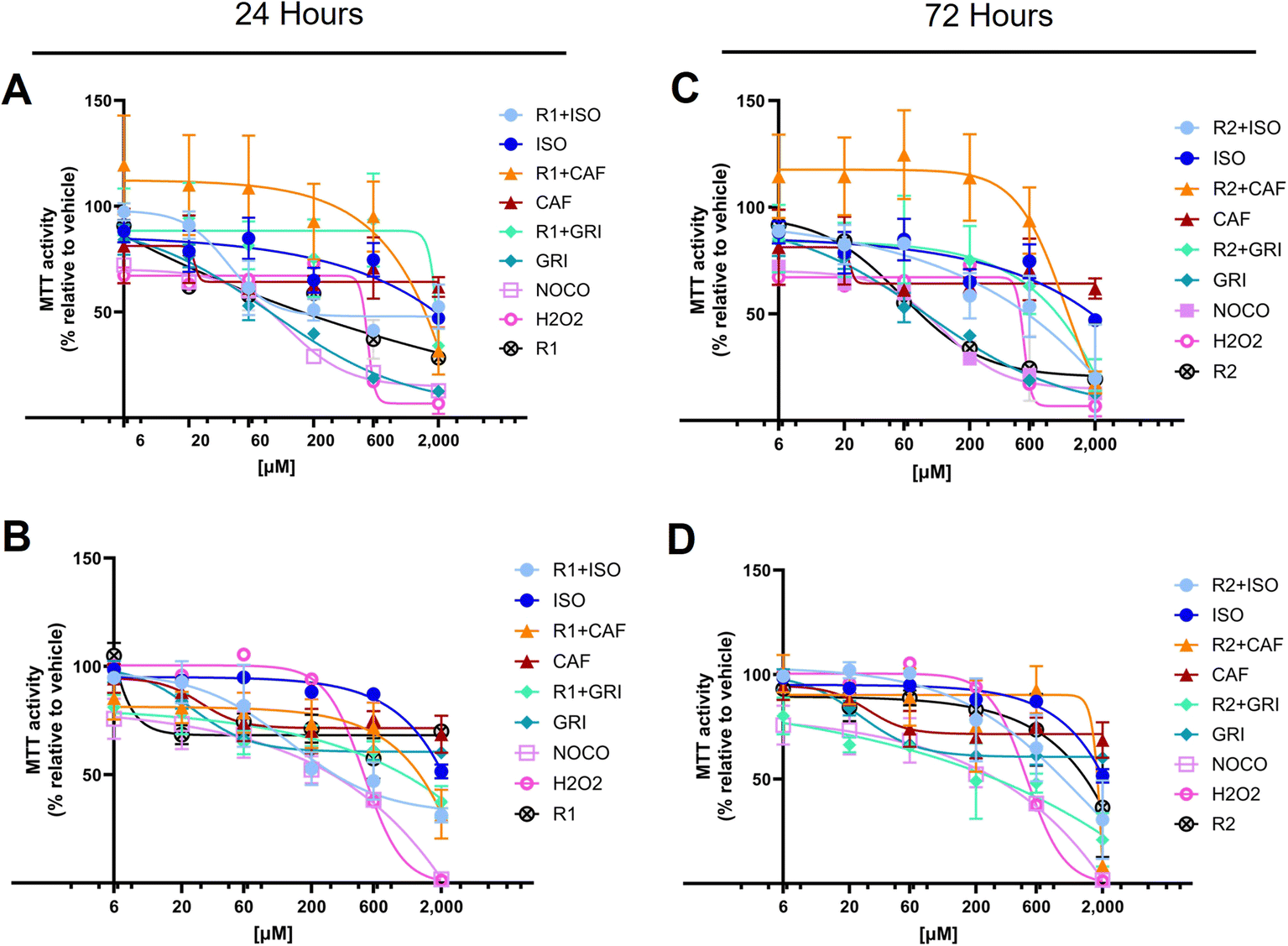 | ||
| Fig. 6 Compounds assayed for inferred cytotoxicity (MTT assay). HEK-293 cells were incubated with a combination of carriers R1 or R2 and caffeine (CAF) or griseofulvin (GRI) or isoniazid (ISO) to infer cytotoxicity via MTT assay. Compounds are assayed at increasing concentrations up to 2000 μM for 24 (A and B) or 72 (C and D) hours. Percentage of MTT activity, shows inferred cytotoxicity relative to the vehicle DMSO that serves as negative control. Nocodazole or H2O2 serve as positive control. Data are representative of at least three biological replicates (mean ± SD), in technical triplicates. Two-way ANOVA Tukey's multiple comparison test. | ||
In literature, it has been shown that caffeine (CAF) decreases cell proliferation in mammalian epithelial MCF-7 cell lines, starting at a lower concentration of 80 μM and reaching a low proliferation rate between 37 and 50% at concentrations equal to 5 mM in a bimodal fashion.61 Our data showed that caffeine alone decreases MTT activity starting at concentrations between 20 and 60 μM but MTT values remain above 50%, both at 24- and 72 hours treatment (Fig. 6A and C). The co-treatment with R1 or R2 and caffeine together (R1 + CAF or R2 + CAF) decreases the MTT activity at concentrations higher than 600 μM, at 24 (Fig. 6A and B) and 72 hours (Fig. 6C and D).
Griseofulvin (GRI), an antifungal treatment, has been reported for its carcinogenicity in murine models62,63 and for treatments up to 100 μM for 24 hours.64,65 Our data confirm GRI cytotoxicity at concentrations lower than 100 μM that are attenuated by the co-treatment of R1 and R2 for up to 24 hours (Fig. 6A and B). It can be inferred that, R1 and R2 alone up to 24 hours significantly decrease the MTT activity in a dose- and time-dependent fashion (Tables 2 and 3). This effect is less evident at an increasing time of up to 72 hours (Tables 2 and 3), likely because of a decreased bioavailability of the compound. Co-treatment with ISO and R1 significantly decreases mitochondrial activity percentage compared to ISO alone at higher concentrations (above 600 μM at 24 hours and 200 μM at 72 hours). ISO, when complexed with R2, affects cells similarly to ISO alone, eventually causing a remarkable cytotoxic effect at concentrations exceeding 600 μM (Tables 2 and 3). CAF, in combination with R1 or R2, is exerting a significant cytotoxic activity between 600 and 2000 μM (Tables 2 and 3). In comparison, CAF alone slightly decreases the MTT activity around 20 to 60 μM to then keep the activity percentages quite constant up to 2000 μM (Tables 2 and 3). Cells treated for 24 hours with GRI alone show a decreased inferred viability in a dose- and time-dependent manner, while at 72 hours, the effect reaches a plateau of around 200 μM (Tables 2 and 3). The cytotoxic effect exerted by GRI in combination with R1 or R2 is markedly increased at prolonged treatment up to 72 hours compared to 24 hours (Tables 2 and 3).
| Tukey's multiple comparison test (24 h, R1) | Summary | Adjusted P value | Tukey's multiple comparison test (72 h, R1) | Summary | Adjusted P value |
|---|---|---|---|---|---|
| 6 μM | 6 μM | ||||
| R1 + ISO vs. ISO | ns | 0.9733 | R1 + ISO vs. ISO | ns | 0.9989 |
| R1 + CAF vs. CAF | *** | 0.0004 | R1 + CAF vs. CAF | ns | 0.7833 |
| R1 + GRI vs. GRI | ns | 0.8149 | R1 + GRI vs. GRI | ns | 0.0834 |
|
|||||
| 20 μM | 20 μM | ||||
| R1 + ISO vs. ISO | ns | 0.8647 | R1 + ISO vs. ISO | ns | >0.9999 |
| R1 + CAF vs. CAF | * | 0.0127 | R1 + CAF vs. CAF | ns | 0.9297 |
| R1 + GRI vs. GRI | ns | 0.4547 | R1 + GRI vs. GRI | ns | 0.9821 |
|
|||||
| 60 μM | 60 μM | ||||
| R1 + ISO vs. ISO | ns | 0.1249 | R1 + ISO vs. ISO | ns | 0.3059 |
| R1 + CAF vs. CAF | **** | <0.0001 | R1 + CAF vs. CAF | ns | 0.9954 |
| R1 + GRI vs. GRI | * | 0.0247 | R1 + GRI vs. GRI | ns | >0.9999 |
|
|||||
| 200 μM | 200 μM | ||||
| R1 + ISO vs. ISO | ns | 0.7657 | R1 + ISO vs. ISO | **** | <0.0001 |
| R1 + CAF vs. CAF | * | 0.0158 | R1 + CAF vs. CAF | ns | 0.9994 |
| R1 + GRI vs. GRI | ** | 0.0013 | R1 + GRI vs. GRI | ns | 0.9526 |
|
|||||
| 600 μM | 600 μM | ||||
| R1 + ISO vs. ISO | ** | 0.0037 | R1 + ISO vs. ISO | **** | <0.0001 |
| R1 + CAF vs. CAF | ns | 0.0992 | R1 + CAF vs. CAF | ns | 0.9987 |
| R1 + GRI vs. GRI | **** | <0.0001 | R1 + GRI vs. GRI | ns | >0.9999 |
|
|||||
| 2000 μM | 2000 μM | ||||
| R1 + ISO vs. ISO | ns | 0.9992 | R1 + ISO vs. ISO | * | 0.0111 |
| R1 + CAF vs. CAF | * | 0.0132 | R1 + CAF vs. CAF | **** | <0.0001 |
| R1 + GRI vs. GRI | ns | 0.2073 | R1 + GRI vs. GRI | ** | 0.0022 |
| Tukey's multiple comparison test (24 h, R2) | Summary | Adjusted P value | Tukey's multiple comparison test (72 h, R2) | Summary | Adjusted P value |
|---|---|---|---|---|---|
| 6 μM | 6 μM | ||||
| R2 + ISO vs. ISO | ns | >0.9999 | R2 + ISO vs. ISO | ns | >0.9999 |
| R2 + CAF vs. CAF | ** | 0.0054 | R2 + CAF vs. CAF | ns | 0.9954 |
| R2 + GRI vs. GRI | ns | >0.9999 | R2 + GRI vs. GRI | ns | 0.3016 |
|
|||||
| 20 μM | 20 μM | ||||
| R2 + ISO vs. ISO | ns | >0.9999 | R2 + ISO vs. ISO | ns | 0.9586 |
| R2 + CAF vs. CAF | ** | 0.0029 | R2 + CAF vs. CAF | ns | 0.9981 |
| R2 + GRI vs. GRI | ns | >0.9999 | R2 + GRI vs. GRI | ns | 0.2851 |
|
|||||
| 60 μM | 60 μM | ||||
| R2 + ISO vs. ISO | ns | >0.9999 | R2 + ISO vs. ISO | ns | 0.9968 |
| R2 + CAF vs. CAF | **** | <0.0001 | R2 + CAF vs. CAF | ns | 0.4858 |
| R2 + GRI vs. GRI | ** | 0.0093 | R2 + GRI vs. GRI | ns | >0.9999 |
|
|||||
| 200 μM | 200 μM | ||||
| R2 + ISO vs. ISO | ns | 0.9977 | R2 + ISO vs. ISO | ns | 0.9091 |
| R2 + CAF vs. CAF | **** | <0.0001 | R2 + CAF vs. CAF | ns | 0.9985 |
| R2 + GRI vs. GRI | ** | 0.0022 | R2 + GRI vs. GRI | ns | 0.7590 |
|
|||||
| 600 μM | 600 μM | ||||
| R2 + ISO vs. ISO | ns | 0.2616 | R2 + ISO vs. ISO | ns | 0.0646 |
| R2 + CAF vs. CAF | ns | 0.1685 | R2 + CAF vs. CAF | ns | 0.3215 |
| R2 + GRI vs. GRI | **** | <0.0001 | R2 + GRI vs. GRI | ns | 0.6855 |
|
|||||
| 2000 μM | 2000 μM | ||||
| R2 + ISO vs. ISO | * | 0.0464 | R2 + ISO vs. ISO | ns | 0.1048 |
| R2 + CAF vs. CAF | **** | <0.0001 | R2 + CAF vs. CAF | **** | <0.0001 |
| R2 + GRI vs. GRI | ns | 0.9818 | R2 + GRI vs. GRI | **** | <0.0001 |
It is usually recommended to use an orthologue approach to confirm the results obtained via MTT assay.66,67 For this purpose, a crystal violet assay has been performed. Cells exposed to cytotoxic compounds lose attachment resulting in a decreased number of cells.68,69 The inferred cell death is directly proportional to the decreased optical density (OD) values.70 Data collected in brightfield qualitatively show the appearance of the cells in the wells before the crystal violet staining (Fig. 7) and support the quantification of the assay (S16).
 | ||
| Fig. 7 Compounds assayed for inferred cytotoxicity (qualitative crystal violet assay). HEK-293 cells were incubated with a combination of carriers R1 or R2 and CAF or GRI or ISO to infer cytotoxicity via crystal violet assay. Cells are incubated with compounds at 6 or 200 μM for 24 (A) or 72 (B) hours. Nocodazole or H2O2 serve as positive control. Data are representative of at least three biological replicates. | ||
Data show that all the compounds at both lower (6 μM) and higher (200 μM) concentrations do not show cytotoxicity compared to DMSO (Fig. 7A and S16A†). However, at a longer exposure (72 hours), GRI alone or combined with both carriers R1 or R2 showed increased cytotoxicity at higher concentrations (200 μM). Carrier R2 alone or combined with ISO at 72 hours of treatment at 200 μM seems to exert a cytotoxic effect on the cells (Fig. 7B). Even though the cells may still be attached to the surface of the plate and retain crystal violet dye (Fig. S16B†), they seem round and clustered (Fig. 7B). This peculiar morphology is usually a clear sign of cell damage.71
Conclusions
The complexation of the drug compounds with the ionic resorcinarenes results in shifts of particle sizes of drug compounds towards that of the macrocycles. Such associations produced significant chemical shifts and binding affinities in the micromolar range, which was sufficient to produce a clearer solution of a drug-resorcinarene mixture compared to pure aqueous solutions of drug compounds. Cytotoxicity assessments revealed that resorcinarenes alone or together with drug compounds yielded non-toxic effects, especially at lower and experimental concentrations, confirming the potential of these poly-ionic resorcinarenes to be used as safe and cheaper drug solubilizing agents in the pharmaceutical industry. Cells treated with all the compounds of interest showed signs of decreased metabolism at higher concentrations and extended treatment time. Orthologue approaches regarding the cytotoxicity studies show that cell damage or cell death is increased when cells are treated with a higher concentration (200 μM) of griseofulvin alone or in combination with carrier R1 or R2, and R2 alone or in combination with isoniazid up to 72 hours treatment. The results show promise in using cavity-containing organic macrocycles as transport and delivery agents for non-water-soluble drugs.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written and edited with contributions from all authors. All authors have approved the final version of the manuscript. Frank Boateng Osei: original concept, NMR, ITC, and DLS measurements. Wrote sections of the manuscript. Kwaku Twum: original concept, NMR, ITC, and DLS measurements. Wrote sections of the manuscript. Barbara Manfredi: cytotoxicity measurements and wrote the section. Mariana Fatohi: undergrad student, 1H NMR, and ITC measurements. Yvonne Bessem Ojong: supervision, wrote, read, and corrected the manuscript. Valance Washington: supervised the cytotoxicity work. Ngong Kodiah Beyeh: supervised the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Acknowledgment is made to the donors of the American Chemical Society Petroleum Research Fund (#65027-DNI10) for support (or partial support), the National Science Foundation (#2236984), the National Institute of Health (R01 HL090933), and Oakland University for this research.Notes and references
- Y. K. Agrawal and R. N. Patadia, Rev. Anal. Chem., 2006, 25, 155–239 CAS.
- P. Ziaja, A. Krogul, T. S. Pawłowski and G. Litwinienko, Thermochim. Acta, 2016, 623, 112–119 CrossRef CAS.
- O. D. Fox, J. Cookson, E. J. S. Wilkinson, M. G. B. Drew, E. J. Maclean, S. J. Teat and P. D. Beer, J. Am. Chem. Soc., 2006, 128, 6990–7002 CrossRef CAS PubMed.
- K. Twum, K. N. Truong, F. B. Osei, C. von Essen, S. Nadimi, J. F. Trant, K. Rissanen and N. K. Beyeh, Cryst. Growth Des., 2022, 23, 1281–1287 CrossRef.
- N. K. Beyeh, D. P. Weimann, L. Kaufmann, C. A. Schalley and K. Rissanen, Chem.–Eur. J., 2012, 18, 5552–5557 CrossRef CAS PubMed.
- R. Kashapov, Y. Razuvayeva, A. Ziganshina, A. Lyubina, S. Amerhanova, A. Sapunova, A. Voloshina, I. Nizameev, V. Salnikov and L. Zakharova, Colliods Surf., A, 2022, 648, 129330–129338 CrossRef CAS.
- A. Halder, D. C. Mukherjee and S. Bhattacharya, J. Solution Chem., 2010, 39, 1327–1340 CrossRef CAS.
- W. C. Geng, J. L. Sessler and D. S. Guo, Chem. Soc. Rev., 2020, 49, 2303–2315 RSC.
- F. Jia, H. V Schrö, L. P. Yang, C. Von Essen, S. Sobottka, B. Sarkar, K. Rissanen, W. Jiang and C. A. Schalley, J. Am. Chem. Soc., 2020, 142, 3306–3310 CrossRef CAS PubMed.
- S. Ohtani, K. Kato, S. Fa and T. Ogoshi, Coord. Chem. Rev., 2022, 462, 214503–214511 CrossRef CAS.
- G. Wenz, Clin. Drug Invest., 2000, 19, 21–25 CrossRef CAS.
- O. Kretschmann, C. Steffens and H. Ritter, Angew. Chem., Int. Ed., 2007, 46, 2708–2711 CrossRef CAS PubMed.
- H. Wen, H. Jung and X. Li, AAPS J., 2015, 17, 1327–1340 CrossRef CAS PubMed.
- C. M. O'Driscoll and B. T. Griffin, Adv. Drug Deliv. Rev., 2008, 60, 617–624 CrossRef PubMed.
- R. Samineni, J. Chimakurthy and S. Konidala, Turk. J. Pharm. Sci., 2022, 19, 706–713 CrossRef CAS PubMed.
- F. Wu, R. Cristofoletti, L. Zhao and A. Rostami-Hodjegan, Biopharm. Drug Dispos., 2021, 42, 118–127 CrossRef CAS PubMed.
- D. C. Vimalson, S. Parimalakrishnan, N. S. Jeganathan and S. Anbazhagan, Asian J. Pharm., 2016, 10, 67–76 Search PubMed.
- D. V. Bhalani, B. Nutan, A. Kumar and A. K. S. Chandel, Biomedicines, 2022, 10, 2055–2087 CrossRef CAS PubMed.
- S. M. Abuzar, S. M. Hyun, J. H. Kim, H. J. Park, M. S. Kim, J. S. Park and S. J. Hwang, Int. J. Pharm., 2018, 538, 1–13 CrossRef CAS PubMed.
- J. S. Kim, H. Park, K. T. Kang, E. S. Ha, M. S. Kim and S. J. Hwang, J. Pharm. Invest., 2022, 52, 353–366 CrossRef CAS.
- R. Jayalakshmy, K. S. Sreethu and C. R. Jayalakshmy, J. Med. Pharm. Allied Sci., 2021, 3, 2763–2771 CrossRef.
- J. B. Ngilirabanga and H. Samsodien, Nano Select, 2021, 2, 512–526 CrossRef CAS.
- S. Sareen, L. Joseph and G. Mathew, Int. J. Pharm. Invest., 2012, 2, 12–17 CrossRef CAS PubMed.
- M. N. S. Patel, M. H. Ahmed, M. Saqib and S. N. Shaikh, J. Drug Delivery Ther., 2019, 9, 542–546 CrossRef CAS.
- A. K. Gupta and C. Drummond-Main, Pediatr. Dermatol., 2013, 30, 1–6 CrossRef PubMed.
- Y. Fujioka, Y. Metsugi, K. I. Ogawara, K. Higaki and T. Kimura, Int. J. Pharm., 2008, 352, 36–43 CrossRef CAS PubMed.
- V. Pittol, K. S. Veras, S. Kaiser, L. J. Danielli, A. M. Fuentefria and G. G. Ortega, Braz. J. Pharm. Sci., 2022, 58, 1–13 Search PubMed.
- R. Kumar, J. Drug. Deliv. Sci. Technol., 2019, 53, 101221–101228 CrossRef CAS.
- D. Yang, R. Kulkarni, R. J. Behme and P. N. Kotiyan, Int. J. Pharm., 2007, 329, 72–80 CrossRef CAS PubMed.
- C. Becker, J. B. Dressman, G. L. Amidon, H. E. Junginger, S. Kopp, K. K. Midha, V. P. Shah, S. Stavchansky and D. M. Barends, J. Pharm. Sci., 2007, 96, 522–531 CrossRef CAS PubMed.
- B. Xuan, S. N. Wong, Y. Zhang, J. Weng, H. H. Y. Tong, C. Wang, C. C. Sun and S. F. Chow, Cryst. Growth Des., 2020, 20, 1951–1960 CrossRef CAS.
- V. Reddy and M. Saharay, J. Phys. Chem. B, 2019, 123, 9685–9691 CrossRef CAS PubMed.
- I. Shumilin, C. Allolio and D. Harries, J. Am. Chem. Soc., 2019, 141, 18056–18063 CrossRef CAS PubMed.
- L. Lomba, A. Polo, J. Alejandre, N. Martínez and B. Giner, J. Drug Delivery Sci. Technol., 2023, 79, 104010–104017 CrossRef CAS.
- D. M. Galindres, D. Cifuentes, L. E. Tinoco, Y. Murillo-Acevedo, M. M. Rodrigo, A. C. F. Ribeiro and M. A. Esteso, Processes, 2022, 10, 684–697 CrossRef CAS.
- A. Karle, K. Twum, N. Sabbagh, A. Haddad, S. M. Taimoory, M. M. Szczęśniak, E. Trivedi, J. F. Trant and N. K. Beyeh, Analyst, 2022, 147, 2264–2271 RSC.
- K. Twum, N. Schileru, B. Elias, J. Feder, L. Yaqoo, R. Puttreddy, M. M. Szczesniak and N. K. Beyeh, Symmetry, 2020, 12, 1–8 CrossRef.
- K. Twum, S. I. Sadraei, J. Feder, S. M. Taimoory, K. Rissanen, J. F. Trant and N. K. Beyeh, Org. Chem. Front., 2022, 9, 1267–1275 RSC.
- P. Singla, O. Singh, S. Sharma, K. Betlem, V. K. Aswal, M. Peeters and R. K. Mahajan, ACS Omega, 2019, 4, 11251–11262 CrossRef CAS PubMed.
- P. C. Griffiths, B. Cattoz, M. S. Ibrahim and J. C. Anuonye, Eur. J. Pharm. Biopharm., 2015, 97, 218–222 CrossRef CAS PubMed.
- E. Vaculikova, A. Cernikova, D. Placha, M. Pisarcik, P. Peikertova, K. Dedkova, F. Devinsky and J. Jampilek, Molecules, 2016, 21, 1005–1012 CrossRef PubMed.
- Á. Sarabia-Vallejo, M. M. Caja, A. I. Olives, M. A. Martín and J. C. Menéndez, Pharmaceutics, 2023, 15, 2345–2397 CrossRef PubMed.
- K. Ueda, K. Higashi, W. Limwikrant, S. Sekine, T. Horie, K. Yamamoto and K. Moribe, Mol. Pharm., 2012, 9, 3023–3033 CrossRef CAS PubMed.
- I. Ghiviriga, Anal. Chem., 2023, 95, 2706–2712 CrossRef CAS PubMed.
- K. Ueda and L. S. Taylor, Mol. Pharm., 2020, 17, 1352–1362 CrossRef CAS PubMed.
- R. Patel, G. Buckton and S. Gaisford, Thermochim. Acta, 2007, 456, 106–113 CrossRef CAS.
- A. I. Rodriguez-Perez, C. Rodriguez-Tenreiro, C. Alvarez-Lorenzo, P. Taboada, A. Concheiro and J. J. Torres-Labandeira, J. Pharm. Sci., 2006, 95, 1751–1762 CrossRef CAS PubMed.
- K. Twum, A. Bhattacharjee, E. T. Laryea, J. Esposto, G. Omolloh, S. Mortensen, M. Jaradi, N. L. Stock, N. Schileru, B. Elias, E. Pszenica, T. M. McCormick, S. Martic and N. K. Beyeh, RSC Med. Chem., 2021, 12, 2022–2030 RSC.
- M. R. Patel, D. A. Lamprou and P. R. Vavia, AAPS PharmSciTech, 2020, 11, 1–16 CAS.
- K. Twum, S. Nadimi, F. B. Osei, R. Puttreddy, Y. B. Ojong, J. J. Hayward, K. Rissanen, J. F. Trant and N. K. Beyeh, Chem. Asian J., 2023, 18, 1308–1320 CrossRef PubMed.
- S. Välimäki, N. K. Beyeh, V. Linko, R. H. A. Ras and M. A. Kostiainen, Nanoscale, 2018, 10, 14022–14030 RSC.
- A. Heding, Expert Rev. Mol. Diagn., 2004, 4, 403–411 CrossRef CAS PubMed.
- L. Ho, C. L. Greene, A. W. Schmidt and L. H. Huang, Cytotechnology, 2004, 45, 117–123 CrossRef CAS PubMed.
- P. L. Tseng, W. H. Wu, T. H. Hu, C. W. Chen, H. C. Cheng, C. F. Li, W. H. Tsai, H. J. Tsai, M. C. Hsieh, J. H. Chuang and W. T. Chang, Sci. Rep., 2018, 8, 3081–3096 CrossRef PubMed.
- A. Kumar, Y. Rai and A. N. Bhatt, Cytotechnology, 2024, 76, 301–311 CrossRef CAS PubMed.
- K. Katsuno, J. N. Burrows, K. Duncan, R. H. V. Huijsduijnen, T. Kaneko, K. Kita, C. E. Mowbray, D. Schmatz, P. Warner and B. T. Slingsby, Nat. Rev. Drug Discovery, 2015, 14, 751–758 CrossRef CAS PubMed.
- S. Bendels, C. Bissantz, B. Fasching, G. Gerebtzoff, W. Guba, M. Kansy, J. Migeon, S. Mohr, J. U. Peters, F. Tillier, R. Wyler, C. Lerner, C. Kramer, H. Richter and S. Roberts, J. Pharmacol. Toxicol. Methods, 2019, 99, 106609–106616 CrossRef CAS PubMed.
- W. K. Kabbara, A. T. Sarkis and P. G. Saroufim, Case Rep. Infect. Dis., 2016, 2016, 3617408–3617413 Search PubMed.
- I. Metushi, J. Uetrecht and E. Phillips, Br. J. Clin. Pharmacol., 2016, 81, 1030–1036 CrossRef PubMed.
- D. Kakkar, A. K. Tiwari, K. Chuttani, A. Khanna, A. Datta, H. Singh and A. K. Mishra, Chem. Biol. Drug Des., 2012, 80, 245–253 CrossRef CAS PubMed.
- A. Kazaks, M. Collier and M. Conley, Curr. Dev. Nutr., 2019, 3, 31–38 Search PubMed.
- P. Aris, Y. Wei, M. Mohamadzadeh and X. Xia, Molecules, 2022, 27, 7034–7041 CrossRef CAS PubMed.
- M. Rustia and P. Shubik, Br. J. Cancer, 1978, 38, 237–249 CrossRef CAS PubMed.
- S. Gupta, A. Kumar and K. K. Tejavath, Mol. Biol. Rep., 2021, 48, 2945–2956 CrossRef CAS PubMed.
- A. B. Petersen, G. Konotop, N. H. M. Hanafiah, P. Hammershøj, M. S. Raab, A. Krämer and M. H. Clausen, Eur. J. Med. Chem., 2016, 116, 210–215 CrossRef CAS PubMed.
- Y. Li, S. Lam, J. C. Mak, C. Zheng and J. C. M. Ho, Lung Cancer, 2013, 81, 354–361 CrossRef PubMed.
- W. Hu, Q. Liu, J. Pan and Z. Sui, Biomed. Pharmacother., 2018, 105, 887–898 CrossRef CAS PubMed.
- F. Wei, S. Wang and X. Gou, Biophys. Rep., 2021, 7, 504–516 CAS.
- M. Feoktistova, P. Geserick and M. Leverkus, Cold Spring Harb. Protoc., 2016, 2016, 343–346 Search PubMed.
- T. V. Berghe, S. Grootjans, V. Goossens, Y. Dondelinger, D. V. Krysko, N. Takahashi and P. Vandenabeele, Methods, 2013, 61, 117–129 CrossRef PubMed.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, copies of 1H NMR, ITC, and DLS data. See DOI: https://doi.org/10.1039/d4ra06682k |
| This journal is © The Royal Society of Chemistry 2024 |