
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, and in vitro and in vivo biological evaluation of triazolopyrimidine hybrids as multitarget directed anticancer agents†
Heba A. Adawya,
Samar S. Tawfik *a,
Abdullah A. Elgazarb,
Khalid B. Selima and
Fatma E. Godaa
*a,
Abdullah A. Elgazarb,
Khalid B. Selima and
Fatma E. Godaa
aDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt. E-mail: drsamarelmasry@mans.edu.eg
bDepartment of Pharmacognosy, Faculty of Pharmacy, Kafrelsheikh University, Kafr El Sheikh, Egypt
First published on 7th November 2024
Abstract
In response to the urgent need for new anti-proliferative agents, four novel series of triazolopyrimidine compounds (7a–e, 9a–d, 11a–f, and 13a–e) were synthesized and evaluated for in vitro anticancer efficacy against HCT116, HeLa, and MCF-7 cell lines. Compound 13c emerged as the most potent, with IC50 values of 6.10, 10.33, and 2.42 μM respectively, while 11e and 7c also showed strong activity. In multi-target suppression tests, 13c exhibited the highest inhibition against EGFR, TOP-II, HER-2, and ARO (IC50: 0.087, 31.56, 0.078, and 0.156 μM, respectively). Flow cytometry revealed 13c's ability to suppress the S-phase cell population in MCF-7 cells. In vivo studies of 13c demonstrated significant tumor growth inhibition, comparable to the positive control. Molecular docking studies supported the experimental findings, confirming the binding of the novel motifs to the target enzymes' active sites. This comprehensive evaluation highlights the potential of these triazolopyrimidine compounds, particularly 13c, as promising anticancer agents, warranting further investigation.
1. Introduction
Estrogen-sensitive cancers, particularly breast and cervical cancers, pose significant health challenges globally. Breast cancer is the most common cancer among women worldwide, with over 2.3 million new cases diagnosed in 2020 alone.1,2 Cervical cancer, while less prevalent in developed countries due to widespread screening and vaccination against human papillomavirus (HPV), remains a major cause of cancer mortality in low- and middle-income countries. These cancers are predicted to continue rising due to aging populations and lifestyle factors. Estrogen-sensitive cancers often show initial responsiveness to hormone therapies; however, the development of resistance remains a critical issue, necessitating more advanced and multifaceted treatment strategies.3–5Epidermal growth factor receptor (EGFR) plays a crucial role in the development and progression of estrogen-sensitive cancers. EGFR is a transmembrane protein that, when activated, triggers signaling pathways promoting cell proliferation, survival, and migration.6,7 Overexpression or mutation of EGFR is associated with poor prognosis and resistance to conventional therapies. Targeting EGFR with tyrosine kinase inhibitors, such as erlotinib and gefitinib, has shown promise in treating various cancers, including breast cancer.8 By inhibiting EGFR signaling, these drugs can reduce tumor growth and enhance the effectiveness of other treatments. However, resistance to EGFR inhibitors can still occur, highlighting the need for combination therapies to overcome this challenge.
HER-2 (human epidermal growth factor receptor 2) is another critical target in estrogen-sensitive cancers, particularly in aggressive forms of breast cancer where it is overexpressed. HER-2 promotes cell proliferation and survival through various signaling pathways.9,10 Targeting HER-2 with monoclonal antibodies like trastuzumab or tyrosine kinase inhibitors such as lapatinib has shown substantial benefits in treating HER-2 positive breast cancers. These therapies can inhibit HER-2 mediated signaling, slowing down or halting tumor growth.11 However, despite their effectiveness, some patients develop resistance, necessitating the integration of HER-2 targeted therapies with other treatment modalities, including EGFR inhibitors, to improve outcomes.
Aromatase plays a crucial role in estrogen-sensitive cancers by catalyzing the conversion of androgens to estrogens. In postmenopausal women, this enzyme is the primary source of estrogen, and its inhibition can significantly reduce estrogen levels, limiting the growth of estrogen-dependent cancer cells.12 Aromatase inhibitors, such as anastrozole and letrozole, are commonly used in the treatment of postmenopausal women with hormone receptor-positive breast cancer. Combining aromatase inhibitors with agents targeting EGFR and HER-2 can provide a multifaceted approach to overcoming resistance.13–15
Topoisomerase II (TOP-II) is an enzyme involved in DNA replication and transcription, playing a pivotal role in cell division. Inhibitors of TOP-II, such as doxorubicin and etoposide, are used to induce DNA damage in cancer cells, leading to apoptosis. These agents could be particularly useful in treating aggressive breast cancers since the expression of TOP-II was linked to poor prognosis.16–18 Hence, the development of therapeutic agents that could target these enzymes concomitantly could provide a comprehensive strategy to manage estrogen-sensitive cancers and improve patient therapeutic outcomes.
Triazolopyrimidines (TPs) have emerged as a versatile privileged class of compounds with a wide range of biological activities, making them valuable scaffolds in drug design. Notably, TPs have demonstrated significant potential in the treatment of several parasites, neurodegenerative diseases and cardiovascular activities.19
Moreover, TPs have been explored for their anticancer properties. For instance, compound I exhibited significant activity against CDK2 with an IC50 of 0.12 μM after applying scaffold hopping modification on Seliciclib which also lead to improved activity towards GSK-3B and cytotoxicity against HCT-116.20 Also, compound II demonstrated activity against tubulin polymerization with an IC50 of 3.8 μM (ref. 21) and potent cytotoxicity against HCT-116 with IC50 = 0.53 μM. Interestingly, its analogue compound III produced pronounced activity against HeLa cells with an IC50 of 0.06 μM.22
In the same context, compound IV displayed potent activity against tyrosyl-DNA phosphodiesterase 2 (TDP2) with an IC50 of 22 μM which could be a promising approach in reversing resistance against TOP-II inhibitors.23 Compound V exhibited remarkable potency against lysine specific demethylase 1 (LSD-1), where it achieved an IC50 of 1.7 μM and was able to reduce the cell viability of A549 cells in dose dependent manner.24,25 Compound VI showed promising inhibitory activity against CA IX after introducing sulfonamide moiety, with an IC50 of 0.0246 μM.
Dawood et al. reported the synthesis of compound VII which showed moderate cytotoxicity against MCF-7 and was able to inhibit the expression of VEGFR-2 in the cells by 63%.26 Compound VIII showed activity against bromodomain-containing protein 4 (BRD-4) with an IC50 of 2.15 μM with strong cytotoxicity against THP-1 with IC50 = 3.86 μM proving to be promising template for new BRD-4 inhibitors. Compound IX was identified using high throughput screening (HTS) as potent inhibitor for cancer cells with highly expressed paired-box gene 2 (PaX-2) and was able to inhibit PaX-2 with an IC50 of 1.5 μM.27
Compound X demonstrated potent activity against methionine aminopeptidase-2 (MetAP-2), with an IC50 of 38 nM as a result of implying scaffold hopping to previously reported purine derivatives.28 Finally, compound XI was developed as ERK-3 inhibitor by HTS, where it was found to fit on the ATP pocket allowing it to achieve IC50 of 38 nM (Fig. 1).
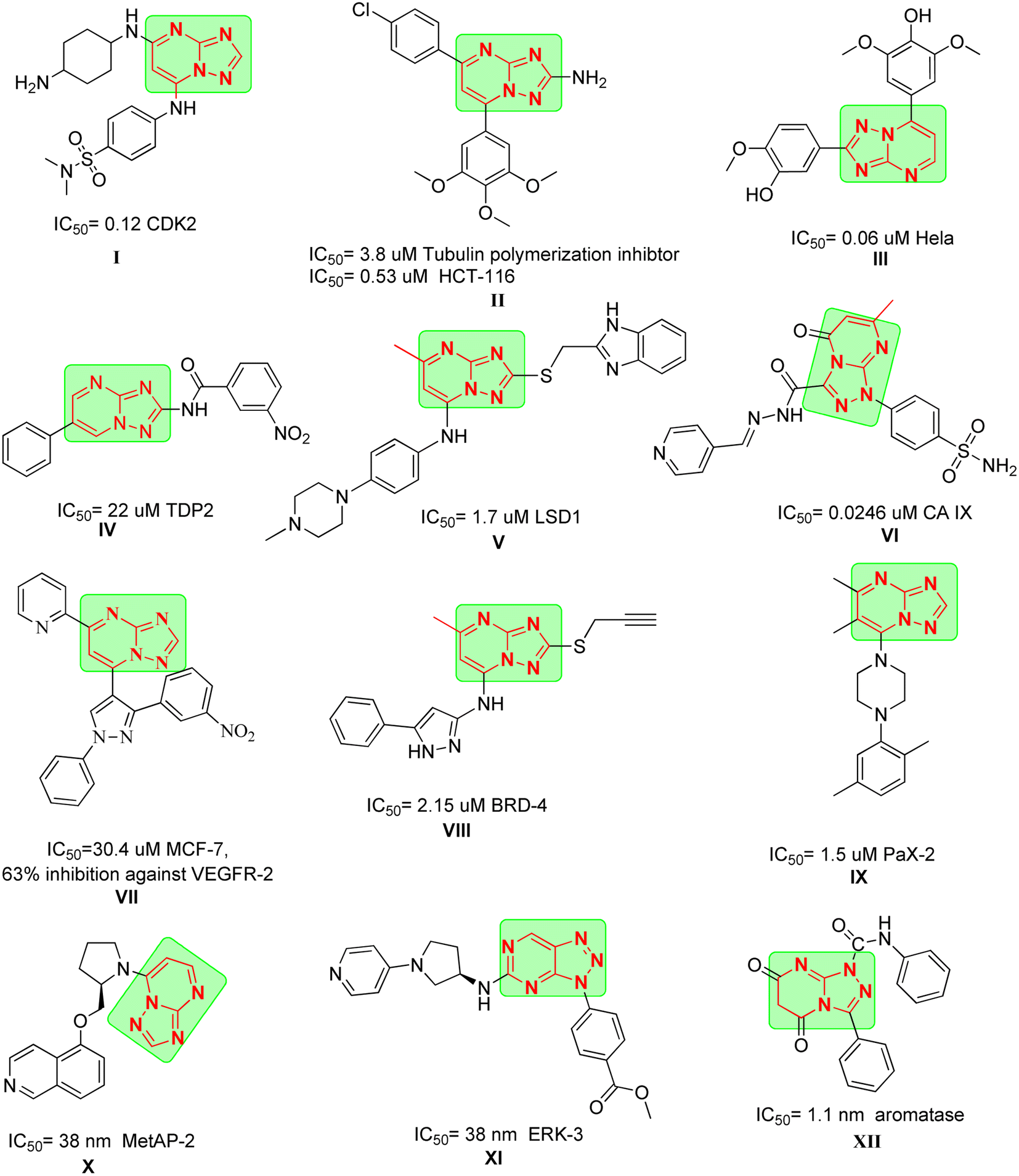 | ||
| Fig. 1 Examples of anticancer agents containing TPs moiety. | ||
These findings underscore the potential of triazolopyrimidine-based compounds as modulable scaffolds, capable of targeting diverse enzymes, cellular processes, and cancer cell lines, with IC50 values spanning from low nanomolar to micromolar ranges. This could be attributed to their ability to act as bioisostere for adenine similar to afatinib and Qap1, allowing the occupation of ATP active site of several enzymes involved in cancer pathogenesis.29
Based on the literature review the development of novel TPs to inhibit multiple targets such as EGFR, Her-2 and TOP-II could be achieved as these enzymes comprise on ATP active sites. In addition, TPs were reported previously as potential aromatase inhibitor30 like compound XII (IC50 of 1.1 nm). Moreover, different scaffolds such as oxadiazole (e.g. compound XIII),31 6-aryl-5-cyanothiouracil (e.g. compound XIV),32 thiadiazole-urea (e.g. compound XV),33 and piperazine (e.g. compound XVI)34 were reported as anticancer activity enhancers. All these findings open a way to introduce multi-target anticancer agents through molecular hybridization (Fig. 2).
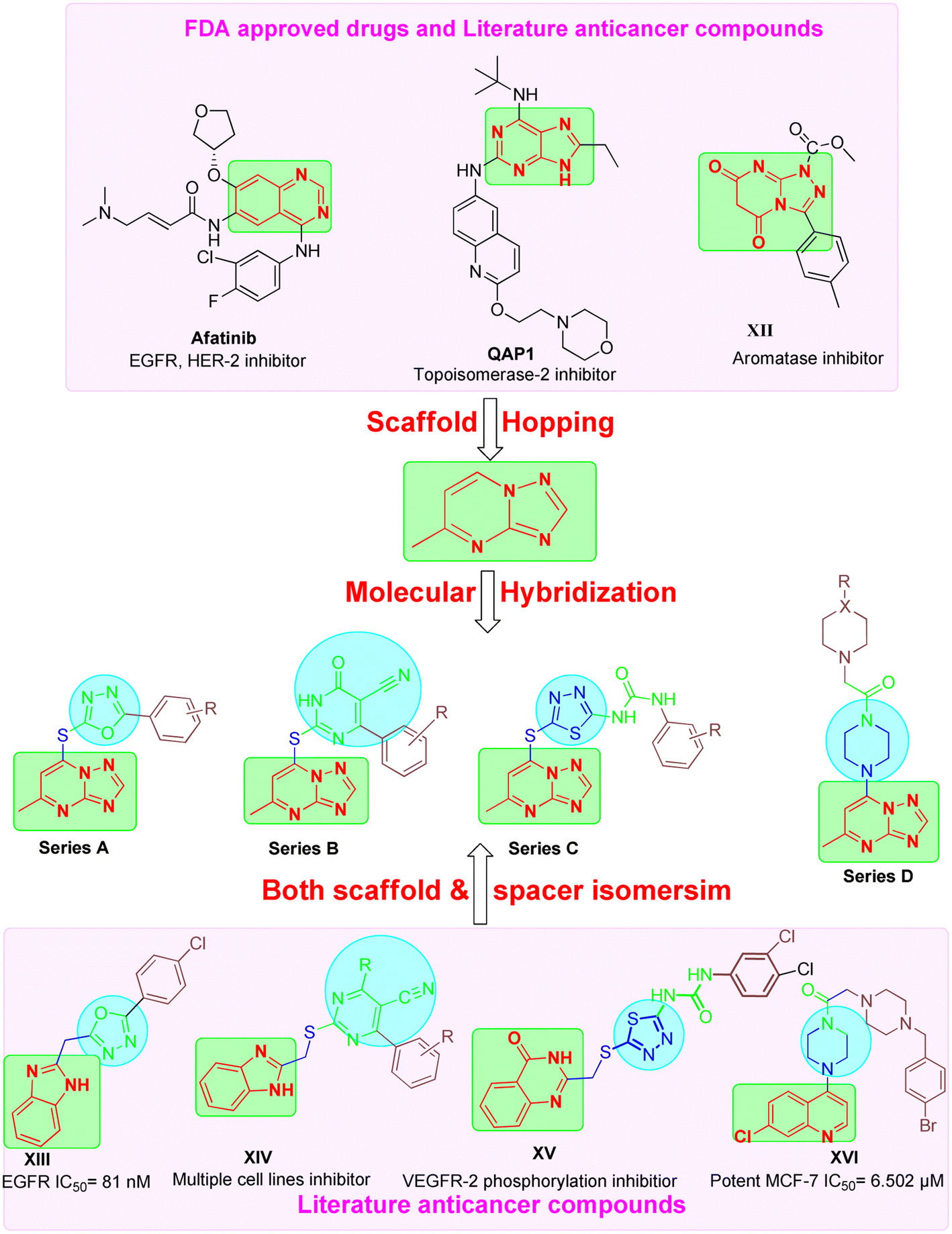 | ||
| Fig. 2 Design strategy of TPs derivatives as multi target anticancer agents. | ||
2. Results and discussion
2.1. Chemistry
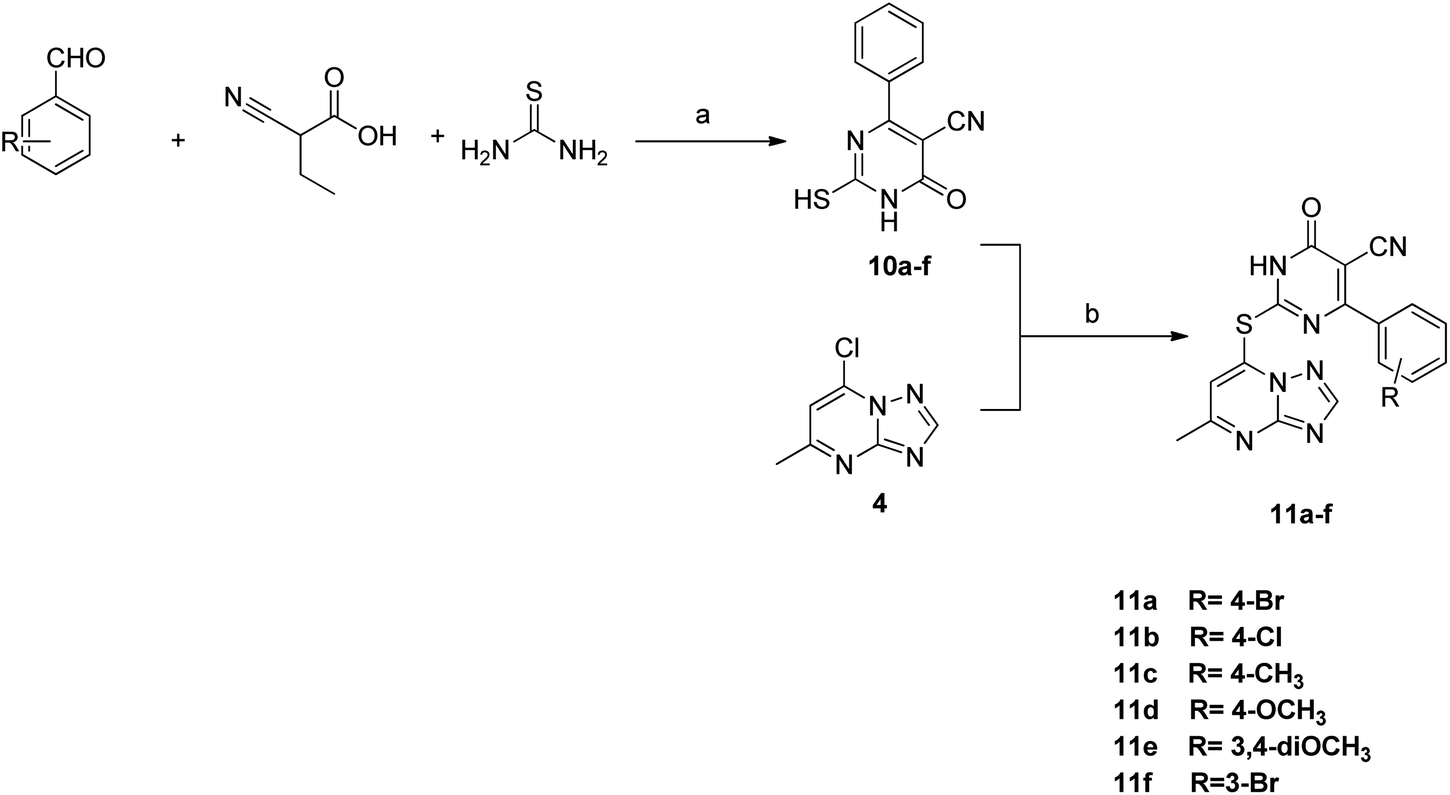
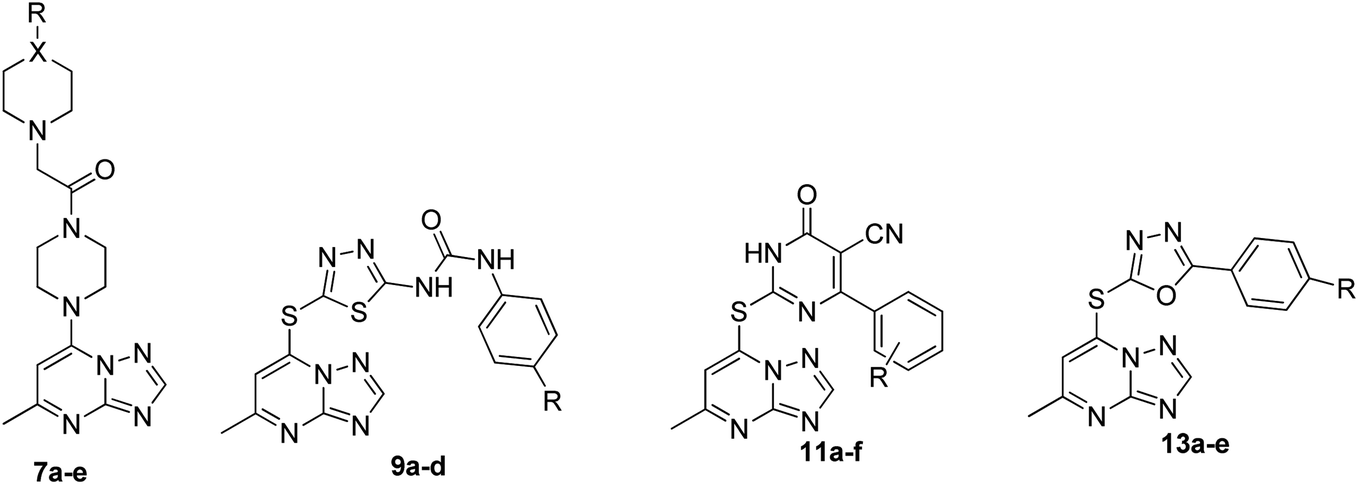
The synthesis of the final compounds 7a–e, 9a–d, 11a–f and 13a–e is illustrated in the following Schemes 1–4. In Scheme 1, the synthesis of 5-methyl-7-(piperazin-1-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 7a–e, was accomplished through the following steps. Aminoguanidine bicarbonate (1) was reacted with formic acid to yield 3-amino-1,2,4-triazole (2). This intermediate (2) was then heated with ethyl acetoacetate in acetic acid to produce 5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (3). The resulting compound was treated with phosphorus oxychloride to obtain the target chloride (4) which serves as a key precursor in the following several reactions.35 The obtained chloride (4) was reacted with piperazine in presence of potassium carbonate (K2CO3) in dioxane. The resulting product (5) was then acylated with chloroacetyl chloride in presence of triethylamine, serving as the base, to yield 4-(2-chloroacetamido)piperazine (6). This intermediate was subsequently treated with various piperazine derivatives, morpholine, and piperidine compounds to produce target compounds 7a–e.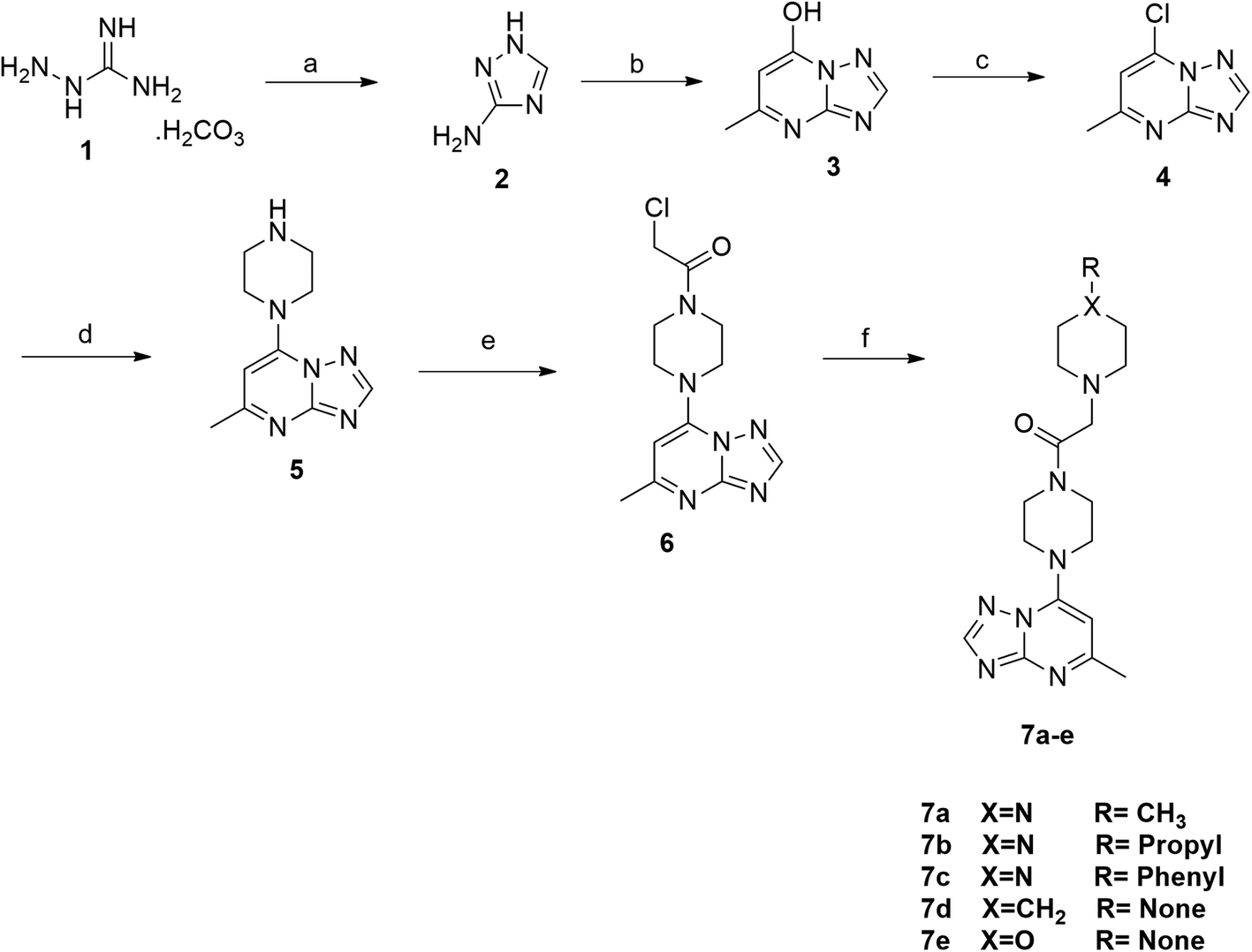 | ||
| Scheme 1 Reagents and conditions: (a) formic acid, reflux; (b) EAA, acetic acid, reflux; (c) POCl3, reflux; (d) piperazine, K2CO3, dioxane, reflux; (e) CAC, TEA, DCM, rt; (f) substituted piperazine, TEA, DCM, reflux. | ||
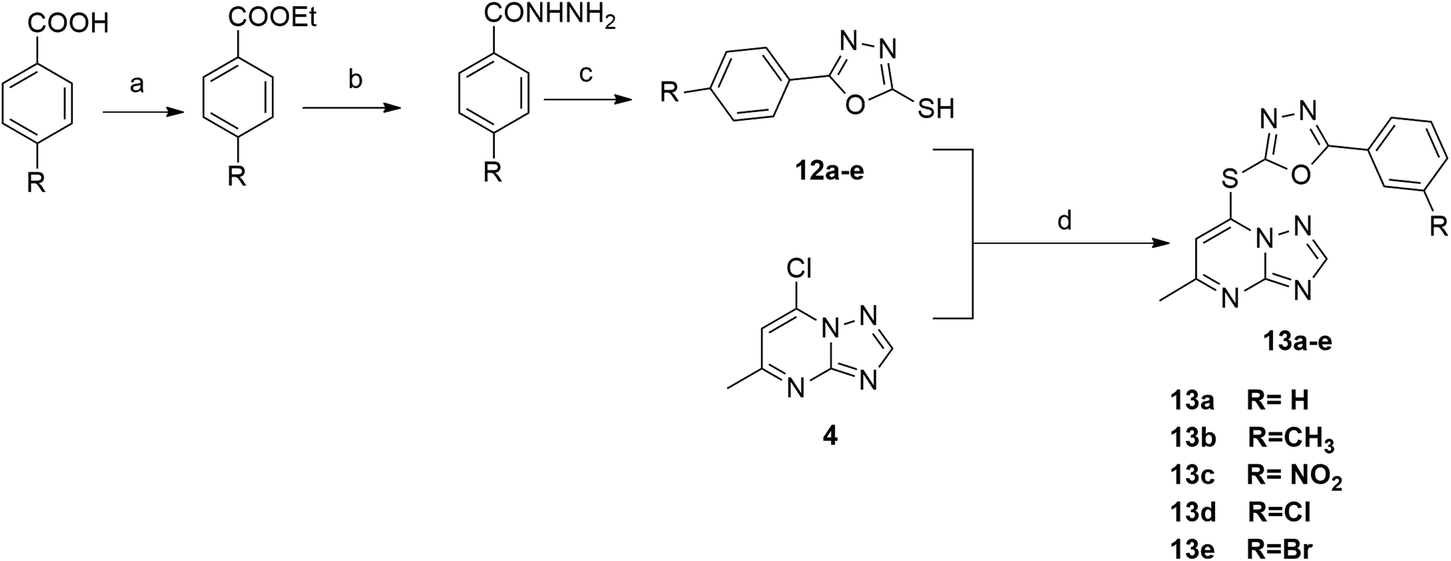
The synthetic strategy for target compounds 1,3,4-thiadiazoles 9a–d, 1,3,4-oxadiazoles 13a–e, and pyrimidines 11a–f involves multiple steps as illustrated in Schemes 2–4. Key intermediates 8a–d were obtained by reacting 5-amino-1,3,4-thiadiazole-2-thiol with various 4-substituted phenyl isocyanates under reflux, using acetonitrile as solvent (Scheme 2).36 Additionally, 6-phenyl-2-thiouracil-5-carbonitrile derivatives 10a–f were synthesized by prolonged heating of different aldehydes with ethyl cyanoacetate and thiourea in ethanol, in presence of K2CO3 (Scheme 3). Finally, compounds 8a–d and 10a–f were refluxed with (4) in acetone/K2CO3 to yield our target compounds 9a–d and 11a–f, respectively (Schemes 2 and 3).
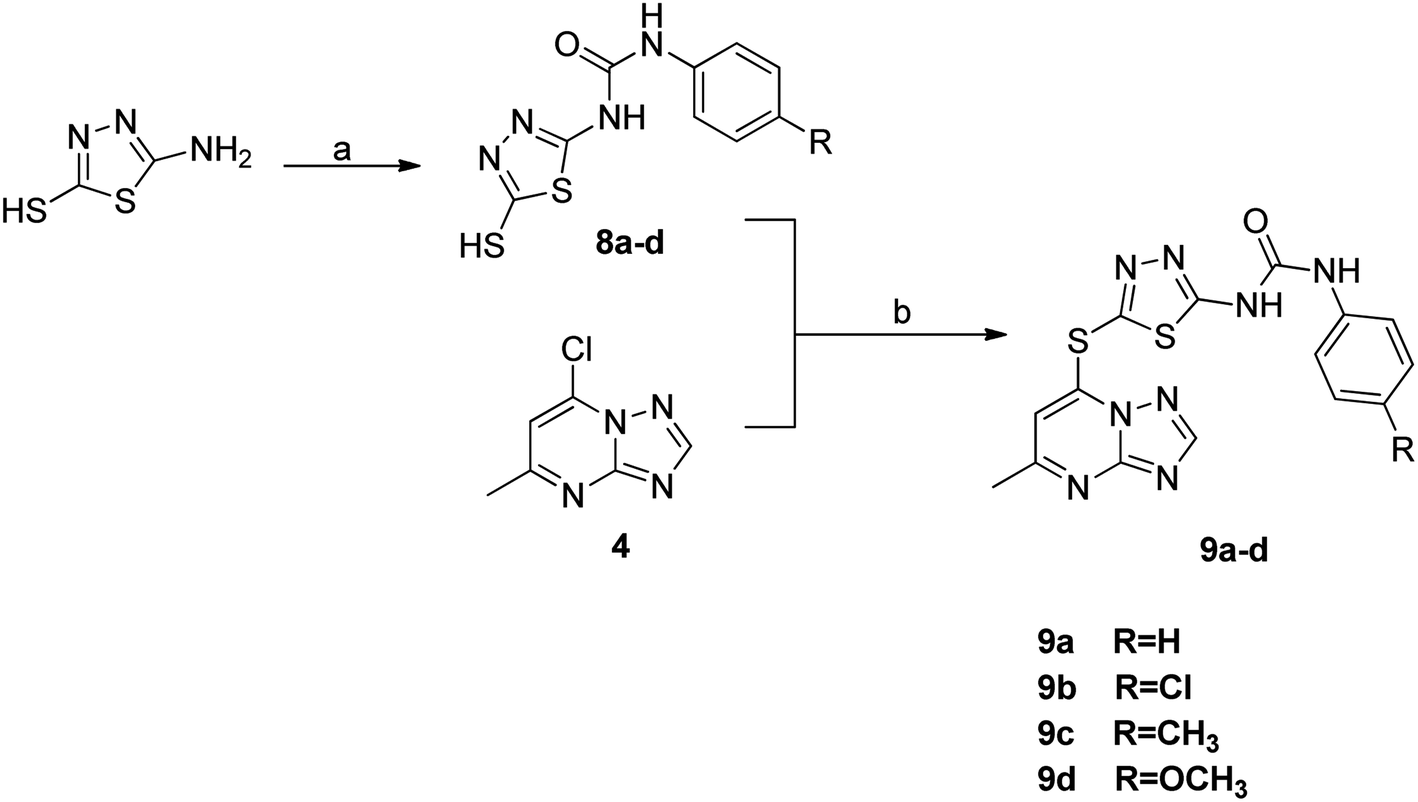 | ||
| Scheme 2 Reagents and conditions: (a) substituted phenyl isocyanate, acetonitrile, reflux; (b) K2CO3, acetone, reflux. | ||
 | ||
| Scheme 3 Reagents and conditions: (a) K2CO3, ethanol, reflux; (b) K2CO3, acetone, reflux. | ||
 | ||
| Scheme 4 Reagents and conditions: (a) C·H2SO4, ethanol, reflux; (b) NH2NH2, ethnaol, reflux; (c) CS2, KOH, ethanol, reflux; (d) K2CO3, dioxane, reflux. | ||
The synthetic pathway for 1,3,4-oxadiazole-2-thiol intermediates 12a–e involved the reaction of substituted benzohydrazide with carbon disulfide. The resulting compounds were then heated with (4) in dioxane to obtain various oxadiazole derivatives 13a–e, as illustrated in Scheme 4.
The newly synthesized derivatives (7a–e, 9a–d, 11a–f and 13a–e) were characterized using different means of melting point, elemental analysis and spectroscopic data (1H-NMR, 13C-NMR, and MS). 1H-NMR spectra for all the newly synthesized target compounds showed two characteristic singlet signals around 6 and 8 ppm corresponding to two aromatic protons of TP ring. Moreover, compounds 9a–d and 11a–f exhibited characteristic singlets corresponding NH protons ranging from 9–11 ppm proving the synthesis of our new targets. In addition, 13C-NMR of compounds 7a–e, 9a–d and 11a–f revealed the appearance of characteristic peak at ∼160–169 ppm equivalent to the carbonyl groups. All final targets exhibited a characteristic peak in the aliphatic region at approximately 25 ppm for the methyl substitution of TP moiety. All other data agreed with the suggested structures. The target compounds' mass spectra displayed the correct molecular ion peaks (M+). The experimental section presents the yields and the synthesis procedures.
2.2. Biological activity
|
|||||
|---|---|---|---|---|---|
| Comp. | R | IC50a (μM) | |||
| HCT-116 | HeLa | MCF-7 | WI-38 | ||
| a The concentration required to prevent 50% of cancer cells from proliferating is known as the IC50 value. The mean ± SD from the dose–response curves of three separate trials is used to illustrate the data. IC50, expressed in μg mL−1, ranges from 1–10 (extremely strong), 11–20 (strong), 21–50 (moderate), and 51–100 (weak). | |||||
| 7a | CH3, X = N | 65.80 ± 3.8 | 52.40 ± 3.4 | 55.96 ± 3.2 | >100 |
| 7b | n-Propyl, X = N | 62.61 ± 3.6 | 45.71 ± 2.4 | 53.85 ± 2.8 | 32.79 ± 2.1 |
| 7c | Phenyl, X = N | 7.03 ± 0.6 | 9.88 ± 0.6 | 6.93 ± 0.4 | 43.93 ± 2.4 |
| 7d | None, X = CH2 | 73.89 ± 4.2 | 59.10 ± 3.3 | 67.34 ± 3.4 | 36.45 ± 2.0 |
| 7e | None, X = O | 88.43 ± 4.5 | 78.54 ± 4.2 | 84.68 ± 4.1 | 26.32 ± 1.9 |
| 9a | H | 44.35 ± 2.6 | 25.19 ± 2.0 | 37.21 ± 2.3 | 48.35 ± 2.6 |
| 9b | Cl | >100 | 87.68 ± 4.4 | >100 | 64.72 ± 3.5 |
| 9c | CH3 | 58.50 ± 3.3 | 51.93 ± 2.8 | 46.05 ± 2.5 | 18.11 ± 1.3 |
| 9d | OCH3 | >100 | 82.39 ± 4.1 | 92.38 ± 4.4 | >100 |
| 11a | 4-Br | 41.03 ± 2.4 | 33.41 ± 2.3 | 30.49 ± 2.1 | 75.94 ± 4.1 |
| 11b | 4-Cl | 12.61 ± 1.0 | 17.52 ± 1.4 | 8.39 ± 0.6 | 56.23 ± 3.0 |
| 11c | 4-CH3 | 21.51 ± 1.5 | 19.24 ± 1.6 | 9.15 ± 0.7 | 45.03 ± 2.4 |
| 11d | 4-OCH3 | 47.87 ± 2.7 | 63.42 ± 3.6 | 34.63 ± 2.3 | 71.23 ± 3.9 |
| 11e | 3,4-diOCH3 | 8.89 ± 0.8 | 13.64 ± 1.1 | 5.92 ± 0.3 | 60.30 ± 3.3 |
| 11f | 3-Br | 31.16 ± 2.2 | 35.88 ± 2.2 | 22.16 ± 1.6 | 77.28 ± 4.3 |
| 13a | H | 15.03 ± 1.2 | 23.59 ± 1.9 | 18.63 ± 1.3 | 91.39 ± 4.9 |
| 13b | CH3 | 29.32 ± 2.0 | 39.01 ± 2.4 | 27.82 ± 2.0 | 83.54 ± 4.5 |
| 13c | NO2 | 6.10 ± 0.5 | 10.33 ± 0.9 | 2.42 ± 0.1 | 38.65 ± 2.2 |
| 13d | Cl | 24.79 ± 1.8 | 42.92 ± 2.5 | 14.21 ± 1.1 | 66.08 ± 3.8 |
| 13e | Br | 55.07 ± 3.5 | 57.41 ± 3.2 | 42.56 ± 2.4 | >100 |
| DOX | 5.23 ± 0.3 | 5.57 ± 0.4 | 4.17 ± 0.2 | 6.72 ± 0.5 | |
| Comp. | IC50 (μM) | |||
|---|---|---|---|---|
| EGFR | TOP-II | HER-2 | ARO | |
| 13c | 0.087 | 31.56 | 0.078 | 0.156 |
| 7c | 0.169 | 48.48 | 0.21 | 0.567 |
| 11e | 0.246 | 36.72 | 0.116 | 0.38 |
| Lapatinib | 0.092 | |||
| Erlotenib | 0.032 | |||
| Doxorubicin | 33.97 | |||
| Ketoconazole | 0.092 | |||
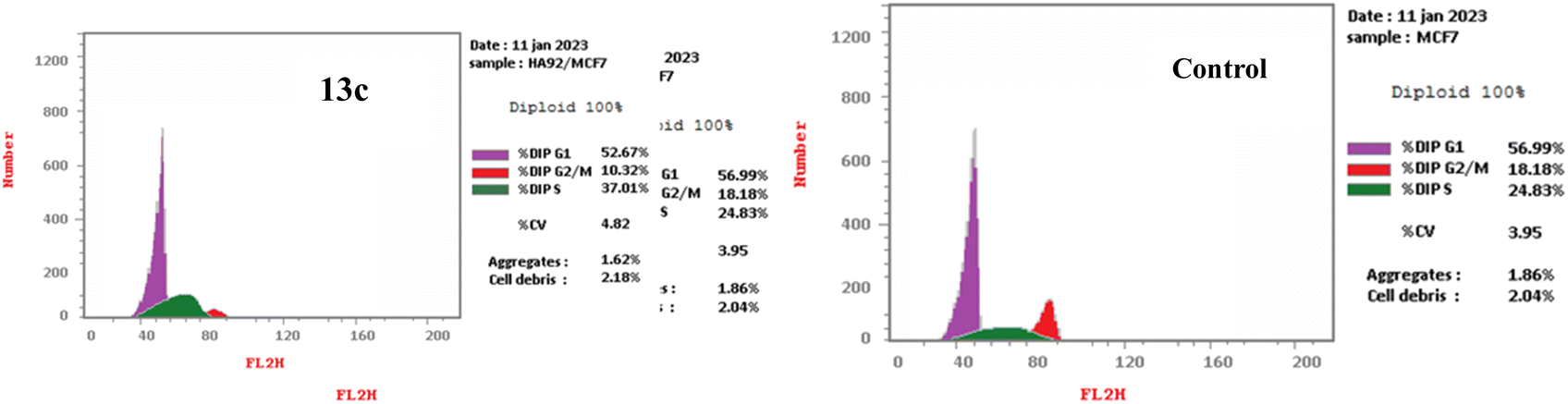 | ||
| Fig. 3 The DNA ploidy in MCF-7 cells was examined using flow cytometry following treatment with compound 13c. | ||
| Comp. no. | Cell cycle distribution (%) | ||
|---|---|---|---|
| G0-G1 | S | G2-M | |
| 13c | 52.67 | 37.01 | 10.32 |
| Control (DMSO) | 56.99 | 24.83 | 18.18 |
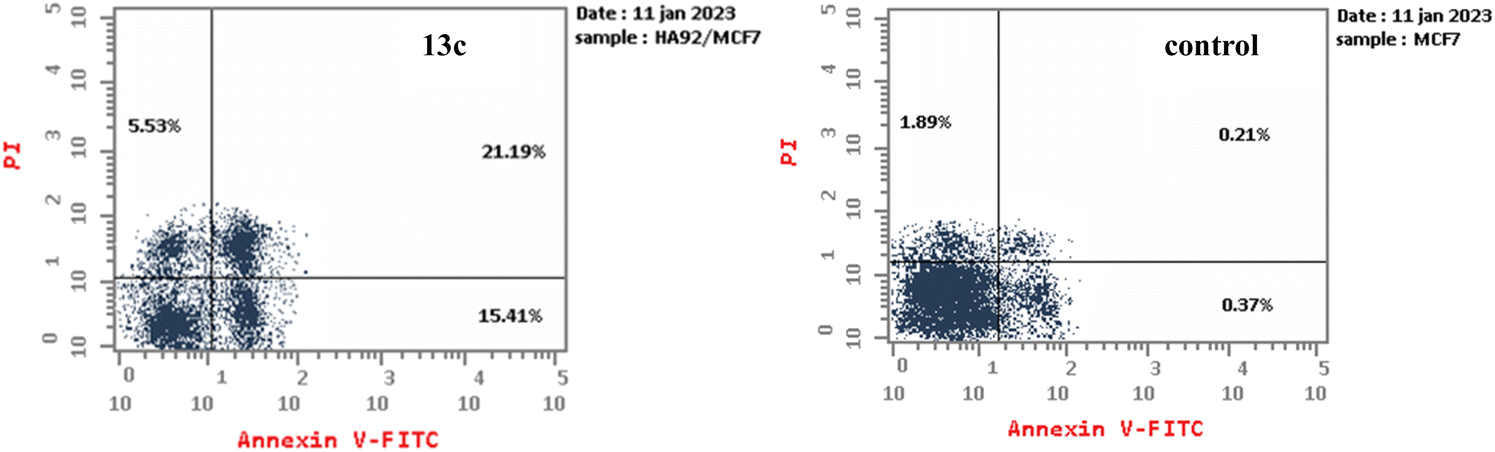 | ||
| Fig. 4 Impact of compound 13c on the percentage of Annexin V-FITC positive staining in MCF-7 cells after 24 hours of incubation, with DMSO used as a control. The four stages of cell death are Q1, necrotic cells; Q2, late apoptosis; Q3, living cells; and Q4, early apoptosis. | ||
Body weight analysis revealed that while the normal control group maintained minimal weight fluctuations, DMBA-treated groups exhibited significant weight gain. Analysis of body weight progression over the 20 days experimental period revealed distinct patterns among the groups. The control group exhibited a steady, moderate increase from 20.51 g to 28.91 g, representing typical growth. In contrast, the DMBA group, starting with the lowest initial weight (18.8 g), demonstrated the most pronounced weight gain, reaching 35.2 g by day 20, significantly surpassing the control. The 13c-treated group, beginning at a weight comparable to the control (20.2 g), showed a more moderate increase, attaining 30.6 g by the study's conclusion. This pattern suggests 13c's potential to mitigate DMBA-induced weight changes. The DOX group, while still exceeding the control's final weight, exhibited a weight gain pattern (19.8 g to 31.8 g) intermediate between the control and DMBA groups. Notably, both 13c and DOX treatments appeared to moderate the excessive weight gain observed in the untreated DMBA group, with 13c demonstrating a slightly more favorable effect in maintaining body weight closer to control levels, particularly in the latter stages of the experiment (Fig. 5a).
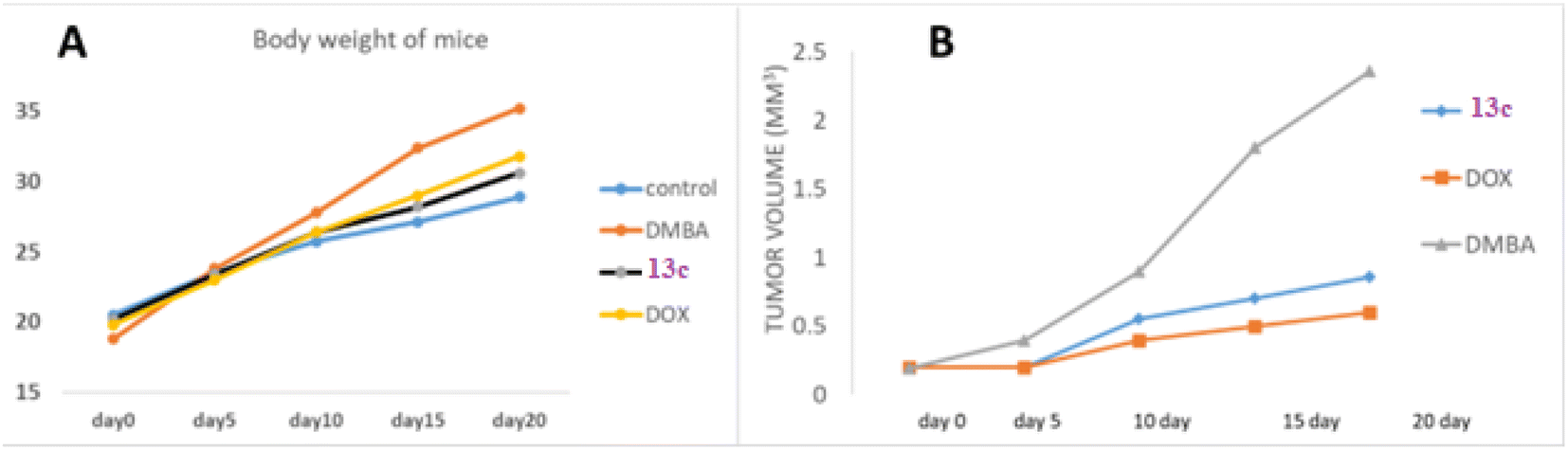 | ||
| Fig. 5 (A) Body weight changes in mice over 20 days following tumor induction with 7,12-dimethylbenz[a]anthracene (DMBA). Groups include control (untreated), DMBA-only, 13c-treated, and doxorubicin (Dox)-treated mice. (B) Tumor volume progression in the same groups over 20 days. Data represents mean values for each group, illustrating the impact of 13c and Dox on body weight and tumor growth in mouse model. | ||
Tumor volume measurements provided crucial insights into treatment efficacy. The untreated DMBA group demonstrated exponential tumor growth, with volumes reaching approximately 2.4 mm3 by day 20. In contrast, the 13c-treated group showed a significantly moderated growth pattern, with final tumor volumes of about 0.9 mm3, representing a substantial 62.5% reduction compared to the untreated group. This reduction was comparable to the effect observed in the positive control group treated with DOX, which achieved a 75% reduction in final tumor volume (approximately 0.6 mm3) relative to the DMBA control.
These findings corroborate the in vitro cytotoxicity results, indicating that 13c possesses significant tumor-suppressive properties in vivo. 13c demonstrated a remarkable ability to inhibit tumor growth, with efficacy approaching that of the established chemotherapeutic agent used as a positive control. This promising performance warrants further investigation of 13c as a potential anticancer agent (Fig. 5b).
The histopathological analysis (Fig. 6A) reveals striking differences across the treatment groups. The control group exhibits normal mammary gland structure with ducts lined by a single, well-organized epithelium. In contrast, the DMBA group shows severe pathological changes, including extensive fibrosis, significant leukocyte infiltration, and marked epithelial disorganization characterized by atypical hyperplasia and increased mitotic figures. The 13c-treated group demonstrates notable improvement over the DMBA group, presenting only mild periductal fibrosis and limited leukocyte infiltration, though some abnormalities persist. Still, it was comparable to the Dox-treated group that exhibited only mild inflammation and largely normalized epithelial ducts.
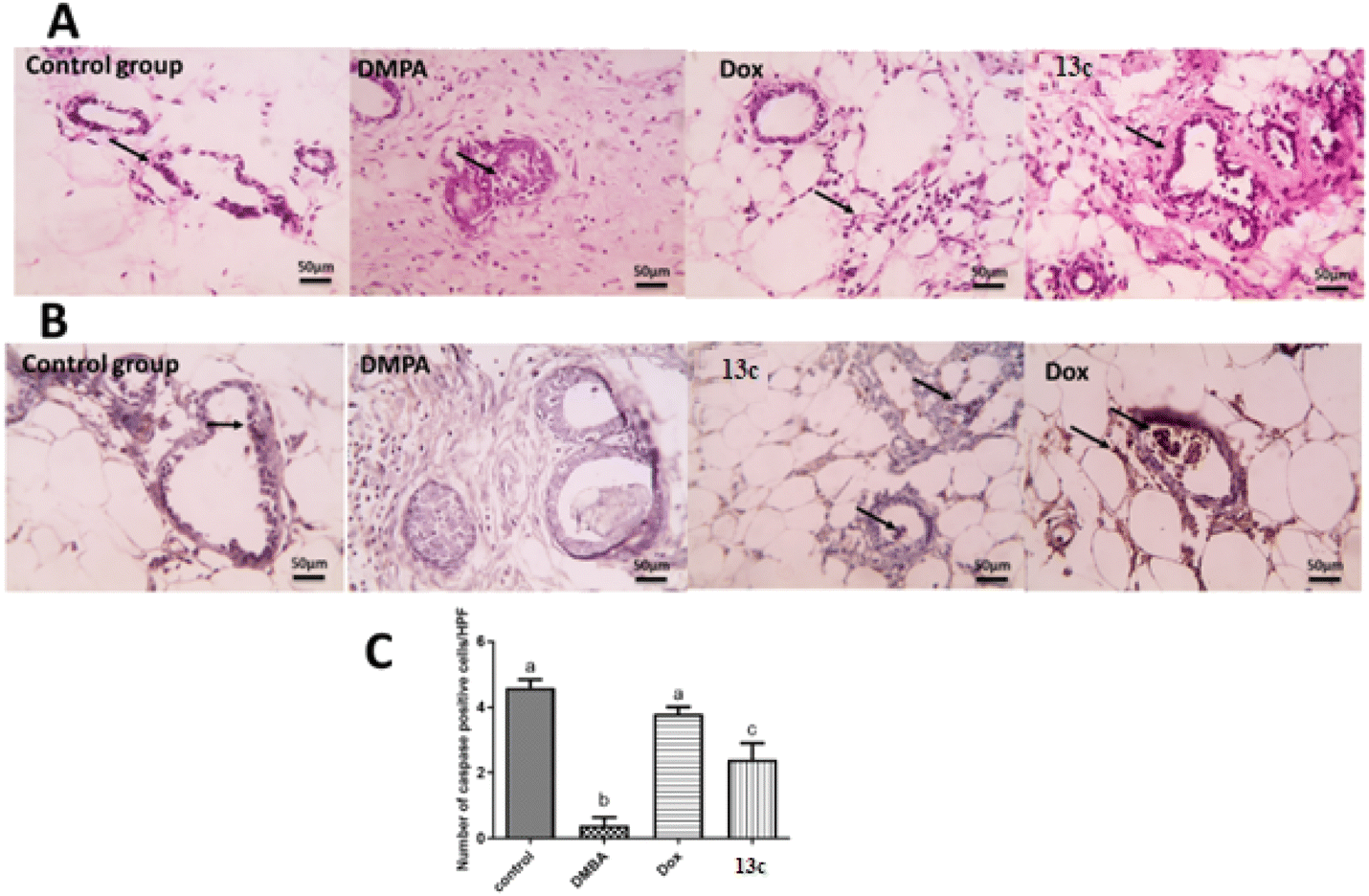 | ||
| Fig. 6 (A) HE-stained sections: the control group shows normal ducts with organized epithelium (black arrows). The DMBA group exhibits disorganized ductal cells with atypical hyperplasia (black arrows). The 13c-treated group shows mild fibrosis with few leukocytes (black arrows), while the Dox-treated group shows mild inflammation and normalized ducts (black arrow). (B) Caspase-3 immunostaining: the control group has positive ductal epithelial cells (black arrows); the DMBA group is mostly negative. The 13c-treated group has fewer positive cells (black arrows) than the Dox-treated group (black arrow). IHC counterstained with Mayer's hematoxylin. (C) Bar charts show a significant decrease in positive cells in the DMBA group and a significant increase in the Dox and 13c groups (P < 0.05). | ||
Interestingly, immunohistochemical analysis for caspase-3 (Fig. 6B) gave insights into the apoptotic activity across groups. The control group displays baseline positive staining in ductal epithelial cells, indicating normal apoptotic function. The DMBA group, however, shows negatively stained ductal epithelial cells, suggesting a severe suppression of apoptosis – a hallmark of cancer progression. The 13c-treated group shows a marked increase in positively stained cells compared to the DMBA group, indicating a partial restoration of apoptotic function which resembles the profile of Dox-treated group.
Quantitative analysis of caspase-3 expression (Fig. 6C) further elucidates these findings. The control group shows 4.5 ± 0.2 caspase-3 positive cells. On the other hand, the DMBA group exhibited a drastic 89% decrease to 0.5 ± 0.1 cells, underscoring the severe suppression of apoptosis in cancer cells. The Dox-treated group demonstrates remarkable efficacy, with 4.0 ± 0.2 cells, representing a 700% increase from the DMBA group and only an 11% decrease from the control. The 13c-treated group shows significant improvement with 2.3 ± 0.3 cells, a 360% increase from the DMBA group.
These results collectively indicate that 13c significantly enhances apoptotic activity in DMBA-induced mammary gland cancer, achieving about 51% of the control's apoptotic activity. Hence, its ability to normalize tissue morphology and significant increasement could be attributed to the increase in caspase-3 activity observed with 13c. Also, the inhibition of several key molecular targets involved in cancer cell survival and proliferation. Inhibition of EGFR (Epidermal Growth Factor Receptor) and HER2 (Human Epidermal Growth Factor Receptor 2) can lead to decreased activation of pro-survival signaling pathways, making cells more susceptible to apoptosis. Aromatase inhibition in hormone-dependent breast cancers can reduce estrogen production, potentially sensitizing cells to apoptotic stimuli. TOP-II inhibition can induce DNA damage, triggering the intrinsic apoptotic pathway. The observed increase in caspase-3 activity with 13c treatment could therefore be a downstream effect of inhibiting one or more of these targets.
2.3. Molecular docking
Since compounds 13c, and 11e and 7c showed promising inhibitory activity against enzymes under the study, we utilized molecular docking to explain their binding mode. Firstly, the evaluation of their binding energy in comparison to co-crystallized ligand revealed the better activity of 13c over other compounds as shown in Table 4. Hence its molecular interaction with the targeted enzyme was further investigated.| No. | Compound | EGFR | HER-2 | TOP-II | Aromatase |
|---|---|---|---|---|---|
| 1 | 13c | −8.0 | −10.4 | −10.7 | −9.9 |
| 2 | 11e | −7.9 | −8.9 | −9.5 | −9.1 |
| 3 | 7c | −7.7 | −10.3 | −10.2 | −10.6 |
| 4 | Co-crystallized ligand in EGFR | −8.1 | — | — | — |
| 5 | Co-crystallized ligand in HER-2 | — | −11.6 | — | — |
| 6 | Co-crystallized ligand in Top-II | — | — | −10.9 | — |
| 7 | Co-crystallized ligand in aromatase | — | — | — | −10.7 |
In the case of EGFR enzyme, compound 13c with TP core appears to be a central anchor, forming hydrogen bonds with LEU A:799 and ASP A:800, which are critical for positioning the ligand within the active site. This interaction contributes significantly to the compound's binding affinity and specificity for EGFR. The 2-mercapto oxadiazole portion of the molecule seems to engage in hydrophobic interactions with VAL A:726 and potentially contributes to the interaction with MET A:766, which could be a pi-stacking interaction.
This interaction with the methionine gatekeeper residue is particularly noteworthy as it often plays a crucial role in inhibitor selectivity. The nitrophenyl group, extending from the core structure, appears to form hydrophobic contacts with LEU A:844 and ALA A:743, further stabilizing the ligand's position. Additionally, this group may be involved in the hydrogen bonding interaction observed with LEU A:798. The compound's ability to engage multiple residues through its diverse functional groups suggests a potentially high affinity and specificity for EGFR.
Moreover, compound 13c also showed significant interaction with the active site of HER-2. A key hydrogen bond is formed between the ligand and ASP A:863. This interaction is complemented by an extensive network of hydrophobic contacts involving residues VAL A:734, LEU A:726, LEU A:852, ALA A:751, LYS A:753, LEU A:800, LEU A:785, and LEU A:796. The triazole moiety seems centrally positioned, possibly engaging with ASP A:863, while the pyrimidine-like structure orients towards VAL A:734 and LEU A:726, suggesting that these interactions may be crucial for binding stability.
Notably, the nitrophenyl group extends into a hydrophobic pocket formed by leucine residues, indicating a snug fit within the active site. This binding profile, with its combination of specific hydrogen bonding and extensive hydrophobic interactions, suggests that the compound could be a potent HER-2 inhibitor. The involvement of residues like VAL A:734 and LEU A:726, potentially part of the ATP-binding region, further emphasizes the ability of the compound to exert good inhibitory activity as shown in the experimental enzyme assay. Fig. 7 illustrates the binding of compound 13c with the active site of EGFR and HER-2.
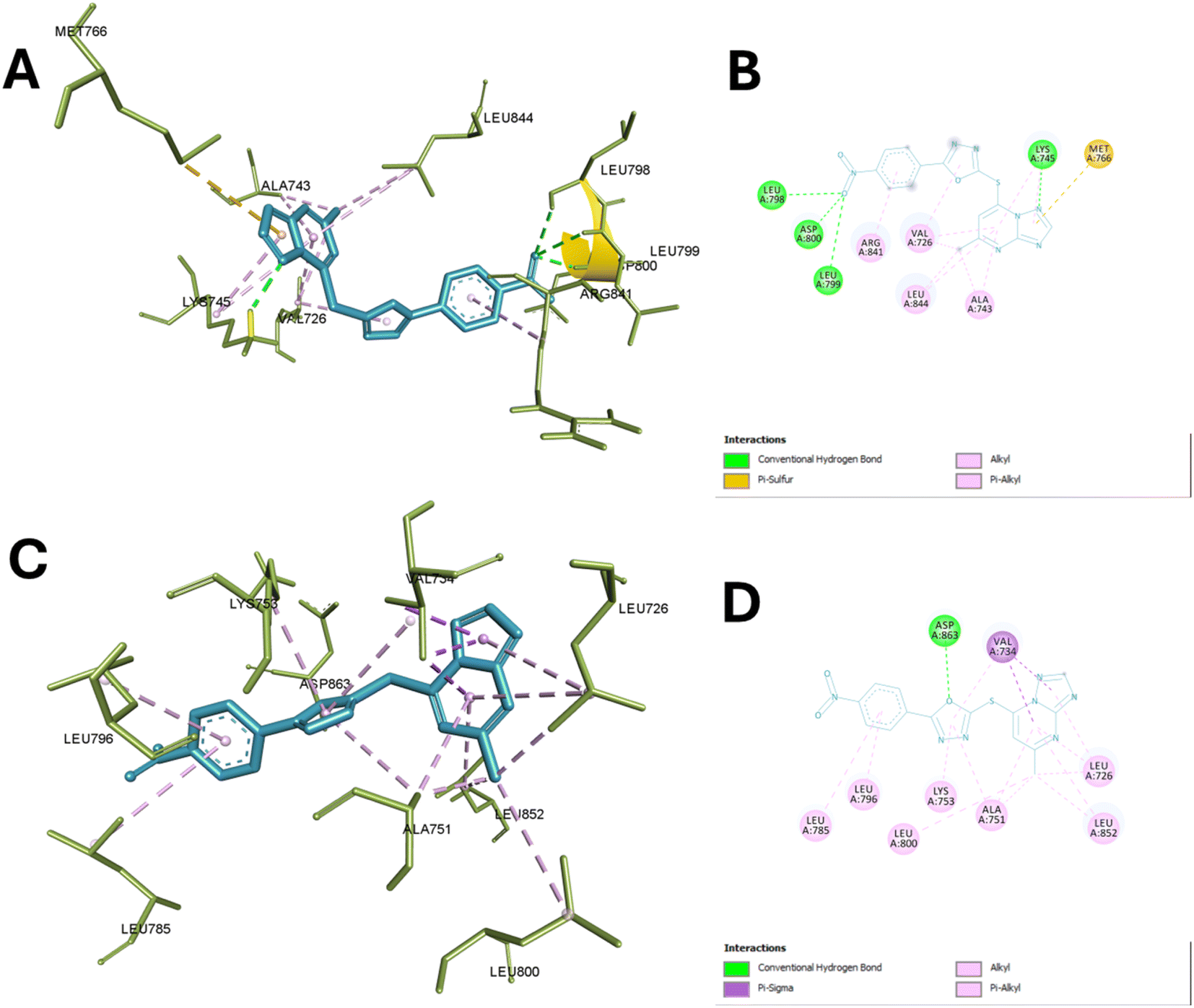 | ||
| Fig. 7 Binding of compound 13c with the active sites of EGFR and HER-2. (A) and (B) Show the 3D and 2D interactions of 13c with the active site of EGFR (PDB ID: 4WKQ), respectively, highlighting hydrogen bonds, hydrophobic contacts, and ionic interactions. (C) and (D) Illustrate the 3D and 2D interactions of 13c with the active site of HER-2 (PDB ID: 3PP0), emphasizing similar key interactions within the active site pockets. | ||
The molecular docking study of 13c with the active site of TOP-II reveals a detailed interaction profile that is crucial for understanding its inhibitory potential. The compound establishes several hydrogen bonds, hydrophobic interactions, and π–π stacking interactions with key amino acid residues within the active site. Specifically, the 2-mercapto oxadiazole moiety formed 2 hydrogen bonds with ASN A:150 and SER A:148, ensuring a stable orientation and strong interaction within the enzyme's active site.
Additionally, it formed hydrophobic interaction with Ile A:141 while, the TP ring also participates in significant π–π stacking interactions with PHE A:142 and TYR B:34, enhancing the binding affinity and stabilization of the compound within the active site. Nitro phenyl fragments were able to interact efficiently with several residues through hydrogen bonding such as Asn A:163, Tyr A:165, Gly A:164, and Gln A:376, further contributing to the compound's binding stability. Hydrophobic interactions are observed with ILE A:141 and, with the latter also involved in π–π stacking with the aromatic ring of the compound, underscoring the combined role of hydrophobic and aromatic interactions in reinforcing the compound's affinity for the enzyme.
These interactions collectively ensure high binding affinity, specificity, and stability, underscoring the compound's potential as a potent enzyme inhibitor by effectively occupying the active site. Specifically, the involvement of these amino acids in the catalytic activity of TOP-II highlights their significance. Asn A:163 and Asn A:150 are essential for stabilizing the DNA cleavage complex, and their interaction with the compound could inhibit this stabilization.
Tyr A:165 and Tyr B:34 are crucial for forming a covalent bond with the DNA phosphate backbone during the cleavage and rejoining process, and their interaction with the compound could block their activity. Gly A:164 and Gln A:376 provide flexibility and stabilization to the active site, and their interaction with the compound could hinder these roles. Ser A:148 is involved in ATP hydrolysis, and its interaction with the compound could disrupt this critical step. Lastly, Ile A:141 and Phe A:142 are important for maintaining the structural integrity of the active site, and their interaction with the compound could alter the enzyme's conformation. Thus, the compound's binding to these key residues effectively inhibits the catalytic activity of Topo II, making it a potent enzyme inhibitor.38
Finally, molecular docking of 13c in the active site of aromatase reveals significant interactions with the active site of aromatase. The nitro group forms conventional hydrogen bonds with ARG A:115, ARG A:145, TRP A:141, and ARG A:435 anchoring this portion of the molecule in the binding pocket. The phenyl group engages in pi–alkyl interactions with ILE A:133, ILE A:133 and ALA A:438. While TP scaffold interacts through pi-sulfur bonds with CYS A:437, THR A:310, and MET A:311 and hydrophobic interaction with ALA A:443 and VAL A:370. A conventional hydrogen bond is also observed between GLY A:439 and the 2-mercapto oxadiazole moiety.
Moreover, it also showed hydrophobic interaction with ALA A:306 and ALA A:438. These interactions collectively contribute to the compound's stability within the aromatase active site. The observed interactions involve several residues crucial for aromatase function. ARG A:115 and ARG A:145 are part of the substrate access channel and play a role in substrate recognition and positioning. TRP A:141 is involved in maintaining the structural integrity of the active site. CYS A:437 is a key residue in the catalytic mechanism, participating in the electron transfer process during substrate oxidation.
The interactions with ILE A:133, ALA A:438, and VAL A:370 are to contribute to defining the hydrophobic environment necessary for optimal substrate binding and orientation. GLY A:439 is a part of the conserved glycine-rich loop that is important for cofactor binding and positioning. Hence, this interactions suggests that this compound may effectively occupy the aromatase active site, potentially interfering with substrate binding and catalysis supporting the experimental findings in the enzyme inhibition assays,39,40 Fig. 8 depict the interaction of compound 13c with aromatase and TOP-II.
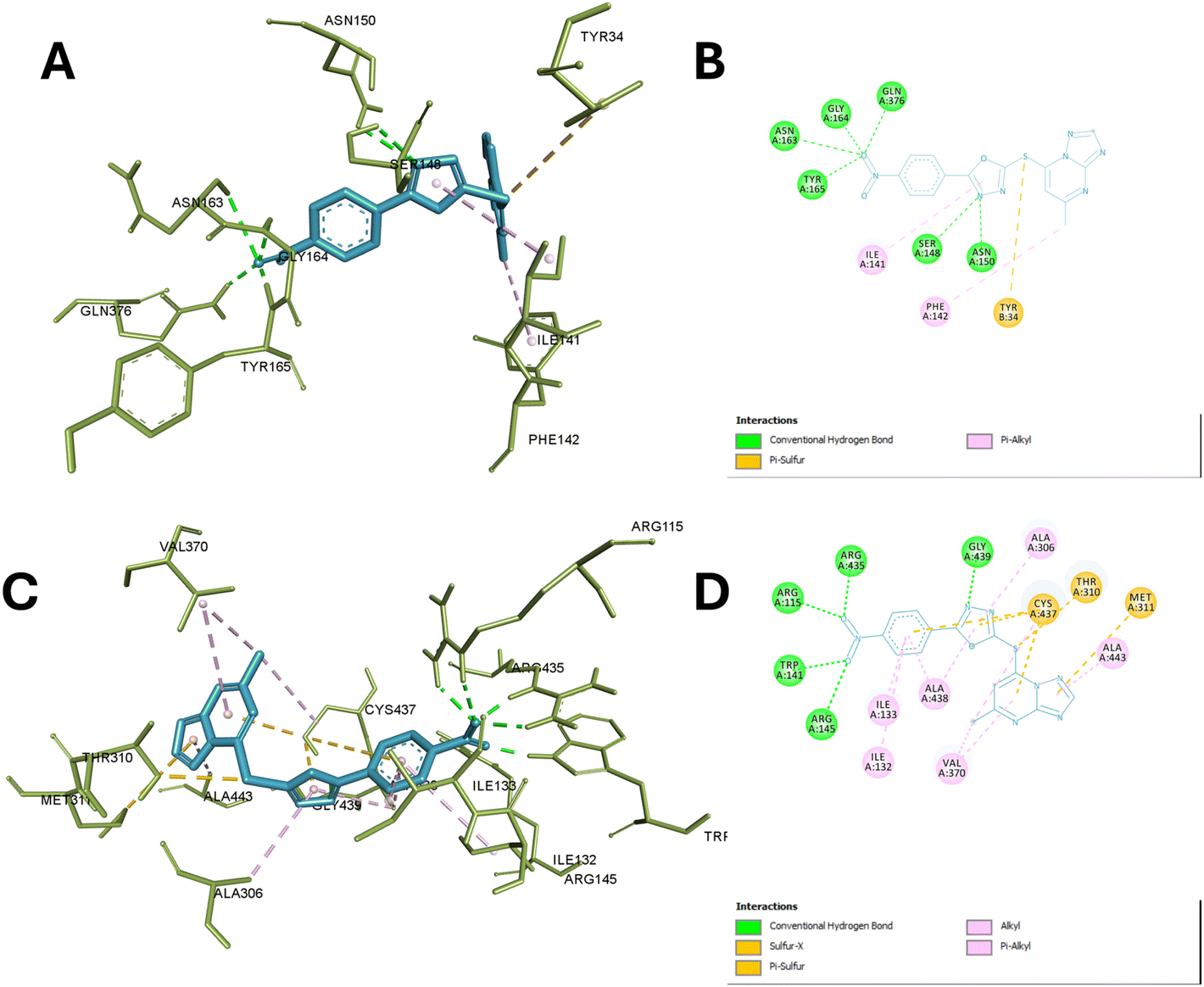 | ||
| Fig. 8 Interaction of compound 13c with aromatase and TOP-II. (A) and (B) Display the 3D and 2D interactions of 13c with the active site of TOP-II (PDB ID: 1ZXM), respectively, detailing hydrogen bonds, hydrophobic contacts. (C) and (D) Present the 3D and 2D interactions of 13c with the active site of aromatase (PDB ID: 3S79), highlighting the key binding interactions, including hydrogen bonds, and hydrophobic interactions. | ||
3. Conclusion
New series of TP compounds 7a–e, 9a–d, 11a–f and 13a–e were synthesized. The structure of the novel hybrids was confirmed using different spectroscopic techniques. They were assessed for their cytotoxic effect vs. three cancer cell lines: HCT116, HeLa & MCF7. 13c showed the best antitumor activity of the tested compounds with IC50 6.10, 10.33 and 2.42 μM in succession. In addition, compound 11e revealed a potent anticancer effect against HCT116 and MCF-7 with IC50 8.89 & 5.92 μM, respectively. Also, compound 7c exhibited very strong activity against HCT116, HeLa & MCF7 with IC50 7.03, 9.88 and 6.93, respectively. The most potent cytotoxic compounds were tested in a multi-target suppressive test, and 13c had the highest inhibition against EGFR, TOP-II, HER2, and ARO with an IC50 of 0.087, 31.56, 0.078 and 0.156 μM, respectively. According to flow cytometric testing, the most effective multi-target inhibitory agent, 13c, was shown to suppress the cell cycle in the S phase in the MCF-7. The compelling in vitro cytotoxic activity of compound 13c prompted further investigation of its efficacy in vivo. 13c demonstrated a remarkable ability to inhibit tumor growth, with efficacy approaching that of the established chemotherapeutic agent used as a positive control. The histopathological analysis reveals 13c-treated group demonstrating notable improvement over the DMBA group, presenting only mild periductal fibrosis and limited leukocyte infiltration. Confirmation of the obtained results was done with the aid of the docking study, which showed that the three motifs might adhere to the enzyme's major active sites, and the results were in good accordance with the experimental inhibitory results.4. Experimental
4.1. Chemistry
All melting points (°C) were recorded on Stuart melting point apparatus (SMP 30) and are uncorrected. 1H-NMR and 13C-NMR spectra were performed in (DMSO-d6 or CDCl3) at 1HNMR (400 MHz), 13CNMR (100 MHz) using an NMR spectrometer (δ ppm), NMR Unit, Faculty of Pharmacy, Mansoura University, using TMS as an internal standard. The following are the abbreviations: (s) for singlet; (d) for doublet; (t) for triplet; (m) for multiplet; and (br) for broad. At Al-Azhar University in Egypt's Regional Center for Mycology and Biotechnology (RCMB), mass spectra were conducted using a Thermo Scientific GCMS type ISQ. For the C, H, and N elements, microanalyses were performed at Cairo University using a PerkinElmer 240 elemental analyzer. The results were obtained within an acceptable range of theoretical values. With a UV lamp set at 254 nm, compounds were found. Every chemical and reagent that was utilized was bought from commercial sources including Aldrich Chemicals Co. in the United States. The key precursor compound 4 and intermediate compounds 8a–d, 10a–f, 12a–e were prepared according to reported procedures in literature.35,36,414.1.2.1 1-(4-(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)piperazin-1-yl)-2-(4-methylpiperazin-1-yl)ethenone (7a). White solid; (0.09 g, 74%). M. p. 160–162 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.50 (s, 1H), 6.67 (s, 1H), 3.91 (d, J = 4.2 Hz, 2H), 3.84 (d, J = 3.9 Hz, 4H), 3.75–3.69 (m, 2H), 3.24 (s, 2H), 2.55 (s, 3H), 2.51–2.27 (m, 8H), 2.23–2.18 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.4, 164.9, 154.3, 149.9, 95.4, 61.1, 55.0, 52.7, 48.4, 47.90, 46.1, 25.0. GC-MS EI m/z (%): 358.34 (M+, 18.22). Anal. calcd for C17H26N8O (358.22): C, 56.96; H, 7.31; N, 31.26. Found: C, 56.88; H, 7.26; N, 31.29%.
4.1.2.2 1-(4-(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)piperazin-1-yl)-2-(4-propylpiperazin-1-yl)ethenone (7b). White solid; (0.1 g, 76%). M. p. 145–147 °C. 1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 6.09 (s, 1H), 3.86–3.76 (m, 4H), 3.69–3.50 (m, 4H), 3.23 (s, 2H), 2.74–2.68 (m, 4H), 2.51 (s, 3H), 2.50–2.42 (m, 4H), 2.38–2.28 (m, 2H), 1.55–1.44 (m, 2H), 0.86 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.2, 163.9, 156.2, 153.2, 149.1, 93.6, 59.4, 59.2, 52.3, 51.4, 46.9, 43.9, 24.2, 18.4, 10.7. GC-MS EI m/z (%): 386.39 (M+, 26.22). Anal. calcd for C19H30N8O (386.25): C, 59.04; H, 7.82; N, 28.99. Found: C, 59.08; H, 7.72; N, 28.91%.
4.1.2.3 1-(4-(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)piperazin-1-yl)-2-(4-phenylpiperazin-1-yl)ethenone (7c). White solid; (0.091 g, 68%). M. p. 190–192 °C. 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.23 (t, J = 7.9 Hz, 2H), 6.86 (t, J = 9.0 Hz, 3H), 6.10 (s, 1H), 3.86 (s, 4H), 3.78–3.66 (m, 4H), 3.43 (s, 2H), 3.17–3.06 (m, 4H), 2.92 (s, 4H), 2.52 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 164.0, 156.1, 153.3, 149.7, 149.0, 148.9, 128.3, 119.8, 115.7, 93.8, 51.1, 49.0, 46.4, 44.4, 40.7, 24.2. GC-MS EI m/z (%): 420.19 (M+, 22.25). Anal. calcd for C22H28N8O (420.24): C, 62.84; H, 6.71; N, 26.65. Found: C, 62.75; H, 6.70; N, 26.57%.
4.1.2.4 1-(4-(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)piperazin-1-yl)-2-(piperidin-1-yl)ethenone (7d). White solid; (0.099 g, 85%). M. p. 185–187 °C. 1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 6.09 (s, 1H), 3.80 (s, 4H), 3.53–3.46 (m, 2H), 3.46–3.37 (m, 2H), 3.21 (s, 2H), 2.71 (d, J = 4.2 Hz, 4H), 2.51 (s, 3H), 1.59 (d, J = 4.7 Hz, 2H), 1.50 (dd, J = 10.8, 4.9 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 167.0, 164.8, 157.2, 154.1, 150.2, 94.6, 60.4, 52.5, 47.9, 46.6, 26.6, 25.2, 24.4. GC-MS EI m/z (%): 343.28 (M+, 26.11). Anal. calcd for C17H25N7O (343.21): C, 59.45; H, 7.34; N, 28.55. Found: C, 59.36; H, 7.30; N, 28.59%.
4.1.2.5 1-(4-(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)piperazin-1-yl)-2-morpholinoethanone (7e). White solid; (0.095 g, 81%). M. p. 164–166 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.45 (s, 1H), 6.63 (s, 1H), 3.99–3.67 (m, 8H), 3.64–3.51 (m, 8H), 3.22 (s, 2H), 2.43 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.9, 164.7, 157.1, 154.4, 145.0, 95.4, 66.6, 61.3, 53.4, 48.4, 45.0, 25.1. GC-MS EI m/z (%): 345.36 (M+, 19.52). Anal. calcd for C16H23N7O2 (345.19): C, 55.64; H, 6.71; N, 28.39. Found: C, 55.49; H, 6.75; N, 28.47%.
4.1.3.1 1-(5-((5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-1,3,4-thiadiazol-2-yl)-3-phenylurea (9a). White solid; (0.082 g, 72%). M. p. 202–204 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.57 (s, 1H), 9.26 (s, 1H), 8.69 (s, 1H), 7.52 (d, J = 7.9 Hz, 2H), 7.36 (t, J = 7.9 Hz, 2H), 7.10 (t, J = 7.4 Hz, 1H), 6.85 (s, 1H), 2.55 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 185.8, 165.8, 165.1, 155.9, 154.6, 147.1, 145.9, 138.5, 129.5, 123.9, 119.6, 108.6, 25.2. GC-MS EI m/z (%): 384.15 (M+, 23.05). Anal. calcd for C15H12N8OS2 (384.06): C, 46.86; H, 3.15; N, 29.15. Found: C, 46.78; H, 3.11; N, 29.19%.
4.1.3.2 1-(4-Chlorophenyl)-3-(5-((5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-1,3,4-thiadiazol-2-yl)urea (9b). White solid; (0.09 g, 71%). M. p. 209–211 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.66 (s, 1H), 9.35 (s, 1H), 8.69 (s, 1H), 7.56 (d, J = 8.6 Hz, 2H), 7.41 (d, J = 8.6 Hz, 2H), 6.86 (s, 1H), 2.56 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 185.8, 165.3, 165.0, 155.8, 154.6, 147.1, 145.9, 138.4, 131.9, 125.3, 123.1, 108.8, 25.2. GC-MS EI m/z (%): 418.25 (M+, 18.21). Anal. calcd for C15H11ClN8OS2 (418.02): C, 43.01; H, 2.65; N, 26.75. Found: C, 43.08; H, 2.51; N, 26.78%.
4.1.3.3 1-(5-((5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-1,3,4-thiadiazol-2-yl)-3-(p-tolyl)urea (9c). White solid; (0.099 g, 83%). M. p. 195–197 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H), 9.08 (s, 1H), 8.69 (s, 1H), 7.39 (d, J = 8.1 Hz, 2H), 7.15 (d, J = 8.1 Hz, 2H), 6.85 (s, 1H), 2.55 (s, 3H), 2.27 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 183.9, 165.7, 165.1, 155.9, 154.6, 147.1, 145.9, 135.8, 133.0, 129.9, 119.7, 108.5, 25.2, 20.9. GC-MS EI m/z (%): 397.92 (M+, 25.22). Anal. calcd for C16H14N8OS2 (398.07): C, 48.23; H, 3.54; N, 28.12. Found: C, 48.22; H, 3.66; N, 28.17%.
4.1.3.4 1-(4-Methoxyphenyl)-3-(5-((5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-1,3,4-thiadiazol-2-yl)urea (9d). White solid; (0.08 g, 65%). M. p. 201–203 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.54 (s, 1H), 9.01 (s, 1H), 8.69 (s, 1H), 7.41 (d, J = 7.9 Hz, 2H), 6.92 (d, J = 6.7 Hz, 2H), 6.84 (s, 1H), 3.74 (s, 3H), 2.55 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 187.6, 165.8, 165.1, 156.0, 155.9, 154.6, 147.2, 145.8, 131.3, 121.6, 114.6, 108.5, 55.7, 25.2. GC-MS EI m/z (%): 414.12 (M+, 19.45). Anal. calcd for C16H14N8O2S2 (414.07): C, 46.37; H, 3.40; N, 27.04. Found: C, 46.25; H, 3.46; N, 27.09%.
4.1.4.1 4-(4-Bromophenyl)-2-((5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (11a). White solid; (0.094 g, 71%). M. p. 229–231 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (s, 1H), 8.05 (s, 1H), 7.68 (d, J = 8.6 Hz, 2H), 7.61 (d, J = 8.6 Hz, 2H), 2.66 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.1, 167.6, 166.7, 164.6, 155.3, 154.9, 143.3, 136.5, 131.7, 130.5, 124.2, 119.5, 115.3, 91.7, 25.2. GC-MS EI m/z (%): 439.15 (M+, 24.19). Anal. calcd for C17H10BrN7OS (438.99): C, 46.38; H, 2.29; N, 22.27. Found: C, 46.22; H, 2.28; N, 22.31%.
4.1.4.2 4-(4-Chlorophenyl)-2-((5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (11b). White solid; (0.088 g, 74%). M. p. 235–237 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (s, 1H), 8.05 (s, 1H), 7.68 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 8.5 Hz, 2H), 2.66 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.1, 167.6, 166.7, 164.6, 155.3, 154.9, 143.3, 136.2, 135.4, 130.3, 128.8, 119.5, 115.3, 91.7, 25.2. GC-MS EI m/z (%): 395.25 (M+, 26.28). Anal. calcd for C17H10ClN7OS (395.04): C, 51.58; H, 2.55; N, 24.77. Found: C, 51.44; H, 2.59; N, 24.63%.
4.1.4.3 2-((5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-6-oxo-4-(p-tolyl)-1,6-dihydropyrimidine-5-carbonitrile (11c). White solid; (0.095 g, 84%). M. p. 220–222 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (s, 1H), 8.08 (s, 1H), 7.54 (d, J = 8.0 Hz, 2H), 7.25 (d, J = 8.0 Hz, 2H), 2.65 (s, 3H), 2.35 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.5, 167.7, 167.5, 164.5, 155.3, 154.9, 143.5, 140.4, 134.6, 129.2, 128.4, 119.8, 115.2, 91.4, 25.1, 21.4. GC-MS EI m/z (%): 375.14 (M+, 17.65). Anal. calcd for C18H13N7OS (375.09): C, 57.59; H, 3.49; N, 26.12. Found: C, 57.41; H, 3.55; N, 26.03%.
4.1.4.4 4-(4-Methoxyphenyl)-2-((5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (11d). White solid; (0.09 g, 77%). M. p. 228–230 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (s, 1H), 8.08 (s, 1H), 7.65 (d, J = 8.7 Hz, 2H), 7.00 (d, J = 8.7 Hz, 2H), 3.82 (s, 3H), 2.66 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 167.4, 167.2, 164.5, 161.3, 155.3, 154.9, 143.6, 130.1, 129.5, 120.0, 115.1, 114.0, 90.8, 55.8, 25.2. GC-MS EI m/z (%): 391.02 (M+, 16.18). Anal. calcd for C18H13N7O2S (391.09): C, 55.23; H, 3.35; N, 25.05. Found: C, 55.11; H, 3.30; N, 25.09%.
4.1.4.5 4-(3,4-Dimethoxyphenyl)-2-((5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (11e). White solid; (0.089 g, 71%). M. p. 236–238 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.54 (s, 1H), 7.81 (s, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.87 (d, J = 1.7 Hz, 1H), 6.78 (dd, J = 8.2, 1.7 Hz, 1H), 5.41 (s, 1H), 4.79 (d, J = 3.4 Hz, 1H), 3.74 (d, J = 1.2 Hz, 6H), 2.15 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 189.5, 176.2, 174.0, 167.3, 161.7, 160.7, 157.1, 151.9, 148.9, 148.3, 139.0, 126.8, 118.6, 112.0, 110.9, 95.0, 57.0, 56.1, 24.6. GC-MS EI m/z (%): 420.92 (M+, 15.78). Anal. calcd for C19H15N7O3S (421.10): C, 54.15; H, 3.59; N, 23.27. Found: C, 54.12; H, 3.48; N, 23.21%.
4.1.4.6 4-(3-Bromophenyl)-2-((5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (11f). White solid; (0.088 g, 67%). M. p. 250–252 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.56 (s, 1H), 7.95 (s, 1H), 7.90 (s, 1H), 7.65 (d, J = 8.1 Hz, 1H), 7.55 (s, 1H), 7.39 (t, J = 7.9 Hz, 1H), 2.65 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.1, 170.1, 167.6, 166.1, 164.5, 161.7, 158.6, 151.9, 139.5, 133.3, 131.1, 127.5, 121.9, 119.3, 115.4, 95.1, 24.6. GC-MS EI m/z (%): 438.85 (M+, 20.36). Anal. calcd for C17H10BrN7OS (438.99): C, 46.38; H, 2.29; N, 22.27. Found: C, 46.36; H, 2.20; N, 22.18%.
4.1.5.1 2-((5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-5-phenyl-1,3,4-oxadiazole (13a). White solid; (0.08 g, 86%). M. p. 260–262 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.12–8.04 (m, 2H), 7.76–7.65 (m, 3H), 7.39 (s, 1H), 2.59 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.3, 165.5, 156.0, 155.5, 154.7, 142.8, 133.2, 130.0, 127.5, 123.4, 110.6, 25.2. GC-MS EI m/z (%): 310.21 (M+, 23.60). Anal. calcd for C14H10N6OS (310.06): C, 54.18; H, 3.25; N, 27.08. Found: C, 54.25; H, 3.20; N, 27.11%.
4.1.5.2 2-((5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-5-(p-tolyl)-1,3,4-oxadiazole (13b). White solid; (0.071 g, 73%). M. p. 240–242 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1H), 7.97 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 7.37 (s, 1H), 2.58 (s, 3H), 2.43 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.4, 165.5, 156.0, 155.2, 154.7, 143.6, 142.9, 130.6, 127.4, 120.7, 110.5, 25.2, 21.7. GC-MS EI m/z (%): 323.95 (M+, 24.31). Anal. calcd for C15H12N6OS (324.08): C, 55.54; H, 3.73; N, 25.91. Found: C, 55.36; H, 3.70; N, 25.88%.
4.1.5.3 2-((5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-5-(4-nitrophenyl)-1,3,4-oxadiazole (13c). White solid; (0.074 g, 70%). M. p. 264–266 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.48 (d, J = 8.9 Hz, 2H), 8.35 (d, J = 8.9 Hz, 2H), 7.45 (s, 1H), 2.59 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.9, 165.5, 156.9, 156.0, 154.7, 150.1, 142.3, 128.9, 128.9, 125.2, 110.9, 25.2. GC-MS EI m/z (%): 355.31 (M+, 19.15). Anal. calcd for C14H9N7O3S (355.05): C, 47.32; H, 2.55; N, 27.59. Found: C, 47.18; H, 2.39; N, 27.52%.
4.1.5.4 2-(4-Chlorophenyl)-5-((5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-1,3,4-oxadiazole (13d). White solid; (0.067 g, 65%). M. p. 258–260 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.10 (d, J = 8.6 Hz, 2H), 7.74 (d, J = 8.6 Hz, 2H), 7.40 (s, 1H), 2.58 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.6, 165.5, 156.0, 155.8, 154.7, 142.7, 138.0, 130.2, 129.3, 122.3, 110.6, 25.2. GC-MS EI m/z (%): 343.92 (M+, 21.22). Anal. calcd for C14H9ClN6OS (344.02): C, 48.77; H, 2.63; N, 24.38. Found: C, 48.70; H, 2.52; N, 24.39%.
4.1.5.5 2-(4-Bromophenyl)-5-((5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)thio)-1,3,4-oxadiazole (13e). White solid; (0.078 g, 67%). M. p. 270–272 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 1H), 8.01 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 8.4 Hz, 2H), 7.40 (s, 1H), 2.59 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.6, 165.3, 156.0, 155.6, 154.2, 142.7, 134.5, 131.0, 128.0, 122.3, 110.6, 25.2. GC-MS EI m/z (%): 387.92 (M+, 19.02). Anal. calcd for C14H9BrN6OS (387.97): C, 43.20; H, 2.33; N, 21.59. Found: C, 43.15; H, 2.3.; N, 21.69%.
4.2. Biological evaluation
4.3. Molecular docking study
To investigate the binding mode of the most active compounds in comparison to the co-crystallized ligand, molecular docking was utilized to obtain insights on their interaction with the active site of EGFR, HER-2, TOP-II and aromatase enzyme. Hence, their crystal structures were obtained from PDB using the codes: 4WKQ, 3PP0, 1ZXM and 3S79 respectively. Using Protein Repair and Analysis Server, the obtained 3D structures were produced by assigning bond ordering, adding missing atoms, optimizing hydrogen bonds, and correcting charges.48 After that, the co-crystallized ligand and water molecules were eliminated. For virtual screening, the protein's produced PDB file was loaded into PyRx software's protein preparation module,49–51 where they were converted to pdbqt files and the active sites were defined as grid box size was 20 × 20 × 20 and the coordinates were for EGFR was X: 2.715 Y: 194.363 Z: 20.975 while for HER-2 was X: 2.715 Y: 194.363 Z: 20.975, in case of TOP-II, X: 65.444 Y: 18.789 Z: 28.34, finally, the coordinates for aromatase were: X: 84.35 Y: 50.9 Z: 43.68.Compounds 13c, 11e and 7c were obtained as mol files using Marvin sketch version 21.17.0, ChemAxon (https://www.chemaxon.com/) which were converted to Pdbqt format using ligand preparation module integrated in PyRx. Autodock vina was used as the engine for the molecular docking study, using default parameters where exhaustiveness was 12 and 3 poses were selected for post-docking analysis based on their binding free energy (ΔG), and the docked poses were subjected to analysis to determine how they bind with the active site using Discovery studio visualizer, where 2D and 3D presentation of ligand protein complexes were generated.52,53
Data availability
Upon reasonable request, the data supporting the conclusions of this study can be obtained from the corresponding author, S. S. T.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors reported there is no funding associated with the work featured in this article.References
- S. Zhang, H. Xu, L. Zhang and Y. Qiao, Chin. J. Cancer Res., 2020, 32, 720 CAS.
- N. L. Dashputre, J. D. Kadam, U. D. Laddha, S. B. Patil, P. B. Udavant and S. P. Kakad, Eur. J. Med. Chem. Rep., 2023, 9, 100116 CAS.
- S. Łukasiewicz, M. Czeczelewski, A. Forma, J. Baj, R. Sitarz and A. Stanisławek, Cancers, 2021, 13, 4287 CrossRef PubMed.
- C. K. Chan, G. Aimagambetova, T. Ukybassova, K. Kongrtay and A. Azizan, J. Oncol., 2019, 2019, 3257939 CrossRef PubMed.
- A. Hamdi, E. Said, A. A Farahat, S. AA El-Bialy and M. AM Massoud, Lett. Drug Des. Discovery, 2016, 13, 912–920 CrossRef CAS.
- M. Oshi, S. Gandhi, Y. Tokumaru, L. Yan, A. Yamada, R. Matsuyama, T. Ishikawa, I. Endo and K. Takabe, Am. J. Cancer Res., 2021, 11, 5094 CAS.
- M. T. Pagano, E. Ortona and M. L. Dupuis, Front. Endocrinol., 2020, 11, 506 CrossRef PubMed.
- X. Li, L. Zhao, C. Chen, J. Nie and B. Jiao, Biochim. Biophys. Acta, 2022, 1877, 188789 CAS.
- E. Rubin, K. S. Shan, S. Dalal, D. U. D. Vu, A. M. Milillo-Naraine, D. Guaqueta and A. Ergle, Int. J. Mol. Sci., 2024, 25, 1064 CrossRef CAS PubMed.
- A. Magnifico, L. Albano, S. Campaner, D. Delia, F. Castiglioni, P. Gasparini, G. Sozzi, E. Fontanella, S. Menard and E. Tagliabue, Clin. Cancer Res., 2009, 15, 2010–2021 CrossRef CAS PubMed.
- P. H. Lin and G. Laliotis, J. Clin. Med., 2022, 11, 5891 CrossRef CAS PubMed.
- D. Molehin, F. Rasha, R. L. Rahman and K. Pruitt, Mol. Cell. Biochem., 2021, 476, 2449–2464 CrossRef CAS PubMed.
- P. Ratre, K. Mishra, A. Dubey, A. Vyas, A. Jain and S. Thareja, Anti-Cancer Agents Med. Chem., 2020, 20, 1994–2004 CrossRef CAS PubMed.
- C. P. Amaro, A. Batra and S. Lupichuk, Curr. Oncol., 2021, 28, 2270–2280 CrossRef PubMed.
- A. M. Farghaly, O. M. AboulWafa, H. H. Baghdadi, H. A. Abd El Razik, S. M. Y. Sedra and M. M. Shamaa, Bioorg. Chem., 2021, 115, 105208 CrossRef CAS PubMed.
- M. Ogino, T. Fujii, Y. Nakazawa, T. Higuchi, Y. Koibuchi, T. Oyama, J. Horiguchi and K. Shirabe, In Vivo, 2020, 34, 3483–3487 CrossRef CAS PubMed.
- Y. Wu, Y. Han, Q. Li, P. Zhang, P. Yuan, Y. Luo, Y. Fan, S. Chen, R. Cai and Q. Li, Breast Cancer Res. Treat., 2022, 193, 381–392 CrossRef CAS PubMed.
- J. Y. Jang, D. Kim and N. D. Kim, Int. J. Mol. Sci., 2023, 24, 8457 CrossRef CAS PubMed.
- K. Oukoloff, B. Lucero, K. R. Francisco, K. R. Brunden and C. Ballatore, Eur. J. Med. Chem., 2019, 165, 332–346 CrossRef CAS PubMed.
- C. M. Richardson, D. S. Williamson, M. J. Parratt, J. Borgognoni, A. D. Cansfield, P. Dokurno, G. L. Francis, R. Howes, J. D. Moore and J. B. Murray, Bioorg. Med. Chem. Lett., 2006, 16, 1353–1357 CrossRef CAS PubMed.
- H. S. Mohamed, N. H. Amin, M. T. El-Saadi and H. M. Abdel-Rahman, Bioorg. Chem., 2022, 121, 105687 CrossRef CAS PubMed.
- X.-S. Huo, X.-E. Jian, J. Ou-Yang, L. Chen, F. Yang, D.-X. Lv, W.-W. You, J.-J. Rao and P.-L. Zhao, Eur. J. Med. Chem., 2021, 220, 113449 CrossRef CAS PubMed.
- C. J. A. Ribeiro, J. Kankanala, J. Xie, J. Williams, H. Aihara and Z. Wang, Bioorg. Med. Chem. Lett., 2019, 29, 257–261 CrossRef CAS PubMed.
- S. Wang, L.-J. Zhao, Y.-C. Zheng, D.-D. Shen, E.-F. Miao, X.-P. Qiao, L.-J. Zhao, Y. Liu, R. Huang and B. Yu, Eur. J. Med. Chem., 2017, 125, 940–951 CrossRef CAS PubMed.
- S. Wang, Z.-R. Li, F.-Z. Suo, X.-H. Yuan, B. Yu and H.-M. Liu, Eur. J. Med. Chem., 2019, 167, 388–401 CrossRef CAS PubMed.
- D. H. Dawood, E. S. Nossier, M. M. Ali and A. E. Mahmoud, Bioorg. Chem., 2020, 101, 103916 CrossRef CAS PubMed.
- S. T. J. Bradford, E. Grimley, A. M. Laszczyk, P. H. Lee, S. R. Patel and G. R. Dressler, Cell Chem. Biol., 2022, 29, 412–422 CrossRef CAS PubMed.
- T. Heinrich, H.-P. Buchstaller, B. Cezanne, F. Rohdich, J. Bomke, M. Friese-Hamim, M. Krier, T. Knöchel, D. Musil, B. Leuthner and F. Zenke, Bioorg. Med. Chem. Lett., 2017, 27, 551–556 CrossRef CAS PubMed.
- X.-J. Dai, L.-P. Xue, S.-K. Ji, Y. Zhou, Y. Gao, Y.-C. Zheng, H.-M. Liu and H.-M. Liu, Eur. J. Med. Chem., 2023, 249, 115101 CrossRef CAS PubMed.
- S. M. Gomha, F. M. Abdelrazek and M. M. Abdulla, J. Chem. Res., 2015, 39, 425–429 CrossRef CAS.
- M. J. Akhtar, A. A. Siddiqui, A. A. Khan, Z. Ali, R. P. Dewangan, S. Pasha and M. S. Yar, Eur. J. Med. Chem., 2017, 126, 853–869 CrossRef CAS PubMed.
- H. T. Abdel-Mohsen, F. A. Ragab, M. M. Ramla and H. I. El Diwani, Eur. J. Med. Chem., 2010, 45, 2336–2344 CrossRef CAS PubMed.
- A. Faraji, R. Motahari, Z. Hasanvand, T. O. Bakhshaiesh, M. Toolabi, S. Moghimi, L. Firoozpour, M. A. Boshagh, R. Rahmani and S. H. Ketabforoosh, Bioorg. Chem., 2021, 108, 104553 CrossRef CAS PubMed.
- M. N. Aboul-Enein, A. M. A. E. S. El-Azzouny, F. A. F. Ragab and M. F. Hamissa, Arch. Pharm., 2017, 350, 1600377 CrossRef PubMed.
- E. Jameel, P. Meena, M. Maqbool, J. Kumar, W. Ahmed, S. Mumtazuddin, M. Tiwari, N. Hoda and B. Jayaram, Eur. J. Med. Chem., 2017, 136, 36–51 CrossRef CAS PubMed.
- W. A. Ewes, S. S. Tawfik, A. M. Almatary, M. Ahmad Bhat, H. W. El-Shafey, A. A. Mohamed, A. Haikal, M. A. El-Magd, A. A. Elgazar and M. Balaha, Molecules, 2024, 29, 3186 CrossRef CAS PubMed.
- M. A. Sabry, M. A. Ghaly, A. R. Maarouf and H. I. El-Subbagh, Eur. J. Med. Chem., 2022, 241, 114661 CrossRef CAS PubMed.
- D. I. A. Othman, A. Hamdi, S. S. Tawfik, A. A. Elgazar and A. S. Mostafa, J. Enzyme Inhib. Med. Chem., 2023, 38, 2166037 CrossRef PubMed.
- M. T. E. Maghraby, T. Mazyad Almutairi, S. Bräse, O. I. A. Salem, B. G. M. Youssif and M. M. Sheha, Molecules, 2023, 28, 7092 CrossRef CAS PubMed.
- K. Pandey, K. Bharat Lokhande, A. Saha, A. Goja, K. V. Swamy and S. Nagar, Curr. Comput.-Aided Drug Des., 2023, 19, 243–257 CrossRef CAS PubMed.
- M. M. Al-Sanea, A. Hamdi, A. A. Mohamed, H. W. El-Shafey, M. Moustafa, A. A. Elgazar, W. M. Eldehna, H. Ur Rahman, D. G. Parambi and R. M. Elbargisy, J. Enzyme Inhib. Med. Chem., 2023, 38, 2166036 CrossRef PubMed.
- N. F. El Hamaky, A. Hamdi, W. A. Bayoumi, A. A. Elgazar and M. N. Nasr, Bioorg. Chem., 2024, 148, 107437 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- S. S. Tawfik, A. Hamdi, A. R. Ali, A. A. Elgazar, H. W. El-Shafey, A. S. El-Azab, A. H. Bakheit, M. M. Hefnawy, H. A. Ghabbour and A.-M. Alaa, RSC Adv., 2024, 14, 26325–26339 RSC.
- F. Tavakolinia, T. Baghipour, Z. Hossaini, D. Zareyee, M. A. Khalilzadeh and M. Rajabi, Nucleic Acid Ther., 2012, 22, 265–270 CrossRef CAS PubMed.
- M. Kusaczuk, R. Krętowski, M. Naumowicz, A. Stypułkowska and M. Cechowska-Pasko, Int. J. Nanomed., 2018, 2279–2294 CrossRef CAS PubMed.
- D. I. Othman, A. Hamdi, W. M. Elhusseiny, A. S. El-Azab, A. H. Bakheit, M. Hefnawy and A.-M. Alaa, Saudi Pharm. J., 2023, 31, 101803 CrossRef CAS PubMed.
- O. S. Nnyigide, T. O. Nnyigide, S.-G. Lee and K. J. J. o. C. I. Hyun, Modeling, 2022, 62, 4232–4246 CAS.
- A. A. Elgazar, R. A. El-Domany, W. M. Eldehna and F. A. Badria, ACS Omega, 2023, 8, 39490–39510 CrossRef CAS PubMed.
- A. Hamdi, M. Yaseen, W. A. Ewes, M. A. Bhat, N. I. Ziedan, H. W. El-Shafey, A. A. B. Mohamed, M. R. Elnagar, A. Haikal, D. I. A. Othman, A. A. Elgazar, A. H. A. Abusabaa, K. S. Abdelrahman, O. M. Soltan and M. M. Elbadawi, J. Enzyme Inhib. Med. Chem., 2023, 38, 2231170 CrossRef PubMed.
- A. A. Elgazar, R. A. El-Domany, W. M. Eldehna and F. A. Badria, RSC Adv., 2023, 13, 25616–25634 RSC.
- F. F. El-Senduny, A. A. Elgazar, H. A. Alwasify, A. Abed, M. Foda, S. Abouzeid, L. Lewerenz, D. Selmar and F. Badria, Planta Med., 2023, 89, 964–978 CrossRef CAS PubMed.
- M. M. Al-Sanea, G. Chilingaryan, N. Abelyan, M. Mamikonyan, H. Gasparyan, S. Hovhannisyan, A. Hamdi, A. R. Ali, S. Selim and A. A. Mohamed, PLoS One, 2022, 17, e0272065 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06704e |
| This journal is © The Royal Society of Chemistry 2024 |