
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient synthesis of polycarbonate ether polyol via copolymerization of CO2 and 1,2-butylene oxide over a layered Zn–Co double metal cyanide catalyst†
Chen Liuab,
Yan Caoa,
Xianqiang Zenga,
Zheng Zhenga,
Ziqiang Hana,
Peng Hea and
Liguo Wang *ac
*ac
aCAS Key Laboratory of Green Process and Engineering, National Engineering Research Center of Green Recycling for Strategic Metal Resources, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: lgwang@ipe.ac.cn
bSchool of Chemistry and Chemical Engineering, University of Jinan, Jinan 250000, China
cSchool of Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 29th October 2024
Abstract
Polycarbonate ether polyol synthesized by the copolymerization of carbon dioxide (CO2) and epoxides is a promising technology for chemically fixing CO2 and manufacturing degradable polymeric materials. However, research on the copolymerization of CO2 and 1,2-butylene oxide (BO) to produce polycarbonate ether polyol is relatively scarce. Herein, we employed layered Zn–Co double metal cyanide (L-DMC) as a catalyst for the copolymerization of CO2 and BO to obtain polycarbonate ether polyol. Under optimized conditions, the L-DMC-mediated copolymerization of CO2 and BO displays excellent activity (catalytic productivity up to 1500 g polymer per g catalyst) and a high CO2 incorporation fraction in polycarbonate ether polyol rate of 35.5%. In situ FTIR spectroscopy experiments showed a short induction period (10 min) in polymerization and competition between the synthesized polycarbonate ether polyol and butylene carbonate (BC). The properties of easy preparation, stability, and excellent catalytic performance of L-DMC suggest that it may be a promising candidate for large-scale production of CO2–BO-based polymers.
1. Introduction
The conversion of carbon dioxide (CO2) into polycarbonate ether polyols is an effective strategy to achieve high value utilization of CO2 and manufacture of degradable materials.1–7 Polycarbonate ether polyols are ideal soft segments for the preparation of high-performance polyurethane materials due to their unique degradability and inoxidizability.8–10 Currently, the synthesis of polycarbonate ether polyols is commonly achieved through the copolymerization of CO2 and propylene oxide (PO).11–15 However, the deficiencies of CO2–PO based polymers in terms of hydrophobicity and heat resistance prevent them from being used in hydrophobic coatings, waterproof film materials and high temperature-resistance adhesives.16–18 Compared with PO-based polymers, BO-based polymers synthesized from 1,2-epoxide butene (BO) with long alkyl side chains have excellent hydrophobicity and heat resistance and have attracted extensive attention. Such polyethers synthesized by ring-opening polymerization of BO have been successfully used in oil-soluble and hydrophobic materials.19,20 To the best of our knowledge, research on the synthesis of CO2–BO-based polymers has been relatively limited to date, resulting in the slow development of such materials.21 Therefore, synthesis of polycarbonate ether polyol by the development of efficient copolymerization methods for CO2 and BO is highly desirable.21Due to the properties of simple preparation and stability, double metal cyanide (DMC) catalysts can be used in the commercial production of polycarbonate ether polyols.22 Since 1966, intense research work have been performed to improve the catalytic efficiency of DMC catalysts.23 Kim et al.24 synthesized Zn–Co DMC catalysts using different dicarbonyl complexing agents which gave polypropylene carbonate polyols with low molecular weights (<4000 g mol−1) and a high CO2 incorporation fraction in polycarbonate ether rate of 46.7%. However, the productivity of the catalysts was lower than 160 g polymer per g DMC. In addition to altering the complexing agent of DMC, the development of novel DMC structures is an effective way to improve its performance. Zhang et al.25 reported a nano-lamellar structure DMC catalyst for the copolymerization reaction between CO2 with PO to obtain polycarbonates with number average molecular weights of >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, but the conversion of PO was only 27.2% in 6 h. Trees et al.26 had successfully synthesized a layered Zn2[Co(CN)6](CH3COO) double metal cyanide catalyst, which exhibited more excellent catalytic performance than conventional DMC for the ring-opening polymerization of epoxides. Experimental characterization results show that the two-dimensional layered structure of L-DMC with high specific surface area can expose more available active sites on the catalyst surface, which is essential for its high catalytic activity. Inspired by this work, we propose that the use of DMC catalysts with a layered structure for the copolymerization of CO2 and BO will not only improve the polymerization efficiency but also increase the CO2 incorporation fraction in the copolymer.
000 g mol−1, but the conversion of PO was only 27.2% in 6 h. Trees et al.26 had successfully synthesized a layered Zn2[Co(CN)6](CH3COO) double metal cyanide catalyst, which exhibited more excellent catalytic performance than conventional DMC for the ring-opening polymerization of epoxides. Experimental characterization results show that the two-dimensional layered structure of L-DMC with high specific surface area can expose more available active sites on the catalyst surface, which is essential for its high catalytic activity. Inspired by this work, we propose that the use of DMC catalysts with a layered structure for the copolymerization of CO2 and BO will not only improve the polymerization efficiency but also increase the CO2 incorporation fraction in the copolymer.
Herein, we synthesize a highly active L-DMC catalyst to achieve copolymerization of CO2 and BO to prepare polycarbonate ether polyol (Fig. 1 and Scheme 1). The L-DMC mediated copolymerization can provide a catalytic productivity of 1553 g polymer per g catalyst and a CO2 incorporation fraction in polycarbonate ether polyol rate of 35.5% at 110 °C and 3 MPa CO2. The high reactivity of L-DMC might be ascribed to its two-dimensional layered structure and high specific surface area, which was confirmed by BET, SEM, TEM, XRD and XPS studies. Moreover, the structure of the polycarbonate ether polyol was determined by FTIR, 1H NMR, 13C NMR and DOSY NMR characterization techniques. In situ FTIR spectroscopy monitored the copolymerization process of CO2 and BO catalyzed, and the relationship between polycarbonate ether polyol and butylene carbonate (BC) during the formation process was revealed. The proposed approach successfully synthesized degradable CO2–BO-based polymer polyols creating better opportunities for preparing high-performance polyurethane materials.
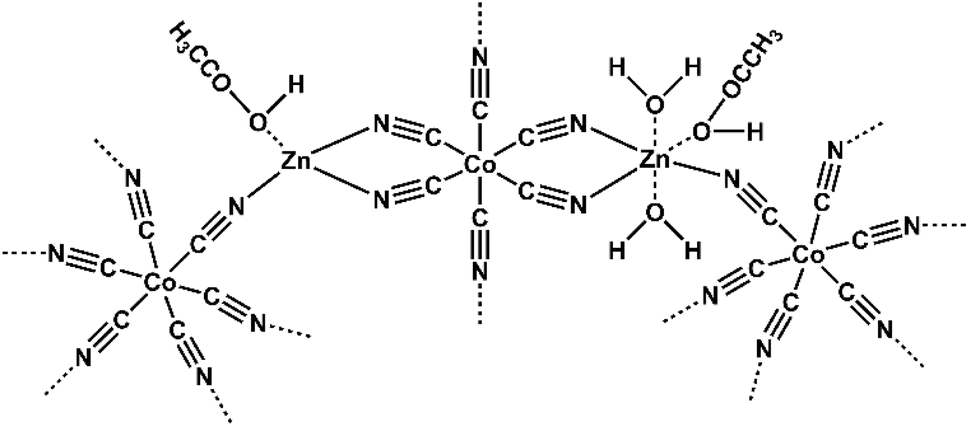 | ||
| Fig. 1 Schematic structure of L-DMC catalyst with acetic acid as ligand. | ||
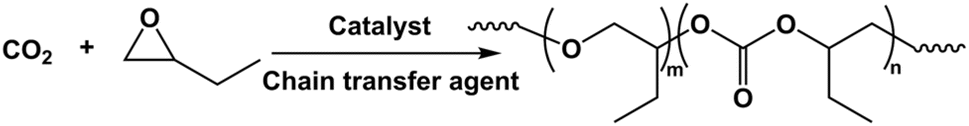 | ||
| Scheme 1 Copolymerization of CO2 and BO to synthesize polycarbonate ether polyol by L-DMC catalyst. | ||
2. Experimental part
2.1 Materials
1,2-Butylene oxide (BO), butane-1,4-diol (1,4-BDO), potassium hexacyanocobaltate(III) (K3[Co(CN)6]), glacial acetic acid dihydrate (Zn(CH3COO)2·2H2O) and tert-butanol (t-BuOH) were purchased from Aladdin Industrial Corporation. Acetic acid (CH3COOH) and zinc chloride (ZnCl2) were provided by Sinopharm Chemical Reagent Co., Ltd. BO was purified through refluxing calcium hydride for 48 h and subsequently kept in a nitrogen environment until needed. 1,4-BDO was dried on a 4 Å molecular sieve for 48 h and also stored under nitrogen conditions for later use.2.2 Preparation of L-DMC catalyst
The L-DMC catalyst was prepared according to the previous literature.25 In order to avoid the influence of K+ on catalyst activity, the required K3[Co(CN)6] in L-DMC synthesis was replaced with H3[Co(CN)6].27 CH3COOH (4.8 g, 0.08 mol) and Zn(CH3COO)2·2H2O (660.0 mg, 3.0 mmol) were dissolved in distilled water (15.0 mL) to prepare solution A. H3[Co(CN)6] (496.0 mg, 1.5 mmol) was dissolved in distilled water (10.0 mL) to prepare solution B. Solution A was placed in an oil bath and heated to 90 °C with stirring, then solution B was added slowly and dropwise. The reaction solution took out from the oil bath right after the drop was finished. The solids that were suspended were gathered through centrifugation and rinsed three times with distilled water, and the resulting precipitate was dried under vacuum at 80 °C for 12 h.2.3 Preparation of DMC catalyst
Preparation of DMC catalyst using methods reported in the literature.28 Solution A was prepared by dissolving H3[Co(CN)6] (1.5 g, 4.8 mmol) in distilled water (20.0 mL). Solution B was prepared by dissolving ZnCl2 (20.0 g, 0.15 mol) in a mixture of deionized water (50.0 mL) and tert-butanol (30.0 mL). Solution B was placed in an oil bath and heated to 50 °C with intense stirring, then solution A was added dropwise and continue stirring for 2 h. The white sediment was recovered by centrifugation and subsequently washed three times using a combined solution of deionized water and tert-butanol (the amount of tert-butanol in the washing solution is progressively increased), and the collected precipitate was dried under vacuum at 60 °C for 24 h.2.4 Copolymerization of CO2 and BO
The copolymerization of BO and CO2 was conducted out in a pre-dehydrated 50 mL stainless steel autoclave reactor system. The catalyst was treated at 80 °C in a vacuum for 12 hours and was subsequently stored in a nitrogen atmosphere for further use. Taking the data from entry 2 in Table 1 as an example, the catalyst (8.0 mg), 1,4-BDO (63.0 mg, 0.7 mmol) and BO (8.0 g, 0.1 mol) were added into the 50 mL autoclave equipped with a magnetic stir. The autoclave was charged with CO2 to reach specified pressure and heated to 110 °C. After cooling down to room temperature and releasing the pressure to obtain the crude product. The crude product was dissolved by methylene chloride, and the catalyst and solvent were removed by filtration and vacuum distillation, respectively. The pure product was obtained by precipitation from methanol and dried to constant weight under vacuum condition at 80 °C.| Entry | Catalyst (mg) | Temperature (°C) | Pressure (MPa) | fCO2d (%) | WBCd (wt%) | Mne (g mol−1) | Đe | Productivityf (g polymer per g catalyst) |
|---|---|---|---|---|---|---|---|---|
| a All copolymerization reactions were carried out in 10 mL BO.b The copolymerization of CO2 and BO was catalyzed by L-DMC.c The copolymerization of CO2 and BO was catalyzed by DMC.d Determined by 1H NMR and formula (1) and (2).e Determined by GPC.f Determined according to Wproduct/Wcat. | ||||||||
| 1b | 7.0 | 110 | 3 | — | — | — | — | — |
| 2 | 8.0 | 110 | 3 | 30.3 | 8.2 | 10700 |
2.8 | 1239 |
| 3 | 10.0 | 110 | 3 | 35.0 | 22.1 | 6800 | 6.6 | 510 |
| 4 | 20.0 | 110 | 3 | 24.7 | 50.3 | — | — | — |
| 5 | 8.0 | 100 | 3 | — | — | — | — | — |
| 6 | 8.0 | 120 | 3 | 23.8 | 12.3 | 8000 | 3.0 | 925 |
| 7 | 8.0 | 140 | 3 | 4.0 | 1.0 | 1400 | 1.8 | — |
| 8 | 8.0 | 110 | 1 | 18.0 | 4.9 | 6300 | 3.1 | 1008 |
| 9 | 8.0 | 110 | 2 | 29.6 | 7.9 | 9700 | 3.5 | 1197 |
| 10 | 8.0 | 110 | 4 | 32.3 | 6.6 | 11100 |
3.9 | 1401 |
| 11 | 8.0 | 110 | 5 | 35.5 | 10.2 | 12600 |
3.2 | 1500 |
| 12c | 8.0 | 110 | 3 | 13.3 | 1.6 | 5000 | 2.5 | 861 |
The CO2 incorporation fraction (fCO2) in polycarbonate ether polyol and the weight ratio of cyclic carbonate (WBC) were determined by integrating the 1H NMR signals at 4.7–4.9 ppm and 4.0–4.3 ppm (representing carbonate units), 3.2–3.7 ppm (indicating ether linkage chain) and 4.0 ppm, 4.5 ppm and 4.6 ppm (corresponding to 1,2-butylene carbonate), in accordance with the formulas (1) and (2) reported by Zhang et al.25,27
| fCO2% = (A4.7–4.9 + A4.0–4.3 − 2A4.5)/[2(A4.7–4.9 + A4.0–4.3 − 2A4.5) + A3.2–3.8] | (1) |
| WBC% = (3A4.5)/(A4.7–4.9 + A4.5 + A4.0–4.3 + A3.2–3.8) | (2) |
3. Results and discussion
3.1 Characterization of L-DMC catalysts and DMC catalysts
Fig. S1† illustrated the powder XRD patterns of DMC and L-DMC catalysts. DMC exhibited broader diffraction peaks from 15° to 21°, whereas L-DMC displayed sharp diffraction peaks from 15° to 35°. This suggests that L-DMC possesses a more integrity crystal structure and exhibits higher crystallinity. Moreover, the layered phases of L-DMC were clearly characterized by reflections at 2θ = 16.9°, 18.4°, 23.6°, 25.6° and 34.3°, which were consistent with the results of previous work.26,29The surface compositions of DMC and L-DMC were analyzed using XPS. The results indicates that Zn, Co, C, N, and O were common components of both catalysts (Fig. S2†). According to previous reports,30 the main role of Co in Zn–Co DMC catalysts is to act as a stabilizer for the cyanide-bridged complexes to ensure that the catalysts remain heterogeneous, while Zn is the main key catalytic active site. Fig. 2 presented the expanded Zn 2p XPS spectra for the DMC and L-DMC catalysts. The binding energies of Zn atoms in both DMC and L-DMC catalyst systems were significantly reduced compared to the binding energy of Zn atoms in ZnCl2 (Ea = 1023.7 eV), which suggests a change in the chemical coordination environment around the active site of Zn(II), thereby influencing its electronic structure properties.31 The DMC exhibited two singlet peaks of the Zn 2p spin–orbits with binding energies at 1022.62/1045.60 eV, and similarly the L-DMC also exhibited two singlet peaks of the Zn 2p spin–orbits with binding energies at 1022.37/1045.36 eV. This result suggests that the coordination between Zn atoms and different complexing agents can affect the binding energy of Zn 2p. CH3COOH as a complexing agent tends to promote the transfer of electrons to the Zn center, which is contributing to the increase of catalytic performance.24
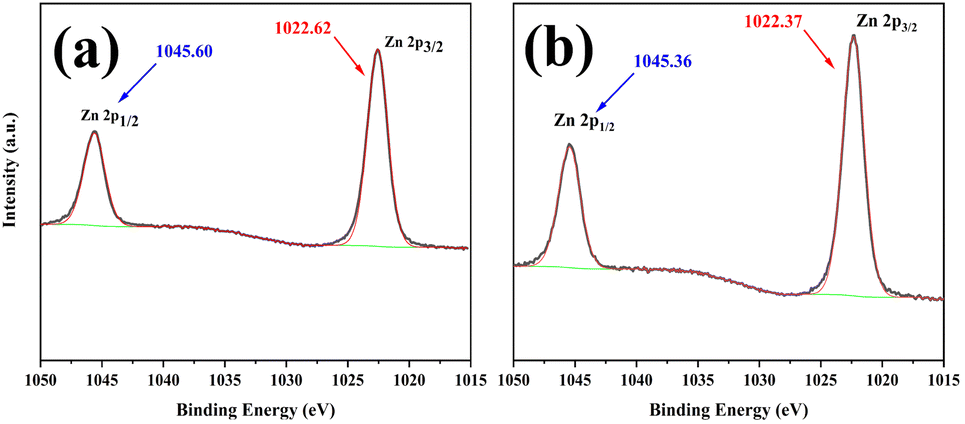 | ||
| Fig. 2 Zn 2p XPS spectra of (a) DMC, (b) L-DMC. | ||
Transmission electron microscopy (TEM) was used to characterize the structural morphology of DMC and L-DMC. As illustrated in Fig. 3(a) and (b), the particle shape of DMC predominantly exhibits a hexagonal configuration with a variation in particle size. In contrast, the particle size of L-DMC, as depicted in Fig. 3(c) and (d), is significantly smaller and exhibits a lamellar structure. These lamellar particles are primarily arranged in a random overlapping manner, leading to a layered structure. Fig. S3† presents the characterization results of the surface morphology as observed through SEM images of the two catalyst samples. As illustrated Fig. S3(a) and (b),† the DMC particles exhibited uneven particle sizes, irregular shapes, and low dispersion, with many particles adhering to one another and not easily separable. In contrast, the surface particles of L-DMC demonstrate greater dispersion, as displayed in Fig. S3(c) and (d).† Furthermore, it is observed that the L-DMC particles are aggregated to form a layered structure, as highlighted in the enlarged image of Fig. S3(d),† which aligns with the findings from TEM characterization.
 | ||
| Fig. 3 TEM images (a and b) DMC, (c and d) L-DMC. | ||
The N2 adsorption isotherm curves for DMC and L-DMC were presented in Fig. S4.† The N2 adsorption isotherm of the DMC catalyst exhibits a typical type V isotherm with a type H3 hysteresis loop,32 indicating a mesoporous structure. On the contrary, the adsorption isotherm of L-DMC catalyst only shows a small hysteresis loop.33 The characteristics of the pore structure properties for both catalysts are presented in Table S1,† where the specific surface area (SBET = 668 m2 g−1) and external specific surface area (Sext = 32 m2 g−1) of L-DMC are significantly higher than those of DMC (SBET = 26 m2 g−1 and Sext = 21 m2 g−1). It has been reported that specific surface area is a crucial factor influencing the activity of the L-DMC catalyst, as the larger specific surface area typically increases the number of active sites exposed on the catalyst surface, thereby improving catalytic activity.34,35 Furthermore, based on the SEM analysis of the samples, it can be inferred that the high specific surface area of L-DMC may be attributed to its smaller particle size and highly dispersed particles without aggregation.
3.2 Influence of chain transfer agent type on the copolymerization of CO2 and BO
Utilizing a chain transfer agent is essential for the synthesis of polycarbonate ether polyol on the copolymerization of CO2 and BO. Research shows that low number average molecular weight PPGs (200–600 g mol−1) alcohols, non-complexing alcohols and carboxylic as chain transfer agents have different effects on the polymerization.13,36–38 This study investigated the effects of polypropylene glycol (PPG-400), butane-1,4-diol (1,4-BDO), and sebacic acid (SA) chain transfer agents on the copolymerization of CO2 and BO. When SA as a chain transfer agent added to the polymerization system (Table S2 (entry 3)†), targeted polymer was not obtained, which is attributed to the deactivation of the catalyst resulted from the strong coordination of SA with the Zn sites of L-DMC.39 Furthermore, as shown in Table S2 (entries 1 and 2),† the employment of PPG-400 as the chain transfer agent, the fCO2 in the product was only 10.9%, which is significantly lower than the desired (fCO2 > 30%). In contrast, the use of 1,4-BDO as a chain transfer agent in the copolymerization of CO2 and BO demonstrated superior performance, achieving an fCO2 of 30.3%, WBC less than 10%, and a catalytic productivity of 1239 g polymer per g L-DMC. This suggested that the coordination of 1,4-BDO activates L-DMC, followed by initiation and propagation with BO. Therefore, 1,4-BDO was selected as the chain transfer agent for further investigations into the copolymerization of CO2 and BO catalyzed by L-DMC. In addition. Table S2 (entry 4)† showed that the copolymerization of CO2 and BO catalyzed by L-DMC can synthesize polycarbonate ether polyol with higher number average molecular weight (Mn = 18300 g mol−1) without the addition of chain transfer agent, which may be the result of adventitious traces of water (such as water present in the monomer) as the chain transfer agent.
3.3 Effect of reaction parameters on the copolymerization of CO2 and BO
The effects of reaction parameters on the copolymerization of CO2 and BO catalyzed by L-DMC are summarized from entries 1–11 in Table 1. Firstly, the effect of the dosage of L-DMC on polymerization was investigated. When the dosage of L-DMC was 7.0 mg (entry 1) under 110 °C and 3 MPa CO2 conditions, no polymer was obtained. It is attributed to the total inactivation of the effective Zn active site of L-DMC after coordination with the chain transfer agent. When the dosage of L-DMC was increased to 8.0 mg (entry 2), copolymerization between CO2 and BO was achieved, yielding 30.3% fCO2 and a productivity of 1239 g polymer per g L-DMC. As the dosage of L-DMC was further increased to 10.0 mg (entry 3), fCO2 rose to 35.0%; however, the weight ratio of BC increased significantly to 22.1% and the dispersity (Đ) value rose to 6.6. This is due to the fact that the number of available zinc active sites increases with the amount of L-DMC, which enhances the monomer conversion. It not only promotes the copolymerization of CO2 with BO, but also facilitates the cycloaddition reaction of BO with CO2. Furthermore, the increased catalyst dosage could lead to polycarbonate ether polyol with high Đ, because the rise in the number of active centers contributes to a higher quantity of initiated chains.40 When the dosage of L-DMC was increased to 20.0 mg (entry 4), WBC further escalated to 50.3%, while fCO2 decreased to 24.7%. This result may be due to the fact that excessive use of the L-DMC accelerated the activation of CO2 in the reaction, leading to the inhibition of copolymerization by a huge amount of activated CO2. While excessive dosage of catalyst increased the number of Zn active sites, improved the conversion of monomer and promoted the formation of BC through the cycloaddition reaction of BO and CO2.The effects of temperature on the copolymerization of CO2 and BO catalyzed by L-DMC were summarized in Table 1 (entries 2 and 5–7). The copolymerization does not take place at 100 °C (entry 5). Data presented in Table 1 (entry 2) indicated that the lowest reaction temperature at which the copolymerization of BO and CO2 taken place catalyzed by L-DMC was 110 °C. The result indicates that the ligands site composed of 1,4-BDO and active sites are more easily replaced by BO to activate L-DMC at higher temperatures. As the reaction temperature increased to 120 °C, the fCO2 in polycarbonate ether polyol decreased from 30.3% to 23.8% while the content of the WBC rose from 8.2% to 12.3% (entry 6). Because higher temperatures promoted the cycloaddition reaction of CO2 and BO to replace part of the copolymerization of CO2 and BO, which is consistent with previous research.41,42 When the reaction temperature was raised to 140 °C (entry 7), fCO2 quickly decreased to 4.0% with only 1.0% of the byproduct BC formed, indicating that the majority of BO underwent homopolymerization into polyether products in a short time. Therefore, temperatures exceeding 120 °C are not conducive to the copolymerization of CO2 and BO. This finding is consistent with the result of previous research, which suggests that lower reaction temperatures are beneficial for the copolymerization catalyzed by L-DMC.43
Furthermore, the influence of CO2 pressure factors on the reaction was investigated. As shown in Table 1 (entries 2 and 8–11), the copolymerization of CO2 and BO catalyzed by L-DMC under different CO2 pressures can synthesize polycarbonate ether polyol with Mn in the range of 6300 g mol−1 to 12600 g mol−1 and Đ in the range of 3.1–3.9. The fCO2 in polycarbonate ether polyol increases from 18.0% to 35.5% with the CO2 pressure rising from 1 to 5 MPa. According to Fig. S5,† the glass transition temperatures (Tg) of the polycarbonate ether polyol are −26.1 °C (fCO2: 18.0%) and −13.5 °C (fCO2: 30.3%), suggesting that Tg increases with the addition of carbonate units. The low glass transition temperature of polycarbonate ether polyol highlights its potential as suitable parts for the synthesis of polyurethane soft segments. This finding indicates that increase of CO2 pressure is an effective way to increase the CO2 incorporation fraction in polycarbonate ether polyols44 In addition, the content of BC also increased with the increase of pressure, indicating that the synthesis of BC was facilitated by the increase in pressure.
Table 1 (entries 2 and 12) presented the catalytic effects of L-DMC and DMC catalysts on the copolymerization of CO2 and BO. Notably, the fCO2 in the polycarbonate ether polyol increased from 13.3% (entry 2) for the DMC catalyst to 30.3% (entry 12) for the synthesized L-DMC catalyst under identical reaction conditions. Furthermore, the catalytic efficiency comparison revealed that L-DMC achieved 1239 g polymer per g L-DMC, surpassing the 861 g polymer per g DMC, thereby indicating the superior catalytic activity of L-DMC. It has been reported that reactions catalyzed by DMC typically occur on the catalyst's surface.34 Compared with DMC catalyst, L-DMC catalyst has a unique layered structure and smaller particle size, while the particles are also better dispersed. These characteristics are beneficial to the increase of the specific surface area of the catalyst, resulting in increased exposure to the available Zn active sites. The characterization results of BET (L-DMC: SBET = 668 m2 g−1, Sext = 32 m2 g−1, DMC: (SBET = 26 m2 g−1, Sext = 21 m2 g−1) also prove the possibility of the above analysis. Therefore, L-DMC showed excellent catalytic activity than DMC on the copolymerization of CO2 and BO.30
3.4 In situ FTIR spectroscopy for the copolymerization of CO2 and BO catalyzed by L-DMC
In situ FTIR three-dimensional stack plot of L-DMC-catalyzed copolymerization of CO2 and BO is presented in Fig. 4(a). The v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and v(C–O–C) characteristic absorption peaks at 1752 cm−1 and 1101 cm−1 were attributed to the carbonate units and the ether linkages of the polycarbonate ether polyol, respectively. Additionally, the characteristic absorption peak at 1819 cm−1 was assigned to cyclic carbonate. In Fig. 4(b), the existence of an induction period for polymerization can be clearly observed, in which no change in the characteristic absorption peaks was observed within 10 min (stage 1) of the polymerization, indicating that the polymerization did not start during this period. This phenomenon indicates that the L-DMC catalyst complys with the coordination polymerization mechanism of conventional DMC catalysts. As the reaction time increases from 10 min to 130 min (stage 2), the characteristic absorption peaks (C–O–C stretching vibration and CO stretching vibration) of the ether linkages and carbonate units at 1101 cm−1 and 1752 cm−1 were gradually intensified. Meanwhile, the characteristic absorption peak (CO stretching vibration) of cyclic carbonate was also gradually intensified. This result suggests that BO and CO2 were rapidly converted to polycarbonate ether polyol and BC after ending the induction period. Notably, the growth rate of the characteristic absorption peaks of ether linkages and cyclic carbonate at the very beginning of polymerization is significantly faster than that of the characteristic absorption peak of carbonate units, indicating that the main formation of ether linkage chain segments and BC. As the reaction proceeds the reaction rate of BC is gradually decreased and the rate of synthesis of carbonate units and ether linkage in the polymer is nearly equal. The production of BC ceased in the reaction after 130 minutes (stage 3), and the synthesis rate of carbonate units and ether linkages in the polymer decreased significantly until BO was nearly fully converted. These findings indicate that the competitive formation between the target product polycarbonate ether polyol and the by-product BC during the copolymerization of CO2 and BO using 1,4-BDO as chain transfer agent and L-DMC as catalyst.
O) and v(C–O–C) characteristic absorption peaks at 1752 cm−1 and 1101 cm−1 were attributed to the carbonate units and the ether linkages of the polycarbonate ether polyol, respectively. Additionally, the characteristic absorption peak at 1819 cm−1 was assigned to cyclic carbonate. In Fig. 4(b), the existence of an induction period for polymerization can be clearly observed, in which no change in the characteristic absorption peaks was observed within 10 min (stage 1) of the polymerization, indicating that the polymerization did not start during this period. This phenomenon indicates that the L-DMC catalyst complys with the coordination polymerization mechanism of conventional DMC catalysts. As the reaction time increases from 10 min to 130 min (stage 2), the characteristic absorption peaks (C–O–C stretching vibration and CO stretching vibration) of the ether linkages and carbonate units at 1101 cm−1 and 1752 cm−1 were gradually intensified. Meanwhile, the characteristic absorption peak (CO stretching vibration) of cyclic carbonate was also gradually intensified. This result suggests that BO and CO2 were rapidly converted to polycarbonate ether polyol and BC after ending the induction period. Notably, the growth rate of the characteristic absorption peaks of ether linkages and cyclic carbonate at the very beginning of polymerization is significantly faster than that of the characteristic absorption peak of carbonate units, indicating that the main formation of ether linkage chain segments and BC. As the reaction proceeds the reaction rate of BC is gradually decreased and the rate of synthesis of carbonate units and ether linkage in the polymer is nearly equal. The production of BC ceased in the reaction after 130 minutes (stage 3), and the synthesis rate of carbonate units and ether linkages in the polymer decreased significantly until BO was nearly fully converted. These findings indicate that the competitive formation between the target product polycarbonate ether polyol and the by-product BC during the copolymerization of CO2 and BO using 1,4-BDO as chain transfer agent and L-DMC as catalyst.
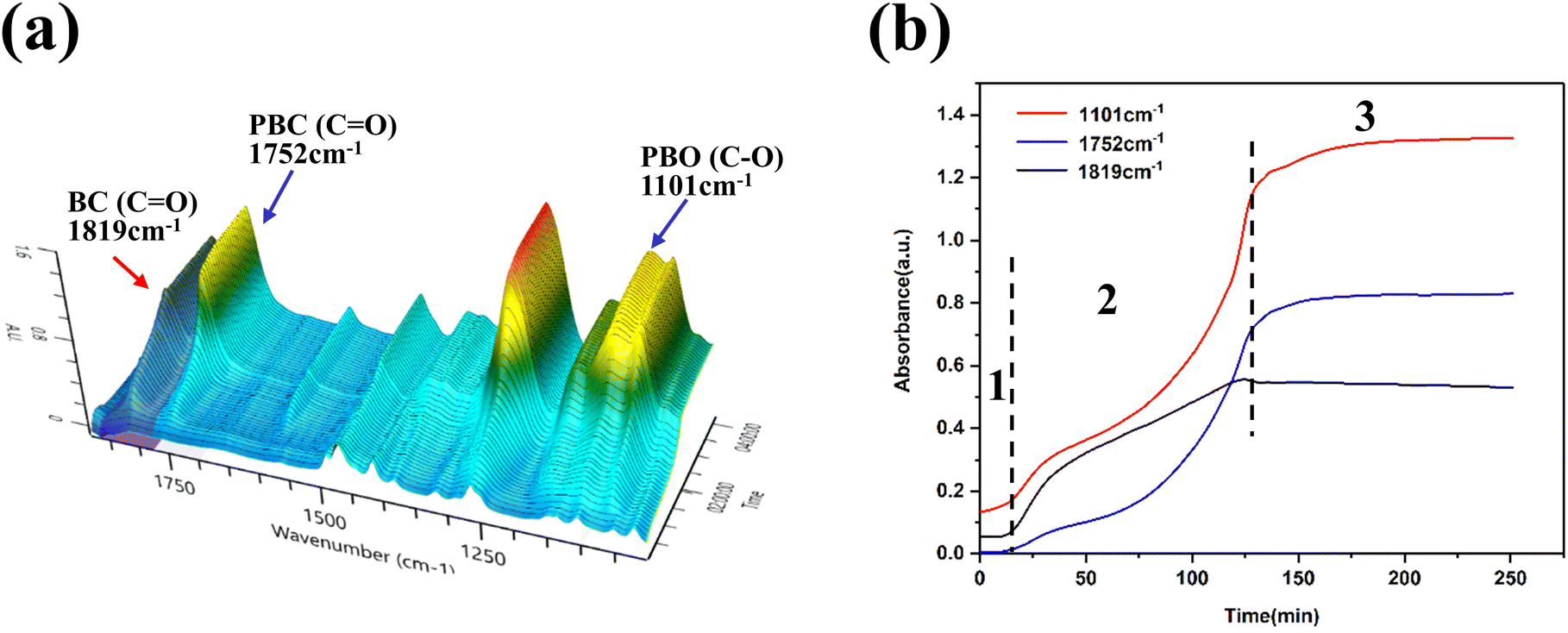 | ||
| Fig. 4 (a) Three-dimensional stack plots of in situ FTIR spectroscopy. (b) Reaction profile monitored by in situ FTIR. Reaction was carried out at 110 °C, 1.5 MPa CO2 pressure. | ||
3.5 Plausible reaction mechanism
Given that the L-DMC catalyst demonstrates high catalytic activity in the copolymerization of CO2 and BO, it is essential to propose a reaction mechanism that elucidates the interactions of its active sites with CO2 and BO. The potential reaction mechanism has been formulated based on the characterization analysis detailed above, as well as previously reported studies on the polymerization processes involving analogous DMC catalysts.24,40 As illustrated in Scheme 2, the process begins with the oxygen atom (O) of the BO monomer and the hydroxyl group of the chain transfer agent replacing the complexing agent of L-DMC, thereby coordinating with the Zn active sites. Subsequently, the hydroxyl group on the chain transfer agent nucleophilically attacks the BO coordinated with the Zn active sites, leading to the opening of its ring and the generation of propagating species. The process of propagation is carried out by the nucleophilic attack of the growing polymer chain on activated BO, while the absorbed CO2 is activated by the Zn active sites and incorporated into the chain until the monomer is fully consumed. The byproduct BC may be generated by the cycloaddition reaction of CO2 and BO.23,43,45 In this context, the acetic acid complexing agent in L-DMC not only interacts with the Zn atoms to influence the activity of catalyst but may also facilitates the absorption of CO2 and enhances the CO2 incorporation fraction in polycarbonate ether polyol.46 This proposed mechanism aligns with the previously mentioned competitive relationship between BC and polycarbonate ether polyol.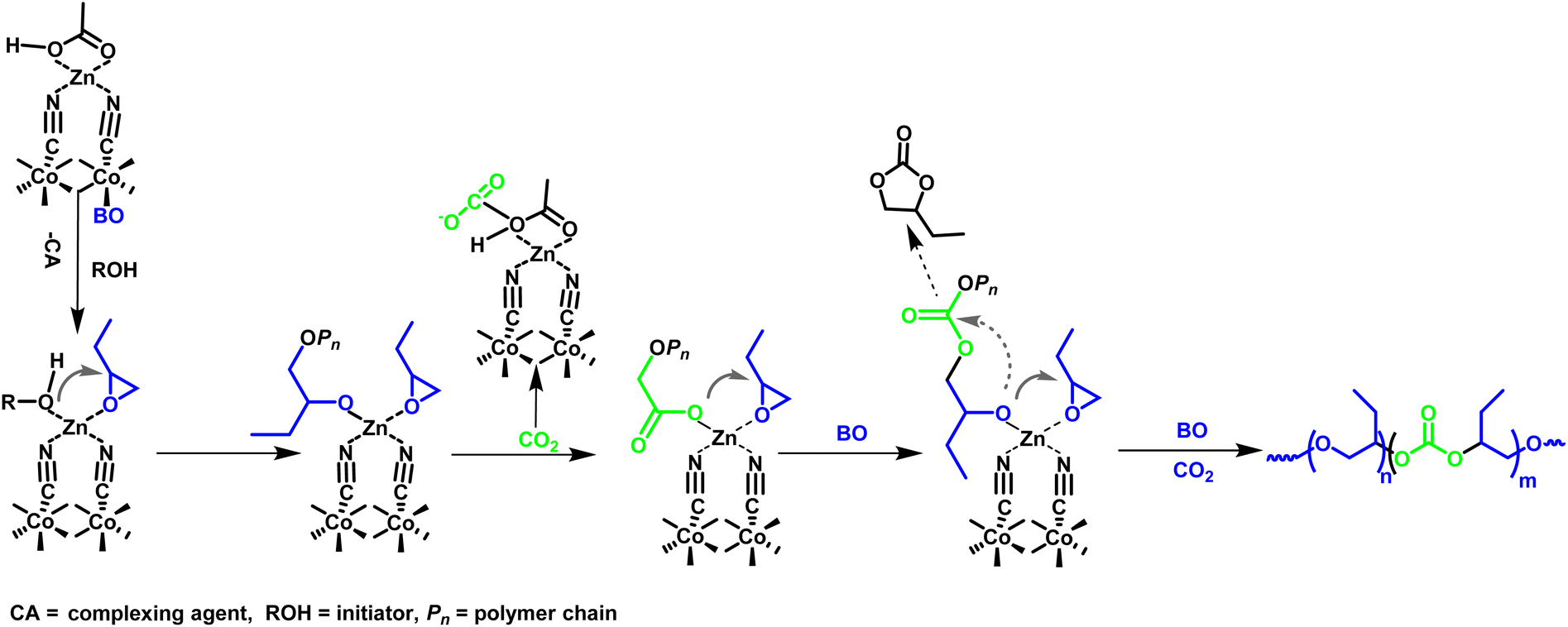 | ||
| Scheme 2 Proposed mechanism for the copolymerization of CO2 and BO using L-DMC catalyst. | ||
3.6 Characterization of polymers
The composition of the polycarbonate ether polyol can be analyzed using FTIR and 1H NMR spectroscopy. It can be observed in the FTIR spectra (Fig. 5(a)), polycarbonate ether polyol exhibits the characteristic v(CO) of the carbonate units and the v(C–O) vibrations associated with the ether units at approximately 1752 cm−1 and 1255 cm−1, respectively. Meanwhile, the stretching vibration of the C–O–C bond in the polyether segment is detected at 1101 cm−1. 1H NMR spectra of copolymer (Fig. 5(c)) exhibited characteristic signals of the methyl to illustrates the composition of the polycarbonate ether polyol. The signals detected in the range of 4.7–4.9 ppm and 4.0–4.3 ppm corresponding to the CH and CH2 groups in the carbonate units, while those at 3.2–3.8 ppm was assigned to the CH and CH2 groups in the ether linkages. Additionally, characteristic signals at 0.9–1.0 ppm and 1.4–1.7 ppm are assigned to the ethyl side chains in the polycarbonate ether polyol. Signals at 4.6 ppm, 4.5 ppm, and 4.0 ppm were ascribed to the aromatic protons of BC. The structure of the polycarbonate ether polyol was obtained through FTIR spectra and 1H NMR spectra, which also provided a basis for calculating the fCO2 and WBC. The 13C NMR spectra of the polycarbonate ether polyol (Fig. S11†) reveals tail-to-tail (T–T) and head-to-tail (H–T) linkages, as evidenced by the typical signals observed at 154.84 ppm, 154.63 ppm, and 154.57 ppm. Consequently, it can be concluded that the BO units in the polycarbonate ether polyol are predominantly connected in the T–T configuration.15,47
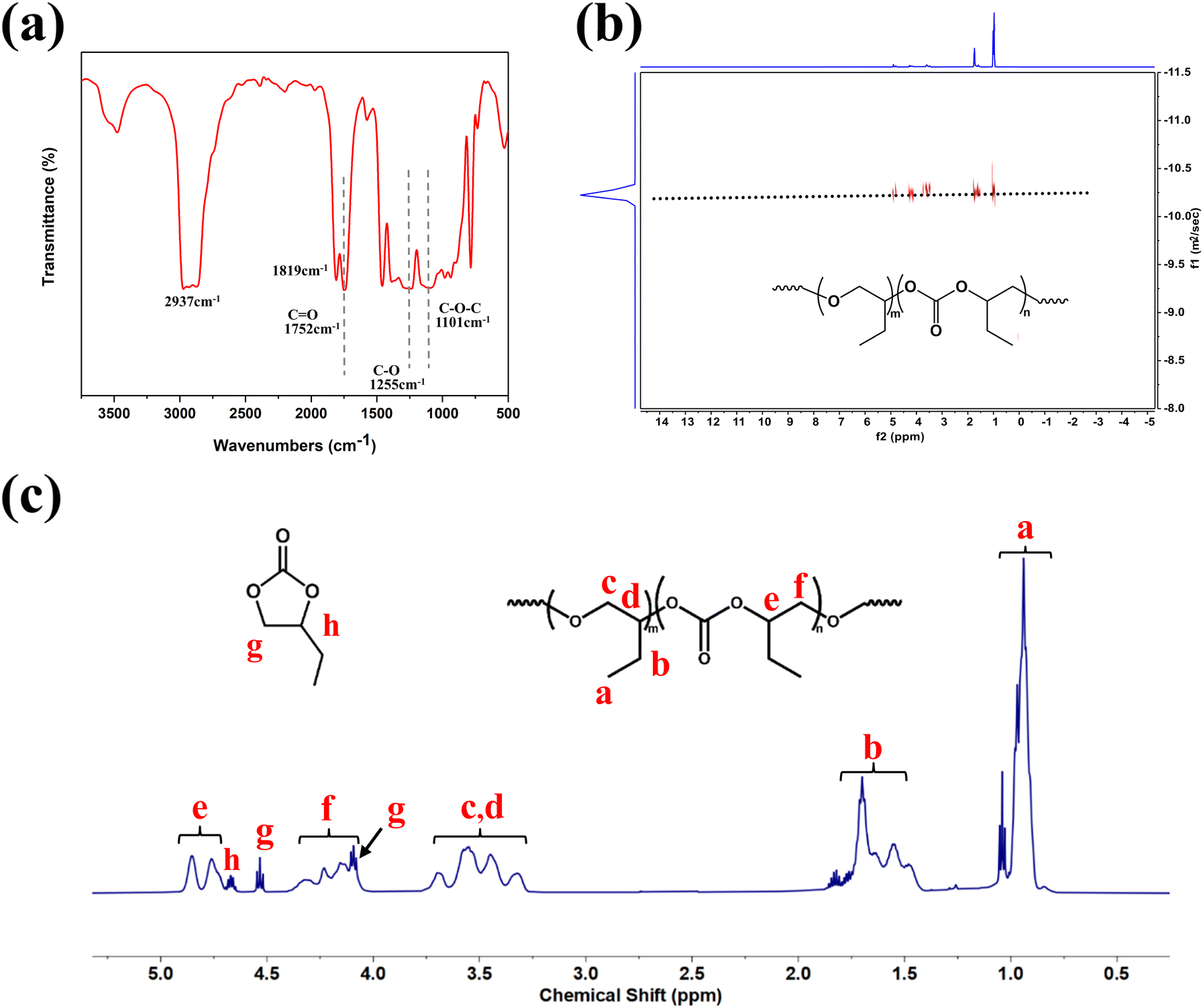 | ||
| Fig. 5 Preparation of the polycarbonate ether polyol (Table 1, entry 2) by the copolymerization of CO2 and BO using L-DMC. (a) FTIR spectrum. (b) DOSY NMR spectrum. (c) 1H-NMR spectrum. | ||
There is only one diffusion coefficient in the DOSY spectra of the CO2–BO copolymer in Fig. 5(b), and this result indicates that the product is a copolymer with mixed carbonate and ether linkages instead of mixture of polycarbonate and polyether. In addition, the MALDI-TOF results (Fig. S17†) showed that the molar masses of the polycarbonate ether polyol can be determined using the formula m/z = 72.1 × (m + n) + 44 × n + 90.1 + 23, where 72.1, 44, 90.1 and 23 represent the respective molar masses of the BO unit, the CO2 unit, 1,4-BDO and sodium ion. For example, the peak at m/z = 11197.3 corresponds to the theoretical mass of the desired 1,4-BDO-initiated polycarbonate ether polyol (calculated for [M + Na]+ = 72.1 × (70 + 52) + 44 × 52 + 90.1 + 23 = 11197.3).
4. Conclusions
In summary, a series of CO2–BO-based polymers were synthesized by high efficiency copolymerization of CO2 and BO. Using L-DMC catalyst, the copolymerization of CO2 and BO can be conducted under 110 °C and 3 MPa CO2 conditions enables the synthesis of polycarbonate ether polyol with number average molecular weights of 6300–12600 g mol−1 with moderate distributions (Đ = 3.1–3.9). The catalytic productivity of L-DMC up to 1500 g polymer per g catalyst and CO2 incorporation fraction (fCO2) up to 35.5%. Compared with the conventional DMC catalysts, the two-dimensional layered structure and high specific surface area of L-DMC resulted in a significant increase in the number of effective Zn active sites facilitating the enhancement of CO2 and BO copolymerization activities. Mechanistic studies revealed that the CH3COOH complexing agent in L-DMC is essential for increasing the CO2 incorporation fraction in polycarbonate ether polyols. 13C NMR spectrum of the polycarbonate ether polyol revealed that BO units are primarily linked in a T–T configuration. In situ FTIR monitoring of the copolymerization process revealed the competitive formation between the target product polycarbonate ether polyol and the byproduct BC and the induction period of the reaction was only 10 min with the use of 1,4-BDO chain transfer agent. Based on the advantages of simple preparation, good stability, and excellent catalytic performance of L-DMC, our next step is to promote its application to the large-scale production of CO2–BO-based polymer materials.Data availability
Data are available on request to the authors.Author contributions
Chen Liu: investigation, methodology, writing – original draft. Yan Cao: writing – review &editing. Xianqiang Zeng: investigation. Zheng Zheng: investigation. Ziqiang Han: investigation. Peng He: investigation. Liguo Wang: conceptualization, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFC3901800), Major Science and Technology Project of Inner Mongolia Autonomous Region (2021ZD0020), Science and Technology Service Network Initiative, Chinese Academy of Sciences (KFJ-STS-QYZD-138).Notes and references
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. Pena Carrodeguas, G. Trott, A. Santmarti, K. Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS.
- C. Chen, Y. Gnanou and X. Feng, Macromolecules, 2023, 56, 892–898 CrossRef CAS.
- X. Zhang, J. Dong, Y. Su, E. G. Lee, Z. Duan, I. Kim and B. Liu, Polym. Chem., 2023, 14, 1263–1274 RSC.
- M. Scharfenberg, S. Hofmann, J. Preis, J. Hilf and H. Frey, Macromolecules, 2017, 50, 6088–6097 CrossRef CAS.
- Y. Li, X. Kou, X. Wang, L. Xia and Z. Li, Polym. Chem., 2024, 15, 2476–2481 RSC.
- Z.-Q. Huang, J.-Q. Chen, X.-Y. Zhang, C.-K. Yuan, P. Wang, Y. Zhao, B.-C. Zhao, W.-X. Qi and W.-Y. Sun, CrystEngComm, 2024, 26, 1701–1709 RSC.
- M. Martínez de Sarasa Buchaca, F. de la Cruz-Martínez, L. F. Sánchez-Barba, J. Tejeda, A. M. Rodríguez, J. A. Castro-Osma and A. Lara-Sánchez, Dalton Trans., 2023, 52, 3482–3492 RSC.
- S. Tang, H. Suo, R. Qu, H. Tang, M. Sun and Y. Qin, Polymers, 2023, 15, 748 CrossRef CAS.
- J. E. Seong, S. J. Na, A. Cyriac, B.-W. Kim and B. Y. Lee, Macromolecules, 2009, 43, 903–908 CrossRef.
- H. I. Mao, C. W. Chen, L. Y. Guo and S. P. Rwei, J. Appl. Polym. Sci., 2022, 139, e52986 CrossRef CAS.
- M. Pohl, E. Danieli, M. Leven, W. Leitner, B. Blümich and T. E. Müller, Macromolecules, 2016, 49, 8995–9003 CrossRef CAS.
- M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef.
- S. Liu, Y. Miao, L. Qiao, Y. Qin, X. Wang, X. Chen and F. Wang, Polym. Chem., 2015, 6, 7580–7585 RSC.
- J. Wang, H. Zhang, Y. Miao, L. Qiao and X. Wang, Green Chem., 2017, 19, 2194–2200 RSC.
- G.-W. Yang, C.-K. Xu, R. Xie, Y.-Y. Zhang, C. Lu, H. Qi, L. Yang, Y. Wang and G.-P. Wu, Nat. Synth., 2022, 1, 892–901 CrossRef.
- S. Liu, K. Yan, P. Ren, X. Shang and X. Zhang, Case Stud. Constr. Mater., 2024, 20, e03136 Search PubMed.
- F. Yu, X. Xu, N. Lin and X. Y. Liu, RSC Adv., 2015, 5, 72544–72552 RSC.
- D. Wang, Z. Huang, S. Shi, J. Ren and X. Dong, J. Appl. Polym. Sci., 2022, 139, e52437 CrossRef CAS.
- X. Zeng, L. Wang, Y. Cao, C. Liu, Z. Han, P. He and H. Li, Eur. Polym. J., 2022, 179, 111483 CrossRef CAS.
- X. Zeng, L. Wang, Y. Cao, C. Liu, P. He and H. Li, Ind. Eng. Chem. Res., 2024, 63, 1215–1227 CrossRef CAS.
- L. Kunze, S.-Y. Tseng, R. Schweins, T. Sottmann and H. Frey, Langmuir, 2019, 35, 5221–5231 CrossRef CAS PubMed.
- D. A. Pyatakov and I. E. Nifant'ev, Pet. Chem., 2024, 63, 1170–1193 CrossRef.
- S. Inoue, K. Matsumoto and Y. Yoshida, Makromol. Chem., 2003, 181, 2287–2292 CrossRef.
- C. H. Tran, S. A. Kim, Y. Moon, Y. Lee, H. M. Ryu, J. H. Baik, S. C. Hong and I. Kim, Catal. Today, 2021, 375, 335–342 CrossRef CAS.
- X.-H. Zhang, R.-J. Wei, X.-K. Sun, J.-F. Zhang, B.-Y. Du, Z.-Q. Fan and G.-R. Qi, Polymer, 2011, 52, 5494–5502 CrossRef CAS.
- C. Marquez, A. Simonov, M. Wharmby, C. Goethem, I. Vankelecom, B. Bueken, A. Krajnc, G. Mali, D. Vos and T. De Baerdemaeker, Chem. Sci., 2019, 10, 4869–4875 RSC.
- Y. H. Seo, Y. B. Hyun, H. J. Lee, J. W. Baek, H. C. Lee, J. H. Lee, J. Lee and B. Y. Lee, J. CO2 Util., 2021, 53, 101755 CrossRef CAS.
- Z. Li, Y. Qin, X. Zhao, F. Wang, S. Zhang and X. Wang, Eur. Polym. J., 2011, 47, 2152–2157 CrossRef CAS.
- J. Sebastian and D. Srinivas, Appl. Catal., A, 2015, 506, 163–172 CrossRef CAS.
- Z. Song, B. Subramaniam and R. V. Chaudhari, ACS Sustainable Chem. Eng., 2019, 7, 5698–5710 CrossRef CAS.
- I. Kim, J.-T. Ahn, C. S. Ha, C. S. Yang and I. Park, Polymer, 2003, 44, 3417–3428 CrossRef CAS.
- H. Yang, L. Wang, S. Xu, Y. Cao, P. He, J. Chen, Z. Zheng and H. Li, Green Energy Environ., 2022, 7, 1361–1376 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- A. Peeters, P. Valvekens, R. Ameloot, G. Sankar, C. E. A. Kirschhock and D. E. De Vos, ACS Catal., 2013, 3, 597–607 CrossRef CAS.
- N. J. Robertson, Z. Qin, G. C. Dallinger, E. B. Lobkovsky, S. Lee and G. W. Coates, Dalton Trans., 2006, 45, 5390–5395 RSC.
- J. K. Varghese, D. S. Park, J. Y. Jeon and B. Y. Lee, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4811–4818 CrossRef CAS.
- S. Liu, Y. Qin, L. Qiao, Y. Miao, X. Wang and F. Wang, Polym. Chem., 2016, 7, 146–152 RSC.
- Y. J. Huang, G. R. Qi and Y. H. Wang, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1142–1150 CrossRef CAS.
- R.-J. Wei, Y.-Y. Zhang, X.-H. Zhang, B.-Y. Du and Z.-Q. Fan, RSC Adv., 2014, 4, 21765–21771 RSC.
- J. Zhao, B. G. Li and H. Fan, Macromol. Theory Simul.c, 2021, 30, 2000101 CrossRef CAS.
- S. Chen, Z. J. Hua, Z. Fang and G. R. Qi, Polymer, 2004, 45, 6519–6524 CrossRef CAS.
- K. Ma, Q. Bai, L. Zhang and B. Liu, RSC Adv., 2016, 6, 48405–48410 RSC.
- Y. Gao, Y. Qin, X. Zhao, F. Wang and X. Wang, J. Polym. Res., 2012, 19, 1–9 CrossRef CAS.
- S. Liu, Y. Qin, X. Chen, X. Wang and F. Wang, Polym. Chem., 2014, 5, 6171–6179 RSC.
- P. Góarecki and W. Kuran, J. Polym. Sci., Polym. Lett. Ed., 2003, 23, 299–304 CrossRef.
- F. Liu, Y. Gu, P. Zhao, J. Gao and M. Liu, ACS Sustainable Chem. Eng., 2019, 7, 5940–5945 CrossRef CAS.
- W. Zhang, Q. Lin, Y. Cheng, L. Lu, B. Lin, L. Pan and N. Xu, J. Appl. Polym. Sci., 2012, 123, 977–985 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06913g |
| This journal is © The Royal Society of Chemistry 2024 |