
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Breaking the blue barrier of nucleobase fluorescence emission with dicyanovinyl-based uracil molecular rotor probes†
Mria Chowdhury,
Akym John and
Robert H. E. Hudson *
*
Department of Chemistry, Western University, London, Ontario N6A 5B7, Canada. E-mail: rhhudson@uwo.ca
First published on 25th November 2024
Abstract
Dicyanovinyl-modified uracil produces fluorescent molecular rotors (FMR) that display massively red-shifted emission and huge Stokes shifts. They are exemplified by DCVSU – an intrinsically fluorescent nucleobase analog (IFNA) with the longest emission wavelength of 592 nm (DMSO) reported thus far which also shows strong polarity sensitivity and large Stokes shift (λ = 181 nm). The IFNAs exhibited typical molecular rotor response to solvent viscosity with brightnesses (ε × φ) of up to 8700 cm−1 M−1. 1H NMR titration confirmed the expected association of the IFNA with the complementary nucleobase adenine-9-ethyl acetate.
Introduction
The search for intrinsically fluorescent nucleobase analogs (IFNAs) that are environmentally sensitive, have comparatively red-shifted emission wavelengths and are bright continues since such analogs can aid the efficient detection of nucleic acid structures, dynamics and functions.1–3 Standard fluorescent tags4 tethered to nucleosides for use in diagnostic assays are limited in their detection ability such as sensitivity towards hybridization with complementary strands, detection of abasic sites and gene mutations.5 Covalent modification of nucleobases such that they are integrated into an autofluorescent moiety while retaining base-pairing ability has the potential to be exploited as a diagnostic and research tool.5,6Fluorescent molecular rotor (FMR) probes have gained increased attention from researchers in recent years and are extensively used in biosensing applications.7–10 Structurally, FMRs typically have electron donor and acceptor moieties separated by a rotatable linker resulting in microenvironmental sensitivity to viscosity,11 hybridization,12 polarity, and pH.13 This provides useful indicators of changes within cells and organelles, protein aggregates, or interactions between DNA–protein or aptamers, polymers and other complex molecular environments.14–16 Intrinsically modified nucleobase FMRs so far fall short in addressing some of the above-mentioned requirements such as insufficient red-shifted emission or low brightness precluding effective use in biological assays.
Previously, we reported the “chimeric strategy”17 that was employed to guide the design of chemically modified nucleobases such that they would possess fluorescence or quenching properties yet retaining their hybridizing ability with complementary nucleobases. In our endeavour to ‘break the blue barrier’ common to most IFNAs and produce red-shifted emission to overcome interference from background auto-fluorescence of the biological milieu, we have taken the chimeric approach of linking part of a well-known fluorescence rotor, dicyanovinyljulolidine (DCVJ, Fig. 1A)18 to uracil nucleobase. The design retains the dicyanovinyl group as the acceptor and replaces the donor julolidine group with uracil (DCVU, Fig. 1B). Previously, the 5-dicyanovinyluracil-based nucleoside analogue was screened for anti-Leishmania activity; however, the photophysical properties were not reported.19 While DCVJ and its analogues like CCVJ (9-(2-carboxy-2-cyanovinyl)julolidine) and FCVJ ((2-carboxy-2-cyanovinyl)-julolidine farnesyl ester) are popularly used FMR dyes used20–22 (Fig. 1A), CCVJ inspired analogues have been tethered to cytosine nucleobase12 and the resulting photophysical properties were not much different from CCVJ itself.
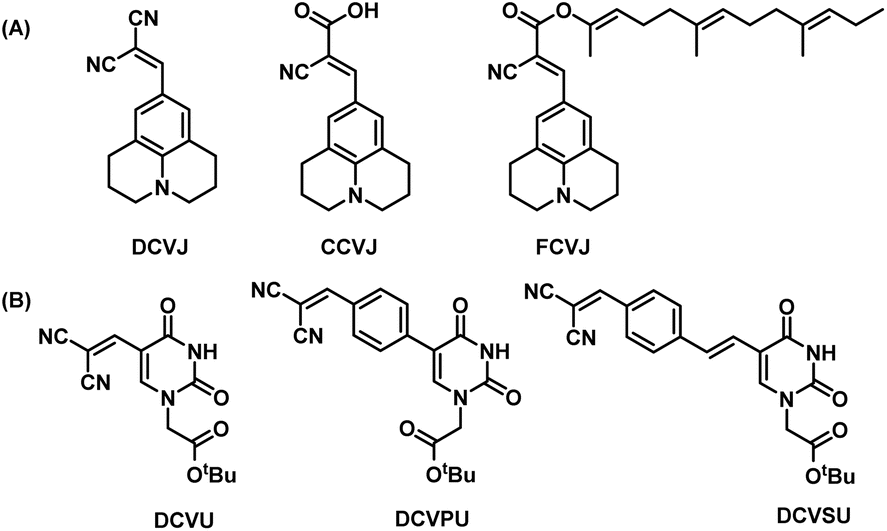 | ||
| Fig. 1 (A) Structures of DCVJ, CCVJ, FCVJ; (B) structures of chimeric nucleobase FMR synthesized for this study DCVU, DCVPU, DCVSU. | ||
Additional structural modifications to influence the fluorescence properties of dicyanovinyl uracil molecular rotors were made by introducing phenyl or styryl linking groups in tandem with the dicyanovinyl (DCV) group. The DCVJ-inspired uracil modifications (Fig. 1B) reported herein have dramatically different emission wavelengths and Stokes shifts as compared to DCVJ and CCVJ.
Results and discussion
Pd-catalyzed coupling reactions are an effective way to introduce various substituents at the C5 position of uracil nucleobase. Suzuki–Miyaura coupling reaction between different boronic acids and 5-halo-uracil/uridine derivatives in the presence of Pd catalyst has been used in the synthesis of a variety of functionalized uracil analogues.23,24 For the present work, uracil was first converted to 5-iodouracil25 followed by regioselective alkylation at the N1 position to yield 1 (see ESI: Scheme SI-1†). Compound 1 served as the coupling partner with commercially available 4-formylphenylboronic acid under Suzuki–Miyaura reaction conditions to yield 2b, full details are given in the ESI.† Alternatively, 1 was reacted with (E)-(4-formylstyryl)boronic acid (Scheme SI-2†) under the same coupling conditions to give 2c. (E)-(4-Formylstyryl)boronic acid was synthesized using Heck coupling reaction between p-bromobenzaldehyde and vinyl-boronic acid pinacol ester. When carried out at an elevated temperature (90 °C) for 48 h, the Heck reaction gave an improved yield of 72% in our hands over the literature procedure (Scheme SI-2†).26Linker-less attachment of the dicyanvinyl group to uracil was achieved using substrate 2a which was synthesized from uracil via 5-hydroxymethyl uracil followed by oxidation according to our previously reported method.17 The dicyanovinyl-functional group was introduced by reaction of the precursor aldehydes 2a–c with malononitrile in ethanol at 50 °C (stirred, overnight) in a Knoevenagel-type reaction without the presence of an exogenous base. In this manner, the DCV-based uracil nucleobase fluorophores 3a (DCVU), 3b (DCVPU), and 3c (DCVSU) were achieved (Scheme 1).
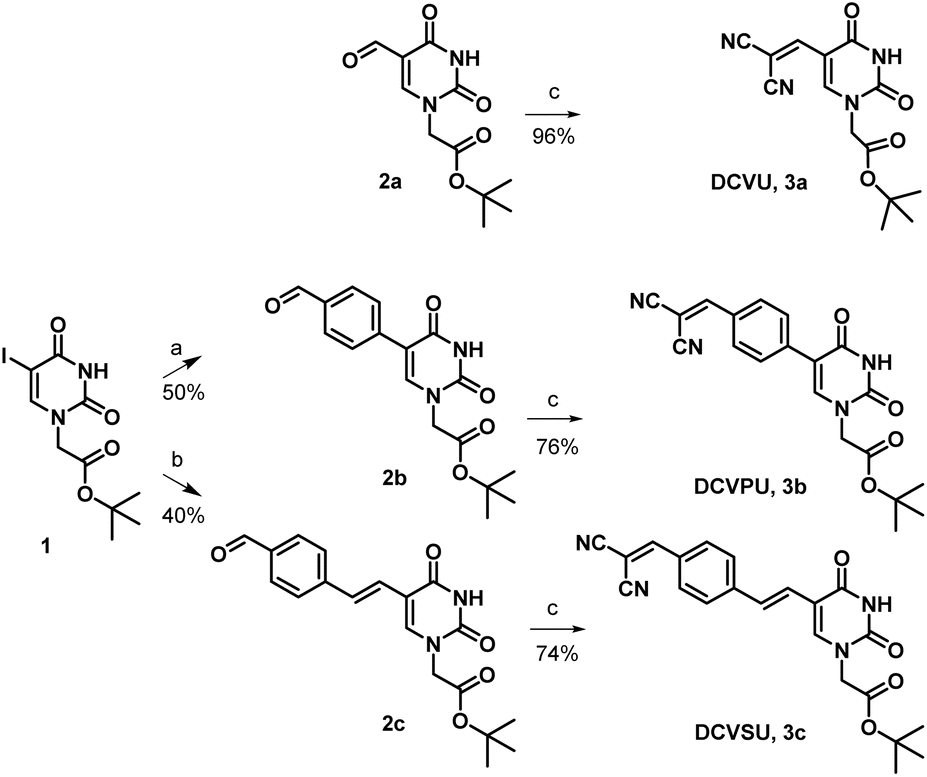 | ||
Scheme 1 Reagents and conditions (a) 4-formylphenylboronic acid, K2CO3, Pd(dppf)Cl2, THF/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 100 °C, 5 h, (b) (4-formylstyryl)boronic acid, K2CO3, Pd(dppf)Cl2 THF/H2O (4:1), 100 °C, 5 h, (c) malononitrile, EtOH, 50 °C, 16 h. :1), 100 °C, 5 h, (b) (4-formylstyryl)boronic acid, K2CO3, Pd(dppf)Cl2 THF/H2O (4:1), 100 °C, 5 h, (c) malononitrile, EtOH, 50 °C, 16 h. | ||
Fluorophores 3a–c and 2c were then subjected to photophysical characterization, Fig. 2 and ESI.† UV-Vis spectroscopy and fluorescence measurements were done for compounds 3a–c and 2c in solvents of varying viscosities and polarities. Data for DCVSU (3c) is shown in Table 1. Complete characterization for 3a–c and 2c is provided in SI-Table 1.†
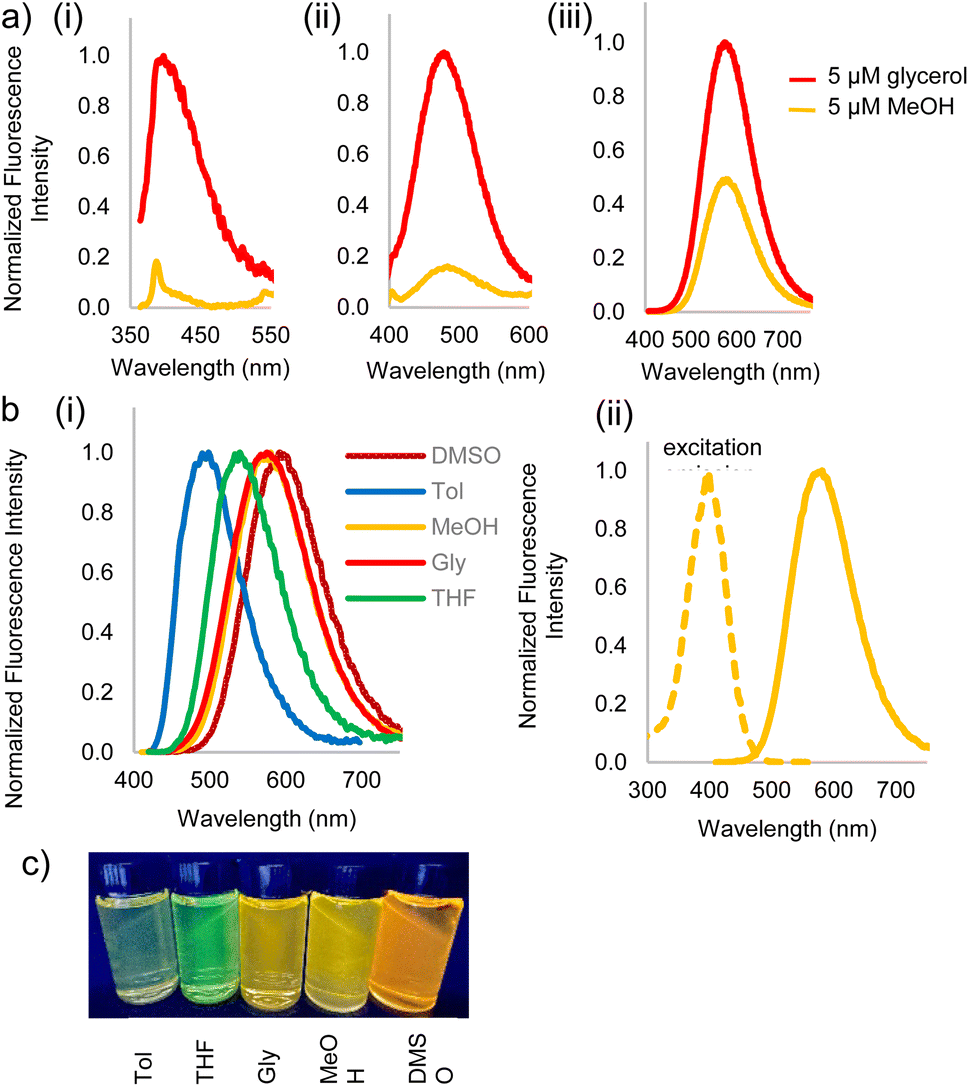 | ||
| Fig. 2 (a) Fluorescence emission spectra in 5 μM solutions of glycerol (with 5% MeOH) and methanol (MeOH) to study viscosity dependence of (i) DCVU 3a, (ii) DCVPU 3b, (iii) DCVSU 3c, (b) (i) emission spectra of 5 μM 3c in DMSO, glycerol, methanol, THF, toluene showing polarity dependence [λex(DMSO) = 592 nm, λex(glycerol) = 573 nm, λex(methanol) = 575 nm, λex(THF) = 537 nm, λex(toluene) = 496 nm], (ii) excitation and emission spectra of 5 μM 3c in methanol showing large Stokes shift, (c) picture of 3c in different solvents: from left-to-right: Tol (toluene), THF (tetrahydrofuran), Gly (glycerol), MeOH (methanol), DMSO (dimethylsulfoxide). | ||
| Property | Solvent | ||||
|---|---|---|---|---|---|
| DMSO | Glya | MeOH | Tola | THF | |
| a Gly = glycerol, Tol = toluene, Δν = Stokes shift in wavenumbers, ε = extinction coefficient, φ = quantum yield, ε × φ = brightness factor. | |||||
| λabs,max (nm) | 411 | 400 | 395 | 402 | 404 |
| λem,max (nm) | 592 | 573 | 575 | 496 | 537 |
| Δλ (nm) (Δν 103 cm−1) | 181 (7.4) | 173 (7.5) | 180 (7.9) | 94 (4.7) | 133 (6.1) |
| ε (104 cm−1 M−1) | 4.2 | 3.7 | 2.7 | 3.1 | 2.4 |
| φ | 0.21 | 0.22 | 0.09 | 0.01 | 0.05 |
| ε × φ (cm−1 M−1) | 8700 | 8000 | 2390 | 310 | 1190 |
As conjugation increases from DCVU to DCVSU, the emission wavelengths undergo a bathochromic shift by approximately 100 nm, going from 389 nm (DMSO) for DCVU to 503 nm (DMSO) for DCVPU to 592 nm (DMSO) for DCVSU, Fig. 2. Such a trend was observed for all the solvents used in this study. To the best of our knowledge, the emission maxima of 573 nm (glycerol), 575 nm (methanol) and 592 nm (DMSO) are the most red-shifted fluorophores reported for any nucleobase FMR or any other intrinsically modified nucleobase fluorophores23 known to date; even more red-shifted than a recently reported “most red-shifted” non-FMR push–pull C-linked 8-(diethylamino)benzo[b][1,8]naphthyridin-2(1H)-one nucleoside (ABN).27
The brightness for DCVSU is nearly as great as some of the recently reported bright modified nucleobase fluorophores27,28 (ε = 2.0–4.0 × 104 measured at the absorption maxima in different solvents, with the highest quantum yield (φ) of 0.21 and 0.22 in DMSO and glycerol, respectively). Quantum yields were measured using fluorescein or rhodamine as standards.
Fluorescence turn-on, ascribed to the molecular rotor behaviour, was observed in a more viscous solvent (glycerol) and low fluorescence (3b, 3c) to complete fluorescence turn-off (3a) was observed in a less viscous solvent (methanol). Isomerization of the pseudo cis–trans of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in dicyanovinyl group in the excited state is likely responsible as the main non-radiative deactivation channel from S1 (excited state) to S0 (ground state), as reported for DCVJ.29 While DCVU (3a) shows complete fluorescence turn-off in a non-viscous solvent, increasing the conjugation length through DCVPU to DCVSU results in a less dramatic turn-off of fluorescence, Fig. 2a. Increased conjugation increases the barrier of rotation of CC in S1 state, thus increased fluorescence is observed and decrease in non-radiative decay.
C bond in dicyanovinyl group in the excited state is likely responsible as the main non-radiative deactivation channel from S1 (excited state) to S0 (ground state), as reported for DCVJ.29 While DCVU (3a) shows complete fluorescence turn-off in a non-viscous solvent, increasing the conjugation length through DCVPU to DCVSU results in a less dramatic turn-off of fluorescence, Fig. 2a. Increased conjugation increases the barrier of rotation of CC in S1 state, thus increased fluorescence is observed and decrease in non-radiative decay.
Significant polarity-dependent solvatochromism was also observed and 100 nm change covering the range from blue to orange region of the visible spectrum was observed, Fig. 2b. Polarity induced solvatochromism can be quantified by Lippert–Mataga plot which draws a relationship between Stokes shift (Δ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) in wavenumber) and orientation polarizability Δf (eqn (1a) and (1b)) derived by Lippert and Mataga.30,31
in wavenumber) and orientation polarizability Δf (eqn (1a) and (1b)) derived by Lippert and Mataga.30,31
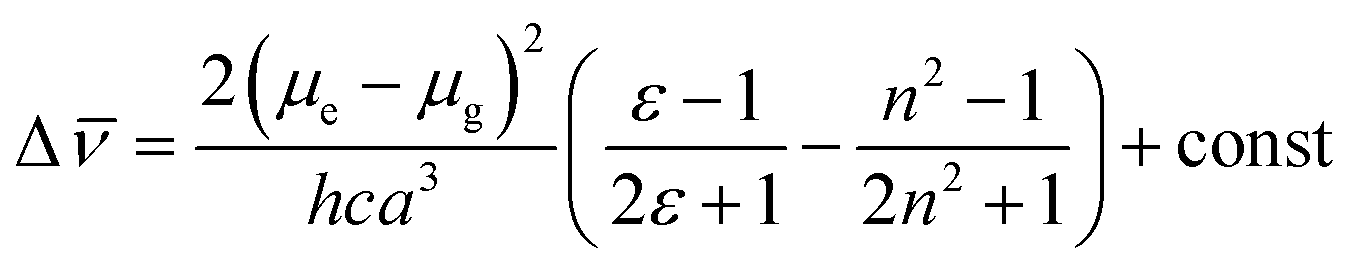 | (1a) |
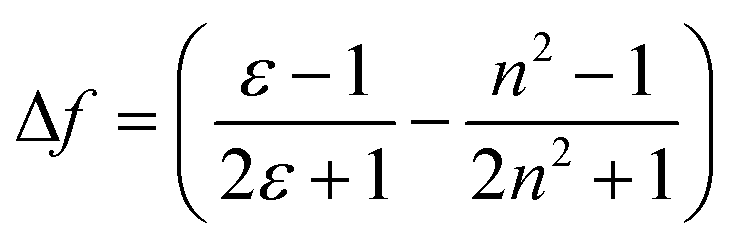 | (1b) |
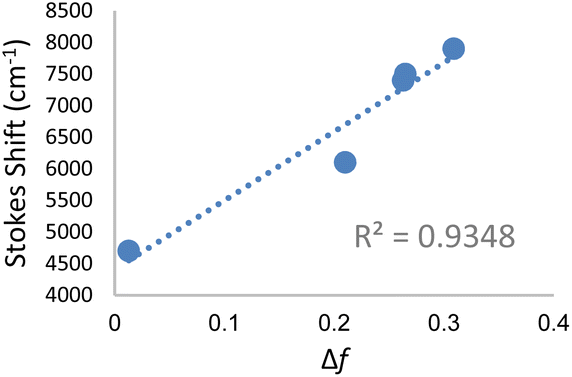 | ||
| Fig. 3 Lippert–Mataga plot for DCVSU (3c) in solvents toluene, THF, methanol, glycerol and DMSO. | ||
According to the conventional description of FMRs, rotors with more donor–acceptor characteristics show more intramolecular charge transfer (ICT) and exhibit polarity sensitivity and not viscosity sensitivity and vice versa for rotors with less ICT behaviour, although deviations from this trend have been observed previously with some dicyanovinyl-based rotors by Zhou et al.29 Similarly, 3a–c exhibit both polarity and viscosity sensitivity making them more environmentally sensitive, as compared to DCVJ.18 Like the archetypal FMR DCVJ, we hypothesize that fluorescence emission of 3a–c occurs from the LE (locally excited) state and not the lower energy TICT (twisted intramolecular charge transfer) state which is dark.36 This is evident from the similarity of emission wavelengths in the low-viscosity solvent methanol and viscous solvent glycerol. If TICT was not dark, then we would see LE emission for modified nucleobase in glycerol and TICT emission for methanol giving us different emission wavelengths. Methanol and glycerol have dramatically different viscosities but similar relative polarity (methanol = 0.76 and glycerol = 0.79); thus, it was deduced that emission was from the LE. Additionally, in DMSO, 3c has a relatively high quantum yield of emission (φ = 0.21), which is possible when the emission is from LE since the increase in the TICT population leads to increased non-radiative decay.
Next, we investigated the modified nucleobases for their ability to base pair with the complementary nucleobase adenine (Fig. 4a). Anhydrous CDCl3 was used to perform titration to measure the association of 3a and adenine-9-ethyl acetate by 1H NMR spectroscopy. This pair was chosen because the base-pairing faces of 3a–c are identical and 3a has better solubility in CDCl3 than 3b or 3c. When 0.5 mM adenine-9-ethyl acetate (host) was titrated with 20 mM of 3a, the association was evident from the downfield shifting of N6H proton of adenine-9-ethyl acetate from 5.54 to 6.40 ppm due to H-bonding interactions. Concomitantly, the C2H peak also shifted downfield from 7.89 to 8.05 ppm owing to pseudo-H bonding37 interactions (Fig. 4b). An association constant, Ka of 155 ± 10 M−1 for N6H⋯O H-bonding (Fig. 4a) and 155 ± 13 M−1 for C2H⋯O interactions (Fig. SI-5†) were calculated. The Ka values were within the range of values observed for 5-substituted uracil and adenine previously in the literature.38,39 The slightly higher association constant compared to A:T may be attributed to the electron-withdrawing nature of the dicyanovinyl group in the C5 position of modified uracil 3a, which is expected to have an acid-strengthening effect on the N3H and consequently increase in H-bonding ability.40
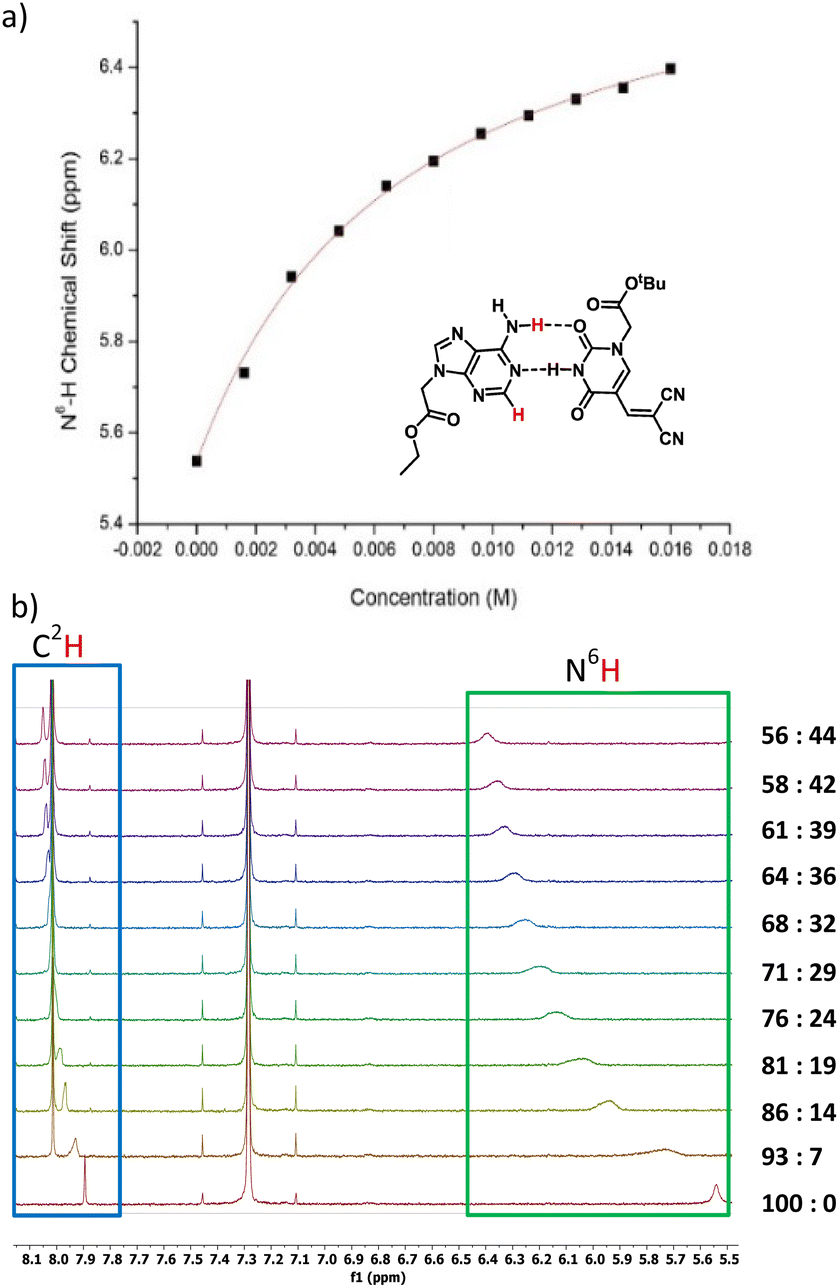 | ||
| Fig. 4 (a) N6H chemical shift of adenine (host) vs. concentration of 3a (guest) plot using non-linear curve fit. Watson–Crick base pair between adenine-9-ethyl acetate and modified uracil 3a, (b) 1H NMR titration of 0.5 mM adenine-9-ethyl acetate (A = host) with 20 mM 3a (guest). A:DCVU represents volume ratios of A and 3a on successive additions of 3a without changing the concentration of A. | ||
Conclusions
We have reported a series of new dicyanovinyl-modified uracil fluorescent nucleobases that exhibited environmentally sensitive fluorescence emissions from the blue to yellow/orange region of the visible spectrum. Such IFNAs have environmental sensitivity which may translate to applications for reporting interactions of nucleic acids with biomolecules such as complementary nucleic acids or proteins. The large Stokes shift and competitive brightness as compared to some of the brightest but “blue” emitting IFNAs recently developed makes these IFNAs great candidates for applications in fluorescence microscopy. The ease of synthesis combined with exceptional fluorescence properties make DCVSU (3c) a much-needed addition to the IFNA FMR toolbox.With evidence of strong binding constant, Ka, research on these chimeric IFNAs is being expanded to incorporate them into peptide nucleic acid (PNA) strands in our laboratories to assess the sequence dependency, hybridization with complementary strands and effect of hybridization on fluorescence.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Funding acquisition, conceptualization, supervision and writing – review & editing (RHEH); investigation, visualization and writing – original draft (MC); investigation (AJ).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Natural Sciences and Engineering Research Council of Canada for funding.Notes and references
- C. S. Eubanks and A. E. Hargrove, Chem. Commun., 2017, 53, 13363–13366 RSC.
- C. Dash, J. W. Rausch and S. F. J. Le Grice, Nucleic Acids Res., 2004, 32, 1539–1547 CrossRef CAS PubMed.
- M. Sholokh, R. Sharma, N. Grytsyk, L. Zaghzi, V. Y. Postupalenko, D. Dziuba, N. P. F. Barthes, B. Y. Michel, C. Boudier, O. A. Zaporozhets, Y. Tor, A. Burger and Y. Mély, Chem.–Eur. J., 2018, 24, 13850–13861 CrossRef CAS PubMed.
- D. L. Ma, H. Z. He, K. H. Leung, H. J. Zhong, D. Shiu-Hin Chan and C. H. Leung, Chem. Soc. Rev., 2013, 42, 3427–3440 RSC.
- B. Y. Michel, D. Dziuba, R. Benhida, A. P. Demchenko and A. Burger, Front. Chem., 2020, 8, 112 CrossRef CAS PubMed.
- N. Klöcker, F. P. Weissenboeck and A. Rentmeister, Chem. Soc. Rev., 2020, 49, 8749–8773 RSC.
- M. Chowdhury and R. H. E. Hudson, Chem. Rec., 2023, 23, e202200218 CrossRef CAS PubMed.
- K. Seio, T. Kanamori and Y. Masaki, Tetrahedron Lett., 2018, 59, 1977–1985 CrossRef CAS.
- R. E. Johnson, M. T. Murray, L. J. Bycraft, S. D. Wetmore and R. A. Manderville, Chem. Sci., 2023, 14, 4832–4844 RSC.
- X. Liu, W. Chi, Q. Qiao, S. V. Kokate, E. P. Cabrera, Z. Xu, X. Liu and Y. T. Chang, ACS Sens., 2020, 5, 731–739 CrossRef CAS PubMed.
- M. E. Nipper, S. Majd, M. Mayer, J. C. M. Lee, E. A. Theodorakis and M. A. Haidekker, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 1148–1153 CrossRef CAS PubMed.
- D. Dziuba, R. Pohl and M. Hocek, Chem. Commun., 2015, 51, 4880–4882 RSC.
- A. Nandi, A. Kushwaha, D. Das and R. Ghosh, Phys. Chem. Chem. Phys., 2018, 20, 7014–7020 RSC.
- T. Z. Cservenyi, A. J. Van Riesen, F. D. Berger, A. Desoky and R. A. Manderville, ACS Chem. Biol., 2016, 11, 2576–2582 CrossRef CAS PubMed.
- L. E. Shimolina, M. A. Izquierdo, I. López-Duarte, J. A. Bull, M. V. Shirmanova, L. G. Klapshina, E. V. Zagaynova and M. K. Kuimova, Sci. Rep., 2017, 7, 41097 CrossRef CAS PubMed.
- M. L. Viriot, M. C. Carré, C. Geoffroy-Chapotot, A. Brembilla, S. Muller and J.-F. Stoltz, Clin. Hemorheol. Microcirc., 1998, 19, 151–160 CAS.
- M. Chowdhury, J. A. Turner, D. Cappello, M. Hajjami and R. H. E. Hudson, Org. Biomol. Chem., 2023, 21, 9463–9470 RSC.
- B. D. Allen, A. C. Benniston, A. Harriman, S. A. Rostron and C. Yu, Phys. Chem. Chem. Phys., 2005, 7, 3035–3040 RSC.
- P. F. Torrence, X. Fan, X. Zhang and P. M. Loiseau, Bioorg. Med. Chem. Lett., 2006, 16, 5047–5051 CrossRef CAS PubMed.
- M. A. Haidekker, N. L'heureux and J. A. Frangos, Am. J. Physiol.: Heart Circ. Physiol., 2000, 278, H1401–H1406 CrossRef CAS PubMed.
- D. L. Wilson and E. T. Kool, J. Am. Chem. Soc., 2019, 141, 19379–19388 CrossRef CAS PubMed.
- M. E. Nipper, S. Majd, M. Mayer, J. C. M. Lee, E. A. Theodorakis and M. A. Haidekker, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 1148–1153 CrossRef CAS PubMed.
- M. Segal and B. Fischer, Org. Biomol. Chem., 2012, 10, 1571–1580 RSC.
- A. V. Ardhapure, V. Gayakhe, S. Bhilare, A. R. Kapdi, S. S. Bag, Y. S. Sanghvi and K. C. Gunturu, New J. Chem., 2020, 44, 14744–14754 RSC.
- R. H. E. Hudson, G. Li and J. Tse, Tetrahedron Lett., 2002, 43, 1381–1386 CrossRef CAS.
- S. K. Stewart and A. Whiting, J. Organomet. Chem., 1994, 482, 293–300 CrossRef CAS.
- G. N. Samaan, M. K. Wyllie, J. M. Cizmic, L. M. Needham, D. Nobis, K. Ngo, S. Andersen, S. W. Magennis, S. F. Lee and B. W. Purse, Chem. Sci., 2021, 12, 2623–2628 RSC.
- A. Karimi, R. Börner, G. Mata and N. W. Luedtke, J. Am. Chem. Soc., 2020, 142, 14422–14426 CrossRef CAS PubMed.
- F. Zhou, J. Shao, Y. Yang, J. Zhao, H. Guo, X. Li, S. Ji and Z. Zhang, Eur. J. Org. Chem., 2011, 25, 4773–4787 CrossRef.
- E. Lippert, Z. Elektrochem., 1957, 61, 962–975 CrossRef CAS.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- R. W. Sinkeldam and Y. Tor, Org. Biomol. Chem., 2007, 5, 2523–2528 RSC.
- H. Chen, L. Liu, K. Qian, H. Liu, Z. Wang, F. Gao, C. Qu, W. Dai, D. Lin, K. Chen, H. Liu and Z. Cheng, Sci. Adv., 2022, 8, 3289 CrossRef PubMed.
- I. Likhotkin, R. Lincoln, M. L. Bossi, A. N. Butkevich and S. W. Hell, J. Am. Chem. Soc., 2023, 145, 1530–1534 CrossRef CAS PubMed.
- Z. Gao, Y. Hao, M. Zheng and Y. Chen, RSC Adv., 2017, 7, 7604–7609 RSC.
- M. K. Kuimova, Chimia, 2012, 66, 159–165 CrossRef CAS PubMed.
- P. Hobza, J. Šponer, E. Cubero, M. Orozco and F. J. Luque, J. Phys. Chem. B, 2000, 104, 6286–6292 CrossRef CAS.
- S. Sivakova and S. J. Rowan, Chem. Soc. Rev., 2005, 34, 9–21 RSC.
- Y. Kyogoku, R. C. Lord and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1967, 57, 250–257 CrossRef CAS PubMed.
- S. I. Kawahara, T. Wada, S. Kawauchi, T. Uchimaru and M. Sekine, J. Phys. Chem. A, 1999, 103, 8516–8523 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and compound characterizations (PDF). See DOI: https://doi.org/10.1039/d4ra07000c |
| This journal is © The Royal Society of Chemistry 2024 |