
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComplexes between 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AAPH) and cucurbit[n]uril hosts modulate the yield and fate of photolytically-generated AAPH radicals†
Angie C. Forero-Girón‡
 a,
Mauricio Oyarzún‡a,
Kevin Droguetta,
Denis Fuentealbaa,
Soledad Gutiérrez-Olivaa,
Barbara Herreraa,
Alejandro Toro-Labbéa,
Eduardo Fuentes-Lemusb,
Michael J. Daviesb,
Camilo López-Alarcón*a and
Margarita E. Aliaga*a
a,
Mauricio Oyarzún‡a,
Kevin Droguetta,
Denis Fuentealbaa,
Soledad Gutiérrez-Olivaa,
Barbara Herreraa,
Alejandro Toro-Labbéa,
Eduardo Fuentes-Lemusb,
Michael J. Daviesb,
Camilo López-Alarcón*a and
Margarita E. Aliaga*a
aDepartmento de Química Física, Escuela de Química, Facultad de Química y de Farmacia, Pontificia Universidad Católica de Chile, Chile. E-mail: mealiaga@uc.cl; clopezr@uc.cl
bDepartment of Biomedical Sciences, Panum Institute, University of Copenhagen, Denmark
First published on 11th November 2024
Abstract
Using theoretical and experimental tools we investigated the recognition of AAPH (2,2′-azobis(2-methylpropionamidine) dihydrochloride), a well-known water-soluble azo-compound employed as a source of peroxyl radicals, by cucurbit[6]uril (CB[6]), and cucurbit[8]uril (CB[8]). Density functional theory calculations and isothermal titration calorimetry experiments demonstrated that AAPH was not included in the cavity of CB[6], however, an exclusion complex was generated. Inclusion of AAPH in the CB[8] cavity was favored, forming stable inclusion complexes at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 stoichiometries; AAPH@CB[8] and 2AAPH@CB[8], respectively. Radical formation upon photolytic cleavage of AAPH was examined theoretically, and by spin trapping with electron paramagnetic resonance. The radical yields detected with uncomplexed (free) AAPH and the AAPH-CB[6] (exclusion) complex were identical, whereas a marked decrease was shown for AAPH@CB[8]. Lower decreases were seen with a bimolecular (2:1) AAPH-CB[8] inclusion complex (2AAPH@CB[8]). This modulation was corroborated by the consumption of pyrogallol red (PGR), an oxidizable dye that does not associate with CB[6] or CB[8]. AAPH-CB[6] and 2AAPH@CB[8] did not significantly modify the initial consumption rate (Ri) of PGR, whereas AAPH@CB[8] decreased this. The oxidative consumption of free Trp, Gly–Trp and Trp–Gly by radicals derived from AAPH in the presence of CB[8] showed a dependence on the association of the targets with CB[8].
:1 and 2:1 stoichiometries; AAPH@CB[8] and 2AAPH@CB[8], respectively. Radical formation upon photolytic cleavage of AAPH was examined theoretically, and by spin trapping with electron paramagnetic resonance. The radical yields detected with uncomplexed (free) AAPH and the AAPH-CB[6] (exclusion) complex were identical, whereas a marked decrease was shown for AAPH@CB[8]. Lower decreases were seen with a bimolecular (2:1) AAPH-CB[8] inclusion complex (2AAPH@CB[8]). This modulation was corroborated by the consumption of pyrogallol red (PGR), an oxidizable dye that does not associate with CB[6] or CB[8]. AAPH-CB[6] and 2AAPH@CB[8] did not significantly modify the initial consumption rate (Ri) of PGR, whereas AAPH@CB[8] decreased this. The oxidative consumption of free Trp, Gly–Trp and Trp–Gly by radicals derived from AAPH in the presence of CB[8] showed a dependence on the association of the targets with CB[8].
1. Introduction
Cucurbit[n]urils (CB[n]) are macrocyclic hosts consisting of n glycoluril monomers bridged by 2n methylene units, resulting in a ‘hollow pumpkin’-like shape. CB[n] have two identical portals (gates) containing hydrophilic carbonyl groups which exhibit partial negative charges in neutral aqueous media. These gates control the entrance to a hydrophobic cavity where guests and solvent molecules can be included.1 The number of glycoluril monomers contained in CB[n] determines the size of the cavity and the type of the guests that can be included within the structure, with medium-sized CB[n], such as CB[6], CB[7], and CB[8], offering advantages over larger and smaller CB[n]. In the case of CB[6] and CB[8], the vertical gate-to-gate distance is ∼9.2 Å, with the cavity diameters being 6.1 and 8.9 Å, respectively (Fig. 1).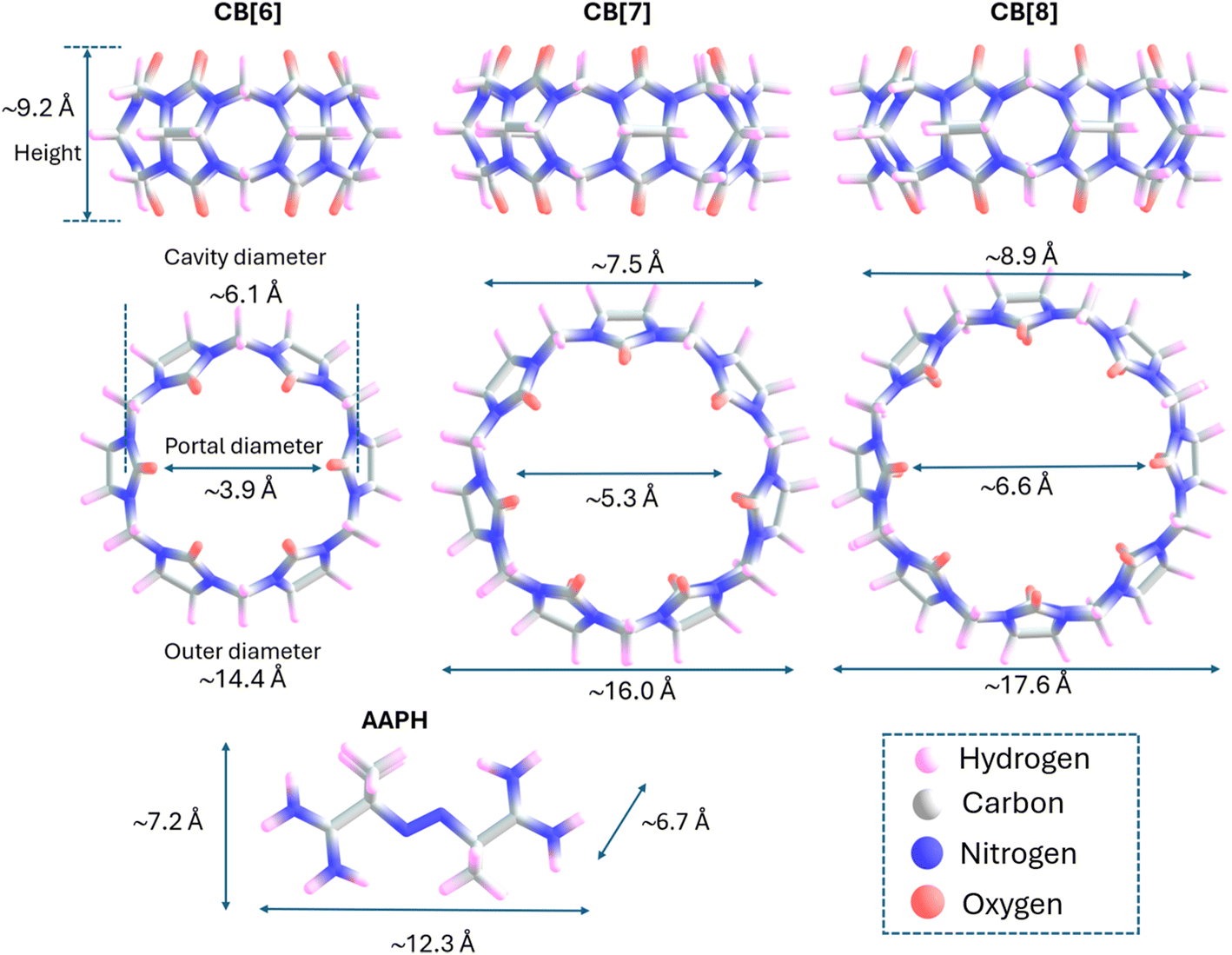 | ||
| Fig. 1 Illustrations of the structures of CB[6], CB[7], CB[8] and AAPH. In each structure dimensions are indicated, which were calculated using Multifwn considering the van der Waals radii (H 1.2, C 1.70, N 1.55, O 1.52). | ||
These hosts have versatile properties and well-known mechanisms for inclusion of hydrophobic guests,2 involving the displacement of high-energy molecules of water improving the association of a variety of guests, as inferred from their reported constants.3,4 The inclusion of species inside CB[6] or CB[8] not only depends on the size and properties of CB[n], but also the size, shape and physicochemical characteristics of the guest compounds.2,5–7 Mecozzi and Rebek8 reported that binding between CB[n] and guests is usually maximal when the guest volume comprises ∼55% of the inner cavity of CB[n]. In line with this, packing coefficients of ∼50% of guests for CB[6], CB[7] and CB[8] complexes, were reported by Nau et al.2 More recently, using computational tools (based on the combination of molecular dynamic simulations and 3-dimensional reference interaction site model theory), Chiangraeng et al.9 investigated the role that water molecules play in the binding of guests to CB[n]. These authors showed that water molecules were released from the cavity of CB[6] as a consequence of guest inclusion, generating a fully dehydrated cavity.9
Inclusion of a variety of compounds has been reported for CB[7],10–12 with these mainly being 1:1 complexes. In contrast, for the larger homologue CB[8], 1:1, 2:1 and 2:2 guest–host stoichiometries have been reported. The 2:1 and 2:2 complexes have been rationalized by the formation of π-stacked or charge transfer pairs in the cavities.12–15 Inclusion complexes with CB[6], which has a smaller cavity, has been reported only for small compounds such as alkyl- and aryl-substituted ammonium ions.16,17 Thus, favorable inclusion of N-butylamine (length, 4.9 Å) inside CB[6] (cavity height 9.1 Å) has been reported.17,18 However, larger derivatives, such as neopentylammonium ion [(CH3)3CCH2NH3+], only associate weakly with CB[6], with this species forming exclusion complexes involving interactions with the external face of the CB[6] gates. Size exclusion effects have also been reported for 2,3-diazabicyclo[2.2.2]oct-2-ene (DBO) which is not included in CB[6], whereas the smaller homologue, 2,3-diazabicyclo[2.2.1]hept-2-ene (DBH), forms an inclusion complex with a binding constant of ∼1300 M−1.19 In contrast, the binding of DBO in the cavity of CB[7] is more than one order of magnitude stronger than DBH.20
The guest–host complexes generated with CB[7] or CB[8] have been used to tune the redox reactions of radicals as a result of stabilization or activation effects.21,22 Recently, the inclusion of AAPH (2,2′-azobis(2-methylpropionamidine) dihydrochloride), a cationic and hydrophilic azo compound, that has been commonly employed as source of peroxyl radicals (ROO˙), into the cavity of CB[7] has been reported.23 A 1:1 supramolecular inclusion complex (AAPH@CB[7]) has been characterized with the generation of this species arising from specific interactions between the amino groups of AAPH, which are protonated (positively charged) at pH 7.4, and the carbonyl functional groups of the CB[7] portals (Kb 2.86 × 105 M−1).23 The height of CB[7] (9.2 Å, Fig. 1) and the distance between the extremes (positive amino groups of AAPH, ∼12.3 Å, Fig. 1) are compatible with noncovalent interactions and formation of the complex. Subsequent homolytic cleavage of the central C–N bonds of AAPH induced by light exposure within the AAPH@CB[7] complex resulted in higher yields of peroxyl radicals when compared to free (uncomplexed) AAPH. This phenomenon was rationalized by theoretical data indicating favorable interactions with AAPH-derived carbon-centered radicals (R˙) within the AAPH@CB[7] complex,23 as well as for other azo compounds,24 opening novel applications of CB[n] in modulating radical reactions.
This study examines whether similar effects occur with CB[6] and CB[8] which have smaller and larger cavities than CB[7] respectively. AAPH-CB[n] interactions, and subsequent photolytically-induced radical formation from AAPH, were studied by isothermal calorimetry titration (ITC), computational methods, electron paramagnetic resonance (EPR) spin trapping, and the oxidation of the redox-sensitive dye pyrogallol red (PGR), free tryptophan (Trp) and Trp-derived peptides. The data obtained indicate that the formation of AAPH-CB[n] complexes can modulate radical yields and allow delicate tuning of radical yields and redox processes in confined environments, with multiple applications.
2. Materials and methods
2.1 Sample preparation
All solvents, chemicals and reagents were obtained from Sigma-Aldrich and used as received. Stock solutions of CB[6] and CB[8] were prepared in ultrapure (Milli-Q) water. Final concentrations of CB[8] were determined by titrating the macrocycle by UV-vis spectroscopy, against known concentrations of the cobaltocenium cation (Cob+).25 Stock solutions of AAPH (0.1–5 mM), pyrogallol red (PGR, 1 mM), free tryptophan (Trp, 2 mM), Gly–Trp and Trp–Gly (1–2 mM) were prepared every day in 10 mM phosphate buffer, pH 7.4.2.2 Isothermal titration calorimetry (ITC)
Microcalorimetric data were obtained using a PEAQ-ITC instrument (Malvern Panalytical), following the procedure described in ref. 23 Briefly, the binding heats between CB[n] and AAPH, PGR, Trp and peptides were studied in 10 mM phosphate buffer (pH 7.4) at 25 °C, and thermodynamic parameters determined with 15–30 injections of 2 μL of titrant solution over the titrate. Injections containing AAPH, PGR, Trp, or peptides (1–2 mM) were added to 0.15 mM CB[6] or 0.06–0.07 mM CB[8]. The heats of reactions were corrected with basal signals obtained by adding the titrated compounds to phosphate buffer.23 Data were processed using software supplied with the PEAQ-ITC instrument and the integrated heat data were further processed using ITC data-fitting software.2.3 Photochemical and spectroscopic experiments
Solutions (1 mL volume) containing 100–200 μM AAPH with or without 100 μM CB[6] or CB[8], were prepared in 10 mM phosphate buffer (pH 7.4), and placed in a quartz cuvette (optical path length 0.3 cm) within a photoreactor (Luzchem Research) equipped with 7 LEDs which generate UV light centered at 365 nm. Solutions were illuminated for 10 min with constant stirring. In specific experiments, PGR (30–500 μM), Trp or peptides (100 μM), were also included. At specific time points, absorption spectra of AAPH, AAPH@CB[8] complexes or PGR were measured using a Hewlett Packard Agilent 8453 spectrometer. Experiments carried out in the presence of Trp and peptides were followed using the intrinsic fluorescence of Trp, with λex 280 nm and emission recorded between 300 and 450 nm using a Fluoromax-4C (Horiba Scientific) instrument. All data were obtained at 25 °C.2.4 EPR measurements
EPR experiments were developed as reported in ref. 23 with minimal changes. Briefly, 200 mM PBN plus 100 or 200 μM AAPH solutions were irradiated at 365 nm (at 25 °C) without or with 100 μM CB[6] or CB[8]. Aliquots were removed at the required time points and placed in capillary tubes, with the EPR spectra of the generated spin adducts recorded using a Bruker EMX spectrometer with the following instrument settings: modulation amplitude 0.1 mT, attenuation 10 dB, microwave power 19 mW, a time constant of 10.24 ms, the center field set to 0.3507 T with a sweep width of 10 mT, and a microwave frequency of 9.84 GHz.2.5 NMR experiments
Solutions of CB[8], AAPH, and a 2:1 (AAPH:CB[8]) mixture, prepared in D2O, were exposed to 365 nm light for 12 min and 1H NMR spectra recorded (62 scans) using a Bruker Avance-200 spectrometer. Non-illuminated solutions were employed as controls and 1H NMR registered under the same setup.
2.6 Computational methods
Structures of CB[6], CB[8], AAPH and the corresponding complexes were optimized using B3LYP-D3(BJ)/6-311G(d,p) level theory in the gas phase, and in water using the Polarizable Continuum Model (PCM) to take account of solvent effects (ε = 78.36).26–30 The B3LYP hybrid function has been widely used to study the structure of CB[n]s and inclusion complexes and has been shown to generate results in agreement with experimental data.23,31,32 The dispersion effects were considered in calculations using the Grimme's dispersion with Becke–Johnson damping D3(BJ).33 The optimized structures of CB[6], CB[7]23 and CB[8] were compared to previous crystallographic data.34,35 The average of root-mean square distances (RMSDs) in all atoms was calculated using Chemcraft software.36 The starting geometries for the complexes were obtained by putting the optimized structure of the guest (AAPH) inside the optimized structure of the macrocycles. Then, the structure of the inclusion complex was optimized and checked with a frequency calculation. The structure of AAPH-derived radicals (R˙) and 2,2,3,3-tetramethylbutane-1,4-bisamidine (the compound generated by self-reaction of two R˙) were investigated as inclusion species inside CB[8]. These structures were obtained after photolytic dissociation of the central C–N bonds of one or two AAPH molecules inside the CB[8] cavity, from either AAPH@CB[8] or 2AAPH@CB[8] complexes (1:1 and 2:1 for the AAPH:CB[8] ratios) and then optimized to give a minimum of energy. All calculations were carried out using the Gaussian 16 software package and atomic charges were obtained from analysis of NBO.37,38 Atomic coordinates of the optimized structures are reported in Table S1.† Analysis of non-covalent interactions (NCI) between CB[n]s and AAPH were obtained using the NCIPLOT software package.39 NCI is an index that identifies favorable and unfavorable non-covalent interactions through a code color. Regions in blue, red, and green represent strong attractive interactions (i.e. dipole–dipole interactions or hydrogen bonds), repulsive interactions (steric effects), and weak interactions (van der Waals), respectively. Definition of a hydrogen bond considered geometrical parameters (the pair-wise distance in the range of 1.84–2.19 Å and the angle between the hydrogen bond donor and the hydrogen bond acceptor from 115° to 167°), as well as the results of the NCI.
2.7 Statistics
Experimental results are given as means (±SD) of data obtained from at least 3 independent experiments each of which was carried out in triplicate. Figures were generated using OriginPro software.3. Results and discussion
3.1 Association of AAPH with CB[6] and CB[8]
Characterization of the inclusion complexes between AAPH and CB[6] and CB[8], was carried out using isothermal titration calorimetry (ITC). The ITC data (Fig. S1†) did not provide evidence of an inclusion complex between AAPH and CB[6], probably due to the small cavity size of this macrocycle. In contrast, the ITC data showed the formation of an inclusion complex with CB[8] with a 2:1 (AAPH:CB[8]) stoichiometry (Fig. 2). The variation point at a molar ratio of 1.0 was reported for the complex AAPH@CB[7];23 while this point was observed at a molar ratio of 0.5 ([CB[8]]/[AAPH] = 0.5) evidencing the formation of a complex with a 2:1 stoichiometry ([AAPH]:[CB[8]], 2AAPH@CB[8]).
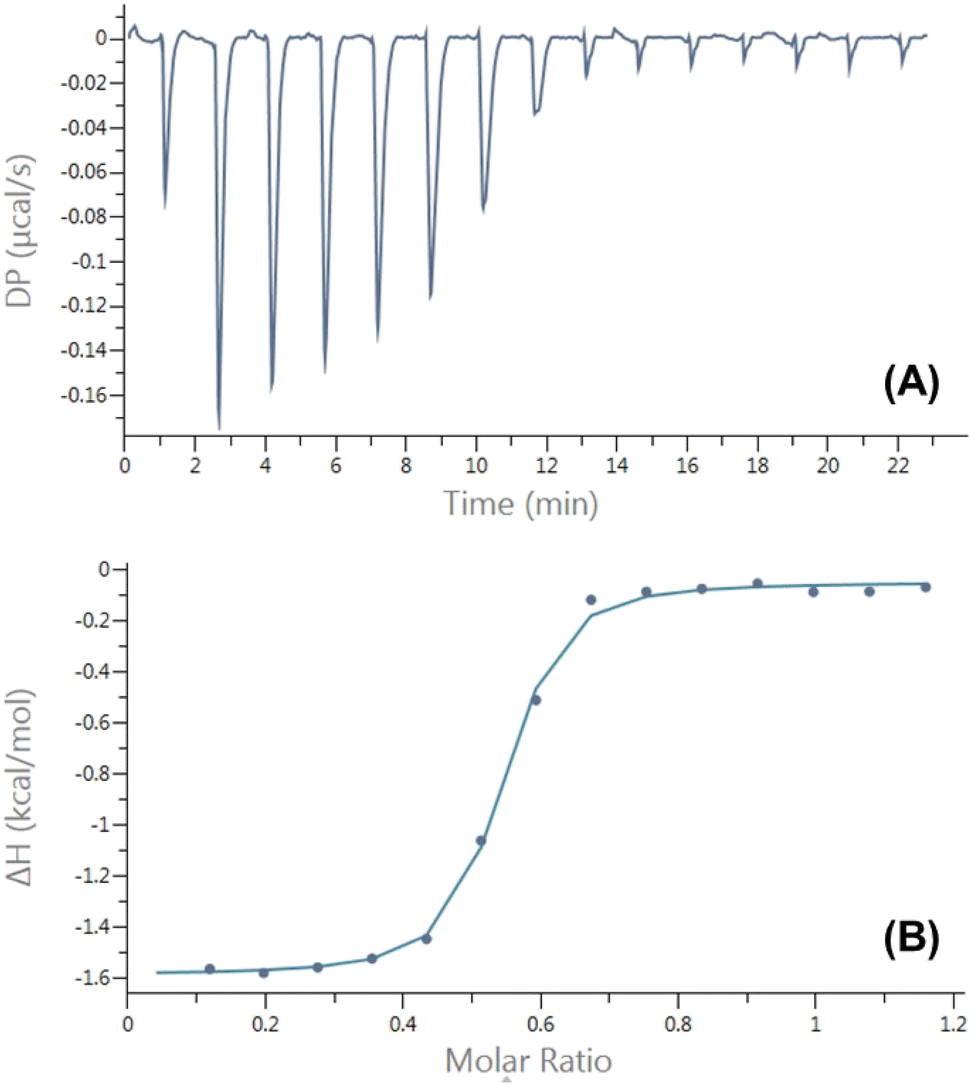 | ||
| Fig. 2 Illustrative enthalpogram for the interaction of AAPH with CB[8] in 10 mM phosphate buffer (pH 7.4) at 25 °C. (panel A) Exothermic heat flows generated as a result of multiple repeated injections of 2 μL of a 2.0 mM AAPH solution into a 100 μM CB[8] solution. (panel B) Plots of the integrated heat data presented as a function of the molar ratio of titrant and titrated components. The control samples consisted of repeat injections of the buffer solution into a CB[8] solution giving heat flows accounting for dilution effects. | ||
The dimensions of the CB[8] cavity (Fig. 1), which is comparable to that of γ-cyclodextrin,40 allow the encapsulation of 2 guest molecules, to form stable (2:1; guest:host) complexes, as reported previously.41 The thermodynamic parameters determined for the inclusion of AAPH and CB[8] were ΔH = (−0.78 ± 0.59) kcal mol−1; TΔS = (7.86 ± 0.53) kcal mol−1, and ΔG = (−8.64 ± 0.27) kcal mol−1. These parameters lead to a binding constant Kb = (3.19 ± 1.28) × 106 M−1; with N = 1.96 ± 0.05 sites. These data are consistent with the binding through an exergonic process with a lesser contribution of enthalpy in comparison to that reported for CB[7].23,40 To obtain a deeper understanding of the inclusion complexes between AAPH and CB[6] and CB[8], computational calculations were performed and compared to those previously reported for the complex between AAPH and CB[7] (AAPH@CB[7]).23 The optimized structures of CB[6], CB[7], and CB[8] isolated presented RMSD values of 0.13 Å, 0.14 Å, and 0.17 Å, respectively, when compared to crystal structure.34,35 In the light of this data, inclusion of AAPH inside the CB[6] and CB[8] was theoretically evaluated using the same level of theory as employed previously for the AAPH@CB[7] complex.23 In agreement with the ITC results, these calculations did not show inclusion of AAPH inside the cavity of CB[6] (Fig. 3A). Only an exclusion complex (AAPH-CB[6]) was found where one of the amidine groups of AAPH interacts with the outside region of the CB[6] portals. The formation of the exclusion complex showed a ΔGf = −10.0 kcal mol−1 in water (ΔGf = −83.7 kcal mol−1 in the gas phase) relative to the independent molecules. This indicates that the exclusion complex has a weak association, which is in line with other data for cations and CB[6].42,43 Inclusion of AAPH in the cavity of CB[8] was also examined theoretically with successful inclusion of 1 and 2 molecules of AAPH to form 1:1 (AAPH@CB[8]) and 2:1 (2AAPH@CB[8]) complexes (Fig. 3B and C). In the former case, the formation of AAPH@CB[8] was favorable by ΔGf = −40.3 kcal mol−1 in water (ΔGf = −135.5 kcal mol−1 in the gas phase) when compared to the independent molecules. In the latter case, the formation of 2AAPH@CB[8] was favorable by ΔGf = −45.1 kcal mol−1 in water (ΔGf = −54.9 kcal mol−1 in the gas phase). The ΔGf values for the formation of the complexes were more positive (less favored) in water than the gas phase, with this attributed to effects of polarization of the implicit solvent increasing the solute–solvent interactions and decreasing the strength of host–guest interactions.
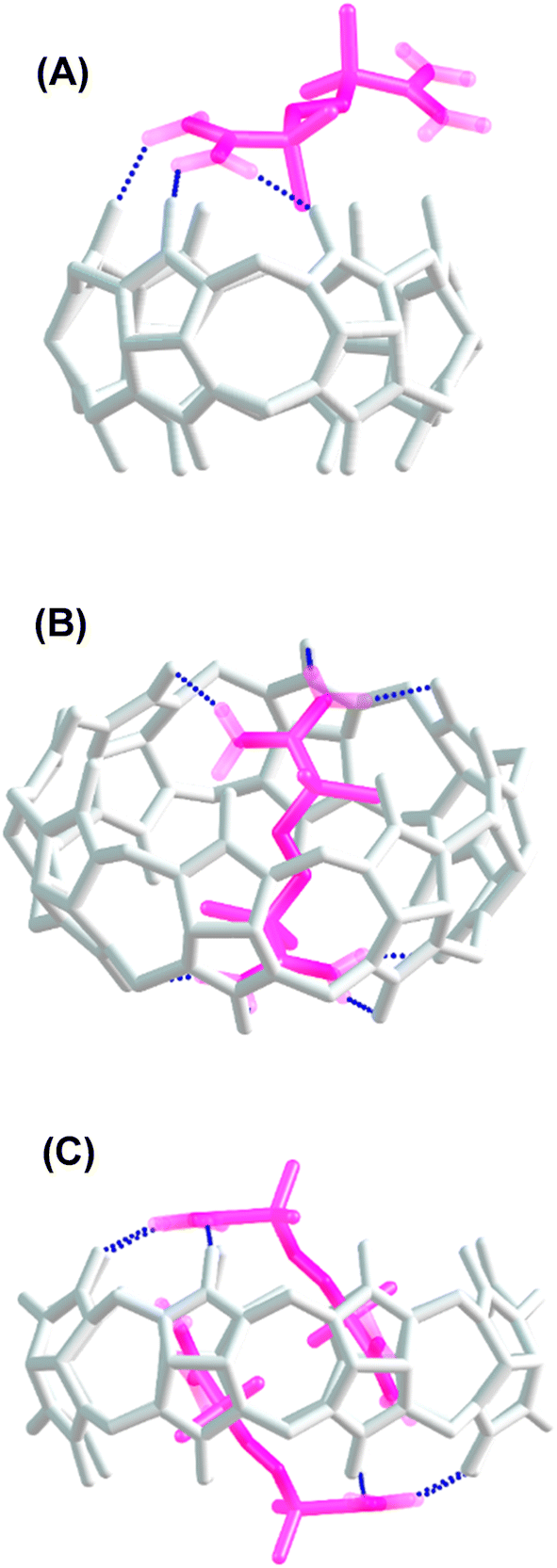 | ||
| Fig. 3 Structure of the complexes formed of AAPH with CB[6] and CB[8]. (panel A) Depicts the exclusion complex between AAPH and CB[6] (AAPH-CB[6]), while (panel B and C) show the inclusion complex of AAPH and CB[8], at 1:1 (AAPH@CB[8]) and 2:1 (2AAPH@CB[8]) stoichiometries, respectively. | ||
In agreement with the thermodynamic data, the formation of the 2AAPH@CB[8] complex in water (ΔGf = −45.1 kcal mol−1) is slightly more favored than the formation of the AAPH@CB[8] complex (ΔGf = −40.3 kcal mol−1) and also the previously reported AAPH@CB[7] complex (ΔGf = −35.2 kcal mol−1).23 This may be related to changes in the geometry adopted by AAPH inside of the cavities of CB[7] versus CB[8]. Since the cavity and portal diameter of CB[8] is greater than CB[7], the AAPH molecule is more relaxed (less rigidly constrained) inside the cavity of CB[8] than CB[7]. This is supported by the calculated conformation energy of AAPH inside CB[8] being 6.6 kcal mol−1 lower than for AAPH within CB[7]. Furthermore, the larger cavity of CB[8] also allowed the formation of a 2AAPH@CB[8] complex. The computational calculations showed that the 2 AAPH molecules were not completely located inside the CB[8] cavity with these situated diagonally when the complex is viewed side-on (Fig. 3C). In this molecular arrangement, one amidine group of each AAPH showed interactions with the portals of CB[8] whilst the other amidine was involved in interactions within the cavity. Additionally, the inclusion of a pair of AAPH molecules inside CB[8] distorted the cavity shape to such extent that this adopted an elliptical shape with dimensions of 20.1 Å × 17.2 Å, compared to the circular shape of isolated CB[8] which has a diameter of ∼17.6 Å.
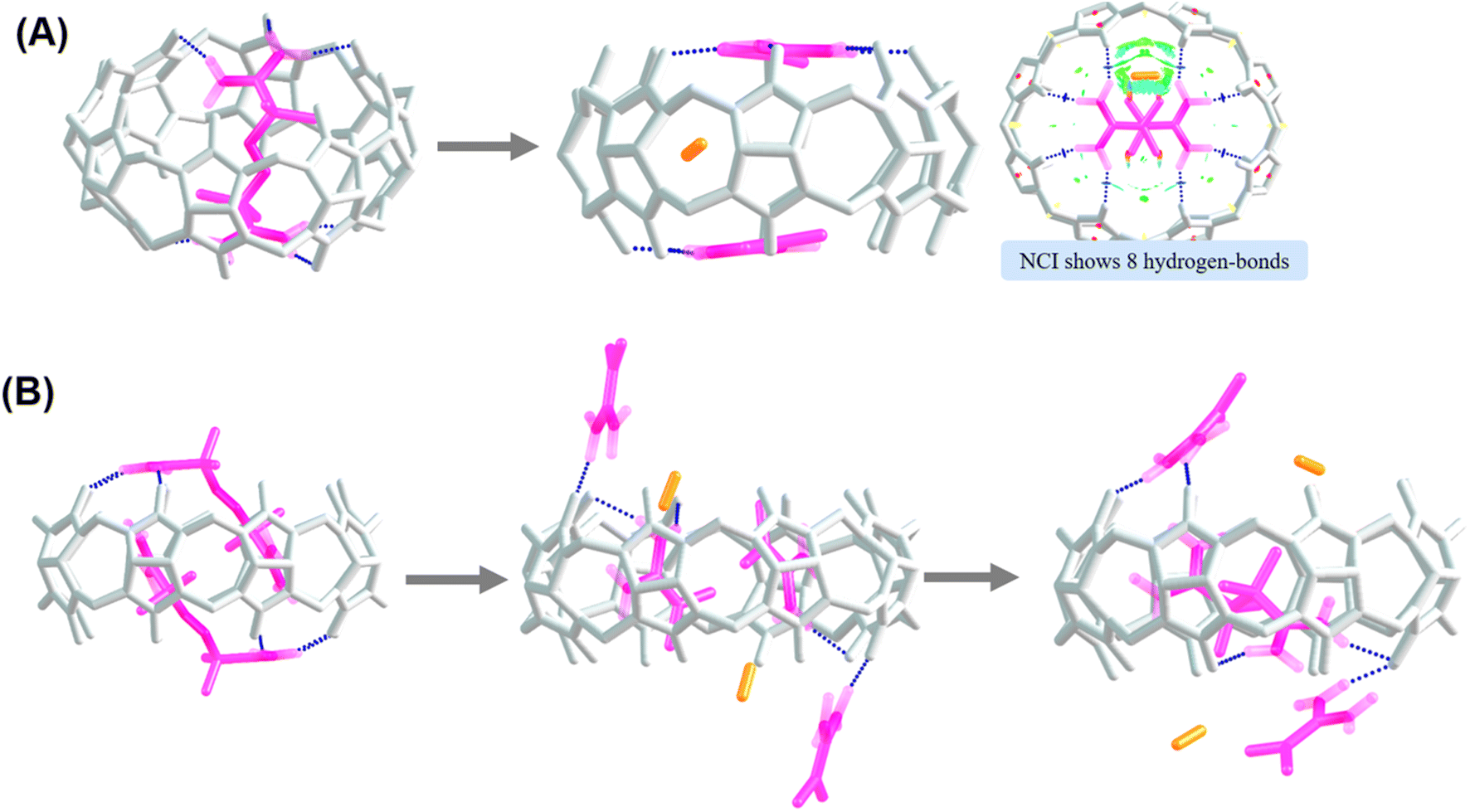
Analysis of noncovalent interactions in the exclusion complex of AAPH and CB[6] indicates that the interactions of AAPH with CB[6] occur via 3 hydrogen bonds (blue regions in Fig. 4A). Conversely, the AAPH@CB[8] complex (Fig. 4B) showed 7 hydrogen bonds involving the (protonated) amino groups of AAPH and the carbonyl moieties of the CB[8] portals; these data are consistent with the 8 hydrogen bonds reported for the AAPH@CB[7] complex.23 Examination of noncovalent interactions in the 2AAPH@CB[8] complex (Fig. 4C), showed that each AAPH molecule interacts with CB[8] through 4 hydrogen bonds between the amidine group of AAPH that is located outside of the cavity of CB[8] and the carbonyl groups of the portal. Within the CB[8] cavity, increased van der Walls interactions involving the two molecules of AAPH were observed (green regions in Fig. 4C).
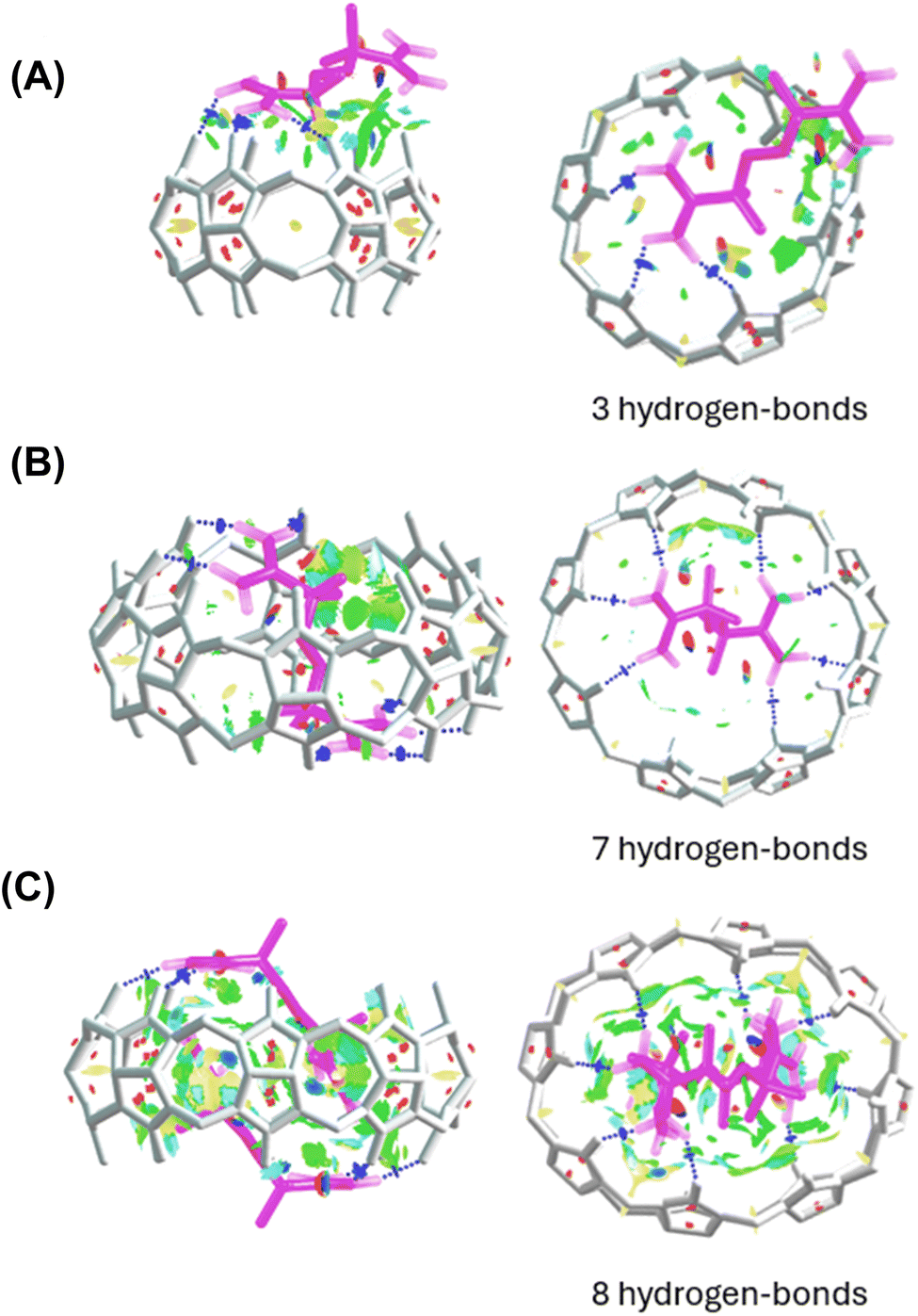 | ||
| Fig. 4 Noncovalent interactions involved in the exclusion complex of AAPH and CB[6] (panel A), and inclusion complexes AAPH@CB[8] (panel B) and 2AAPH@CB[8] (panel C). Each panel depicts side-on (left) and top (right) views of each complex. | ||
The AAPH-CB[6] exclusion complex, and the inclusion species AAPH@CB[8] and 2AAPH@CB[8], were also studied by mapping the molecular electrostatic potential (MEP). CB[6], CB[8] and AAPH were individually characterized through their MEP, elucidating their respective contributions to the electronic density of AAPH-CB[6], AAPH@CB[8], and 2AAPH@CB[8] complexes. The results are presented in Fig. S2† with the red and blue areas indicating high and low electron density zones, respectively. Similar to the data reported for CB[7],23 higher electron density zones (yellow), located near the carbonyl groups of the portals of CB[6] and CB[8], were determined (Fig. S2A and S2B,† respectively). In the case of the optimized structure of AAPH, two zones of low electron density (blue) associated with the two positively charged terminal amino groups, were observed (Fig. S2C†). In the MEP of AAPH-CB[6], AAPH@CB[8] and 2AAPH@CB[8], the individual zones of electron density of AAPH and CB[8] were partially neutralized. The light blue regions indicated for AAPH-CB[6] and AAPH@CB[8], emphasize the stabilization associated with complex formation (Fig. S2D and S2E†). In the 2AAPH@CB[8] complex, blue regions around the whole complex were observed, indicating low electron density due to the four positive charges of two AAPH molecules distributed in the 2AAPH@CB[8] complex (Fig. S2F†). The stabilization of the electrostatic interactions in the AAPH@CB[8] and 2AAPH@CB[8] complexes can be also analyzed from the atomic charges of the carbon and oxygen atoms of the carbonyl moieties of CB[8] and the hydrogen and nitrogen atoms of the amino groups of AAPH. The results depicted in Fig. S3,† with the color code designating green and red for positive and negative charges, respectively, show that the average atomic charges of carbon (qC) and oxygen (qO) atoms in the carbonyl groups of CB[8] are 0.862 and −0.694 in the AAPH@CB[8] complex, respectively. While in the 2AAPH@CB[8] complex, qC = 0.862 and qO = −0.691 were determined. The charge of the hydrogen atoms of the amidine groups of AAPH was 0.433 for AAPH@CB[8] and 0.436 for 2AAPH@CB[8]. These values were similar to those reported for CB[7] (qC ≈ 0.863|e|, qO ≈ −0.697|e| and qH ≈ 0.432|e|)23 providing evidence of similar electrostatic interactions involving the amidine groups of AAPH and the carbonyls of the CB[8] and CB[7] portals.
As a whole, these results could suggest that thermodynamic data obtained by ITC (ΔH = −0.78 ± 0.59 kcal mol−1) for the 2AAPH@CB[8] complex can be explained by a mixture of effects including a significant, but partial, release of water molecules from the CB[8] cavity, repulsive interactions between AAPH molecules, and distortions of the shape of CB[8] in the complex. Although there are multiple factors affecting the formation of the complex, the slightly negative value of ΔH determined by ITC would indicate its favorable formation, probably explained by favorable non-covalent interactions. TΔS and ΔH values obtained from DFT calculations were −18.6 and −59.0 kcal mol−1 for the AAPH@CB[8] complex, and −42.9 and −88.0 kcal mol−1 for 2AAPH@CB[8], respectively. The more negative values of ΔH obtained by the DFT analysis than those determined by ITC for 2AAPH@CB[8] could be explained by disparities in the consideration of the factors affecting the formation of the complex.
3.2 Generation of radicals by photolysis of AAPH
As the AAPH@CB[7] complex has been reported to generate an enhanced flux of radicals on photolysis at 365 nm,23 we explored the influence of CB[6] and CB[8] on the photolytic generation of AAPH-derived radicals. Solutions containing 100 and 200 μM AAPH in the presence and absence of CB[8], were irradiated at 365 nm for 10 min. When CB[8] was absent, solutions of 100 or 200 μM AAPH induced an increase in the intensity of the absorption at 240 nm (Fig. S4†), which is ascribed to product(s) generated from the photochemical cleavage of AAPH. Photolysis of the AAPH@CB[8] complex gave a similar pattern of changes at 240 nm (Fig. S4A,† insert).The production of AAPH-derived radicals was examined using EPR spectroscopy in conjunction with the spin trapping agent α-phenyl-N-t-butylnitrone (PBN, 200 mM). In the absence of CB[8], irradiation of solutions containing 100 or 200 μM AAPH and 200 mM PBN for 5 min at 365 nm gave EPR signals (Fig. 5, black spectra in panels A and B, respectively) interpreted as a triplet (1:1:1; aN 1.5 mT) of doublets (1:1; aH 0.4 mT). Spectra with these types of coupling constants have been previously assigned to alkoxyl radicals (RO˙),44 but the large aH coupling is more consistent with the trapping of alkyl radicals,45 and therefore these spectra are assigned to adducts of R˙ formed on photolysis of AAPH. Fig. 5, shows EPR signals arising from the photolysis (5 min at 365 nm) of the AAPH@CB[8] and 2AAPH@CB[8] complexes (red spectra in panels A and B, respectively) in the presence of 200 mM PBN. The parameters of the signals are identical to those determined in the absence of CB[8] (i.e. are ascribed to R˙ adducts) but the signals were less intense (i.e. lower radical adduct concentrations), with the AAPH@CB[8] complex generating ∼45% of the intensity determined with free AAPH (Fig. 5A). The 2AAPH@CB[8] complex also yielded lower radical adduct intensities than free AAPH, with a decrease of ∼30%. These data are interpreted in terms of a lower extent of radical trapping by PBN, particularly in the case of the AAPH@CB[8], with the CB[8] complex shielding the AAPH radicals from reaction with PBN. For the corresponding AAPH-CB[6] exclusion complex, the same pattern and signal intensities were detected as for free AAPH (Fig. S5†).
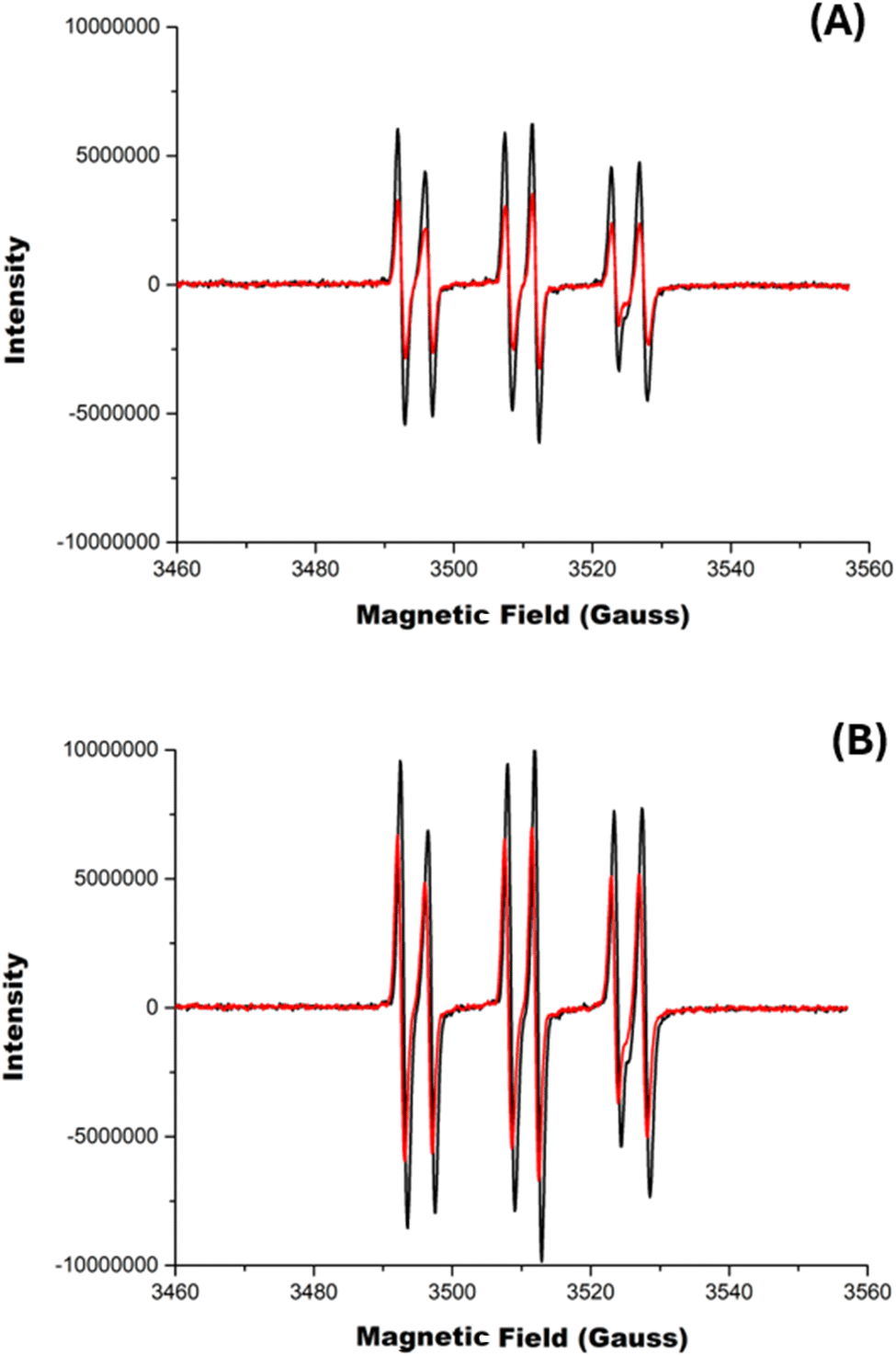 | ||
| Fig. 5 Electron paramagnetic resonance (EPR) spectra detected on reaction of the spin trap PBN with carbon centered radicals (R˙) arising from photolysis of AAPH in the presence or absence of CB[8]. Phosphate buffer solutions containing 200 mM PBN and 100 μM (panel A) and 200 μM (panel B) of AAPH were irradiated for 5 min at 365 nm in the presence (red spectrum) or absence (black spectrum) of 100 μM CB[8]. Samples from the reaction mixtures were removed and analyzed immediately by EPR spectroscopy (15 scans). | ||
3.3 Oxidation of pyrogallol red (PGR) elicited by radicals generated from photolysis of AAPH
PGR is a dye molecule that is readily oxidized by AAPH-derived radicals via a known reaction mechanism,46 but does not associate with CB[8], allowing the consumption and kinetics of oxidation by AAPH-derived species to be assessed quantitatively by visible spectrophotometry. This was determined for both free AAPH and the AAPH@CB[8] and 2AAPH@CB[8] complexes. The kinetic profile of PGR consumption (followed at 560 nm) was altered for AAPH@CB[8] (Fig. 6A) when compared to free AAPH, with a decrease in the initial PGR consumption rate (Ri) from ∼53 μM min−1 to ∼17 μM min−1.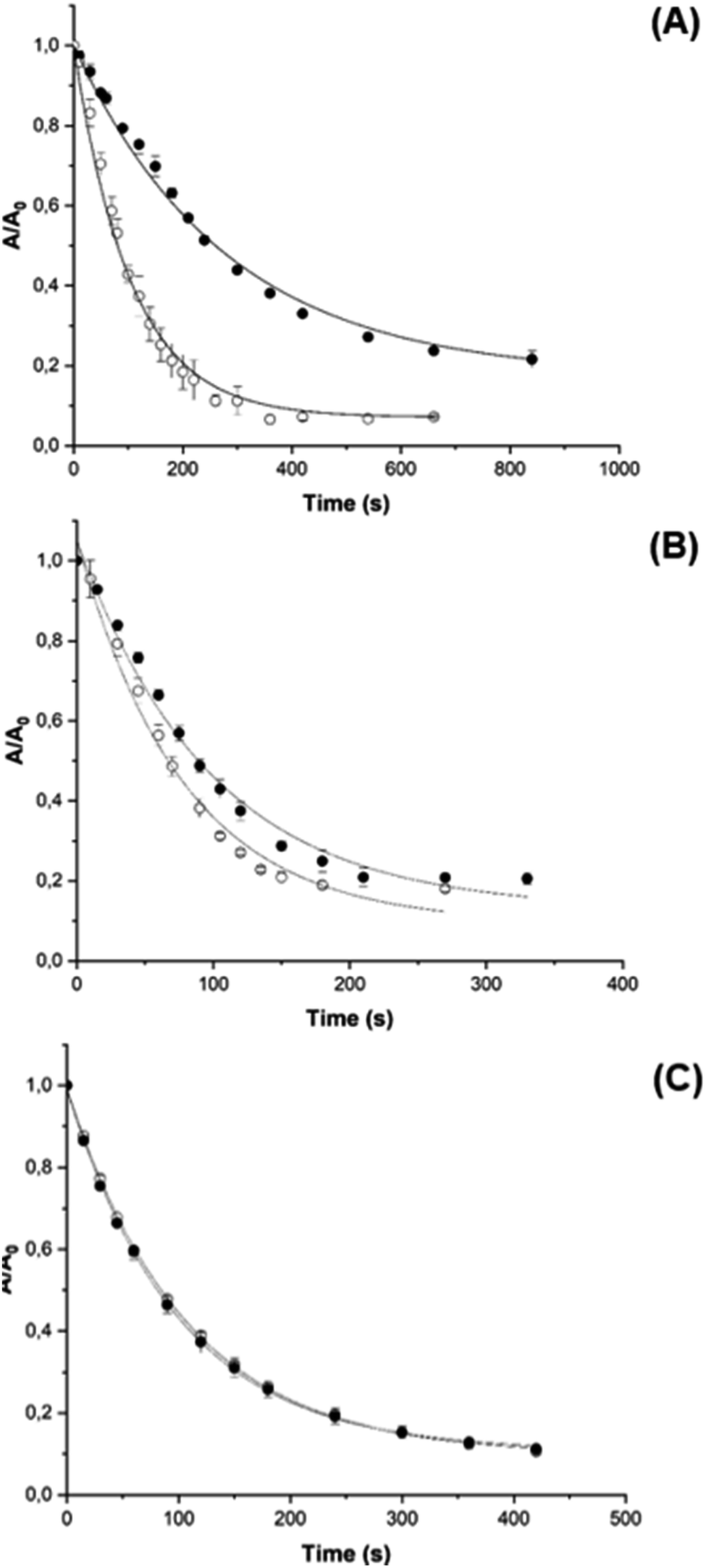 | ||
| Fig. 6 Kinetics of PGR oxidation induced by radicals generated by photolysis of AAPH, in the absence (open circles) and in the presence of 100 μM CB[8] or CB[6] (black circles). Solutions of PGR (100 μM) and AAPH (100 or 200 μM) with or without CB[8] or CB[6] were irradiated at 365 nm for 10 min. At defined times absorption spectra were recorded and the intensity at 560 nm plotted versus irradiation time. (panel A) Shows the consumption of PGR induced by solutions containing AAPH@CB[8], while (panels B and C) show 2AAPH@CB[8], and AAPH-CB[6] complexes, respectively. | ||
This decrease is consistent with a decreased flux of radicals (∼33%) available to react with PGR, if it is assumed that the PGR consumes all the radicals generated from AAPH.46 For the 2AAPH@CB[8] complex (Fig. 6B), Ri was not markedly affected, with a value of ∼56 μM min−1, compared to 74 μM min−1 for 200 μM free AAPH. These data imply that the 2AAPH@CB[8] complex mimics the effect of a solvent cage, where half of the R˙, generated on cleavage of the central C–N bonds in AAPH, escape to react with O2 producing ROO˙.47 The AAPH-CB[6] exclusion complex did not affect the kinetic profile of PGR consumption, giving similar values of Ri to that detected in the absence of the complex (Fig. 6C). These data are in line with the EPR spin trapping data (Fig. S5†), corroborating that AAPH is not included within the CB[6] cavity.
Although a similar effect on the consumption of PGR was observed for the AAPH-CB[6] exclusion complex, and the inclusion complex 2AAPH@CB[8], the origin of both results is believed to be different. In the former case, PGR consumption is triggered by radicals generated in bulk solution, with only the solvent cage modulating the yield of radicals available to react with PGR. For the 2AAPH@CB[8] complex, the cavity of CB[8] mimics this cage, with data suggesting that two of the radicals generated within the complex self-react to give non-radical products, whilst the other two radicals (presumed to be those generated at the portals of CB[8]) are available to react with PGR. These data agree with the experimental ITC and EPR results and the computational calculations. A ‘pull-off’ effect on the AAPH-generated R˙ has been proposed to explain the increased consumption of PGR and Trp residues elicited on photolysis of the AAPH@CB[7] complex.23 This effect does not appear to occur for the complexes formed between AAPH and CB[8].
Computational studies were also carried out to explore the fate of the AAPH derived radicals in the AAPH@CB[8] and 2AAPH@CB[8] complexes. For such purpose, the C–N bonds of AAPH molecules included in both complexes were separated, the multiplicity changed, and the structures optimized. The structures of the two R˙ generated from the AAPH@CB[8] complex are shown in Fig. 7A. As shown, once the R˙ are generated, these are stabilized by interactions with the carbonyls of the CB[8] portals. This is consistent with the lack of a ‘pull-off’ effect in the CB[8] based system where the dimensions of the portals (which are greater than for CB[7]) allow stabilization of the R˙. The stabilization is linked with non-covalent interactions involving 8 hydrogen bonds between the amino groups present on the R˙ with the carbonyl functions of CB[8]. The difference between the free energy of the structure of R˙ interacting with of CB[8] and the AAPH@CB[8] complex is ΔG = −12.7 kcal mol−1 in water (ΔG = −40.3 kcal mol−1 in the gas phase) supporting a favorable stabilization of the interactions of R˙ with CB[8]. The structure of the four R˙ generated by homolysis of the central C–N bonds of the two AAPH molecules of the 2AAPH@CB[8] complex is shown in Fig. 7B. The optimized structure for the four R˙ showed that two R˙ were located inside the CB[8] cavity, while the other two R˙, together with the two molecules of N2, are present outside the cavity. The difference in the free energies between the four R˙ interacting with CB[8] and the 2AAPH@CB[8] complex is ΔG = ∼−30 kcal mol−1 in water and ΔG = −36.1 kcal mol−1 in gas phase. The stabilization inferred by these values is consistent with the presence of two R˙ inside the CB[8] cavity, while the other two R˙ are released into the bulk solvent with only weak interactions detected (Fig. 7B). It is important to note that this complex also lacks of a pull-off effect of the R˙ since the AAPH molecules which generate the released R˙, were originally exposed to the solvent. As the two R˙ located inside the CB[8] cavity are likely to self-react to give 2,2,3,3-tetramethylbutane-1,4-bisamidine, we studied the interactions of this species within the CB[8] cavity. The optimized structure for this compound within the cavity of CB[8], yielded a free energy difference between this structure and the 2AAPH@CB[8] complex of ΔG = −59.1 kcal mol−1 in water, and ΔG = −63.4 kcal mol−1 in the gas phase (Fig. 7B). These values confirm that recombination of two AAPH-derived R˙ within the 2AAPH@CB[8] complex is thermodynamically viable.
 | ||
| Fig. 7 Formation of AAPH-derived R˙ from AAPH@CB[8] (panel A), and 2AAPH@CB[8] (panel B). The nitrogen molecule is represented in orange color. (panel B) Also shows the formation of 2,2,3,3-tetramethylbutane-1,4-bisamidine from 2AAPH-derived R˙ in the 2AAPH@CB[8] complex. | ||
Together, these results reveal that the R˙ arising from the AAPH@CB[8] and 2AAPH@CB[8] complexes on photolysis have different fates, while R˙ would be stabilized in the former complex, two R˙ would be released, and two neutralized inside the cavity, for the 2AAPH@CB[8] complex. Aimed to get new insights about this, we carried out 1H-NMR analysis on the 2AAPH@CB[8] complex before and after illumination at 365 nm. As presented in Fig. S6,† the spectrum of AAPH showed a singlet signal at 1.55 ppm corresponding to the methyl protons. While the spectrum of CB[8] showed two doublets (between 5.86 and 4.21 ppm), and a singlet signal at 5.55 ppm, corresponding to the –CH2, and the –CH protons, respectively. Spectra obtained for the mixture of AAPH and CB[8] evidenced a clear displacement of the singlet signal of AAPH at 0.83 ppm reflecting inclusion of AAPH inside the cavity of CB[8]. Nonetheless, the singlet of AAPH at 1.55 ppm was also detected in the presence of CB[8] showing the presence of free AAPH in the solution. This suggest that, in spite of solutions were prepared in a 2:1 molar ratio (AAPH:CB[8]), the low solubility of CB[8] in deuterium water would limit the stoichiometric formation of the complexes. After illumination the singlet signal of AAPH (free and included in CB[8]) was not detected reflecting its light-mediated consumption. Irrespective of the presence of CB[8], new signals were detected demonstrating the photolytic formation of AAPH products.
3.4 Oxidation of Trp residues mediated by radicals derived from AAPH photolysis
As the AAPH@CB[8] complex showed a lower extent of consumption of PGR than free AAPH, we examined whether this effect also occurs for targets with different binding constants with CB[8]. Free Trp, Gly–Trp and Trp–Gly were examined as these show variable binding affinities with CB[8], varying from non-binding for Gly–Trp to binding constants of ∼103 to ∼105 M−1 for Trp and Trp–Gly, respectively (ITC data presented in Fig. S7† and Table 1). These findings are coherent with an absence of binding of Gly–Trp to CB[7] (Table 1 and other reports)23 and the association of Trp (1:1) with CB[8] with a binding constant of approximately 104 M−1.48–50 Table 1 also provides corresponding ITC data for the CB[6] system. In agreement with the smaller cavity of CB[6], and previous data obtained using X-ray crystal diffraction, the interactions between CB[6] and Gly–Trp occur mostly outside the CB[6] cavity.51
| Kb (M−1) | N (sites) | ΔH | −TΔS | ΔG | |
|---|---|---|---|---|---|
| CB[6] | |||||
| AAPH | No binding | ||||
| Trp | No binding | ||||
| Trp–Gly | No binding | ||||
| Gly–Trp | No binding | ||||
|
|||||
| CB[7] | |||||
| AAPH23 | 2.51 ± 0.76 × 105 | 1.075 ± 0.01 | −2.27 ± 0.11 | −5.10 ± 0.17 | −7.37 ± 0.02 |
| Trp23 | 7.94 ± 1.07 × 102 | 1 | −3.22 ± 0.46 | −0.73 ± 0.53 | −3.95 ± 0.01 |
| Trp–Gly23 | 3.48 ± 1.55 × 104 | 0.67 ± 0.052 | −11.50 ± 1.12 | −5.47 ± 1.29 | −6.02 ± 0.25 |
| Gly–Trp23 | No binding | ||||
|
|||||
| CB[8] | |||||
| AAPH | (3.19 ± 1.28) × 106 | 1.96 ± 0.05 | −0.78 ± 0.59 | −7.86 ± 0.53 | −8.64 ± 0.27 |
| PGR | No binding | ||||
| Trp | (5.18 ± 1.19) × 103 | 2.01 ± 0.09 | −7.89 ± 1.46 | −2.83 ± 1.60 | −5.06 ± 0.14 |
| Trp–Gly | (1.49 ± 0.29) × 105 | 2.00 ± 0.12 | −6.48 ± 0.32 | −0.58 ± 0.43 | −7.05 ± 0.11 |
| Gly–Trp | No binding | ||||
Consumption of free Trp and Gly–Trp and Trp–Gly, elicited by AAPH photolysis, either free or from the AAPH@CB[8] complex, was studied by examining changes in the fluorescence intensity arising from the indole ring of Trp. When solutions of free Trp, or Trp-containing peptides, were irradiated in the presence of 100 μM AAPH, a constant decrease in Trp fluorescence was observed (Fig. 8A, B), with this decreasing to ∼60% of the initial intensity after 4 min.
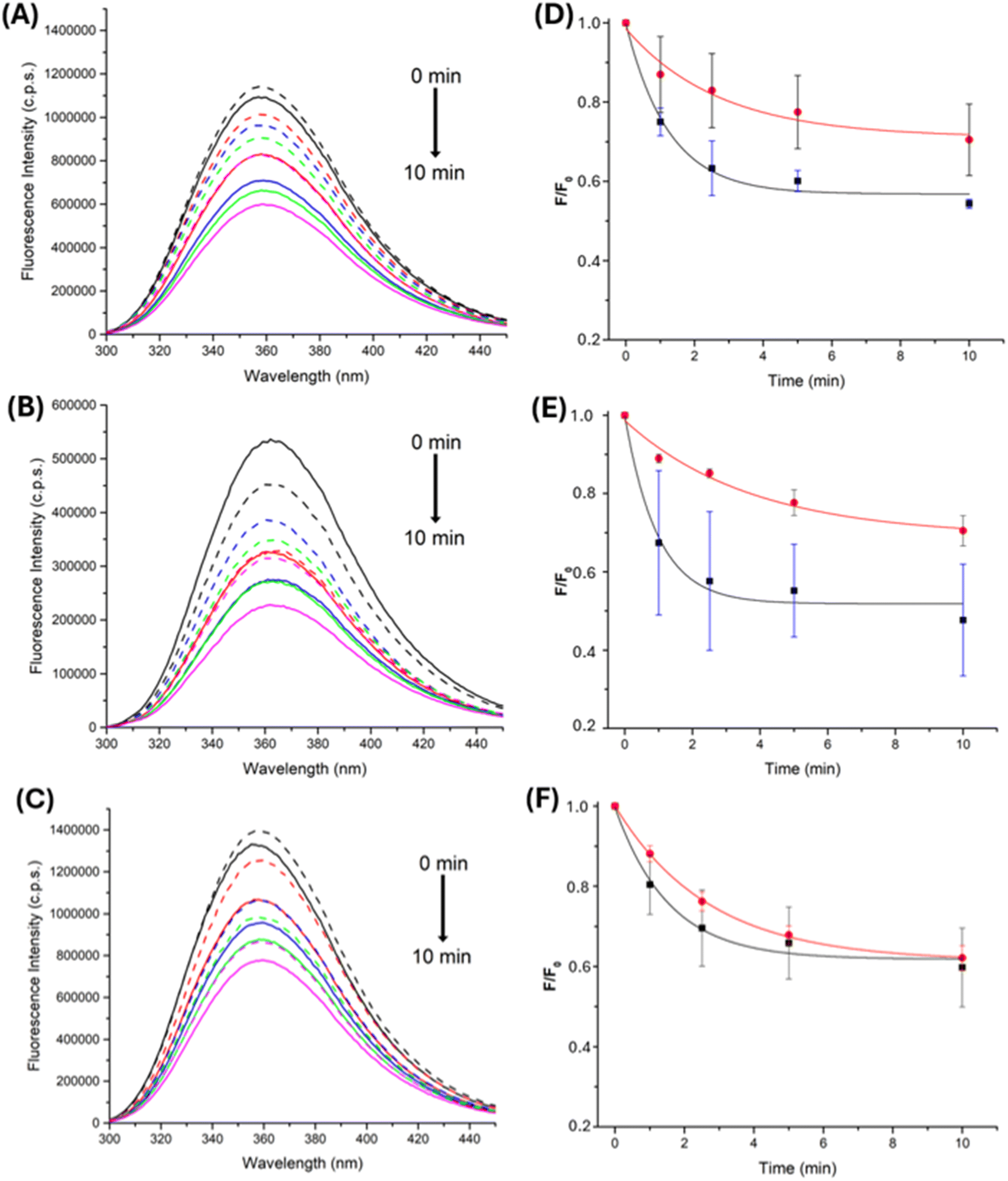 | ||
| Fig. 8 Fluorescence spectra were recorded from solutions containing 100 μM of (A) Trp, (B) Gly–Trp and (C) Trp–Gly and AAPH (100 μM) without (solid lines) or with (dashed lines) 100 μM CB[8], registered at 1, 2.5, 5 and 10 min (black, red, blue, orange, and purple lines, respectively) irradiations at 365 nm. Emission spectra were obtained using λex 280 nm. (panels D–F) Dependence of the fluorescence intensity over time (λem 360 nm) during the irradiation of 100 μM AAPH and 100 μM Trp (D) Gly–Trp (E) and Trp–Gly (F) in the absence (black square symbols) and presence (red circles) of 100 μM CB[8]. | ||
Similar experiments, with the AAPH@CB[8] complex, showed different effects, with decreased consumption for of Trp and Gly–Trp, and minimal effects on Trp–Gly (Fig. 8D–F). These results are interpreted in terms of association of each target with CB[8] and the ability of the complex to retain the generated R˙. As free Trp has a binding constant approximately three order of magnitude lower than AAPH ((3.19 ± 1.28) × 106 M−1, Table 1), and Gly–Trp does not associate with CB[8] (Table 1), reactions of these targets with radicals would be more hindered than for Trp–Gly which has a binding constant only one order of magnitude lower than AAPH (Table 1), with the latter probably being more exposed to AAPH-generated radicals. The consumption data obtained for free Trp, Gly–Trp and Trp–Gly, on photolysis of the 2AAPH@CB[8] complex, showed minimal changes to that detected for free AAPH, suggesting R˙ release with a similar efficiency to the solvent cage.
4. Conclusions
The association between AAPH with the medium-sized cucurbiturils, CB[6] and CB[8], is modulated by specific interactions, and the dimensions of the cavity and portals of the hosts. This results in the generation of an exclusion complex with CB[6] (AAPH-CB[6]) and 1:1 and 2:1 inclusion complexes with CB[8]; AAPH@CB[8] and 2AAPH@CB[8], respectively. The yield of radicals generated by homolytic cleavage of AAPH inside the CB[8] cavity is affected by the complex stoichiometry, with the yield decreased for AAPH@CB[8] and minimally affected for 2AAPH@CB[8]. The latter system mimics the effects of a solvent cage on radical production as inferred from EPR spin trapping of the radicals, and consumption of pyrogallol red and Trp residues. Photolysis of AAPH@CB[8] resulted in decreased pyrogallol red consumption, and mediated the oxidation of Trp, Gly–Trp, and Trp–Gly. The data show that the effects of the cucurbiturils not only depends on its own properties, as association of the targets with CB[8] also plays a pivotal role. These results indicate that complexation of oxidant-generating systems, such as AAPH, by cucurbiturils can be used to modulate redox processes. This approach may be of use in different fields where oxidation of specific substrates is desired, or necessary (e.g. to remove pollutants in biological systems). However, interaction of the host cucurbituril with the oxidizable target also needs to be considered.
Data availability
The dataset employed in this research is incorporated in the manuscript itself and in the ESI.†Author contributions
AFG and MO carried out theoretical and experimental work (DFT, ITC, fluorescence, photolysis). DF designed ITC experiments and collaborated in discussion of the results and KD carried out experimental work (EPR, ITC and fluorescence, photolysis). AFG, SG, BH and ATL designed theoretical experiments, collaborated in discussion of the results. MJD and EFL contributed to the analysis of results and manuscript preparation. MEA and CLA conceived the project, provided overall direction and coordination, contributed to manuscript preparation and obtained financial support. All authors contributed to writing the paper, reviewed the data, and approved submission of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Fondecyt grants no. 1220459 (to CLA), 1210751 (to MEA), 1241224 (ATL) and Postdoctoral funding 3240525 (to AFG). Authors also thank FONDEQUIP (EQM170120, EQM180103) for equipment grants. KDM acknowledges the ANID Doctoral Fellowship Folio No. 21210424. MJD is supported by the Novo Nordisk Foundation (grant: NNF20SA0064214).Notes and references
- C. Marquez and W. M. Nau, Angew. Chem., Int. Ed., 2001, 40, 4387–4390, DOI:10.1002/1521-3773(20011203)40:23<4387::AID-ANIE4387>3.0.CO;2-H.
- W. M. Nau, M. Florea and K. I. Assaf, Isr. J. Chem., 2011, 51, 559–577, DOI:10.1002/ijch.201100044.
- F. Biedermann, V. D. Uzunova, O. A. Scherman, W. M. Nau and A. De Simone, J. Am. Chem. Soc., 2012, 134, 15318–15323, DOI:10.1021/ja303309e.
- F. Biedermann, W. M. Nau and H. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11158–11171, DOI:10.1002/anie.201310958.
- T.-C. Lee, E. Kalenius, A. I. Lazar, K. I. Assaf, N. Kuhnert, C. H. Grün, J. Jänis, O. A. Scherman and W. M. Nau, Nat. Chem., 2013, 5, 376–382, DOI:10.1038/nchem.1618.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418, 10.1039/C4CS00273C.
- K. I. Assaf, M. Florea, J. Antony, N. M. Henriksen, J. Yin, A. Hansen, Z. Qu, R. Sure, D. Klapstein, M. K. Gilson, S. Grimme and W. M. Nau, J. Phys. Chem. B, 2017, 121, 11144–11162, DOI:10.1021/acs.jpcb.7b09175.
- S. Mecozzi and J. Rebek Jr, Chem.–Eur. J., 1998, 4, 1016–1022, DOI:10.1002/(SICI)1521-3765(19980615)4:6<1016::AID-CHEM1016>3.0.CO;2-B.
- N. Chiangraeng, H. Nakano, P. Nimmanpipug and N. Yoshida, J. Phys. Chem. B, 2023, 127, 3651–3662, DOI:10.1021/acs.jpcb.3c00343.
- S. S. R. Kommidi and B. D. Smith, J. Org. Chem., 2023, 88, 8431–8440, DOI:10.1021/acs.joc.3c00423.
- F. Zatloukal, E. Achbergerová, D. Gergela, M. Rouchal, L. Dastychová, Z. Prucková and R. Vícha, Dyes Pigm., 2021, 192, 109420, DOI:10.1016/j.dyepig.2021.109420.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980, DOI:10.1021/cr200213s.
- F. Tian, D. Jiao, F. Biedermann and O. A. Scherman, Nat. Commun., 2012, 3, 1207, DOI:10.1038/ncomms2198.
- F. Zhou, J. Wang, Y. Zhang, Q. Wang, C. Guo, F. Wang and H. Zhang, Theor. Chem. Acc., 2019, 138, 50, DOI:10.1007/s00214-019-2447-9.
- J. Mohanty, S. Dutta Choudhury, H. P. Upadhyaya, A. C. Bhasikuttan and H. Pal, Chem.–Eur. J., 2009, 15, 5215–5219, DOI:10.1002/chem.200802686.
- W. L. Mock and N. Y. Shih, J. Am. Chem. Soc., 1988, 110, 4706–4710, DOI:10.1021/ja00222a031.
- W. L. Mock and N. Y. Shih, J. Org. Chem., 1986, 51, 4440–4446, DOI:10.1021/jo00373a018.
- I. Osaka, M. Kondou, N. Selvapalam, S. Samal, K. Kim, M. V. Rekharsky, Y. Inoue and R. Arakawa, J. Mass Spectrom., 2006, 41, 202–207, DOI:10.1002/jms.978.
- C. Márquez, R. R. Hudgins and W. M. Nau, J. Am. Chem. Soc., 2004, 126, 5806–5816, DOI:10.1021/ja0319846.
- A. L. Koner, C. Márquez, M. H. Dickman and W. M. Nau, Angew. Chem., Int. Ed., 2011, 50, 545–548, DOI:10.1002/anie.201005317.
- Y. Jiao, W.-L. Li, J.-F. Xu, G. Wang, J. Li, Z. Wang and X. Zhang, Angew. Chem., 2016, 128, 9079–9083, DOI:10.1002/ange.201603182.
- B. Tang, W.-L. Li, Y. Jiao, J.-B. Lu, J.-F. Xu, Z. Wang, J. Li and X. Zhang, Chem. Sci., 2018, 9, 5015–5020, 10.1039/C8SC01434E.
- A. C. Forero-Girón, D. Fuentealba, N. Mariño-Ocampo, S. Gutiérrez-Oliva, B. Herrera, A. Toro-Labbé, E. Fuentes-Lemus, M. J. Davies, M. E. Aliaga and C. López-Alarcón, J. Mol. Liq., 2023, 389, 122840, DOI:10.1016/j.molliq.2023.122840.
- A. C. Forero-Girón, S. Gutiérrez-Oliva, C. López-Alarcón, B. Herrera and M. E. Aliaga, J. Mol. Model., 2024, 30, 337, DOI:10.1007/s00894-024-06132-7.
- S. Yi and A. E. Kaifer, J. Org. Chem., 2011, 76, 10275–10278, DOI:10.1021/jo2018312.
- J. Andzelm, C. Kölmel and A. Klamt, J. Chem. Phys., 1995, 103, 9312–9320, DOI:10.1063/1.469990.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104, DOI:10.1063/1.3382344.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222, DOI:10.1007/BF00533485.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261, DOI:10.1063/1.1677527.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652, DOI:10.1063/1.464913.
- T. Pooventhiran, M. Cheriet, U. Bhattacharyya, A. Irfan, R. Puchta, S. Sowrirajan and R. Thomas, Polycyclic Aromat. Compd., 2022, 42, 5443–5455, DOI:10.1080/10406638.2021.1937238.
- S. Gadde, E. K. Batchelor, J. P. Weiss, Y. Ling and A. E. Kaifer, J. Am. Chem. Soc., 2008, 130, 17114–17119, DOI:10.1021/ja807197c.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465, DOI:10.1002/jcc.21759.
- Y.-M. Jeon, J. Kim, D. Whang and K. Kim, J. Am. Chem. Soc., 1996, 118, 9790–9791, DOI:10.1021/ja962071x.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541, DOI:10.1021/ja993376p.
- G. A. Zhurko, and D. A. Zhurko, Chemcraft, Version 1.8s, 2009 Search PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter, and F. Weinhold, NBO, Version 3.1. University of Wisconsin, Madison, 2001 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, GAUSSIAN 16 (Revision B.01), Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632, DOI:10.1021/ct100641a.
- D.-S. Guo, V. D. Uzunova, K. I. Assaf, A. I. Lazar, Y. Liu and W. M. Nau, Supramol. Chem., 2016, 28, 384–395, DOI:10.1080/10610278.2015.1105374.
- W. S. Jeon, H.-J. Kim, C. Lee and K. Kim, Chem. Commun., 2002, 1828–1829, 10.1039/B202082C.
- H.-J. Buschmann, K. Jansen, C. Meschke and E. Schollmeyer, J. Solution Chem., 1998, 27, 135–140, DOI:10.1023/A:1022605306651.
- S. Zhang, L. Grimm, Z. Miskolczy, L. Biczók, F. Biedermann and W. M. Nau, Chem. Commun., 2019, 55, 14131–14134, 10.1039/C9CC07687E.
- M. Minetti, D. Pietraforte, A. M. Michela Di Stasi and C. Mallozzi, Biochem. J., 1996, 319, 369–375, DOI:10.1042/bj3190369.
- G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259–303, DOI:10.1016/S0891-5849(87)80033-3.
- E. Atala, G. Velásquez, C. Vergara, C. Mardones, J. Reyes, R. A. Tapia, F. Quina, M. A. Mendes, H. Speisky, E. Lissi, M. S. Ureta-Zañartu, A. Aspée and C. López-Alarcón, J. Phys. Chem. B, 2013, 117, 4870–4879, DOI:10.1021/jp400423w.
- C. López-Alarcón, E. Fuentes-Lemus, J. D. Figueroa, E. Dorta, C. Schöneich and M. J. Davies, Free Radical Biol. Med., 2020, 160, 78–91, DOI:10.1016/J.FREERADBIOMED.2020.06.021.
- J. N. Martins, J. C. Lima and N. Basílio, Molecules, 2021, 26, 106, DOI:10.3390/MOLECULES26010106.
- M. E. Bush, N. D. Bouley and A. R. Urbach, J. Am. Chem. Soc., 2005, 127, 14511–14517, DOI:10.1021/ja0548440.
- F. Biedermann, U. Rauwald, M. Cziferszky, K. A. Williams, L. D. Gann, B. Y. Guo, A. R. Urbach, C. W. Bielawski and O. A. Scherman, Chem.–Eur. J., 2010, 16, 13716–13722, DOI:10.1002/chem.201002274.
- O. Danylyuk, CrystEngComm, 2017, 19, 3892–3897, 10.1039/C7CE00881C.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07150f |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |