
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrasound-assisted synthesis of novel 2-aryl-3-ethoxy-5-methyl-3-oxido-2H-thiazolo[2,3-e][1,4,2]diazaphosphole-6-carboxylates and their anticancer efficacy in inducing apoptosis and autophagy and targeting cell cycle progression†
Wafa A. Bawazira,
Tarik E. Ali *bc,
Mohammed A. Assiric,
Ali A. Shatid,
Mohammad Y. Alfaifid and
Serag E. I. Elbehairid
*bc,
Mohammed A. Assiric,
Ali A. Shatid,
Mohammad Y. Alfaifid and
Serag E. I. Elbehairid
aChemistry Department, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
bCentral Labs, King Khalid University, AlQuraa, Abha, Saudi Arabia. E-mail: tarik_elsayed1975@yahoo.com; tismail@kku.edu.sa
cDepartment of Chemistry, Faculty of Science, King Khalid University, AlQuraa, Abha, Saudi Arabia
dDepartment of Biology, Faculty of Science, King Khalid University, AlQuraa, Abha, Saudi Arabia
First published on 25th November 2024
Abstract
A novel class of ethyl 2-aryl-3-ethoxy-5-methyl-3-oxido-2H-thiazolo[2,3-e][1,4,2]diazaphosphole-6-carboxylates (2a–j) were synthesized via a one-pot, three-component method. This reaction utilized ethyl 2-amino-4-methylthiazole-5-carboxylate (1) with different aromatic aldehydes and ethyl dichlorophosphite in THF under ultrasonic irradiation, with triethylamine as an efficient catalyst at 50 °C. The reaction provided the desired products 2a–j in high yields within a short timeframe. The cytotoxic effects of the synthesized compounds were assessed against two human cancer cell lines, lung cancer (A549) and renal cancer (TK-10), using the sulforhodamine B (SRB) assay. This evaluation revealed that products 2e, 2h and 2j demonstrated significantly higher cytotoxicity against the studied cancer cells than the standard drug doxorubicin. These bioactive products notably increased the late apoptosis rate in both cell lines and demonstrated a promising high ability to arrest the cell cycle at different phases in renal TK-10 and lung A549 cancer cells. Additionally, compounds 2e, 2h and 2j displayed potential for inducing autophagy.
1. Introduction
Thiazole-based compounds exhibit a wide range of effects in medicinal chemistry and are present in numerous commercially available medicines, displaying a variety of pharmacological behaviors.1 The thiazole moiety plays a critical role as a pharmacophore, a fundamental building block for various biological compounds. Notably, it is essential for vitamin B1 (thiamine) synthesis, which supports the nervous system by enabling acetylcholine production.2 Thiazole derivatives are present in a wide array of natural products and medications with numerous health benefits (Fig. 1). These versatile molecules can lower blood pressure, fight infections, control seizures, combat various cancers, reduce inflammation, and exhibit promising effects against viruses, oxidative damage, pain, HIV, and malaria.3–12 The diverse biological activities of thiazole derivatives make them a fascinating area for research with significant potential to impact numerous areas of human health.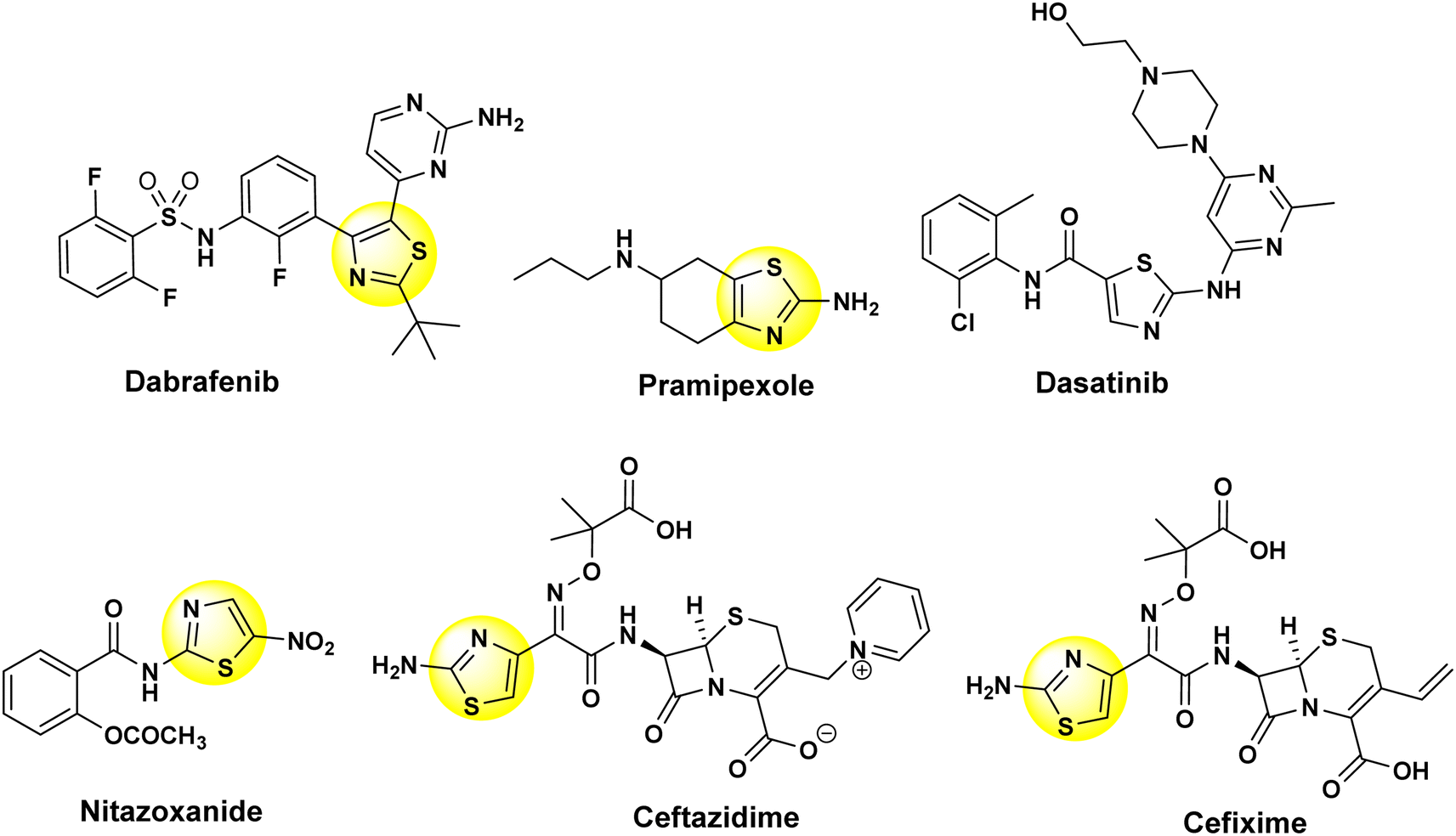 | ||
| Fig. 1 Thiazole derivatives that are found in a remarkable variety of commercial drugs. | ||
Phosphorus-containing heterocycles are interesting molecular frameworks for the synthesis of pharmacological drugs in medicinal chemistry due to their structural diversity and biocidal effects13–15 (Fig. 2). In particular, heterophospholes are a broad family of five-membered ring molecules with a unique structure (one phosphorus and two other elements), showing significant potential in drug development. Certain isomers, such as 1,4,2- and 1,2,3-diazaphospholes, exhibit valuable bioactivities, including anti-inflammatory, antimicrobial, and anticancer properties.16–20
 | ||
| Fig. 2 Some phosphorus-containing heterocycles used in commercial drugs. | ||
As part of our studies on heterophospholes, we recently synthesized a promising series of some 1,4,2- and 1,2,3-diazaphosphole compounds, including 1,2,4-triazolo[3,4-e][1,2,3]diazaphospholes (A),21 1,2,4-triazolo[5,1-e][1,4,2]diazaphospholes (B),22 and 5-pyrazolyl-1,4,2-diazaphosphole (C)23 (Fig. 3). According to our literature survey, molecular frameworks that combine 1,3-thiazole and 1,4,2-diazaphosphole rings are rare.24–28 These structures were constructed through Hantzsch-type 3 + 2 cyclocondensation of 2-amino-thiazoles with phosphorus halides in the presence of triethylamine (Fig. 4).
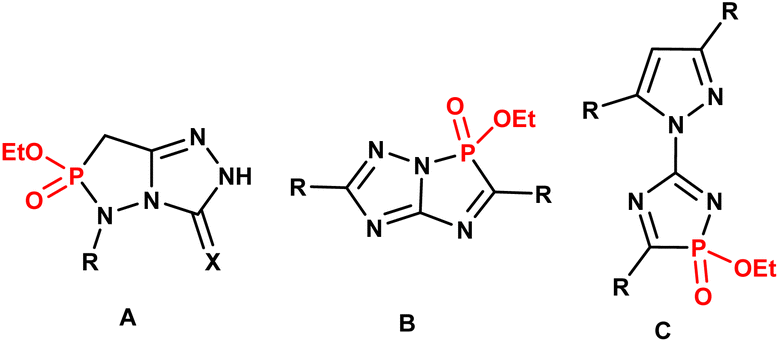 | ||
| Fig. 3 Some of our recently prepared 1,4,2- and 1,2,3-diazaphosphole compounds. | ||
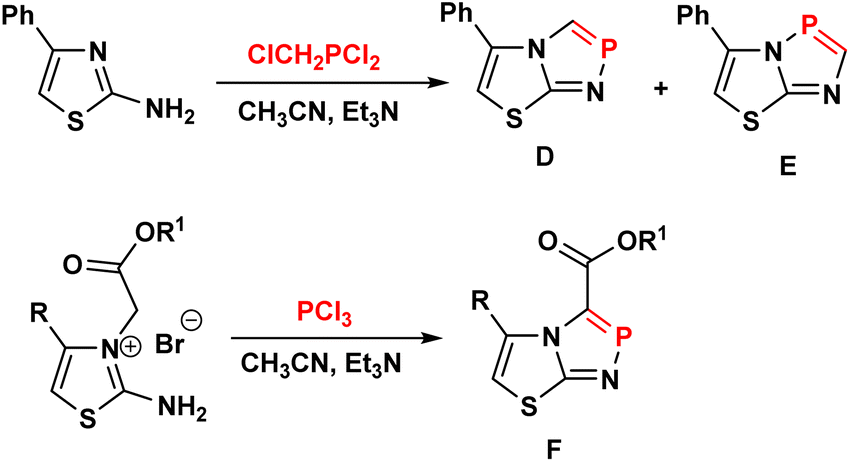 | ||
| Fig. 4 Hantzsch-type 3 + 2 cyclocondensation of 2-aminothiazoles with phosphorus halides. | ||
Building on the promising bioactivity observed in our exploration of novel phosphorus heterocycles,29–32 we developed a simple and effective methodology to club the 1,4,2-diazaphosphole ring with the thiazole moiety, generating a novel class of thiazolo[2,3-e][1,4,2]diazaphospholes. These compounds were evaluated for their antiproliferative activities in vitro against two specific human cancer cell lines, lung cancer (A549) and kidney cancer (TK-10), to determine their therapeutic potential. The motivation for investigating these novel products arises from the ongoing need for new anticancer agents that can overcome the limitations of the existing therapies, such as drug resistance and toxicity. Additionally, structure–activity relationship (SAR) studies could provide insights into the optimal structural features for maximizing their anticancer potency and selectivity. Ultimately, these findings aimed to inform the optimization of these compounds, leading to the development of potentially life-saving synthetic drugs.
2. Results and discussion
The synthetic strategy for designing novel ethyl 2-aryl-3-ethoxy-5-methyl-3-oxido-2H-thiazolo[2,3-e][1,4,2]diazaphosphole-6-carboxylates (2a–j) was achieved via a two-step, one-pot synthesis. The first step involved the condensation of ethyl 2-amino-4-methylthiazole-5-carboxylate (1)33 with various aromatic aldehydes, forming a Schiff base. In the second step, this Schiff base reacted with ethyl dichlorophosphite under Pudovik reaction conditions. To optimize the experimental conditions, a model reaction was conducted to produce 3-ethoxy-5-methyl-2-phenyl-3-oxido-2H-thiazolo[2,3-e][1,4,2]diazaphosphole-6-carboxylate (2a) by reacting ethyl 2-amino-4-methylthiazole-5-carboxylate (1) with benzaldehyde, followed by ethyl dichlorophosphite (Scheme 1). | ||
| Scheme 1 Model reaction for synthesis of the target product 2a. | ||
Initially, the reaction conducted under catalyst-free and solventless conditions produced no yield, even after 10 h at 25 °C and 60 °C (Table 1, entries 1 and 2). When basic solvents, such as DMF and pyridine, were used, the target product 2a was formed with yields of 15% and 18%, respectively (Table 1, entries 3 and 4). We then explored the effect of various non-polar solvents, including DCM, benzene, toluene, dioxane, and THF, in the presence of equimolar amounts of various catalysts, such as K2CO3, DBU, and Et3N and (Table 1, entries 5–19). Among the catalysts tested, Et3N worked effectively, yielding the desired product 2a in good quantities across all solvents (Table 1, entries 7, 10, 13, 16 and 19). The optimal outcome was achieved in THF, where a 75% yield was obtained with 1.0 mmol Et3N catalyst at 60 °C (Table 1, entry 19). Increasing the amount of Et3N to 2.0 mmol did not improve the yield further (Table 1, entry 20), but extending the reaction time from 5 to 10 h increased the yield slightly, from 75% to 78% (Table 1, entry 21). Subsequently, we aimed to perform the reaction in shorter reaction times and improved yields by using ultrasonic irradiation. Ultrasonic irradiation enhances chemical reactions by promoting cavitation, which generates high-energy microbubbles that collapse, creating localized extreme conditions (high temperature and pressure).34,35 This accelerates reaction rates, increases the reactant surface area, and facilitates bond cleavage. It is widely used in organic synthesis, catalysis, and polymerization, offering benefits such as faster reactions, higher yields, and milder conditions.36 Therefore, when the synthesis of product 2a was carried out in the presence of Et3N (1.0 mmol) under ultrasonication at room temperature, it took about 0.5 h to afford the product in a 62% yield (Table 1, entry 22). However, when the reaction was repeated under the same conditions at 50 °C for 1.5 and 3 h, it produced the final product 2a with an improved yield of 82% (Table 1, entries 23 and 24).
| Entrya | Catalyst | Solvent | Time (h) | Temp (°C) | Yieldb |
|---|---|---|---|---|---|
| a Reagents: amine (1) (1.0 mmol) with benzaldehyde (1.0 mmol) then ethyl dichlorophosphite (1.0 mmol).b Yields for the isolated product. | |||||
| 1 | — | — | 10 | 25 | — |
| 2 | — | — | 10 | 60 | — |
| 3 | — | DMF | 5 | 100 | 15% |
| 4 | — | Pyridine | 5 | 100 | 18% |
| 5 | K2CO3 (1.0 mmol) | DCM | 5 | 40 | 9% |
| 6 | DBU (1.0 mmol) | DCM | 5 | 40 | 11% |
| 7 | Et3N (1.0 mmol) | DCM | 5 | 40 | 19% |
| 8 | K2CO3 (1.0 mmol) | Benzene | 5 | 75 | 15% |
| 9 | DBU (1.0 mmol) | Benzene | 5 | 75 | 18% |
| 10 | Et3N (1.0 mmol) | Benzene | 5 | 75 | 21% |
| 11 | K2CO3 (1.0 mmol) | Toluene | 5 | 100 | 18% |
| 12 | DBU (1.0 mmol) | Toluene | 5 | 100 | 20% |
| 13 | Et3N (1.0 mmol) | Toluene | 5 | 100 | 28% |
| 14 | K2CO3 (1.0 mmol) | Dioxane | 5 | 100 | 24% |
| 15 | DBU (1.0 mmol) | Dioxane | 5 | 100 | 54% |
| 16 | Et3N (1.0 mmol) | Dioxane | 5 | 100 | 63% |
| 17 | K2CO3 (1.0 mmol) | THF | 5 | 60 | 25% |
| 18 | DBU (1.0 mmol) | THF | 5 | 60 | 52% |
| 19 | Et3N (1.0 mmol) | THF | 5 | 60 | 75% |
| 20 | Et3N (2.0 mmol) | THF | 5 | 60 | 75% |
| 21 | Et3N (1.0 mmol) | THF | 10 | 60 | 78% |
| 22 | Et3N (1.0 mmol) | THF | 0.5 | US, 25 °C | 62% |
| 23 | Et3N (1.0 mmol) | THF | 1.5 | US, 50°C | 82% |
| 24 | Et3N (1.0 mmol) | THF | 3 | US, 50 °C | 82% |
After determining the optimal reaction conditions, an equimolar amount of triethylamine was used as a catalyst to promote the reaction, allowing all the components to couple and form the final products (2a–j) within 60–120 min at 50 °C in THF under ultrasonic irradiation (Scheme 2). Commercially available aldehydes with various activating or deactivating substituents were employed to yield the corresponding 2-aryl-3-ethoxy-5-methyl-3-oxido-2H-thiazolo[2,3-e][1,4,2]diazaphosphole-6-carboxylate compounds (2a–j). Details of the structures, yields, and reaction times for 2a–j are provided in Scheme 2.
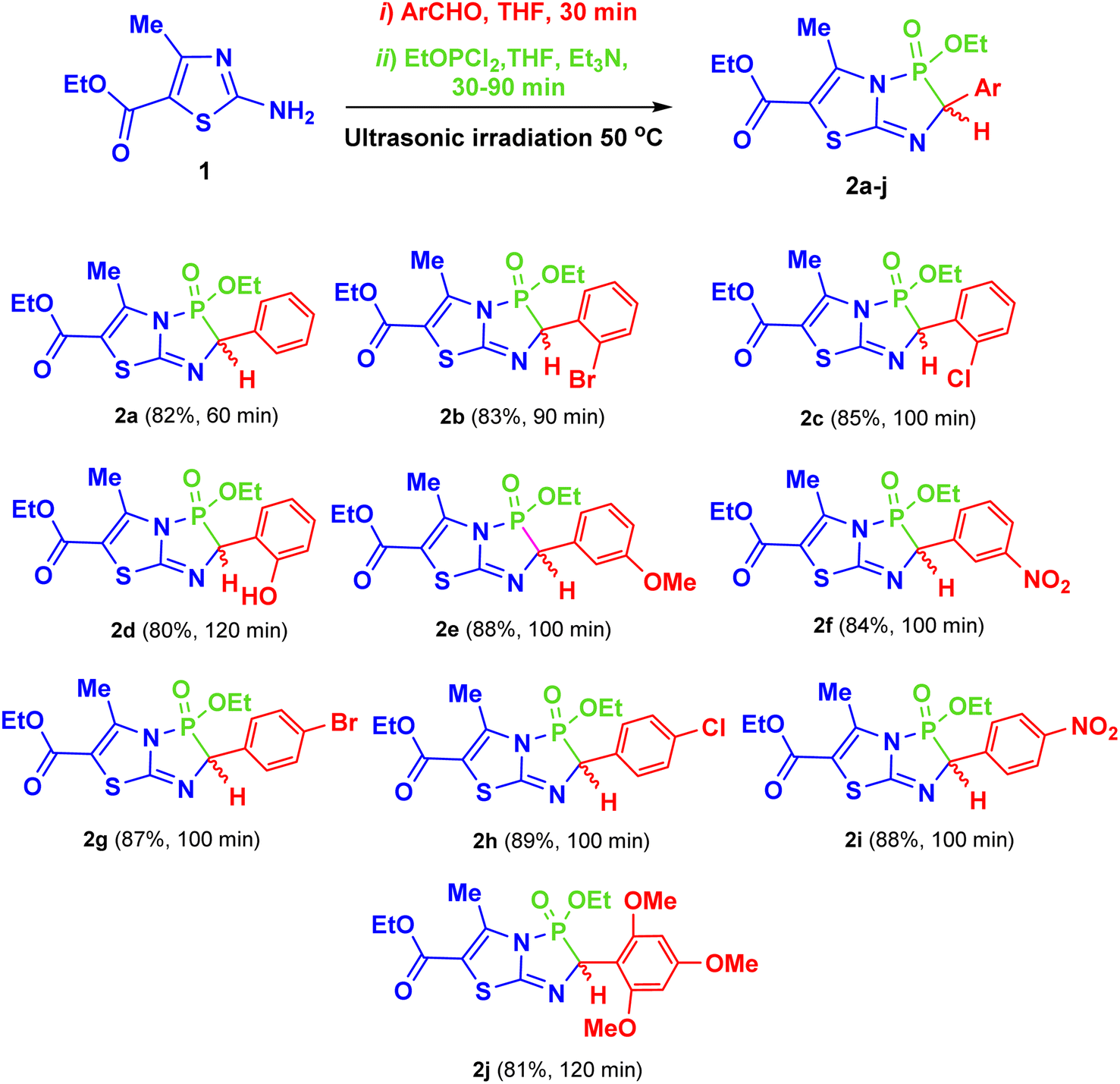 | ||
| Scheme 2 Synthesis of the target products 2a–j, and their chemical structures, reaction times and yields. | ||
The proposed pathway for constructing products 2a–j is illustrated in Scheme 3. The reaction of amine 1 with an aromatic aldehyde produces intermediate A, as a Schiff base, through the elimination of a water molecule. Ethyl dichlorophosphite then undergoes partial hydrolysis, forming ethyl phosphonochloridate (B) upon the removal of water. Subsequent nucleophilic attack by intermediate B at the azomethine bond of A, catalyzed by Et3N, leads to the formation of species D, which can undergo intermolecular protonation to yield the intermediate α-aminophosphonate E. Then, Et3N activates the NH group of the thiazole ring in the tautomeric form F, facilitating an attack on the phosphorus atom and removal of a HCl molecule, ultimately affording the target products 2a–j (Scheme 3).37
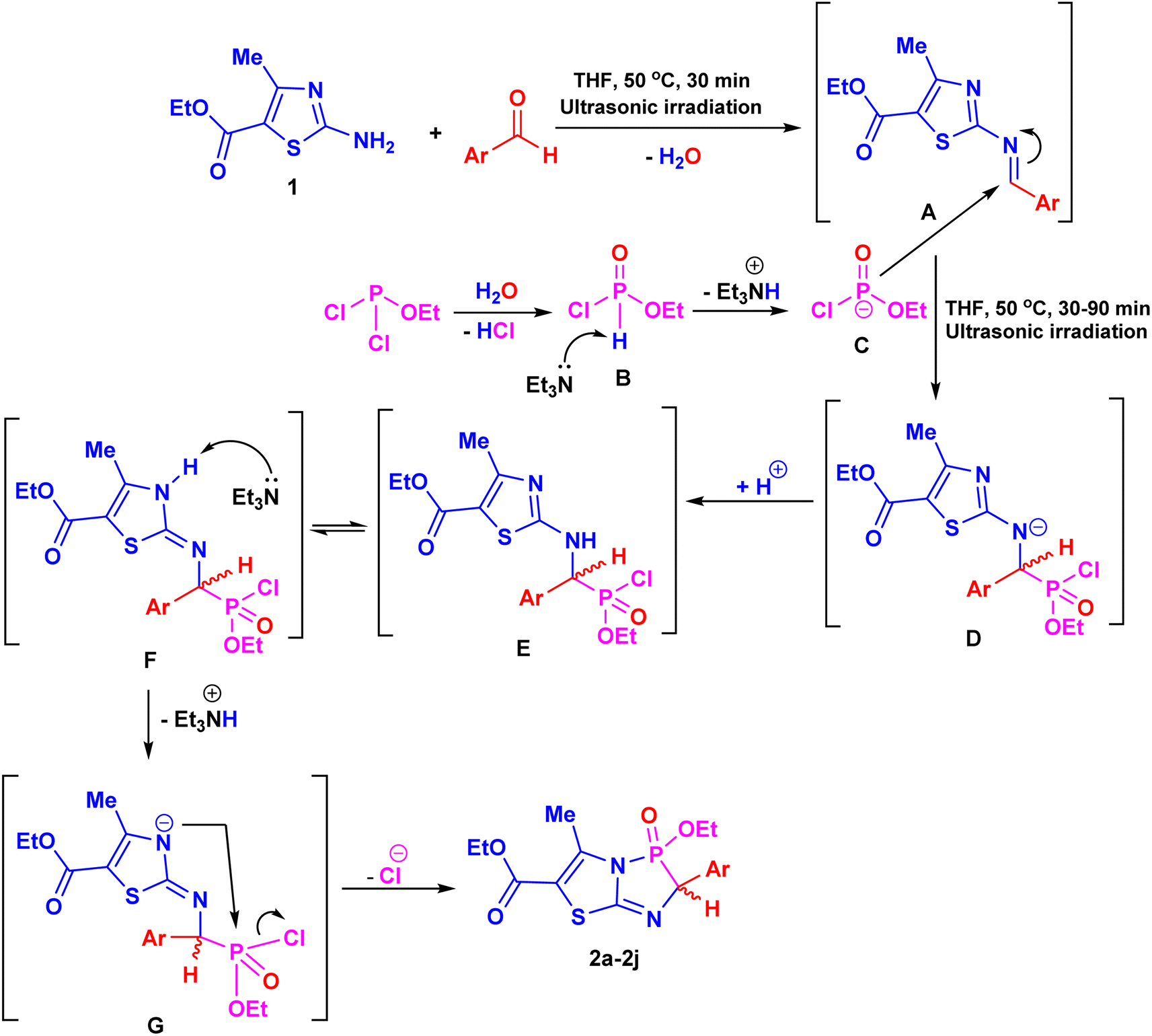 | ||
| Scheme 3 The suggested reaction mechanism for the formation of the target products 2a–2j. | ||
We confirmed the identity and structure of the target compounds 2a–j using various physical and spectral techniques, including IR, NMR, and mass spectrometry. The IR spectra showed no NH absorption bands but displayed specific absorption bands in the regions of 1694–1707, 1263–1272, and 1014–1097 cm−1, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, PO, and P–O–C functionalities, respectively. In the 1H-NMR spectra, doublet chemical shifts in the range of δ 5.68–5.93 ppm were attributed to CH–P protons, with coupling constants of 17.6–22.8 Hz.38 Quartet signals at δ 3.89–4.24 ppm were assigned to OCH2 protons of ethoxy groups, while the singlets at δ 2.12–2.37 ppm corresponded to CH3 protons. Additionally, triplets in the range of δ 1.03–1.29 ppm confirmed the presence of CH3 protons in ethoxy groups. In the 13C-NMR spectra, signals in the ranges of δ 13.0–15.7, 59.0–63.3, 47.5–49.8 (JPC = 143–157.8 Hz), 162.8–164.9, and 166.5–168.7 ppm verified the presence of CH3, OCH2, CH–P, C-7a, and CO carbons, respectively.38 The 31P-NMR chemical shifts for the synthesized compounds were within a narrow range of δ 19.52–22.52 ppm,39 indicating the similar electronic environments for the phosphorus atoms across these compounds.
O, PO, and P–O–C functionalities, respectively. In the 1H-NMR spectra, doublet chemical shifts in the range of δ 5.68–5.93 ppm were attributed to CH–P protons, with coupling constants of 17.6–22.8 Hz.38 Quartet signals at δ 3.89–4.24 ppm were assigned to OCH2 protons of ethoxy groups, while the singlets at δ 2.12–2.37 ppm corresponded to CH3 protons. Additionally, triplets in the range of δ 1.03–1.29 ppm confirmed the presence of CH3 protons in ethoxy groups. In the 13C-NMR spectra, signals in the ranges of δ 13.0–15.7, 59.0–63.3, 47.5–49.8 (JPC = 143–157.8 Hz), 162.8–164.9, and 166.5–168.7 ppm verified the presence of CH3, OCH2, CH–P, C-7a, and CO carbons, respectively.38 The 31P-NMR chemical shifts for the synthesized compounds were within a narrow range of δ 19.52–22.52 ppm,39 indicating the similar electronic environments for the phosphorus atoms across these compounds.
2.1 Antiproliferative properties
| Compounds | IC50 (μg mL−1) | |
|---|---|---|
| TK-10 | A549 | |
| 2a | ≥100 | ≥100 |
| 2b | ≥100 | 66.9 ± 0.7 |
| 2c | 11.7 ± 2.3 | 33.4 ± 0.8 |
| 2d | ≥100 | ≥100 |
| 2e | 1.0 ± 0.1 | 2.4 ± 0.3 |
| 2f | ≥100 | ≥100 |
| 2g | 5.1 ± 0.7 | 10.9 ± 1.2 |
| 2h | 2.3 ± 0.1 | 4.7 ± 0.2 |
| 2i | ≥100 | 44.7 ± 3.6 |
| 2j | 1.4 ± 0.1 | 1.0 ± 0.1 |
| Doxorubicin | 1.4 ± 0.4 | 1.9 ± 0.7 |
• The existence of any substituent at the phenyl ring in most compounds 2b–j caused cytotoxic effects in comparison with compound 2a against the two cancer cell lines.
• The presence of ortho-substituted groups in compounds 2b (2-Br), 2c (2-Cl), and 2d (2-HO) generally exhibited lower IC50 values against A549 cells. This suggested that steric effects in the ortho position may hinder effective binding.
• The presence of meta-substituted groups, as in compound 2e (3-MeO), significantly enhanced the bioactivity. Compound 2e had among the lowest IC50 values (2.4 μg mL−1 for A549 cells and 1.0 μg mL−1 for TK-10 cells), indicating that a meta-positioned electron-donating group (MeO) is highly favorable for potency.
• The presence of para-substituted halogens in compounds 2g (4-Br) and 2h (4-Cl) also improved the activity, although they were less potent than meta-substituted 2e (3-MeO).
• The existence of nitro groups in any position, as in both products 2f and 2i, appeared to reduce the cytotoxic effects, as these compounds had IC50 values ≥ 100 μg mL−1 for at least one of the cell lines.
• Halogen-substituted compounds, such as 2b, 2c, 2g, and 2h, may be effective in improving the cytotoxic effect, especially when they are in the para-position.
• Both products 2e and 2j showed IC50 values that were close to doxorubicin, indicating their strong anticancer activities. This suggested that a single meta-positioned methoxy or multiple methoxy groups may be effective modifications to optimize the potency.
• We can hypothesize the potential mechanisms based on the structural features. The different aromatic rings and planar structures in these products can often intercalate into DNA, disrupting replication and transcription processes, which is common in anticancer agents. In addition, the products that have halogen and methoxy substituents could interact with specific cell receptors or enzymes, potentially inhibiting pathways critical to cancer cell survival and proliferation.
 | ||
| Fig. 5 Apoptosis/necrosis assessment of the products 2e, 2h and 2j against the TK-10 cell lines. | ||
 | ||
| Fig. 6 Apoptosis/necrosis assessment of the products 2e, 2h and 2j against the A549 cell lines. | ||
 | ||
| Fig. 7 Cell cycle distribution determined using DNA cytometry analysis of the products 2e, 2h and 2j against the TK-10 cell lines. | ||
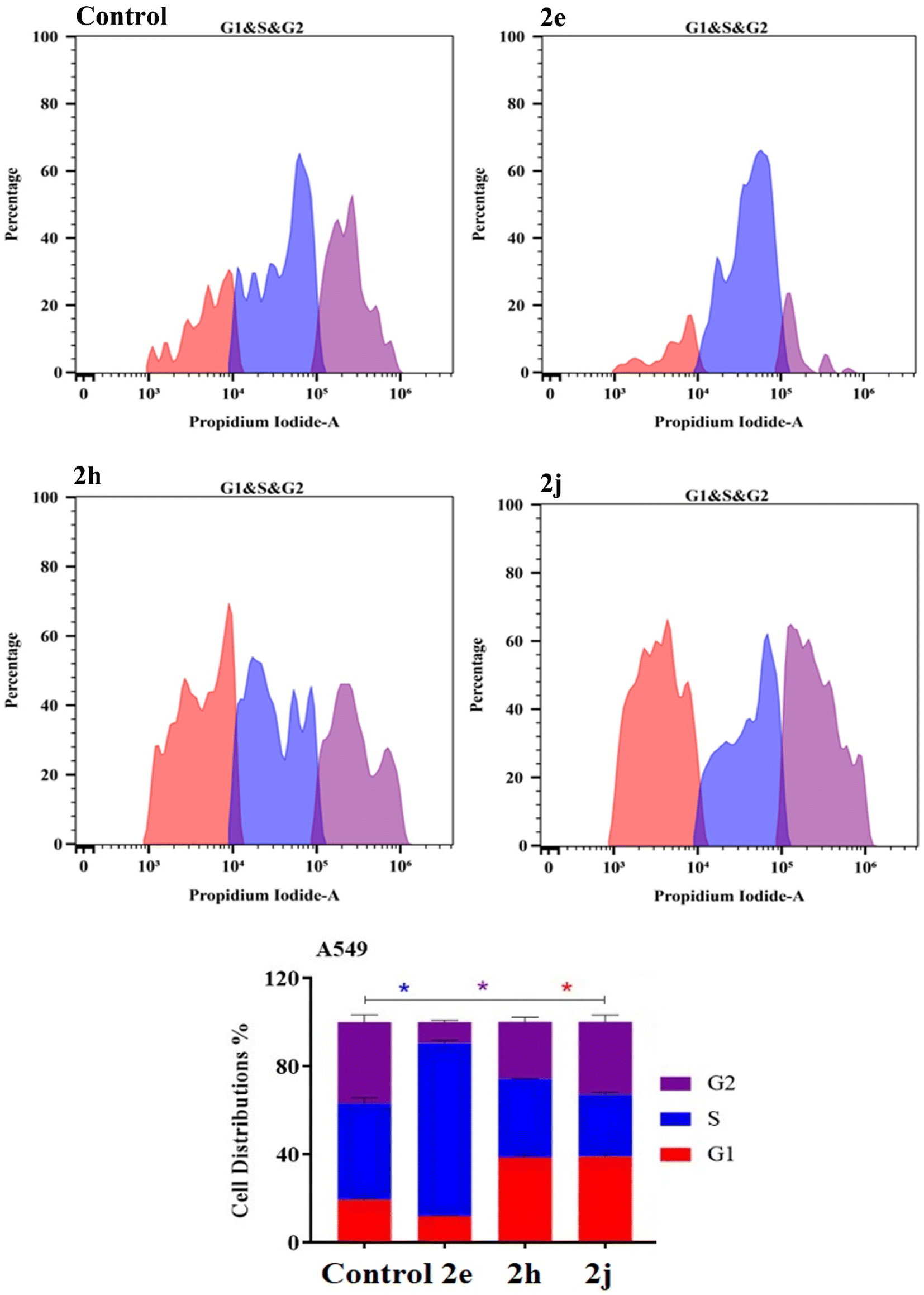 | ||
| Fig. 8 Cell cycle distribution determined using DNA cytometry analysis of the products 2e, 2h and 2j against the A549 cell lines. | ||
 | ||
| Fig. 9 Autophagic cell death assessment in TK-10 cell lines to compounds 2e, 2h and 2j. | ||
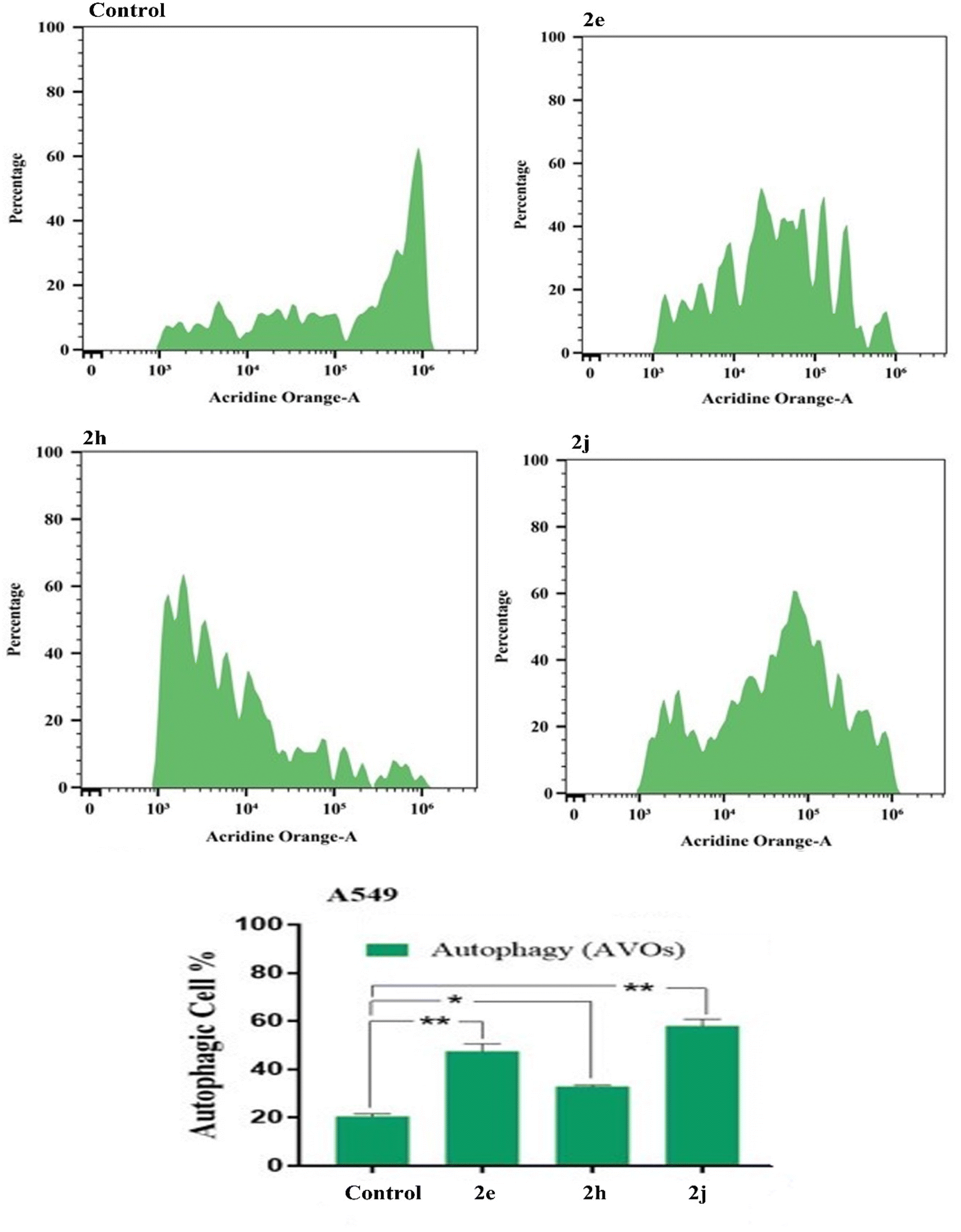 | ||
| Fig. 10 Autophagic cell death assessment in TK-10 cell lines to compounds 2e, 2h and 2j. | ||
3. Experimental
The melting points were measured using a digital Stuart SMP-3 apparatus in open capillary tubes. Infrared spectroscopy (FT-IR) was performed using a Nicolet iS10 spectrophotometer. Nuclear magnetic resonance (NMR) analysis, comprising 1H, 13C, and 31P NMR spectroscopies, was performed on a Bruker 400 MHz spectrometer in deuterated dimethyl sulfoxide (DMSO-d6) at 400, 100, and 162 MHz, respectively, with tetramethyl silane (TMS) as the internal standard. Mass spectrometry was performed using a single quadrupole mass analyzer (Thermo Scientific GCMS) with a direct probe controller inlet. The purity of the synthesized compounds was confirmed using thin-layer chromatography (TLC), while their elemental composition was determined by analysis with a PerkinElmer 2400II instrument. For ultrasonic irradiation, an Elmasonic S 60 (H) (ultrasonic frequency 37 MHz, power 60 W) ultrasound bath was used, and the reaction flask was located in the ultrasonic bath containing water.3.1 Syntheses of 2-phenyl-3-ethoxy-5-methyl-3-oxido-2H-thiazolo[2,3-e][1,4,2]diazaphosphole-6-carboxylate (2a–2j)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) elution system, yielding the purified product.:2) elution system, yielding the purified product.O), 1268 (PO), 1088, 1043, 1017 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.06 (t, 3H, J = 7.2 Hz, CH3), 1.26 (t, 3H, J = 7.2 Hz, CH3), 2.33 (s, 3H, CH3), 3.99 (q, 2H, J = 7.2 Hz, OCH2), 4.21 (q, 2H, J = 7.2 Hz, OCH2), 5.82 (d, 1H, JPCH = 20.4 Hz, P–CH), 7.15 (d, 2H, J = 7.2 Hz, Ph–H), 7.37–7.45 (m, 3H, Ph–H). 13C-NMR (100 MHz, DMSO-d6): 13.0 (CH3), 14.2 (CH3), 15.1 (CH3), 47.5 (d, JPC = 143.0 Hz, C-2), 62.3 (OCH2), 63.3 (OCH2), 125.4 (C-4phenyl), 129.4 (C-3,5phenyl), 130.1 (C-2,6phenyl), 137.0 (C-1phenyl), 139.6 (C-6), 153.7 (C-5), 164.9 (C-7a), 168.4 (CO). 31P-NMR (162 MHz, DMSO-d6): 22.25 ppm. MS (m/z, I%): 366 (M+, 18%). Anal. calcd for C16H19N2O4PS (366.37): C, 52.45%; H, 5.23%; N, 7.65%; S, 8.75%. Found: C, 52.34%; H, 5.16%; N, 7.49%; S, 8.61%.O), 1269 (PO), 1097, 1038 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.06 (t, 3H, J = 7.2 Hz, CH3), 1.23 (t, 3H, J = 7.6 Hz, CH3), 2.20 (s, 3H, CH3), 3.92 (q, 2H, J = 7.2 Hz, OCH2), 4.13 (q, 2H, J = 6.8 Hz, OCH2), 5.79 (d, 1H, JPCH = 17.6 Hz, P–CH), 7.39–7.7.47 (m, 2H, Ar–H), 7.72 (t, 1H, J = 7.2 Hz, Ar–H), 7.90 (d, 1H, J = 7.2 Hz, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 13.1 (CH3), 15.3 (CH3), 16.1 (CH3), 48.5 (d, JPC = 148.7 Hz, C-2), 59.0 (OCH2), 60.4 (OCH2), 124.3 (C-2aryl), 126.9 (C-5aryl), 127.7 (C-4aryl), 131.3 (C-6aryl), 132.1 (C-3aryl), 137.9 (C-6), 141.5 (C-1aryl), 155.5 (C-5), 164.1 (C-7a), 167.5 (CO). 31P-NMR (162 MHz, DMSO-d6): 20.25 ppm. MS (m/z, I%): 444 (M+, 27%) and 446 (M + 2, 18%). Anal. calcd for C16H18BrN2O4PS (445.27): C, 43.16%; H, 4.07%; N, 6.29%; S, 7.20%. Found: C, 43.04%; H, 3.92%; N, 6.13%; S, 7.10%.O), 1268 (PO), 1082, 1042 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.07 (t, 3H, J = 6.4 Hz, CH3), 1.23 (t, 3H, J = 6.8 Hz, CH3), 2.21 (s, 3H, CH3), 3.93 (q, 2H, J = 7.2 Hz, OCH2), 4.24 (q, 2H, J = 6.4 Hz, OCH2), 5.82 (d, 1H, JPCH = 20.8 Hz, P–CH), 7.42 (t, 1H, J = 7.2 Hz, Ar–H), 7.50 (t, 1H, J = 7.2 Hz, Ar–H), 7.91 (d, 1H, J = 7.6 Hz, Ar–H), 8.04 (d, 1H, J = 8.0 Hz, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 13.5 (CH3), 15.6 (CH3), 16.3 (CH3), 48.4 (d, JPC = 148.6 Hz, C-2), 60.5 (OCH2), 61.6 (OCH2), 125.8 (C-5aryl), 126.9 (C-4aryl), 128.1 (C-3aryl), 130.5 (C-6aryl), 133.3 (C-2aryl), 135.5 (C-1aryl), 138.4 (C-6), 154.6 (C-5), 164.4 (C-7a), 168.7 (CO). 31P-NMR (162 MHz, DMSO-d6): 20.03 ppm. MS (m/z, I%): 399 (M+, 9%) and 401 (M+, 3%). Anal. calcd for C16H18ClN2O4PS (400.81): C, 47.95%; H, 4.53%; N, 6.99%; S, 8.00%. Found: C, 47.83%; H, 4.42%; N, 6.89%; S, 7.85%.O), 1267 (PO), 1091, 1016 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.16 (t, 3H, J = 6.8 Hz, CH3), 1.22 (t, 3H, J = 7.2 Hz, CH3), 2.39 (s, 3H, CH3), 4.02 (q, 2H, J = 7.2 Hz, OCH2), 4.21 (t, 2H, J = 6.8 Hz, OCH2), 5.93 (d, 1H, JPCH = 20.4 Hz, P–CH), 6.82 (t, 2H, J = 7.2 Hz, Ar–H), 7.11 (d, 1H, J = 7.2 Hz, Ar–H), 7.43 (d, 1H, J = 7.6 Hz, Ar–H), 9.07 (brs, 1H, OH). 13C-NMR (100 MHz, DMSO-d6): 13.5 (CH3), 15.6 (CH3), 16.4 (CH3), 48.1 (d, JPC = 151.4 Hz, C-2), 60.4 (OCH2), 62.2 (OCH2), 116.5 (C-3aryl), 121.2 (C-5aryl), 125.8 (C-1aryl), 128.8 (C-4aryl), 129.0 (C-6aryl), 138.4 (C-6), 154.6 (C-5), 157.5 (C-2aryl), 163.6 (C-7a), 167.0 (CO). 31P-NMR (162 MHz, DMSO-d6): 20.02 ppm. MS (m/z, I%): 382 (M+, 6%). Anal. calcd for C16H19N2O5PS (382.37): C, 50.26%; H, 5.01%; N, 7.33%; S, 8.38%. Found: C, 50.08%; H, 4.92%; N, 7.18%; S, 8.22%.O), 1268 (PO), 1083, 1041 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.03 (t, 3H, J = 6.4 Hz, CH3), 1.23 (t, 3H, J = 7.2 Hz, CH3), 2.12 (s, 3H, CH3), 3.71 (s, 3H, OCH3), 3.92 (q, 2H, J = 7.2 Hz, OCH2), 4.21 (q, 2H, J = 6.8 Hz, OCH2), 5.82 (d, 1H, JPCH = 21.2 Hz, P–CH), 7.43 (t, 1H, J = 7.2 Hz, Ar–H), 7.71 (d, 1H, J = 8.00 Hz, Ar–H), 7.91 (d, 1H, J = 7.6 Hz, Ar–H), 8.43 (s, 1H, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 13.7 (CH3), 14.8 (CH3), 15.9 (CH3), 48.0 (d, JPC = 152.6 Hz, C-2), 54.3 (OCH3), 59.3 (OCH2), 61.9 (OCH2), 115.5 (C-4aryl), 116.6 (C-2aryl), 121.5 (C-6aryl), 123.4 (C-5aryl), 137.1 (C-1aryl), 139.5 (C-6), 156.0 (C-5), 159.0 (C-3aryl), 162.8 (C-7a), 166.7 (CO). 31P-NMR (162 MHz, DMSO-d6): 19.52 ppm. MS (m/z, I%): 396 (M+, 43%). Anal. calcd for C17H21N2O5PS (396.40): C, 51.51%; H, 5.34%; N, 7.07%; S, 8.09%. Found: C, 51.34%; H, 5.31%; N, 6.95%; S, 7.96%.O), 1270 (PO), 1097, 1044, 1014 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.10 (t, 3H, J = 6.8 Hz, CH3), 1.14–1.23 (m, 3H, CH3), 2.38 (s, 3H, CH3), 3.89–4.16 (m, 4H, OCH2), 5.72 (dd, 1H, JPCH = 22.0 and 9.2 Hz, P–CH), 7.69 (t, 1H, J = 7.6 Hz, Ar–H), 8.18 (d, 1H, J = 7.2 Hz, Ar–H), 8.33 (d, 1H, J = 7.2 Hz, Ar–H), 8.39 (s, 1H, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 13.2 (CH3), 14.3 (CH3), 15.2 (CH3), 47.9 (d, JPC = 157.8 Hz, C-2), 60.1 (OCH2), 61.9 (OCH2), 125.1 (C-2aryl), 128.5 (C-4aryl), 129.4 (C-5aryl), 135.6 (C-6aryl), 137.4 (C-6), 139.4 (C-1aryl), 143.5 (C-3aryl), 154.2 (C-5), 163.1 (C-7a), 167.4 (CO). 31P-NMR (162 MHz, DMSO-d6): 20.06 ppm. MS (m/z, I%): 411 (M+, 51%). Anal. calcd for C16H18N3O6PS (411.37): C, 46.72%; H, 4.41%; N, 10.21%; S, 7.79%. Found: C, 46.58%; H, 4.31%; N, 10.09%; S, 7.67%.O), 1271 (PO), 1097, 1045, 1015 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.10 (t, 3H, J = 7.2 Hz, CH3), 1.29 (t, 3H, J = 7.2 Hz, CH3), 2.22 (s, 3H, CH3), 4.01–4.08 (m, 4H, OCH2), 5.83 (d, 1H, JPCH = 20.4 Hz, P–CH), 7.11 (d, 2H, J = 8.0 Hz, Ar–H), 8.04 (d, 2H, J = 7.6 Hz, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 13.8 (CH3), 15.0 (CH3), 15.9 (CH3), 48.5 (d, JPC = 155.4 Hz, C-2), 59.5 (OCH2), 60.6 (OCH2), 121.4 (C-4aryl), 130.8 (C-2,6aryl), 131.7 (C-3,5aryl), 136.7 (C-1aryl), 138.3 (C-6), 153.1 (C-5), 163.9 (C-7a), 168.2 (CO). 31P-NMR (162 MHz, DMSO-d6): 22.52 ppm. MS (m/z, I%): 444 (M+, 25%) and 446 (M + 2, 23%). Anal. calcd for C16H18BrN2O4PS (445.27): C, 43.16%; H, 4.07%; N, 6.29%; S, 7.20%. Found: C, 43.01%; H, 3.93%; N, 6.11%; S, 7.04%.O), 1268 (PO), 1091, 1018 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.10 (t, 3H, J = 6.8 Hz, CH3), 1.19 (t, 3H, J = 6.4 Hz, CH3), 2.33 (s, 3H, CH3), 3.95 (q, 2H, J = 6.4 Hz, OCH2), 4.19 (q, 2H, J = 6.8 Hz, OCH2), 5.85 (d, 1H, JPCH = 22.0 Hz, P–CH), 7.18 (d, 2H, J = 8.4 Hz, Ar–H), 8.05 (d, 2H, J = 7.6 Hz, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 14.1 (CH3), 15.1 (CH3), 15.9 (CH3), 49.2 (d, JPC = 154.0 Hz, C-2), 60.6 (OCH2), 61.4 (OCH2), 128.5 (C-3,5aryl), 131.3 (C-4aryl), 132.2 (C-2,6aryl), 137.3 (C-6), 135.2 (C-1aryl), 154.6 (C-5), 163.2 (C-7a), 168.6 (CO). 31P-NMR (162 MHz, DMSO-d6): 20.04 ppm. MS (m/z, I%): 399 (M+, 29%) and 401 (M + 2, 9%). Anal. calcd for C16H18ClN2O4PS (400.81): C, 47.95%; H, 4.53%; N, 6.99%; S, 8.00%. Found: C, 47.81%; H, 4.40%; N, 6.86%; S, 7.86%.O), 1272 (PO), 1097, 1045, 1016 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.10 (t, 3H, J = 6.8 Hz, CH3), 1.22 (t, 3H, J = 6.8 Hz, CH3), 2.37 (s, 3H, CH3), 3.92 (q, 2H, J = 6.8 Hz, OCH2), 4.13 (q, 2H, J = 6.8 Hz, OCH2), 5.68 (dd, 1H, JPCH = 22.8 and 9.6 and Hz, P–CH), 8.16 (d, 2H, J = 8.4 Hz, Ar–H), 8.41 (d, 2H, J = 8.4 Hz, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 13.3 (CH3), 15.1 (CH3), 16.0 (CH3), 49.2 (d, JPC = 154.0 Hz, C-2), 59.8 (OCH2), 61.3 (OCH2), 123.4 (C-3,5aryl), 129.5 (C-2,6aryl), 137.1 (C-6), 142.0 (C-1aryl), 143.3 (C-4aryl), 153.8 (C-5), 163.6 (C-7a), 168.3 (CO). 31P-NMR (162 MHz, DMSO-d6): 21.01 ppm. MS (m/z, I%): 411 (M+, 35%). Anal. calcd for C16H18N3O6PS (411.37): C, 46.72%; H, 4.41%; N, 10.21%; S, 7.79%. Found: C, 46.56%; H, 4.28%; N, 10.06%; S, 7.64%.O), 1263 (PO), 1090, 1014 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.10–1.19 (m, 6H, CH3), 2.36 (s, 3H, CH3), 3.72 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 4.00–4.15 (m, 4H, OCH2), 5.85 (d, 1H, JPCH = 20.4 Hz, P–CH), 7.17 (s, 1H, Ar–H), 7.78 (s, 1H, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 14.0 (CH3), 15.7 (CH3), 16.0 (CH3), 49.8 (d, JPC = 149.1 Hz, C-2), 54.1 (OCH3), 55.8 (2 OCH3), 60.9 (OCH2), 62.3 (OCH2), 93.5 (C-3,5aryl), 106.7 (C-1aryl), 138.7 (C-6), 154.7 (C-5), 160.2 (C-2,6aryl), 161.5 (C-4aryl), 163.4 (C-7a), 166.5 (CO). 31P-NMR (162 MHz, DMSO-d6): 20.05 ppm. MS (m/z, I%): 456 (M+, 100%). Anal. calcd for C19H25N2O7PS (456.45): C, 50.00%; H, 5.52%; N, 6.14%; S, 7.02%. Found: C, 49.87%; H, 5.42%; N, 6.03%; S, 6.90%.
:2) elution system, yielding the purified product.:2) elution system, yielding the purified product.O), 1268 (PO), 1088, 1043, 1017 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.06 (t, 3H, J = 7.2 Hz, CH3), 1.26 (t, 3H, J = 7.2 Hz, CH3), 2.33 (s, 3H, CH3), 3.99 (q, 2H, J = 7.2 Hz, OCH2), 4.21 (q, 2H, J = 7.2 Hz, OCH2), 5.82 (d, 1H, JPCH = 20.4 Hz, P–CH), 7.15 (d, 2H, J = 7.2 Hz, Ph–H), 7.37–7.45 (m, 3H, Ph–H). 13C-NMR (100 MHz, DMSO-d6): 13.0 (CH3), 14.2 (CH3), 15.1 (CH3), 47.5 (d, JPC = 143.0 Hz, C-2), 62.3 (OCH2), 63.3 (OCH2), 125.4 (C-4phenyl), 129.4 (C-3,5phenyl), 130.1 (C-2,6phenyl), 137.0 (C-1phenyl), 139.6 (C-6), 153.7 (C-5), 164.9 (C-7a), 168.4 (CO). 31P-NMR (162 MHz, DMSO-d6): 22.25 ppm. MS (m/z, I%): 366 (M+, 18%). Anal. calcd for C16H19N2O4PS (366.37): C, 52.45%; H, 5.23%; N, 7.65%; S, 8.75%. Found: C, 52.34%; H, 5.16%; N, 7.49%; S, 8.61%.O), 1269 (PO), 1097, 1038 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.06 (t, 3H, J = 7.2 Hz, CH3), 1.23 (t, 3H, J = 7.6 Hz, CH3), 2.20 (s, 3H, CH3), 3.92 (q, 2H, J = 7.2 Hz, OCH2), 4.13 (q, 2H, J = 6.8 Hz, OCH2), 5.79 (d, 1H, JPCH = 17.6 Hz, P–CH), 7.39–7.7.47 (m, 2H, Ar–H), 7.72 (t, 1H, J = 7.2 Hz, Ar–H), 7.90 (d, 1H, J = 7.2 Hz, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 13.1 (CH3), 15.3 (CH3), 16.1 (CH3), 48.5 (d, JPC = 148.7 Hz, C-2), 59.0 (OCH2), 60.4 (OCH2), 124.3 (C-2aryl), 126.9 (C-5aryl), 127.7 (C-4aryl), 131.3 (C-6aryl), 132.1 (C-3aryl), 137.9 (C-6), 141.5 (C-1aryl), 155.5 (C-5), 164.1 (C-7a), 167.5 (CO). 31P-NMR (162 MHz, DMSO-d6): 20.25 ppm. MS (m/z, I%): 444 (M+, 27%) and 446 (M + 2, 18%). Anal. calcd for C16H18BrN2O4PS (445.27): C, 43.16%; H, 4.07%; N, 6.29%; S, 7.20%. Found: C, 43.04%; H, 3.92%; N, 6.13%; S, 7.10%.O), 1268 (PO), 1082, 1042 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.07 (t, 3H, J = 6.4 Hz, CH3), 1.23 (t, 3H, J = 6.8 Hz, CH3), 2.21 (s, 3H, CH3), 3.93 (q, 2H, J = 7.2 Hz, OCH2), 4.24 (q, 2H, J = 6.4 Hz, OCH2), 5.82 (d, 1H, JPCH = 20.8 Hz, P–CH), 7.42 (t, 1H, J = 7.2 Hz, Ar–H), 7.50 (t, 1H, J = 7.2 Hz, Ar–H), 7.91 (d, 1H, J = 7.6 Hz, Ar–H), 8.04 (d, 1H, J = 8.0 Hz, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 13.5 (CH3), 15.6 (CH3), 16.3 (CH3), 48.4 (d, JPC = 148.6 Hz, C-2), 60.5 (OCH2), 61.6 (OCH2), 125.8 (C-5aryl), 126.9 (C-4aryl), 128.1 (C-3aryl), 130.5 (C-6aryl), 133.3 (C-2aryl), 135.5 (C-1aryl), 138.4 (C-6), 154.6 (C-5), 164.4 (C-7a), 168.7 (CO). 31P-NMR (162 MHz, DMSO-d6): 20.03 ppm. MS (m/z, I%): 399 (M+, 9%) and 401 (M+, 3%). Anal. calcd for C16H18ClN2O4PS (400.81): C, 47.95%; H, 4.53%; N, 6.99%; S, 8.00%. Found: C, 47.83%; H, 4.42%; N, 6.89%; S, 7.85%.O), 1267 (PO), 1091, 1016 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.16 (t, 3H, J = 6.8 Hz, CH3), 1.22 (t, 3H, J = 7.2 Hz, CH3), 2.39 (s, 3H, CH3), 4.02 (q, 2H, J = 7.2 Hz, OCH2), 4.21 (t, 2H, J = 6.8 Hz, OCH2), 5.93 (d, 1H, JPCH = 20.4 Hz, P–CH), 6.82 (t, 2H, J = 7.2 Hz, Ar–H), 7.11 (d, 1H, J = 7.2 Hz, Ar–H), 7.43 (d, 1H, J = 7.6 Hz, Ar–H), 9.07 (brs, 1H, OH). 13C-NMR (100 MHz, DMSO-d6): 13.5 (CH3), 15.6 (CH3), 16.4 (CH3), 48.1 (d, JPC = 151.4 Hz, C-2), 60.4 (OCH2), 62.2 (OCH2), 116.5 (C-3aryl), 121.2 (C-5aryl), 125.8 (C-1aryl), 128.8 (C-4aryl), 129.0 (C-6aryl), 138.4 (C-6), 154.6 (C-5), 157.5 (C-2aryl), 163.6 (C-7a), 167.0 (CO). 31P-NMR (162 MHz, DMSO-d6): 20.02 ppm. MS (m/z, I%): 382 (M+, 6%). Anal. calcd for C16H19N2O5PS (382.37): C, 50.26%; H, 5.01%; N, 7.33%; S, 8.38%. Found: C, 50.08%; H, 4.92%; N, 7.18%; S, 8.22%.O), 1268 (PO), 1083, 1041 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.03 (t, 3H, J = 6.4 Hz, CH3), 1.23 (t, 3H, J = 7.2 Hz, CH3), 2.12 (s, 3H, CH3), 3.71 (s, 3H, OCH3), 3.92 (q, 2H, J = 7.2 Hz, OCH2), 4.21 (q, 2H, J = 6.8 Hz, OCH2), 5.82 (d, 1H, JPCH = 21.2 Hz, P–CH), 7.43 (t, 1H, J = 7.2 Hz, Ar–H), 7.71 (d, 1H, J = 8.00 Hz, Ar–H), 7.91 (d, 1H, J = 7.6 Hz, Ar–H), 8.43 (s, 1H, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 13.7 (CH3), 14.8 (CH3), 15.9 (CH3), 48.0 (d, JPC = 152.6 Hz, C-2), 54.3 (OCH3), 59.3 (OCH2), 61.9 (OCH2), 115.5 (C-4aryl), 116.6 (C-2aryl), 121.5 (C-6aryl), 123.4 (C-5aryl), 137.1 (C-1aryl), 139.5 (C-6), 156.0 (C-5), 159.0 (C-3aryl), 162.8 (C-7a), 166.7 (CO). 31P-NMR (162 MHz, DMSO-d6): 19.52 ppm. MS (m/z, I%): 396 (M+, 43%). Anal. calcd for C17H21N2O5PS (396.40): C, 51.51%; H, 5.34%; N, 7.07%; S, 8.09%. Found: C, 51.34%; H, 5.31%; N, 6.95%; S, 7.96%.O), 1270 (PO), 1097, 1044, 1014 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.10 (t, 3H, J = 6.8 Hz, CH3), 1.14–1.23 (m, 3H, CH3), 2.38 (s, 3H, CH3), 3.89–4.16 (m, 4H, OCH2), 5.72 (dd, 1H, JPCH = 22.0 and 9.2 Hz, P–CH), 7.69 (t, 1H, J = 7.6 Hz, Ar–H), 8.18 (d, 1H, J = 7.2 Hz, Ar–H), 8.33 (d, 1H, J = 7.2 Hz, Ar–H), 8.39 (s, 1H, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 13.2 (CH3), 14.3 (CH3), 15.2 (CH3), 47.9 (d, JPC = 157.8 Hz, C-2), 60.1 (OCH2), 61.9 (OCH2), 125.1 (C-2aryl), 128.5 (C-4aryl), 129.4 (C-5aryl), 135.6 (C-6aryl), 137.4 (C-6), 139.4 (C-1aryl), 143.5 (C-3aryl), 154.2 (C-5), 163.1 (C-7a), 167.4 (CO). 31P-NMR (162 MHz, DMSO-d6): 20.06 ppm. MS (m/z, I%): 411 (M+, 51%). Anal. calcd for C16H18N3O6PS (411.37): C, 46.72%; H, 4.41%; N, 10.21%; S, 7.79%. Found: C, 46.58%; H, 4.31%; N, 10.09%; S, 7.67%.O), 1271 (PO), 1097, 1045, 1015 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.10 (t, 3H, J = 7.2 Hz, CH3), 1.29 (t, 3H, J = 7.2 Hz, CH3), 2.22 (s, 3H, CH3), 4.01–4.08 (m, 4H, OCH2), 5.83 (d, 1H, JPCH = 20.4 Hz, P–CH), 7.11 (d, 2H, J = 8.0 Hz, Ar–H), 8.04 (d, 2H, J = 7.6 Hz, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 13.8 (CH3), 15.0 (CH3), 15.9 (CH3), 48.5 (d, JPC = 155.4 Hz, C-2), 59.5 (OCH2), 60.6 (OCH2), 121.4 (C-4aryl), 130.8 (C-2,6aryl), 131.7 (C-3,5aryl), 136.7 (C-1aryl), 138.3 (C-6), 153.1 (C-5), 163.9 (C-7a), 168.2 (CO). 31P-NMR (162 MHz, DMSO-d6): 22.52 ppm. MS (m/z, I%): 444 (M+, 25%) and 446 (M + 2, 23%). Anal. calcd for C16H18BrN2O4PS (445.27): C, 43.16%; H, 4.07%; N, 6.29%; S, 7.20%. Found: C, 43.01%; H, 3.93%; N, 6.11%; S, 7.04%.O), 1268 (PO), 1091, 1018 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.10 (t, 3H, J = 6.8 Hz, CH3), 1.19 (t, 3H, J = 6.4 Hz, CH3), 2.33 (s, 3H, CH3), 3.95 (q, 2H, J = 6.4 Hz, OCH2), 4.19 (q, 2H, J = 6.8 Hz, OCH2), 5.85 (d, 1H, JPCH = 22.0 Hz, P–CH), 7.18 (d, 2H, J = 8.4 Hz, Ar–H), 8.05 (d, 2H, J = 7.6 Hz, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 14.1 (CH3), 15.1 (CH3), 15.9 (CH3), 49.2 (d, JPC = 154.0 Hz, C-2), 60.6 (OCH2), 61.4 (OCH2), 128.5 (C-3,5aryl), 131.3 (C-4aryl), 132.2 (C-2,6aryl), 137.3 (C-6), 135.2 (C-1aryl), 154.6 (C-5), 163.2 (C-7a), 168.6 (CO). 31P-NMR (162 MHz, DMSO-d6): 20.04 ppm. MS (m/z, I%): 399 (M+, 29%) and 401 (M + 2, 9%). Anal. calcd for C16H18ClN2O4PS (400.81): C, 47.95%; H, 4.53%; N, 6.99%; S, 8.00%. Found: C, 47.81%; H, 4.40%; N, 6.86%; S, 7.86%.O), 1272 (PO), 1097, 1045, 1016 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.10 (t, 3H, J = 6.8 Hz, CH3), 1.22 (t, 3H, J = 6.8 Hz, CH3), 2.37 (s, 3H, CH3), 3.92 (q, 2H, J = 6.8 Hz, OCH2), 4.13 (q, 2H, J = 6.8 Hz, OCH2), 5.68 (dd, 1H, JPCH = 22.8 and 9.6 and Hz, P–CH), 8.16 (d, 2H, J = 8.4 Hz, Ar–H), 8.41 (d, 2H, J = 8.4 Hz, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 13.3 (CH3), 15.1 (CH3), 16.0 (CH3), 49.2 (d, JPC = 154.0 Hz, C-2), 59.8 (OCH2), 61.3 (OCH2), 123.4 (C-3,5aryl), 129.5 (C-2,6aryl), 137.1 (C-6), 142.0 (C-1aryl), 143.3 (C-4aryl), 153.8 (C-5), 163.6 (C-7a), 168.3 (CO). 31P-NMR (162 MHz, DMSO-d6): 21.01 ppm. MS (m/z, I%): 411 (M+, 35%). Anal. calcd for C16H18N3O6PS (411.37): C, 46.72%; H, 4.41%; N, 10.21%; S, 7.79%. Found: C, 46.56%; H, 4.28%; N, 10.06%; S, 7.64%.O), 1263 (PO), 1090, 1014 (P–O–C and O–C). 1H-NMR (400 MHz, DMSO-d6): 1.10–1.19 (m, 6H, CH3), 2.36 (s, 3H, CH3), 3.72 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 4.00–4.15 (m, 4H, OCH2), 5.85 (d, 1H, JPCH = 20.4 Hz, P–CH), 7.17 (s, 1H, Ar–H), 7.78 (s, 1H, Ar–H). 13C-NMR (100 MHz, DMSO-d6): 14.0 (CH3), 15.7 (CH3), 16.0 (CH3), 49.8 (d, JPC = 149.1 Hz, C-2), 54.1 (OCH3), 55.8 (2 OCH3), 60.9 (OCH2), 62.3 (OCH2), 93.5 (C-3,5aryl), 106.7 (C-1aryl), 138.7 (C-6), 154.7 (C-5), 160.2 (C-2,6aryl), 161.5 (C-4aryl), 163.4 (C-7a), 166.5 (CO). 31P-NMR (162 MHz, DMSO-d6): 20.05 ppm. MS (m/z, I%): 456 (M+, 100%). Anal. calcd for C19H25N2O7PS (456.45): C, 50.00%; H, 5.52%; N, 6.14%; S, 7.02%. Found: C, 49.87%; H, 5.42%; N, 6.03%; S, 6.90%.3.2 In vitro cytotoxicity
The American type of culture collection (ATCC) provided human cell lines for the human cancer cells TK-10 and A549. A humidified, 5% (v/v) CO2 atmosphere was used to culture the cells at 37 °C in RPMI-1640 supplemented with (100 μg mL−1), penicillin (100 units/mL), and heat-inactivated fetal bovine serum (10% v/v). Using the sulforhodamine B (SRB) assay, the cytotoxicity of the synthesized compounds against TK-10 and A549 human tumor cells was assessed before being treated with the synthesized compounds.403.3 Apoptosis analysis
The TK-10 and A549 cancer cells were treated for 48 h with the products 2e, 2h, and 2j before being trypsinized and subjected to two PBS washes. According to the manufacturer, apoptosis was evaluated using Alexa Fluor-488/PI staining apoptosis.413.4 Cell cycle analysis
The IC50 values for the products 2e, 2h, and 2j were pre-calculated and administered to TK-10 and A549 cells for 48 h. For the distribution of the cell cycle phases for each sample, FACS analysis was completed using a Cytek® Northern Lights 2000 spectral flow cytometer (Cytek Biosciences, Fremont, CA, USA).423.5 Autophagy assessment
Autophagic cell death was quantitatively assessed using acridine orange lysosomal staining coupled with flow cytometric analysis. After treatment with the test compounds for the specified duration, cells (105 cells) were collected by trypsinization and washed twice with ice-cold PBS (pH 7.4). The cells were then stained with acridine orange (10 μM) and incubated in the dark at 37 °C for 30 min. After staining, the cells were then subjected to FACS analysis using a Cytek® Northern Lights 2000 spectral flow cytometer and SpectroFlo™ software version 2.2.0.3 (Cytek Biosciences, Fremont, CA, USA).434. Conclusion
In summary, we have reported a straightforward protocol for the construction of ethyl 2-aryl-3-ethoxy-5-methyl-3-oxido-2H-thiazolo[2,3-e][1,4,2]diazaphosphole-6-carboxylates. The main features of this work are the protocol is less time-consuming, inexpensive, offers a simple procedure and easy handling, and the derivatives can be obtained in high yields. Compounds that have methoxy groups and chlorine atoms displayed promising cytotoxic activities.Data availability
All data are available in the manuscript and ESI.†Author contributions
Data curation: AAS. Formal analysis: AAS. Investigation: TEA, WAB, MYA and SEIE. Writing-original draft: TEA, MAA and WAB. Conceptualization: TEA, MAA and WAB. Supervision: TEA. Resources: TEA, WAB and SEIE. Software: TEA and SEIE. Methodology: WAB and SEIE. Writing – review & editing: TEA, MAA, WAB and SEIE. All authors read and approved the final version of the manuscript.Conflicts of interest
Authors declare that they have no conflict of interest.Acknowledgements
The authors extend their appreciation to University Higher Education Fund for funding this research work under Research Support Program for Central Labs at King Khalid University through the Project Number CL/PRI/B/6.References
- S. J. Kashyap, V. J. Garg, P. K. Sharma, N. Kumar, R. Dudhe and J. K. Gupta, Thiazoles: having diverse biological activities, Med. Chem. Res., 2012, 21, 2123 CrossRef CAS.
- C. A. Calderón-Ospina and M. O. Nava-Mesa, B Vitamins in the nervous system: current knowledge of the biochemical modes of action and synergies of thiamine, pyridoxine, and cobalamin, CNS Neurosci. Ther., 2020, 6, 5 CrossRef PubMed.
- M. Bagheri, M. Shekarchi, M. Jorjani, M. H. Ghahremani, M. Vosooghi and A. Shafiee, Synthesis and Antihypertensive Activity of 1-(2-Thiazolyl)-3,5-disubstituted-2-Pyrazolines, Arch. Pharm. Chem. Life Sci., 2004, 337, 25–34 CrossRef CAS PubMed.
- G. M. Reddy, J. R. Garcia, V. H. Reddy, A. M. Andrade Jr, A. Camilo, R. A. P. Ribeiro and S. R. Lazaro, Synthesis, antimicrobial activity and advances in structure-activity relationships (SARs) of novel tri-substituted thiazole derivatives, Eur. J. Med. Chem., 2016, 123, 508–515 CrossRef CAS PubMed.
- N. Siddiqui and W. Ahsan, Triazole incorporated thiazoles as a new class of anticonvulsants: design, synthesis and in vivo screening, Eur. J. Med. Chem., 2010, 45, 1536–1543 CrossRef CAS PubMed.
- S. Anuradha, R. Patel, P. Patle, A. Parameswaran and J. A. Shard, Design, computational studies, synthesis and biological evaluation of thiazole-based molecules as anticancer agents, Eur. J. Pharm. Sci., 2019, 134, 20–30 CrossRef PubMed.
- M. H. M. Helal, M. A. Salem, M. S. A. El-Gaby and M. Aljahdali, Synthesis and biological evaluation of some novel thiazole compounds as potential anti-inflammatory agents, Eur. J. Med. Chem., 2013, 65, 517–526 CrossRef CAS PubMed.
- P. Singh, S. Gupta and S. Kumar, Thiazole Compounds as Antiviral Agents: An Update, Med. Chem., 2020, 16, 4–23 CrossRef PubMed.
- V. Jaishree, N. Ramdas, J. Sachin and B. Ramesh, In vitro antioxidant properties of new thiazole derivatives, J. Saudi Chem. Soc., 2012, 16, 371–376 CrossRef CAS.
- R. G. Kalkhambkar, G. M. Kulkarni, H. Shivkumar and R. N. Rao, Synthesis of novel triheterocyclic thiazoles as anti-inflammatory and analgesic agents, Eur. J. Med. Chem., 2007, 42, 1272–1276 CrossRef CAS PubMed.
- T. K. Venkatachalam, E. A. Sudbeck, C. Mao and F. M. Uckun, Anti-HIV activity of aromatic and heterocyclic thiazolyl thiourea compounds, Bioorg. Med. Chem. Lett., 2001, 11, 523–528 CrossRef CAS PubMed.
- F. Delmas, A. Avellaneda, C. D. Giorgio, M. Robin, E. D. Clercq, P. Timon-David and J. P. Galy, Synthesis and antileishmanial activity of (1,3-benzothiazol-2-yl)amino-9-(10H)-acridinone derivatives, Eur. J. Med. Chem., 2004, 39, 685–690 CrossRef CAS PubMed.
- K. Moonen, I. Laureyn and C. V. Stevens, Synthetic Methods for Azaheterocyclic Phosphonates and their Biological Activity, Chem. Rev., 2004, 104, 6177–6216 CrossRef CAS PubMed.
- V. Iaroshenko and S. Mkrtchyan, Phosphorus Heterocycles, Organophosphorus Chem., 2019, 295–456 Search PubMed.
- G. Keglevich, Newer Developments in the Synthesis of P-Heterocycles, Curr. Org. Chem., 2019, 23, 1342–1355 CrossRef CAS.
- A. Schmidpeter, Heterophospholes, Phosphorus-Carbon Heterocyclic Chemistry, 2001, pp. 363–461 Search PubMed.
- V. R. Katla, R. Syed, C. S. Kuruva, H. K. Kuntrapakam and N. R. Chamarthi, Synthesis of Novel Phosphorylated Guanidine Derivatives from Cyanamide and their Anti-inflammatory Activity, Chem. Pharm. Bull., 2013, 61, 25–32 CrossRef CAS PubMed.
- M. Golla, S. R. Devineni, R. Syed, B. K. Rayalacheru, R. Maram and N. R. Chamarthi, New substituted 2-aminomethyl-2-oxo-2λ5-perhydro-[1,3,2]oxazaphospholo[3,4-a] pyridine: design, synthesis and antimicrobial activity, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 794–798 CrossRef CAS.
- W. M. Abdou, M. D. Khidre, A. A. Kamel and G. E. A. Awad, Facile regioselective synthesis and antimicrobial activity of heterocycle-phosphor esters, Monatsh. Chem., 2014, 145, 675–682 CrossRef CAS.
- T. E. Ali, M. A. Assiri, H. M. El-Shaaer, S. M. Abed-Kariem, W. R. Abdel-Monem, S. M. El-Edfawy, N. M. Hassanin, A. A. Shati, M. Y. Alfaifi and S. E. I. Elbehairi, Synthesis and Biological Activities of Some New Phosphorus Compounds Containing Pyranopyrazole Moiety, Heterocycles, 2021, 102, 1119–1137 CrossRef CAS.
- T. E. Ali and M. A. Assiri, One-Pot synthesis and antimicrobial of novel 6-ethoxy-6-oxido-3-oxo(thioxo)(imino)-5-substituted-2,7-dihydro-1,2,4-triazolo[3,4-e][1,2,3]diazaphospholes, Phosphorus, Sulfur Silicon Relat. Elem., 2021, 196, 965–969 CrossRef CAS.
- T. E. Ali and M. A. Assiri, Novel 5-Ethoxy-5H-5λ5-[1,2,4]triazolo[5,1-e][1,4,2] diazaphosphol-5-ones: One-Pot Synthesis, Characterization and Antimicrobial Activities, Russ. J. Org. Chem., 2022, 58, 94–98 CrossRef CAS.
- T. E. Ali, M. A. Assiri, A. Y. Alzahrani and M. A. Salem, Simple One-Pot Synthesis of Novel 5-(3,5-Dialkyl-1H-pyrazol-1-yl)-2-ethoxy-1,4,2λ5-diazaphosphole 2-Oxides, Russ. J. Org. Chem., 2023, 59, 285–289 CrossRef.
- R. K. Bansal, K. Karaghiosoff and A. Schmidpeter, Anellated heterophospholes, Tetrahedron, 1994, 50, 7675–7745 CrossRef CAS.
- R. K. Bansal, R. Mahnot, D. C. Sharma and K. Karaghiosoff, Synthesis of New Heterocycles with Dicoordinated Phosphorus: 3-Substituted Thiazolo[3,2-d][1,4,2]diazaphosphole and its 5,6-Dihydro and Benzo Derivatives, Synthesis, 1992, 267–269 CrossRef CAS.
- C. Hettstedt, W. Betzl and K. Karaghiosoff, Synthesis and Characterization of (–)-Menthyl Containing N-Alkyl Cycloimmonium Salts, Z. Anorg. Allg. Chem., 2012, 638, 377–382 CrossRef CAS.
- K. Karaghiosoff, R. Mahnot, C. Cleve, N. Gandhi, R. K. Bansal and A. Schmidpeter, 1,4,2-Diazaphospholothiazoles and -pyridines by a Hantzsch-Type Condensation Using Chloromethyldichlorophosphane, Chem. Ber., 1995, 128, 581–587 CrossRef CAS.
- W. Betzl, C. Hettstedt and K. Karaghiosoff, New anellated 4H-1,4,2-diazaphospholes, New J. Chem., 2013, 37, 481–487 RSC.
- T. E. Ali, M. A. Assiri and N. M. Hassanin, Reaction of 1-(2-Hydroxyphenyl)-3-Phenylpropane-1,3-Dione with Some Phosphorus Halides: A Simple Synthesis of Novel 1,2-Benzoxaphosphinines, Synth. Commun., 2022, 52, 2334–2343 CrossRef CAS.
- T. E. Ali, M. A. Assiri and N. M. Hassanin, One-Pot Synthesis, Antimicrobial Activities and Drug-Likeness Analysis of Some Novel 1,2-Benzoxa-phosphinines, Phospholo-benzofuran, and Chromonyl/Coumarinyl/Indenonyl Phosphonate, Synth. Commun., 2022, 52, 1967–1980 CrossRef CAS.
- T. E. Ali and M. A. Ultrasound, Assisted One Pot, Three-Component Reaction For Facile Design Of Novel Assiri, 1,2,3-Diazaphosphole, 1,5,2-Diazaphosphinine And 1,5,2-Diazaphosphepine Compounds Containing Coumarin Ring, Heterocycles, 2022, 104, 1573–1583 CrossRef CAS.
- D. A. Bakhotmah, T. E. Ali, M. A. Assiri and I. S. Yahia, Synthesis of some novel 2-{pyrano[2,3-c]pyrazoles-4-ylidene}malononitrile fused with pyrazole, pyridine, pyrimidine, diazepine, chromone, pyrano[2,3-c]pyrazole and pyrano[2,3-d]pyrimidine systems as anticancer agents, Polycyclic Aromat. Compd., 2022, 42, 2136 CrossRef CAS.
- M. Narender, M. S. Reddy, V. P. Kumar, B. Srinivas, R. Sridhar, Y. V. D. Nageswar and K. R. Rao, Aqueous-Phase One-Pot Synthesis of 2-Aminothiazole- or 2-Aminoselenazole-5-carboxylates from β-Keto Esters, Thiourea or Selenourea, and N-Bromo-succinimide under Supramolecular Catalysis, Synthesis, 2007, 3469–3472 CAS.
- B. Banerjee, Recent developments on ultrasound assisted catalyst-free organic synthesis, Ultrason. Sonochem., 2017, 35, 1–14 CrossRef CAS PubMed.
- B. Banerjee, Recent developments on ultrasound-assisted synthesis of bioactive N-heterocycles at ambient temperature, Aust. J. Chem., 2017, 70, 872–888 CrossRef CAS.
- Q. Q. Zhang and R. C. Jin, The Application of Low-Intensity Ultrasound Irradiation in Biological Wastewater Treatment: A Review, Crit. Rev. Environ. Sci. Technol., 2015, 45, 2728–2761 CrossRef.
- E. Bálint, Á. Tajti, A. Ádám, I. Csontos, K. Karaghiosoff, M. Czugler, P. Ábrányi-Balogh and G. Keglevich, The synthesis of α-aryl-α-aminophosphonates and α-aryl-α-aminophosphine oxides by the microwave-assisted Pudovik reaction, Beilstein J. Org. Chem., 2017, 13, 76–86 CrossRef PubMed.
- A. J. Rao, P. V. Rao, V. K. Rao, C. Mohan, C. N. Raju and C. S. Reddy, Microwave Assisted One-pot Synthesis of Novel α-Aminophosphonates and their Biological Activity, Bull. Korean Chem. Soc., 2010, 31, 1863–1868 CrossRef CAS.
- S. Hkiri, M. Mekni-Toujani, E. Üstün, K. Hosni, A. Ghram, S. Touil, A. Samarat and D. Sémeril, Synthesis of Novel 1,3,4-Oxadiazole-Derived α-Aminophosphonates/α-Aminophosphonic Acids and Evaluation of their In vitro Antiviral Activity against the Avian Coronavirus Infectious Bronchitis Virus, Pharmaceutics, 2023, 15, 114 CrossRef CAS PubMed.
- A. Mahmoud, A. M. Al-Abd, D. A. Lightfoot and H. A. El-Shemy, Anticancer characteristics of mevinolin against three different solid tumor cell lines was not solely p53-dependent, J. Enzyme Inhib. Med. Chem., 2012, 27, 673–679 CrossRef CAS PubMed.
- S. W. Fesik, Promoting apoptosis as a strategy for cancer drug discovery, Nat. Rev. Cancer, 2005, 5, 876–885 CrossRef CAS PubMed.
- R. Nunez, DNA measurement and cell cycle analysis by flow cytometry, Curr. Issues Mol. Biol., 2001, 3, 67–70 CAS.
- P. Czarny, E. Pawlowska, J. Bialkowska-Warzecha, K. Kaarniranta and J. Blasiak, Autophagy in DNA Damage Response, Int. J. Mol. Sci., 2015, 16, 2641–2662 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07173e |
| This journal is © The Royal Society of Chemistry 2024 |