DOI:
10.1039/D4RA07241C
(Paper)
RSC Adv., 2024,
14, 34643-34660
Novel carboranyl-BODIPY conjugates: design, synthesis and anti-cancer activity†
Received
8th October 2024
, Accepted 23rd October 2024
First published on 29th October 2024
Abstract
A series of four carboranyl-BODIPY conjugates (o-CB-10, m-CB-15, Me-o-CB-28, and Me-o-CB-35) and one phenylene-BODIPY conjugate (PB-20) were synthesized. The carboranyl-BODIPY conjugates incorporate boron clusters, specifically ortho- and meta-carboranes, covalently linked to BODIPY fluorophores while the phenylene-BODIPY conjugate features a phenylene ring covalently linked to BODIPY fluorophore. The newly synthesized conjugates were characterized by 1H NMR, 13C NMR, 11B NMR, 19F NMR, FT-IR, and high-resolution mass spectral analysis. In vitro cytotoxicity of the synthesized conjugates has been evaluated against the HeLa cervical cancer cell line. The study reveals that o-CB-10 shows a maximum cell death potential at lower concentrations (12.03 μM) and inhibited cell proliferation and migration in cancer (HeLa) cells. Additionally, flow cytometry study reveals that o-CB-10 and Me-o-CB-28 arrest the cell cycle at the S phase. The results indicate that the carboranyl-BODIPY conjugates have the potential to be effective anticancer agents.
1. Introduction
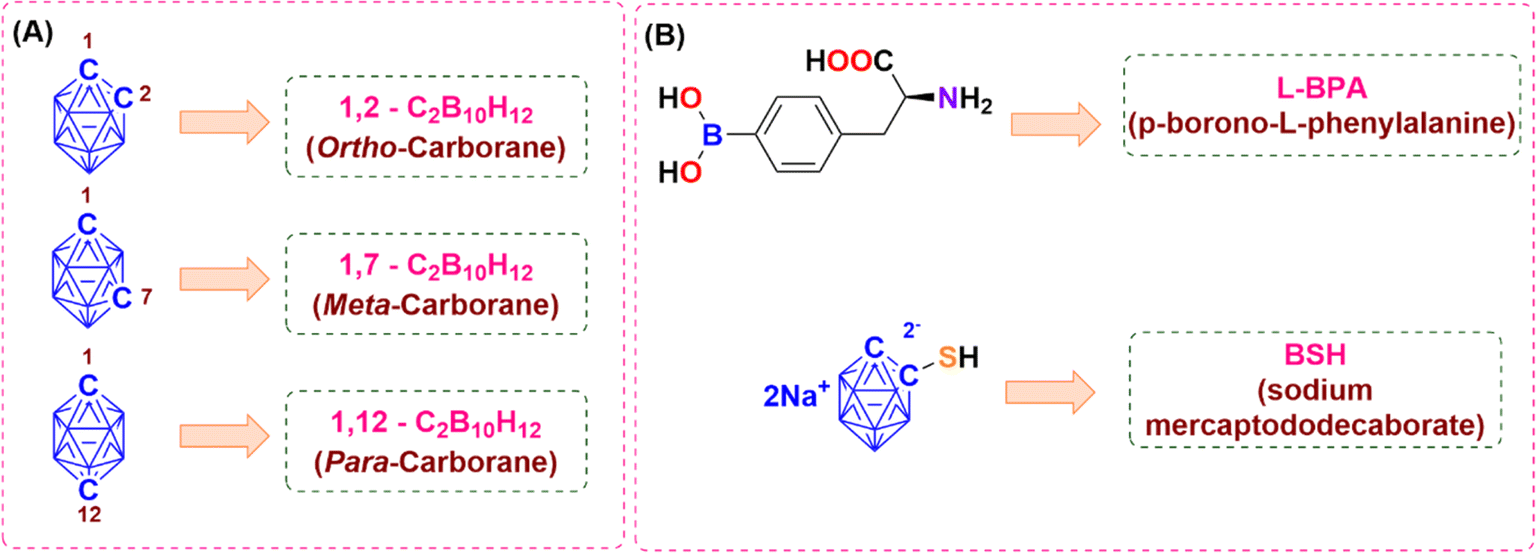
Icosahedral carboranes are often regarded as three-dimensional analogues of benzene.1 They exist in three isomers such as ortho-, meta- and para-carborane distinguished by the relative positions of the two carbon atoms within the icosahedral cage (Fig. 1). They display distinctive electronic and molecular structures,2 which have been applied to a variety of applications such as in medicinal chemistry,3–12 material science13–29 and organometallic/coordination chemistry.30–36 Derivatives of icosahedral carboranes showed cytotoxicity towards several cancer cells. Continuous efforts have been devoted by researchers to conjugate carboranes with several tumour targeting moieties such as amino acids,37–39 receptor-specific membrane antigens,40–43 porphyrins,44–46 micelles,47–49 polymeric nanoparticles,50–53 peptides,54 liposomes,55 dendrimers,56,57 carbohydrates,58–60 and BODIPY,61,62 etc. to enhance the therapeutic effect of the drugs for selective and effective treatment of cancer in addition to their application in boron neutron capture therapy (BNCT). Two boron-based drugs, sodium mercaptododecaborate (BSH) and boronophenylalanine (BPA), have clinically demonstrated efficacy as BNCT agents63 (Fig. 1). Moreover, derivatives of icosahedral carboranes have also shown cytotoxicity towards glioma cells.64,65
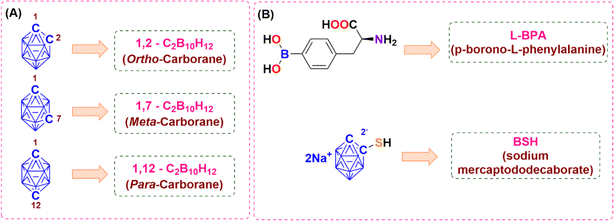 |
| | Fig. 1 (A) Three isomers of icosahedral carborane. (B) Clinically approved boron-based drugs as anti-cancer agents. | |
4,4-Difluoro-4-bora-3a,4a-diaza-s-indacene, commonly referred to as BODIPY, is a compound in which BF2 is chelated to dipyrromethene, the half form of porphyrin. BODIPY-based compounds have garnered significant interest due to their versatility across various applications, including drug administration,66–69 bioimaging,70–79 photodynamic treatment80–82 (PDT), and artificial photosynthetic systems.83–85 Notably, these compounds exhibit a preferential accumulation in tumor tissue, creating favorable tumor-to-healthy tissue ratios, thereby rendering them promising candidates for boron neutron capture therapy (BNCT).86 Moreover, BODIPY can be easily modified to include biological capabilities without affecting its inherent features.
Despite significant progress in therapy, cancer continues to be a prominent cause of death worldwide. Existing therapeutic methods, such as chemotherapy, frequently have limited effectiveness because of the occurrence of unwanted side effects and the emergence of drug resistance.87 These issues arise from the inability to specifically target cancer cells while maintaining the integrity of healthy tissues. Tumorigenesis is a dynamic process characterized by genetic, epigenetic, and metabolic changes that disturb the normal cell cycle.88,89 Cancer cells avoid programmed cell death (apoptosis) and alter cell cycle checkpoints, resulting in uncontrolled replication.90 In the cell cycle, a sequence of events leading to cell division is typically controlled by checkpoints during the G1/S, S, and G2/M phases. Cancer cells evade these regulatory checkpoints, allowing uncontrolled proliferation. Targeting proteins involved in the course of the cell cycle has become a viable technique for treating cancer.91
Multiple chemicals have been formulated to disrupt the course of the cell cycle. Flavopiridol, for example, hinders cyclin-dependent kinases (CDKs), resulting in the halting of the cell cycle in both the G1 and G2 stages.92 Indisulam (E7070), a compound derived from sulfonamide derivative, causes cells to stop in the G1 phase of the cell cycle and slows down the transition from G1 to the S phase, and finally results in cells being arrested in the G2 phase and undergoing apoptosis.93 Additional drugs, including AZD5438, SNS-032, bryostatin-1, seliciclib, PD 0332991, and SCH 727965, demonstrate inhibitory effects on the cell cycle at various phases.94
So, developing novel chemotherapeutic medications that can specifically target and eliminate cancer cells while limiting harm to healthy organs is crucial. In 2020, our research group developed triazine-cored dendritic molecules containing multiple o-carborane clusters and investigated their cytotoxicity against breast cancer cell lines.4 In the same year, we reported the synthesis of a series of carborane-appended glycoconjugates with selective cytotoxicity towards MCF-7 breast cancer cells and A431 skin cancer cells over healthy cells.6 Additionally, we also developed star-shaped phenylene BODIPY and conducted toxicity and biocompatibility evaluations on zebrafish.66 In 2023, dendritic glycoconjugates containing multiple o-carborane clusters were synthesized, and their anti-cancer efficacy was studied against MCF-7 breast cancer cell lines.56 These findings inspired us to continue the bioactivity exploration of carborane and BODIPY-based molecules. Herein, we report the synthesis of four carboranyl-BODIPY conjugates: ortho-carboranyl BODIPY 10 (o-CB-10), meta-carboranyl BODIPY 15 (m-CB-15), methylene bridged ortho-carboranyl BODIPY 28 (Me-o-CB-28) and methylene bridged ortho-carboranyl BODIPY 35 containing two carborane moieties (Me-o-CB-35) along with one phenylene-BODIPY conjugate 20 (PB-20). Biological evaluation of these compounds was carried out against HeLa cervical cancer cell lines to explore the ability of the synthesized carboranyl BODIPYs as anti-cancer therapeutics.
2. Results and discussion
2.1 Synthesis
Four carboranyl-BODIPY conjugates (o-CB-10, m-CB-15, Me-o-CB-28, and Me-o-CB-35) and one phenylene-BODIPY conjugate (PB-20) were designed and successfully synthesized following several reaction conditions. Synthetic methods like Sonogashira cross-coupling reaction, Suzuki cross-coupling reaction, decaborane insertion, PCC mediated oxidation, base promoted substitution reaction, etc. were involved in addition to TFA catalyzed preparation of dipyrromethane, DDQ mediated formation of dipyrromethene and subsequent BF3. OEt2 promoted the formation of BODIPY in the presence of a base like Et3N. All the newly synthesized compounds were well characterized through FT-IR, NMR, and mass spectral analysis (see ESI†).
2.1.1 Synthesis of ortho-carboranyl BODIPY 10 (o-CB-10). Synthesis of ortho-carboranyl BODIPY 10 (o-CB-10) was carried out starting from commercially available compound 1-bromo-4-iodobenzene 1 as shown in Scheme 1. The reaction of compound 1 with trimethylsilyl acetylene 2 under Sonogashira cross-coupling reaction conditions afforded compound 3 with excellent yield. K2CO3 promoted deprotection of the TMS group in compound 3, which yielded alkynylated compound 4 in good yield. Ortho-carborane appended derivative 5 was synthesized upon decaborane insertion reaction of compound 4 in acetonitrile. Subsequently, the ortho-carborane appended aldehyde 7 was formed via Suzuki coupling reaction in the presence of mild base Cs2CO3. The condensation of compound 7 with compound 8 in the presence of trifluoroacetic acid (TFA) produced o-carboranyl dipyrromethane 9. The resultant o-carboranyl dipyrromethane subsequently oxidized using 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) to afford the stable ortho-carborane appended dipyrromethene which undergone deprotonation in basic conditions using Et3N before being exposed to excess BF3·OEt2and formed the desired o-CB-10 with 41% yield. Substituted pyrrole 8 used in the synthesis of dipyrromethene derivatives was prepared in three steps in accordance with the reported procedure.95
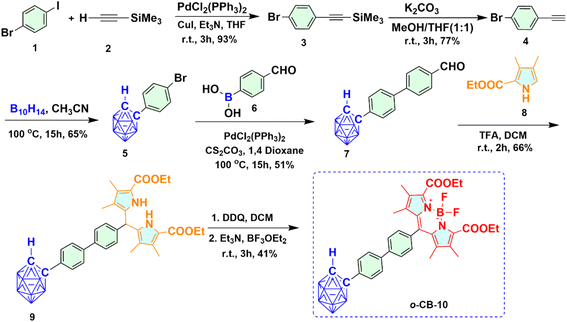 |
| | Scheme 1 Synthetic route for ortho-carboranyl BODIPY 10 (o-CB-10). | |
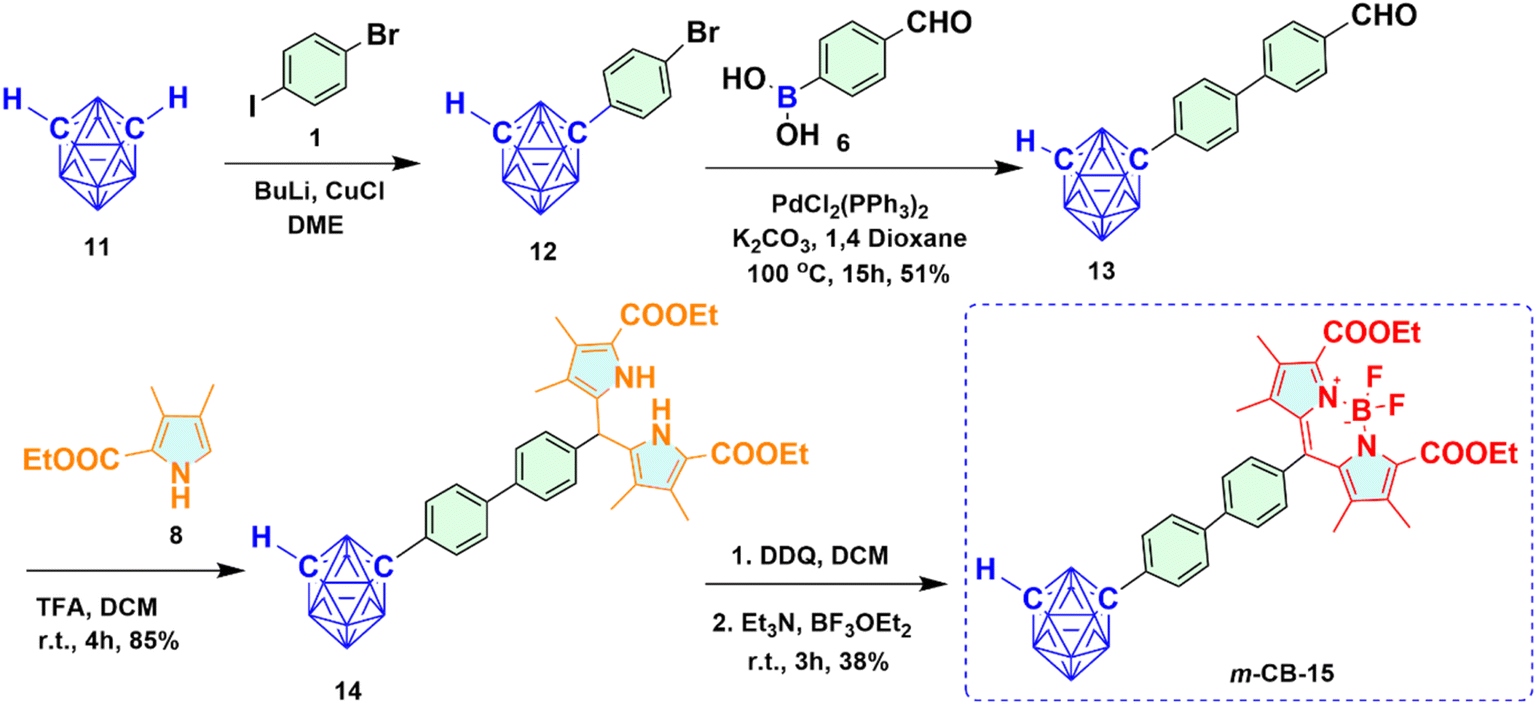
2.1.2 Synthesis of meta-carboranyl BODIPY 15 (m-CB-15). Starting from BuLi mediated reaction of meta-carborane 11, m-CB-15 was prepared as depicted in Scheme 2. At first, the C-arylated mono-bromo derivative of m-carborane 12 was synthesized from the reaction of meta-carborane 11 with compound 1 via reported Cu(I) mediated approach in the presence of strong base like BuLi followed by purification using column chromatography and sublimation. Upon reaction of compound 12 with 4-formyl phenyl boronic acid 6 under Suzuki cross-coupling reaction conditions, meta-carboranyl C-aryl aldehyde 13 was prepared in good yield. Trifluoroacetic acid (TFA) catalyzed condensation of compound 13 with compound 8 produced meta-carboranyl dipyrromethane 14 in decent yield as a white solid. The resultant m-carboranyl dipyrromethane 14 subsequently oxidized to m-carboranyl dipyrromethene derivative using DDQ, and upon addition of Et3N followed by BF3·OEt2 to the same reaction mixture, m-CB-15 was synthesized as red solid with 38% yield.
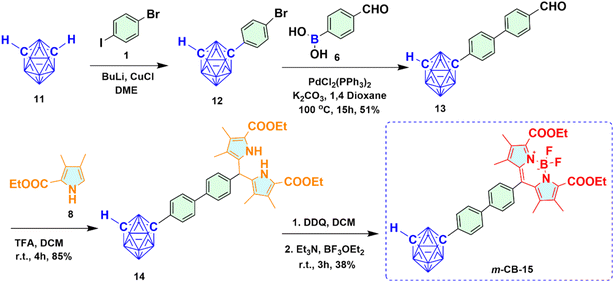 |
| | Scheme 2 Synthetic route for meta-carboranyl BODIPY 15 (m-CB-15). | |
2.1.3 Synthesis of phenylene BODIPY 20 (PB-20). To find out the effect of o- and meta-carborane on the properties of BODIPY derivatives, PB-20 was synthesized by starting from the Suzuki coupling reaction of compound 1 with phenylboronic acid 16 as presented in Scheme 3. The reaction of compound 1 with compound 16 catalysed by PdCl2(PPh3)2 in presence of Na2 CO3 afforded compound 17 in 81% yield. Again, under the Suzuki coupling condition, the corresponding aldehyde 18 was prepared upon reaction of compound 17 with 4-formylphenyl boronic acid 6 with 65% yield. Phenylene dipyrromethane 19 was synthesized with 64% yield upon treatment of compound 18 with substituted pyrrole 8 catalyzed by trifluoroacetic acid (TFA). Finally, in one pot reaction, oxidation of dipyrromethane 19 was carried out using DDQ to form phenylene dipyrromethene derivative, which upon treatment with Et3N and BF3·OEt2 afforded PB-20 as red solid in 42% yield.
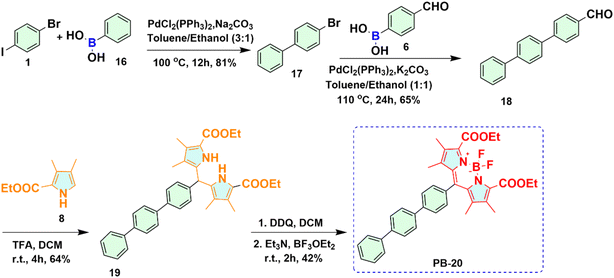 |
| | Scheme 3 Synthetic route for phenylene BODIPY 20 (PB-20). | |
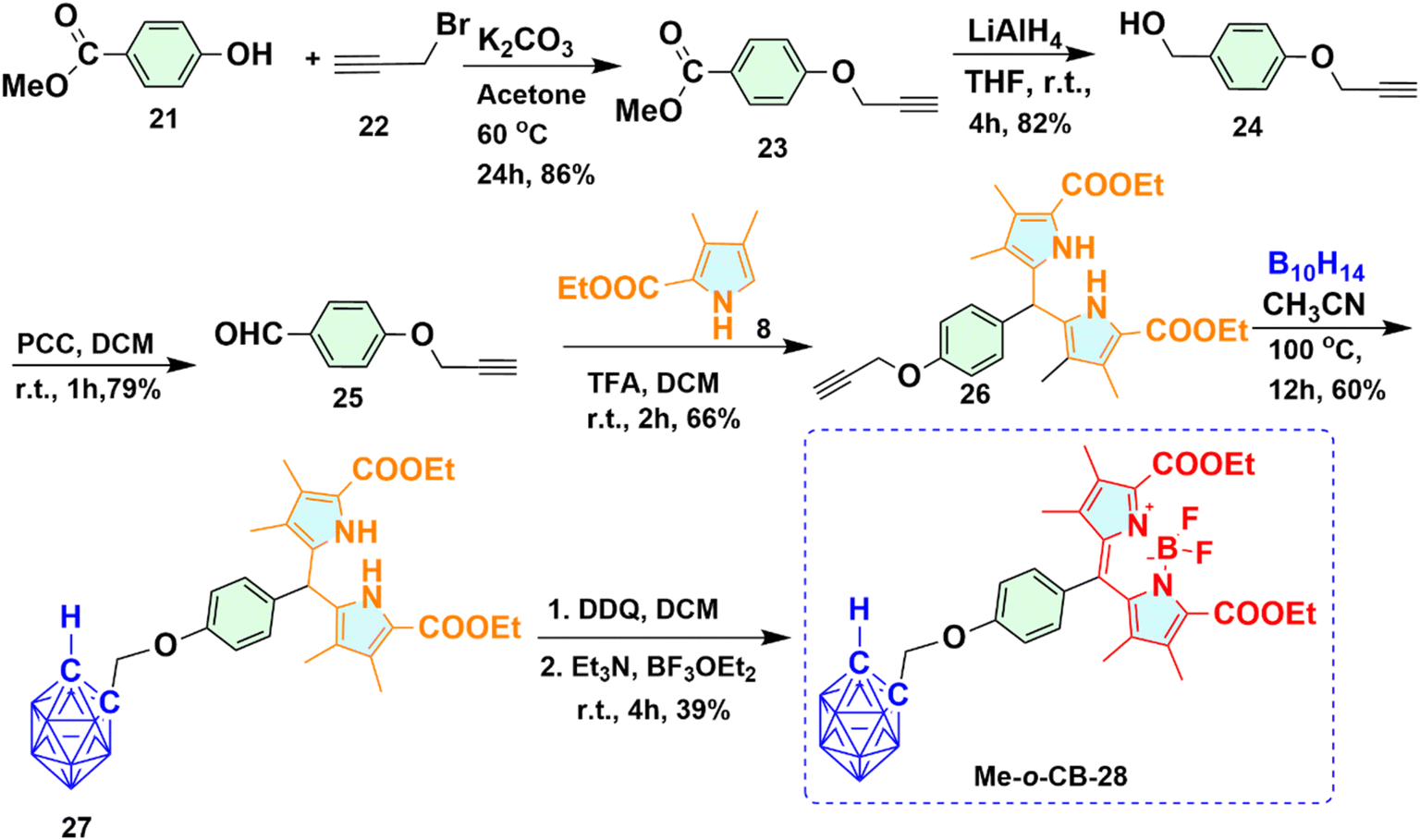
2.1.4 Synthesis of methylene bridged ortho-carboranyl BODIPY 28 (Me-o-CB-28). Synthetic routes used to obtain Me-o-CB-28 is shown in Scheme 4. Synthesis of BODIPY 28 containing one ortho-carborane moiety was achieved starting from a base-promoted nucleophilic substitution reaction. Commercially available methyl-4-hydroxybenzoate 21 and propargyl bromide 22 underwent a nucleophilic substitution reaction in the presence of base K2CO3 to get alkyne derivative 23 with 86% yield. 4-(Prop-2-ynyloxy) phenyl methanol 24 was obtained upon reduction of the ester group of compound 23 with LiAlH4. The primary alcohol group of compound 23 oxidized to 4-(Prop-2-ynyloxy) benzaldehyde 25 using pyridinium chlorochromate (PCC) as an oxidizing agent. To prevent the reduction of the aldehyde group to primary alcohol in the presence of decaborane, compound 25 was first converted to dipyrromethane 26, and then the o-carborane cage was formed upon refluxing with decaborane. Trifluoroacetic acid (TFA) catalyzed the reaction of compound 25 with substituted pyrrole 8 in dichloromethane, afforded dipyrromethane 26 with 66% yield. Ortho-carboranyl dipyrromethane conjugate 27 was prepared from compound 26 upon refluxing with decaborane. In a one-pot reaction system, the resultant o-carboranyl dipyrromethane 27 underwent an oxidation reaction using 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ), yielding o-carboranyl dipyrromethene monitored by TLC. To the same reaction mixture, subsequent addition of triethyl amine followed by BF3·OEt2 provided the desired Me-o-CB-28 in 39% yield as a red solid.
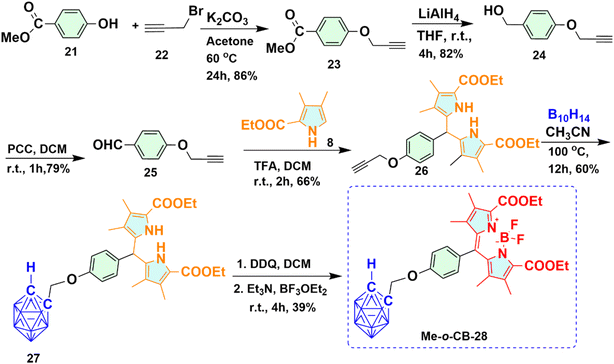 |
| | Scheme 4 Synthetic route for methylene bridged ortho-carboranyl BODIPY 28 (Me-o-CB-28). | |
2.1.5 Synthesis of methylene bridged ortho-carboranyl BODIPY 35 containing two carborane moieties (Me-o-CB-35). Under a similar approach as described in the preparation of Me-o-CB-28, carboranyl BODIPY 35 containing two ortho-carborane cages was prepared as deep red solid shown in Scheme 5. The reaction of commercially available methyl-3,5-dihydroxybenzoate 29 with propargyl bromide 22 in the presence of base K2CO3 yielded alkyne derivative 30, which upon subsequent reduction with LiAlH4 provided compound 31 with a decent yield. PCC-mediated oxidation of compound 31 afforded formyl compound 32 with 85% yield. Trifluoroacetic acid (TFA) catalyzed the reaction of compound 32 with substituted pyrrole 8, afforded dipyrromethane 33 with 66% yield, which, upon refluxed with decaborane compound 34 containing two carborane moieties, was prepared. The resultant ortho-carboranyl dipyrromethane 34 underwent oxidation with DDQ forming o-carboranyl dipyrromethene checked by TLC. Subsequent addition of triethyl amine and BF3·OEt2 provided the desired Me-o-CB-35 in 43% yield.
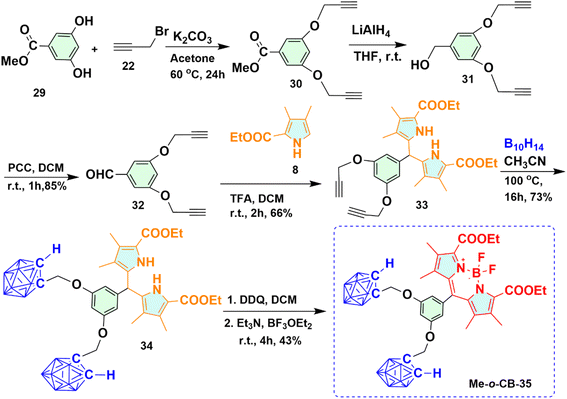 |
| | Scheme 5 Synthetic route for methylene bridged carboranyl BODIPY 35 (Me-o-CB-35) having two ortho-carborane moieties. | |
The final BODIPY conjugates o-CB-10, m-CB-15, Me-o-CB-28, Me-o-CB-35, and PB-20 were confirmed from 1H NMR, 13C NMR, 11B NMR, 19F NMR, FT-IR and high-resolution mass spectral evidence. All conjugates exhibited solubility in DMSO solvent. In all BODIPY compounds, the disappearance of the –NH proton peak around δ = 8.43–9.26 ppm and meso proton around δ = 5.5 ppm in 1H NMR spectra indicated the formation of BODIPY conjugates. The presence of the BF2 group in BODIPY conjugates was evident from 11B NMR and 19F NMR spectral data. The presence of fluorine was confirmed from 19F NMR spectral analysis, which showed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 quartet in the range δ = −140 to δ = −141 ppm. In proton decoupled 11B NMR spectra of BODIPY conjugates, a triplet peak is observed in the range δ = −0.23 to δ = −0.85, corresponding to BF2 boron. In 13C NMR spectra appearance of peaks around δ = 60 ppm and δ = 75 ppm for two cage carbons and in proton decoupled 11B NMR spectra resonance of peaks in the range δ = 0 ppm to δ = −15 ppm confirms the presence of carborane cage/s in carboranyl BODIPY conjugates. FT-IR spectral analysis showed strong bands around νstr = 2600 cm−1 corresponding to B–H stretching in all carboranyl BODIPY conjugates. The anti-cancer efficacy of the synthesized BODIPYs against HeLa cervical cancer cells were investigated through in vitro cytotoxicity, cell proliferation activity, cell migration activity, and p53 expression study. Flow cytometry study was also carried out to explore at which phase the synthesized BODIPYs able to inhibit the HeLa cell cycle progression.
:1:1:1 quartet in the range δ = −140 to δ = −141 ppm. In proton decoupled 11B NMR spectra of BODIPY conjugates, a triplet peak is observed in the range δ = −0.23 to δ = −0.85, corresponding to BF2 boron. In 13C NMR spectra appearance of peaks around δ = 60 ppm and δ = 75 ppm for two cage carbons and in proton decoupled 11B NMR spectra resonance of peaks in the range δ = 0 ppm to δ = −15 ppm confirms the presence of carborane cage/s in carboranyl BODIPY conjugates. FT-IR spectral analysis showed strong bands around νstr = 2600 cm−1 corresponding to B–H stretching in all carboranyl BODIPY conjugates. The anti-cancer efficacy of the synthesized BODIPYs against HeLa cervical cancer cells were investigated through in vitro cytotoxicity, cell proliferation activity, cell migration activity, and p53 expression study. Flow cytometry study was also carried out to explore at which phase the synthesized BODIPYs able to inhibit the HeLa cell cycle progression.
2.2 Biological evaluation
2.2.1 Cytotoxicity of BODIPY conjugates (o-CB-10, m-CB-15, Me-o-CB-28, Me-o-CB-35, and PB-20) on HeLa cell viability. The anti-cancer efficacy of the BODIPY conjugates (o-CB-10, m-CB-15, Me-o-CB-28, Me-o-CB-35, and PB-20) against Human Cervical cancer cells (HeLa cells) were investigated. Fig. 2A illustrates the results of in vitro cytotoxicity studies conducted with BODIPY conjugates against HeLa cells after 72 hours, and dose-dependent cytotoxic effects were observed. The half-maximal inhibitory concentration (IC50) values were calculated and are presented in Fig. 2B. In vitro cytotoxicity assays revealed that the carboranyl-BODIPY conjugates exhibited varying levels of toxicity against HeLa cervical cancer cells. Notably, o-CB-10 demonstrated the highest cytotoxicity, with an IC50 value of 12.03 μM, indicating its potent anti-cancer activity. Conversely, m-CB-15 displayed intermediate cytotoxicity (IC50 = 28.74 μM), while the phenylene BODIPY (PB-20) exhibited significantly lower cytotoxicity (IC50 = 48.35 μM). Surprisingly, the methylene-bridged ortho-carboranyl BODIPY conjugates, Me-o-CB-28 and Me-o-CB-35, exhibited notably lower cytotoxicity, with IC50 values of 48.99 μM and 42.38 μM, respectively. These findings suggest that the conjugation of carborane moieties to BODIPY enhances cytotoxicity compared to phenylene-based conjugates with ortho-carborane showing greater potency than meta-carborane (Fig. 3). Furthermore, the presence of a methylene spacer between the carborane and BODIPY moieties appears to significantly diminish cytotoxicity, possibly due to disruption of the electronic conjugation between these groups.
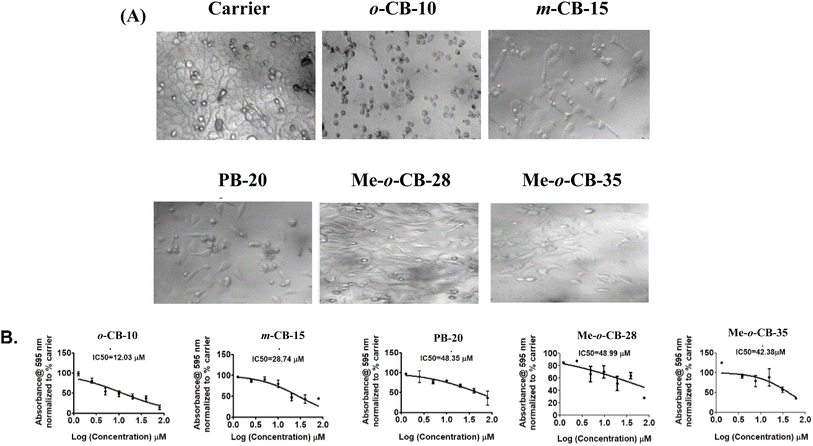 |
| | Fig. 2 (A) Representative images of the HeLa cells treated with carrier (DMSO) or 10 μM concentrations of each of the compounds o-CB-10, m-CB-15, PB-20, Me-o-CB-28 and Me-o-CB-35 for 72 h on HeLa cells. The compound o-CB-10 shows highest degree of cell death in comparison to others (B) Represents the dose inhibitory curve determined by MTT cell viability assay. The IC50 values are indicated in the graph. Results are expressed as the mean ± standard error. | |
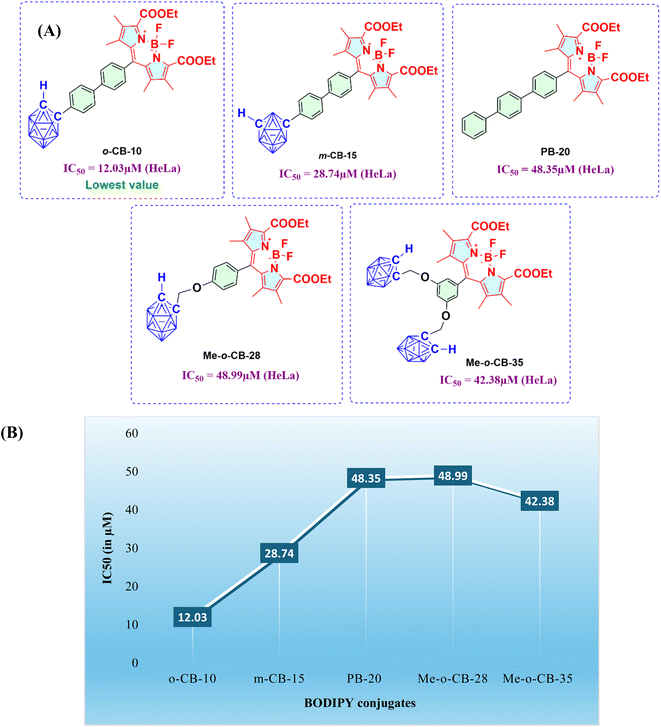 |
| | Fig. 3 (A) Shows the IC50 values of compounds o-CB-10, m-CB-15, PB-20, Me-o-CB-28, Me-o-CB-35. (B) Table showing trends in IC50 values BODIPY-conjugates. | |
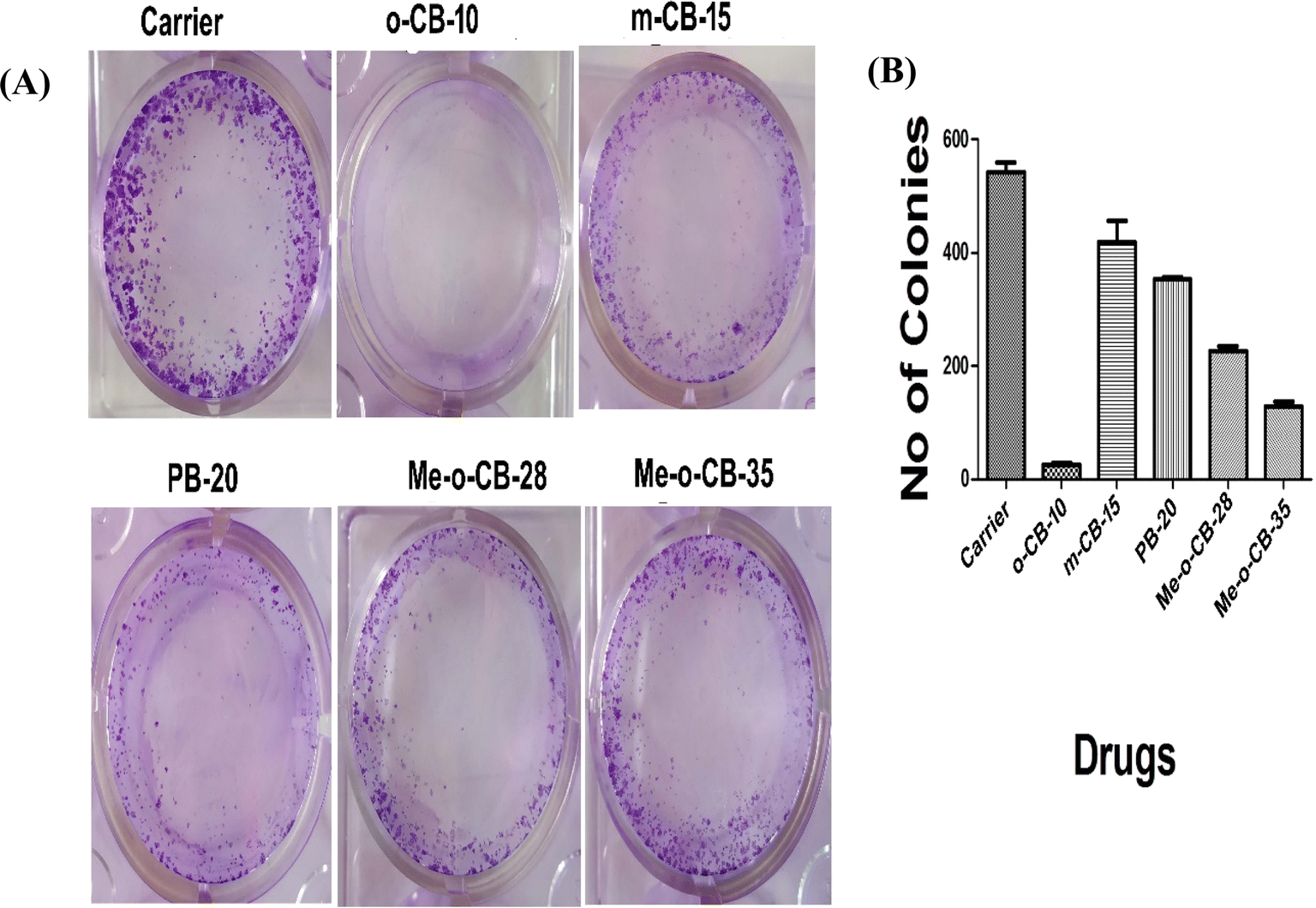
2.2.2 Effect of BODIPY conjugates on HeLa cell proliferation. The anti-cancer effects of BODIPY conjugates can further be assessed form the inhibition of cell proliferation. The study evaluates the reproductive potential of cells for unrestricted division and colony formation by employing the cell survival assay to examine the capacity of an individual cell to develop into a colony.96 HeLa Cells were treated with a fixed sub-lethal concentration (10 μM) of the BODIPY conjugates (o-CB-10, m-CB-15, PB-20, Me-o-CB-28 and Me-o-CB-35) along with the carrier. The colonies were stained with crystal violet, and the photograph was taken. The representative images of crystal violet stained colonies are shown in Fig. 4A. The colonies were counted and shown in graph Fig. 4B. The result indicates that o-CB-10 is more effective in inhibiting HeLa cell proliferation compared to other tested compounds. Significant anti-proliferative activity was observed in the case of ortho-carboranyl BODIPY o-CB-10, while other compounds under study were found to be less efficient in inhibiting HeLa cell proliferation.
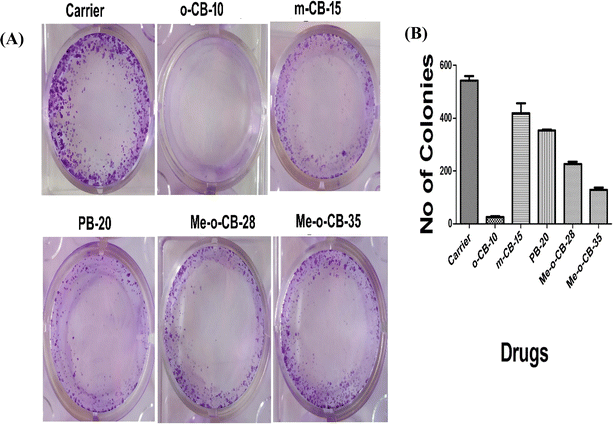 |
| | Fig. 4 (A) Colony forming assay of HeLa cells. Cells were treated with carrier or 10 μM each of BODIPY conjugates in 12 well plates and incubated for 14 days. Cells were fixed and stained with crystal violet. (B) The number of colonies was quantified by using ImageJ software and the graph was plotted by using Graph pad Prism. | |
2.2.3 Effects of BODIPY conjugates on HeLa cell migration. The development of tumors to metastasis is a multistage process wherein malignant cells spread from the main tumor to inhabit distant locations.97 To investigate the potential of BODIPY conjugates (o-CB-10, m-CB-15, Me-o-CB-28, Me-o-CB-35, and PB-20) to inhibit cell migration, a wound-healing assay was employed. This in vitro model mimics the migratory behavior of cells observed in vivo during wound repair. By creating a “wound” in a cell monolayer and monitoring its closure over time, the rate of cell migration can be quantified. The wound was made using a P10 pipette tip and treated with carrier (DMSO) as well as 10 μM concentration of each of the synthesized BODIPY conjugates for 24 h. To observe the scratch, distance images were collected at 0 hours and 24 hours under an inverted microscope and are shown in Fig. 5A. Cell migration was analyzed as reported earlier98 and plotted in graph as depicted in Fig. 5B. The result indicate that o-CB-10 is highly significant in inhibiting cell migration in comparison to the carrier and other tested compounds.
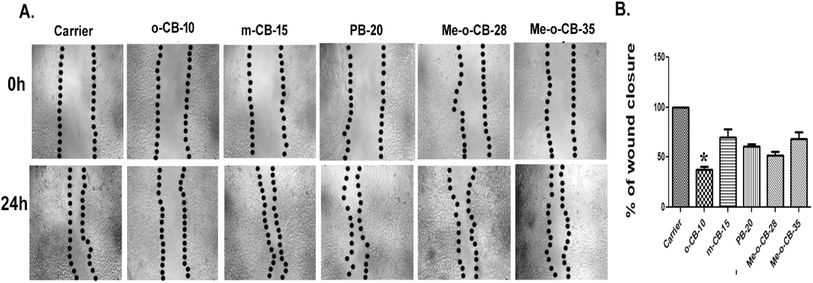 |
| | Fig. 5 (A) Photomicrograph of HeLa cells treated with carrier or 10 μM concentrations of each of the compounds at 0 h, and 24 h. (B) Graph represents the closure of wound area for carrier and compound treated cells at 0 h, and 24 h respectively. The compound o-CB-10 has least wound closure than carrier and others. Statistical analysis was carried out using ANOVA and the p < 0.05. | |
2.2.4 Flow cytometry study of BODIPY conjugates. To investigate the effects of BODIPY conjugates on cell cycle regulation, HeLa cells were treated with a fixed sub-lethal concentration (10 μM) of each BODIPY conjugate for 24 hours. The percentage of cells in the G1, S, and G2/M phases of the cell cycle was determined based on the distribution of PI fluorescence intensity (FL2-A channel). Forward scatter (FSC–H) was used to exclude cell debris and aggregates. The results depicted in Fig. 6 demonstrate that treatment with o-CB-10 and Me-o-CB-28 induced a significant arrest of HeLa cells in the S phase of the cell cycle. In contrast, the remaining compounds, m-CB-15, PB-20, and Me-o-CB-35, primarily caused a G1 phase arrest. These findings suggest that the BODIPY conjugates exert distinct effects on cell cycle progression, with o-CB-10 and Me-o-CB-28 specifically targeting S phase cell cycle arrest. S phase arrest serves as a protective mechanism that facilitates DNA repair, prevents mutation accumulation, and enhances therapeutic strategies against cancer by promoting apoptosis and enabling synchronized cellular responses.99
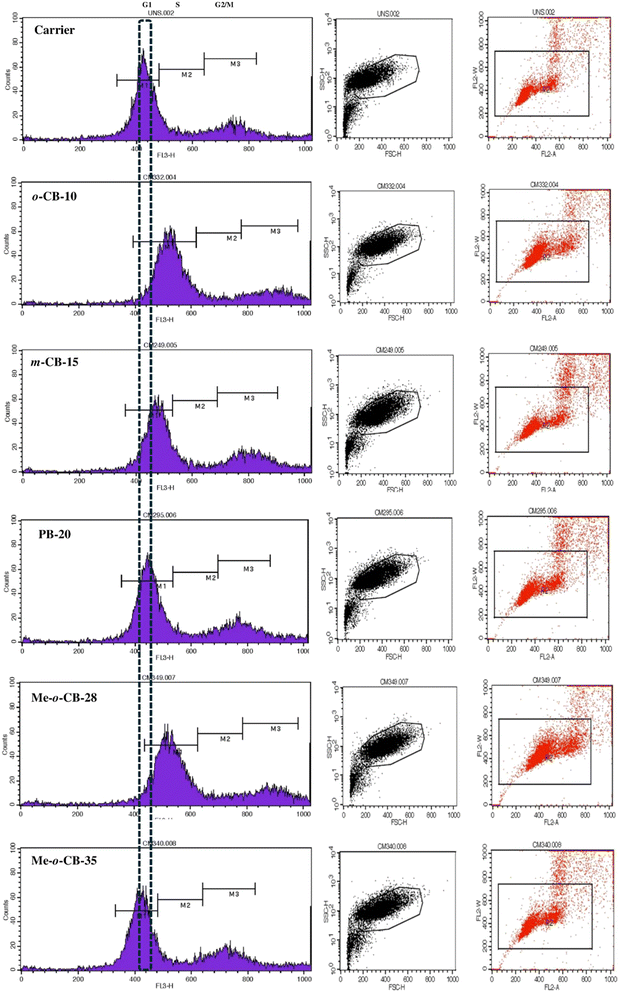 |
| | Fig. 6 Effect of BODIPY conjugates on the cell cycle distribution of HeLa cells. The compounds o-CB-10 and Me-o-CB-28 treated cell are arrested in S phase of cell cycle. | |
2.2.5 Effects of BODIPY compounds on the expression of p53 on HeLa cells. p53, a crucial tumor suppressor, safeguards genomic integrity by regulating cell cycle, DNA repair and apoptosis. Activated by DNA damage, p53 halts the cell cycle for repair or initiates apoptosis to prevent cancerous cells from spreading.100,101 HeLa cells typically contain wild-type p53 alleles alongside HPV (human papillomaviruses) DNA.102 Research shows that HPV E6 can inhibit p53's transcriptional activity without degrading it, which is crucial for understanding how HPV contributes to cervical carcinogenesis.103 The expression levels and activity of p53 can serve as biomarkers for predicting responses to therapies. Elevated p53 levels have been associated with increased sensitivity to treatments that induce apoptosis.104 We evaluated the p53 expression of HeLa cells treated with BODIPY conjugates (10 μM) using immunocytochemistry. The cells were stained with Hoechst 33342, and fluorescence images were merged using the ImageJ program, as shown in Fig. 7A. The images were quantified using an ImageJ plotted graph, and statistical analysis was carried out using Graph pad Prism with a p-value of 0.05, as shown in Fig. 7B. The data reveal that p53 expression was significantly higher in the cells treated with Me-o-CB-28. However, the other drugs did not significantly alter the expression of p53 remarkably (Fig. 8).
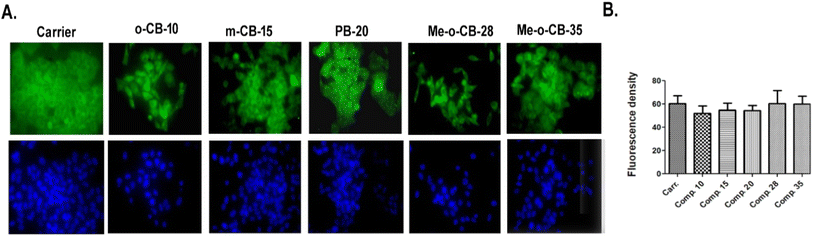 |
| | Fig. 7 p53 immunofluorescence analysis of HeLa cells treated with BODIPY conjugates. (A) Fluorescence images of the treated cells stained with Hoechst 33342 (B) statistical analysis using Graph pad Prism with p value 0.05. | |
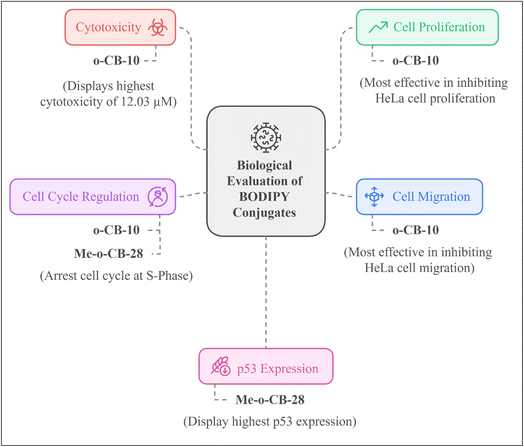 |
| | Fig. 8 Summary of biological evaluation of BODIPY conjugates. | |
3. Conclusion
In summary, four carboranyl-BODIPY conjugates (o-CB-10, m-CB-15, Me-o-CB-28, and Me-o-CB-35) and one phenylene-BODIPY conjugate (PB-20) were synthesized. The anti-cancer efficacy of the synthesized compounds were evaluated against HeLa cervical cancer cells. Among them, ortho-carboranyl BODIPY compound 10 (o-CB-10) exhibited higher cytotoxicity, efficiently inhibiting HeLa cell proliferation and migration, and causing cell cycle arrest at the S phase. The methylene-bridged ortho-carboranyl BODIPY conjugate 28 (Me-o-CB-28) was found to restore p53 protein in HeLa cells without affecting EGFR expression, while also inducing cell cycle arrest at the S phase, as shown by flow cytometry studies.
Although compound o-CB-10 demonstrated potent anticancer activity, further studies are needed to clarify the underlying mechanisms of toxicity, particularly regarding the role of ortho-carborane. The observation that compounds Me-o-CB-28, which contain one ortho-carborane moiety and Me-o-CB-35, which contain two ortho-carborane moiety, do not exhibit proportionally greater toxicity suggests that factors such as cellular internalization or other mechanisms may influence the differences in cytotoxicity. Therefore, the role of ortho-carborane in the observed biological activity remains inconclusive.
These findings highlight the potential of carboranyl-BODIPY conjugates as anticancer agents. The effectiveness of the conjugates can be attributed to their ability to generate reactive oxygen species (ROS).105 When the conjugates react chemically with cancer cells, the generated ROS can induce oxidative stress within cancer cells, leading to DNA damage, protein oxidation, and lipid peroxidation.106 This oxidative stress can trigger apoptotic pathways, ultimately resulting in cell death. Moreover, the conjugates also interact with cellular targets involved in cell cycle regulation or signaling pathways, disrupting normal cellular processes and promoting apoptosis. The combined effects of these mechanisms contribute to the observed anticancer activity of the carboranyl-BODIPY conjugates. But further research involving other cancer cell lines, in vivo models, and mechanistic studies is required to better understand their efficacy, specificity, and potential side effects. Ongoing work is addressing these aspects.
4. Experimental section
4.1 Materials and methods
Reactions were generally performed under argon atmosphere in oven-dried flasks. Solvents and reagents were added using syringes. Solvents were dried and distilled using standard procedures. Reagents were purchased from commercial suppliers and were used as received without further purification. All compounds were purified by column chromatography on silica gel (60–120 mesh). Yields of the products refer to analytically pure samples. Progress of the reaction was monitored by TLC on silica gel 60 F254-coated TLC plates and visualized by Short-UV light at 254 nm. 1H and 13C NMR spectra were recorded on Bruker Fourier Transform multinuclear NMR spectrometer at 400 MHz and 100 MHz respectively. Chemical shifts are reported relative to TMS (1H: δ 0.00 ppm), CDCl3 (13C: δ = 77.0 ppm) and coupling constants are given in Hertz (Hz). 11B NMR spectra are proton-decoupled and recorded at 128 MHz relative to BF3·Et2O. 19F NMR spectra were recorded at 376 MHz. FT-IR spectra were recorded on a Thermo Scientific Nicolet iS5 FT-IR spectrophotometer. Melting points were measured with optics digital melting point apparatus and are uncorrected.
4.2 Cell culture
Dulbecco's modified eagle medium (DMEM), fetal bovine serum (FBS), trypsin 0.25% EDTA, penicillin–streptomycin (cell clone, Genetix India), ITS – Insulin Transferrin Selenium (cell clone). Human cervical cancer (HeLa cells) obtained from National Centre for Cell Sciences, Pune, India and were cultured in complete Dulbecco's modified eagle medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) and 1% penicillin–streptomycin (PS) in a humidified incubator with 5% CO2 and in atmospheric oxygen.
4.3 Cell viability assay
Cells were seeded in 96 well plates (2 × 103 per well) incubated overnight for attachment and treated with different concentrations (1.25 μM, 2.51 μM, 5.02 μM, 10.05 μM, 20.11 μM, 40.23 μM and 80 μM) of BODIPY conjugates (o-CB-10, m-CB-15, PB-20, Me-o-CB-28, and Me-o-CB-35) for 72 h. Then 50 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg mL−1 in DMEM w/o phenol red) was added to each well and the cells were further incubated for next 4 h. After that, the medium was aspirated, and the purple formazan crystals were dissolved by adding 80 μl DMSO and kept in a shaker for 30 minutes. Then absorbance was measured at 595 nm by a microplate reader.
4.4 Colony forming assay
Colony Forming Assay was performed by taking approximately 500 numbers of cells per well. They were seeded in a 12 well plate and treated with 10 μM concentrations of drugs and carrier. The next day the drug was discarded, and new complete media was added and was left for 14 days. The media was changed at regular intervals. After that we fixed the cells with methanol and acetic acid at 1:3 ratios and left it for 30 minutes. Then 200–300 μl of crystal violet was added for further 15 minutes and the plate was washed with tap water. The colony of cells was counted in bare eyes and the graph was plotted.
4.5 Wound healing assay
For wound healing assay cells were seeded in 12 well plates (105 cells per well) and incubated at 37 °C in 5% CO2 incubator. After 24 hours of cell seeding, they were treated with 10 μM concentrations of synthetic compounds along with the carrier. Then a scratch wound was made on the confluent monolayer of HeLa cells with P10 pipette tip. Then the cells were observed under an inverted microscope at ‘0’ hours and ‘24’ hours and the image was captured. All the images were quantified using ImageJ and the graph was plotted by using GraphPad prism against wound size on the ‘X’ axis and time on the ‘Y’ axis.
4.6 Immunocytochemistry
Cells were cultured in 96 well plates and treated with carrier and respective BODIPY conjugated compounds (10 μM) for 24 h. The cells were fixed by 4% formaldehyde for 30 minutes, permeabilized using 0.25% Triton X-100, washed three times with ice-cold PBS each for 5 minutes. The cells were blocked with 1% BSA in PBST for 1 hour, incubated with diluted antibody (1:300 dilution) in blocking solution overnight at 4 °C. After overnight incubation, the cells were washed three times in PBS, 5 minutes in each wash, followed by incubated with diluted secondary antibody (1:300 dilution) for three hours at room temperature in dark and washed three times with PBS for 5 minutes each in dark. Nuclei were stained with diluted Hoechst solution for 30 minutes and images were taken in fluorescence microscope.
4.7 Flow cytometry
Subconfluent cells (4 × 105 no of cells) were incubated in 6 mm well plates for 24 h and treated with the carrier and respective BODIPY conjugated compound such 5 μM and 10 μM. After 24 h of treatment, the cells were harvested with Trypsin/EDTA, centrifuged for getting the plate. These plates were properly mixed with 75% ethanol for preservation. The cells were finally resuspended in PI solution (50 μL ml−1) and Rnase A (10 mg mL−1) and incubated for 30 min at 25 °C. Finally, the distribution of cells in various phases of the cell cycle was estimated using BD Accuri E6 flow cytometer at 10000 cells.
4.8 Preparation and analytical data of compounds
4.8.1 Compound 3. Commercially available 1-bromo-4-iodobenzene 1 (3.0 g, 10.6 mmol) was dissolved in anhydrous THF (50 mL). To that solution triethylamine (6.0 mL), cuprous iodide (81 mg, 0.424 mmol), PdCl2(PPh3)2 (149 mg, 0.212 mmol) and trimethyl silyl acetylene (1.47 mL, 10.6 mmol) were added and stirred at room temperature for 3 hours. The reaction mixture was filtered through short silica gel pad and evaporated. The crude product obtained was purified by silica gel column chromatography using hexane as eluent to obtain 2.5 g of pure compound 3 as colorless solid. Yield: 93%. Mp: 60 °C.1H NMR (400 MHz, CDCl3, δ ppm): 7.24 (d, J = 8.0 Hz, 2H), 7.13 (d, J = 8.0 Hz, 2H), 0.05 (s, 9H, –CH3). 13C NMR (100 MHz, CDCl3, δ ppm): 133.5, 131.6, 122.9, 122.2, 104.0, 95.7, 0.01. HRMS (m/z): calcd for C11H13BrSi: 251.9964, found 251.9961 [M+].
4.8.2 Compound 4. To a solution of compound 3 (4.0 g, 15.79 mmol) in mixed solvents (80 mL) (THF:MeOH = 1:1), K2CO3 (3.27 g, 23.7 mmol) was added and stirred at room temperature for 3 hours. Filtered the reaction mixture over short silica pad and concentrated. The crude product was purified by silica gel column chromatography using hexane as eluent to obtain 2.2 g of pure compound 4 as a colorless solid. Yield: 77%. Mp: 65 °C.1H NMR (400 MHz, CDCl3, δ ppm): 7.38 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 3.04 (s, 1H alkyne-H). 13C NMR (100 MHz, CDCl3, δ ppm): 133.6, 131.6, 123.2, 121.1, 82.6, 78.4. IR (KBr): 2924, 2853, 2108, 1484, 821 cm−1. HRMS (m/z): calcd for C8H5Br: 180.9647, found 180.9646 [M+].
4.8.3 Compound 5. Solution of decaborane (1.42 g, 11.6 mmol) in anhydrous CH3CN (14 mL) was refluxed at 100 °C for 2 hours to obtain a white precipitate. To that precipitate compound 4 (1.4 g, 7.73 mmol) dissolved in anhydrous acetonitrile (5 mL) was added and refluxed at 100 °C for another 12 hours. After completion of reaction, MeOH (2 mL) was added to quench the excess decaborane and concentrated in rotary evaporator. The crude product was purified by silica gel column chromatography using hexane as eluent to obtain pure compound 5 as white solid. Yield: 65%. Mp: 135 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 7.46 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 12.0 Hz, 2H), 3.91 (s, 1H, Ccage–H). 13C NMR (100 MHz, CDCl3, δ ppm): 132.6, 132.1, 129.3, 124.7, 75.6 (Ccage–C), 60.2 (Ccage–C). 11B NMR (128 MHz, CDCl3, proton decoupled): −2.23, −3.39, −9.19, −10.39, −13.0. IR (KBr): 3063, 2958, 2921, 2565 (B–H), 1633, 1491, 1397, 1070, 830 cm−1. HRMS (m/z): calcd for C8H15B10Br: 298.1355, found 298.1344 [M+].
4.8.4 Compound 7. To a solution of compound 5 (200 mg, 0.664 mmol) and 4-formylphenylboronic acid 6 (199 mg, 1.328 mmol) in anhydrous 1,4-dioxane (15 mL), Cs2CO3 (234 mg, 0.664 mmol) was added. Refluxed the reaction mixture at 100 °C for 15 hours. After completion of reaction, filtered the reaction mixture through short silica gel pad and concentrated. The crude product was purified by silica gel column chromatography using 10% ethyl acetate in hexane as eluent to obtain pure compound 7 as white solid. Yield: 51%. Mp: 170 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 10.05 (s, 1H, –CHO), 7.96 (d, J = 8.0 Hz, 2H), 7.71 (d, J = 8.0 Hz 2H), 7.59 (s, 4H), 4.03 (s, 1H, Ccage–H). 13C NMR (100 MHz, CDCl3, δ ppm): 191.8, 145.3, 141.4, 135.9, 133.7, 130.5, 128.3, 127.8, 127.8, 76.1 (Ccage–C), 60.3 (Ccage–C). 11B NMR (128 MHz, CDCl3, proton decoupled): −2.31, −3.48, −9.19, −10.39, −12.41. IR (KBr): 3029, 2962, 2921, 2580 (B–H), 1693 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1605, 1390, 1170, 1072, 1003, 820 cm−1. HRMS (m/z): calcd for C15H20B10O: 325.2590, found 325.2588 [M+].
O), 1605, 1390, 1170, 1072, 1003, 820 cm−1. HRMS (m/z): calcd for C15H20B10O: 325.2590, found 325.2588 [M+].
4.8.5 Compound 9. To a solution of compound 7 (200 mg, 0.613 mmol) and compound 8 (225 mg, 1.349 mmol) in anhydrous dichloromethane (20 mL),TFA (0.5 mL, 6.13 mmol) was added at 0 °C and stirred at room temperature for 2 hours. After completion of reaction, distilled water (10 mL) was added to quench the reaction. Extracted the crude product using dichloromethane, washed with water (3 times), dried the combined organic layers over anhydrous Na2SO4 and concentrated. The crude product was purified by silica gel column chromatography using 12% ethyl acetate in hexane as eluent to obtain 260 mg of pure compound 9 as white solid. Yield: 66%. Mp: 137 °C.1H NMR (400 MHz, CDCl3, δ ppm): 8.61 (s, 2H, –NH), 7.52 (d, J = 8.0 Hz, 4H), 7.47 (d, J = 8.0 Hz, 2H),7.16 (d, J = 8.0 Hz, 2H), 5.55 (s, 1H, meso-H), 4.21 (q, J = 8.0 Hz, 4H, –CH2ester), 4.0 (s, 1H, Ccage–H), 2.23 (s, 6H, –CH3pyrrole), 1.81 (s, 6H, –CH3pyrrole), 1.29 (t, J = 8.0 Hz, 6H, –CH3ester). 13C NMR (100 MHz, CDCl3, δ ppm): 162.1, 142.2, 139.3, 138.5, 132.6, 131.6, 129.0, 128.2, 127.7, 127.4, 118.2, 118.1, 76.4 (Ccage–C), 60.0, 53.5 (Ccage–C), 40.6 (meso-C), 14.6, 10.8, 9.0. 11B NMR (128 MHz, CDCl3, proton decoupled): −2.25, −3.57, −9.27, −10.64, −12.25. IR (KBr): 3307, 3054, 2978, 2924, 2858, 2594 (B–H), 1670, 1498, 1439, 1387, 1249, 1146, 1097, 944 cm−1. HRMS (m/z): calcd for C33H44B10N2O4: 641.4377, found 641.4375 [M+].
4.8.6 Compound o-CB-10. To a solution of compound 9 (230 mg, 0.358 mmol) in anhydrous dichloromethane (30 mL), DDQ (98 mg, 0.429 mmol) was added and stirred at room temperature for 1 hour. After complete consumption of starting material 9 monitored by TLC, triethylamine (0.5 mL, 3.58 mmol) was added to it followed by BF3·OEt2 (0.9 mL, 7.16 mmol) at 0 °C and stirred at room temperature for another 2 hours. After completion of reaction distilled water (10 mL) was added to the reaction mixture at 0 °C to quench the reaction. Extracted the crude product using dichloromethane (3 times). The combined organic layers were dried over anhydrous Na2SO4 and concentrated. The crude product was purified by neutral silica gel column chromatography using 15% ethyl acetate in hexane as eluent to obtain pure compound o-CB-10 as red solid. Yield: 41%. Mp: 127 °C.1H NMR (400 MHz, CD2Cl2, δ ppm): 7.61 (d, J = 8.0 Hz, 2H), 7.54 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 4.13 (q, J = 8.0 Hz, 4H, –CH2ester), 4.04 (s, 1H, Ccage–H), 2.15 (s, 6H, –CH3pyrrole), 1.55 (s, 6H, –CH3pyrrole), 1.32 (t, J = 8.0 Hz, 6H, –CH3ester). 13C NMR (100 MHz, CD2Cl2, δ ppm): 161.5, 141.4, 139.2, 133.6, 128.1, 128.1, 127.5, 127.4, 127.3, 127.2, 118.0, 117.6, 76.6 (Ccage–C), 74.4, 61.9 (Ccage–C), 59.9, 14.2, 10.2, 9.2. 11B NMR (128 MHz, CD2Cl2, proton decoupled): 0.23 (t, J = 28.2 Hz, BF2), −2.16, −8.94, −10.78(Ccage–B). 19F NMR (376 MHz, CD2Cl2, δ ppm): −141.09 (q, J = 26.3 Hz). IR (KBr): 2925, 2854, 2595 (B–H), 1664, 1443, 1385, 1243, 1102, 1032, 818 cm−1. HRMS (m/z): calcd for C33H41B11F2N2O4: 686.4125, found 686.4207 [M+].
4.8.7 Compound 13. To a solution of compound 12 (500 mg, 1.66 mmol) in mixed solvents (THF:toluene = 1:1) (30 mL), 4-formylphenylboronic acid (498 mg, 3.32 mmol), PdCl2(PPh3)2 (58 mg, 5 mol%), a solution of K2CO3 (687 mg, 4.98 mmol) in 3 mL of distilled water were added and refluxed at 110 °C for 24 hours. Extracted the crude product using dichloromethane (3 times), dried the combined organic layer over anhydrous Na2SO4 and concentrated. The crude product was purified by silica gel column chromatography using 5% ethyl acetate in hexane as eluent to obtain pure compound 13 as white solid. Yield: 63%. Mp: 140 °C.1H NMR (400 MHz, CDCl3, δ ppm): 9.98 (s, 1H, –CHO), 7.78 (d, J = 8.0 Hz, 2H), 7.63 (d, J = 8.0 Hz, 2H), 7.50 (s, 4H), 3.03 (s, 1H, Ccage–H). 13C NMR (100 MHz, CDCl3, δ ppm): 191.8, 145.8, 140.1, 135.6, 135.4, 130.3, 128.5, 127.6, 127.3, 76.7 (Ccage–C), 55.2 (Ccage–C). 11B NMR (128 MHz, CDCl3, proton decoupled): −3.79, −10.31, −13.29, −15.15. IR (KBr): 3061, 2959, 2926, 2851, 2734, 2604 (B–H), 1701 (CO), 1605, 1492, 1215, 1170, 1005, 838 cm−1. HRMS (m/z): calcd for C15H20B10O: 325.2599, found 325.2587 [M+].
4.8.8 Compound 14. To a solution of compound 13 (250 mg, 0.766 mmol) and compound 8 (281 mg, 1.687 mmol) in anhydrous dichloromethane (30 mL), TFA (0.23 mL, 3.064 mmol) was added at 0 °C and stirred at room temperature for 4 hours. After completion of reaction, quenched the reaction using distilled water. Extracted the crude product using dichloromethane (3 times), washed with water, dried the combined organic layers over anhydrous Na2SO4 and concentrated. The crude product was purified by silica gel column chromatography using 7% ethyl acetate in hexane as eluent to obtain pure compound 14 as colorless solid. Yield: 85%. Mp: 92 °C.1H NMR (400 MHz, CD2Cl2, δ ppm): 8.86 (s, 2H, –NH), 7.36–7.42 (dd, J = 8.0 Hz, 6H), 7.07 (d, J = 8.0 Hz, 2H), 5.55 (s, 1H, meso-H), 4.07 (q, J = 8.0 Hz, 4H, –CH2ester), 3.09 (s, 1H, Ccage–H), 2.11 (s, 6H, –CH3pyrrole), 1.76 (s, 6H, –CH3pyrrole), 1.16 (t, J = 8.0 Hz, 6H, –CH3ester). 13C NMR (100 MHz, CD2Cl2, δ ppm): 161.8, 140.8, 139.4, 138.5, 134.2, 131.8, 128.7, 128.3, 127.3, 126.8, 118.2, 118.0, 78.2 (Ccage–C), 59.8, 55.5 (Ccage–C), 40.2 (meso-C), 14.2, 10.4, 8.6. 11B NMR (128 MHz, CD2Cl2, proton decoupled): −2.36, −3.5, −9.27, −10.53, −11.04, −12.38. IR (KBr): 3310 (N–H), 3032, 2924, 2832, 2740, 2598 (B–H), 1696, 1604, 1520, 1483, 1393, 1306, 1213, 1167, 1005, 840 cm−1. HRMS (m/z): calcd for C33H44B10N2O4: 641.4262, found 641.4376 [M+].
4.8.9 Compound m-CB-15. To a solution of compound 14 (250 mg, 0.389 mmol) in anhydrous dichloromethane (30 mL), DDQ (97 mg, 0.428 mmol) was added and stirred at room temperature for 1 hour. After complete consumption of starting material 14 checked by TLC, triethylamine (0.27 mL, 1.945 mmol) was added to the reaction mixture followed by BF3·OEt2 (0.49 mL, 3.89 mmol) at 0 °C and stirred at room temperature for another 2 hours. After completion of reaction, quenched the reaction by using distilled water (10 mL) at 0 °C and extracted with dichloromethane (3 times). Combined organic layers were dried over anhydrous Na2SO4 and concentrated. The crude product was purified by neutral silica gel column chromatography using 10% ethyl acetate in hexane as eluent to obtain pure compound m-CB-15 as red solid. Yield: 38%. Mp: 230 °C.1H NMR (400 MHz, CD2Cl2, δ ppm): 7.70 (d, J = 8.0 Hz, 2H), 7.53 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 4.33 (q, J = 8.0 Hz, 4H, –CH2ester), 3.11 (s, 1H, Ccage–H), 1.90 (s, 6H, –CH3pyrrole), 1.31 (s, 6H, –CH3pyrrole), 1.31 (t, J = 8.0 Hz, 6H, –CH3ester). 13C NMR (100 MHz, CD2Cl2, δ ppm): 162.1, 141.1, 140.2, 135.0, 133.9, 133.0, 128.5, 128.3, 128.0, 127.0, 118.0, 117.5, 78.0 (Ccage–C), 59.9, 55.5 (Ccage–C), 31.9, 13.8, 12.2, 9.2. 11B NMR (128 MHz, CD2Cl2, proton decoupled): 0.23 (t, J = 29.4 Hz, BF2), −3.93, −10.43, −13.27, −15.07(Ccage–B). 19F NMR (376 MHz, CD2Cl2, δ ppm): −141.09 (q, J = 26.3 Hz). IR (KBr): 3060, 2926, 2848, 2604 (B–H), 1719, 1670, 1647, 1530, 1443, 1392, 1267, 1183, 1121, 1075, 1005 cm−1. HRMS (m/z): calcd for C33H41B11F2N2O4: 686.4125, found 686.4106 [M+].
4.8.10 Compound 17. To a solution of commercially available 1-bromo-4-iodobenzene 1 (1.5 g, 5.3 mmol) in mixed solvents (toluene:ethanol = 3:1) (20 mL), phenylboronic acid (646 mg, 5.3 mmol), PdCl2 (PPh3)2 (149 mg, 4 mol%), a solution of K2CO3 in distilled water (2 mL) were added and refluxed at 100 °C for 12 hours. Extracted the crude product using dichloromethane (3 times). Dried the combined organic layers were over anhydrous Na2SO4 and concentrated. The crude product was purified by silica gel column chromatography using hexane as eluent to obtain pure compound 17 as white solid. Yield: 81%. Mp: 92 °C.1H NMR (400 MHz, CDCl3, δ ppm): 7.51 (d, J = 8.0 Hz, 2H), 7.45–7.48 (dd, J = 8.0 Hz, 2H), 7.33–7.37 (m, J = 8.0 Hz, 4H), 7.27 (t, J = 8.0, Hz, 1H). 13C NMR (100 MHz, CDCl3, δ ppm): 131.9, 129.0, 128.8, 127.7, 127.3, 127.2, 127.0, 121.6. HRMS (m/z): calcd for C12H9Br: 233.1039, found 250.82 [M+ + H2O].
4.8.11 Compound 18. To a solution of compound 17 (700 mg, 3.0 mmol) in mixed solvents (THF:toluene = 1:1) (40 mL), 4-formylphenylboronic acid 6 (675 mg, 4.5 mmol), PdCl2(PPh3)2 (84 mg, 4 mol%), a solution of K2CO3 (1.24 g, 9.0 mmol) in distilled water (2 mL) were added and refluxed at 110 °C for 24 hours. Extracted the crude product using dichloromethane (3 times). Dried the combined organic layers were over anhydrous Na2SO4 and concentrated. The crude product was purified by silica gel column chromatography using 10–20% ethyl acetate in hexane as eluent to obtain pure compound 18 as white solid. Yield: 65%. Mp: > 200 °C.1H NMR (400 MHz, CDCl3, δ ppm): 9.98 (s, 1H, –CHO), 7.89 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 8.0 Hz, 2H), 7.64 (s, 4H), 7.57 (d, J = 8.0 Hz, 2H), 7.39 (t, J = 8.0 Hz, 2H), 7.30 (t, J = 8.0 Hz, 1H). 13C NMR (100 MHz, CDCl3, δ ppm): 192.0, 146.7, 141.4, 140.3, 138.5, 135.2, 130.4, 128.9, 127.8, 127.8, 127.7, 127.6, 127.1. IR (KBr): 2921, 2831, 2741, 1703 (CO), 1602, 1483, 1385, 1005, 820 cm−1. FTMS (m/z): calcd for C19H14O: 258.3139, found 259.11 [M+ + H].
4.8.12 Compound 19. To a solution of compound 18 (350 mg, 1.35 mmol) and 8 (497 mg, 2.98 mmol)in anhydrous dichloromethane (30 mL), TFA (1.0 mL, 13.5 mmol) was added at 0 °C and stirred at room temperature for 4 hours. After completion of reaction monitored by TLC, distilled water (10 ML) was added to quench the reaction. Extracted the crude product using dichloromethane (3 times). Dried the combined organic layers were over anhydrous Na2SO4 and concentrated. The crude product was purified by silica gel column chromatography using 10% ethyl acetate in hexane as eluent to obtain 500 mg of pure compound 19 as white solid. Yield: 64%. Mp: 197 °C.1H NMR (400 MHz, CD2Cl2, δ ppm): 9.26 (s, 2H, –NH), 7.57 (dd, J = 8.0 Hz, 6H), 7.46 (d, J = 8.0 Hz, 2H), 7.36 (t, J = 8.0 Hz, 2H), 7.26 (t, J = 8.0 Hz, 1H), 7.09 (d, J = 8.0 Hz, 2H), 5.56 (s, 1H, meso-H), 4.03 (q, J = 8.0 Hz, 4H, –CH2ester), 2.10 (s, 6H, –CH3pyrrole), 1.80 (s, 6H, –CH3pyrrole), 1.14 (t, J = 8.0 Hz, 6H, –CH3ester). 13C NMR (100 MHz, CD2Cl2, δ ppm): 162.0, 140.5, 140.1, 139.3, 139.2, 128.9, 128.7, 127.4, 127.3, 127.1, 126.9, 118.2, 118.0, 59.9, 40.0 (meso-C), 14.3, 10.5, 8.7. IR (KBr): 3349 (N–H), 2983, 2921, 2856, 1666, 1485, 1432, 1280, 1250, 1164, 1097, 761 cm−1. HRMS (m/z): calcd for C37H38N2O4: 574.7086, found 573.34 [M+ − H].
4.8.13 Compound PB-20. To a solution of compound 19 (330 mg, 0.574 mmol) in anhydrous dichloromethane (30 mL), DDQ (156 mg, 0.688 mmol) and stirred at room temperature for 2 hours. After complete consumption of starting material 19 checked by TLC, triethylamine (0.4 mL, 2.87 mmol) was added to it followed by BF3·OEt2 (0.7 mL, 5.74 mmol) at 0 °C and stirred at room temperature for another 2 hours. Quenched the reaction using distilled water (10 mL) at 0 °C and extracted the crude product using dichloromethane (3 times). Dried the combined organic layers were over anhydrous Na2SO4 and concentrated. The crude product was purified by silica gel column chromatography using 10% ethyl acetate in hexane as eluent to obtain pure compound PB-20 as red solid. Yield: 42%. Mp: 110 °C.1H NMR (400 MHz, CD2Cl2, δ ppm): 7.62 (s, 4H), 7.57–7.60 (m, J = 8.0 Hz, 4H), 7.38 (t, J = 8.0 Hz, 4H), 7.29 (d, J = 8.0 Hz, 1H), 4.14 (q, J = 8.0 Hz, 4H, –CH2ester), 2.17 (s, 6H, –CH3pyrrole), 1.58 (s, 6H, –CH3pyrrole), 1.22 (t, J = 8.0 Hz, 6H, –CH3ester). 13C NMR (100 MHz, CD2Cl2, δ ppm): 161.5, 141.4, 140.5, 140.4, 140.3, 139.1, 128.8, 127.4, 127.4, 127.3, 127.1, 126.9, 118.0, 117.5, 74.5, 59.9, 14.2, 10.2, 9.3. 11B NMR (128 MHz, CD2Cl2, proton decoupled): 0.25 (t, J = 28.2 Hz). 19F NMR (376 MHz, CD2Cl2, δ ppm): −141.09 (q, J = 26.3 Hz). IR (KBr): 3023, 2926, 2864, 1669, 1645, 1484, 1445, 1268, 1187, 765 cm−1. HRMS (m/z): calcd for C37H35BF2N2O4: 621.2731, found 621.2727 [M+].
4.8.14 Compound 23. To a solution of commercially available methyl-4-hydroxybenzoate 21 (1.5 g, 9.86 mmol) in anhydrous acetone (30 mL), K2CO3 (2.99 g, 21.67 mmol), propargyl bromide 22 (1.05 mL, 11.8 mmol) were added and refluxed at 60 °C for 24 hours. After completion of reaction, the reaction mixture was filtered through short silica gel pad and concentrated. The crude product was purified by silica gel column chromatography using 10% ethyl acetate in hexane as eluent to obtain 1.6 g of pure compound 23 as colorless solid. Yield: 86%. Mp: 78 °C.1H NMR (400 MHz, CDCl3, δ ppm): 7.99 (d, J = 8.0 Hz, 2H), 6.98 (d, J = 8.0 Hz, 2H), 4.73 (s, 2H, –CH2), 3.87 (s, 3H, –CH3ester), 2.54 (t, J = 4.0 Hz, 1H, alkyne-H). 13C NMR (100 MHz, CDCl3, δ ppm): 166.7, 161.2, 131.6, 123.5, 114.6, 77.9, 76.2, 55.9, 52.0. IR (KBr): 3246, 2958, 2913, 2860, 2128, 1706, 1608, 1511, 1440, 1289, 1176, 1106, 1024, 853 cm−1. HRMS (m/z): calcd for C11H10O3: 190.1953, found 191.05 [M+ + H].
4.8.15 Compound 24. LiAlH4 (1.59 g, 42.0 mmol) was suspended in dry THF (40 mL) and the reaction flask was cooled to 0 °C. Then compound 23 (2 g, 10.5 mmol) was dissolved in 10 mL of dry THF and added to the reaction mixture at 0 °C. The reaction mixture was stirred at 0 °C for 1 hour and then stirred at room temperature for another 4 hours. At 0 °C, water was added dropwise to the reaction mixture to quench the LiAlH4, and reaction mixture was extracted with dichloromethane. The organic layers were washed with water, dried over anhydrous Na2SO4 and solvent was evaporated in rotary evaporator. The crude reaction mixture was purified by silica gel column chromatography using 30% ethyl acetate in hexane to obtain 1.4 g of compound 24 as colorless liquid. Yield: 82%. 1H NMR (400 MHz, CDCl3, δ ppm): 7.28 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 8.0 Hz, 2H), 4.68 (d, J = 4.0 Hz, 2H, –CH2), 4.59 (s, 1H, –OH), 2.51 (t, J = 4.0 Hz, 1H, alkyne-H). 13C NMR (100 MHz, CDCl3, δ ppm): 157.2, 134.2, 128.7, 115.1, 78.6, 75.7, 65.0, 55.9. IR (KBr): 3287, 2924, 2872, 2120, 1883, 1610, 1587, 1510, 1456, 1376, 1216, 1027, 814 cm−1.
4.8.16 Compound 25. To a solution of compound 24 (1 g, 6.749 mmol) in anhydrous dichloromethane (40 mL), PCC (2.9 g, 13.489 mmol) was added at 0 °C and stirred at room temperature for 1 hour. Filtered the reaction mixture through short silica gel pad and concentrated. The crude product was purified by silica gel column chromatography using 15% ethyl acetate in hexane as eluent to obtain 850 mg of pure compound 25 as colourless solid. Yield: 79%. Mp: 90 °C.1H NMR (400 MHz, CDCl3, δ ppm): 9.87 (s, 1H, –CHO), 7.83 (d, J = 8.0 Hz, 2H), 7.06 (d, J = 8.0 Hz, 2H), 4.76 (d, J = 4.0 Hz, 2H, –CH2), 2.56 (t, J = 4.0 Hz, 1H, alkyne-H). 13C NMR (100 MHz, CDCl3, δ ppm): 190.8, 162.4, 132.0, 130.7, 115.3, 77.6, 76.5, 56.0 IR (KBr): 3211, 3057, 2929, 2832, 2122, 1681 (CO), 1603, 1577 cm−1. HRMS (m/z): calcd for C10H8O2: 161.0597, found 161.0596 [M+].
4.8.17 Compound 26. To a solution of compound 25 (400 mg, 2.497 mmol) and compound 8 (915.65 mg, 5.49 mmol) in anhydrous dichloromethane (30 mL), TFA (3.8 mL, 49.94 mmol) was added at 0 °C and stirred at room temperature for 2 hours. After completion of reaction, quenched the reaction at 0 °C using distilled water (10 mL). Extracted the crude product using dichloromethane (3 times), washed with water, dried the combined organic layers over anhydrous Na2SO4 and concentrated. The crude product was purified by silica gel column chromatography using 10% ethyl acetate in hexane to obtain 758 mg of pure compound 26 as colorless solid. Yield: 64%. Mp: 100 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 8.52 (s, 2H, –NH), 7.0 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.0 Hz, 2H), 5.46 (s, 1H, meso-H), 4.65 (s, 2H, –CH2), 4.20 (q, J = 8.0 Hz, 4H, –CH2ester), 2.52 (t, J = 4.0 Hz, 1Halkyne), 2.23 (s, 6H, –CH3pyrrole), 1.78 (s, 6H, –CH3pyrrole), 1.28 (t, J = 8.0 Hz, 6H, –CH3ester). 13C NMR (100 MHz, CDCl3, δ ppm): 162.0, 156.9, 132.1, 132.1, 129.4, 127.7, 118.0, 117.9, 115.4, 78.5, 75.8, 59.9, 55.9, 40.1 (meso-C), 14.6, 10.7, 8.9. IR (KBr): 3284 (N–H), 2978, 2924, 2860, 2120, 1652, 1508, 1441, 1245, 1146, 1099, 1026 cm−1. HRMS (m/z): calcd for C28H32N2O5: 477.2384, found 477.2386 [M+].
4.8.18 Compound 27. Solution of decaborane (205 mg, 1.678 mmol) in anhydrous acetonitrile (3 mL) and resulting solution was refluxed at 100 °C for 2 hours. To it a solution of compound 26 (400 mg, 0.839 mmol) in anhydrous acetonitrile (5 mL) was added and refluxed at 100 °C for another 12 hours. After completion of reaction, MeOH (2 mL) was added to quench the excess decaborane and concentrated. The crude product was purified by silica gel column chromatography using 15% ethyl acetate in hexane to obtain 300 mg of compound 27 as white solid. Yield: 60%. Mp: 156 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 8.44 (s, 2H, –NH), 7.01 (d, J = 8.0 Hz, 2H), 6.76 (d, J = 8.0 Hz, 2H), 5.45 (s, 1H, meso-H), 4.38 (s, 2H, –CH2), 4.23 (q, J = 8.0 Hz, 4H, –CH2ester), 4.08 (s, 1H, Ccage-H), 2.23 (s, 6H, –CH3pyrrole), 1.76 (s, 6H, –CH3pyrrole), 1.30 (t, J = 8.0 Hz, 6H, –CH3ester). 13C NMR (100 MHz, CDCl3, δ ppm): 162.0, 156.3, 133.4, 131.7, 129.8, 127.7, 118.1, 117.9, 115.3, 71.4 (Ccage–C), 69.3, 60.0, 57.8 (Ccage–C), 40.3 (meso-C), 14.6, 10.8. 11B NMR (128 MHz, CDCl3, proton decoupled): −3.10, −4.29, −9.38, −10.56, −11.91, −13.19. IR (KBr): 3322 (N–H), 2983, 2921, 2860, 2585 (B–H), 1678, 1655, 1508, 1441, 1241, 1142, 1021, 911 cm−1. HRMS (m/z): calcd for C28H42B10N2O5: 595.4170, found 595.4179 [M+].
4.8.19 Compound Me-o-CB-28. To a solution of compound 27 (200 mg, 0.343 mmol) in anhydrous dichloromethane (20 mL), DDQ (117 mg, 0.514 mmol) was added at 0 °C and stirred at room temperature for 2 hours. Then after complete consumption of compound 27 checked by TLC, triethylamine (0.24 mL, 1.715 mmol) was added to it followed by BF3·OEt2 (0.43 mL, 3.43 mmol) at 0 °C and stirred at room temperature for another 2 hours. The reaction was quenched using distilled water (10 mL) at 0 °C and extracted the crude product using dichloromethane (3 times). Dried the combined organic layers were over anhydrous Na2SO4 and concentrated. The crude product was purified by neutral silica gel column chromatography using 20% ethyl acetate in hexane as eluent to obtain 85 mg of pure compound Me-o-CB-28 as red solid. Yield: 39%. Mp: > 200 °C. 1H NMR (400 MHz, CD2Cl2, δ ppm): 7.20 (d, J = 8.0 Hz, 2H), 7.02 (d, J = 8.0 Hz, 2H), 4.51 (s, 2H, –CH2), 4.39 (q, J = 8.0 Hz, 4H, –CH2ester), 4.16 (s, 1H, Ccage–H), 1.96 (s, 6H, –CH3pyrrole),1.37 (t, J = 8.0 Hz, 6H, –CH3ester), 1.34 (s, 6H, –CH3pyrrole). 13C NMR (100 MHz, CD2Cl2, δ ppm): 162.2, 158.2, 147.9, 145.2, 142.3, 133.3, 129.4, 129.1, 128.4, 115.8, 71.6 (Ccage–C), 69.1, 62.0, 58.5 (Ccage–C), 13.9, 12.4, 9.3. 11B NMR (128 MHz, CD2Cl2, proton decoupled): −0.80 (t, J = 28.2 Hz, BF2), −3.14, −4.36, −9.54, −10.71, −13.19(Ccage–B). 19F NMR (376 MHz, CD2Cl2, δ ppm): −141.06 (q, J = 26.3 Hz). IR (KBr): 3081, 2980, 2925, 2570 (B–H), 1747, 1713, 1610, 1537, 1470, 1387, 1225, 1181, 1118, 1014, 856 cm−1. HRMS (m/z): calcd for C28H39B11N2F2O5: 640.3918, found 640.4022 [M+].
4.8.20 Compound 32. To a solution of compound 31 (1 g, 4.625 mmol) in anhydrous dichloromethane (40 mL), PCC (1.99 g, 9.25 mmol) was added at 0 °C and stirred at room temperature for 1 hour. Filtered the reaction mixture through short silica gel pad and concentrated. The crude product was purified by silica gel column chromatography using 15% ethyl acetate in hexane as eluent to obtain 840 mg of pure compound 32 as colorless solid. Yield: 85%. Mp: 70 °C.1H NMR (400 MHz, CDCl3, δ ppm): 9.89 (s, 1H, –CHO), 7.10 (d, J = 0 Hz, 2H), 6.84 (t, J = 4.0 Hz, 1H), 4.71 (d, J = 0 Hz, 4H, –CH2), 2.54 (t, J = 4.0 Hz, 2H, alkyne-H). 13C NMR (100 MHz, CDCl3, δ ppm): 191.6, 159.2, 138.5, 109.0, 108.8, 77.8, 76.3, 56.2. IR (KBr): 2936, 2872, 2754, 2114, 1702 (CO), 1601, 1459, 1340, 1280, 1155, 1061, 953, 895. HRMS (m/z): calcd for C13H10O3: 215.0703, found 215.0702 [M+].
4.8.21 Compound 33. To a solution of compound 32 (600 mg, 2.801 mmol) and 8 (1.03 g, 6.162 mmol) in anhydrous dichloromethane (40 mL), TFA (2.14 mL, 28.01 mmol) was added at 0 °C and stirred at room temperature for 2 hours. After completion of reaction, distilled water (10 mL) was added to quench the reaction. Extracted the crude product using dichloromethane, washed with water (3 times), dried the combined organic layers over anhydrous Na2SO4 and concentrated. The crude product was purified by silica gel column chromatography using 20–30% ethyl acetate in hexane to obtain 1.0 g of pure compound 33 as light pink solid. Yield: 67%. Mp: 155 °C. 1H NMR (400 MHz, CDCl3, δ ppm): 8.49 (s, 2H, –NH), 6.52 (t, J = 4.0 Hz, 1H), 6.34 (d, J = 0 Hz, 2H), 5.42 (s, 1H, meso-H), 4.59 (d, J = 0 Hz, 4H, –CH2), 4.21 (q, J = 8.0 Hz, 4H, –CH2ester), 2.52 (t, J = 4.0 Hz, 2Halkyne–H), 2.23 (s, 6H, –CH3pyrrole), 1.78 (s, 6H, –CH3pyrrole), 1.29 (t, J = 8.0 Hz, 6H, –CH3ester). 13C NMR (100 MHz, CDCl3, δ ppm): 161.9, 159.1, 141.7, 131.3, 127.7, 118.1, 118.1, 108.4, 101.1, 78.1, 76.1, 60.0, 56.0, 41.2 (meso-C), 14.6, 10.8, 8.9. IR (KBr): 3320 (N–H), 2974, 2924, 2868, 2118, 1651, 1597, 1441, 1260, 1150, 1065, 943, 824, 776 cm−1. HRMS (m/z): calcd forC31H34N2O6: 531.2490, found 531.2483 [M+].
4.8.22 Compound 34. Solution of decaborane (461 mg, 3.769 mmol) in anhydrous acetonitrile (5 mL) was refluxed at 100 °C for 2 hours to obtain a white precipitate. To it a solution of compound 33 (500 mg, 0.942 mmol) in anhydrous acetonitrile (6 mL) was added and refluxed at 100 °C for another 16 hours. After completion of reaction, MeOH (2 mL) was added to quench the excess decaborane and concentrated. The crude product was purified by silica gel column chromatography using 15% ethyl acetate in hexane to obtain 530 mg of pure compound 34 as white solid. Yield: 73%. Mp: >200 °C.1H NMR (400 MHz, CDCl3, δ ppm): 8.43 (s, 2H, –NH), 6.25 (d, J = 0 Hz, 2H), 6.23 (t, J = 4.0 Hz, 1H), 5.41 (s, 1H, meso-H), 4.32 (s, 4H, –CH2), 4.23 (q, J = 8.0 Hz, 4H, –CH2ester), 3.99 (s, 2H, Ccage–H), 2.24 (s, 6H, –CH3pyrrole), 1.78 (s, 6H, –CH3pyrrole), 1.31 (t, J = 8.0 Hz, 6H, –CH3ester). 13C NMR (100 MHz, CDCl3, δ ppm): 162.2, 158.7, 143.1, 130.7, 127.7, 118.5, 118.2, 108.6, 100.8, 71.4 (Ccage–C), 69.2, 60.2, 58.0 (Ccage–C), 41.1 (meso-C), 14.6, 10.8. 11B NMR (128 MHz, CD2Cl2, proton decoupled): −2.95, −4.13, −9.34, −10.52, −13.13. IR (KBr): 3336 (N–H), 3048, 2980, 2926, 2852, 2593 (B–H), 1698, 1648, 1596, 1442, 1384, 1266, 1164, 1092, 1018, 944777 cm−1. HRMS (m/z): calcd for C31H54B20N2O6: 767.6079, found 767.6077 [M+].
4.8.23 Compound Me-o-CB-35. To a solution of compound 34 (400 mg, 0.5194 mmol) in anhydrous dichloromethane (30 mL), DDQ (130 mg, 0.571 mmol) was added at 0 °C and stirred at room temperature for 2 hours. After complete consumption of compound 34 checked by TLC, triethylamine (0.36 mL, 2.597 mmol) was added to it followed by BF3·OEt2 (0.65 mL, 5.194 mmol) at 0 °C and stirred at room temperature for another 2 hours. The reaction was quenched using distilled water (10 mL) at 0 °C and extracted the crude product using dichloromethane (3 times). Dried the combined organic layers were over anhydrous Na2SO4 and concentrated. The crude product was purified by neutral silica gel column chromatography using 15% ethyl acetate in hexane to obtain 180 mg of pure compound Me-o-CB-35 as red solid. Yield: 43%. Mp: >200 °C. 1H NMR (400 MHz, CD2Cl2, δ ppm): 6.55 (t, J = 4.0 Hz, 1H), 6.51 (d, J = 4.0, Hz, 2H), 4.42 (s, 4H, –CH2), 4.39 (q, J = 8.0 Hz, 4H, –CH2ester), 4.07 (s, 2H, Ccage–H), 1.98 (s, 6H, –CH3pyrrole), 1.42 (s, 6H, –CH3pyrrole), 1.37 (t, J = 8.0 Hz, 6H, –CH3ester). 13C NMR (100 MHz, CD2Cl2, δ ppm): 162.0, 159.4, 146.0, 145.8, 142.1, 137.1, 132.5, 129.4, 108.1, 103.2, 71.3 (Ccage–C), 69.6, 62.1, 58.6 (Ccage–C), 13.9, 12.2, 9.3. 11B NMR (128 MHz, CD2Cl2, proton decoupled): −0.85 (t, J = 28.2 Hz, BF2), −3.08, −4.30, −9.51, −10.69, −13.25(Ccage–B). 19F NMR (376 MHz, CD2Cl2, δ ppm): −141.25 (q, J = 26.3 Hz). IR (KBr): 2987, 2925, 2848, 2593 (B–H), 1664, 1438, 1385, 1243, 1163, 1020, 773 cm−1. HRMS (m/z): calcd for C31H51B21N2F2O6: 812.5820, found 812.5617 [M+].
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
The authors report no conflict of interest.
Acknowledgements
Author RS would like to thank SERB-POWER, Science & Engineering Research Board, India (grant numbers: SPG/2021/ 002579), the Council of Scientific & Industrial Research, India (grant number: 02(0308)/17/EMR-II), the Higher Education Department, Government of Odisha, grant number: 26913/HED/HEPTC-WB-02-17 (OHEPEE), Science and Technology Department, Government of Odisha (grant no. ST-SCST-MISC. 0036-2023) and LSRB (grant number: LSRB-431/BTB/2024) for their financial support. The author BPD is thankful to Odisha State Higher Education Council for the extramural research funding under MRIP-2023 (grant number: 23EM/CH/16) for financial support. The author RS also acknowledges DST, FIST-New Delhi (SR/FST/CSI-243/2012) for providing NMR facility to Ravenshaw University, Cuttack.
References
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed.
- R. N. Grimes, in Carboranes, Elsevier, 2016, pp. 283–502 Search PubMed.
- B. Bhusan Jena and R. Satapathy, ChemistrySelect, 2023, 8, e202302310 CrossRef CAS.
- B. B. Jena, S. R. Jena, B. R. Swain, C. S. Mahanta, L. Samanta, B. P. Dash and R. Satapathy, Appl. Organomet. Chem., 2020, 34, e5754 CrossRef CAS.
- B. P. Dash, R. Satapathy, B. P. Bode, C. T. Reidl, J. W. Sawicki, A. J. Mason, J. A. Maguire and N. S. Hosmane, Organometallics, 2012, 31, 2931–2935 CrossRef CAS.
- B. R. Swain, C. S. Mahanta, B. B. Jena, S. K. Beriha, B. Nayak, R. Satapathy and B. P. Dash, RSC Adv., 2020, 10, 34764–34774 RSC.
- M. F. Hawthorne, Angew Chem. Int. Ed. Engl., 1993, 32, 950–984 CrossRef.
- P. Stockmann, M. Gozzi, R. Kuhnert, M. B. Sárosi and E. Hey-Hawkins, Chem. Soc. Rev., 2019, 48, 3497–3512 RSC.
- A. Marfavi, P. Kavianpour and L. M. Rendina, Nat. Rev. Chem, 2022, 6, 486–504 CrossRef PubMed.
- F. Ali, N. S Hosmane and Y. Zhu, Molecules, 2020, 25, 828 CrossRef CAS PubMed.
- N. S. Hosmane and R. Eagling, Handbook of Boron Science: With Applications in Organometallics, Catalysis, Materials and MedicineVolume 1: Boron in Organometallic Chemistry, World Scientific, Europe, 2018, vol. 1 Search PubMed.
- Y. Chen, F. Du, L. Tang, J. Xu, Y. Zhao, X. Wu, M. Li, J. Shen, Q. Wen, C. H. Cho and Z. Xiao, Mol. Ther.–Oncolytics, 2022, 24, 400–416 CrossRef CAS PubMed.
- B. P. Dash, R. Satapathy, E. R. Gaillard, K. M. Norton, J. A. Maguire, N. Chug and N. S. Hosmane, Inorg. Chem., 2011, 50, 5485–5493 CrossRef CAS PubMed.
- B. P. Dash, R. Satapathy, E. R. Gaillard, J. A. Maguire and N. S. Hosmane, J. Am. Chem. Soc., 2010, 132, 6578–6587 CrossRef CAS PubMed.
- B. B. Jena, L. Satish, C. S. Mahanta, B. R. Swain, H. Sahoo, B. P. Dash and R. Satapathy, Inorg. Chim. Acta, 2019, 491, 52–58 CrossRef CAS.
- P. Bauduin, S. Prevost, P. Farràs, F. Teixidor, O. Diat and T. Zemb, Angew. Chem., Int. Ed., 2011, 50, 5298–5300 CrossRef CAS PubMed.
- A. M. Cioran, A. D. Musteti, F. Teixidor, Ž. Krpetić, I. A. Prior, Q. He, C. J. Kiely, M. Brust and C. Viñas, J. Am. Chem. Soc., 2012, 134, 212–221 CrossRef CAS PubMed.
- J. J. Schwartz, A. M. Mendoza, N. Wattanatorn, Y. Zhao, V. T. Nguyen, A. M. Spokoyny, C. A. Mirkin, T. Baše and P. S. Weiss, J. Am. Chem. Soc., 2016, 138, 5957–5967 CrossRef CAS PubMed.
- C. J. Villagómez, T. Sasaki, J. M. Tour and L. Grill, J. Am. Chem. Soc., 2010, 132, 16848–16854 CrossRef PubMed.
- E. A. Qian, A. I. Wixtrom, J. C. Axtell, A. Saebi, D. Jung, P. Rehak, Y. Han, E. H. Moully, D. Mosallaei, S. Chow, M. S. Messina, J. Y. Wang, A. T. Royappa, A. L. Rheingold, H. D. Maynard, P. Král and A. M. Spokoyny, Nat. Chem., 2017, 9, 333–340 CrossRef CAS PubMed.
- A. Saha, E. Oleshkevich, C. Vinas and F. Teixidor, Adv. Mater., 2017, 29, 1704238 CrossRef PubMed.
- H. Jude, H. Disteldorf, S. Fischer, T. Wedge, A. M. Hawkridge, A. M. Arif, M. F. Hawthorne, D. C. Muddiman and P. J. Stang, J. Am. Chem. Soc., 2005, 127, 12131–12139 CrossRef CAS PubMed.
- M. Koshino, T. Tanaka, N. Solin, K. Suenaga, H. Isobe and E. Nakamura, Science, 2007, 316, 853 CrossRef CAS PubMed.
- L. Liang, A. Rapakousiou, L. Salmon, J. Ruiz, D. Astruc, B. P. Dash, R. Satapathy, J. W. Sawicki and N. S. Hosmane, Eur. J. Inorg. Chem., 2011, 2011, 3043–3049 CrossRef CAS.
- L. Wu, M. Holzapfel, A. Schmiedel, F. Peng, M. Moos, P. Mentzel, J. Shi, T. Neubert, R. Bertermann, M. Finze, M. A. Fox, C. Lambert and L. Ji, Nat. Commun., 2024, 15, 3005 CrossRef CAS PubMed.
- A. V. Zaitsev, S. S. Kiselev, A. F. Smol'yakov, Y. V. Fedorov, E. G. Kononova, Y. A. Borisov and V. A. Ol'shevskaya, Org. Biomol. Chem., 2023, 21, 4084–4094 RSC.
- C. Bellomo, D. Zanetti, F. Cardano, S. Sinha, M. Chaari, A. Fin, A. Maranzana, R. Núñez, M. Blangetti and C. Prandi, Dyes Pigm., 2021, 194, 109644 CrossRef CAS.
- E. Berksun, I. Nar, A. Atsay, İ. Özçeşmeci, A. Gelir and E. Hamuryudan, Inorg. Chem. Front., 2018, 5, 200–207 RSC.
- C. Bellomo, M. Chaari, J. Cabrera-González, M. Blangetti, C. Lombardi, A. Deagostino, C. Viñas, N. Gaztelumendi, C. Nogués, R. Nuñez and C. Prandi, Chem.–Eur. J., 2018, 24, 15622–15630 CrossRef CAS PubMed.
- Z. Xie, Coord. Chem. Rev., 2002, 231, 23–46 CrossRef CAS.
- Z. Xie, Acc. Chem. Res., 2003, 36, 1–9 CrossRef CAS PubMed.
- Z.-J. Yao and G.-X. Jin, Coord. Chem. Rev., 2013, 257, 2522–2535 CrossRef CAS.
- Z. Qiu, S. Ren and Z. Xie, Acc. Chem. Res., 2011, 44, 299–309 CrossRef CAS PubMed.
- J. Estrada and V. Lavallo, Angew. Chem., Int. Ed., 2017, 56, 9906–9909 CrossRef CAS PubMed.
- J. C. Axtell, K. O. Kirlikovali, P. I. Djurovich, D. Jung, V. T. Nguyen, B. Munekiyo, A. T. Royappa, A. L. Rheingold and A. M. Spokoyny, J. Am. Chem. Soc., 2016, 138, 15758–15765 CrossRef CAS PubMed.
- Y.-P. Zhou, S. Raoufmoghaddam, T. Szilvási and M. Driess, Angew. Chem.,
Int. Ed., 2016, 55, 12868–12872 CrossRef CAS PubMed.
- S. Kimura, S. Masunaga, T. Harada, Y. Kawamura, S. Ueda, K. Okuda and H. Nagasawa, Bioorg. Med. Chem., 2011, 19, 1721–1728 CrossRef CAS PubMed.
- Y. Hattori, S. Kusaka, M. Mukumoto, K. Uehara, T. Asano, M. Suzuki, S. Masunaga, K. Ono, S. Tanimori and M. Kirihata, J. Med. Chem., 2012, 55, 6980–6984 CrossRef CAS PubMed.
- T. He and R. A. Musah, ACS Omega, 2019, 4, 3820–3826 CrossRef CAS.
- S. M. Hillier, K. P. Maresca, F. J. Femia, J. C. Marquis, C. A. Foss, N. Nguyen, C. N. Zimmerman, J. A. Barrett, W. C. Eckelman, M. G. Pomper, J. L. Joyal and J. W. Babich, Cancer Res., 2009, 69, 6932–6940 CrossRef CAS PubMed.
- T. Maurer, M. Eiber, M. Schwaiger and J. E. Gschwend, Nat. Rev. Urol., 2016, 13, 226–235 CrossRef CAS PubMed.
- M. Eder, M. Schäfer, U. Bauder-Wüst, W.-E. Hull, C. Wängler, W. Mier, U. Haberkorn and M. Eisenhut, Bioconjugate Chem., 2012, 23, 688–697 CrossRef CAS PubMed.
- S. Wang, C. Blaha, R. Santos, T. Huynh, T. R. Hayes, D. R. Beckford-Vera, J. E. Blecha, A. S. Hong, M. Fogarty, T. A. Hope, D. R. Raleigh, D. M. Wilson, M. J. Evans, H. F. VanBrocklin, T. Ozawa and R. R. Flavell, Mol. Pharmaceutics, 2019, 16, 3831–3841 CrossRef CAS.
- G. H. Vicente and M. Sibrian-Vazquez, in Handbook of Porphyrin Science (Volume 4) with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine, World Scientific, 2010 Search PubMed.
- N. V. S. D. K. Bhupathiraju and M. G. H. Vicente, in Applications of Porphyrinoids, ed. R. Paolesse, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, vol. 34, pp. 31–52 Search PubMed.
- N. V. S. D. K. Bhupathiraju and M. G. H. Vicente, Bioorg. Med. Chem., 2013, 21, 485–495 CrossRef CAS.
- D.-Q. Wu, B. Lu, C. Chang, C.-S. Chen, T. Wang, Y.-Y. Zhang, S.-X. Cheng, X.-J. Jiang, X.-Z. Zhang and R.-X. Zhuo, Biomaterials, 2009, 30, 1363–1371 CrossRef CAS PubMed.
- A. A. D'Souza and P. V. Devarajan, J. Controlled Release, 2015, 203, 126–139 CrossRef PubMed.
- T. Zhang, G. Li, S. Li, Z. Wang, D. He, Y. Wang, J. Zhang, J. Li, Z. Bai, Q. Zhang, B. Liu, Q. Zhao, Y. Liu and H. Zhang, Colloids Surf., B, 2019, 182, 110397 CrossRef CAS PubMed.
- V. V. Mody, A. Cox, S. Shah, A. Singh, W. Bevins and H. Parihar, Appl. Nanosci., 2014, 4, 385–392 CrossRef CAS.
- X. Chen, X. Bao, J.-C. Zhao and S. G. Shore, Experimental and Computational Study of the Formation Mechanism of the Diammoniate of Diborane, https://pubs.acs.org/doi/pdf/10.1021/ja203648w, accessed July 28, 2024.
- H. Kuang, S. Wu, Z. Xie, F. Meng, X. Jing and Y. Huang, Biomacromolecules, 2012, 13, 3004–3012 CrossRef CAS PubMed.
- H. Xiong, D. Zhou, Y. Qi, Z. Zhang, Z. Xie, X. Chen, X. Jing, F. Meng and Y. Huang, Biomacromolecules, 2015, 16, 3980–3988 CrossRef CAS PubMed.
- I. Nakase, M. Katayama, Y. Hattori, M. Ishimura, S. Inaura, D. Fujiwara, T. Takatani-Nakase, I. Fujii, S. Futaki and M. Kirihata, Chem. Commun., 2019, 55, 13955–13958 RSC.
- H. Koganei, M. Ueno, S. Tachikawa, L. Tasaki, H. S. Ban, M. Suzuki, K. Shiraishi, K. Kawano, M. Yokoyama, Y. Maitani, K. Ono and H. Nakamura, Bioconjugate Chem., 2013, 24, 124–132 CrossRef CAS PubMed.
- B. R. Swain, S. R. Jena, S. K. Beriha, C. S. Mahanta, B. B. Jena, T. Prasanth, L. Samanta, R. Satapathy and B. P. Dash, New J. Chem., 2023, 47, 10296–10308 RSC.
- R. Dubey, S. Kushal, A. Mollard, L. Vojtovich, P. Oh, M. D. Levin, J. E. Schnitzer, I. Zharov and B. Z. Olenyuk, Bioconjugate Chem., 2015, 26(1), 78–89 CrossRef CAS PubMed.
- R. Satapathy, B. P. Dash, C. S. Mahanta, B. R. Swain, B. B. Jena and N. S. Hosmane, J. Organomet. Chem., 2015, 798, 13–23 CrossRef CAS.
- A. R. Genady and M. E. El-Zaria, Appl. Organomet. Chem., 2008, 22, 227–232 CrossRef CAS.
- S. Stadlbauer, P. Welzel and E. Hey-Hawkins, Inorg. Chem., 2009, 48, 5005–5010 CrossRef CAS PubMed.
- J. H. Gibbs, H. Wang, N. V. S. D. K. Bhupathiraju, F. R. Fronczek, K. M. Smith and M. G. H. Vicente, J. Organomet. Chem., 2015, 798, 209–213 CrossRef CAS PubMed.
- S. Xuan, N. Zhao, Z. Zhou, F. R. Fronczek and M. G. H. Vicente, J. Med. Chem., 2016, 59, 2109–2117 CrossRef CAS PubMed.
- M. Shirakawa, A. Zaboronok, K. Nakai, Y. Sato, S. Kayaki, T. Sakai, T. Tsurubuchi, F. Yoshida, T. Nishiyama, M. Suzuki, H. Tomida and A. Matsumura, Cells, 2021, 10, 3421 CrossRef CAS PubMed.
- M. Couto, M. F. García, C. Alamón, M. Cabrera, P. Cabral, A. Merlino, F. Teixidor, H. Cerecetto and C. Viñas, Chem.–Eur. J., 2018, 24, 3122–3126 CrossRef CAS PubMed.
- M. Couto, I. Mastandrea, M. Cabrera, P. Cabral, F. Teixidor, H. Cerecetto and C. Viñas, Chem.–Eur. J., 2017, 23, 9233–9238 CrossRef CAS PubMed.
- C. S. Mahanta, S. Aparna, S. K. Das, B. B. Jena, B. R. Swain, M. Patri, B. P. Dash and R. Satapathy, ChemistrySelect, 2020, 5, 8429–8434 CrossRef CAS.
- E. Antina, N. Bumagina, Y. Marfin, G. Guseva, L. Nikitina, D. Sbytov and F. Telegin, Molecules, 2022, 27, 1396 CrossRef CAS PubMed.
- V. I. Martynov and A. A. Pakhomov, Russ. Chem. Rev., 2021, 90, 1213–1262 CrossRef.
- D. Dai, G. Lian, X. He, J. Feng and G. Jin, Photochem. Photobiol. Sci., 2022, 21, 185–194 CrossRef CAS PubMed.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC.
- P. Kaur and K. Singh, J. Mater. Chem. C, 2019, 7, 11361–11405 RSC.
- S. Kolemen and E. U. Akkaya, Coord. Chem. Rev., 2018, 354, 121–134 CrossRef CAS.
- A. N. Kursunlu, Z. E. Koc, A. Y. Obalı and E. Güler, J. Lumin., 2014, 149, 215–220 CrossRef CAS.
- J. L. Donnelly, D. Offenbartl-Stiegert, J. M. Marín-Beloqui, L. Rizzello, G. Battaglia, T. M. Clarke, S. Howorka and J. D. Wilden, Chem.–Eur. J., 2020, 26, 863–872 CrossRef CAS PubMed.
- E. R. Thapaliya, Y. Zhang, P. Dhakal, A. S. Brown, J. N. Wilson, K. M. Collins and F. M. Raymo, Bioconjugate Chem., 2017, 28, 1519–1528 CrossRef CAS PubMed.
- Y. Ni and J. Wu, Org. Biomol. Chem., 2014, 12, 3774 RSC.
- J. Zhang, L. Wang and Z. Xie, ACS Biomater. Sci. Eng., 2018, 4, 1969–1975 CrossRef CAS PubMed.
- V. Nguyen, Y. Yim, S. Kim, B. Ryu, K. M. K. Swamy, G. Kim, N. Kwon, C. Kim, S. Park and J. Yoon, Angew. Chem., Int. Ed., 2020, 59, 8957–8962 CrossRef CAS PubMed.
- Y. Liu, N. Song, L. Chen, S. Liu and Z. Xie, Chem.–Asian J., 2018, 13, 989–995 CrossRef CAS PubMed.
- M. A. Masood, Y. Wu, Y. Chen, H. Yuan, N. Sher, F. Faiz, S. Yao, F. Qi, M. I. Khan, M. Ahmed, N. Mushtaq, W. He and Z. Guo, Dyes Pigm., 2022, 202, 110255 CrossRef CAS.
- Y. Qin, X. Liu, P.-P. Jia, L. Xu and H.-B. Yang, Chem. Soc. Rev., 2020, 49, 5678–5703 RSC.
- R. Prieto-Montero, A. Prieto-Castañeda, R. Sola-Llano, A. R. Agarrabeitia, D. García-Fresnadillo, I. López-Arbeloa, A. Villanueva, M. J. Ortiz, S. De La Moya and V. Martínez-Martínez, Photochem. Photobiol., 2020, 96, 458–477 CrossRef CAS PubMed.
- L. Collado, T. Naranjo, M. Gomez-Mendoza, C. G. López-Calixto, F. E. Oropeza, M. Liras, J. Marugán and V. A. De La Peña O'Shea, Adv. Funct. Mater., 2021, 31, 2105384 CrossRef CAS.
- K. Ladomenou, V. Nikolaou, G. Charalambidis, A. Charisiadis and A. G. Coutsolelos, C. R. Chim., 2017, 20, 314–322 CrossRef CAS.
- J.-Y. Liu, X.-N. Hou, Y. Tian, L. Jiang, S. Deng, B. Röder and E. A. Ermilov, RSC Adv., 2016, 6, 57293–57305 RSC.
- G. Kalot, A. Godard, B. Busser, J. Pliquett, M. Broekgaarden, V. Motto-Ros, K. D. Wegner, U. Resch-Genger, U. Köster, F. Denat, J.-L. Coll, E. Bodio, C. Goze and L. Sancey, Cells, 2020, 9, 1953 CrossRef CAS PubMed.
- M. Prieto-Vila, R. Takahashi, W. Usuba, I. Kohama and T. Ochiya, Int. J. Mater. Sci., 2017, 18, 2574 Search PubMed.
- J. L. Griffin and J. P. Shockcor, Nat. Rev. Cancer, 2004, 4, 551–561 CrossRef CAS PubMed.
- J. S. You and P. A. Jones, Cancer Cell, 2012, 22, 9–20 CrossRef CAS PubMed.
- G. H. Williams and K. Stoeber, J. Pathol., 2012, 226, 352–364 CrossRef CAS PubMed.
- T. Otto and P. Sicinski, Nat. Rev. Cancer, 2017, 17, 93–115 CrossRef CAS PubMed.
- U. Raju, E. Nakata, K. A. Mason, K. K. Ang and L. Milas, Cancer Res., 2003, 63, 3263–3267 CAS.
- C. Dittrich, M. Gneist, A. Zandvliet, C. Johnston and M. Yule, Eur. J. Cancer Suppl., 2004, 2, 129 CrossRef.
- M. A. Dickson and G. K. Schwartz, Curr. Oncol., 2009, 16, 36–43 CrossRef CAS PubMed.
- Q. Chen, T. Wang, Y. Zhang, Q. Wang and J. Ma, Synth. Commun., 2002, 32, 1031–1040 CrossRef CAS.
- A. G. Joseph, M. Biji, V. P. Murali, D. R. Sherin, A. Valsan, V. P. Sukumaran, K. V. Radhakrishnan and K. K. Maiti, RSC Med. Chem., 2024, 15, 3444–3459 RSC.
- A. E. Mercier, A. M. Joubert, R. Prudent, J. Viallet, A. Desroches-Castan, L. De Koning, P. Mabeta, J. Helena, M. S. Pepper and L. Lafanechère, Cancers, 2024, 16, 2941 CrossRef CAS PubMed.
- T. Gebäck, M. M. P. Schulz, P. Koumoutsakos and M. Detmar, BioTechniques, 2009, 46, 265–274 CrossRef PubMed.
- C. Bertoli, J. M. Skotheim and R. A. M. De Bruin, Nat. Rev. Mol. Cell Biol., 2013, 14, 518–528 CrossRef CAS PubMed.
- H. Wang, M. Guo, H. Wei and Y. Chen, Signal Transduction Targeted Ther., 2023, 8, 92 CrossRef CAS PubMed.
- T. Ozaki and A. Nakagawara, Cancers, 2011, 3, 994–1013 CrossRef CAS PubMed.
- F. Hoppe-Seyler and K. Butz, J. Virol., 1993, 67, 3111–3117 CrossRef CAS PubMed.
- A. K. Ajay, A. S. Meena and M. K. Bhat, Cell Biosci., 2012, 2, 2 CrossRef CAS PubMed.
- S. A. Mujib, F. Alamsyah and W. P. Taruno, Integr. Med. Int., 2017, 4, 161–170 CrossRef.
- A. V. Zaitsev, E. G. Kononova, A. A. Markova, A. V. Shibaeva, A. A. Kostyukov, A. E. Egorov, V. A. Kuzmin, A. A. Shtil and V. A. Ol'shevskaya, Dyes Pigm., 2022, 207, 110711 CrossRef CAS.
- G. Barrera, ISRN Oncol., 2012, 2012, 1–21 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Barada Prasanna Dash
a,
Barada Prasanna Dash