
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalcium-atom-modified boron phosphide (BP) biphenylene as an efficient hydrogen storage material†
Yusuf Zuntu Abdullahi ab,
Ikram Djebablia*cd,
Tiem Leong Yoone and
Lim Thong Lengf
ab,
Ikram Djebablia*cd,
Tiem Leong Yoone and
Lim Thong Lengf
aDepartment of Physics, Aydin Adnan Menderes University, Aydin 09010, Turkey
bDepartment of Physics, Faculty of Science, Kaduna State University, P.M.B. 2339, Kaduna State, Nigeria
cRadiation and Matter Physics Laboratory, Matter Sciences Department, Mohamed-Cherif Messaadia University, P.O. Box 1553, Souk-Ahras, 41000, Algeria. E-mail: ik.djebablia@univ-soukahras.dz
dPhysics Laboratory at Guelma, Faculty of Mathematics, Computing and Material Sciences, University 8 May 1945 Guelma, P.O. Box 401, Guelma 24000, Algeria
eSchool of Physics, Universiti Sains Malaysia, 11800 Penang, Malaysia
fFaculty of Engineering and Technology, Multimedia University, Jalan Ayer Keroh Lama, 75450 Melaka, Malaysia
First published on 12th December 2024
Abstract
Porous nanosheets have attracted significant attention as viable options for energy storage materials because of their exceptionally large specific surface areas. A recent study (Int. J. Hydrogen Energy, 2024, 66, 33–39) has demonstrated that Li/Na-metalized inorganic BP-biphenylene (b-B3P3) and graphenylene (g-B6P6) analogues possess suitable functionalities for hydrogen (H2) storage. Herein, we evaluate the H2 storage performance of alkaline earth metal (AEM = Be, Mg, Ca)-decorated b-B3P3 and g-B6P6 structures based on first-principles density functional theory (DFT) calculations. Our investigations revealed that individual Be and Mg atoms are not stable on pure b-B3P3 and g-B6P6 sheets, and the formation of aggregates is favored due to their low binding energy to these surfaces. However, the binding energy improves for Ca-decorated b-B3P3 (b-B3P3(mCa)) and g-B6P6 (g-B6P6(nCa)) structures, forming stable and uniform mCa(nCa) (m and n stand for the numbers of Ca atom) coverages on both sides. Under maximum hydrogenation, the b-B3P3(8Ca) and g-B6P6(16Ca) structures exhibited the ability to adsorb up to 32H2 and 48H2 molecules with average adsorption energy (Ea) values of −0.23 eV per H2 and −0.25 eV per H2, respectively. Gravimetric H2 uptakes of 7.28 wt% and 5.56 wt% were found for b-B3P3(8Ca)@32H2 and g-B6P6(16Ca)@48H2 systems, exceeding the target of 5.50 wt% set by the US Department of Energy (DOE) to be reached by 2025. Our findings indicate the importance of these b-B3P3 and g-B6P6 sheets for H2 storage technologies.
1 Introduction
Growing concerns over environmental pollution and the limited supply of fossil fuels have driven the pursuit of clean energy sources.1,2 Hydrogen (H2) is regarded as a sustainable and alternative energy source capable of eradicating the use of fossil fuels, especially in transportation applications.3–5 However, one of the main hindrances to the advancement of H2 technology is creating small, light, safe, and affordable storage systems with large storage capacity under practical conditions.3,6 Solid-state H2 storage materials, which store H2 via the physisorption or chemisorption method, have been extensively investigated as a promising approach to overcome the challenges associated with gas or liquid H2 storage, which requires very high pressure (350–700 bar).3,7,8 According to the US Department of Energy (DOE), by 2025 an ideal H2 storage material should attain gravimetric and volumetric capacity greater than 5.5 wt% and 30 g L−1, with adsorption energy (Ea) between 0.2 and 0.6 eV per H2.9–11 By meeting these minimum thresholds, an adsorbent material will allow suitable reversible H2 storage under ambient conditions.To date, a wide range of materials, including carbon-based materials,12–14 MXenes,15,16 metal–organic-frameworks (MOFs)17–19 covalent organic frameworks (COFs)20–22 and related low-dimensional materials23–32 have attracted a significant number of studies in several research areas, including spintronics, batteries, optoelectronics and catalysis. Porous two-dimensional (2D) materials, in particular organic/inorganic-based biphenylene (BPN) or graphenylene (BPC) sheets, are ideally suited for H2 storage applications.33–44 However, the weak interaction between these surfaces and H2 molecules (due to low Ea values) reduces their H2 storage capacities. Extensive theoretical studies have shown that the metal-atom decoration technique45–47 can be effective for improving the Ea, resulting in higher H2 uptake. For instance, Denis et al.33 studied the H2 storage performance of BPN with adsorbed Li-adatoms, and obtained a H2 uptake of 7.4 wt%, with an averaged Ea of 0.20 eV per H2. In addition, ultrahigh H2 storage capacity values of 11.9 wt% and 11.63% for K- and Ca-decorated BPN structures was predicted by Mahamiya et al.34 Singh et al.48 have found 11.07 wt% H2 uptake near ambient temperature for a Sc-decorated BPN structure. Kaewmaraya et al.35 reported 6.66 wt% and 6.76 wt% H2 storage capacities for Li/Na-decorated divacancy BPN. Moreover, H2 storage analysis was carried out on Li-decorated BPN and Li-decorated N-doped BPN by Zhang et al.38 They reported H2 uptakes of 9.581 wt% and 10.588 wt% for Li-decorated BPN and Li-decorated N-doped BPN, respectively. In another separate study, adsorption of H2 on a Li-decorated B-doped BPN structure has been explored by Ma et al.40 Their analysis indicated that the H2 storage capacity value changed from 6.30 wt% to 19.22 wt% as a function of Li-ion concentration, which increased from 7.69% to 25.00%. On the other hand, Hussain et al.36 used light metals to enhance the H2 storage capacity value of BPC sheets. Their results revealed that the BPC can accommodate 20 H2 molecules with storage capacity values between 4.90 wt% and 6.14 wt% under operating conditions. Additionally, Boezar et al.49 evaluated the H2 adsorption behavior of transition metal (Fe, Sc and Ti) decorated BPC structures. Their studies indicate that these structures can take up to 20 H2 molecules with average Ea values higher than 0.2 eV per H2. Structures with Li(Na)-atom-decorated inorganic graphenylene (IGP) based on SiC (Li(Na)@IGP-SiC) have been investigated by Martins et al.37 for H2 storage performance. Their calculations revealed that Li(Na)@IGP-SiC can take up to 48 H2 molecules, yielding an enhanced H2 storage capacity value of 8.27 wt% (6.78 wt%) for Li(Na)@IGP-SiC structures. Following this use of IGP sheets, Djebablia et al.39 recently studied the H2 performance of b-BP(Li, Na, K)/g-BP(Li, Na, K) structures. They found that each adatom binds strongly on the host b-BP/g-BP surfaces and adsorbs multiple H2 molecules, resulting in uptakes of 9.05% and 6.99% for b-BP(Li) and g-BP(Li) structures, respectively, under practical conditions.
Motivated by the results mentioned above, we investigate the effect of alkaline earth metal (AEM = Be, Mg, Ca)-decorated b-B3P3 and g-B6P6 sheets for H2 storage applications, using spin-polarized density functional theory (DFT). The obtained results reveal that Be and Mg atoms bind weakly to the b-B3P3 and g-B6P6 sheets. In contrast, Ca atoms exhibit a strong binding energy, making them suitable hosts for adsorbing a considerable number of H2 molecules. Meanwhile, addition of H2 molecules on the Ca-decorated b-B3P3 (b-B3P3(Ca)) and g-B6P6 (g-B6P6(Ca)) structures was analyzed to fully understand their H2 storage performance. Moreover, the b-B3P3(Ca) and g-B6P6(Ca) structures reached storage capacities of 7.28 wt% and 5.56 wt%, respectively, exceeding the target of 5.50 wt% set by the US DOE to be attained by 2025. For the sake of convenience, b-B3P3(Ca) and g-B6P6(Ca) with adsorbed p(q)H2 molecules are named b-B3P3(mCa)@pH2 and g-B6P6(nCa)@qH2, respectively (p/q and n/m correspond to the numbers of adsorbed H2 molecules and Ca adatoms, respectively).
2 Computational details
The spin-polarized DFT50 calculations on the ground-state properties of all studied structures have been implemented in the Vienna ab initio simulation package (VASP).51 The projector augmented-wave (PAW) approach52 was used to describe ion–electron interactions. The exchange–correlation functional was treated using the generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE) functional.52 The van der Waals (vdW) interaction correction was added through the DFT-D2 approach.53 It should be noted that the PBE-D2 vdW correction is prone to overestimating adsorption energy values in certain cases, which could have an impact on the conclusions drawn.54 During the structural optimization calculations, the Brillouin zone (BZ) of all studied structures was sampled using the Monkhorst–Pack approach55 with (6 × 6 × 1) and (12 × 12 × 1) grids for self-consistency and total density-of-state computations, respectively. We used a plane-wave basis set with a kinetic energy cut-off of 500 eV to expand the wave functions. We set a vacuum layer at least 18 Å thick perpendicularly to the cell dimension for all calculations. The convergence criteria for energy and force for all calculations were set at 0.001 eV Å−1 and 10−5 eV, respectively. To assess the thermal stability of all studied b-B3P3(Ca) and g-B6P6(Ca) structures, ab initio molecular dynamics (AIMD) simulations have been performed at room temperature (300 K).56 In the NVT ensemble, 1 fs for 5000 iterations and the Nosé thermostat were used for the MD simulation parameters. Bader analysis57 was employed to study the charge transfer mechanism. With the aid of VESTA software,58 the charge distribution and structural analysis were obtained.Considering the b-B3P3(Ca) case, the average adsorption energy (Ea) is defined as:
| Ea = (Eb-B3P3(mCa) − Eb-B3P3 − mECa)/m | (1) |
The average Ea for the b-B3P3(mCa)@pH2 structure is computed as:
| Ea = (Eb-B3P3(8Ca)@pH2) − Eb-B3P3(8Ca) − pEH2)/p | (2) |
The H2 molecule storage capacity (wt%) has been evaluated by employing the following expression:
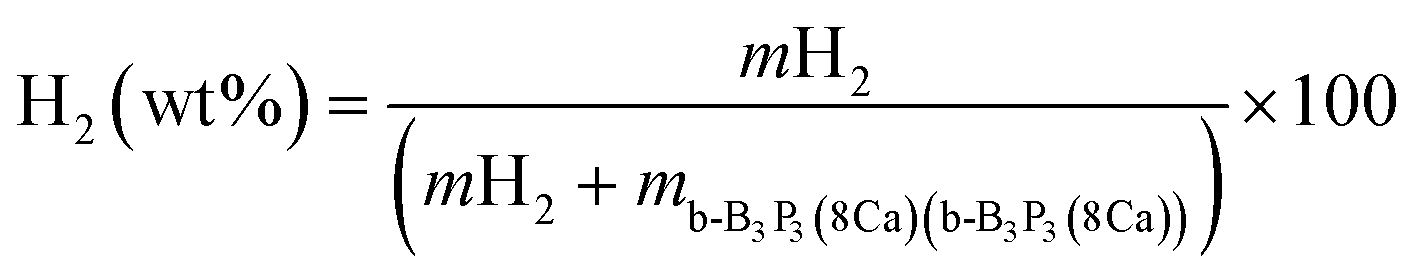 | (3) |
The average desorption temperature (Td) of the b-B3P3(8Ca)@pH2 and g-B6P6(16Ca)@qH2 systems has been estimated by employing the van't Hoff equation,59 expressed as:
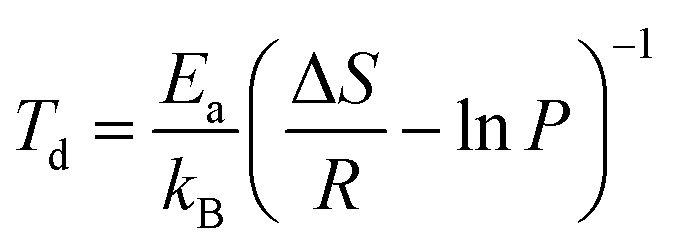 | (4) |
3 Results and discussion
3.1 Adsorbent structures
This paper expands on the recently reported H2 storage on b-B3P3(Li) and g-B6P6(Li) structures,39 by investigating the H2 storage performance of b-B3P3(Mg, Be, Ca) and g-B6P6(Mg, Be, Ca) structures. To begin with, we construct b-B3P3 and g-B6P6 supercells in (2 × 2 × 1) dimensions, as depicted in Fig. 1. Each (2 × 2 × 1) b-B3P3/g-B6P6 sheet consists of 24 B, 24 P, and 4 Ca atoms. Henceforth, we shall consider the (2 × 2 × 1) supercell of b-B3P3/g-B6P6 as b-B12P12X4/g-B24P24X4 sheets. The optimized lattice constants were evaluated to be a = 11.80 Å and b = 9.75 Å, and a = b = 17.61 Å, respectively, for b-B3P3 and g-B6P6 sheets, in good agreement with a recently reported paper.60 It is well-known that pure 2D materials24,33–36,39,61–63 form weak interactions with H2 molecules, which reduces their storage capacities. However, metalization of these b-B3P3 and g-B6P6 sheets with AEM (Be, Mg, Ca) adatoms is an efficient route to improve the H2 chemical activity on their surfaces. We have examined eight distinct adsorption sites on the b-B3P3 and g-B6P6 sheets to determine the preferred location of single AEM atoms. Fig. 1 illustrates these adsorption sites, which are at the top of the boron (TB) and phosphorus (TP) atoms, the octagonal/decagonal cavity (A1), the square (A2) and hexagonal (A3) rings, and the bridging sites (A4, A5 and A6).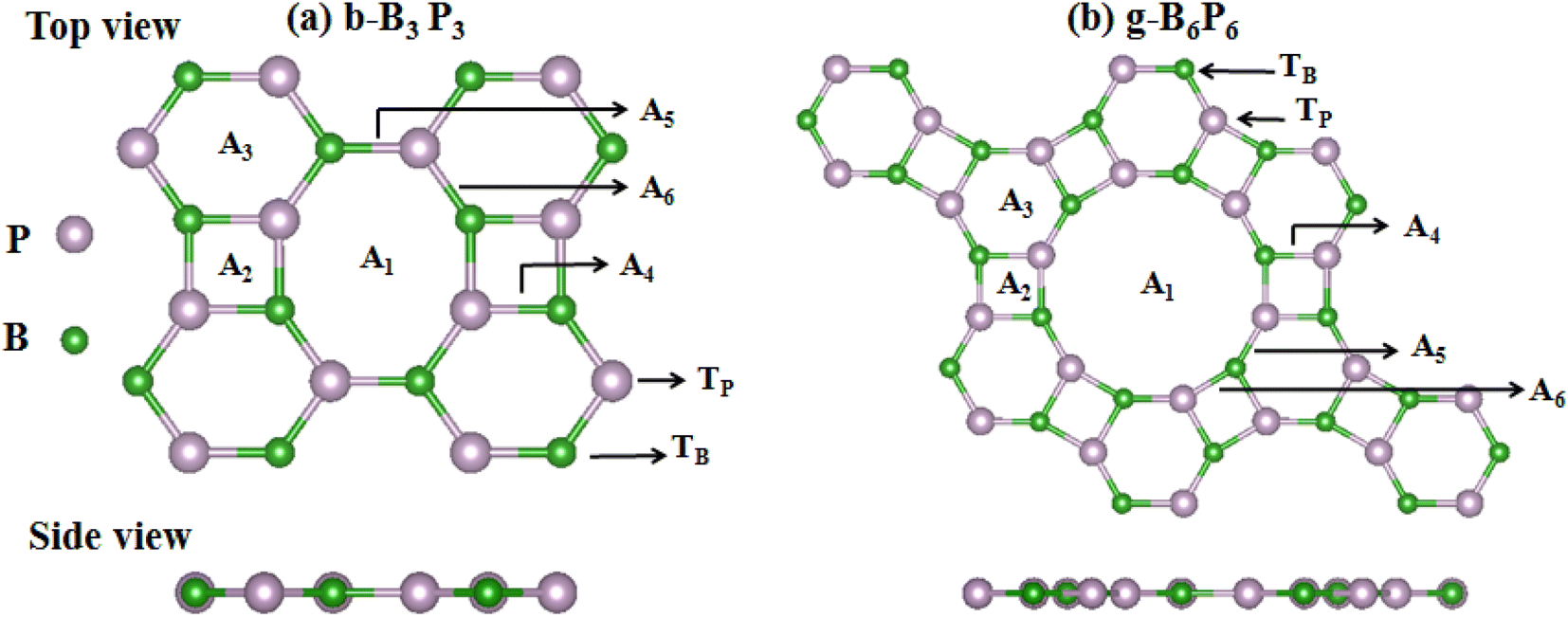 | ||
| Fig. 1 Geometric structures of (a) b-B3P3 and (b) g-B6P6 sheets: top and side views with illustration of the available binding sites (A1, A2, A3, A4, A5, A6, TB and TP). | ||
To achieve a uniform distribution, each AEM adatom must bind firmly to these b-B3P3 and g-B6P6 sheets. A negative Ea value signifies a stable configuration. Conversely, a positive Ea value suggests an unfavorable adsorption process. The calculated Ea values of single AEM adatoms at the most stable b-B3P3 and g-B6P6 sites are presented in Fig. 2. We have found that the Be and Mg adatoms bind on the b-B3P3 and g-B6P6 sheets with very small Ea values. Additionally, their cohesive energy (Ec) is notably greater than their Ea values on the these surfaces. Hence, these metals would rather form clusters than bind separately to the b-B3P3 and g-B6P6 sheets. Therefore, we have not considered b-B3P3(Be), b-B3P3(Mg), g-B6P6(Be) and g-B6P6(Mg) structures for the H2 adsorption calculations (see Fig. 2). However, the Ea values of Ca-decorated b-B3P3 and g-B6P6 sheets are found to be −2.82 eV and −2.62 eV, respectively, larger than the Ec value of the bulk Ca atom (1.84 eV).64 Bader charge (Q) analysis57 confirms that there was a significant transfer of 1.40 e− from the Ca+ ion to the b-B3P3/g-B6P6 monolayer, as compared with 1.32/1.02 e− from Be+/Mg+ ions, respectively. This shows a favorable ionic bonding between Ca adatoms and the b-B3P3/g-B6P6 surfaces. The obtained Ea values correlate with the estimated electron transfer values. Hence, these stable b-B3P3(Ca) and g-B6P6(Ca) structures are considered for further studies. Because they have a large surface area, the b-B3P3 and g-B6P6 sheets are expected to possess the ability of binding multiple Ca adatoms. This could significantly improve their H2 storage performance. Consequently, we introduced several Ca adatoms to investigate their binding properties. Our findings revealed that eight Ca adatoms can be adsorbed on the b-B3P3 sheet, with an Ea value of −2.46 eV per Ca. The g-B6P6 sheet can take up to 16Ca adatoms with Ea values of −2.47 eV per Ca. We have also carried out another analysis to illustrate the stability of dispersed Ca adatoms on b-B3P3 and g-B6P6 surfaces compared to Ca dimer formation or aggregation into clusters. This is done by comparing the single Ca atom and dimer Ea values for adsorption on these surfaces. As we know, if the Ca dimer binding on the surface becomes less stable compared to single-atom binding, cluster formation can be avoided. Hence, the clustering energy (Ecluster) was determined by comparing the Ea of the dimer to that of the single Ca adsorbed on the b-B3P3/g-B6P6 surfaces. If the Ecluster has a positive (negative) value, it means that the Ca atoms are susceptible to cluster formation (dispersion). The obtained Ecluster values are −0.23 and −0.79 eV for b-B3P3 and g-B6P6 monolayers, respectively. Clearly, all Ca atoms possess negative clustering energy values on these b-B3P3 and g-B6P6 surfaces. It can be inferred that the Ca atoms that are dispersed on the b/g-BP surfaces are generally stable and Ca cluster formation can be avoided. Additionally, Fig. S4 in the ESI File† confirms the dynamic stability of these b-B3P3 and g-B6P6 structures with adsorbed Ca adatoms.
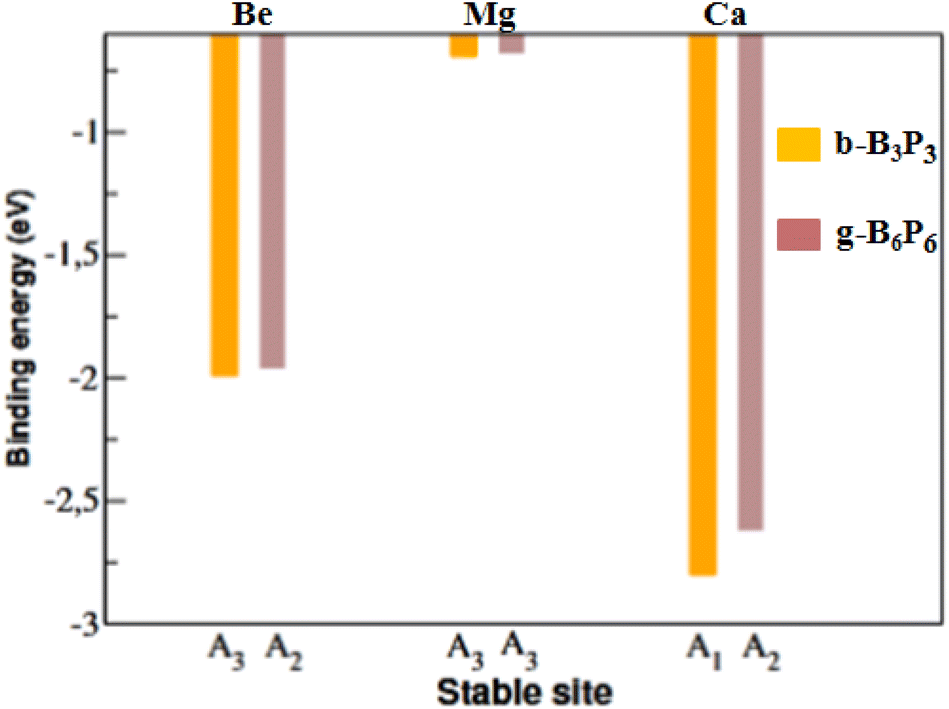 | ||
| Fig. 2 The Ea values of b-B3P3(Be, Mg, Ca) and g-B6P6(Be, Mg, Ca) structures. | ||
Table 1 lists the calculated Ea values, the average heights of Ca adatoms from the b-B3P3(g-B6P6) sheets (bond lengths, dCa–b-B3P3(g-B6P6)) and the amounts of charge released by the Ca adatoms. The top and side views of the optimized b-B3P3(8Ca) and g-B6P6(16Ca) geometries are shown in Fig. S1.† For the b-B3P3(8Ca) and g-B6P6(16Ca) structures, there was an absence of any indication of structural reconstruction after optimization. Additionally, there is no tendency for Ca adatoms to cluster on the b-B3P3 and g-B6P6 surfaces.
| System | m/n | Ea | dCa–b-B3P3(g-B6P6) (Å) | Q (e−) |
|---|---|---|---|---|
| b-B3P3(mCa) | 1 | −2.80 | 1.23 | 1.39 |
| 4 | −2.31 | 1.54 | 1.31 | |
| 8 | −2.46 | 1.81 | 1.11 | |
| g-B6P6(nCa) | 1 | −2.62 | 1.79 | 1.39 |
| 8 | −2.35 | 2.28 | 1.15 | |
| 16 | −2.47 | 1.98 | 1.09 |
We provide charge analysis calculations for the b-B3P3(mCa) and g-B6P6(nCa) structures to illustrate the charge transferred from the Ca adatoms to the b-B3P3(g-B6P6) sheets (see Table 1). There is a significant charge transfer from Ca+ ions to the b-B3P3 and g-B6P6 sheets for all structures. As a result, Ca adatoms and these surfaces form an ionic bond, as confirm by the ELF plots in Fig. S3.† It should be noted that the obtained Ea values agree with the obtained electron transfer values. The averaged bond length along the z-axis between Ca adatoms and all the B/P atoms in the b-B3P3 and g-B6P6 surfaces corresponds well with the Ea values listed in Table 1. It shows that the stronger the interaction, the lower the bond length, which affects the values of Ea. Also, the thermal stability of the b-B3P3(Ca) and g-B6P6(Ca)) structures was evaluated through AIMD at 300 K for a duration of 5 ps. In Fig. S4,† we have shown the oscillation range of total energy against time step at 300 K. According to the figure, the energy oscillations for each atom fluctuate within a fixed range on the order of meV. In addition, in Fig. S4† we give images of the side and top views of the b-B3P3(Ca) and g-B6P6(Ca)) structures. It is clear that these structures maintain their structural integrity at room temperature without any visible structural reconstructions. It is evident that these b-B3P3(Ca) and g-B6P6(Ca)) structures are suitable ones for H2 storage applications under ambient conditions.
3.2 H2 adsorption on b-B3P3(8Ca) and g-B6P6(16Ca) structures
Firstly, the H2 molecule is introduced at a given height above the stable adsorption site of the b-B3P3(8Ca) and g-B6P6(16Ca) structures. Then, the b-B3P3(Ca)@H2 and g-B6P6(Ca)@H2 systems are fully relaxed without any geometry restriction. Subsequently, a number of H2 molecules was incrementally added until the maximum adsorption limit was reached. According to the Ea values obtained for b-B3P3(8Ca) and g-B6P6(16Ca), there is strong physisorption and chemisorption of H2 molecules. These structures provide a better binding of H2 molecules than those obtained from the pristine b-B3P3@H2 and g-B6P6@H2 systems.39 Fig. 4 displays all the remaining estimated Ea values. It is revealed that the b-B3P3(8Ca) structure can adsorb up to 32 H2 molecules, with an average Ea value of −0.23 eV per H2. On the other hand, the g-B6P6(16Ca) structure can retain 48H2 molecules with an average Ea value of −0.25 eV per H2. It should be noted that these estimated Ea values show the tendency to accommodate more H2 molecules on the b-B3P3(8Ca) and g-B6P6(16Ca) structures. In comparison with the earlier results based on DFT calculations, our estimated moderate H2 Ea values are better than or comparable with those reported for metallized boron monoxide, boron hydride and Me–C8B5 monolayers.47,65,66 The significantly negative Ea value means a stronger interaction of H2 molecules on b-B3P3(8Ca) and g-B6P6(16Ca) structures. The Ea value we obtained for the maximum of adsorbed H2 molecules is within the above-mentioned acceptable range required for a H2 storage material. This means that the H2 molecule has been strongly physisorbed onto these b-B3P3(8Ca) and g-B6P6(16Ca) structures. Fig. 3 shows the relaxed structures of the b-B3P3(8Ca)@32H2 and g-B6P6(16Ca)@48H2 structures, whereas the remaining b-B3P3(8Ca)@pH2 and g-B6P6(16Ca)@qH2 structures are illustrated in Fig. S5 and S6.† We also provide, in Table 2, the numbers (p,q) of H2 molecules adsorbed on the b-B3P3(8Ca) and g-B6P6(16Ca) structures, the Ea (eV per H2), the average H–H bond length (dH–H) and the corresponding H2 capacity (wt%).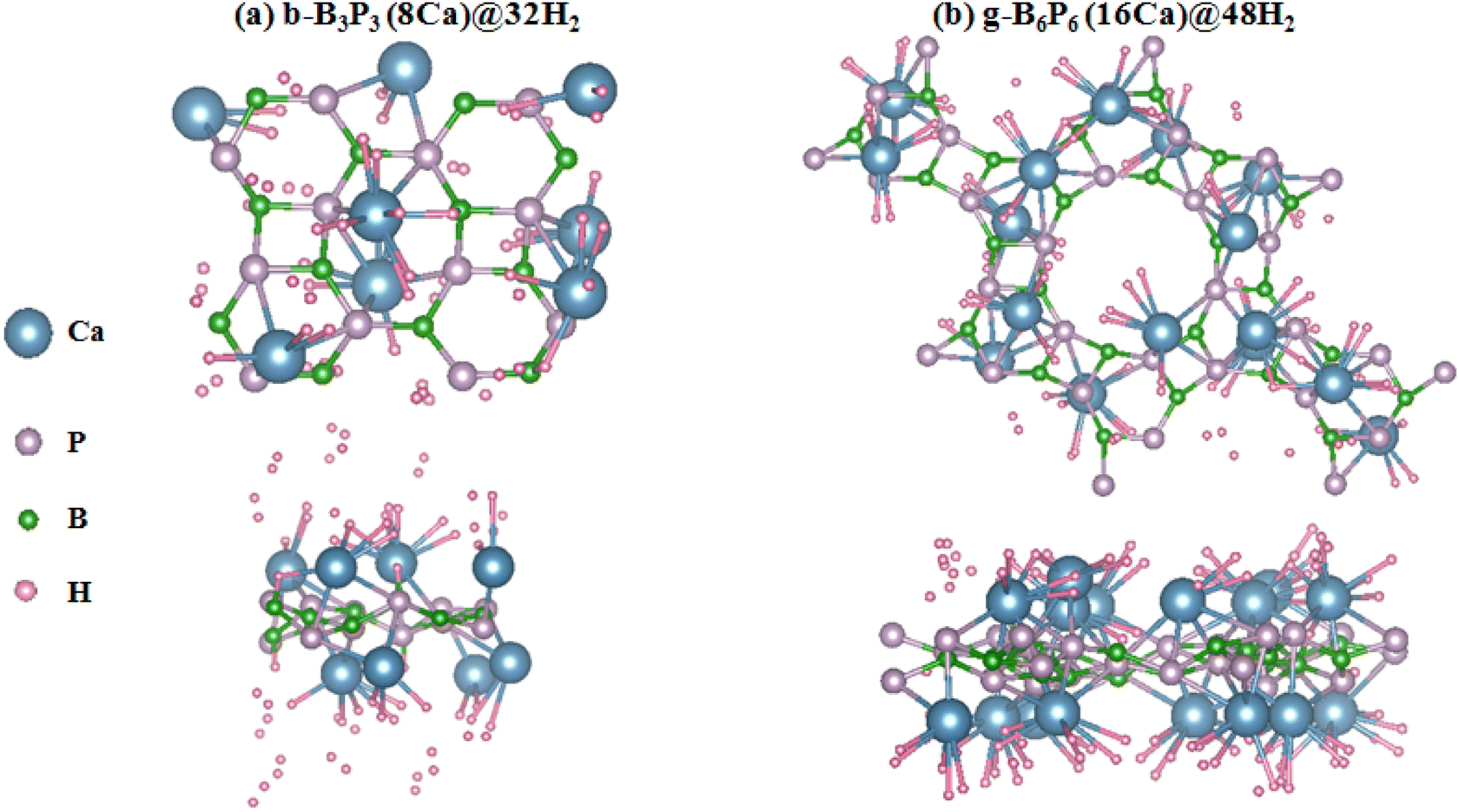 | ||
| Fig. 3 The optimized structures with maximum adsorbed H2 molecules: (a) b-B3P3(8Ca) and (b) g-B6P6(16Ca) structures. | ||
| System | Number of H2 | Ea (eV per H2) | d(H–H) | TD (K) | W (wt%) |
|---|---|---|---|---|---|
| b-B3P3(8Ca) | 1 | −0.38 | 0.762 | 486.21 | 0.24 |
| 4 | −0.16 | 0.763 | 204.72 | 0.97 | |
| 8 | −0.35 | 0.970 | 447.82 | 1.92 | |
| 12 | −0.19 | 0.762 | 243.10 | 2.86 | |
| 16 | −0.20 | 0.869 | 255.90 | 3.78 | |
| 32 | −0.23 | 0.973 | 294.28 | 7.28 | |
| g-B6P6(16Ca) | 1 | −1.35 | 0.769 | 1727.32 | 0.12 |
| 8 | −0.34 | 0.770 | 435.03 | 0.97 | |
| 16 | −0.51 | 1.081 | 652.54 | 1.92 | |
| 24 | −0.35 | 0.947 | 447.82 | 2.86 | |
| 48 | −0.25 | 0.840 | 319.87 | 5.56 |
It well-known that the interaction of an adsorbent surface with a H2 molecule can be attributed to the electronic properties of the host material and the vdW forces between the H2 and the surface. For weak vdW forces, the H2 molecule can be physisorbed on the surface. Conversely, the H2 molecule may dissociate into double H atoms and form a strong chemical bond with the surface atoms via a chemisorption mechanism, as previously reported.47,63,67 It is noted from the optimized structures that 4 H2 and 12 H2 molecules are adsorbed through a physisorption process on the b-B3P3(8Ca) structure. However, we found that for adsorption of 8 H2 and 16 H2 molecules, one H2 molecule is dissociated into 2H atoms, and four 4 H2 molecules show a tendency to dissociate into 8 H atoms. In these cases, we found that the average H–H bond length is extended from 0.74 Å (gas-phase H–H distance) to 0.87 and 0.97 Å, showing that the adsorption of H2 molecules on the b-B3P3(8Ca) structure arises through a strong physisorption process (see Fig. 4).
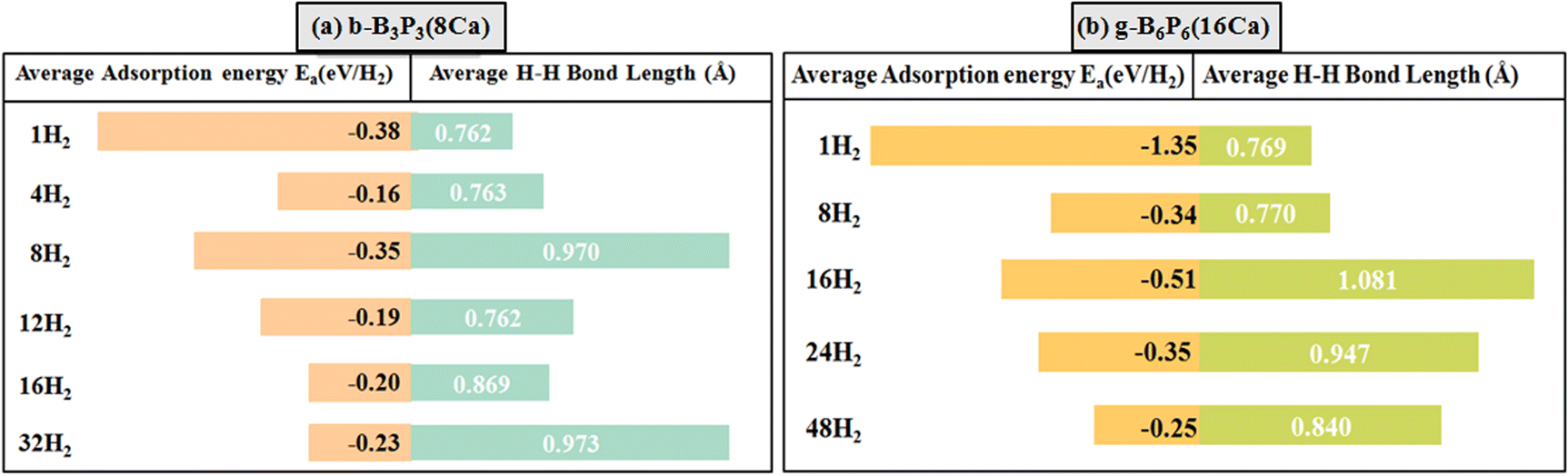 | ||
| Fig. 4 The average Ea values (eV per H2) and average bond lengths (H–H) of H2 molecules for (a) b-B3P3(8Ca) and (b) g-B6P6(16Ca). The lengths of the bars correspond to the Ea values. | ||
On the other hand, for the g-B6P6(16Ca) structure, the adsorption of 8 H2 molecules occurs through physisorption (molecular form). However, 3 H2 molecules are dissociated into 6 H atoms in the case of 16 adsorbed H2 molecules, while 2 H2 molecules are dissociated into 4 H atoms in the cases of adsorption of 24 H2 and 48 H2 molecules. Their corresponding H–H bond lengths range between 0.84–1.08 Å. The tendency towards dissociation is attributed to the larger Ea values, which lead to an expanded dH–H value for H2 molecules (see Fig. 4). According to eqn (3), the estimated H2 storage capacities are 7.28% and 5.56% for the b-B3P3(8Ca) and g-B6P6(16Ca) structures, respectively, exceeding the US DOE requirements. Within the limit of the theoretical method used in the current study, these obtained values are either similar to or higher than those of previously investigated metal-decorated 2D materials,36,68,69 including the recently reported metallized boron monoxide, boron hydride and Me–C8B5 monolayers.47,65,66 According to the PDOS plots, all the b-B3P3(8Ca)@32H2 and g-B6P6(16Ca)@48H2 structures exhibit metallic properties (see Fig. S4†). All the structures show evidence of asymmetric spin states around the Fermi level and beyond, which implies the existence of magnetic moments. The states are mainly from the p orbital of the Ca atom, while the s orbital of the H atom makes a small contribution in the vicinity of the Fermi level. Furthermore, there is a relative overlap between the Ca p orbital and the B p and P p orbitals beyond the Fermi level, except for H atoms. This emphasizes the nature of Ca’s interactions with the surroundings, specifically the s orbital of the H atom.
To achieve practical H2 storage on these b-B3P3(8Ca) and g-B6P6(16Ca) surfaces, it is necessary to determine the average desorption temperature (TD). Using eqn (4), the obtained TD values for the b-B3P3(8Ca)@pH2 and g-B6P6(16Ca)@qH2 structures are summarised in Table 2. The obtained TD values for the b-B3P3(8Ca)@pH2 and g-B6P6(16Ca)@qH2 structures are found to be in the range of 204–486 K and 319–1727 K, respectively. The TD values decrease as the number of H2 molecules increases, while the Ea value decreases. The TD values for 32 H2 and 48 H2 molecules adsorbed on the b-B3P3(8Ca) and g-B6P6(16Ca) structures are estimated as 294 K and 319 K, respectively. The TD value is over 9 times larger than the critical point of hydrogen (33 K). The obtained results clearly illustrate the correlation between bond lengths, Ea values, and TD values. Table 2 reveals the TD values that correspond to the average lengths of the H–H bonds. The higher TD values suggest that some molecules are adsorbed through strong physisorption, and the greater H–H bond lengths may lead to dissociation at room temperature. It is worth mentioning that magnetic properties have been proven to be an effective way to regulate H2's desorption temperature.70 It is expected that these studied b-B3P3(8Ca) and g-B6P6(16Ca) structures will enable the storage of H2 under suitable conditions and have good reversibility.
4 Conclusions
In brief, spin-polarized DFT calculations are employed to investigate the performance of b-B3P3 and g-B6P6 sheets with adsorbed AEM (Be, Mg, Ca) adatoms for hydrogen storage. We find that Be and Mg adatoms are prone to cluster formation on b-B3P3 and g-B6P6 surfaces due to their low Ea values. However, the high Ea values for b-B3P3(Ca) and g-B6P6(Ca) structures show that the Ca adatom is stable and can prevent the segregation of Ca on the b-B3P3 and g-B6P6 surfaces, even at high concentrations. In addition, It has been demonstrated that the b-B3P3(Ca) and g-B6P6(Ca) structures are thermally stable at 300 K. Bader charge analysis reveals that Ca transfers an average of 1.11 (1.09) electrons to the b-B3P3(Ca) (g-B6P6(Ca)) sheets, making the b-B3P3(Ca) (g-B6P6(Ca)) surfaces suitable for enhanced H2 molecule storage. In particular, the b-B3P3(8Ca) structure can store up to 32 H2 molecules with an average Ea value of −0.23 eV per H2. Meanwhile, the g-B6P6(16Ca) structure can take up 48 H2 molecules with an average Ea value of −0.25 eV per H2. The H2 molecule storage capacities of the b-B3P3(8Ca)@32H2 and g-B6P6(16Ca)@48H2 structures are 7.28 wt% and 5.56 wt%, respectively. The calculated wt% values are higher than the 5.50 wt% target that needs to be reached by 2025. According to these findings, the b-B3P3(8Ca) and g-B6P6(16Ca) structures possess the potential to be reversible hydrogen storage media.Data availability
Data will be made available on request.Author contributions
Yusuf Zuntu Abdullahi: conceptualization, formal analysis, investigation, software, validation, writing – reviewing and editing, supervision, project administration. Ikram Djebablia: formal analysis, investigation, visualization, validation, writing – reviewing and editing. Tiem Leong Yoon: data curation, formal analysis, writing – reviewing. Lim Thong Leng: data curation, formal analysis, writing – reviewing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The calculations were performed at TUBITAK ULAKBIM, High Performance and Grid Computing Center (TR-Grid e-Infrastructure).Notes and references
- M. S. Dresselhaus and I. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- G. W. Crabtree, M. S. Dresselhaus and M. V. Buchanan, Phys. Today, 2004, 57, 39–44 CrossRef CAS.
- C.-J. Winter and J. Nitsch, Hydrogen as An Energy Carrier: Technologies, Systems, Economy, Springer Science & Business Media, 2012 Search PubMed.
- A. W. van den Berg and C. O. Areán, Chem. Commun., 2008, 668–681 RSC.
- P. Jena, J. Phys. Chem. Lett., 2011, 2, 206–211 CrossRef CAS.
- C. Tarhan and M. A. Çil, J. Energy Storage, 2021, 40, 102676 CrossRef.
- S. K. Bhatia and A. L. Myers, Langmuir, 2006, 22, 1688–1700 CrossRef CAS.
- Y.-H. Kim, Y. Zhao, A. Williamson, M. J. Heben and S. Zhang, Phys. Rev. Lett., 2006, 96, 016102 CrossRef PubMed.
- R. C. Lochan and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 1357–1370 RSC.
- T. Rimza, S. Saha, C. Dhand, N. Dwivedi, S. S. Patel, S. Singh and P. Kumar, ChemSusChem, 2022, 15, e202200281 CrossRef CAS PubMed.
- M. Mohan, V. K. Sharma, E. A. Kumar and V. Gayathri, Energy Storage, 2019, 1, e35 CrossRef CAS.
- X. Wu, Y. Chen, Z. Xing, C. W. K. Lam, S.-S. Pang, W. Zhang and Z. Ju, Adv. Energy Mater., 2019, 9, 1900343 CrossRef.
- M. Hu, H. Zhang, T. Hu, B. Fan, X. Wang and Z. Li, Chem. Soc. Rev., 2020, 49, 6666–6693 RSC.
- P. Kumar, S. Singh, S. Hashmi and K.-H. Kim, Nano Energy, 2021, 85, 105989 CrossRef CAS.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed.
- L. Zhu, X.-Q. Liu, H.-L. Jiang and L.-B. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef CAS PubMed.
- S. Ma and H.-C. Zhou, Chem. Commun., 2010, 46, 44–53 RSC.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 Search PubMed.
- J. Wang and S. Zhuang, Coord. Chem. Rev., 2019, 400, 213046 CrossRef CAS.
- X. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- H. Jiang, W. Shyy, M. Liu, L. Wei, M. Wu and T. Zhao, J. Mater. Chem. A, 2017, 5, 672–679 RSC.
- N. Khossossi, Y. Benhouria, S. R. Naqvi, P. K. Panda, I. Essaoudi, A. Ainane and R. Ahuja, Sustainable Energy Fuels, 2020, 4, 4538–4546 RSC.
- T. Zhang, Y. Ma, B. Huang and Y. Dai, ACS Appl. Mater. Interfaces, 2019, 11, 6104–6110 CrossRef CAS.
- A. Kara, H. Enriquez, A. P. Seitsonen, L. L. Y. Voon, S. Vizzini, B. Aufray and H. Oughaddou, Surf. Sci. Rep., 2012, 67, 1–18 CrossRef CAS.
- V. Eswaraiah, Q. Zeng, Y. Long and Z. Liu, Small, 2016, 12, 3480–3502 CrossRef CAS PubMed.
- Y. Yong, H. Cui, Q. Zhou, X. Su, Y. Kuang and X. Li, Appl. Surf. Sci., 2019, 487, 488–495 CrossRef CAS.
- Y. Z. Abdullahi, F. Ersan, Z. D. Vatansever, E. Aktürk and O. Ü. Aktürk, J. Appl. Phys., 2020, 128, 113903 CrossRef CAS.
- T. Li, C. He and W. Zhang, J. Mater. Chem. A, 2019, 7, 4134–4144 RSC.
- Y. Z. Abdullahi, A. Tigli and F. Ersan, Phys. Rev. Appl., 2023, 19, 014019 CrossRef CAS.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271–279 CrossRef CAS.
- P. A. Denis and F. Iribarne, Comput. Theor. Chem., 2015, 1062, 30–35 CrossRef CAS.
- V. Mahamiya, A. Shukla and B. Chakraborty, Int. J. Hydrogen Energy, 2022, 47, 41833–41847 CrossRef CAS.
- T. Kaewmaraya, N. Thatsami, P. Tangpakonsab, R. Kinkla, K. Kotmool, C. Menendez, K. Aguey-Zinsou and T. Hussain, Appl. Surf. Sci., 2023, 629, 157391 CrossRef CAS.
- T. Hussain, M. Hankel and D. J. Searles, J. Phys. Chem. C, 2017, 121, 14393–14400 CrossRef CAS.
- N. F. Martins, A. S. Maia, J. A. Laranjeira, G. S. Fabris, A. R. Albuquerque and J. R. Sambrano, Int. J. Hydrogen Energy, 2024, 51, 98–107 CrossRef CAS.
- X. Zhang, F. Chen, B. Jia, Z. Guo, J. Hao, S. Gao, G. Wu, L. Gao and P. Lu, Int. J. Hydrogen Energy, 2023, 48, 17216–17229 CrossRef CAS.
- I. Djebablia, Y. Z. Abdullahi, K. Zanat and F. Ersan, Int. J. Hydrogen Energy, 2024, 66, 33–39 CrossRef CAS.
- L.-J. Ma, Y. Sun, J. Jia and H.-S. Wu, Fuel, 2024, 357, 129652 CrossRef CAS.
- Y. Fu, J. Xiang, Y. Xie, X. Gong, Y. Xu, Q. Zhao, Y. Liu, J. Xu and W. Liu, Comput. Mater. Sci., 2024, 238, 112951 CrossRef CAS.
- X. Gong, Y. Fu, Y. Xu, W. Liu and J. Xu, Mater. Today Commun., 2024, 40, 109626 CrossRef CAS.
- Y. Xie, L. Chen, J. Xu and W. Liu, RSC Adv., 2022, 12, 20088–20095 RSC.
- Y. Xu, Y. Fu, X. Gong, J. Xu and W. Liu, Mater. Today Commun., 2024, 41, 110394 CrossRef CAS.
- P. Panigrahi, A. Kumar, A. Karton, R. Ahuja and T. Hussain, Int. J. Hydrogen Energy, 2020, 45, 3035–3045 CrossRef CAS.
- P. Panigrahi, M. Desai, M. K. Talari, H. Bae, H. Lee, R. Ahuja and T. Hussain, Int. J. Hydrogen Energy, 2021, 46, 7371–7380 CrossRef CAS.
- J. I. Jason, Y. Pal, P. Anees, H. Lee, T. Kaewmaraya, T. Hussain and P. Panigrahi, Int. J. Hydrogen Energy, 2024, 50, 455–463 CrossRef.
- M. Singh, A. Shukla and B. Chakraborty, Sustainable Energy Fuels, 2023, 7, 996–1010 CAS.
- K. Boezar, A. Reisi-Vanani and M. Dehkhodaei, Int. J. Hydrogen Energy, 2021, 46, 38370–38380 CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- D. Nazarian, P. Ganesh and D. S. Sholl, J. Mater. Chem. A, 2015, 3, 22432–22440 RSC.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- R. F. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- E. Durgun, S. Ciraci and T. Yildirim, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 085405 CrossRef.
- Y. Z. Abdullahi and F. Ersan, Comput. Mater. Sci., 2024, 242, 113103 CrossRef CAS.
- K. Alhameedi, T. Hussain, H. Bae, D. Jayatilaka, H. Lee and A. Karton, Carbon, 2019, 152, 344–353 CrossRef CAS.
- A. Hashmi, M. U. Farooq, I. Khan, J. Son and J. Hong, J. Mater. Chem. A, 2017, 5, 2821–2828 RSC.
- A. M. Satawara, G. A. Shaikh, S. K. Gupta, A. N. Andriotis, M. Menon and P. Gajjar, Int. J. Hydrogen Energy, 2023, 48, 25438–25449 CrossRef CAS.
- C. Kittel and P. McEuen, Introduction to Solid State Physics, John Wiley & Sons, 2018 Search PubMed.
- W. Othman, W. Alfalasi, T. Hussain and N. Tit, J. Energy Storage, 2024, 98, 113014 CrossRef.
- Y. Gong, D. Chen, B. Guo, S. Chen, Z. Zhu and M. Cheng, Int. J. Hydrogen Energy, 2024, 82, 384–397 CrossRef CAS.
- S. Nachimuthu, P.-J. Lai, E. G. Leggesse and J.-C. Jiang, Sci. Rep., 2015, 5, 16797 CrossRef CAS PubMed.
- S. R. Naqvi, T. Hussain, P. Panigrahi, W. Luo and R. Ahuja, RSC Adv., 2017, 7, 8598–8605 RSC.
- L. Yuan, L. Kang, Y. Chen, D. Wang, J. Gong, C. Wang, M. Zhang and X. Wu, Appl. Surf. Sci., 2018, 434, 843–849 CrossRef CAS.
- A. Yadav, B. Chakraborty, A. Gangan, N. Patel, M. Press and L. M. Ramaniah, J. Phys. Chem. C, 2017, 121, 16721–16730 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Snapshots of the molecular dynamics simulations, Electron Localization Function (ELF) and total density of states for optimized structures of b-B3P3(8Ca)@32H2 and g-B3P3(8Ca)@48H2 structures. See DOI: https://doi.org/10.1039/d4ra07271e |
| This journal is © The Royal Society of Chemistry 2024 |