
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromoting photoswitching in mismatching mixed-linker multivariate Zr6 MOFs†
Carmen Rosales-Martínez a,
Diego López-Alcaláa,
Marcelo Assisb,
Celia Castillo-Blasc,
José J. Baldovía and
Isabel Abánades Lázaro*a
a,
Diego López-Alcaláa,
Marcelo Assisb,
Celia Castillo-Blasc,
José J. Baldovía and
Isabel Abánades Lázaro*a
aInstituto de Ciencia Molecular, Universitat de Valencia, Paterna, 46980, Spain. E-mail: isabel.abanades@uv.es
bBiomaterials and Bioengineering Lab, Translational Research Centre San Alberto Magno, Universidad Catolica de Valencia San Vicente Mártir (UCV), Valencia, 46002, Spain
cDepartment of Materials Science and Metallurgy, University of Cambridge, CB3 0FS, UK
First published on 28th November 2024
Abstract
Multivariate metal–organic frameworks (MTV MOFs) have emerged as promising materials due to their ability to combine properties that enhance features beyond those of their pristine counterparts. Despite the potential for tailoring electronic properties through structural distortions and defects introduced by linkers of variable lengths, examples remain scarce, and information on the electronic structure is limited. Here, we present the multivariate mismatching linker approach to generate photoswitching nanoparticulated MOFs with variable lattice parameters and porosity features controlled by mixed-linker composition. Structural defects, such as dangling linkers, are generated due to mismatching crystal lattices, tuning the electronic structure. Combining biphenyl and azobenzene ditopic linkers promotes cis–trans photoswitching of dangling azobenzene linkers, which is constrained in Zr6-azobenzene MOFs. Moreover, introducing low quantities of azobenzene drastically reduces the bandgap of the materials due to the contribution of the azo group, which is supported by first-principles calculations. This paves the way for new photo-responsive materials for photo-switching applications.
Introduction
In the quest to replicate the structural complexity of biological systems, multivariate (MTV) metal–organic frameworks (MOFs) have garnered tremendous attention,1–3 allowing for the generation of pores with dual, opposing properties, leading to enhanced applications.4–10 However, aside from sequential linker installation in asymmetric frameworks,11,12 or pillared MOFs,13–15 most MTV studies focus on linkers of similar length, as matching the crystal lattice has been deemed essential in developing mixed-linker multivariate MOFs.Among the few examples of linker mismatching MTV MOFs,16–21 the continuous variation of lattice dimension within Zr6-dicarboxylate MTV MOFs has demonstrated the control of the material's porosity.21 However, despite the potential effect of rising structural distortions on electronic properties, this information is elusive in the literature.
Photoswitching in MOFs has gained notable attention for its ability to dynamically control the materials' properties,22–26 as switching materials have potential applications in optoelectronics, energy storage, memory devices, photocatalysis, sensing, gas separation and stimuli-controlled drug delivery among other fields.
However, the photoisomerization of multitopic linkers is often hindered in the solid state due to the constraints of the rigid frameworks. In this regard, combining the linker mismatching strategy in MTV MOFs with photoswitching linkers could result in unique properties arising from structural distortions.
For instance, induced defects, such as dangling linkers, could promote or enhance the photoswitching of moieties within linkers by providing greater conformational freedom.
Among photoswitching linkers, azobenzene moieties are of particular interest due to their photochromic properties, undergoing reversible cis–trans isomerization upon light exposure, with molecular changes of 3.5 Å.27,28 This property makes azobenzene a focal point in photophysics and photochemistry studies. Consequently, azobenzene moieties are extensively explored for photoresponsive systems, from drug delivery to photocatalysis or photo-driven molecular machines.29–31
However, although the azobenzene dicarboxylate linker bound to metal clusters within Zr6 MOF systems has been proven to be flexible through dynamic disorder,32 its cis–trans photoisomerization is prevented in MOFs due to the geometric restrictions provided by bonding to metal clusters through both ends of the azobenzene moiety.26,28,32 This transformation can only be achieved if azobenzene groups are present as side chains, reducing porosity.
Herein, we present the linker mismatching approach as a new method to synthesise azobenzene photoresponsive nanoparticulate MTV MOFs based on dicarboxylate azobenzene linkers, with the structural distortions originated by the accommodation of linkers of different lengths results in defects such as dangling linkers, which enable the photoisomerization of the, otherwise constrained, azobenzene dicarboxylate linkers.
By fine-tuning the synthetic conditions, controllable composition, lattice parameters, porosity and electronic properties are achieved, drastically reducing the bandgap even at low azobenzene concentrations.
Results and discussion
Synthetic control of composition and cell parameters
To synthesize linker mismatching MTV Zr6-MOFs, two dicarboxylate linkers – biphenyl-4,4′-dicarboxylic acid with a molecular size of 11 Å and azobenzene-4,4′-dicarboxylic Acid with a molecular size of 13 Å, denoted as BPDC and AzDC respectively, represented in Fig. 1a – were introduced during synthesis. The percentage of AzDC linker was varied systematically from 0 to 25, 50, 75, and 100% within excess of linkers added compared to Zr (linker: metal = 2), denoting the samples as the percentage of AzDC added to the synthesis. In contrast to the previously reported syntheses of single crystals of similar materials,21 the excess of total linker shall result in the formation of nanoparticles, which is beneficial for several applications, and shall promote the formation of defects.33 Acetic acid was introduced as pH-regulating acid in a 0.05 ratio compared to the solvent (DMF) to avoid fast nucleation and crystallization leading to amorphous samples, but with a concentration low enough to avoid single crystals (see ESI S.2, Table S1† for detailed synthetic conditions).33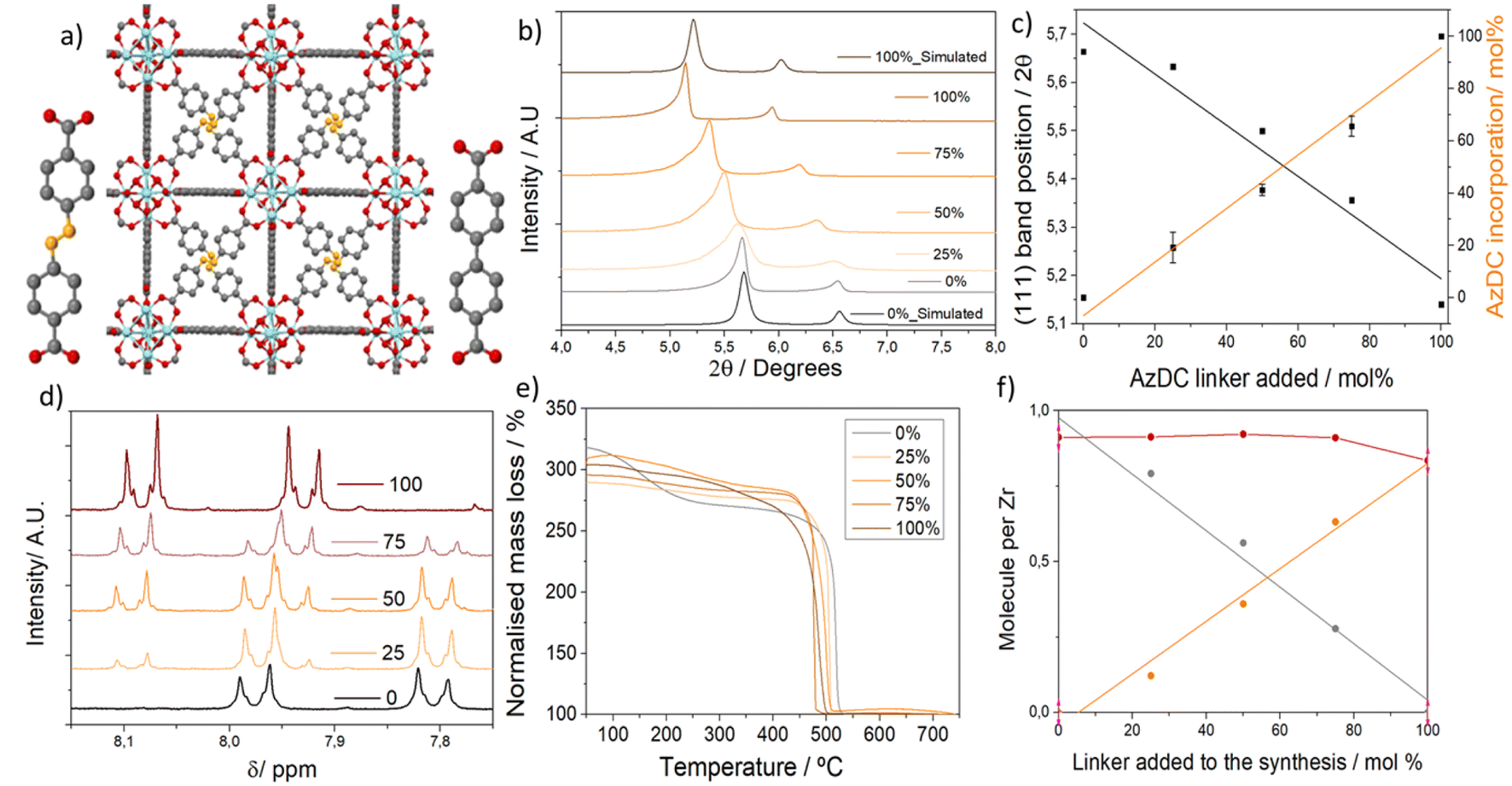 | ||
| Fig. 1 (a) Schematic representation of an MTV mismatching MOF alongside the two linkers. (b) PXRD showing the concomitant variation of Bragg reflection bands. (c) 2 theta position of the first reflection band and molar percentage of AzDC linker incorporated as a function of the molar percentage of AzDC linker added. (d) Amplification of 1H NMR profiles showing the variation of linkers. (e) Thermal decomposition profiles of the samples, with residue normalised to 100%. (f) Composition of the frameworks per Zr metal, as characterised by the combination of 1H NMR and TGA. | ||
The formation of multivariate mismatching fcu frameworks was confirmed by powder X-ray diffraction (PXRD), showing the concomitant linear variation of Bragg reflection bands with the increased percentage of longer ligand (AzDC) added to the synthesis (Fig. 1b). This indicates that AzDC was incorporated as a multivariate framework rather than as separate structures, as confirmed by Pawley refinements (see ESI S.3.1, Fig. S4–S8 and Table S2†).21 The presence of a second component in the 25–75% samples around 5.5° is attributed to asymmetry within the frameworks.
The position of the reflection bands and the cell lattice parameters are related to the percentage of ligands added (Fig. 1c), which is itself linearly related to the molar % of ligands incorporated (Fig. 1c and d), as determined by acid-digested 1H NMR. The molar percentage incorporation of AzDC ligand compared to BPDC is slightly lower than the percentage of addition in all cases (Table 1), indicating slower nucleation in agreement with its lower acidity.33 1H NMR profiles also prove that frameworks' activation (complete DMF removal) is achieved upon stirring in MeOH for 24 hours. This procedure also replaces AcO− defect compensating species by MeO−, as previously observed for other Zr-based MOF systems,34 but leaves unaltered the mixed-linker ratios (see ESI S.3.2, Fig. S9–S13 and Tables S3, S4† for detailed analysis).
| % AzDC added | AzDC incorporated (mol%) | Particle size (nm) | Lattice parameter (Å) | Experimental band gap (eV) | Calculated band gap (eV) |
|---|---|---|---|---|---|
| 0 | 0 | 173.22 ± 22.40 | a = 26.806 (8) | 3.441 | 2.7 |
| 25 | 19.22 ± 5.91 | 283.40 ± 24.32 | a = 26.966 (7) | 2.205 | 1.75 |
| 50 | 41.24 ± 2.23 | 633.32 ± 68.29 | a = 27.396 (13) | 2.199 | 1.75 |
| 75 | 65.57 ± 3.85 | 621.11 ± 57.44 | a = 29.383 (18) | 2.206 | 1.65 |
| 100 | 100 | 675.96 ± 60.84 | a = 29.5466 (26) | 2.174 | 1.6 |
Lattice parameters were determined by Pawley refinements (Table S2 and Fig. S4–S8†). Asymmetry corrections were applied regarding the asymmetric Bragg peaks of these nanocrystallites due to linker mismatching. Analyzing the lattice parameters determined by Pawley refinements as a function of the molar fraction of linker incorporated into the structure (Zr6O4OH4[BPDCXAZDC1−X]Y) shows that these multivariate structures follow the Vegard's Law,35 with a linear correlation between the amount of AzDC and the lattice parameters, which vary from 26.806 to 29.546 Å (Fig. S14–S16†). This indicates that synthetic control of the composition within these nanoparticulate MTV MOFs and unit cell parameters can be achieved in a one-pot synthesis.
To explore the nature of the observed changes in the MTV series upon introducing AzDC, we performed systematic density functional theory (DFT) calculations to elucidate the structural modifications that the different ligand ratios can create on the Zr6 MOF (see ESI S.3.3† for computational details). In agreement with experimental results, the introduction of AzDC resulted in an increase in the lattice parameters and related features (Table S5 and Fig. S17–S26†). However, the calculated lattice increase is slightly more pronounced than the experimental (Fig. S27 and S28†), which can be attributed to the flexibility of the dihedral angle of biphenyl rings36 and the presence of defects in the framework. In fact, in the calculated structures, a gradual variation in the AzDC length as a consequence of the reduction in the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C angle is observed across the series (Fig. S29†).
N–C angle is observed across the series (Fig. S29†).
We observe a linear increase of the lattice parameters upon 25% and 100% AzDC linker addition, in agreement with experimental results. In contrast, an abrupt increase takes place between the 0% and 25% AzDC MOF. Experimentally, we do not observe such an abrupt increase, but linear trends across the whole series, matching well for the 0 and 100% AzDC pristine samples. In this regard, we hypothesize that part of the AzDC ligands could act as dangling linkers in the MTV MOFs, with only one carboxylate end being coordinated to Zr6 clusters, thus reducing the experimental lattice expansion, especially at lower AzDC concentrations (25 and 50% of AzDC), for which experimental and calculated cell parameters differ more significantly.
Thermal decomposition profiles (Fig. 1d) confirmed the thermal stability of the samples but showed a linear gradual decrease in thermal decomposition temperature of the final step with the increased incorporation of AzDC (from ca. 475 to 419 °C). Instead of two decomposition steps corresponding to separate frameworks, this palatine decrease in stability further confirms the formation of MTV frameworks (Table S6 and Fig. S31, S32†). Combining thermal decomposition profiles (Fig. S33–S37†) with 1H NMR data allowed us to estimate the MOFs' composition and defectivity (see ESI S.3.4† for calculation details, Table S7 and Fig. S38, S39†).37 The ratio between BPDC and Zr linearly decreases as the AzDC/Zr linearly increases (Fig. 1f), confirming the synthetic control of the MTV MOFs' composition, which is related to the experimental unit cell parameters (Fig. S40 and S41†). Within the structures, the total number of linkers (BPDC + AzDC) is relatively constant (0.92 to 0.83 per Zr), displaying moderate defectivity, with ca. 8% molar percentage of missing linkers for the 0% and MTV samples (ca. 11-connected Zr6 clusters), and ca. 16% for the 100% AzDC sample (10-connected Zr6 clusters). The estimated MeO− defect compensating species based on the molar ratio extracted from acid-digested 1H NMR profiles (and acetate before full activation) is higher than the decrease in the linker-to-metal ratio for the 25% and 50% AzDC MOFs (Table S7†), supporting the presence of AzDC dangling linkers for low AzDC concentrations.37
Control over porosity
N2 adsorption and desorption isotherms, represented in Fig. 2a, were recorded at 77 K to provide information about the porosity of the MTV materials (see ESI S.3.5† for a detailed analysis). The surface areas and pore volumes of the pristine MOFs were in agreement with literature reports38,39 (SBET 2586 m2 g−1 and 3511 m2 g−1 and pore volumes from 1.01 and 1.24 cm3 g−1 for 0% and 100% AzDC respectively, Table S8†), although the 100% AzDC had slightly bigger pores (ca. 15 Å) than reported (ca. 13 Å), possibly due to its defectivity.21 The MTV materials displayed high porosity (Fig. S42 and S43†), which was intermediate to the pristine MOFs and linearly related to the molar percentage of linkers incorporated into the MTV framework (Fig. S44–S49†), apart from the 75%, which displays a decrease in porosity. However, it is still more porous than the 0% sample. This could be expected if dangling linkers were to occupy pore space, given that 1H NMR and TGA showed good sample activation.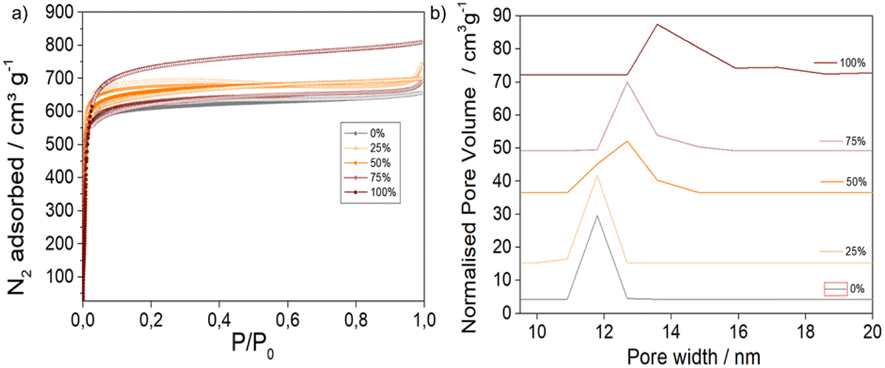 | ||
| Fig. 2 (a) N2 adsorption and desorption isotherms. (b) Pore size distributions, N2 NLDFT model. | ||
In agreement with the fine-tuning of unit cell parameters, the pore size distributions (Fig. S50–S52†) revealed a gradual increase in pore size upon introducing AzDC (Fig. 2b). These results are compatible with the findings of Yuan et al., on single crystals of similar materials,21 showing that the formation of nanoparticles, more suitable for applications, does not alter the fine-tuning of porosity through the linker mismatching approach.
Interestingly, the 0% and 100% AzDC pristine MOFs exhibit different N2 adsorption profiles in the micropore region, which is more evident in the logarithmic representation of the isotherms (Fig. S53 and S54†). In concordance with their MTV nature, the mixed-linker samples exhibit intermediate micropore-filling isotherm profiles that vary with the percentage of AzDC linker incorporated. The adsorption kinetics of MTV MOFs are also intermediate to the 0% and 100% AzDC pristine samples (Fig. S55–59†).
Particle size and aggregation
The MOFs have octahedral morphology, as determined by scanning electron microscopy (SEM) (Fig. 3a), with particle size increasing with the addition of the less acidic linker (AzDC) from ca. 175 nm to ca 675 nm (see ESI S.3.6, Fig. S60–S62 and Table S9†), agreeing with the slower nucleation and crystallization, as observed by its lower incorporation. The increase in particle size is not linear and reaches a plateau at ca. 50% of AzDC addition (Fig. 3b), indicating that AzDC has a key role in nucleation and crystallization kinetics. The particle size distribution of the MTV MOFs is homogeneous within each sample (Fig. S63–S66†), indicating together with previous techniques, that the structures are MTV fcu MOFs and no two separate MOFs.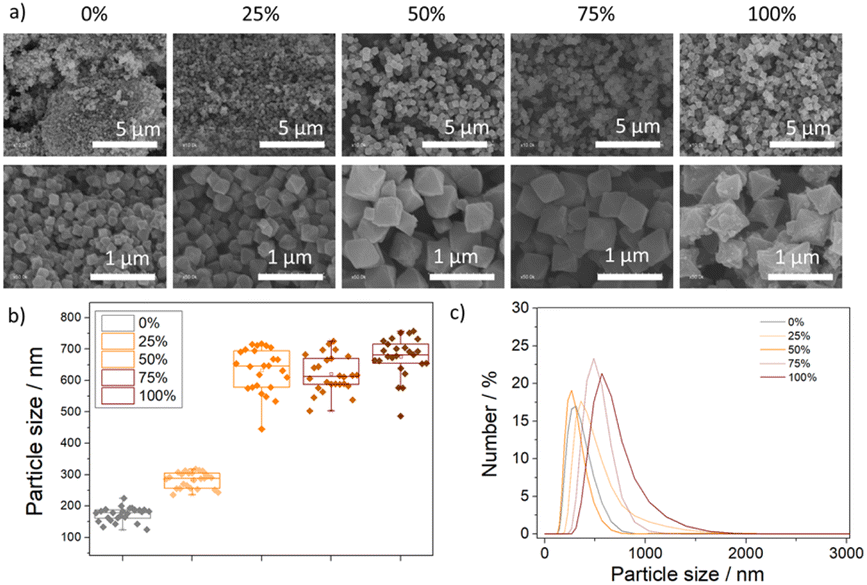 | ||
| Fig. 3 (a) Scanning electron microscope images of the samples. (b) Particle size distributions. (c) Hydrodynamic diameter extracted from DLS measurements of the samples dispersed in water. | ||
Z-Potential was measured in water to evaluate the surface charge of the particles (see ESI S.3.7, Fig. S67–S75 and Table S10†). The pristine samples displayed negative Z-potentials (ca. −22 eV and −11 eV for 0% and 100% AzDC respectively), whereas the MTV samples exhibited positive Z-potentials (ca. 3 eV and 16 eV for the 25% and 50% samples), with exception of the 75% AzDC, with a negative Z-potential of ca. −8 eV.
The samples were well-dispersed in water (see ESI S.3.8†), as measured by dynamic light scattering (DLS) and represented in Fig. 3c, with hydrodynamic diameters similar to the particle sizes determined by SEM, negligible aggregation over three consecutive measurements (Fig. S76–S83, Table S11†) and correlograms showing the absence of big aggregates (Fig. S84–S98†). The good colloidal stability of the MTV MOFs highlights their potential for selected applications, as MOFs typically aggregate in water.40,41
Structural distortions
Despite the particle size increase upon AzDC addition, the Bragg reflection bands of the MTVM MOFs are broader than their pristine counterparts. Thus, an important structural distortion within the MTV mismatching frameworks must occur, which is not typically seen for MTV MOFs with similar-length linkers.Considering the diffraction of the main peak (111), the lattice strain (ε) was calculated for each sample (see ESI S.3.9†),42 reflecting how the proportion of substitution of BPDC linkers with AzDC affects the MTV MOFs' structure. As observed by PXRD in Fig. 1a and ESI S.3.1,† increasing the proportion of AzDC results in an expansion of the crystalline cell, which is less significant than in calculated structures. Additionally, the FWHM also varies with the incorporation of AzDC, initially showing a significant expansion with 25% AzDC, followed by a linear reduction up to the sample with 100% AzDC. Specifically, the sample with only BPDC ligands presents an ε of 0.604. Replacing 25% of the ligands with AzDC drastically increases the ε value to 1.622. With higher proportions of AzDC, there is a reduction in ε values: 1.280 for 50%, 0.988 for 75%, and 0.529 for 100% AzDC. These results, represented in Fig. 4a, suggest that the larger size of AzDC compared to BPDC initially induces a high strain in the fcu crystalline structure due to the structural adjustments necessary for incorporating this linker. As the concentration of AzDC increases, the crystal lattice adjusts better, reducing the structural strain and stress (Fig. S89 and S90†). These trends are compatible with our calculations, for which we observe a linear decrease between 25 and 100% AzDC linker, whereas an abrupt reduction takes place when 25% AzDC linker is incorporated in comparison to when the structures are composed exclusively of BPDC linker.
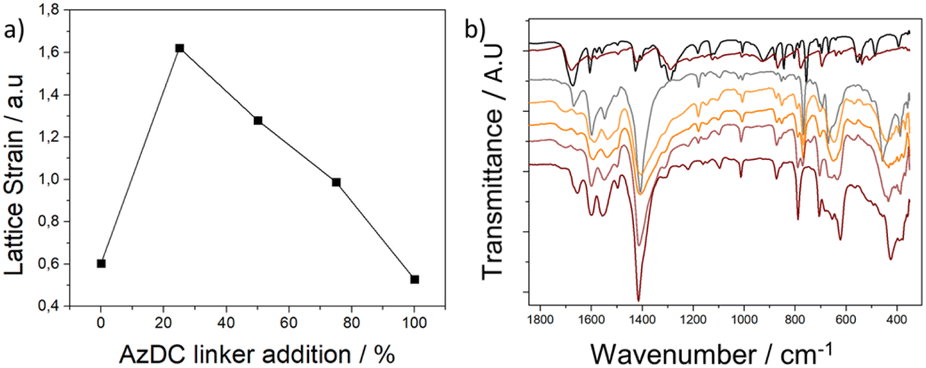 | ||
| Fig. 4 (a) Lattice strain of the samples as a function of the percentage of AzDC linker added. (b) FT-IR of the samples showing the broadening of bands and presence of free carboxylates and the cluster-related structural distortions of the MTV materials. | ||
Fourier transform infrared spectroscopy (FT-IR) showed significant variations between the pristine MOFs and their MTV offspring, such as a general broadening of the bands which is related to their defective nature and structural distortions (Fig. 4b). The signal at ca. 460 nm−1 in the 0% sample, attributed to μ3-OH stretching (in-phase),43 is shifted and appears as a broad band in the MTV structures and similarly occurs for other cluster-related vibration bands, as represented in Fig. 4c (see ESI S.10, Fig. S91–S94†). This suggests cluster deformation within the MTV structures upon the co-existence of linkers with different lengths, as Zr atoms shall have slightly different positions to accommodate the linkers. Accordingly, the calculated structures exhibit changes in the Zr–Zr and Zr–O distances (Table S12†), with the 25% sample having the shortest Zr–Zr distances and then increasing for 50% and 75% AzDC below pristine values, which aligns with trends in lattice strain (Fig. S95†). Moreover, new vibration bands in the carboxylate region that do not correspond to free linkers are observable in the FT-IR profiles of the MTV MOFs (Fig. 4c), supporting the presence of dangling linkers to accommodate the lattice mismatching.
As a complementary analysis to FTIR, Raman scattering analyses were performed to obtain additional information about the vibrational modes of the samples (see ESI S.3.10†).
Unfortunately, the bands assigned to cis and trans azobenzene in the literature44 are masked by the O–C–O symmetric and asymmetric stretches and other MOF-related bands.45 Although it does not allow for determination, the interconversion from 0% to 100% AzDC can be observed across the MTV series (Fig. S96–101†).
Tuning the photoelectronic properties
Despite containing less photoactive linker incorporated, the MTV samples displayed a brighter colour than the pristine sample (100% AzDC), suggesting that the MTV configuration leads to differences in electronic configuration, which is elusive in the literature.21 Solid-UV-vis spectra showed in agreement, that the absorbance (Fig. 5a), and hence reflectance (Fig. 5b), of the MTV MOFs is not intermediate from their pristine counterparts, but shifted compared to 100% AzDC (see ESI S.11†).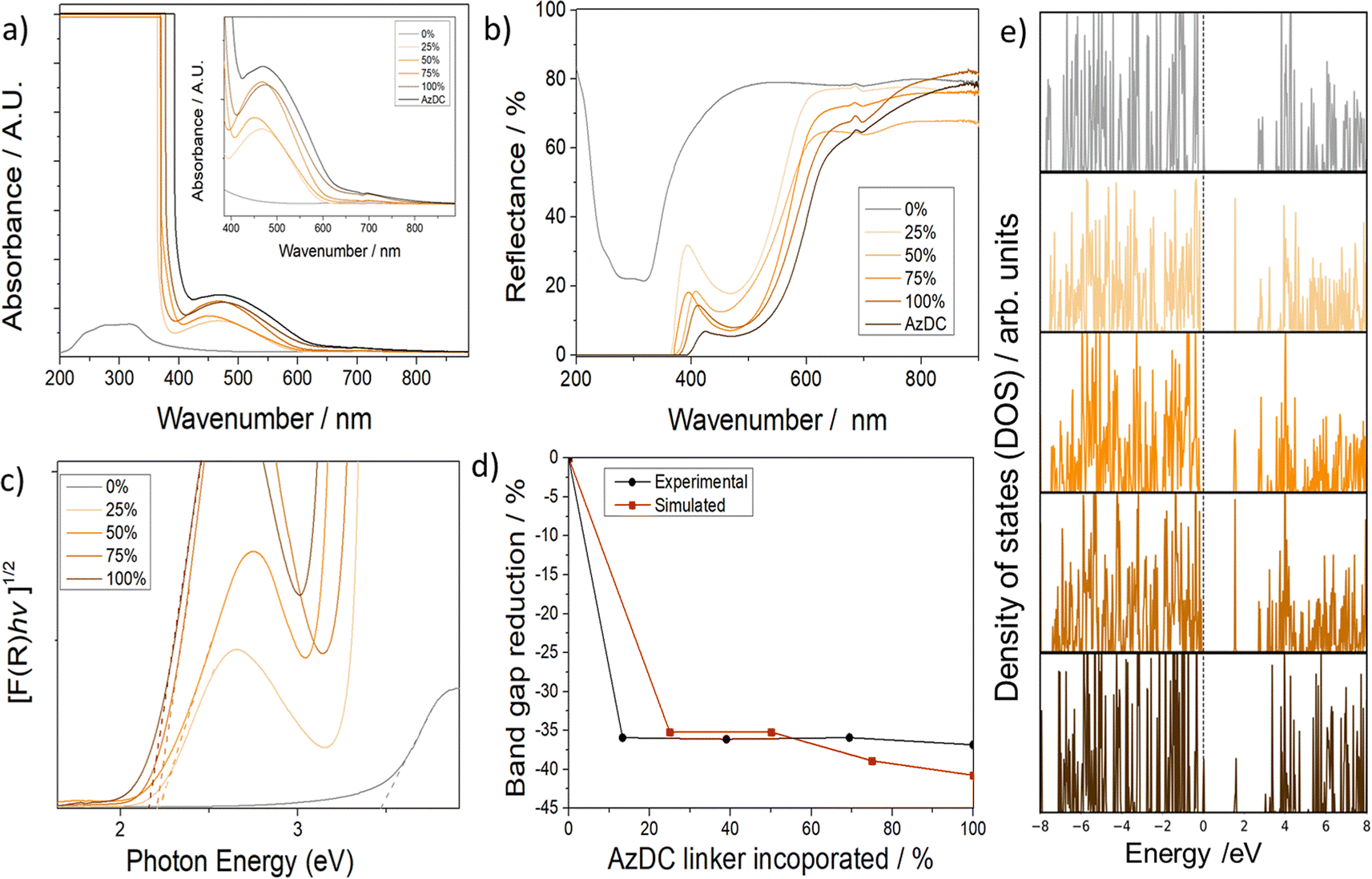 | ||
| Fig. 5 (a) Solid-state UV-vis profiles of the samples. (b) Reflectance profiles of the samples. (c) Tauc plots of the samples. (d) Comparison of the percentage of decrease in experimental and calculated band gap values of the MTV MOFs upon the introduction of AzDC. (e) Total projected density of states of the MTV series. | ||
The solid-state UV-vis shows that the π–π* transition (the most intense absorption band in azobenzene, at ca. 320–340 nm) is saturated and does not allow for evaluation. The absorption band at ca. 460 nm is attributed to the n–π* transition of the trans azobenzene chromophore, arising from the excitation of a non-bonding electron on the nitrogen to the π* orbital,27,28 and displays important changes within the samples (Fig. 5a). The MTV MOFs have a minor shift in the n–π* band towards lower wavelengths, which is more pronounced for the 25% and 50% samples. This blue shift indicates more energetic transitions for the MTV samples. Interestingly, in the cis isomer, the n–π* transition is slightly shifted towards shorter wavelengths due to the reduced overlap between the p orbitals of the nitrogen atoms and the π* system of the phenyl rings due to the bending of the azobenzene moieties, which can cause a slight increase in the energy of the n orbital.27,28 Moreover, in the cis isomer, the n–π* band is more intense, and the changes in relative intensities of the n–π* and π–π* bands are typically used to explore cis–trans photoisomerization. Thus, if the ca. 450 nm band was to only correspond to trans isomers, the intensity should be related to the AzDC incorporation percentage, which is not the case. Therefore, our results suggest an unprecedentedly favourable partial cis configuration within the mismatching MTV MOFs, which shall be the result of dangling linkers.
The diffuse reflectance was used to estimate the optical bandgap of the materials (Table 1 and Fig. 5c, d). The 0% AzDC (UiO-67) sample has an estimated optical band gap of ca. 3.44 eV, while the 100% AzDC of ca. 2.17 eV. The MTV structures range from 2.20 to 2.22 eV, suitable as semiconductor materials (Fig. S102†). This proves that the mismatching MTV approach alters the electronic structure even at the lowest concentrations of the photoactive linker.
In order to support these findings, we performed systematic DFT calculations on the MTV series to analyse the electronic structure of the corresponding MOF systems and to elucidate the nature of this decrease in the band gap upon the addition of AzDC ligand (see ESI S.3.3 and S.3.11† for computational details). Although GGA-PBE is known to underestimate the band gap of solid semiconductors, it offers a good compromise between accuracy and calculation efficiency that is suitable for studying the variation of the structural and electronic properties of the MTV series, since it has been used in theoretical studies on similar systems with consistent results.46,47
Fig. 5e shows the total projected density of states (PDOS) of the MTV series, where it is observed that the reduction of the band gap is mainly due to the appearance of a state at ∼1.7 eV caused by the AzDC ligand due to the main N contribution (see ESI S.3.11b, Fig. S103,† for atomic contributions to DOS). Here, one can also observe an abrupt decrease in the band gap when introducing the minimal amount of AzDC (25%), which is close to the 100% sample, due to the presence of the new azobenzene states that narrow the band gap, as observed in the experimental measurements (Fig. S104 and S105†). The significant reduction in the band gap upon low AzDC incorporation could be beneficial from the industrial point of view, given that AzDC is more expensive than BPDC.
In fact, calculating the percentage of band gap reduction compared to the 0% sample shows comparable values for both experimental and calculated bandgaps, as represented in Fig. 5d. Moreover, the PODS suggests that the energy needed to transfer an excited electron from the linker to the cluster node is reduced upon introducing AzDC, which, together with the bandgap reduction, could suggest enhanced photocatalytic activity but deserves further analysis.
Although the cis–trans azobenzene photoisomerization is prevented when bound to metal clusters in MOFs through both carboxylates,26,32 it could take place at the surface or defect sites of the crystals, such as dangling linkers. Thus, we measured the solid-state UV-vis spectra after one hour of light irradiation in the solid state (see ESI S.3.11b and S.3.11c, Fig. S106–S108†). Although the changes were moderate, and so were for free AzDC, an increase in the intensity of the n–π* band, more significant for 25% and 50% AzDC samples (Fig. 6a), was observed for the MTV MOFs, whereas the changes were almost insignificant for the 100% AzDC pristine sample, in alignment with photoisomerization dangling molecules resulting from the multivariate linker mismatching approach, since ditopic bonding leads to constrained isomerization in MOFs.26,32 A slight reduction in the band gap, was consequently observed for the MTV MOFs (Fig. S109–S117 and Table S13†).
 | ||
| Fig. 6 (a) Solid-state UV-vis of 25% and 50% AzDC before and after light irradiation. (b) UV-vis of 50% AzDC and free AzDC dispersed in water before and after light irradiation. (c) PL spectra of the MOFs. (d) Normalised PL spectra of the MOFs. (e) Percentage of contribution of deconvoluted PL signals as a function of AzDC incorporated in molar percentage. (f) Maximum intensity of the 574 nm and 629 nm signals a function of the AzDC incorporated in molar percentage. | ||
To further evaluate the cis–trans isomerization, the UV-vis was then collected for dispersions of the MOFs in water. Free AzDC dissolved in water displays several adsorption bands, centered at ca. 194 nm, 231 nm, 330 nm, and 430 nm (Fig. S118†). Since the MOFs are dispersed in water rather than dissolved, we also measured the dispersion of AzDC in water (Fig. S119 and S120†), which showed similar bands, but less defined and slightly shifted. Upon light irradiation, dispersed AzDC showed an increase in the ca. 430 nm band (cis n–π*). Moreover, a new band at ca. 289 nm appeared, presumably due to the π–π* transition of cis azobenzene (Fig. 6b).
The UV-vis of dispersed MTV MOFs shows a similar profile to dispersed AzDC, with a wide absorption band centred at ca. 395 nm with contributions from trans and cis azobenzene species (Fig. S121–S124†). In agreement with solid-state UV-vis, the MTV MOFs exhibit a significant increase in the cis AzDC contribution after light irradiation (Fig. S125–S132†), as presented in Fig. 6b for 50% AzDC, whereas this does not occur for the pristine 100% AzDC.
To complement the analysis of the electronic structure, photoluminescence (PL) emission spectra were measured for the samples, excited at 350 nm for 2 minutes (see ESI S.3.12, Fig. S133 and S134†). PL provides insights into the impact of AzDC linker on the pristine structure, as it depends on the structural defects of the sample and the efficiency of metal–ligand charge transfer.48,49
The lower conjugation of BPDC linkers, due to the simple C–C bond between the benzene rings, may limit the efficiency of electronic transfer between the metal center and the linkers. However, the AzDC ligand is well-known for its low fluorescence quantum yield, intrinsic to photoisomerization mechanisms.32 Thus, despite UiO-67 with BPDC linker (0% AzDC) not having extremely high emission, introducing AzDC linkers to form mismatched MTV Zr6-MOFs is expected to result in decreased PL emission. This reflects the nature of the linker and the structural defects caused in the UiO-67 framework due to its inclusion.
The UiO-67 sample, comprising solely BPDC linkers, demonstrates the highest luminescence intensity, as represented in Fig. 6c, characterised by a peak emission at 472 nm (∼2.63 eV).50 The introduction of small quantities of the AzDC linker shows a notable decrease in luminescence emission, aligning with expectations. Additionally, PL analysis provides insight into the photo-induced charge separation efficiency of MOFs.51 Hence, a lower PL intensity indicates greater charge separation efficiency.52 Consequently, samples containing AzDC ligands might exhibit enhanced efficiency for photocatalytic applications due to the more effective separation of electron–hole pairs and the reduced likelihood of recombination.
Detailed analysis of the samples with AzDC linkers reveals three distinct emission peaks that do not coincide with the initial peak observed for BPDC linkers, centered around ∼410 nm (∼3.06 eV), ∼574 nm (∼2.16 eV) and ∼629 nm (∼1.97 eV). In the normalised spectra provided in Fig. 6d, it can be observed that the proportion of these peaks changes with increasing AzDC linker concentration: the higher the AzDC concentration, the lower the emission at ∼410 nm and the greater the contribution of the emission at ∼574 nm (see ESI S.12, Fig. S135–S140,† for deconvoluted spectra). The ca. 629 nm contribution percentage increases with AzDC incorporation but reaches a plateau at ca. 50% incorporation, as represented in Fig. 6d. This phenomenon is intriguing and may originate from the isomerization of the AzDC linker, given that the excitation wavelength promotes photoisomerization.53 The information on the differences in emission spectra of cis and trans azobenzene isomers is limited, possibly due to their non-radiative decay pathways resulting in low photoluminescence emission,32,53,54 together with the ease of identification of cis–trans isomers through UV-vis.
The planar structure of the trans isomer facilitates extensive delocalization of π-electrons over the entire molecule, which results in a higher energy transition (shorter wavelength), whereas the reduced conjugation in the cis form causes a less efficient overlap of the π-orbitals, leading to a lower energy transition (longer wavelength).55 Based on this information, the contribution attributed to the trans isomer (ca. 574 nm) increases with AzDC incorporation. In contrast, the cis isomer (ca. 629 nm), also present in all the samples, reaches its maximum at ca. 50% AzDC, which agrees with previous insights into their higher composition of dangling linkers within the MTV MOFs (Fig. 6d). In alignment, the maximum intensity at longer wavelengths, (ca.500–650), which is not present in 0%AzDC, follows a tendency that is not related to the percentage of AzDC introduced and varies for the ca. 574 nm and ca. 629 nm contributions (75% > 50% > 100% ∼25% and 50% > 75% > 25% > 100%), as shown in Fig. 6f.
Therefore, it may be that the linker excess conditions to synthesize nanoparticulate MOFs and the mismatched MTV Zr6-MOFs with AzDC and BPDC linkers can partially control the isomerism of AzDC linkers within the fcu framework through dangling linker promotion. This isomeric relationship is responsible for the alterations in micro- and macro-structure, which can directly impact the electronic properties of the MTV Zr6-MOFs.
Experimental
All experimental details are available in the ESI.†Conclusions
We have applied the linker mismatching strategy to form multivariate MOF nanoparticles of Zr-BPDC/AzDC, with variable lattice parameters and porosity, controllable through their linker ratios. Importantly, these nanoparticles display homogeneous sizes and are well-dispersed in water, a must for their applicability.Through this work, we show that the multivariate mixed-linker strategy tunes the electronic properties of the MTV materials beyond their pristine counterparts. Even at the lowest concentrations of photoactive AzDC linkers, the band gap is tuned from ca. 3.4 eV to ca. 2.2 eV, proven to be due to the contribution of N atoms from the AzDC ligand.
Besides, we show that the multivariate mismatching linker approach leads to significant structural distortions within the framework, which are more pronounced with lower AzDC linker additions. Thus, this approach promotes the formation of defects, including dangling AzDC linkers that enable cis–trans photoisomerization, which is constrained when the ditopic AzDC linker is fully bonded to metal clusters. In this regard, despite the lower AzDC incorporation, MTV structures show more significant PL contributions in the azobenzene region than 100% AzDC. This approach opens the doors to new photo-switching nano-particulate MOFs with variable lattice parameters and porosity, which shall be beneficial for several applications, such as photocatalysis or drug delivery among others, as this methodology could be applied to other systems, including other MOFs, metal–organic cages, covalent organic frameworks or hybrid perovskites.
Data availability
Data for this article, including full characterisation, are available at Zenodo at https://doi.org/10.5281/zenodo.14186373.Author contributions
IAL conceptualised, designed, and supervised the project, and acquired funding for it. CRM synthesised and characterised the materials. DLA performed the DFT calculations under JJB supervision. CCB performed Pawley refinements. MA collected and interpreted the PL and Raman data. IAL and CMR wrote the first draft of the manuscript with input from all authors. All the authors participated in the revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project that gave rise to these results received the support of a fellowship from ‘‘la Caixa” Foundation (LCF/BQ/PR23/11980041). This publication is part of the I+D project PID2023-148580NA-I00 financed by MCIN/AEI/10.13039/501100011033/ and FEDER ‘a way to make Europe’. IAL and CRM thank” la Caixa” Foundation and the University of Valencia. IAL and MAacknowledge MICINN for Ramón y Cajal (RYC2022-036868-I) and Juan de la Cierva (JDC2022-049934-I) fellowships. CCB thanks Leverhulme Trust Research Project Grant (RPG-2020-005). DLA and JJB acknowledge funding from the European European Union (ERC-2021-StG101042680 2D-SMARTiES) and the Generalitat Valenciana (grantCIDEXG/2023/1).Notes and references
- A. Helal, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Natl. Sci. Rev., 2017, 4, 296 CrossRef CAS.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846 CrossRef CAS PubMed.
- M. Viciano-Chumillas, X. Liu, A. Leyva-Pérez, D. Armentano, J. Ferrando-Soria and E. Pardo, Coord. Chem. Rev., 2022, 451, 214273 CrossRef CAS.
- Y. Wang, H. Lv, E. S. Grape, C. A. Gaggioli, A. Tayal, A. Dharanipragada, T. Willhammar, A. K. Inge, X. Zou, B. Liu and Z. Huang, J. Am. Chem. Soc., 2021, 143, 6333 CrossRef CAS PubMed.
- A. Valverde, D. Payno, L. Lezama, J. M. Laza, S. Wuttke and R. F. de Luis, Adv. Sustainable Syst., 2022, 6, 2200024 CrossRef CAS.
- A. Ma, H. Cong and H. Deng, Green Energy Environ., 2022, 7, 575 CrossRef CAS.
- I. A. Lázaro, C. J. R. Wells and R. S. Forgan, Angew. Chem., Int. Ed., 2020, 59, 5211 CrossRef PubMed.
- I. A. Lázaro, J. Mater. Chem. A, 2022, 10, 10466 RSC.
- N. Hanikel, X. Pei, S. Chheda, H. Lyu, W. Jeong, J. Sauer, L. Gagliardi and O. M. Yaghi, Science, 2021, 374, 454 CrossRef CAS PubMed.
- Z. Dong, Y. Sun, J. Chu, X. Zhang and H. Deng, J. Am. Chem. Soc., 2017, 139, 14209 CrossRef CAS PubMed.
- S. Yuan, W. Lu, Y.-P. Chen, Q. Zhang, T.-F. Liu, D. Feng, X. Wang, J. Qin and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 3177 CrossRef CAS PubMed.
- Y. Hu, X. Zhang, R. S. H. Khoo, C. Fiankor, X. Zhang and J. Zhang, J. Am. Chem. Soc., 2023, 145, 13929 CrossRef CAS PubMed.
- S. Yuan, J.-S. Qin, L. Zou, Y.-P. Chen, X. Wang, Q. Zhang and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6636 CrossRef CAS PubMed.
- Z. Zhu, Z. Wang, Q.-H. Li, Z. Ma, F. Wang and J. Zhang, Dalton Trans., 2023, 52, 4309 RSC.
- X. Li, H. Bian, W. Huang, B. Yan, X. Wang and B. Zhu, Coord. Chem. Rev., 2022, 470, 214714 CrossRef CAS.
- T. T. M. Nguyen, H. M. Le, Y. Kawazoe and H. L. Nguyen, Mater. Chem. Front., 2018, 2, 2063 RSC.
- L. Feng, S. Yuan, J.-L. Li, K.-Y. Wang, G. S. Day, P. Zhang, Y. Wang and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 1719 CrossRef CAS PubMed.
- K. Hemmer, S. M. Kronawitter, N. Grover, B. Twamley, M. Cokoja, R. A. Fischer, G. Kieslich and M. O. Senge, Inorg. Chem., 2024, 63, 2122 CrossRef CAS PubMed.
- H. Kim, D. Kim, D. Moon, Y. N. Choi, S. B. Baek and M. S. Lah, Chem. Sci., 2019, 10, 5801 RSC.
- V. Guillerm and D. Maspoch, J. Am. Chem. Soc., 2019, 141, 16517 CrossRef CAS PubMed.
- S. Yuan, L. Huang, Z. Huang, D. Sun, J.-S. Qin, L. Feng, J. Li, X. Zou, T. Cagin and H.-C. Zhou, J. Am. Chem. Soc., 2020, 142, 4732 CrossRef CAS PubMed.
- G. C. Thaggard, K. C. Park, J. Lim, B. K. P. M. Kankanamalage, J. Haimerl, G. R. Wilson, M. K. McBride, K. L. Forrester, E. R. Adelson, V. S. Arnold, S. T. Wetthasinghe, V. A. Rassolov, M. D. Smith, D. Sosnin, I. Aprahamian, M. Karmakar, S. K. Bag, A. Thakur, M. Zhang, B. Z. Tang, J. A. Castaño, M. N. Chaur, M. M. Lerch, R. A. Fischer, J. Aizenberg, R. Herges, J.-M. Lehn and N. B. Shustova, Nat. Commun., 2023, 14, 7556 CrossRef CAS PubMed.
- C. R. Martin, K. C. Park, R. E. Corkill, P. Kittikhunnatham, G. A. Leith, A. Mathur, S. L. Abiodun, A. B. Greytak and N. B. Shustova, Faraday Discuss., 2021, 231, 266 RSC.
- A. B. Grommet, L. M. Lee and R. Klajn, Acc. Chem. Res., 2020, 53, 2600 CrossRef CAS PubMed.
- F. Bigdeli, C. T. Lollar, A. Morsali and H. Zhou, Angew. Chem., Int. Ed., 2020, 59, 4652 CrossRef CAS PubMed.
- A. Modrow, D. Zargarani, R. Herges and N. Stock, Dalton Trans., 2011, 40, 4217 RSC.
- J. Griffiths, Chem. Soc. Rev., 1972, 1, 481 RSC.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2011, 41, 1809 RSC.
- Z. Wang, K. Müller, M. Valášek, S. Grosjean, S. Bräse, C. Wöll, M. Mayor and L. Heinke, J. Phys. Chem. C, 2018, 122, 19044 CrossRef CAS.
- H. Cheng, S. Zhang, J. Qi, X. Liang and J. Yoon, Adv. Mater., 2021, 33, e2007290 CrossRef PubMed.
- I. Aprahamian, ACS Cent. Sci., 2020, 6, 347 CrossRef CAS PubMed.
- A. Sussardi, R. J. Marshall, S. A. Moggach, A. C. Jones and R. S. Forgan, Chem.–Eur. J., 2021, 27, 14871 CrossRef CAS PubMed.
- C. R. Marshall, S. A. Staudhammer and C. K. Brozek, Chem. Sci., 2019, 10, 9396 RSC.
- J. Marreiros, C. Caratelli, J. Hajek, A. Krajnc, G. Fleury, B. Bueken, D. E. D. Vos, G. Mali, M. B. J. Roeffaers, V. V. Speybroeck and R. Ameloot, Chem. Mater., 2019, 31, 1359 CrossRef CAS.
- A. R. Denton and N. W. Ashcroft, Phys. Rev. A:At., Mol., Opt. Phys., 1991, 43, 3161 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062 RSC.
- I. A. Lázaro, Eur. J. Inorg. Chem., 2020, 2020, 428 CrossRef.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643 CrossRef CAS PubMed.
- A. Schaate, S. Dühnen, G. Platz, S. Lilienthal, A. M. Schneider and P. Behrens, Eur. J. Inorg. Chem., 2012, 2012, 790 CrossRef CAS.
- I. A. Lázaro and R. S. Forgan, Coord. Chem. Rev., 2019, 380, 230 CrossRef.
- W. Morris, S. Wang, D. Cho, E. Auyeung, P. Li, O. K. Farha and C. A. Mirkin, ACS Appl. Mater. Interfaces, 2017, 9, 33413 CrossRef CAS PubMed.
- K. L. Patrocinio, J. R. Santos, L. I. Granone, M. A. Ponce, M. S. Churio, L. K. Ribeiro, M. D. Teodoro, R. Llusar, J. Andrés, E. Longo and M. Assis, Dalton Trans., 2023, 52, 14982 RSC.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700 CrossRef CAS.
- C. M. Stuart, R. R. Frontiera and R. A. Mathies, J. Phys. Chem. A, 2007, 111, 12072 CrossRef CAS PubMed.
- K. I. Hadjiivanov, D. A. Panayotov, M. Y. Mihaylov, E. Z. Ivanova, K. K. Chakarova, S. M. Andonova and N. L. Drenchev, Chem. Rev., 2021, 121, 1286 CrossRef CAS PubMed.
- A. D. Vos, K. Hendrickx, P. V. D. Voort, V. V. Speybroeck and K. Lejaeghere, Chem. Mater., 2017, 29, 3006 CrossRef PubMed.
- K. Hendrickx, D. E. P. Vanpoucke, K. Leus, K. Lejaeghere, A. V. Y.-D. Deyne, V. V. Speybroeck, P. V. D. Voort and K. Hemelsoet, Inorg. Chem., 2015, 54, 10701 CrossRef CAS PubMed.
- H. Zhu, J. Huang, Q. Zhou, Z. Lv, C. Li and G. Hu, J. Lumin., 2019, 208, 67 CrossRef CAS.
- M. Li, Y. Liu, F. Li, C. Shen, Y. V. Kaneti, Y. Yamauchi, B. Yuliarto, B. Chen and C.-C. Wang, Environ. Sci. Technol., 2021, 55, 13209 CAS.
- R. Verma, G. Dhingra, G. Singh, J. Singh, N. Dureja and A. K. Malik, J. Fluoresc., 2023, 1, 1631 Search PubMed.
- K. M. Batoo, E. Ali, S. A. Hussein, S. Chandra, A. S. Abdulwahid, S. H. Kareem, M. F. Ijaz, A. A. Omran, M. K. Abid and A. Alawadi, J. Mol. Struct., 2024, 1312, 138492 CrossRef CAS.
- A. E. B. Lima, M. Assis, A. L. S. Resende, H. L. S. Santos, L. H. Mascaro, E. Longo, R. S. Santos, L. S. Cavalcante and G. E. Luz, J. Solid State Electrochem., 2022, 26, 997 CrossRef CAS.
- T. Fujino, S. Y. Arzhantsev and T. Tahara, J. Phys. Chem. A, 2001, 105, 8123 CrossRef CAS.
- M. Yamauchi, M. Okaji, N. Aratani, H. Yamada and S. Masuo, ChemPhotoChem, 2022, 6, e202100301 CrossRef CAS.
- H. Bisle and H. Rau, Chem. Phys. Lett., 1975, 31, 264 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and full characterisation of materials. See DOI: https://doi.org/10.1039/d4ra07366e |
| This journal is © The Royal Society of Chemistry 2024 |