
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormulating abiraterone acetate-HPMCAS-based amorphous solid dispersions: insights into in vitro and biorelevant dissolution assessments and pharmacokinetic evaluations†
Manisha Choudharia,
Shantanu Damleb,
Ranendra Narayan Sahac,
Sunil Kumar Dubeyd and
Gautam Singhvi *a
*a
aDepartment of Pharmacy, Birla Institute of Technology and Science, Pilani Vidya Vihar, Pilani Campus, Rajasthan 333031, India. E-mail: gautam.singhvi@pilani.bits-pilani.ac.in
bColorcon Asia Pvt. Ltd, Verna Industrial Estate, Verna, Goa 403722, India
cBiophore Group of Companies, Hyderabad, Telangana, India
dR&D Healthcare Division Emami Ltd, 13, BT Road, Belgharia, Kolkata 700056, India
First published on 5th December 2024
Abstract
Abiraterone acetate (ABTA) is used as a primary treatment for metastatic castration-resistant prostate cancer. Its low aqueous solubility results in inadequate dissolution and poor oral bioavailability (<10%), necessitating the consumption of large doses of ABTA (1000 mg per day) for desired efficacy. The aim of this study is to enhance the solubility, dissolution, and bioavailability of ABTA through amorphous solid dispersions (SDs). ABTA-SD was prepared via a solvent granulation method with different grades of hydroxypropyl methylcellulose acetate succinate (HPMCAS 716 and 912). The theoretical solubility parameter between ABTA and HPMCAS was below 7 MPa1/2, indicating miscibility between the drug and the polymer according to the Hansen solubility parameter. HPMCAS showed a remarkable recrystallization inhibition of up to 180 min compared to the free drug (10 min), maintaining the soluble drug in supersaturation state and exhibiting the “spring and parachute” phenomenon. ABTA-SD exhibited a higher solubility (1.16-fold to 52-fold) in different media than free ABTA. The results of DSC, PXRD, ATR-FTIR, and FE-SEM indicated that the crystallinity of ABTA was completely transformed to an amorphous form and maintained in the SD formulation. In vitro and bio-relevant dissolution behavior of ABTA was studied in various dissolution media, indicating the higher dissolution of ABTA-SD than that of free ABTA. The pharmacokinetic study conducted in Wistar rats revealed that Cmax and AUC0–t of the optimized ABTA-SD formulation were significantly enhanced by 1.92-fold and 2.87-fold, respectively, compared to those of free ABTA.
Introduction
Abiraterone acetate (ABTA) is used for the treatment of metastatic castration-resistant prostate cancer.1,2 ABTA acts as a prodrug that undergoes in vivo transformation into abiraterone (ABT), an inhibitor of androgen biosynthesis.1,3 It has poor water solubility and low permeability. ABTA is a weakly basic drug with a pKa of 5.19 and exhibits pH-dependent solubility.4,5 During gastric transition, solubilized ABTA from the gastric fluid to the intestine region leads to precipitation in the intestinal fluid because of its pH-dependent solubility.6 The precipitation diminishes the solubility, impedes the dissolution of API, and hinders drug absorption across the intestinal membrane.6–8 The marketed product of ABTA is orally administrated at a high daily dosage of 1000 mg, necessitating four 250 mg tablets, owing to its limited water solubility.9 The commercial drug product needs to be taken at least 2 h after a meal to avoid overexposure, as taking it with food can increase the AUC up to 10-fold and Cmax up to 17-fold, which may lead to toxicity risk and inconsistent dosing due to increased absorption.9–12 This may cause inter-subject and intra-subject pharmacokinetic variability for the patient and promote non-compliance (with food or without food), ultimately affecting the therapeutic benefits.13Therefore, it would be highly beneficial to develop a formulation that can enhance the solubility and bioavailability of ABTA. Additionally, that would allow the elimination of the food impact and possibly dose reduction because such a formulation would adhere to higher criteria for patient convenience and safety. Formulation researchers have employed various strategies, such as particle size reduction, prodrugs, amorphous solid dispersions (SD), and self-emulsifying systems, to improve the solubility, dissolution, and absorption of BCS Class IV drug.14–20 However, enhancing solubility or dissolution alone often falls short in significantly improving bioavailability.21
A few approaches have been used to enhance ABTA's solubility and bioavailability, exploring both bottom-up (nano-amorphous ABTA) and top-down (wet-milled ABTA) methods to reduce the particle size. Nano-amorphous ABTA, produced via continuous flow precipitation, achieved particle size d50 and d90 values of 186 nm and 254 nm, respectively, showing much better dissolution than wet-milled crystalline ABTA (d50: 497 nm, d90: 1050 nm). This nano-form demonstrated an 8.3-fold and 2.9-fold increase in solubility in fasted-state and fed-state simulated intestinal fluids, due to its smaller particle size and amorphous structure.7 Apart from that, SoluMatrix fine particle technology, developed by iCeutica Inc., improves the dissolution and bioavailability of poorly water-soluble drugs such as ABTA by reducing the particle size (200–800 nm).
Among the numerous ways to increase the solubility by amorphization, SD is the effective strategy in which API is homogenously distributed in a polymeric matrix,22 though it carries a risk of recrystallization. Several strategies are employed in SD formulations to counteract the risk of re-crystallization. Consequently, choosing an appropriate polymer for SDs becomes a crucial step, as it not only enhances the API solubility and the dissolution of SDs but also hinders undesirable API precipitation.23–25 Previous studies have revealed that cellulosic polymers play a vital role in maintaining supersaturation in SDs by inhibiting crystallization and keeping the drug molecules in a dissolved state for a prolonged period.8,26 Furthermore, cellulosic polymer chains interact with the API molecules, forming hydrogen bonds, reducing their mobility, and preventing them from precipitating out of the solution.26 The utilization of a high-energy form of the API and enhanced release properties, when combined with a hydrophilic/amphiphilic cellulosic polymer, are possibilities for the improved in vivo performance of SDs.21
Hydroxypropyl methyl cellulose acetate succinate (HPMCAS), the cellulose succinate derivative chosen for this study, has been utilized as a carrier in the SD. It enhances solubility and improves the bioavailability of the low aqueous-soluble API.27 HPMCAS is amphiphilic, available in 3 distinct grades, i.e., HPMCAS 716, HPMCAS 912, and HPMCAS 126.23 Additionally, HPMCAS aids in sustaining the drug's supersaturation by inhibiting precipitation, thereby enhancing dissolution and extent in aqueous environments.23,28
This present study aimed to enhance the solubility and dissolution of ABTA using amphiphilic polymer HPMCAS. For that, the Hansen solubility parameter and Flory–Huggins drug–polymer interaction approach were employed to forecast the miscibility between ABTA and the selected polymer, HPMCAS. SDs of ABTA were developed by a solvent granulation method employing different grades of HPMCAS.29 Moreover, the ability of HPMCAS to impede crystallization was investigated via the precipitation inhibition study. The free ABTA and its SDs were studied for saturation solubility and in vitro release testing in different dissolution media. The physicochemical characterizations of SDs were evaluated by differential scanning calorimetry (DSC), attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy, powder X-ray diffraction (PXRD), thermogravimetric analysis (TGA), and scanning electron microscopy (SEM). Stability studies of the developed formulations were also conducted. The pharmacokinetic study was performed in Wistar rats to study the bioavailability of prepared ABTA-SDs.
Materials and methods
Materials
ABTA with 98% purity (CAS no. 154229-18-2) was generously gifted by Biophore India Pharmaceuticals Pvt. Ltd, India. HPMCAS 716, HPMCAS 912, and Startab were supplied as gift samples from Colorcon Asia Pvt. Ltd, India. Soya lecithin and sodium taurocholate were procured from the SRL. Mannitol and citric acid were purchased from Hi-Media. Lactose monohydrate and magnesium stearate were procured from CDH. Analytical grade HPLC solvents such as acetone, methanol, dichloromethane, and acetonitrile were procured from Merck.Biorelevant media composition
Drug–polymer miscibility predictions
Drug–polymer miscibility is a critical consideration in the preparation of SD. The Flory–Huggins interaction parameter (χ) was employed to assess drug–polymer miscibility and solubility by utilizing key thermodynamic parameters, such as the free energy of mixing. Hansen solubility parameter predictions were utilized to forecast drug–polymer miscibility for various drugs and polymers, which was determined via the van Krevelen and Hoftyzer group contribution method. This approach was utilized to evaluate χ.30–33Hansen solubility parameter approach
The van Krevelen and Hoftyzer group contribution method34 was used to determine the Hansen solubility parameter (δ) of drug (ABTA) and polymer (HPMCAS) using their chemical structure, as shown in Fig. 1.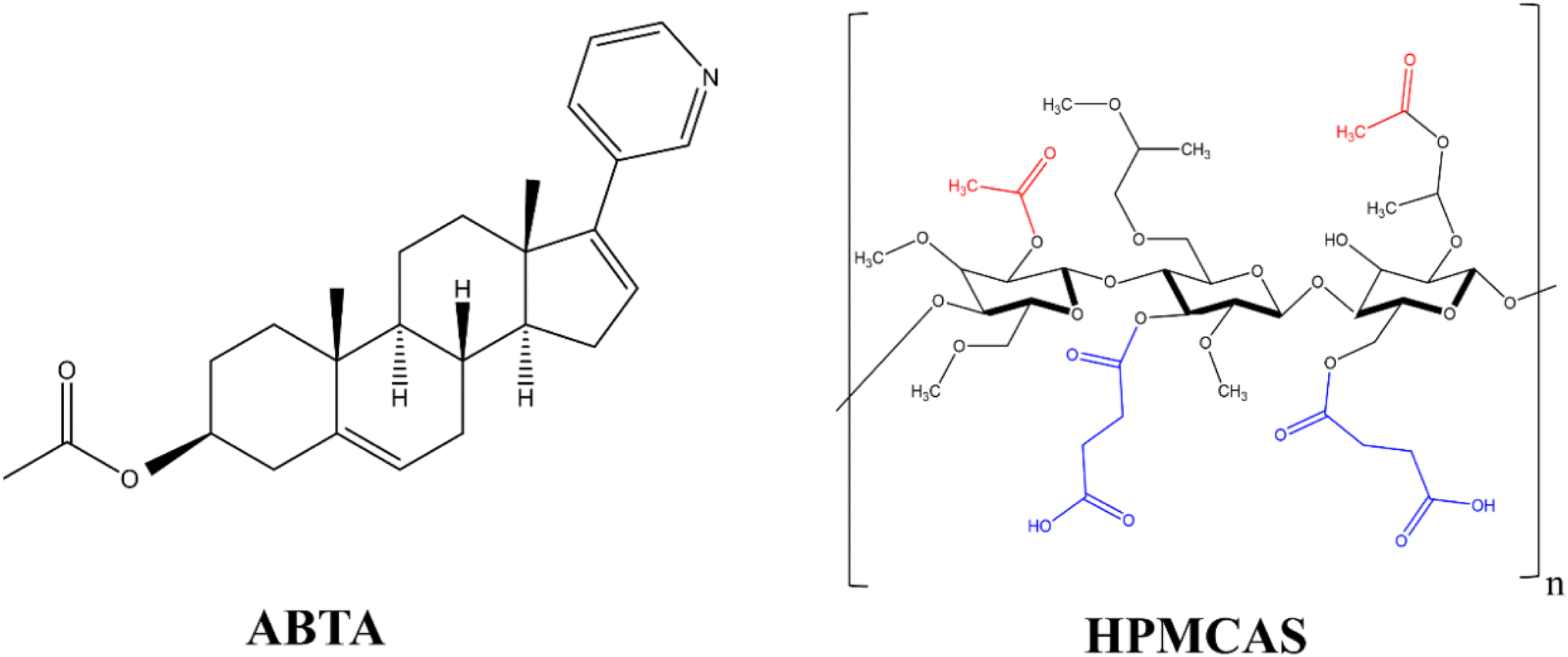 | ||
| Fig. 1 Chemical structure of ABTA and HPMCAS. | ||
The three primary Hansen parameters were calculated for each molecule: (i) dispersion forces amongst the molecule (δd), (ii) dipolar intermolecular forces amongst the molecule (δp), and (iii) hydrogen bonding energy amongst the molecule (δh). The overall solubility parameter (δt), typically expressed in MPa0.5, was determined by aggregating all three Hansen parameters as depicted in eqn (1):
| δt2 = δd2 + δp2 + δh2 | (1) |
These forces can be determined using eqn (2). In contrast, Fdi, Fpi, and Fh are the group contributions to the dispersion forces, dipolar intermolecular forces, and hydrogen bonding energy. V is the group contribution to the molar volume respectively.
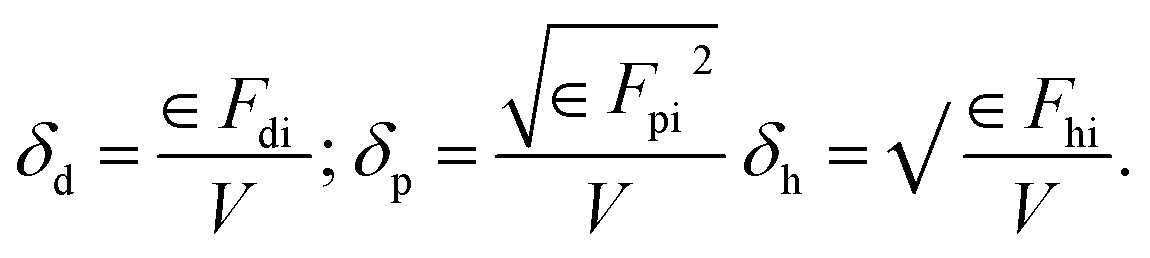 | (2) |
Flory–Huggins drug–polymer interaction approach
Flory–Huggins parameter (χ) for drug–polymer interactions was determined by the difference between the solubility parameters of drugs and polymers, calculated using eqn (3). In this equation, Vsite represents the hypothetical lattice volume, while T denotes the temperature, R stands for the gas constant, and δdrug and δpolymer signify the solubility parameters of the drug and polymer, respectively.35
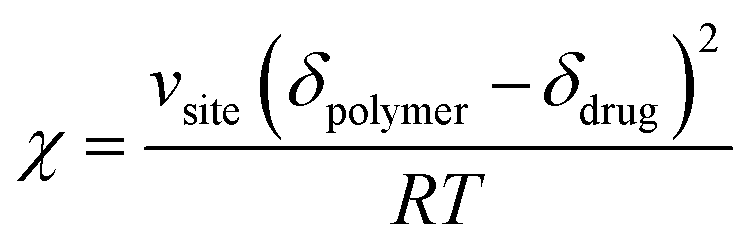 | (3) |
Preparation and optimization of ABTA-SD formulation
ABTA-SD formulations were prepared by the solvent granulation method. The components of the ABTA-SD formulations are shown in Table 1. Solvent granulation involved dissolving the polymer (HPMCAS 716 or 912) in an organic solvent system, i.e., acetone, followed by the addition of ABTA. This solution was then gradually added to lactose monohydrate and thoroughly mixed, allowing the solvent to evaporate at room temperature (25 °C). The resulting SD was collected, sieved, and stored in desiccators for further analysis. Additionally, physical mixtures (PMs) of ABTA were formulated via geometric mixing of different ratios of the drug and polymer in the mortar and pestle with less force, confirming that no grinding was employed during the process. The formulated SD was collected, sieved, and stored in desiccators until further analysis.29| Formulation | P1 | P2 | P3 | P4 | P5 | P6 | F1 | F2 | F3 | F4 | F5 | F6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a P1–P6: physical mixture of ABTA with HPMCAS 716 and 912; F1–F6: solid dispersion of ABTA using HPMCAS 716 and 912. | ||||||||||||
| ABTA (mg) | 166.66 | 125 | 100 | 166.66 | 125 | 100 | 166.66 | 125 | 100 | 166.66 | 125 | 100 |
| HPMCAS716 (mg) | 166.66 | 250 | 300 | — | — | — | 166.66 | 250 | 300 | — | — | — |
| HPMCAS912 (mg) | — | — | — | 166.66 | 250 | 300 | — | — | — | 166.66 | 250 | 300 |
| Lactose monohydrate | 166.66 | 125 | 100 | 166.66 | 125 | 100 | 166.66 | 125 | 100 | 166.66 | 125 | 100 |
| Acetone (mL) | — | — | — | — | — | — | 5 | 5 | 5 | 5 | 5 | 5 |
| Total (mg) | 500 | 500 | 500 | 500 | 500 | 500 | 500 | 500 | 500 | 500 | 500 | 500 |
Saturation solubility studies
To conduct the saturation solubility studies, excessive quantities of free ABTA, PM of HPMCAS 716 and 912 (P1–P6), and SD of HPMCAS 716 or 912 prepared from solvent granulation methods (F1–F6) were introduced into vials containing distilled water, pH 6.8, pH 1.2, fasted-state simulated intestinal fluid (FaSSIF, pH 6.5) and fed-state simulated intestinal fluid (FeSSIF, pH 5.0). These vials were placed on an orbital shaker and continuously stirred for 6 h at a controlled temperature of 37 ± 0.5 °C, allowing equilibrium to be reached. After 6 h, 2 mL aliquots of the samples were extracted. A 0.22 μm nylon syringe filter was used to filter the supernatant and subsequently subjected to analysis using RP-HPLC.36 The analysis was performed using a Hypersil Gold C18 column (50 × 4.6 mm × 5 μm) using a mobile phase of acetonitrile and 0.01 mM potassium phosphate buffer (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20, v/v%), at pH 6.5. The flow rate was 1.0 mL min−1, with a 20 μL injection volume, detection at 254 nm, and the column temperature maintained at 40 °C.
:20, v/v%), at pH 6.5. The flow rate was 1.0 mL min−1, with a 20 μL injection volume, detection at 254 nm, and the column temperature maintained at 40 °C.
Precipitation inhibition assay
For the precipitation inhibition study, supersaturation was attained by incorporating 2 mL of ABTA solution (6 mg mL−1 in methanol) to 100 mL of FaSSIF, pH 6.5 media. While incorporating the ABTA solution, the magnetic stirrer continued to stir at 200 rpm at 37 °C ± 0.5 °C. To explore the impact of HPMCAS 716 or 912 polymers, they were pre-dissolved in the precipitation media (FaSSIF) to evaluate the effect. In contrast, precipitation media without polymer was employed as a control.8,37 The ABTA concentration was measured at predetermined time intervals, i.e., 5, 10, 15, 30, 45, 60, 90, 120, and 180 min in the presence and absence of HPMCAS 716 and 912 and quantified using RP-HPLC. For ABTA precipitation inhibition, HPMCAS 716 or 912 was investigated at a weight ratio of 1:2 (ABTA/HPMCAS) in a binary mixture. This ratio was chosen based on the maximum solubility of ABTA.
Dynamic light scattering
The particle size of the precipitate was assessed through dynamic light scattering (DLS) using a Malvern Zeta Sizer (Nano ZS, Malvern Instruments, UK) at a temperature of 25 °C as part of the precipitation inhibition study. Samples were removed from the precipitation inhibition study after 5, 10, 15, 30, 45, 60, 120, and 180 min.37Characterization of solid dispersions
Determination of the ABTA content
The concentration of ABTA in the SD was quantified by RP-HPLC (Shimadzu) analysis using a Hypersil Gold C18 column (50 × 4.6 mm × 5 μm) with a mobile phase consisting of acetonitrile and dibasic potassium phosphate (0.01 mM) in a ratio of 80:20 (%v/v). The mobile phase pH was set to 6.5, at a flow rate of 1.0 mL min−1 and an injection volume of 20 μL. Detection was conducted at a wavelength of 254 nm, and the column temperature was maintained at 40 °C.41
In vitro dissolution of the ABTA-SD formulation
In vitro dissolution studies of ABTA were performed in various dissolution media, such as pH 1.2, pH 6.8, and pH 4.5, to evaluate the rate and extent of the drug from the dissolution of SD, and this was compared with the pure drug dissolution. Additionally, the dissolution behavior pattern in these various media was investigated. Specifically, for the pH 4.5 dissolution media, 0.25% sodium lauryl sulphate was incorporated, as per its specification in the official pharmacopeia. The USP II apparatus was employed for the dissolution testing. In our investigations, USP II dissolution apparatus with a media volume of 300 mL and a speed of 150 rpm was utilized to examine drug dissolution from both the SD and free drug. In brief, 10 mg of ABTA and ABTA-SD (F2 and F5) (equivalent to 10 mg of ABTA) sample was added to 250 mL media of pH 1.2, pH 6.8, and pH 4.5 with 0.25% SLS maintained at 37 °C and stirred at 150 rpm. Samples were collected at predetermined intervals (5, 10, 15, 30, 45, 60, and 120 min), and an equal volume of fresh media was replenished. The collected samples were filtered using a 0.45 μm syringe filter and analyzed by RP-HPLC.41Bio-relevant dissolution of the ABTA-SD formulation
Biorelevant dissolution media are used to replicate the physiological conditions of the gastrointestinal tract, thus aiding in the prediction of a drug's behavior and dissolution within the human body. These studies play a pivotal role in forecasting in vivo performance and can offer valuable insights into how a formulation behaves in the gastrointestinal tract. However, it is essential to understand that biorelevant dissolution studies offer a closer simulation of in vivo conditions than traditional dissolution tests. Moreover, bio-relevant dissolution studies can be critical when a drug shows a food effect. The food effects refer to the influence of the food on the absorption and pharmacokinetics of a drug after oral administration.Given that ABTA exhibits a food effect, it is imperative to investigate its dissolution in fasted-state simulated intestinal fluid (FaSSIF, pH 6.5) and fed-state simulated intestinal fluid (FeSSIF, pH 5.0) media.42 In brief, 10 mg of ABTA and ABTA-SD (F2 and F5) (equivalent to 10 mg of ABTA) sample was added to 250 mL of FaSSIF (pH 6.5) and FeSSIF (pH 5.0) media maintained at 37 °C and stirred at 150 rpm. Samples were collected at scheduled intervals (5, 10, 15, 30, 45, 60, and 120 min), followed by the addition of an equal volume of fresh media. The collected samples were filtered through a 0.45 μm syringe filter before RP-HPLC analysis.43
Mathematical analysis of in vitro and bio-relevant dissolution data
The dissolution profiles of both free ABTA and the optimized formulation ABTA-SD (F2 and F5) were assessed using several parameters: percent dissolution efficiency (DE) and mean dissolution time (MDT). Additionally, the DD Solver software conducted the mathematical modeling of the dissolution profiles. Various models were employed to determine which one best represented the dissolution data, utilizing R2 (correlation coefficient), MSC (model selection criteria), and AIC (Akaike information criterion), as criteria for selection.44Stability studies
Optimized ABTA-SD formulations were stored in sealed amber glass bottles for three months (90 days) in stability chambers under accelerated conditions (40 ± 2 °C and 75 ± 5% RH). The stability study was conducted by examining the physical characteristics (color), DSC, and PXRD both initially and after three months under accelerated conditions.45In vivo pharmacokinetic study
All experimental protocols were approved by the Institutional Animal Ethics Committee (IAEC) before the start of the work (protocol no. IAEC/RES/32/01). Healthy Wistar rats were selected for pharmacokinetic studies of free ABTA and ABTA-SD formulations. Rats with an average weight of 200–250 gm (6–8 weeks old) were obtained from Central Animal Facility (CAF), BITS, Pilani, Pilani Campus, Rajasthan, India. Animals were kept in a standard plastic cage maintained under controlled conditions (23 ± 2 °C, 60 ± 5% RH, and 12 h darkness-12 h light cycle) and provided standard laboratory pellet food with water ad libitum. Rats were acclimated to the study environment for at least five days before the study began. Throughout the study, all animals were monitored for general conditions, unusual signs, toxicity, and mortality.Pharmacokinetic studies were conducted on 3 groups (n = 3) that received different oral formulations under fasting conditions. The rats were divided into three groups and given oral suspensions as follows: group 1 was administered the free drug ABTA; group 2 was administered ABTA-SD (with HPMCAS 716: F2); group 3 was administered ABTA-SD with HPMCAS 912 (F5). Prior to the start of the experiments, animals were given free access to water and allowed to fast overnight for 12 h. The dose was administered orally with the suspension of ABTA, F2, and F5, by gavage as a single dose of 200 mg kg−1 of the drug. Blood samples (0.5 mL) were collected from the retro-orbital venous plexus at specific time intervals (0.5 h, 1 h, 2 h, 4 h, 6 h, 8 h, and 12 h) post-administration, respectively. Collected blood samples were transferred into a labeled microcentrifuge tube containing 20 μL of 10% EDTA as an anticoagulant and mixed well. The plasma samples were separated by centrifugation at 8000 rpm for 10 min at 4 °C. The supernatant was obtained and stored at −20 °C till further analysis. The RP-HPLC bioanalytical method was used to quantify the concentration of ABTA. This analysis was carried out using a Hypersil gold C18 HPLC column (50 mm × 4.6 mm, particle size = 5 μm), the mobile phase mixture comprised acetonitrile and 0.01 mM K2HPO4 buffer (pH 3.0) in a ratio of 34:66 (v/v%), 1 mL min−1 flow rate, 50 μL injection volume, and 30 °C column temperature.
The obtained chromatographic peak was calculated for plasma drug concentration. To understand the plasma profile, plasma drug concentration vs. time was plotted. The maximum observed plasma concentration (Cmax) was recorded directly from the individual plasma concentration versus time profiles. Various pharmacokinetic parameters such as area under the curve (AUC0–t), Tmax, and MRT were estimated by the non-compartmental analysis (NCA) method using Phoenix WinNolin Certera™ (Pharsight, U.S.A; Version: 8.0). The relative bioavailability (Frel) of SD formulations was calculated using AUC with respect to the same for free ABTA suspension administered.46
Results and discussion
Drug-solubility parameter approach
When formulating the amorphous-based solid dispersion, information on the solubility and miscibility of the drug within the polymeric matrix is required. A critical aspect of formulating stabilized SDs involves achieving a single homogeneous phase wherein the drug and polymer are thermodynamically compatible. The thermodynamic miscibility of ABTA with the specified polymers (HPMCAS 716 and 912) was investigated by determining Hansen solubility parameters, employing the van Krevelen and Hoftyzer group's contribution method. Due to its relative simplicity, this method of calculating solubility parameters and applying them to SDs remains widely utilized. In this method, when the solubility parameter between a drug and a polymer differs by less than 7 MPa1/2, a system is said to be miscible, or when both compounds have similar Δδ values. However, a system is immiscible if Δδ is more than 10 MPa1/2.30 The estimation of the solubility parameter of HPMCAS 716 and 912 and ABTA using the Hansen group contribution theory is shown in Tables 1S and 2S.†As indicated in Table 2, the Δδ value between ABTA and HPMCAS was below 7 MPa1/2, suggesting favorable miscibility. Using these solubility parameter values for the drug–polymer system, the Flory–Huggins interaction parameter (χ) was calculated using eqn (3). This equation illustrates that if the solubility characteristics of the drug and polymer are similar, the interaction parameters will be close to zero. A low value of χ indicates a smaller enthalpy of mixing and more negative free energy, facilitating mixing; thus, a value closer to zero signifies a more vital interaction between the drug and the polymer.32 However, the interaction parameter value for the ABTA–HPMCAS system, as shown in Table 2, is close to zero, indicating miscibility between them.
| Drug/excipient | Hansen solubility parameter (MPa0.5) | Δδ (MPa1/2) (δHPMCAS − δABTA) | Flory–Huggins drug–polymer interaction parameter (χ) |
|---|---|---|---|
| ABTA | 19.38 | — | |
| HPMCAS 716 | 23.67 | 4.29 | 2.00 |
| HPMCAS 912 | 23.22 | 3.85 | 1.61 |
Saturation solubility studies
The solubility results demonstrate a significant improvement in ABTA solubility when formulated as SDs compared to both the free ABTA and the PM across all tested media. The PM (P1–P6) of ABTA with the same carriers in the same proportion exhibited a slight enhancement in solubility in various media compared to free ABTA. The SDs and PMs were prepared using different grades of HPMCAS polymers, specifically 716 and 912, at various ABTA ratios. As shown in Fig. 2A, the solubility of free ABTA in water was approximately 1.012 μg mL−1. Among all the formulations (F1–F6), the SDs prepared with HPMCAS 716 and 912 combined with lactose in a 1/2/1 ratio (F2 and F5) exhibited the highest solubility in water better than that of both the PMs and free ABTA. The SD, i.e., F2 and F5 formulation in distilled water, exhibited 26-fold and 21-fold higher solubility, respectively, than free ABTA, as shown in Fig. 2A. The solubility of ABTA decreased after specific ratios of HPMCAS (1/3/1); this might be due to the increased viscosity of the solution, which slows down the diffusion of water molecules into the drug–polymer matrix, hindering the drug's dissolution. Additionally, at higher HPMCAS ratios, the solution may reach a saturation point where it can no longer effectively solubilize additional drug, further limiting ABTA solubility.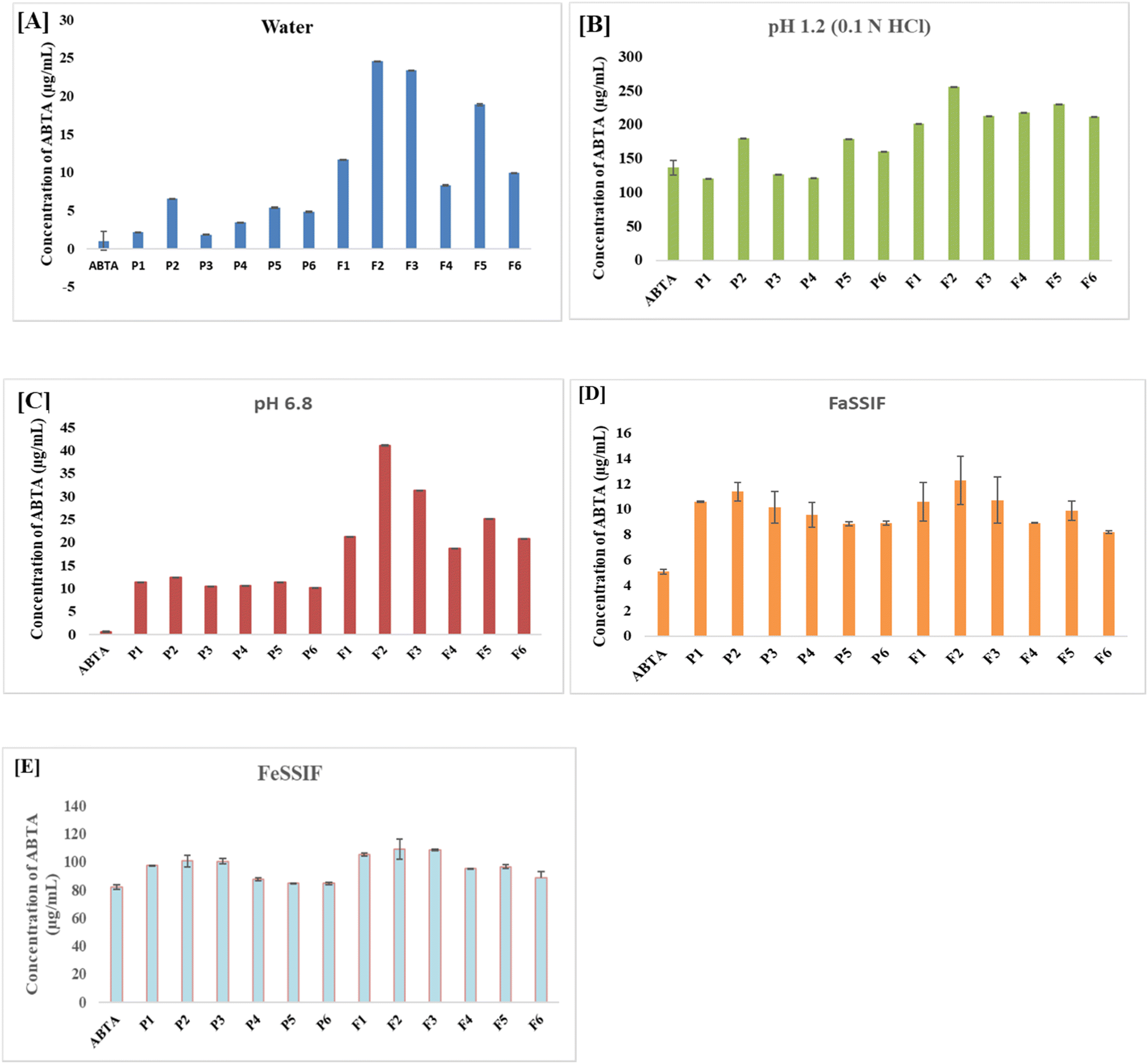 | ||
| Fig. 2 Solubility profiles of all ABTA-SD formulations and physical mixture (PM) in different media: [A] water; [B] pH 1.2; [C] pH 6.8; [D] FaSSIF and [E] FeSSIF. (P1–P6: physical mixture of ABTA with HPMCAS 716 and 912; F1–F6: solid dispersion of ABTA using HPMCAS 716 and 912). | ||
A similar trend was observed in all media, i.e., pH 6.8 and pH 1.2, FaSSIF, and FeSSIF. The solubility of SD compared to free drug in pH 1.2 (Fig. 2B), pH 6.8 (Fig. 2C), FaSSIF (Fig. 2C), and FeSSIF (Fig. 2D) were found to be 26-fold, 52-fold, 1.72-fold, 2.53-fold, 1.33-fold respectively for F2 formulation and F5 showed 21-fold, 38-fold, 1.6-fold, 2.35-fold, and 1.16 respectively higher solubility.
The SD with HPMCAS 716 (F2) showed a higher solubility than that of HPMCAS 912, indicating the substantial impact of acetate and succinate substitution in the polymer. HPMCAS 716, a cellulose succinate derivative containing 14–18% succinate and 5–9% acetate group, imparts greater hydrophilicity. In contrast, HPMCAS 912 polymer with 10–14% succinate group and 7–11% acetate group exhibits hydrophobicity. An increase in the acetate group corresponds to hydrophobicity, while an increase in the succinate group is correlated with hydrophilicity.23 The enhanced solubility of ABTA in the HPMCAS 716 polymer can be attributed to its higher hydrophilic nature than that of HPMCAS 912.47 The solubility profiles of all ABTA-SD formulations and PM are shown in Fig. 2.
Precipitation inhibition effect
To assess the effectiveness of cellulose succinate derivatives in sustaining the supersaturation state of ABTA, the soluble concentration of ABTA was determined in the solutions of HPMCAS 716 and 912 within a pre-dissolved FaSSIF (pH 6.5) buffer. As depicted in Fig. 3A, the concentration of ABTA initially declined and continued to decrease over time. However, the HPMCAS 716 and 912 polymers demonstrated a notable recrystallization inhibition effect, as the concentration of ABTA in the media remained elevated throughout the 180 min period. In this study, ABTA was chosen with either HPMCAS 716 or 912 in a weight ratio of 1/2. This ratio was selected based on the maximum solubility of ABTA observed in different media.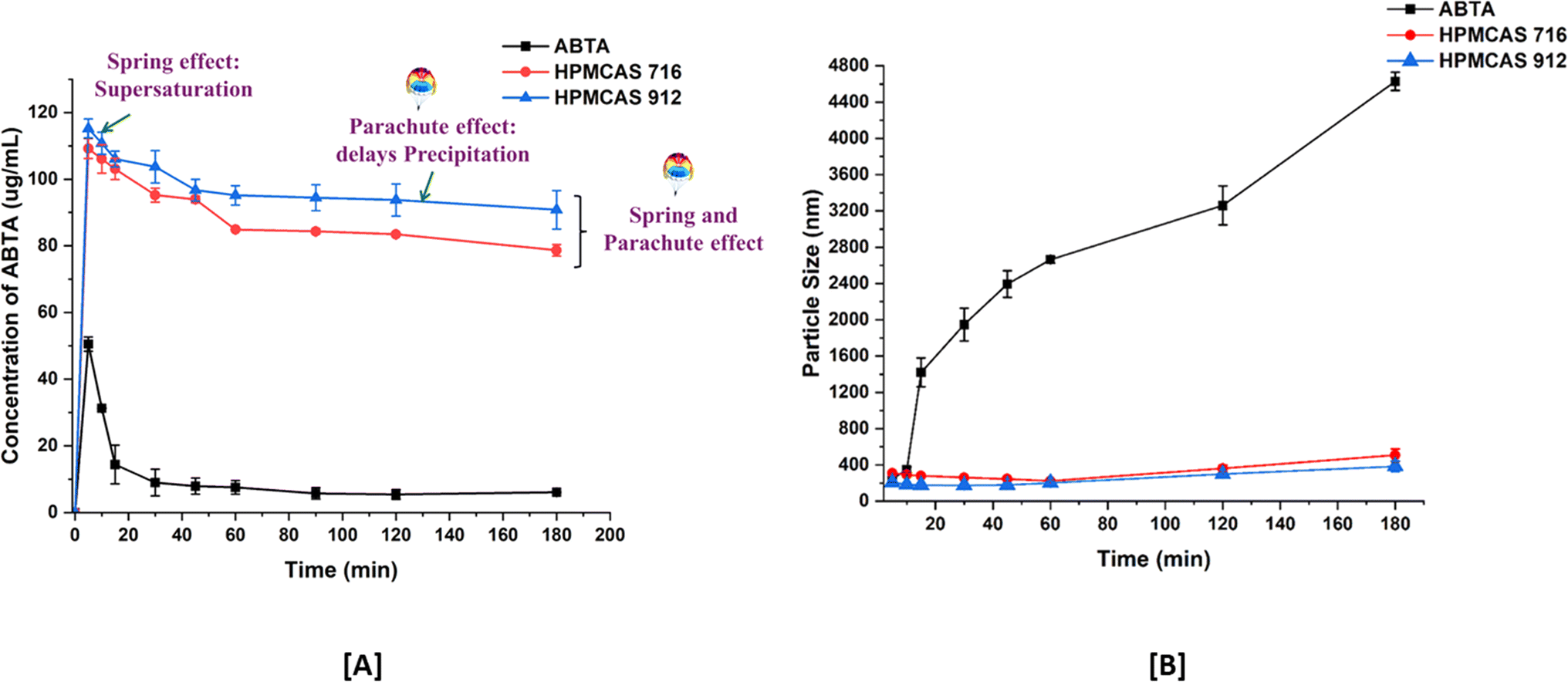 | ||
| Fig. 3 Precipitation inhibition assay [A]; and particle size profiles [B] of supersaturated ABTA in FaSSIF, pH 6.5, in the presence of pre-dissolved HPMCAS 716 and 912 (mean ± S.D, n = 3). | ||
Upon inducing supersaturation of ABTA in the absence of any polymer, an instantaneous and complete precipitation was observed; this phenomenon is attributed to the “spring” effect. The presence of pre-dissolved HPMCAS 716 and 912 polymers provided a pronounced and stable supersaturation effect. This study exhibited that the HPMCAS 716 and 912 polymers acted as “parachute” to impede crystal nucleation and growth. Therefore, HPMCAS is characterized as an amphiphilic molecule encompassing both hydrophilic and hydrophobic groups, demonstrating a recrystallization inhibition effect.48,49 Furthermore, it was elaborated that the adsorption of hydrophobic groups from these polymers onto the crystal surface of hydrophobic APIs may impede the crystal growth of the drug. Consequently, polymers with hydrophobic characteristics, such as HPMCAS, exhibited a strong affinity with hydrophobic drugs, leading to their higher recrystallization inhibition effect. However, the HPMCAS 912 polymer, possessing a higher hydrophobic nature and having a higher substitution of acetate group, showed greater effectiveness in inhibiting precipitation than HPMCAS 716.47,50
The aforementioned conclusion was confirmed through pre-dissolved HPMCAS 716 and 912 particle size monitoring. The size of precipitates formed during induced precipitation was assessed using dynamic light scattering (DLS). As depicted in Fig. 3A, in the absence of pre-dissolved polymer, ABTA exhibited recrystallization within the first 10 min, evident from a notable increase in particle size up to 4500 nm. Conversely, in the presence of pre-dissolved HPMCAS, 716 and 912 maintained a particle size of around 150–400 nm (Fig. 3B) without substantial growth throughout the precipitation inhibition study.40
Characterization of the developed formulations
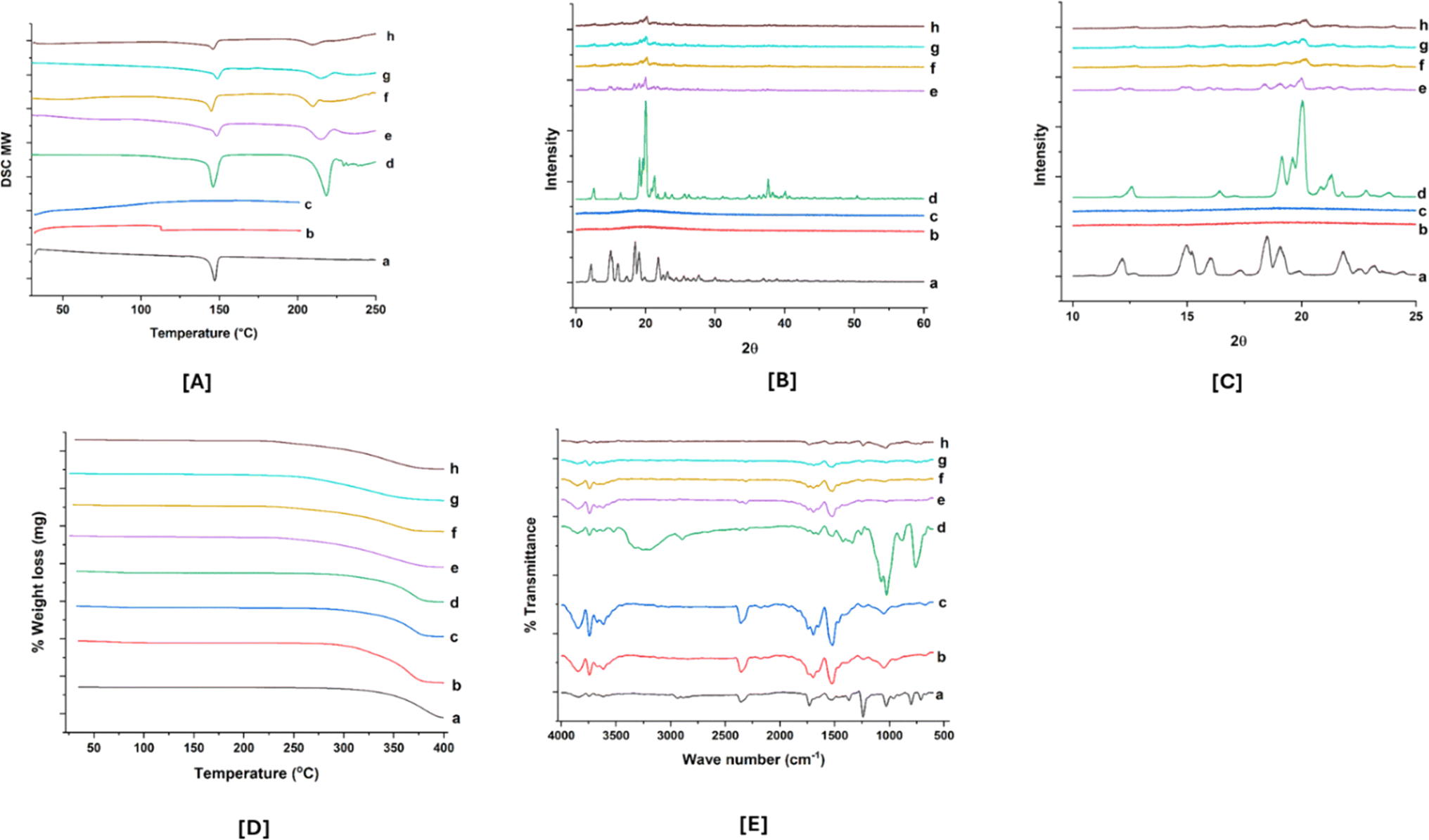 | ||
| Fig. 4 Characterizations of ABTA and ABTA-SD: [A] DSC thermogram; [B] PXRD spectrum; [C] PXRD spectrum (magnified view); [D] TGA spectra; and [E] FTIR spectra. [a] ABTA; [b] HPMCAS 716; [c] HPMCAS 912; [d] lactose monohydrate; [e] P2 (physical mixture); [f] P5 (physical mixture); [g] F2 (solid dispersion with HPMCAS 716); and [h] F5 (solid dispersion with HPMCAS 912). | ||
However, the F2 and F5 formulations showed that the endothermic peak corresponded to lactose monohydrate, not ABTA. Additionally, ABTA-SD prepared with HPMCAS 716 and 912, excluding lactose monohydrate, exhibited the absence of the ABTA endothermic peak in the thermogram, as illustrated in Fig. 1S.† This confirms that the endothermic peak observed for the F2 and F5 formulations originated from lactose monohydrate. Generally, reducing the crystallinity of a drug enhances its dissolution and solubility since less energy is needed to disrupt the crystal lattice.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching (pyridine ring vibration), CO stretching, and aromatic C–H stretching vibrations, respectively. The IR spectra of HPMCAS 716, which resembles HPMCAS 912, showed characteristics peaks at 3606 cm−1, 2358 cm−1, 1745 cm−1, 1646 cm−1, and 1455 cm−1, which are ascribed to the O–H stretching, C–O stretching, CO stretching, CC stretching, and CH3 stretching vibrations respectively.40 Meanwhile, the IR spectra of lactose monohydrate displayed a peak at 3280 cm−1 assigned to O–H stretching (broad peak) and doublet peak at 1070 cm−1 and 1030 cm−1, respectively, attributed to C–C stretching. PM (P2 and P5) of ABTA/HPMCAS 716/912/lactose monohydrate presented all the leading bands of the three compounds. Furthermore, the decrease in intensity and the disappearance of the ABTA peak in the IR spectrum of ABTA-SD suggest molecular interactions, potentially involving hydrogen bonding between ABTA and HPMCAS.
N stretching (pyridine ring vibration), CO stretching, and aromatic C–H stretching vibrations, respectively. The IR spectra of HPMCAS 716, which resembles HPMCAS 912, showed characteristics peaks at 3606 cm−1, 2358 cm−1, 1745 cm−1, 1646 cm−1, and 1455 cm−1, which are ascribed to the O–H stretching, C–O stretching, CO stretching, CC stretching, and CH3 stretching vibrations respectively.40 Meanwhile, the IR spectra of lactose monohydrate displayed a peak at 3280 cm−1 assigned to O–H stretching (broad peak) and doublet peak at 1070 cm−1 and 1030 cm−1, respectively, attributed to C–C stretching. PM (P2 and P5) of ABTA/HPMCAS 716/912/lactose monohydrate presented all the leading bands of the three compounds. Furthermore, the decrease in intensity and the disappearance of the ABTA peak in the IR spectrum of ABTA-SD suggest molecular interactions, potentially involving hydrogen bonding between ABTA and HPMCAS. | ||
| Fig. 5 FE-SEM images of [a] ABTA; [b] F2; and [c] F5. | ||
In vitro dissolution
In vitro dissolution experiments were conducted to examine the improved drug-dissolution patterns of the SD formulation compared to crystalline ABTA. Assessing the dissolution behavior of the SD formulation provides insights into their potential performance in vivo. The in vitro dissolution profiles of crystalline ABTA and ABTA-SD were evaluated across different pH conditions, namely pH 1.2, pH 6.8, and pH 4.5 with 0.25% SLS, which are shown in Fig. 6. Crystalline ABTA showed an inadequate dissolution with only 1.14% in pH 6.8 and 56.41% in pH 1.2 after 120 min. Furthermore, according to the monograph, the in vitro dissolution profile of ABTA was specified at pH 4.5 with 0.25% SLS; however, only 58.39% of ABTA was dissolved after 120 min. These findings suggest potential factors such as poor wettability and particle agglomeration contributing to this poor dissolution behavior, as evidenced by the observed floating of the drug powder on the media's surface. Therefore, improving the dissolution behavior of ABTA is imperative to facilitate rapid and high absorption. Fig. 6 depicts that ABTA-SDs have considerably increased the dissolution of ABTA compared to the crystalline ABTA in all the buffer media. In pH 1.2 dissolution media, F2 and F5 SD formulations demonstrated 89.54% and 84.88% drug release within 120 min, suggesting that drug dissolution increased by 1.58 and 1.50 times compared to crystalline ABTA, as shown in Fig. 6A. In pH 6.8 dissolution media, the dissolution of ABTA from F2 and F5 SD formulations was 10.22% and 6.33%, respectively, as shown in Fig. 6B, suggesting that drug dissolution increased by 8.97 and 5.61 times compared to its crystalline form.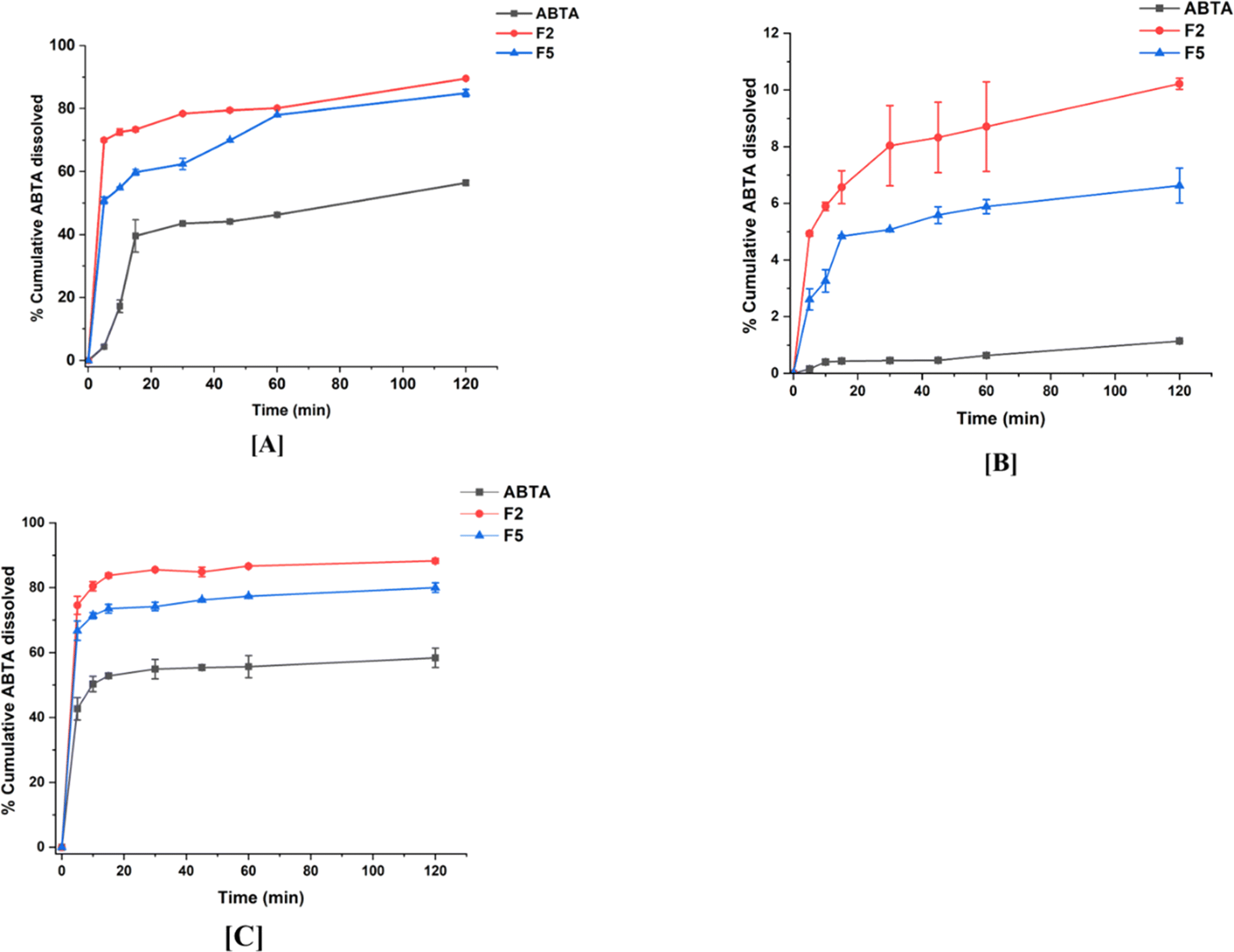 | ||
| Fig. 6 In vitro dissolution profile of ABTA and its SD formulations (F2 and F5) in different buffer media: [A] pH 1.2; [B] pH 6.8; and [C] pH 4.5. | ||
A similar trend was observed when the dissolution was performed at pH 4.5 with 0.25% SLS buffer; rapid dissolution of the drug from the SD formulation occurred, reaching an initial plateau within 5 min, as shown in Fig. 6C. The F2 formulation exhibited superior drug dissolution compared to the F5 formulation, attributed to the variance in the HPMCAS polymer grades. Higher dissolution of ABTA was noted with HPMCAS 716 compared to HPMCAS 912, highlighting the significant influence of acetate and succinate substitution. The degree of succinate substitution in HPMCAS directly correlates with dissolution; thus, higher succinate substitution yields increased dissolution.47 HPMCAS 716, with 14–18% succinate substitution, demonstrated enhanced dissolution relative to HPMCAS 912, which contains 10–14% succinate substitution.
Biorelevant dissolution studies
Bio-relevant dissolution studies can be critical when a drug shows a food effect. The food effects refer to the influence of the food on the absorption and pharmacokinetics of a drug after oral administration. Given that ABTA exhibits a food effect, it is imperative to investigate its dissolution under different food conditions. Here, biorelevant dissolution profiles of the crystalline ABTA and ABTA-SD were evaluated in FaSSIF (pH 6.5) and FeSSIF (pH 5.0), as shown in Fig. 7A and B. In the case of FaSSIF dissolution media, SD formulations F2 and F5 revealed the increase in drug release by 4.13 and 3.07 times compared to the crystalline ABTA. Furthermore, in FeSSIF dissolution media, drug release was increased by 1.68 and 1.58 times from F2 and F5 compared to the crystalline ABTA respectively. However, due to the succinyl substitution, the F2 formulation shows higher drug dissolution in both the media FaSSIF and FeSSIF than that of the F5 formulation.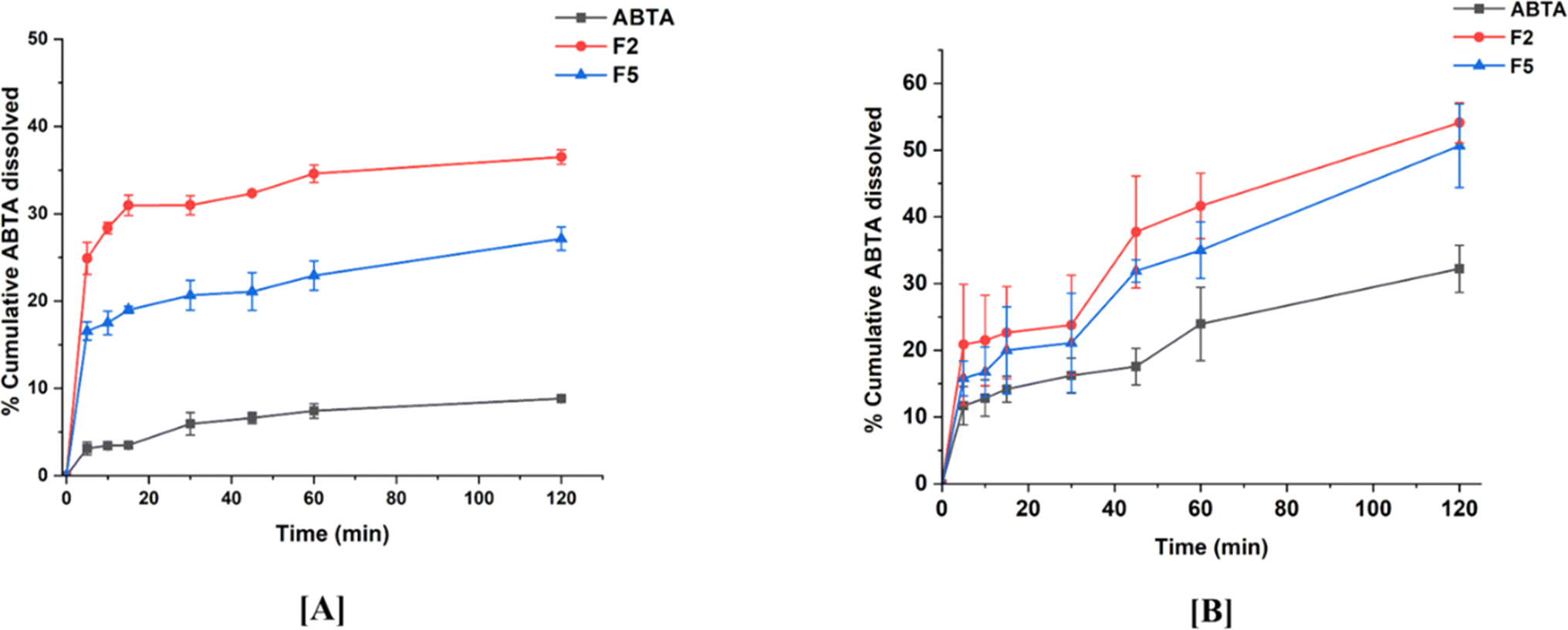 | ||
| Fig. 7 In vitro dissolution profile of ABTA and SD formulations [F2 and F5] in different buffer media: [A] FaSSIF (pH 6.5) and [B] FeSSIF (pH 5.0). | ||
Mathematical analysis of in vitro and bio-relevant dissolution data
The dissolution parameters such as R2 adjusted, kKP, n, DE, MDT, AIC, and MSC were calculated using the Microsoft Excel add-in DDSolver for both ABTA and the optimized formulation ABTA-SD (F2 and F5) across various dissolution media. The results are brief in Tables 3 and 3S.† It was observed that the dissolution models for the free ABTA and optimized ABTA-SD formulations (F2 and F5) demonstrated a better fit with the Korsmeyer–Peppas (KP) models in pH 1.2, pH 4.5, pH 6.8, FaSSIF, and FeSSIF respectively, as detailed in Table 3. Furthermore, in these dissolution media, the DE for the optimized ABTA-SD formulation (F2 and F5) was higher than that of pure ABTA. In contrast, MDT for the optimized formulation of ABTA-SD (F2 and F5) had a faster dissolution than free ABTA, as illustrated in Table 3.| Formulation | Model | R2 adjusted | kKP | n | MDT | DE |
|---|---|---|---|---|---|---|
| Dissolution media | pH 1.2 | |||||
| ABTA | KP | 0.8284 | 9.72 | 0.384 | 27 | 0.437 |
| F2 | KP | 0.9961 | 60.77 | 0.075 | 13.85 | 0.792 |
| F5 | KP | 0.9839 | 36.74 | 0.173 | 19.42 | 0.711 |
| Dissolution media | pH 4.5 with 0.25% SLS | |||||
| ABTA | KP | 0.9963 | 46.96 | 0.07 | 7.91 | 0.599 |
| F2 | KP | 0.9966 | 73.57 | 0.04 | 5.96 | 0.860 |
| F5 | KP | 0.9988 | 64.30 | 0.05 | 7.73 | 0.768 |
|
||||||
| Dissolution media | pH 6.8 | |||||
| ABTA | KP | 0.8979 | 0.075 | 0.570 | 50.98 | 0.007 |
| F2 | KP | 0.9672 | 3.622 | 0.218 | 22.31 | 0.083 |
| F5 | KP | 0.9661 | 2.062 | 0.254 | 20.87 | 0.055 |
|
||||||
| Dissolution media | FaSSIF (pH 6.5) | |||||
| ABTA | KP | 0.9549 | 1.962 | 0.330 | 22.17 | 0.072 |
| F2 | KP | 0.9935 | 21.82 | 0.109 | 12.66 | 0.326 |
| F5 | KP | 0.9907 | 12.26 | 0.157 | 21.97 | 0.222 |
|
||||||
| Dissolution media | FeSSIF (pH 5.0) | |||||
| ABTA | KP | 0.8903 | 5.059 | 0.392 | 29.95 | 0.242 |
| F2 | KP | 0.9764 | 5.247 | 0.488 | 25.02 | 0.402 |
| F5 | KP | 0.9648 | 13.693 | 0.308 | 28.45 | 0.341 |
Stability studies of the developed formulations
Physical stability is a significant challenge in the development of SD. Ensuring good miscibility between the drug and the polymer, as well as enhancing drug–polymer interactions, is crucial for improving SD stability. After 90 days of stability studies, selected ABTA-SD formulations (F2 and F5) were analyzed using physical characteristics, drug content, DSC, and PXRD, with results compared to the initial results shown in Table 4. The optimized formulation showed no change in color over the 90 days compared to the initial results. The DSC thermograms and PXRD patterns at 0 days and 90 days under accelerated conditions are shown in Fig. 8A and B. These stability studies confirmed that ABTA did not recrystallize and the ABTA-SD (F2 and F5) formulations remained in an amorphous form for the entire 90 days period under accelerated conditions, as illustrated in Fig. 8.| Formulation | F2 | F5 | ||
| Time | 0th day | 90th day | 0th day | 90th day |
| Temperature | RT | 40 °C/75% RH | RT | 40 °C/75% RH |
| Drug content | 98.89 ± 0.112 | 98.01 ± 0.245 | 97.45 ± 0.321 | 97.11 ± 0.398 |
| Color | White color and intact throughout the study period | |||
| DSC | No endothermic peak was observed of ABTA, which remained in an amorphous form | |||
| PXRD | No diffraction peak pattern was observed in ABTA, which remained in an amorphous form | |||
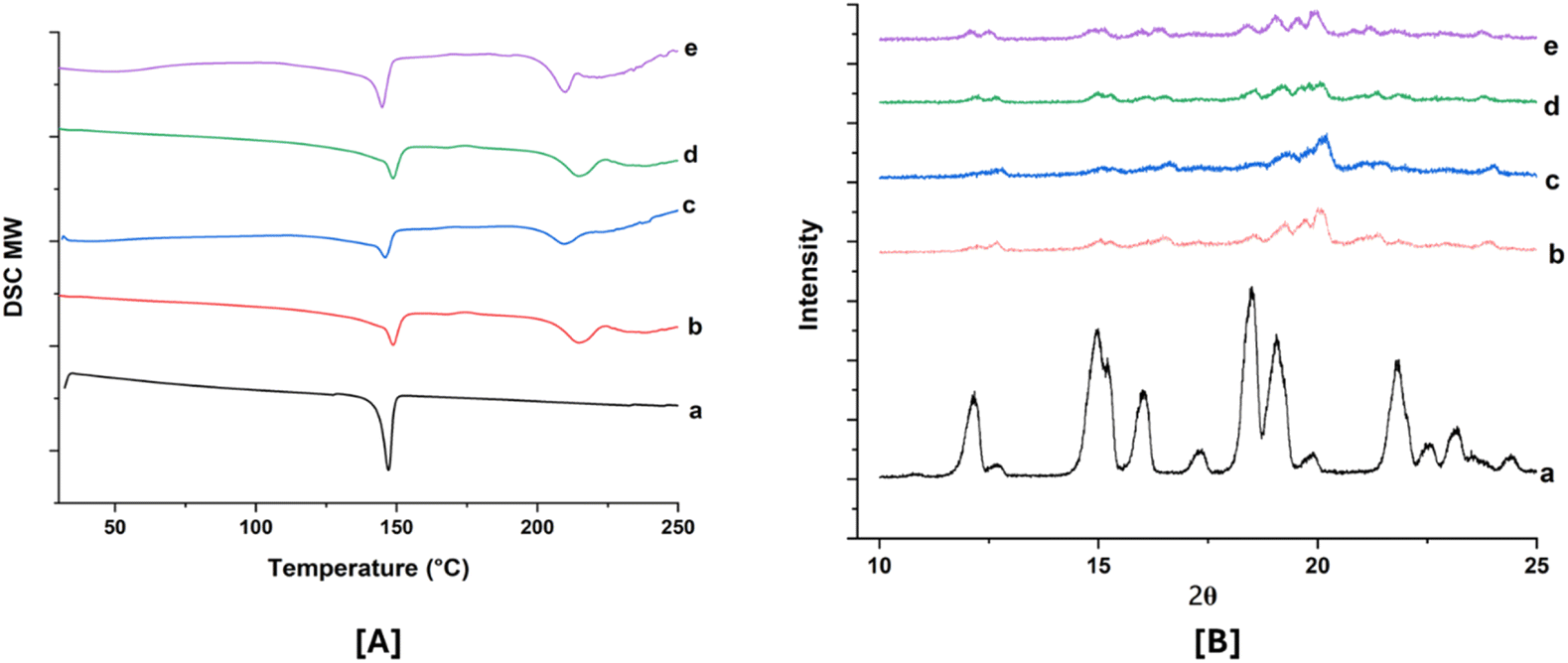 | ||
| Fig. 8 DSC thermogram [A] and PXRD patterns (magnified view) [B]: (a) ABTA; (b) F2 (0 days); (c) F5 (0 days); (d) F2 (90 days); and (e) F5 (90 days). | ||
In vivo pharmacokinetic study
ABTA, as a prodrug, undergoes rapid metabolism to the active metabolite ABT upon oral administration. The pharmacokinetic profiles of both pure ABTA and ABTA-SD (Fig. 2S†) were studied in Wistar rats, with the pertinent pharmacokinetic parameters detailed in accompanying Table 5. The results indicated that ABTA-SD, i.e., F2 and F5 formulations, was absorbed rapidly in vivo. Here, free ABTA showed low Cmax and AUC0–4h values, which were 243.27 ± 24.40 ng mL−1 and 1494.08 ± 191.40 ng mL−1 h−1 due to the reduced exposure attributed to the larger crystalline particles of pure ABTA, which undergo slow dissolution and consequently exhibit limited absorption. Comparatively, the Cmax values of ABTA in the F2 and F5 batches were 466.76 ± 14.75 ng mL−1 and 306.32 ± 6.22 ng mL−1 respectively, exhibiting 1.92-fold and 1.26-fold increase compared to free ABTA, whereas the AUC0–12h values of ABTA in the F2 batch and AUC0–8h of ABTA in the F5 batch were 4299.1 ± 66.7 ng mL−1 h−1 and 1910.4 ± 12.97 ng mL−1 h−1, respectively, increased by 2.87-fold and 1.28-fold, respectively, when compared to those of free ABTA.| Parameters | Unit | Free ABTA | F2 | F5 |
|---|---|---|---|---|
| Tmax | h | 3.33 ± 0.67 | 6.67 ± 0.67 | 2.7 ± 0.67 |
| Cmax | ng mL−1 | 243.27 ± 24.40 | 466.76 ± 14.75 | 306.30 ± 6.22 |
| AUC0–h | ng mL−1 h−1 | 1494.08 ± 191.40 | 4299.10 ± 66.70 | 1910.40 ± 12.97 |
| AUC0–∞ | ng mL−1 h−1 | 3913.84 ± 1205.40 | 6508.17 ± 1140.90 | 3707.90 ± 547.65 |
| Vd/F | L kg−1 | 1356 ± 62.22 | 264.41 ± 43.80 | 531.22 ± 17.25 |
| CL/F | L h−1 kg−1 | 56.14 ± 24.04 | 32.88 ± 5.29 | 56.75 ± 9.60 |
| Ke | h−1 | 0.11 ± 0.074 | 0.14 ± 0.04 | 0.11 ± 0.06 |
| MRT | h | 4.00 ± 0.12 | 5.82 ± 0.22 | 4.00 ± 0.11 |
| Frel | — | 1.00 | 2.87 | 1.28 |
The mean residence time (MRT) for all the SD formulations was found to be higher than that of free ABTA, suggesting that more drug was absorbed, and the drug remained in the body for a longer period. This extended residence time can improve the therapeutic effectiveness and patient compliance, and potentially reduce the frequency of dosing. Moreover, the bioavailability of ABTA-SD in the F2 and F5 batches increased by 2.87-fold and 1.28-fold compared to those of free ABTA. These improved exposures demonstrate the positive influences of micronized ABTA, the transformation of a crystalline state to an amorphous state, and the capability of HPMCAS to prevent precipitation, thereby maintaining the supersaturated state of ABTA.
The pharmacokinetic profile of ABTA-SD showed delayed Cmax when compared to the unformulated ABTA due to the pH-responsive nature of HPMCAS. Due to the tendency of API to release more when a pH shift is there, this HPMCAS may exhibit delayed dissolution or swelling under certain pH conditions, which in turn delays the release and absorption of the drug. When comparing the F2 and F5 ABTA-SD formulations, the F2 batch showed a higher Cmax value than that of the F5 batch. This is because of the different substitution patterns of the succinate and acetate groups in HPMCAS 716 and HPMCAS 912.
Conclusions
The formulation of SDs has emerged as a promising approach to enhancing the pharmaceutical properties of poorly water-soluble active pharmaceutical ingredients. A stable SD of ABTA was effectively developed using a HPMCAS polymer. Given its favorable dissolution characteristics, the formulation containing ABTA/HPMCAS/lactose monohydrate in a ratio of 1/2/1 is anticipated to serve as a novel formulation for oral absorption. The Hansen solubility parameters suggest that the solubility parameter (Δδ) between ABTA and HPMCAS was below 7 MPa1/2, suggesting good miscibility amongst them. HPMCAS is one of the best precipitation inhibitors for maintaining the supersaturation of ABTA, demonstrating the spring and parachute phenomenon. The DSC, PXRD, and FTIR spectroscopy studies confirmed the amorphization of ABTA during the SD formation. The dissolution of ABTA-SDs was enhanced in all media compared to pure ABTA, whereas the pharmacokinetic study revealed that ABTA-SD significantly enhanced Cmax and AUC0–t compared to ABTA. The current investigation elucidated the ability of SDs to promote the greater absorption of ABTA and offer the great advantage of reducing the drug dose, which helps lower the cost of the formulation. This study offers a new approach to addressing prostate cancer treatment.Abbreviations
| ABTA: | Abiraterone acetate |
| BCS | Biopharmaceutical classification system |
| API | Active pharmaceutical ingredients |
| SD | Amorphous solid dispersion |
| HPMCAS | Hydroxy propyl methyl cellulose acetate succinate |
| HPMC | Hydroxy propyl methyl cellulose |
| ATR-FTIR | Attenuated total reflectance-Fourier transform infrared |
| PXRD | Powder X-ray diffraction |
| DSC | Differential scanning calorimetry |
| FE-SEM | Field emission scanning electron microscopy |
| TGA | Thermogravimetric analysis |
| PM | Physical mixture |
| FaSSIF | Fasted state simulated intestinal fluid |
| FeSSIF | Fed simulated intestinal fluid |
| RP-HPLC | Reverse phase-high-performance liquid chromatography |
| DLS | Dynamic light scattering |
| SLS | Sodium lauryl sulphate |
| Cmax | Maximum concentration |
| AUC | Area under the curve |
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Manisha Choudhari: material preparation, data collection, and analysis, writing – original draft, Shantanu Damle: writing – review and editing, Ranendra Narayan Saha, Sunil Kumar Dubey: conceptualization, resources; Gautam Singhvi: conceptualization, writing – review and editing, project administration, supervision, validation, visualization. All authors read and approved the final manuscript.Conflicts of interest
The authors have no conflicts of interest to declare that they are relevant to the content of this article.Acknowledgements
The authors sincerely acknowledge the invaluable support from Colorcon Asia Pvt. Ltd, Goa, India. The authors are thankful to Biophore India Pharmaceuticals Pvt. Ltd, India, for providing generous gift sample of Abiraterone acetate. Special thanks are extended to the Department of Pharmacy, BITS Pilani, Pilani campus for research facilities, the Central Analytical Facility (CAF) for granting access to ATR-FTIR, DSC, PXRD, and DSC equipment and to the Sophisticated Instrumentation Facility (SIF) for the use of FE-SEM. Furthermore, the author is grateful to the Central Animal Facility for providing the necessary resources for conducting animal studies.References
- C. J. Ryan, M. R. Smith, L. Fong, J. E. Rosenberg, P. Kantoff, F. Raynaud, V. Martins, G. Lee, T. Kheoh, J. Kim, A. Molina and E. J. Small, J. Clin. Oncol., 2010, 28, 1481–1488 CrossRef CAS PubMed.
- A. Rawlinson, A. Mohammed, J. Beatty, R. Bell and M. Miller, Expert Rev. Anticancer Ther., 2012, 12, 429–437 CrossRef CAS PubMed.
- M. Acharya, A. Bernard, M. Gonzalez, J. Jiao, R. De Vries and N. Tran, Cancer Chemother. Pharmacol., 2012, 69, 1583–1590 CrossRef CAS PubMed.
- T. Solymosi, F. Tóth, J. Orosz, O. Basa-Dénes, R. Angi, T. Jordán, Z. Ötvös and H. Glavinas, J. Chem. Eng. Data, 2018, 63, 4453–4458 CAS.
- H. B. Schultz, T. R. Meola, N. Thomas and C. A. Prestidge, Int. J. Pharm., 2020, 577, 119069 CrossRef CAS PubMed.
- J. Bevernage, J. Brouwers, M. E. Brewster and P. Augustijns, Int. J. Pharm., 2013, 453, 25–35 CrossRef CAS PubMed.
- T. Solymosi, Z. Ötvös, R. Angi, B. Ordasi, T. Jordán, S. Semsey, L. Molnár, S. Ránky, G. Filipcsei, G. Heltovics and H. Glavinas, Int. J. Pharm., 2017, 532, 427–434 CrossRef CAS PubMed.
- A. A. Ravikumar, P. K. Kulkarni, R. A. M. Osmani, U. Hani, M. Ghazwani, A. Al Fatease, A. H. Alamri and D. V. Gowda, Polymers, 2022, 14, 4977 CrossRef CAS PubMed.
- G. Attard, A. H. M. Reid, R. J. Auchus, B. A. Hughes, A. M. Cassidy, E. Thompson, N. B. Oommen, E. Folkerd, M. Dowsett, W. Arlt and J. S. de Bono, J. Clin. Endocrinol. Metab., 2012, 97, 507–516 CrossRef CAS PubMed.
- U. Gala, D. Miller and R. O. Williams, Pharmaceutics, 2020, 12, 1–24 CrossRef PubMed.
- K. N. Chi, J. Spratlin, C. Kollmannsberger, S. North, C. Pankras, M. Gonzalez, A. Bernard, H. Stieltjes, L. Peng, J. Jiao, M. Acharya, T. Kheoh, T. W. Griffin, M. K. Yu, C. Chien and N. P. Tran, J. Clin. Pharmacol., 2015, 55, 1406–1414 CrossRef CAS PubMed.
- S. Geboers, J. Stappaerts, R. Mols, J. Snoeys, J. Tack, P. Annaert and P. Augustijns, J. Pharm. Sci., 2016, 105, 2974–2981 CrossRef CAS PubMed.
- R. Chennuru, R. Devarapalli, P. Rengaraj, P. L. Srinivas, S. Dey and C. M. Reddy, Cryst. Growth Des., 2020, 20, 5018–5030 CrossRef CAS.
- Y. Kawabata, K. Wada, M. Nakatani, S. Yamada and S. Onoue, Int. J. Pharm., 2011, 420, 1–10 CrossRef CAS PubMed.
- H. P. Patel, P. J. Patel, B. V Desai, R. V Patel, D. T. Desai, B. A. Vyas, M. D. P. Willcox and F. A. Maulvi, Colloids Surf., A, 2024, 690, 133786 CrossRef CAS.
- M. R. Donthi, S. R. Munnangi, K. V. Krishna, S. A. Marathe, R. N. Saha, G. Singhvi and S. K. Dubey, AAPS PharmSciTech, 2022, 23, 1–15 CrossRef PubMed.
- M. F. Emam, A. A. El-Ashmawy, N. M. Mursi and L. H. Emara, AAPS PharmSciTech, 2022, 23, 248 CrossRef CAS PubMed.
- L. Liu, F. Ouyang, T. Li, M. Wen, G. Zha, L. Chen, X. Fu and L. qing Zhu, Pharm. Chem. J., 2024, 57, 1627–1636 CrossRef CAS.
- R. H. Müller, C. Jacobs and O. Kayser, Adv. Drug Delivery Rev., 2001, 47, 3–19 CrossRef PubMed.
- T. Boleslavská, O. Rychecký, M. Krov, P. Žvátora, O. Dammer, J. Beránek, P. Kozlík, T. Křížek, J. Hořínková, P. Ryšánek, J. Roušarová, N. K. Canová, M. Šíma, O. Slanař and F. Štěpánek, AAPS J., 2020, 22(6), 1–12 Search PubMed.
- D. K. Jha, D. S. Shah and P. D. Amin, Carbohydr. Polym. Technol. Appl., 2021, 2, 100137 CAS.
- S. Baghel, H. Cathcart and N. J. O'Reilly, J. Pharm. Sci., 2016, 105, 2527–2544 CrossRef CAS PubMed.
- K. Ueda, K. Higashi, K. Yamamoto and K. Moribe, Int. J. Pharm., 2014, 464, 205–213 CrossRef CAS PubMed.
- A. R. Nair, Y. D. Lakshman, V. S. K. Anand, K. S. N. Sree, K. Bhat and S. J. Dengale, AAPS PharmSciTech, 2020, 21, 309 CrossRef CAS PubMed.
- M. J. Choi, M. R. Woo, H. G. Choi and S. G. Jin, Int. J. Mol. Sci., 2022, 23, 9491 CrossRef CAS PubMed.
- D. B. Warren, H. Benameur, C. J. H. Porter and C. W. Pouton, J. Drug Targeting, 2010, 18, 704–731 CrossRef CAS PubMed.
- K. Ueda, K. Higashi, K. Yamamoto and K. Moribe, Mol. Pharm., 2013, 10, 3801–3811 CrossRef CAS PubMed.
- H. Al-Obaidi and G. Buckton, AAPS PharmSciTech, 2009, 10, 1172–1177 CrossRef CAS PubMed.
- N. S. Trasi, S. V. Bhujbal, D. Y. Zemlyanov, Q. (Tony) Zhou and L. S. Taylor, Int. J. Pharm.:X, 2020, 2, 100052 CAS.
- M. F. Simões, A. Pereira, S. Cardoso, S. Cadonau, K. Werner, R. M. A. Pinto and S. Simões, Mol. Pharm., 2020, 17, 554–568 Search PubMed.
- D. J. Greenhalgh, A. C. Williams, P. Timmins and P. York, J. Pharm. Sci., 1999, 88, 1182–1190 CrossRef CAS PubMed.
- S. Mukesh, P. Joshi, A. K. Bansal, M. C. Kashyap, S. K. Mandal, V. Sathe and A. T. Sangamwar, Mol. Pharm., 2021, 18, 2334–2348 CrossRef CAS PubMed.
- A. Forster, J. Hempenstall, I. Tucker and T. Rades, Int. J. Pharm., 2001, 226, 147–161 CrossRef CAS PubMed.
- D. W. van Krevelen, Properties of Polymers, 3rd edn, 1997, pp. 189–225 Search PubMed.
- Y. Zhao, P. Inbar, H. P. Chokshi, A. W. Malick and D. S. Choi, J. Pharm. Sci., 2011, 100, 3196–3207 CrossRef CAS PubMed.
- M. Patel, D. Mori, K. Dudhat, S. Shah, J. Chavda and A. Patel, J. Pharm. Innov., 2022, 17, 1148–1159 CrossRef.
- A. Butreddy, S. Sarabu, M. Almutairi, S. Ajjarapu, P. Kolimi, S. Bandari and M. A. Repka, Int. J. Pharm., 2022, 615, 121471 CrossRef CAS PubMed.
- F. Frizon, J. de O. Eloy, C. M. Donaduzzi, M. L. Mitsui and J. M. Marchetti, Powder Technol., 2013, 235, 532–539 CrossRef CAS.
- S. S. Gupta, N. Solanki and A. T. M. Serajuddin, AAPS PharmSciTech, 2016, 17, 148–157 CrossRef CAS PubMed.
- J. Liu, Y. Li, W. Ao, Y. Xiao, M. Bai and S. Li, ACS Omega, 2022, 7, 39907–39912 CrossRef CAS PubMed.
- M. Choudhari, M. Donthi, S. Damle, G. Singhvi, R. Saha and S. Dubey, Curr. Chromatogr., 2022, 09, 78–94 CrossRef CAS.
- J. H. Fagerberg, O. Tsinman, N. Sun, K. Tsinman, A. Avdeef and C. A. S. Bergström, Mol. Pharm., 2010, 7, 1419–1430 CrossRef CAS PubMed.
- M. El Sayed, A. Alhalaweh and C. A. S. Bergström, Mol. Pharm., 2021, 18, 4079–4089 CrossRef CAS PubMed.
- N. Castro, I. González-Hernández, L. Mayet-Cruz, J. Becerril-Vega, S. Soto-Romo and H. Jung-Cook, Braz. J. Pharm. Sci., 2023, 59, 1–10 Search PubMed.
- A. Butreddy, S. Sarabu, M. Almutairi, S. Ajjarapu, P. Kolimi, S. Bandari and M. A. Repka, Int. J. Pharm., 2022, 615, 121471 CrossRef CAS PubMed.
- J. S. Choi, S. E. Lee, W. S. Jang, J. C. Byeon and J. S. Park, Mater. Sci. Eng., C, 2018, 90, 387–396 CrossRef CAS PubMed.
- K. Ueda, K. Higashi, K. Yamamoto and K. Moribe, Int. J. Pharm., 2014, 464, 205–213 CrossRef CAS PubMed.
- C. Liu, Z. Chen, Y. Chen, J. Lu, Y. Li, S. Wang, G. Wu and F. Qian, Mol. Pharm., 2016, 13, 599–608 CrossRef CAS PubMed.
- V. R. Wilson, X. Lou, D. J. Osterling, D. A. F. Stolarik, G. J. Jenkins, B. L. B. Nichols, Y. Dong, K. J. Edgar, G. G. Z. Zhang and L. S. Taylor, Sci. Rep., 2020, 10, 1–12 CrossRef PubMed.
- Q. Zhang, Y. Zhao, Y. Zhao, Z. Ding, Z. Fan, H. Zhang, M. Liu, Z. Wang and J. Han, Colloids Surf., B, 2018, 172, 118–126 CrossRef CAS PubMed.
- Z. Wang, M. Sun, T. Liu, Z. Gao, Q. Ye, X. Tan, Y. Hou, J. Sun, D. Wang and Z. He, Asian J. Pharm. Sci., 2019, 14, 95–103 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08163c |
| This journal is © The Royal Society of Chemistry 2024 |