
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinary mixture of subcritical water and acetone: a hybrid solvent system towards the production of lignin nanoparticles†
Philip S.
McMichael
ab,
Mahfuzul
Hoque
ab,
Fernanda Brito
dos Santos
ab,
Victoria
French
ab and
E. Johan
Foster
 *ab
*ab
aDepartment of Chemical and Biological Engineering, University of British Columbia, 2385 East Mall, Pulp and Paper Centre, V6T 1Z4, Canada. E-mail: johan.foster@ubc.ca
bBioproducts Institute, 2385 East Mall Vancouver, BC V6T 1Z4, Canada
First published on 3rd January 2024
Abstract
Valorization of technical lignin is crucial for the circular bioeconomy – this can be achieved by the transformation of lignin to high-value nanomaterials. However, the majority of lignin nanoparticle (LNP) production methods require high volumes of organic solvent, and specialized equipment, or fail in certain cases to produce a morphologically uniform product with superior physicochemical properties to technical lignin. Herein, a binary solvent system of subcritical water and acetone based on the principles of solvent–antisolvent (SAS) precipitation of lignin is proposed. Spherical LNPs with a high degree of uniformity in the chemical structure are produced via a batch reactor from kraft lignin demonstrating high yield (88–92%), colloidal stability in water, thermal stability, and low mean particle size – these are desirable for advanced biocomposite and biomedical applications; achieved in this study by using lower volumes of recyclable acetone (4–7 times less) than previously reported.
Introduction
Chemical industries are the third largest producers of global carbon dioxide emissions owing to their reliance on fossil fuels derived feedstocks, necessitating their reinvention via innovative reaction engineering solutions. Fueled by the global climate change crisis (global warming), and the acute health concerns stemming from petroleum-derived plastics,1 there has been an upsurge in preference for renewable and “earth-friendly” materials and products.One such renewable chemical is lignin, an aromatic biopolymer found in the cell walls of all vascular plants, which is produced in large quantities worldwide2 and comprises 20–30% of the total mass of most commercial tree species.3 The pulp and paper industry are the chief producers of purified lignin, which is primarily extracted from the by-product (black liquor) of the pulping processes including kraft pulping. Industrial lignin makes up 35–45% of the solids content of black liquor and can be extracted through a variety of established methods (kraft, organosolv, and lignosulfonate, to name a few) resulting in amorphous lignin with different structural features (i.e., formation of recalcitrant C–C bonds) to that of native lignin, hence, it is called technical lignin. Now, although nearly 70 million tons of kraft lignin (a type of softwood-derived technical lignin) is produced annually,4,5 over 98% is burned for energy recovery in pulp and paper mills6 – an opportunity for chemical reaction engineering and process development to change the paradigm of lignin valorization.
Interestingly, the production of LNPs has emerged as a means of improving the morphological and chemical uniformity of lignin – owing to an increased surface area to volume ratio, they can demonstrate unique properties, including improved UV protection, antibacterial, and anti-oxidant properties.7,8 Also, this leads to higher reactivity (of phenolic and aliphatic hydroxyl groups9), which affords the development of advanced functional materials, including CO2/N2-switchable Pickering emulsions.10 Furthermore, improved miscibility in water as well as in polymers (i.e., high-density polyethylene11) broadens the application portfolio of lignin, as a drug carrier (i.e., curcumirin12), for adhesives,13 composites,14 and coatings.15
The reaction chemistry to produce LNPs involves predominantly wet chemistry and hydrodynamics-based approaches, which includes pH and solvent shifting, antisolvent precipitation, polymerization (e.g., graft) and spray drying, electrospinning, self-assembly and more.16,17 Details about these methods can be found elsewhere.18 Now, most methods suffer from various drawbacks, including dependence on high volumes of organic solvents, low process yields, multi-step procedures, and expensive equipment designed for laboratory environments.19
In this work, we present a process for the synthesis of LNPs using a novel system that relies on the solvent–antisolvent (SAS) method combined with the inherent properties of water at subcritical conditions (Fig. 1). The term scH2O refers to water above its atmospheric pressure boiling point of 100 °C, at elevated pressure to maintain its liquid phase, but at conditions below its critical point (374 °C with a vapor pressure of 22 MPa).20 At subcritical conditions, water can reach a relative permittivity (dielectric constant) near that of common organic solvents like dimethyl sulfoxide, methanol, and acetone. ScH2O functions as a solvent for certain organic non-polar compounds, including lignin, effective for hydrolysis and fractionation and production of bio-oils from lignocellulosic biomasses.21–24 However, no present work has demonstrated its use to produce LNPs from technical lignin.
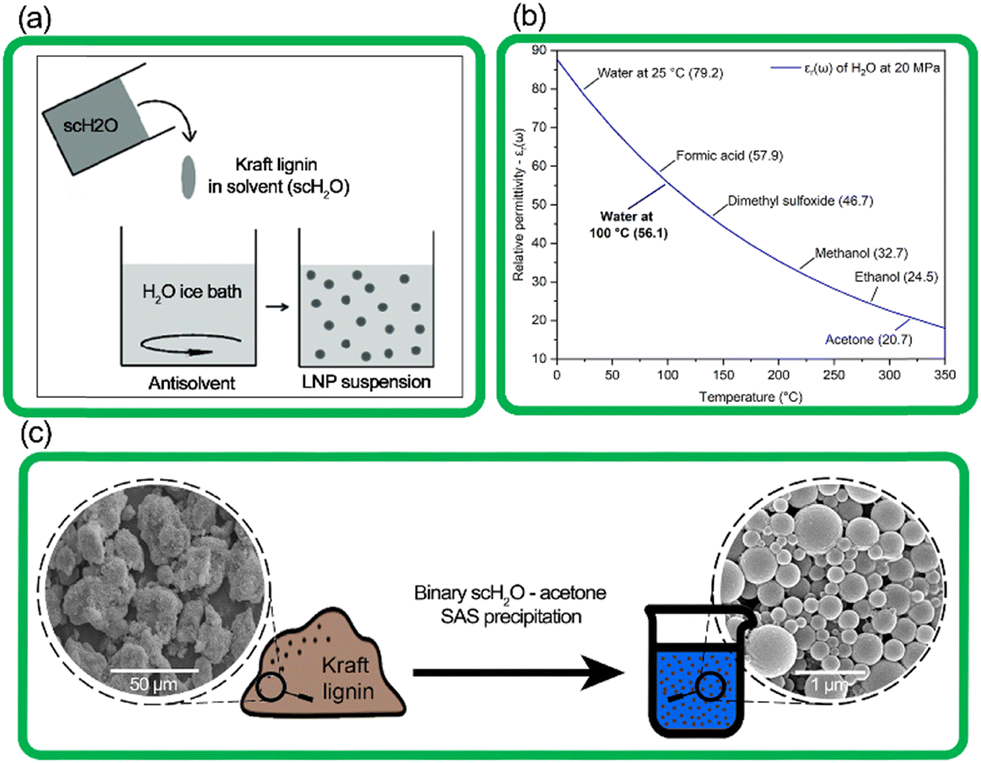 | ||
| Fig. 1 (a) The simplified process of SAS precipitation of nanoparticles,25 (b) the relative permittivity (εr(ω)) of water at increasing temperature, with constant pressure of 20 MPa, and εr(ω) of common solvents for reference (Uematsu et al. 1980),26 (c) transition from amorphous kraft lignin to spherical lignin nanoparticles, as seen by SEM images. | ||
We note that the SAS precipitation method to produce nanoparticles has been utilized for a range of nanoparticle suspensions, including to produce germanium and silver nanoparticles,27 semiconductor nanoparticles,28 and nanopharmaceutical applications.29,30Via SAS, production of LNPs works by mixing dry lignin powder with a solvent (most commonly tetrahydrofuran or acetone, though ethylene glycol,13 ethanol,31 γ-valerolactone,32 and other solvents have been used), which provides a completely solubilized lignin solution, followed by mixing with an antisolvent; a chemical (commonly water for kraft lignin) that lignin is completely insoluble in. Thus, owing to the increase in the chemical potential (caused by the antisolvent), the rapid supersaturation leads to spontaneous homogenous nucleation of particles (crystalline/amorphous),33 forming stable colloidal suspensions in the resultant SAS mixture. This self-assembly process driven by solvent–solvent interactions ensure uniform spherical morphology of the amorphous LNPs. However, large volumes of organic solvents are needed to produce relatively low amounts of LNPs.16,19,34
In this study, scH2O was rapidly expelled from the high-pressure reactor and rapidly mixed with water below room temperature, in the form of an ice bath, to ensure that spontaneous nucleation of new nanoparticles was favored over growth of particle aggregates, according to prior literature which suggests a clear correlation between faster SAS mixing rates and smaller mean particle diameter.35,36 This hybrid scH2O is assisted by low volumes of recyclable acetone (several orders lower than previously reported), effectively solubilizing lignin to produce chemically and morphologically uniform LNPs, and the synthesized LNPs were investigated for morphological, chemical, and particle size uniformity, as well as thermal and colloidal stability.
Methods
Materials
Kraft lignin produced through the LignoBoost® process, from a chip furnish of 30% interior (white/Engelmann) spruce, 60% lodgepole pine, and 10% subalpine fir was provided by Canfor Corporation (Vancouver, BC, Canada). Reagent-grade acetone (>99% purity) was purchased from Sigma-Aldrich (St. Louis, MO, USA). A 500 mL volume high-pressure/high-temperature batch reactor, model 4575 from Parr Instrument Company (Moline, IL, USA) was used, and pressurized with N2 gas (99.999% purity) from Airgas, Inc. (Radnor, PA, USA).Production of LNPs
Kraft lignin on a dry weight basis was weighted out based on a concentration of 5 grams per liter and added to solutions of acetone (based on a volume by total volume percentage) and completed with water to a total volume of 250 mL. Three samples of 5 g L−1 kraft lignin were prepared, 2.5% acetone with 97.5% deionized (DI, Milli-Q®) H2O (labeled 2.5%A LNPs), 5% acetone with 95% DI H2O (labeled 5%A LNPs), and 10% acetone with 90% DI H2O (labeled 10%A LNPs). An additional sample of 5 g L−1 kraft lignin in 250 mL of DI H2O, without acetone, was prepared (labeled 0%A lignin). Each sample was then stirred using an IKA (Staufen, DE) T-25 Ultra-Turrax homogenizer for 5 minutes at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. A 50 mL aliquot of each sample was then taken and stored for further characterization and comparison.
000 rpm. A 50 mL aliquot of each sample was then taken and stored for further characterization and comparison.
Each 200 mL sample was then placed in the Parr 4575 reactor, which was then sealed and pressurized to 15 MPa with N2 gas. A heating rate of 2.5 °C per minute was used to heat the samples from 25 °C to 100 °C, which raised internal reactor pressure to 20 MPa. Due to the higher vapor pressure of acetone than water, samples with higher concentrations of acetone had slightly higher pressures at 100 °C. After heating, the pressure of these samples was decreased to 20 MPa, using a gas outlet valve, to ensure a standard pressure for all tests. Samples were held at 100 °C and 20 MPa for 1 hour, with continuous stirring at 200 rpm. After 1 hour, the sample outlet valve was opened and the solution was rapidly expelled from the reactor into 400 mL of DI water mixed with crushed ice, allowing for rapid mixing of the solvent and anti-solvent. Each acetone-containing sample then underwent rotary evaporation at 65 °C for two hours, to ensure total removal of acetone. 200 mL of each sample was then set aside to be frozen for lyophilization, while the remaining sample was stored in solution at 4 °C. Fig. 2 outlines the LNP production using the high-pressure reactor.
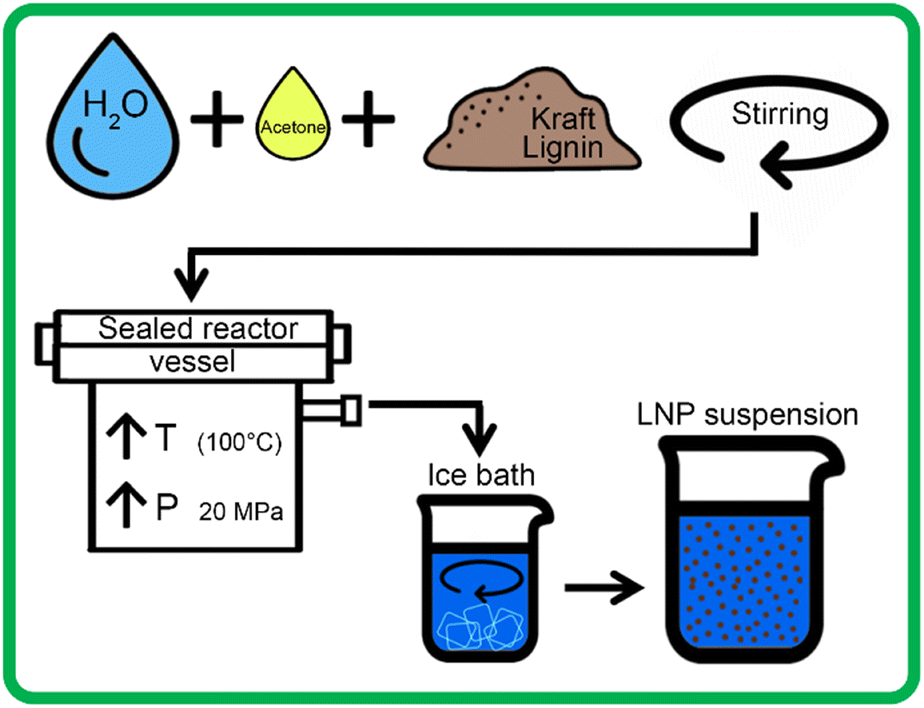 | ||
| Fig. 2 Illustration of process of synthesizing LNPs using SAS process with scH2O and acetone, showing general conditions in high pressure reactor, followed by rapid mixing with ice bath, to generate colloidally stable LNP suspension, to be followed by rotary evaporation. The size of the droplets (top-left side) corresponding to water (blue-colored) and the acetone (yellow-colored) signify the relative proportion of the volume used to produce LNPs. | ||
Analytical methods
Lignin samples for morphological analysis with scanning electron microscopy (SEM) (Helios NanoLab 650, FEI, Hillsboro, OR, USA) were prepared using a 0.1 wt% and 0.01 wt% dilution of prepared LNPs in DI water. Carbon tape was placed on SEM pin stubs, and 100 μL of the diluted LNP solution was pipetted onto the surface of the tape and dried at 60 °C until excess water was evaporated. The procedure of pipetting and drying was repeated three times, to ensure that enough LNP sample was present on the SEM stub. ImageJ image processing software was used to analyze SEM images and estimate particle size based on SEM imagery, with a sample size of 200 particles for each particle size distribution histogram generated. The particle size distribution of lignin particles was also studied by use of a laser diffraction particle size analyzer (Mastersizer 3000, Malvern Panalytical, UK). Span values of particle size distribution were calculated by eqn (1), with D90, D50, and D10 being the points in the size distribution that 90%, 50% and 10% of total material volume is contained within (smaller than), respectively.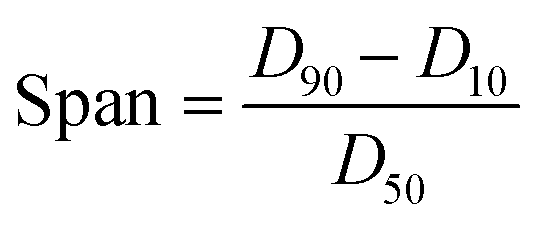 | (1) |
The undiluted liquid sample was sonicated at 80% amplitude for 60 seconds using the built-in equipment sonicator and measured at a set obscuration of 5%. Thermal degradation of lyophilized lignin samples was analyzed with thermogravimetric analysis (TGA) equipment (TGA 5500, TA Instruments, USA), using a heating ramp rate of 10 °C per minute, up to 800 °C. OriginPro Software (2022 Version, OriginLab, USA) was used to analyze the derivative thermogravimetry (DTG) of the samples. The onset temperature of thermal degradation, shown in Table 1, was calculated according to procedures outlined in ISO 11358-1. Sample yield was calculated based on the total dry weight of the sample extracted after lyophilization, as a fraction of the initial dry weight of the kraft lignin sample mixed with acetone/water solutions, given a lignin concentration of 5 grams per liter. Zeta potential analysis was conducted with Malvern Zetasizer Nano DLS equipment (Malvern Panalytical, UK), with sample concentrations in DI water set at 0.01 g L−1 for all measurements. Hansen solubility parameters and spheres were calculated with assistance of the HSP Excel Spreadsheet provided by https://www.hansen-solubility.com. Lignin was mixed at a concentration of 0.5 g L−1 in 20 common solvents, (coordinates given in Fig. S5†) stirred overnight, and allowed to settle for 24 hours before solubility assessments were made.
| Sample name | Yield | T onset (°C) | Span | Zeta potential (mV) |
|---|---|---|---|---|
| 0%A lignin | 71% | 240 | 127.8 | −20.7 ± 1.7 |
| 2.5%A LNPs | 88% | 226 | 11.4 | −37.5 ± 4.9 |
| 5%A LNPs | 91% | 283 | 25.4 | −50.3 ± 2.3 |
| 10%A LNPs | 92% | 292 | 1.6 | −52.6 ± 0.8 |
Results and discussion
Uniform LNP synthesis with scH2O and acetone treatment
To maximize the yield of LNPs (>90%), total lignin de-aggregation, followed by complete solubilization, is necessary. As such, a variety of reactor conditions were initially evaluated to determine optimal conditions for LNP synthesis. At temperatures at and above 250 °C, the burning of lignin was evident in the reactor system, while at temperatures above 150 °C, thermal degradation of the lignin was still evident (confirmed through TGA) with increased aggregation of lignin particles in the reactor. A temperature of 100 °C was found to be sufficient for LNP synthesis while limiting thermal degradation and particle aggregation in the reactor. At this temperature, a pressure greater than 0.364 MPa (3.58 atm) is necessary to maintain acetone/water solutions in a purely liquid phase, given the vapor pressure of acetone at 100 °C. However, reactions at higher pressures were found to be more effective at decreasing particle size, compared with reactions at pressures only slightly above 0.364 MPa. The effect of pressure has been previously found to have a negligible effect on the solubilities of most organic compounds in scH2O.37 There exists the possibility that high pressure may aid this unique process of nanoparticle synthesis in several ways, including preventing aggregation of subcritical-water-insoluble portions of lignin, which may function as nucleation sites during the SAS precipitation step, and in turn may encourage excessively large sphere growth, rather than new sphere formation.Additionally, higher reactor pressures translated to more rapid and complete expulsion of the sample from the high-pressure reactor into the ice bath, allowing for a greater portion of the sample to undergo SAS nucleation. Faster rates of solvent and antisolvent mixing have been shown to directly correlate to smaller particle sizes, due to high supersaturation favoring new sphere formation. If an environment of high supersaturation can be quickly achieved, the unfavorable growth of existing spheres, rather than nucleation of new spheres, can be avoided.38–40 Exploratory tests found that a reactor pressure of 20 MPa was found to produce a higher yield of nanoparticles compared with pressures of 15 MPa and 10 MPa. After the temperature ramp-up, all samples were held at 20 MPa and 100 °C for 60 minutes. Preliminary tests showed that increasing holding time from 60 minutes to 120 and 180 minutes had negligible effects on the properties of the lignin particles.
ScH2O, while effectively acting as a polar organic solvent to solubilize low molecule weight units within the lignin macromolecule, is ineffective at solubilizing higher molecular weight lignin units. These insoluble units function as sites for undesirable heterogeneous nucleation and aggregation of solubilized lignin species, upon SAS mixing. As such, a small volume of solvent was added to the scH2O system, to ensure complete solubilization in the high-pressure reactor, and ensure homogenous nucleation upon mixing. Acetone, an easily recyclable organic solvent, is sufficient to serve this purpose, at much lower volumes compared with existing methods for solvent-based synthesis of LNPs. Having a relative permittivity of 20.7 at 25 °C, acetone/water mixtures will have a reduced εr compared to pure water systems. It is important to note that no known hazardous reaction exists between acetone and water,41 and while acetone/water systems have been assessed for changes in their flammability hazard at elevated conditions, acetone concentrations below 25% and 150 °C were assessed as having an explosion class rating of St-0 (no explosion).42 Several different concentrations of acetone (2.5%, 5%, and 10% v/v) were evaluated for their effectiveness at producing morphologically and chemically uniform and thermally stable LNPs.
Particle morphology analysis with SEM
SEM was performed on the dried scH2O lignin samples to elucidate the morphology of the lignin particle structure (Fig. 3). Without any acetone to improve solubility and prevent agglomeration during cooling, lignin aggregates inside the reactor vessel and upon quenching. Nanoparticles that do form are aggregated within micrometer-scale particle clumps. A portion of lignin may remain insoluble at the tested subcritical conditions. While subcritical and supercritical water have previously been found to effectively solubilize lignin, prior studies have utilized harsher conditions in order to initiate fractionation and depolymerization of the organic polymer.21–24 The conditions tested in this work, chosen to reduce thermal degradation of the kraft lignin, may not fully solubilize the lignin, and larger particles may serve as nucleation sites upon SAS mixing, which would explain the clumping of particles evident in SEM of the 0%A lignin sample. Upon the addition of acetone, the formation of uniform nanoparticles is evident. As the percentage of acetone increases from 2.5% up to 10% of the total sample volume, average particle size decreases, while span decreases most significantly for 10%A LNP. Importantly, all three acetone-containing samples demonstrate synthesis of spherical nanoparticles, with few irregular or elongated shapes present for the 10%A LNP sample. Previous research has demonstrated the efficacy of acetone/water cosolvent mixtures to produce LNPs, though these works utilize a much higher volume of acetone (i.e., 70% acetone, 30% water) for the effective synthesis of nanoparticles.43Fig. 4 shows selected studies that have utilized solvents for LNP synthesis, and calculated solvent volumes required for treatment of 1 gram of lignin for nanoparticle production.31,35,43–58 At a concentration of 5 g L−1, sample 10%A LNP uses 20 mL of acetone per treatment of 1 gram of kraft lignin, while comparable studies rely on volumes several times greater.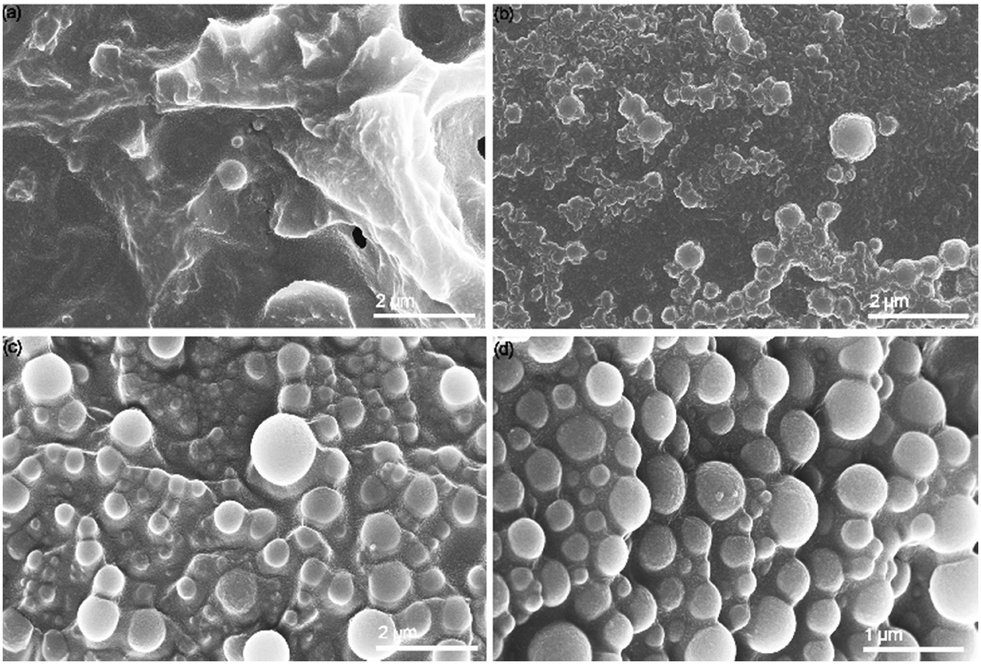 | ||
| Fig. 3 SEM images of (a) 0%A lignin, showing non-uniform, irregular structure (b) 2.5%A LNPs, showing partial nucleation of nanoparticles (c) 5%A LNPs showing increased volume of nanoparticles (d) 10%A LNPs, showing further increased volume of nanoparticles. | ||
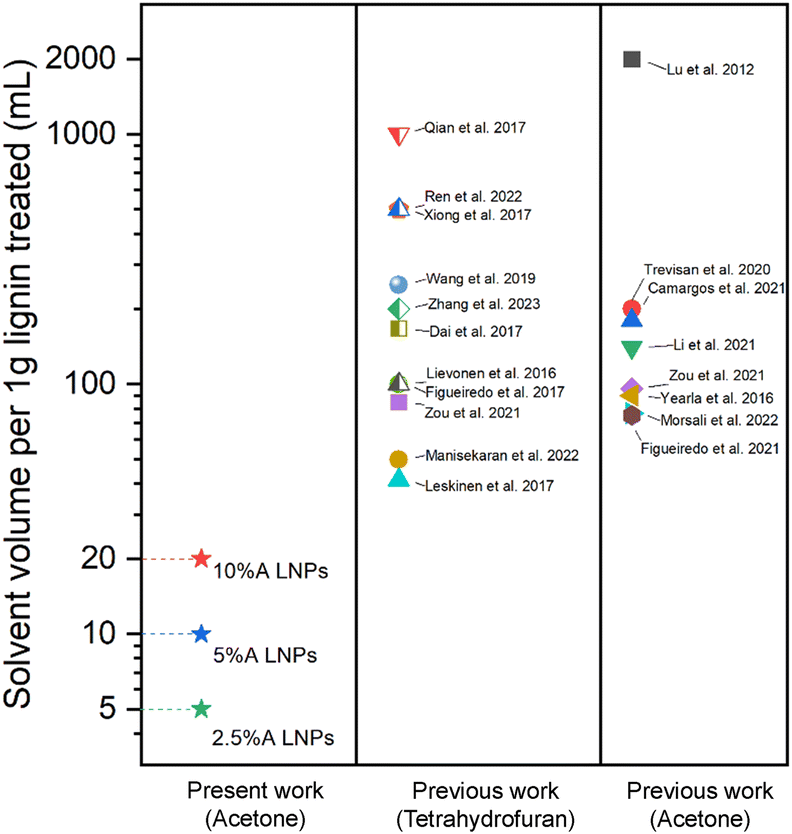 | ||
| Fig. 4 Selected comparison of solvent volumes required for production of LNPs in prior research utilizing SAS precipitation technique with a variety of solvents.31,35,43–58 Tetrahydrofuran and acetone are compared, being the two most used solvents for SAS precipitation of LNPs. For studies that used a range of lignin-in-solvent concentrations, the highest concentrations (least volume of solvent used) were shown here. | ||
Fig. 5 shows sample 10%A LNP with higher dilution to better clarify the morphology of the synthesized nanoparticles, revealing no amorphous regions or non-spherical structures in this sample. This further demonstrates the enhanced uniformity that the self-assembly process imbues on the particles. Lignin self-assembles into spheres by molecular rearrangement, with initial nucleation driven by π–π stacking interactions between the hydrophobic aromatic rings.59 These high molecular weight structures form the interior core of the nuclei. Lower molecular-weight and more hydrophilic units in the form of primarily methoxyl, carboxyl, and hydroxyl groups absorb onto the surface of the nuclei.58,60 These ionized surface groups, especially ionized carboxylic acid groups, provide electrostatic stability in solution, and prevent aggregation.61 The presence of charged functional groups on the surface of each spherical nanoparticle, evidenced by increasingly negative zeta potential correlated with smaller mean particle diameter (Table 1), can improve theoretical yield of functionalized lignin products, as these processes rely on chemical interactions with the functional groups of lignin rather than the stable aromatic ring structures of the molecule.62 Formation of spherical shapes, rather than other nonuniform structures (i.e., rough edges or elongated structures), is due to the understood affinity for molecular nucleation to favor minimization of surface energy by minimizing surface area to volume ratio, resulting in a perfect sphere shape being preferred during particle formation.
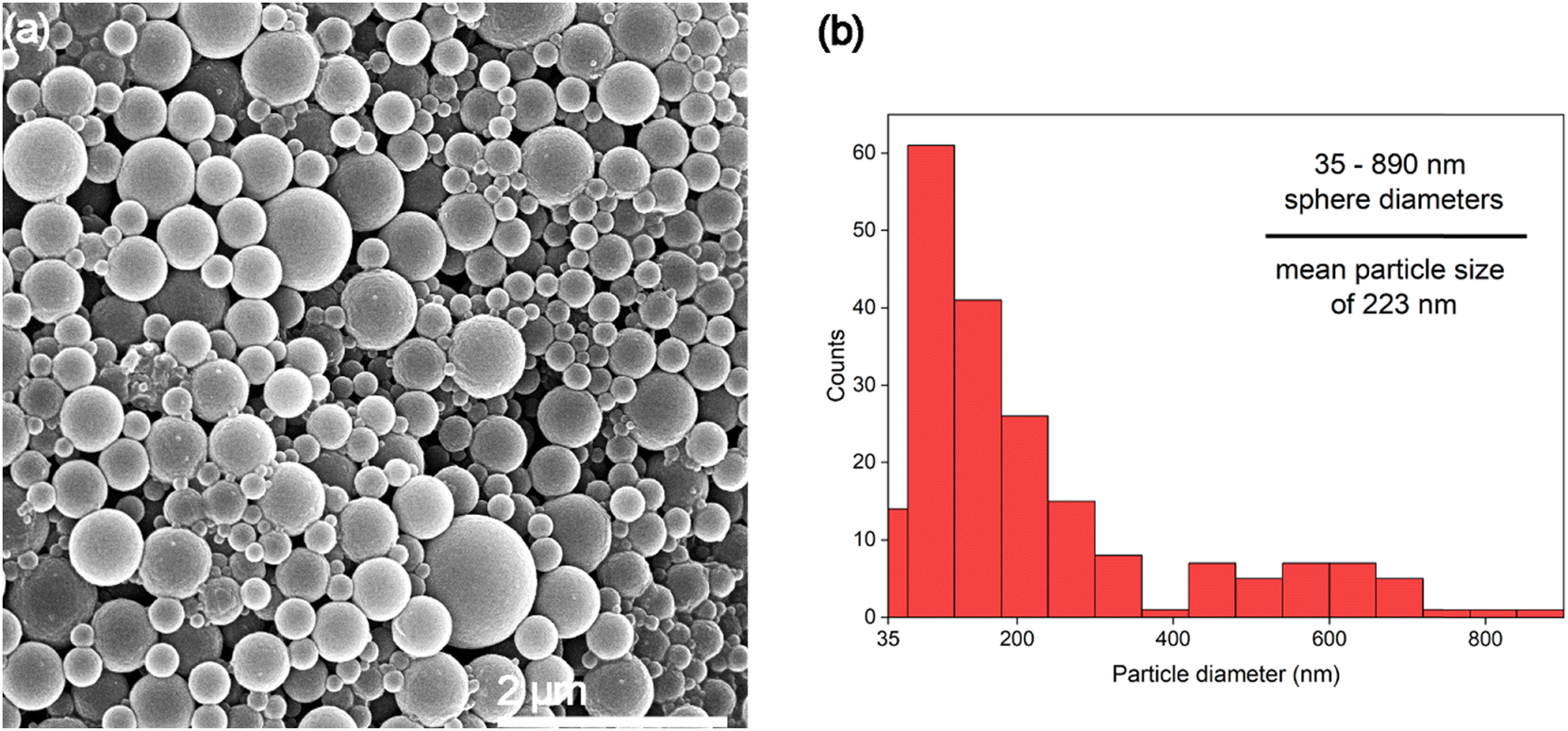 | ||
| Fig. 5 (a) SEM image of highly diluted (0.01 wt%) 10%A LNPs, showing high yield of spherical nanoparticles without evidence of aggregation or irregular shape formation (b) particle size histogram (calculated using ImageJ analysis software), of image Fig. 5a. | ||
Laser diffraction particle size analysis
To investigate the particle size distribution of produced samples, laser diffraction analysis was used, to gain a better understanding of the polydispersity of samples. Particles below 10 nm in diameter cannot be reliably measured with laser diffraction analysis, though this method of analysis provides a more comprehensive picture of sample makeup compared with dynamic light scattering (DLS).63 DLS performs well with relatively monodisperse nanoparticle samples, but poorly with more polydisperse samples. Laser diffraction analysis gives a more complete picture of the partially or totally homogenous nucleation of LNPs and allows us to use span values as a measure of the breadth of particle diameter distribution (Table 1). Fig. 6a shows that 0%A lignin samples had a negligible volume of particles below 1000 nanometers, and a similar volume density of micrometer-sized particles as untreated kraft lignin dispersed in water, which may be due to in-reactor aggregation of the lignin. Acetone in increasing concentrations showed increasing volumes of nanoparticles, and conversely, a decreasing volume of particles above 1000 nanometers. Higher volumes of acetone increase the dissolution of all lignin compounds in the scH2O/acetone mixtures, which reduces the chance of heterogeneous nucleation sites existing in the form of insoluble lignin particles. While the highest volume of nanoparticles was identified as sample 10%A LNPs (maximum volume density at 156 nm), sample 5%A LNPs achieved a slightly lower volume of nanoparticles (maximum volume density at 177 nm).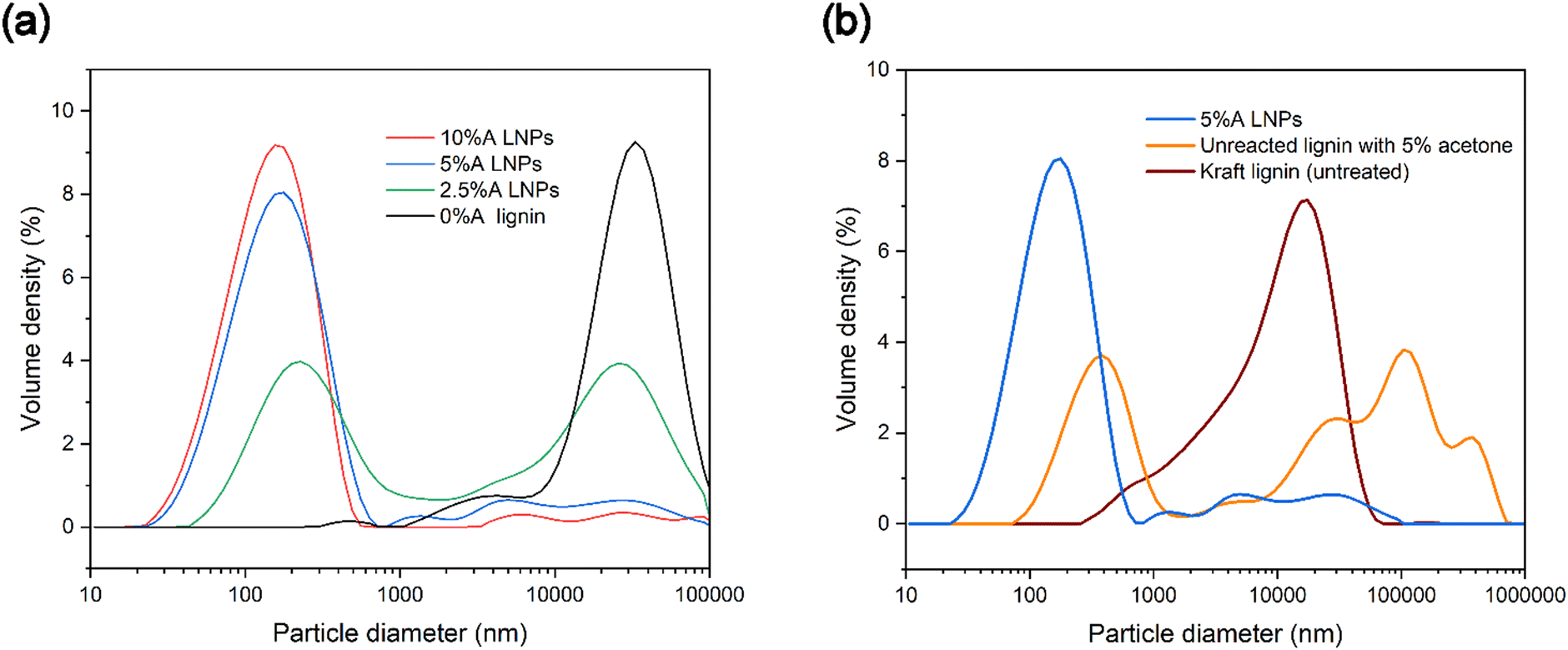 | ||
| Fig. 6 (a) Particle size distribution graph of lignin particle and nanoparticle suspensions, showing a correlation between increasing acetone concentration and decreasing mean particle size. (b) Comparison of an untreated 5%A LNP sample with a scH2O-treated samples, demonstrating increased yield when treated. | ||
Additionally, a sample identical to 5%A LNP, excluding treatment at subcritical conditions or SAS precipitation, was tested to clarify the importance of the novel addition of subcritical treatment for high-yield nanoparticle production (Fig. 6b). Importantly, the LNP sample produced through the novel scH2O synthesis method had a higher volume of nanoparticles compared with purely acetone/water mixtures without scH2O treatment (labeled as “unreacted lignin with 5% acetone”).
Thermal treatment from scH2O
Thermal stability and rate of thermal decomposition were analyzed by use of TGA under a nitrogen atmosphere. Similarly with chemical composition changes, decreased thermal stability was evident for samples that were treated at elevated temperatures above 150 °C. Samples that were produced through the studied conditions of 100 °C and 20 MPa, with a 1-hour residence time at these conditions, displayed varying thermal degradation characteristics correlated with the particle size of the resultant lignin material. Thermogravimetry curve analysis for all synthesized LNP samples revealed expected degradation steps, consistent with that of untreated kraft lignin (Fig. 7a).64 These samples followed the expected degradation steps of softwood lignin, with three distinctive steps of thermal degradation.65 The initial step, occurring from temperatures near 120 °C up to 250–275 °C, represents removal of trace moisture bound in the lignin molecule, followed by dehydration of hydroxyl group side chains of the lignin. The second step, representing the step with the most rapid rate of degradation, occurs from approximately 250 °C to 500 °C. In this temperature range, functional side groups, including carbonyl, carboxyl, methoxyl, and sulfonic acid groups, are separated and eliminated through oxidative reactions. The third step, above 500 °C, represents decomposition through cleavage of the remaining aromatic ring clusters, followed by eventual carbonization.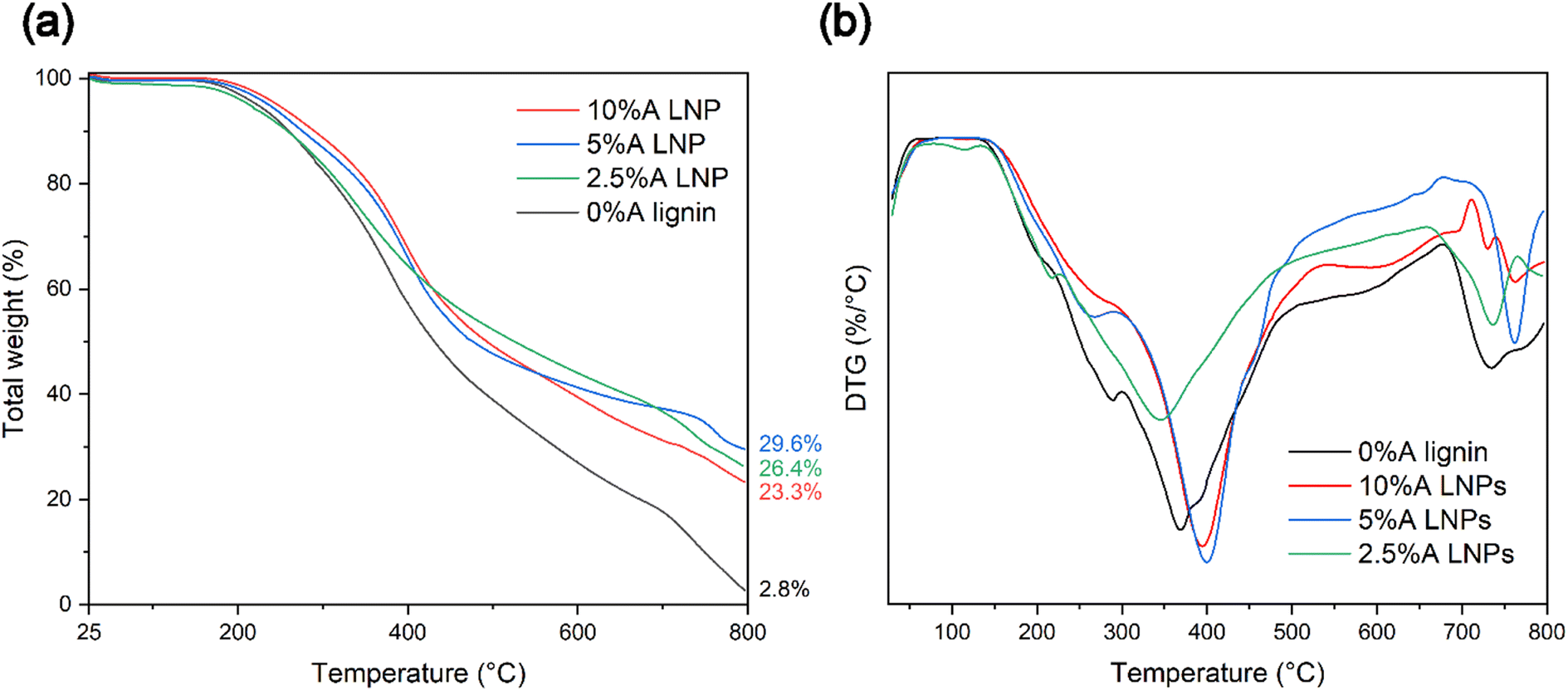 | ||
| Fig. 7 (a) TGA curves of oven-dried lignin samples, under nitrogen atmosphere, with ramp rates of 10 °C per minute, up to 800 °C, showing improved thermal stability for LNP samples, and (b) DTG graphs representing rate of mass loss per heating rate for oven-dried lignin samples, showing thermal degradation steps consist with unmodified kraft lignin. | ||
Samples 2.5%A LNP, 5%A LNP and 10%A LNP, which produced nanoparticles after synthesis, demonstrated thermal stability equivalent to that of unmodified kraft lignin, with temperatures (Tonset) of 282.9 °C and 291.5 °C, for 5%A LNPs and 10%A LNPs, respectively. Thermal stability of processed kraft LNPs, compared with raw kraft lignin, is consistent with previous literature,66,67 as well as for LNP-composites,68 which may be due to higher amounts of phenolic hydroxyl groups. Heat treatment of lignin, which leads to condensation of the lignin macromolecule, and crosslinking of phenolic hydroxyl groups, can also improve the thermal stability of lignin.69,70 However, higher temperatures than the conditions of this study may be necessary to initiate this crosslinking,71 providing an intriguing avenue for further research.
Additionally, the high volume of π–π stacking interactions between higher molecular-weight aromatic ring units has been shown to increase thermal stability of polymers by limiting molecular mobility.72 These interactions are not present in such high density in samples that failed to either entirely or partially self-assemble into nanoparticles upon SAS mixing (samples 0%A lignin and 2.5%A LNP), and these samples had more rapid onset of thermal degradation (Fig. 7b). Sample 2.5%A LNP, having partial nanoparticle self-assembly, but retaining a more amorphous structure compared with samples 5%A LNP and 10%A LNP, did demonstrate similar thermal decomposition (after Tonset, from a weight fraction standpoint) to those samples, showing that in-reactor thermal decomposition was reduced for all acetone-assisted samples. The low ash content of 0%A lignin compared with other samples is due to the scH2O-insoluble, high Mw aromatic groups in lignin that agglomerated onto the interior of the reactor vessel during treatment, contributing to the lower yield of this sample (71%). Given that the majority of this lost mass was most likely more thermally-stable aromatic rings, this caused sample 0%A lignin to have reduced thermal stability compared with other samples when analysed by TGA.
Sample properties
Lignin sample yield increased with higher acetone concentration, due to lower amounts of aggregates sticking to the interior body of the reactor vessel. While the true yield of nanoparticles is difficult to accurately assess, based on SEM imagery, 10%A LNP samples were comprised almost entirely of particles below 900 nm in size, with mean particle sizes of 223 nm, and likely had decreasing yield of microparticles with increasing acetone concentrations, evidenced by laser diffraction particle size analysis. High sample yield is especially important for considering LNP usage in high-value applications that seek to replace existing, high-yield manufacturing of polymers or fillers with LNPs.Suspension stability and solubility
Fig. 8 shows photographs taken of the produced lignin and LNPs in solution at 1 wt% concentration, after 5 minutes of sonication. In contrast with the acetone-free sample (0%A lignin), which demonstrated rapid settling over a period of just 15 minutes, all acetone-containing samples demonstrated minimal settling over a period of 24 hours. The increased particle–particle repulsion caused by the rearrangement of less-hydrophilic carboxylic acid groups on the exterior of the particle causes repulsive forces that resist settling and imbue the LNP solution with colloidal stability in water.44,61 Additionally, Hansen solubility parameters (HSPs) for unmodified kraft lignin and our 10%A LNP sample were determined (Fig. S5†). Kraft lignin was found to have HSPs of δD: 17.6, δP: 13.7, δH: 18.1, and RO of 17, while 10%A LNPs were found to have HSPs of δD: 17, δP: 13.4, δH: 16.5, and RO of 20.75. Essentially, out of 20 common solvents tested, both samples were found to be fully soluble in only dimethyl sulfoxide, ethylene glycol, N,N-dimethyl acetamide, and N,N-dimethyl formamide, while 10%A LNP was found to additionally be fully soluble in acetone, as would be expected. Partial solubility in 1,4-dioxane, ethanol, methanol, ethyl acetate, and tetrahydrofuran remained unchanged between unmodified kraft lignin and 10%A LNPs. These results indicate that while LNPs may have improved colloidal stability in water systems, their solubility in organic solvents is not greatly altered by this treatment process.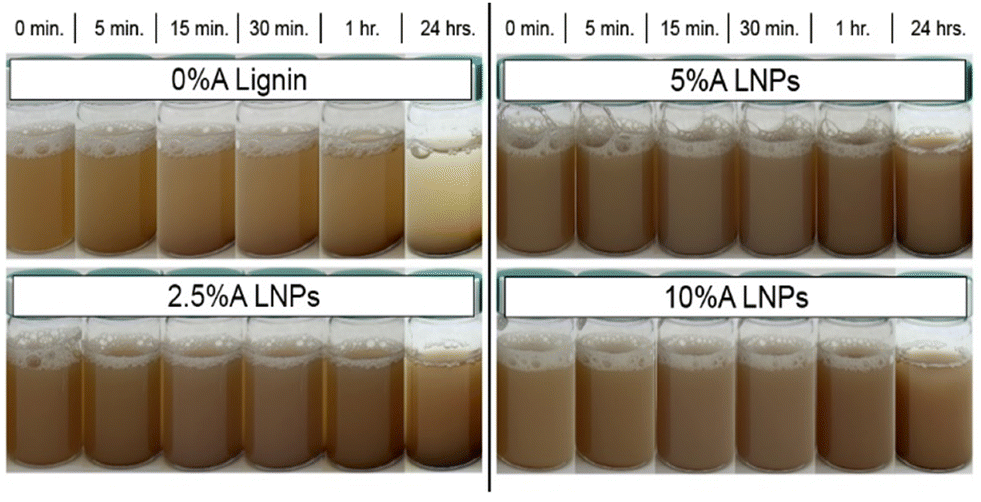 | ||
| Fig. 8 Images of sample settling over time. All samples demonstrated improved stability compared with 0%A lignin, with 10%A LNPs showing the least settling over 24 hours. | ||
Conclusions
Uniform, spherical LNPs were synthesized by a novel process, using subcritical H2O and low volumes of organic solvent. The SAS approach was successfully replicated by using a subcritical H2O solution with recyclable acetone as the solvent phase for kraft lignin, and an ice water bath as the antisolvent phase. The produced nanoparticles at a concentration of 10% acetone showed minimal aggregation, with individualized, well-defined lignin spheres (diameter, d < 1000 nm). Importantly, acetone-assisted nanoparticle samples showed smaller mean particle size, higher end-product yield, improved suspension stability in water, and higher thermal stability, compared with pure-scH2O samples (0%A lignin). The proposed organic solvent lean system can be considered a more sustainable (vs. traditional SAS approach), and efficient method to produce a renewable, bio-based material in the form of LNPs. Such a chemically and morphologically uniform lignin nanomaterials have potential use in a wide range of high-value applications, including Pickering emulsion stabilization, polymer additives, and functionalized lignin products.Author contributions
E. J. F.: conceptualization, resources, writing – review & editing, supervision, and funding acquisition. P. S. M.: methodology, formal analysis, investigation, data curation, writing – original draft preparation. M. H.: methodology, writing – review and editing. F. B. S.: investigation, writing – review and editing. V. F.: investigation, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support provided by the NSERC Canfor Industrial Research Chair in Advanced Bioproducts, (# 553449 - 19), NSERC Discovery Grant (RGPIN-2021-03172), the Canada Foundation for Innovation (Project number 022176) and Pacific Economic Development Canada (PacifiCan). UBC Centre for High-Throughput Phenogenomics is acknowledged for providing SEM imaging facilities. R. Osei-Bonsu is gratefully acknowledged for AFM imaging.Notes and references
- N. R. Maddela, B. Ramakrishnan, D. Kakarla, K. Venkateswarlu and M. Megharaj, RSC Adv., 2022, 12, 12396–12415 RSC.
- L. A. Zevallos Torres, A. Lorenci Woiciechowski, V. O. de Andrade Tanobe, S. G. Karp, L. C. Guimarães Lorenci, C. Faulds and C. R. Soccol, J. Cleaner Prod., 2020, 263, 121499 CrossRef CAS.
- T. D. H. Bugg, J. J. Williamson and G. M. M. Rashid, Curr. Opin. Chem. Biol., 2020, 55, 26–33 CrossRef CAS PubMed.
- P. Bajpai, Biermann's handbook of pulp and paper: raw material and pulp making, Elsevier, Amsterdam, Netherlands, Cambridge, MA, 3rd edn, 2018 Search PubMed.
- G. A. Smook, Handbook for pulp & paper technologists, Tappi Press, Peachtree Corners, GA, 4th edn, 2016 Search PubMed.
- A. Boarino and H.-A. Klok, Biomacromolecules, 2023, 24, 1065–1077 CrossRef CAS PubMed.
- Y. Zhang and M. Naebe, ACS Sustainable Chem. Eng., 2021, 9, 1427–1442 CrossRef CAS.
- X. Lu, X. Gu and Y. Shi, Int. J. Biol. Macromol., 2022, 210, 716–741 CrossRef CAS PubMed.
- L. E. Low, K. C. Teh, S. P. Siva, I. M. L. Chew, W. W. Mwangi, C. L. Chew, B.-H. Goh, E. S. Chan and B. T. Tey, Environ. Nanotechnol., Monit. Manage., 2021, 15, 100398 CAS.
- Y. Qian, Q. Zhang, X. Qiu and S. Zhu, Green Chem., 2014, 16, 4963–4968 RSC.
- Y. Qian, X. Qiu, X. Zhong, D. Zhang, Y. Deng, D. Yang and S. Zhu, Ind. Eng. Chem. Res., 2015, 54, 12025–12030 CrossRef CAS.
- M. S. Alqahtani, A. Alqahtani, A. Al-Thabit, M. Roni and R. Syed, J. Mater. Chem. B, 2019, 7, 4461–4473 RSC.
- T. Zou, M. H. Sipponen, A. Henn and M. Österberg, ACS Nano, 2021, 15, 4811–4823 CrossRef CAS PubMed.
- B. Zhao, M. Borghei, T. Zou, L. Wang, L.-S. Johansson, J. Majoinen, M. H. Sipponen, M. Österberg, B. D. Mattos and O. J. Rojas, ACS Nano, 2021, 15, 6774–6786 CrossRef CAS PubMed.
- J. Wang, W. Chen, D. Yang, Z. Fang, W. Liu, T. Xiang and X. Qiu, ACS Nano, 2022, 16, 20705–20713 CrossRef CAS PubMed.
- S. Beisl, A. Miltner and A. Friedl, Int. J. Mol. Sci., 2017, 18, 1244 CrossRef CAS PubMed.
- S. Iravani and R. S. Varma, Green Chem., 2020, 22, 612–636 RSC.
- A. Lourenço and J. Gominho, in Lignin, ed. A. Sand and J. Tuteja, IntechOpen, Rijeka, 2023, ch. 7 Search PubMed.
- W. Gao and P. Fatehi, Can. J. Chem. Eng., 2019, 97, 2827–2842 CrossRef CAS.
- Y. Pu, J.-X. Wang, D. Wang, N. R. Foster and J.-F. Chen, Chem. Eng. Process., 2019, 140, 36–42 CrossRef CAS.
- M. S. Celiktas, M. Yaglikci and F. Khosravi Maleki, Polym. Test., 2019, 77, 105918 CrossRef.
- A. Martínez-Abad, N. Giummarella, M. Lawoko and F. Vilaplana, Green Chem., 2018, 20, 2534–2546 RSC.
- Y. Qian, C. Zuo, J. Tan and J. He, Energy, 2007, 32, 196–202 CrossRef CAS.
- J. Lappalainen, D. Baudouin, U. Hornung, J. Schuler, K. Melin, S. Bjelić, F. Vogel, J. Konttinen and T. Joronen, Energies, 2020, 13, 3309 CrossRef CAS.
- F. de Boer, PhD Thesis, Utrecht University, 2019 Search PubMed.
- M. Uematsu and E. U. Franck, J. Phys. Chem. Ref. Data, 1980, 9, 1291–1306 CrossRef CAS.
- B. M. Nolan, E. K. Chan, X. Zhang, E. Muthuswamy, K. van Benthem and S. M. Kauzlarich, ACS Nano, 2016, 10, 5391–5397 CrossRef CAS PubMed.
- M. Meyns, S. Willing, H. Lehmann and C. Klinke, ACS Nano, 2015, 9, 6077–6087 CrossRef CAS PubMed.
- D. Rudolph, N. Redinger, K. Schwarz, F. Li, G. Hädrich, M. Cohrs, L. A. Dailey, U. E. Schaible and C. Feldmann, ACS Nano, 2023, 17, 9478–9486 CrossRef CAS PubMed.
- H. K. Valo, P. H. Laaksonen, L. J. Peltonen, M. B. Linder, J. T. Hirvonen and T. J. Laaksonen, ACS Nano, 2010, 4, 1750–1758 CrossRef CAS PubMed.
- P. Figueiredo, K. Lintinen, A. Kiriazis, V. Hynninen, Z. Liu, T. Bauleth-Ramos, A. Rahikkala, A. Correia, T. Kohout, B. Sarmento, J. Yli-Kauhaluoma, J. Hirvonen, O. Ikkala, M. A. Kostiainen and H. A. Santos, Biomaterials, 2017, 121, 97–108 CrossRef CAS PubMed.
- L. Chen, Y. Shi, B. Gao, Y. Zhao, Y. Jiang, Z. Zha, W. Xue and L. Gong, ACS Sustainable Chem. Eng., 2020, 8, 714–722 CrossRef CAS.
- V. Dighe, P. K. R. Podupu, P. Coliaie and M. R. Singh, Cryst. Growth Des., 2022, 22, 3119–3127 CrossRef.
- I. Pylypchuk and M. H. Sipponen, Green Chem., 2022, 24, 8705–8715 RSC.
- A. Manisekaran, P. Grysan, B. Duez, D. F. Schmidt, D. Lenoble and J.-S. Thomann, J. Colloid Interface Sci., 2022, 626, 178–192 CrossRef CAS PubMed.
- C. Liu, Y. Li and Y. Hou, Eur. Polym. J., 2019, 112, 15–23 CrossRef CAS.
- G. Carr, R. Mammucari and N. R. Foster, Chem. Eng. J., 2011, 172, 1–17 CrossRef.
- J. Zhang, Z. Tian, X. Ji and F. Zhang, Polymer, 2022, 14, 4196 CAS.
- M.-W. Park and S.-D. Yeo, Chem. Eng. Res. Des., 2012, 90, 2202–2208 CrossRef CAS.
- Y. Wang, J. Song, S. F. Chow, A. H. L. Chow and Y. Zheng, Int. J. Pharm., 2015, 494, 479–489 CrossRef CAS PubMed.
- H. M. Yusoff, L. K. Li, S. Izhar and M. S. Md Said, Case Stud. Therm. Eng., 2021, 28, 101631 CrossRef.
- Y. Chang, J. Lee, S. Wu, C.-C. Chen and C. Shu, J. Therm. Anal. Calorim., 2009, 95, 525–534 CrossRef CAS.
- X. Li, J. Shen, B. Wang, X. Feng, Z. Mao and X. Sui, ACS Sustainable Chem. Eng., 2021, 9, 5470–5480 CrossRef CAS.
- M. Lievonen, J. J. Valle-Delgado, M.-L. Mattinen, E.-L. Hult, K. Lintinen, M. A. Kostiainen, A. Paananen, G. R. Szilvay, H. Setälä and M. Österberg, Green Chem., 2016, 18, 1416–1422 RSC.
- Q. Lu, M. Zhu, Y. Zu, W. Liu, L. Yang, Y. Zhang, X. Zhao, X. Zhang, X. Zhang and W. Li, Food Chem., 2012, 135, 63–67 CrossRef CAS.
- S. R. Yearla and K. Padmasree, J. Exp. Nanosci., 2016, 11, 289–302 CrossRef CAS.
- P. Figueiredo, M. H. Lahtinen, M. B. Agustin, D. M. de Carvalho, S.-P. Hirvonen, P. A. Penttilä and K. S. Mikkonen, ChemSusChem, 2021, 14, 4718–4730 CrossRef CAS PubMed.
- C. H. M. Camargos and C. A. Rezende, Int. J. Biol. Macromol., 2021, 193, 647–660 CrossRef CAS PubMed.
- M. Morsali, A. Moreno, A. Loukovitou, I. Pylypchuk and M. H. Sipponen, Biomacromolecules, 2022, 23, 4597–4606 CrossRef CAS PubMed.
- B. Wang, D. Sun, H.-M. Wang, T.-Q. Yuan and R.-C. Sun, ACS Sustainable Chem. Eng., 2019, 7, 2658–2666 CrossRef CAS.
- I. Dai, R. Liu, L.-Q. Hu, Z.-F. Zou and C.-L. Si, ACS Sustainable Chem. Eng., 2017, 5, 8241–8249 CrossRef.
- T. Leskinen, M. Smyth, Y. Xiao, K. Lintinen, M.-L. Mattinen, M. A. Kostiainen, P. Oinas and M. Österberg, Nord. Pulp Pap. Res. J., 2017, 32, 586–596 CrossRef CAS.
- Y. Qian, X. Zhong, Y. Li and X. Qiu, Ind. Crops Prod., 2017, 101, 54–60 CrossRef CAS.
- C. Ren, M. Li, W. Huang, Y. Zhang and J. Huang, J. Mater. Sci., 2022, 57, 18356–18369 CrossRef CAS.
- H. Trevisan and C. A. Rezende, Ind. Crops Prod., 2020, 145, 112105 CrossRef CAS.
- F. Xiong, Y. Han, S. Wang, G. Li, T. Qin, Y. Chen and F. Chu, Ind. Crops Prod., 2017, 100, 146–152 CrossRef CAS.
- J. Zhang, Z. Tian, X.-X. Ji and F. Zhang, Int. J. Biol. Macromol., 2023, 231, 123244 CrossRef CAS PubMed.
- T. Zou, N. Nonappa, M. Khavani, M. Vuorte, P. Penttilä, A. Zitting, J. J. Valle-Delgado, A. M. Elert, D. Silbernagl, M. Balakshin, M. Sammalkorpi and M. Österberg, J. Phys. Chem. B, 2021, 125, 12315–12328 CrossRef CAS PubMed.
- I. V. Pylypchuk, P. A. Lindén, M. E. Lindström and O. Sevastyanova, ACS Sustainable Chem. Eng., 2020, 8, 13805–13812 CrossRef CAS.
- M. E. Jawerth, C. J. Brett, C. Terrier, P. T. Larsson, M. Lawoko, S. V. Roth, S. Lundmark and M. Johansson, ACS Appl. Polym. Mater., 2020, 2, 668–676 CrossRef CAS.
- A. Moreno, J. Liu, R. Gueret, S. E. Hadi, L. Bergström, A. Slabon and M. H. Sipponen, Angew. Chem., Int. Ed., 2021, 60, 20897–20905 CrossRef CAS PubMed.
- A. Moreno, J. Liu, M. Morsali and M. H. Sipponen, in Micro and Nanolignin in Aqueous Dispersions and Polymers, ed. D. Puglia, C. Santulli and F. Sarasini, Elsevier, 2022, pp. 385–431 Search PubMed.
- P. Bowen, J. Dispersion Sci. Technol., 2002, 23, 631–662 CrossRef CAS.
- S. Kubo and J. F. Kadla, J. Wood Chem. Technol., 2008, 28, 106–121 CrossRef CAS.
- M. Poletto, Maderas: Cienc. Tecnol., 2017, 19, 63–74 CAS.
- Z. Zhang, C. Belda Marín, M. Lefebvre, C. Lefebvre, V. Terrasson and E. Guénin, Int. J. Biol. Macromol., 2022, 222, 1830–1839 CrossRef CAS PubMed.
- A. Myint, H. W. Lee, B. Seo, W.-S. Son, J. Yoon, T. J. Yoon, H. J. Park, J. Yu, J. Yoon and Y.-W. Lee, Green Chem., 2016, 18, 2129–2146 RSC.
- S. Nair, S. Sharma, Y. Pu, Q. Sun, S. Pan, J. Y. Zhu, Y. Deng and A. Ragauskas, ChemSusChem, 2014, 7, 3513–3520 CrossRef CAS PubMed.
- J.-Y. Kim, E.-J. Shin, I.-Y. Eom, K. Won, Y. H. Kim, D. Choi, I.-G. Choi and J. W. Choi, Bioresour. Technol., 2011, 102, 9020–9025 CrossRef CAS PubMed.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 4862–4887 RSC.
- J.-Y. Kim, H. Hwang, S. Oh, Y.-S. Kim, U.-J. Kim and J. W. Choi, Int. J. Biol. Macromol., 2014, 66, 57–65 CrossRef CAS PubMed.
- Y. Shi, M. J. van Steenbergen, E. A. Teunissen, L. Novo, S. Gradmann, M. Baldus, C. F. van Nostrum and W. E. Hennink, Biomacromolecules, 2013, 14, 1826–1837 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional AFM images, CHNS Elemental Analysis of samples, Fourier-transform infrared spectroscopy and accompanying discussion, 7 pages. See DOI: https://doi.org/10.1039/d3re00424d |
| This journal is © The Royal Society of Chemistry 2024 |