
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous flow synthesis of the ionizable lipid ALC-0315†
Jakob B.
Wolf
 ab,
Ju Weon
Lee
c,
Matthew B.
Plutschack
a,
Dario
Cambié
a,
Andreas
Seidel-Morgenstern
cd and
Peter H.
Seeberger
*ab
ab,
Ju Weon
Lee
c,
Matthew B.
Plutschack
a,
Dario
Cambié
a,
Andreas
Seidel-Morgenstern
cd and
Peter H.
Seeberger
*ab
aDepartment of Biomolecular Systems, Max-Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: Peter.Seeberger@mpikg.mpg.de
bDepartment of Chemistry and Biochemistry, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany
cDepartment of Physical and Chemical Foundations of Process Engineering, Max-Planck-Institute for Dynamics of Complex Technical Systems, Sandtorstraße 1, 39106 Magdeburg, Germany
dInstitute of Process Engineering, Otto von Guericke University Magdeburg, Universitätsplatz 2, 39106 Magdeburg, Germany
First published on 8th January 2024
Abstract
The ionizable lipid ALC-0315 is the major component of the lipid nanoparticles used to encapsulate the mRNA in the Biontech–Pfizer COVID-19 vaccine. In early 2021, difficulties encountered in scaling up the production and purification of this lipid impeded vaccine manufacturing, leading to delays in vaccination campaigns designed to combat the global coronavirus pandemic. Flow chemistry has impacted synthetic capabilities on all scales. This work describes a proof of principle continuous flow process comprised of four telescoped steps for the production of ALC-0315 with an overall yield of 20% and a productivity of 7 mmol per hour.
Introduction
During the COVID-19 pandemic, mRNA-based vaccines lived up to their promise as they can be developed rapidly, are highly potent, and safe.1,2 Like other RNA-based therapeutics, vaccine stability is limited due to the ubiquitous presence of RNases. RNA interference (RNAi)-based therapeutics have addressed this issue through the development of lipid nanoparticles (LNPs) as delivery systems. These LNPs play a crucial role in incorporating and protecting the negatively charged RNA payload, facilitating its penetration and release.3 The approval of the first RNAi-based drug in 2018 was a milestone but until 2021 RNA-based therapeutics were limited to low-volume products for the treatment of rare diseases. Consequently, the components required to assemble LNPs were produced on small scale. Key components driving the formation of these LNPs are cationic or ionizable cationic lipids, such as ALC-0315 (1), that balance the negative charges of the encapsulated nucleic acid (see Fig. 1). | ||
| Fig. 1 a) Illustration depicting the role of ionizable lipids in lipid nanoparticles (LNPs) for mRNA delivery.4,5 b) Structure of ALC-0315, a key component in the formulation of Pfizer–BioNTech COVID-19 vaccine. | ||
The need for mRNA-based vaccines during the COVID-19 pandemic required large-scale production to address the global health crisis. Robust and scalable processes are needed to synthesize the ionizable lipids constituting the LNPs used as delivery systems for these vaccines. We focused on ALC-0315,6 the ionizable lipid utilized in the Pfizer COVID-19 vaccine,7 since in early 2021 its production was a bottleneck for vaccine procurement.8
In pursuit of an improved, scalable synthetic route for ALC-0315, we decided to investigate a telescoped flow-based approach to reduce purification steps and explore continuous purification methods like simulated moving bed chromatography (SMBC). Continuous-flow synthesis is an enabling technology in the pharmaceutical industry that allows for improved safety, precise reaction control, high efficiency, and scalability.9–12 To expedite the optimization process, we used in-line analytics within the continuous-flow synthesis setup. Real-time monitoring and analysis of key parameters could be developed into PAT for large-scale production. This would be attractive, since rapid process control and prompt shutdown in the event of any deviations minimize material loss.13 ALC-0315 is an ionizable, liquid amino lipid with two degradable hexyldecanoate ester tails. The retrosynthetic analysis (Fig. 1b) presents clear disconnections, and the patented process for this compound uses inexpensive starting materials that are available on scale.6 However, the patented procedure employs halogenated solvents, toxic reagents (e.g., Cr(IV) in pyridinium chlorochromate (PCC)), and necessitates chromatographic purification at each step. Our goal was to devise an intensified and more sustainable process, substituting halogenated solvents, and ideally using the same solvent throughout the entire process.14 Furthermore, we sought to avoid the use of toxic reagents and aimed to avoid chromatographic purification steps by using in-line purification such as simple liquid–liquid extractions. Since ionizable lipids used as excipients in mRNA-encapsulating LNPs usually require a chromatographic purification of the final product,15 we aimed to develop a flow synthesis that in principle is amenable to continuous purification to potentially circumnavigate batch processing bottlenecks.
At the outset of our investigation, syntheses of ALC-0315 were exclusively disclosed as patents.6,16–18 Since then, several alternative synthetic routes have also been published (Scheme 1).19–21 The more recent syntheses proceed via an amine alkylation rather than a reductive amination as in the original patents.22 For example, in the patent by Dong and Tang, 6-bromohexanol (Scheme 1a) was acylated with 2-hexyldecanoic acid 2 using catalytic 4-methylbenzene-1-sulfonic acid (TsOH) to yield 7 in 76% yield, enabling a di-alkylation of 4-aminobutanol to produce ALC-0315 in 87% yield (Scheme 1d).19,22 Saadati et al. recently reported the same synthetic approach, albeit with different conditions for the acylation (86%) and amine di-alkylation (55%) (Scheme 1b and e).23 Yet another amine-alkylation approach starts with the alkylation of 2-hexyldecanoic acid 2 with 1,6-dibromohexane (Scheme 1c) to yield 7 in 85% yield, with the subsequent amine di-alkylation yielding ALC-0315 in 56% yield (Scheme 1f).20
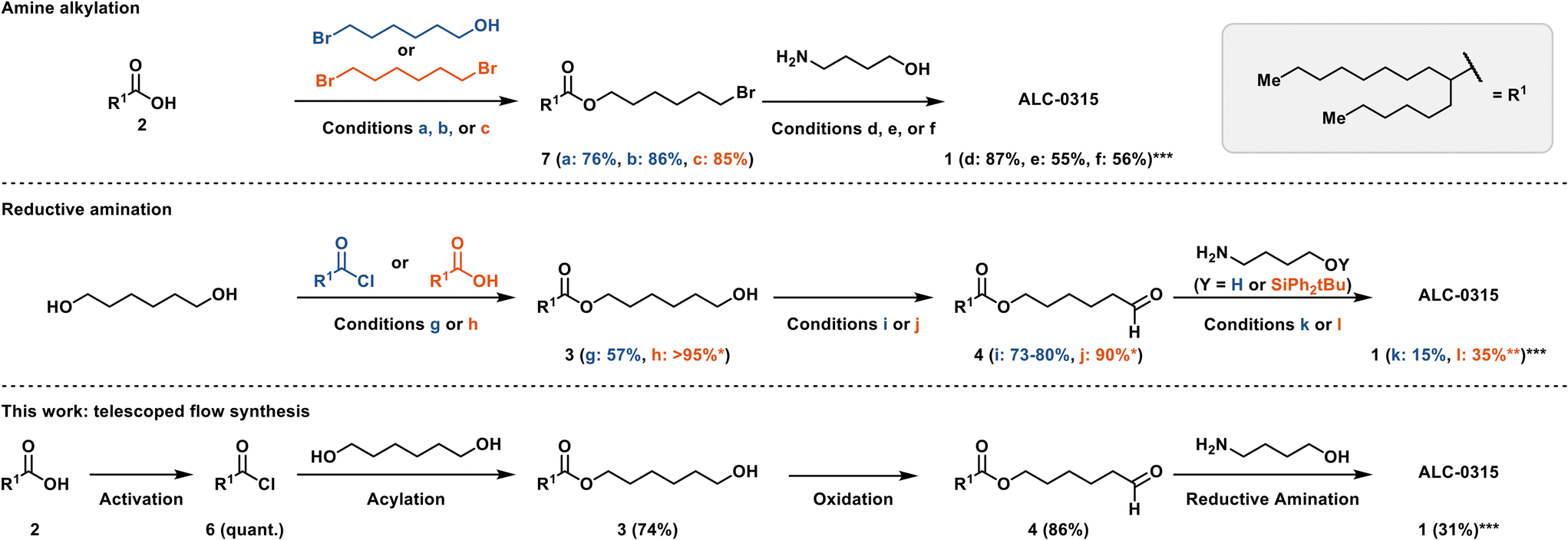 | ||
| Scheme 1 Detailed synthetic conditions representing prior art and this work. a: Acid 2 (1.1 equiv.), TsOH (2.2 mol%), bromohexanol (1 equiv.), toluene, Dean–Stark.19 b: Acid 2 (1.1 equiv.), bromohexanol (1 equiv.), H2SO4 (cat.), cyclohexane, Dean–Stark, 48 h.23 c: Acid 2 (1 equiv.), dibromohexane (4 equiv.), K2CO3 (1.5 equiv.), DMF, 72 h.20 d: Bromide 7 (2.1 equiv.), 4-aminobutanol (1 equiv.), K2CO3 (1 equiv.) or KOH (2 equiv.), KI (9.4 mol%), NH4Cl (0.2 equiv.) benzyltriethylammonium chloride (4.4 mol%), toluene.19 e: Bromide 7 (2.3 equiv.), 4-aminobutanol (1 equiv.), Na2CO3 (2 equiv.), NaI (0.55 equiv.), acetonitrile, 75 °C, sealed reactor, 20 h.23 f: Bromide 7 (2.2 equiv.), 4-aminobutanol (1 equiv.), K2CO3 (2.2 equiv.), 72 h, DMF.20 g: Acyl chloride 6 (1 equiv.), 1,6-hexandiol (2.3 equiv.), triethylamine (2 equiv.), THF/CH2Cl2, 1 h.16,17,24 h: Acid 2 (1 equiv.), DCC (1.1 equiv.), DMAP (1.2 equiv.), CH2Cl2, 72 h.6,18,24 i: Ester 3 (1 equiv.), PCC (1.16 equiv.), CH2Cl2, 2 h.6,18 j: Ester 3 (1 equiv.) TEMPO (0.2 equiv.), bleach, NaHCO3 aq. (sat.), CH2Cl2, 2 h.21 k: Aldehyde 4 (2.6 equiv.), 4-aminobutanol (1 equiv.), sodium triacetoxyborohydride (2.4 equiv.), acetic acid (2.1 equiv.), CH2Cl2, 2 h.6,16–18,24 l: Aldehyde 4 (2.3 equiv.), sodium tripropionyloxy borohydride (3 equiv.), acetic acid (cat.), 4-aminobutanol (1 equiv.), CH2Cl2, 0 °C → RT, 3 h, deprotection: HF-pyridine (4 equiv.), THF, 0 °C, overnight.21 See ESI† S14 for a comparison of overall yields. *No purification reported, crude contains diacetylated ester; **yield includes deprotection step; ***yield is calculated based on compound 4/7. | ||
The original patents, however, described a synthetic approach based on a reductive amination strategy (Scheme 1). For example, 1,6-hexanediol could either be acylated using 2-hexyldecanoyl chloride 6 (Scheme 1g) to yield 3 in 57% yield16,17,24 or directly coupled with the free acid 2via a Steglich-type esterification (Scheme 1h) to yield crude 3 in quantitative yield.6,18,24 In the Acuitas patent, the resulting monoester 3 was oxidized with PCC (Scheme 1i) to yield aldehyde 4 in 73–80% yield.6,18 The subsequent reductive amination used sodium triacetoxyborohydride (STAB-H) to produce ALC-0315 in 15% yield (Scheme 1k).6,16–18,24 The article by Saadati et al. replaced PCC with an Anelli-like oxidation (Scheme 1j) and used the silylether protected 4-aminobutanol in the reductive amination to improve the yield to 35% (Scheme 1l).21 The patent procedures in particular, offered robust batch reaction conditions as a starting point for further exploration in flow on a laboratory scale.
Results and discussion
Solvents to carry through all steps were screened to facilitate successful telescoping of the overall process, and attempts were made to exclude toxic solvents (ex. CH2Cl2), reactive solvents (ex. alcohols and ketones) as well as solvents that complicate biphasic separations. 2-Methyltetrahydrofuran (2-MeTHF) emerged as the most attractive candidate since it can be produced from renewable resources,25 is considered a green solvent,14 has a higher boiling point (80 °C) than THF and requires lower pressure for superheating.26Acid activation
Two routes for the monoesterification of 1,6-hexanediol rely on the use of the corresponding acyl chloride 6 (Scheme 1g), that is derived from the acid 2, which can alternatively directly be activated with N,N′-dicyclohexylcarbodiimide (DCC, Scheme 1h)6 to produce crude monoester 3. We initially investigated the acyl chloride route, as the homogeneous reaction conditions offered, in contrast to DCC, higher atom-economy, lower cost, facile separation and a straightforward conversion to flow.27 A telescoped process of chlorination–esterification would afford the desired monoester rapidly and with high productivity.To activate the acid, we explored the use of thionyl chloride, carbonyldiimidazole and bis(trichloromethyl)carbonate (BTC). Inexpensive thionyl chloride is commonly used for acyl chloride formation with good atom economy. Thionyl chloride was particularly attractive for our flow approach, since it is liquid and we could pump it neat, resulting in higher reaction concentrations, thereby avoiding a significant drop in productivity (Scheme 2). Preliminary batch experiments showed complete conversion of the acid to the corresponding chloride overnight at room temperature. Heating to 80 °C in flow resulted in complete reaction after 20 min (see page S4 ESI† for more details on optimization). Without proper isolation of the thionyl chloride delivery unit, pumping neat thionyl chloride resulted in corrosion damage to nearby electronic devices such as syringe pumps (see ESI† for details). The use of DMF is discouraged in the EU due to concerns about reproductive toxicity and only is allowed if sufficient measure is taken to limit exposure of staff.28 Carbonyldiimidazole, a milder alternative, that fully activated the acid in batch, was too poorly soluble in 2-MeTHF for use in flow. Reaction of the imidazole-activated ester resulted in low conversions even under superheating conditions and prolonged reaction times. Attempts to use BTC as a solid phosgene equivalent to avoid the reactive sulphur-based byproducts observed with thionyl chloride presented safety risks. Notably, an off-white solid accumulated on the PTFE-tubing that was likely the result of BTC diffusing through the tubing walls.29 Despite the necessity to use DMF with SOCl2, further reagent exploration was halted, as proper isolation of the SOCl2 pumping module from the rest of the equipment easily resolved the electronics issue and other reagents posed a more acute safety risk in our opinion.
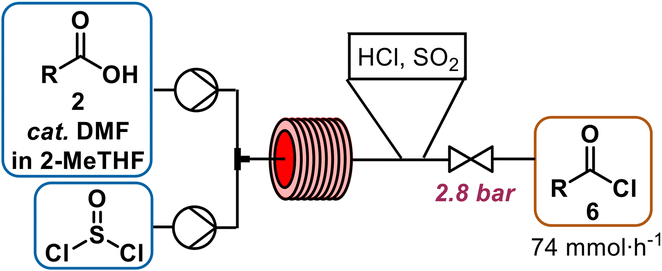 | ||
| Scheme 2 Activation of carboxylic acid 2 in 2-MeTHF (1.4 M) with cat. DMF (5 mol%) by transformation to acyl chloride 6 at 80 °C for 20 min with 1.1 equiv. of neat thionylchloride. | ||
An in-line infrared (IR) spectrometer was used to optimize the temperature, residence time, SOCl2 and N,N-dimethylformamide (DMF) equivalents for the new flow conditions. The distinct difference in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O-stretches of the carboxylic acid (1700 cm−1) and the acid chloride (1797 cm−1) allowed for real-time optimization (refer to ESI† for details on PAT use).‡ Temperatures higher or equal to 100 °C resulted in a brown reaction mixture, indicating decomposition. Acyl chloride was obtained in quantitative yield within 20 min residence time at 80 °C, when thionyl chloride was used in slight excess (1.1 equiv.). Full conversion was very important to avoid residual free acid 2 (Scheme 2), because unreacted carboxylic acid is deprotonated in the downstream oxidation conditions, leading to the formation of a difficult-to-pump viscous emulsion. While not implemented in our telescoped process, this complication combined with the rapid sampling rate of IR monitoring renders it as a potentially useful PAT for production.13
O-stretches of the carboxylic acid (1700 cm−1) and the acid chloride (1797 cm−1) allowed for real-time optimization (refer to ESI† for details on PAT use).‡ Temperatures higher or equal to 100 °C resulted in a brown reaction mixture, indicating decomposition. Acyl chloride was obtained in quantitative yield within 20 min residence time at 80 °C, when thionyl chloride was used in slight excess (1.1 equiv.). Full conversion was very important to avoid residual free acid 2 (Scheme 2), because unreacted carboxylic acid is deprotonated in the downstream oxidation conditions, leading to the formation of a difficult-to-pump viscous emulsion. While not implemented in our telescoped process, this complication combined with the rapid sampling rate of IR monitoring renders it as a potentially useful PAT for production.13
Acylation
Initially, the subsequent esterification (Scheme 3) was attempted with a base to neutralize the HCl generated (2 equiv.) in the activation step. N-Methylimidazole was tested to not only catalyse the esterification by formation of an acyl imidazolium intermediate,30 but also neutralize the reaction mixture. In order to optimize the mono![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diester selectivity, we screened various stoichiometric amounts of 1,6-hexanediol. Equimolar amounts of acyl chloride and 1,6-hexanediol resulted in significant formation of diester, and increasing the equivalents of diol did not sufficiently improve the selectivity toward the desired monoacylated product (see ESI† for further information). A biphasic mixture was present at the reactor outlet, with the denser phase containing 1,6-hexanediol and methylimidazolium chloride (1H-NMR). Therefore, we hypothesized that monoacylation selectivity could be mixing dependent, with insufficient mixing of diol and acyl chloride as a result of the phase separation, resulting in more undesired diacetylated side product. Other amines such as diisopropylethylamine and triethylamine led to reactor fouling due to the low solubility of the corresponding hydrochlorides in 2-MeTHF. The use of tributylamine prevented precipitation, however, quantitative conversion was achieved as well by heating the acyl chloride and 1,6-hexanediol in the absence of base at 100 °C for 4 min with no detectable decomposition. These conditions did not result in a biphasic mixture and different equivalents of 1,6-hexanediol could more easily be screened to optimize selectivity (see Table 1). Ultimately, three equivalents was an optimal trade-off between selectivity and excessively high amounts of 1,6-hexanediol (Table 1).
:diester selectivity, we screened various stoichiometric amounts of 1,6-hexanediol. Equimolar amounts of acyl chloride and 1,6-hexanediol resulted in significant formation of diester, and increasing the equivalents of diol did not sufficiently improve the selectivity toward the desired monoacylated product (see ESI† for further information). A biphasic mixture was present at the reactor outlet, with the denser phase containing 1,6-hexanediol and methylimidazolium chloride (1H-NMR). Therefore, we hypothesized that monoacylation selectivity could be mixing dependent, with insufficient mixing of diol and acyl chloride as a result of the phase separation, resulting in more undesired diacetylated side product. Other amines such as diisopropylethylamine and triethylamine led to reactor fouling due to the low solubility of the corresponding hydrochlorides in 2-MeTHF. The use of tributylamine prevented precipitation, however, quantitative conversion was achieved as well by heating the acyl chloride and 1,6-hexanediol in the absence of base at 100 °C for 4 min with no detectable decomposition. These conditions did not result in a biphasic mixture and different equivalents of 1,6-hexanediol could more easily be screened to optimize selectivity (see Table 1). Ultimately, three equivalents was an optimal trade-off between selectivity and excessively high amounts of 1,6-hexanediol (Table 1).
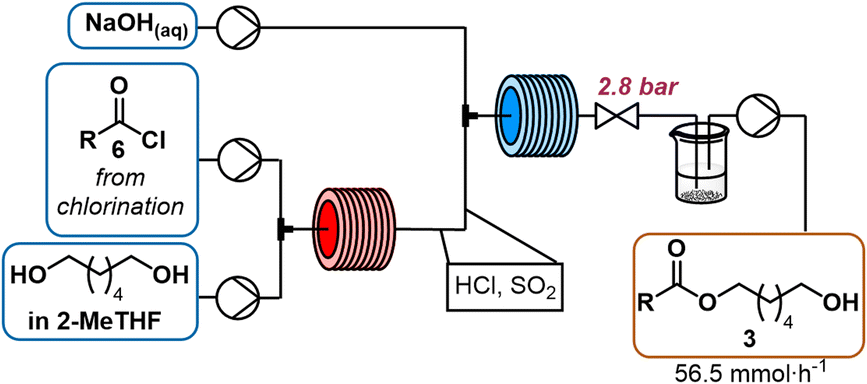 | ||
| Scheme 3 Flow scheme for the acylation of 1,6-hexanediol in 2-MeTHF (3 equiv., 2.5 M) at 100 °C for 4 min with fatty acyl chloride 6 (1 equiv., crude from previous reaction) and subsequent neutralisation with aqueous NaOH (1 M, 4.4 equiv.) at 0 °C for 2 min to obtain monoester 3. | ||
Oxidation
Various large scale strategies31 focus on high productivity and avoiding chlorinated solvents for alcohol oxidation. The copper-mediated aerobic oxidation32 that had been successfully employed for the SST-01 synthesis by Fujiwara et al.,33 failed to achieve acceptable productivity (2.5 mmol h−1, Fujiwara et al. report 3.3 mmol h−1, normalised to a 10 mL reactor, see ESI† for details). This result is in line with reports that aliphatic substrates react sluggishly compared to benzylic alcohols.34,§ As an alternative, an Anelli-oxidation35 in 2-MeTHF provided a satisfactory yield (70%) at a residence time of 50 s at room temperature, corresponding to 45.9 mmol h−1 of aldehyde and thereby 13-fold increase over the reported aerobic oxidation.36 Telescoping this synthesis of aldehyde 4 by adding sodium carbonate to neutralize the acylation stream reduced conversion to 10% (1H-NMR). Even with excess base, the reactor outlet remained acidic, and the yield was not improved. The instability of technical bleach at pH 9.5 (ref. 37) resulted in varying OCl− concentration over time, complicating reaction optimization. Using NaOCl pentahydrate in a sodium carbonate buffer (pH ∼12)38 to stabilize hypochlorite, improved the conversion, but not sufficiently to telescope the entire synthesis. The Anelli oxidation is strongly pH dependent, pH ∼9.5 is ideal, a higher pH slows down the reaction while lower pH accelerates NaOCl decomposition.37,39 The sulphur dioxide byproduct from thionyl chloride activation may not only consume the oxidant, but also acidify the reaction. Additional evidence supporting this hypothesis was obtained when the crude esterification outlet was concentrated under vacuum to remove volatile SO2, redissolved in 2-MeTHF, and subjected to Anelli conditions, resulting in 71% yield of aldehyde (based on 1H-NMR analysis). Consequently, a porous PTFE membrane (AF2400) in a tube-in-tube setup40 was used to degas the outlet stream by applying vacuum, flowing basified water through the outer tube or bubbling nitrogen through the outlet solution of the acylation reactor. None of these approaches improved the yield for the telescoped process significantly above 10%. Liquid–liquid extraction of the effluent with a saturated NaHCO3 solution in a separation funnel yielded 70% of the aldehyde in subsequent oxidation. The best continuous solution utilized inline extraction with aqueous sodium hydroxide after the acylation reactor (Scheme 3) to afford aldehyde 4 in 70% yield in the subsequent oxidation (Scheme 4). Quenching and separation after acylation not only facilitated telescoped aldehyde synthesis, but also removed excess 1,6-hexanediol, thus preventing the formation of additional aldehydes that could lead to side products in the reductive amination step. | ||
| Scheme 4 Oxidation of primary alcohol 3 to aldehyde 4. Reaction conditions: ester 3 in 2-MeTHF (0.72 M, 1 equiv., from previous step), TEMPO in 2-MeTHF (7 mol%, 0.5 M), 2.3 equiv. aqueous hypochlorite (NaOCl, 2 M and Na2CO3, 0.3 M in water) and 0.35 equiv. aqueous potassium bromide (KBr, 0.25 M and NaHCO3, 0.7 M in water) are reacted at room temperature for 2.3 min. | ||
Purification as a guide for process development
We made efforts to optimize the purification of ALC-0315 early on in this study. ALC-0315 is highly soluble in apolar solvents, while its ionizable polar head and carboxylic esters have strong affinity to polar adsorbent. For normal phase chromatography41 gradient elution was required to isolate the target compound since the reaction mixture contains excess polar and nonpolar reagents as well as byproducts, complicating the design of a continuous chromatographic separation process. Simple gradient elution is preferable ideally involving a single step gradient with two different mobile phases.42A mixture of n-hexane (n-Hex) and ethanol (EtOH) served as a mobile phase with a silica column to isolate the target compound. Since precipitation in the eluent upon contact with the reaction mixture would cause column fouling, a solvent switch to n-Hex relied on solvent removal under reduced pressure to reduce polar impurities.
The target compound was resolved in chromatographic batches to illustrate that continuous chromatography will be possible during further scale-up. For good resolution of non-polar impurities and the target compound, the mobile phase composition was set to n-Hex/EtOH = 97/3 (vol%), and the amount of EtOH was increased to 95 vol% after the target compound eluted to flush strongly retained polar impurities off the column. Prior to the next injection, the column was re-equilibrated to the initial mobile phase composition. This batch separation was performed 50 times and the proposed step-gradient was reproducible (Fig. 2), affording ALC-0315 with purity >98% (according to 1H-NMR analysis). These findings illustrate that it will be possible to telescope the continuous flow process with continuous purification and provided information for simplifying the purification. The secondary amine was retained stronger than the tertiary amine and full conversion during the reductive amination was important as secondary amine impurities would necessitate a gradient separation.
 | ||
| Fig. 2 Batch separation chromatograms demonstrating the reproducibility of the separation protocol over 25 and 50 separations. | ||
Reductive amination
The Acuitas patent served as starting point for optimization of the reductive amination6 and two challenges became immediately apparent: 1) a low yield of tertiary amine (only 15% based on the aldehyde, see ESI† for further details),6 and 2) the insolubility of STAB-H in 2-MeTHF posed difficulties for use in continuous flow.43 However, STAB-H offered high selectivity since it avoided competitive reduction of the aldehyde (Scheme 1i).44 To address these challenges, potential solvent combinations as well as the use of tetramethylammonium salts of triacetoxyborohydride (TMATAB-H) were considered. These ammonium salts are more readily soluble in organic solvents, such as N-methylpyrrolidin-2-one (NMP). Methanol (MeOH) was used as a co-solvent to accelerate iminium-formation.33 Mindful of the purification experiments outline in Fig. 2, special precaution was directed at reducing the quantity of secondary amine in the final product mixture. For this reason, excess aldehyde was employed to fully consume the secondary amine and form the tertiary amine exclusively (refer to ESI† for further details). Different residence times, temperatures and aldehyde equivalents were screened. Higher temperatures did not accelerate tertiary amine formation, likely due to the enhanced degradation of the reducing agent. Ultimately, three equivalents of aldehyde and a residence time of 16 minutes at room temperature were necessary to effectively consume secondary amine to afford tertiary amine (Scheme 5). These reductive amination conditions in flow doubled the yield over the original patent procedure and reached a comparable yield to Saadati's published reductive amination conditions that utilized a protecting group (Scheme 1l).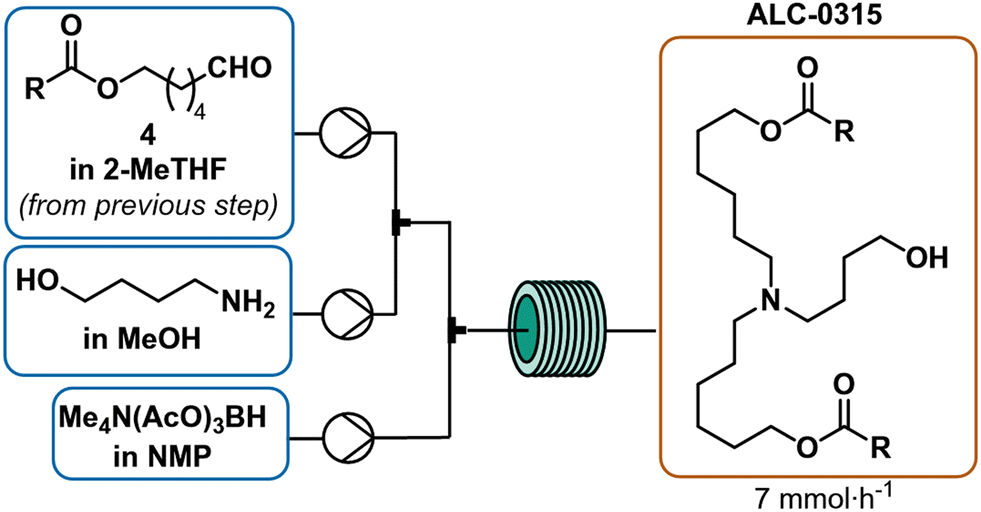 | ||
| Scheme 5 Continuous reductive amination process for the synthesis of the target tertiary amine 1. Reaction conditions: aldehyde 4 in 2-MeTHF (3 equiv., 0.55 M), 4-aminobutanol in methanol (1 equiv., 0.2 M) and tetramethylammonium triacetoxyborohydride in NMP (3 equiv., 0.45 M) are reacted at room temperature for 16 min. | ||
Overall process
Using the continuous flow process (Scheme 6), 7 mmol h−1 (5.4 g h−1) of ALC-0315 can be produced without intermediate purification. Process conditions were selected to eventually enable a fully continuous regime by coupling continuous synthesis with a continuous purification method such as SMBC. Since the process is conducted continuously in flow, it can potentially be scaled more easily than a comparable batch process by leveraging strategies like numbering up, running multiple reactors in parallel, or potentially “sizing-up”, by increasing reactor dimensions and flow rates.45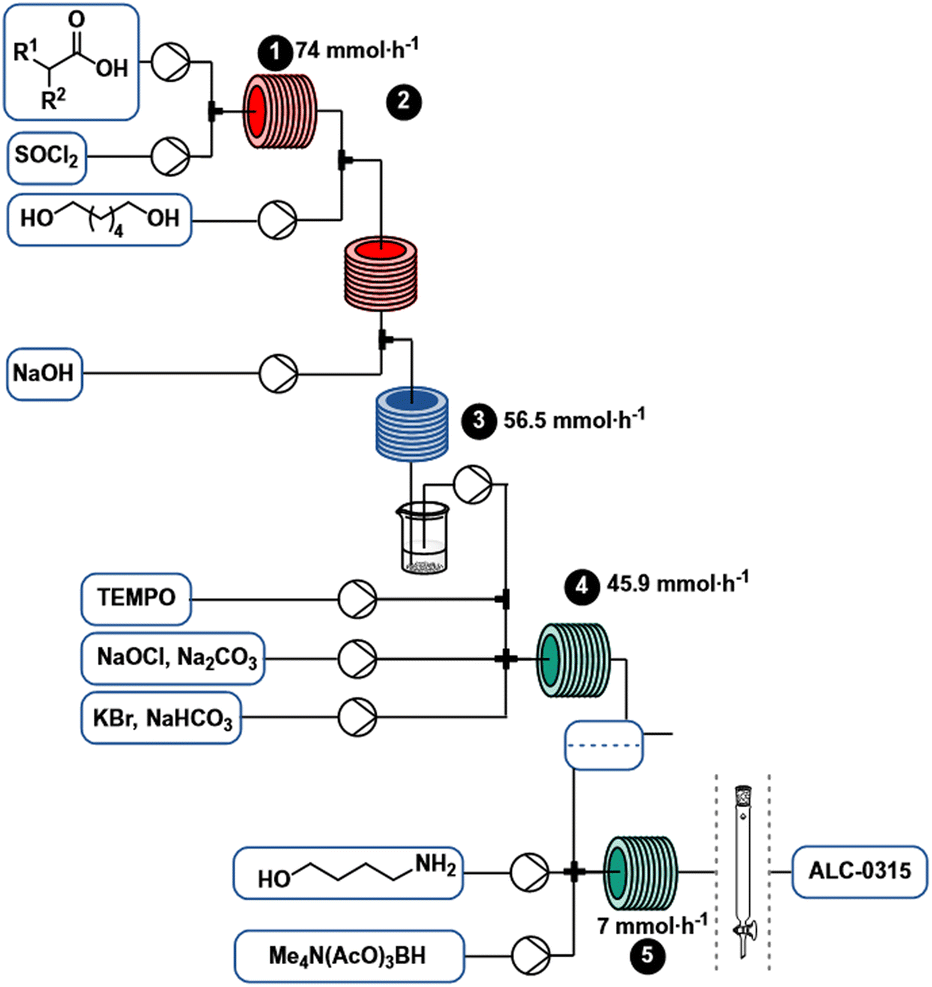 | ||
| Scheme 6 Complete process scheme for the synthesis of ALC-0315. | ||
Experimental
2-Hexyldecanoyl chloride
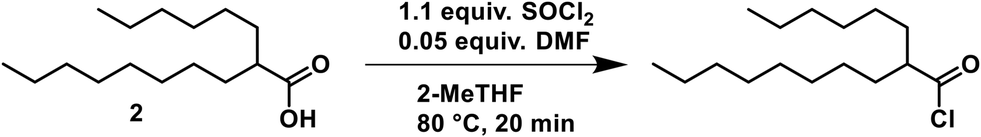 A solution of 2 (1.374 M) and DMF (5 mol%) in 2-MeTHF was pumped via a HPLC-pump (0.901 mL min−1) and mixed in a T-piece with neat thionyl chloride (1.1 equiv.) that was delivered from a continuous syringe pump (0.099 mL min−1). The resulting stream was heated at 80 °C with a residence time of 20 min (20 mL reactor volume).
A solution of 2 (1.374 M) and DMF (5 mol%) in 2-MeTHF was pumped via a HPLC-pump (0.901 mL min−1) and mixed in a T-piece with neat thionyl chloride (1.1 equiv.) that was delivered from a continuous syringe pump (0.099 mL min−1). The resulting stream was heated at 80 °C with a residence time of 20 min (20 mL reactor volume).
6-Hydroxyhexyl-2-hexyldecanoate (3)
 A solution of 1,6-hexanediol in 2-MeTHF (2.50 M) was delivered by a HPLC pump (1.5 mL min−1) and combined with the acid chloride stream via a T-mixer. The resulting stream was heated at 100 °C for 4 min (10 mL reactor). Aqueous sodium hydroxide (1 M) was pumped by an HPLC pump (5.5 mL min−1) and joined with the acylation stream via a T-mixer. The resulting flow was cooled at 0 °C with a residence time of 1.25 min (10 mL reactor). The entire system was held under constant pressure by a backpressure regulator (2.8 bar). The outlet was collected in a beaker where phases separated.
A solution of 1,6-hexanediol in 2-MeTHF (2.50 M) was delivered by a HPLC pump (1.5 mL min−1) and combined with the acid chloride stream via a T-mixer. The resulting stream was heated at 100 °C for 4 min (10 mL reactor). Aqueous sodium hydroxide (1 M) was pumped by an HPLC pump (5.5 mL min−1) and joined with the acylation stream via a T-mixer. The resulting flow was cooled at 0 °C with a residence time of 1.25 min (10 mL reactor). The entire system was held under constant pressure by a backpressure regulator (2.8 bar). The outlet was collected in a beaker where phases separated.
6-Oxohexyl 2-hexyldecanoate (4)
 The organic phase obtained from the acylation was pumped by an HPLC pump (1.6 mL min−1) and mixed in a T-mixer with a solution of TEMPO in 2-MeTHF (0.50 M) at a flow rate of 0.16 mL min−1. The resulting stream was combined in a crossmixer with a solution of NaOCl·5H2O and Na2CO3 in water (2 M and 0.3 M respectively) delivered from a continuous syringe pump (1.33 mL min−1) and a solution of KBr and NaHCO3 in water (0.25 M and 0.7 M respectively) pumped with a HPLC pump (1.6 mL min−1). The resulting continuous biphasic stream was reacted at room temperature with a residence time of 2.3 min (11 mL reactor immersed in a water bath). The reaction mixture color turned from red (near the cross mixer) to almost colorless (toward the end) through the reactor.
The organic phase obtained from the acylation was pumped by an HPLC pump (1.6 mL min−1) and mixed in a T-mixer with a solution of TEMPO in 2-MeTHF (0.50 M) at a flow rate of 0.16 mL min−1. The resulting stream was combined in a crossmixer with a solution of NaOCl·5H2O and Na2CO3 in water (2 M and 0.3 M respectively) delivered from a continuous syringe pump (1.33 mL min−1) and a solution of KBr and NaHCO3 in water (0.25 M and 0.7 M respectively) pumped with a HPLC pump (1.6 mL min−1). The resulting continuous biphasic stream was reacted at room temperature with a residence time of 2.3 min (11 mL reactor immersed in a water bath). The reaction mixture color turned from red (near the cross mixer) to almost colorless (toward the end) through the reactor.
After reaching steady state following two residence times, the biphasic outlet was collected for 10 min and manually separated. An aliquot of the organic phase was analyzed by quantitative NMR with 1,3,5-trimethoxybenzene as internal standard. Collection over 10 minutes gave 13.8 mL of organic phase, with 0.55 M (avg. of three experiment) aldehyde concentration, corresponding to 64% overall yield over three steps.
ALC-0315 ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (1)
 The organic phase from the previous step was combined at a flow rate of 78 μL min−1 in a crossmixer with TMATAB-H in 1-NMP (0.45 M) at a flow rate of 98.5 μL min−1 and 4-aminobutan-1-ol in MeOH (0.2 M) at 74 μL min−1. The resulting stream was reacted in a 4 mL reactor at room temperature (16 min residence time). After reaching steady state (two residence times), the outlet was collected for 50 min and quenched with water, hexanes were added, the biphasic solution was basified to pH 9.5 and the phases separated. The organic phase was dried over Na2SO4, to yield 1.265 g of crude red oil after solvent removal.
The organic phase from the previous step was combined at a flow rate of 78 μL min−1 in a crossmixer with TMATAB-H in 1-NMP (0.45 M) at a flow rate of 98.5 μL min−1 and 4-aminobutan-1-ol in MeOH (0.2 M) at 74 μL min−1. The resulting stream was reacted in a 4 mL reactor at room temperature (16 min residence time). After reaching steady state (two residence times), the outlet was collected for 50 min and quenched with water, hexanes were added, the biphasic solution was basified to pH 9.5 and the phases separated. The organic phase was dried over Na2SO4, to yield 1.265 g of crude red oil after solvent removal.
An aliquot of crude product (106 mg) was purified on a plug of silica, by sequential washes with hexanes (with 0.1 v% of TFA), hexanes/ethyl acetate (4:1, with 0.1 v% of TFA) and pure ethyl acetate (with 0.1 v% of TFA). A slightly yellow oil (22 mg, yield 47% based on 4-aminobutanol, 31% based on aldehyde) was obtained¶ and identity confirmed by 1H- and 13C-NMR and HRMS (see ESI†).
Conclusions
Successful continuous synthesis of ALC-0315 highlights several advantages of flow chemistry. While acylation and reductive amination exhibit satisfactory performance in batch mode, the oxidation step benefits greatly from flow conditions, due to improved heat and mass transfer. The yield of final compound in the reductive amination was doubled compared to the original patent procedure, combined with an outline for simplified purification. This flow synthesis of ALC-0315 offers a proof-of-principle example that is more amenable to scaling and allows for the production of affordable materials on a laboratory scale.|| Small, decentralized production facilities capable of rapid start-up and shutdown could enable emergency production of essential APIs during supply chain disruptions.Conflicts of interest
A patent (WO2022269041A1) dealing with the continuous flow process for the production of cationic lipids has been published on December 29, 2022 with the International Search Report under Publication # WO2022269041A1 with J. W., M. P., D. C., and P. S. as potential beneficiaries from royalty payments.Acknowledgements
We thank the Max-Planck Society and the DFG InCHeM (FOR 2177) for generous financial support. We also thank Biogeneral, Inc. for their kind supply of a sample of Teflon™ AF-2400 tubing. Open Access funding provided by the Max Planck Society.Notes and references
- N. Chaudhary, D. Weissman and K. A. Whitehead, Nat. Rev. Drug Discovery, 2021, 20, 817–838 CrossRef CAS PubMed.
- N. Pardi, M. J. Hogan, F. W. Porter and D. Weissman, Nat. Rev. Drug Discovery, 2018, 17, 261–279 CrossRef CAS PubMed.
- Y. Zhang, C. Sun, C. Wang, K. E. Jankovic and Y. Dong, Chem. Rev., 2021, 121, 12181–12277 CrossRef CAS PubMed.
- A. K. Leung, I. M. Hafez, S. Baoukina, N. M. Belliveau, I. V. Zhigaltsev, E. Afshinmanesh, D. P. Tieleman, C. L. Hansen, M. J. Hope and P. R. Cullis, J. Phys. Chem. C, Nanomater. Interfaces, 2012, 116, 18440–18450 CrossRef CAS PubMed.
- P. R. Cullis and M. J. Hope, Mol. Ther., 2017, 25, 1467–1475 CrossRef CAS PubMed.
- S. M. Ansell and X. Du, (Acuitas Therapeutics Inc), WO2017075531A1, 2017.
- D. M. Hinton, 2021, Pfizer-BioNTech COVID-19 Vaccine EUA Letter of Authorization, https://www.fda.gov/media/144412/download, accessed on 29/6/2023.
- M. McCoy, Chem. Eng. News, 2021, 99(6), 4 CrossRef.
- D. L. Hughes, Org. Process Res. Dev., 2020, 24, 1850–1860 CrossRef CAS.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2015, 20, 2–25 CrossRef.
- A. R. Bogdan and A. W. Dombrowski, J. Med. Chem., 2019, 62, 6422–6468 CrossRef CAS PubMed.
- C. N. Talicska, E. C. O'Connell, H. W. Ward, A. R. Diaz, M. A. Hardink, D. A. Foley, D. Connolly, K. P. Girard and T. Ljubicic, React. Chem. Eng., 2022, 7, 1419–1428 RSC.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- K. Rajappan, S. P. Tanis, S. Roberts, P. Karmali, P. Chivukula, J. Zhuang, M. Xin, M. Zheng and I. Edmonds, Org. Process Res. Dev., 2021, 25, 1383–1390 CrossRef CAS.
- S. M. Ansell and X. Du, (Acuitas Therapeutics Inc), WO2015199952A1, 2015.
- D. Weissman, N. Pardi, Y. Tam and M. Hope, (Univ Pennsylvania; Acuitas Therapeutics Inc; Weissman Drew; Pardi Norbert), WO2018081638A1, 2018.
- M. Hope, B. Mui, P. Lin, C. Barbosa, T. Madden, S. Ansell and X. Du, (Acuitas Therapeutics Inc), WO2018081480A1, 2018.
- Y. Dong and X. Tang, (Silicon Yu Science and Tech Shanghai Limited Company), CN114249662A, 2022.
- I. A. Boldyrev, V. P. Shendrikov, A. G. Vostrova and E. L. Vodovozova, Russ. J. Bioorg. Chem., 2023, 49, 412–415 CrossRef CAS PubMed.
- F. Saadati, S. Cammarone and M. A. Ciufolini, Chem. – Eur. J., 2022, 28, e202200906 CrossRef CAS PubMed.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS PubMed.
- F. Saadati, N. D. P. Atmuri and M. A. Ciufolini, (Nanovation Therapeutics Inc), WO2023173203A1, 2023.
- P. Lin and Y. Tam, (Acuitas Therapeutics Inc), WO2018191719A1, 2018.
- V. Pace, P. Hoyos, L. Castoldi, P. Dominguez de Maria and A. R. Alcantara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- D. F. Aycock, Org. Process Res. Dev., 2006, 11, 156–159 CrossRef.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- Commission Regulation (EU) 2021/2030 of 19 November 2021 amending Annex XVII to Regulation (EC) No 1907/2006 of the European Parliament and of the Council concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) as regards N,N-dimethylformamide, 32021R2030, E. Union, https://eur-lex.europa.eu/eli/reg/2021/2030/oj, 14.12.2023, 2021.
- L. Cotarca, T. Geller and J. Répási, Org. Process Res. Dev., 2017, 21, 1439–1446 CrossRef CAS.
- H. Nakatsuji, M. Morimoto, T. Misaki and Y. Tanabe, Tetrahedron, 2007, 63, 12071–12080 CrossRef CAS.
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed.
- J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901–16910 CrossRef CAS PubMed.
- K. Fujiwara, H. Ishitani and S. Kobayashi, Org. Process Res. Dev., 2020, 24, 1988–1995 CrossRef CAS.
- J. F. Greene, J. M. Hoover, D. S. Mannel, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2013, 17, 1247–1251 CrossRef CAS.
- P. Lucio Anelli, C. Biffi, F. Montanari and S. Quici, J. Org. Chem., 2002, 52, 2559–2562 CrossRef.
- R. H. Yu, R. P. Polniaszek, M. W. Becker, C. M. Cook and L. H. L. Yu, Org. Process Res. Dev., 2007, 11, 972–980 CrossRef CAS.
- E. Fritz-Langhals, Org. Process Res. Dev., 2005, 9, 577–582 CrossRef CAS.
- T. V. Bommaraju, Water Qual. Res. J., 1995, 30, 339–361 CrossRef CAS.
- J. M. Bobbitt, C. Brückner and N. Merbouh, in Organic Reactions, John Wiley & Sons, Ltd, 2010, pp. 103–424 Search PubMed.
- M. Brzozowski, M. O'Brien, S. V. Ley and A. Polyzos, Acc. Chem. Res., 2015, 48, 349–362 CrossRef CAS PubMed.
- J. V. Amari, P. R. Brown, C. M. Grill and J. G. Turcotte, J. Chromatogr., 1990, 517, 219–228 CrossRef CAS PubMed.
- Z. Horváth, E. Horosanskaia, J. W. Lee, H. Lorenz, K. Gilmore, P. H. Seeberger and A. Seidel-Morgenstern, Org. Process Res. Dev., 2015, 19, 624–634 CrossRef.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed.
- C. A. Hone and C. O. Kappe, Chem.: Methods, 2021, 1, 454–467 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00630a |
| ‡ Concentration of 2 higher than 1.0 M resulted in the appearance of a second broad band, centered at 1648 cm−1, assigned to the CO stretch of the acid dimer. |
| § The slightly lower productivities we observed compared to Kobayashi are attributed to the slug flow regime obtained in our case due to the use of pure oxygen as gaseous phase, while Kobayashi's use of 5% O2 in N2 translated in higher gas flowrates and resulted in an annular flow regime which is known to have a higher mass transfer coefficient. |
| ¶ Overall productivity was extrapolated from small-scale productivity in the last step since the required reactor size of 67 mL was not available and would have required excessive amounts of tubing and aldehyde (85 m w 1 mm I.D.). |
| || Despite the ready availability of ALC-0315 from chemical suppliers, the price remains elevated when considering the required quantity for use as an encapsulating lipid, with a current cost of 2220 € per g from Sigma Aldrich (as of 29/06/2023). |
| This journal is © The Royal Society of Chemistry 2024 |