
Selective production of para-xylene from biomass-derived 2,5-dimethylfuran through tandem Diels–Alder/dehydration reactions with a bifunctional Ga,Al-zeolite catalyst†
Jaeyul
Kim
 ,
Sungmin
Han‡
and
Jeffrey D.
Rimer
*
,
Sungmin
Han‡
and
Jeffrey D.
Rimer
*
William A. Brookshire Department of Chemical and Biomolecular Engineering, University of Houston, Houston, TX 77204, USA. E-mail: jrimer@central.uh.edu
First published on 24th September 2024
Abstract
Here we demonstrate that Ga,Al-*BEA zeolites are effective bifunctional catalysts for para-xylene (p-xylene) production from bio-derived 2,5-dimethylfuran (DMF) through tandem Diels–Alder/dehydration reactions. A series of catalysts was synthesized via direct (one-pot) and post-synthesis techniques to introduce Brønsted and Lewis acid sites. The synthesis approach employed in this study avoids cost- and time-intensive processes typically associated with the preparation of metal-substituted *BEA zeolite catalysts for p-xylene production. Our findings reveal that Ga,Al-*BEA catalysts enhance DMF conversion and p-xylene selectivity in comparison to Al-*BEA zeolite. The pairing of Ga and Al in a single catalyst yields fewer byproducts, such as 2,5-hexanedione, 1-methyl-4-propyl-benzene, alkylated products, and oligomers. Comparisons of zeolites prepared with different Ga content reveal a higher turnover frequency for DMF conversion to p-xylene over Ga,Al-*BEA catalysts prepared by a one-pot synthesis compared to Al-*BEA catalysts. We observed a correlation between Ga content and p-xylene selectivity and yield, which is attributed to the Brønsted acidity of Ga framework sites (with reduced acid strength compared to Al sites) and to the Lewis acidity of extra-framework Ga species. The latter contribution was confirmed by analysis of Ga-impregnated Al-*BEA zeolites, which are less active than framework species but have a positive effect on p-xylene selectivity. Our collective findings indicate that tuning zeolite acidity by optimizing the amount of heteroatom incorporation in the crystal framework to tailor the speciation and strength of acid sites is beneficial to maximize p-xylene production from renewable resources.
1. Introduction
Para-xylene (p-xylene) is a key building precursor in the production of terephthalic acid which is a feedstock material for polyethylene terephthalate (PET).1 Increasing demand for p-xylene from 50 million tons in 2021 with a 5% compound annual growth rate projected until 2028 motivates global manufacturers to actively seek alternative methods of production.2,3 Instead of revamping existing aromatic facilities using naphtha feedstock, a renewable p-xylene production process using sustainable biomass-based molecule feedstocks is an appealing option to circumvent petroleum-based feedstocks for fuel and chemical formation.4 One promising route that has been considered is a Diels–Alder/dehydration reaction involving the cycloaddition of biomass-derived 2,5-dimethylfuran (DMF) with ethylene, followed by the dehydration of oxanorbornene cycloadduct (1,4-dimethyl-7-oxabicyclo[2,2,1]hept-2-ene) to p-xylene (Scheme 1).5,6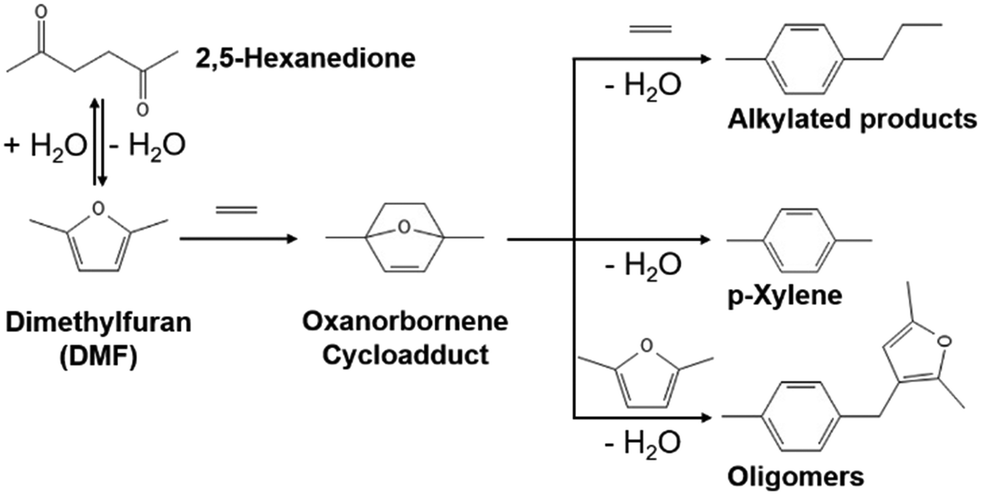 | ||
| Scheme 1 Diels–Alder/dehydration reactions of DMF with ethylene7 where oxanorbornene forms via the cycloaddition of DMF with ethylene, p-xylene forms via the dehydration of oxanorbornene, and side reactions generate alkylated products and oligomers. | ||
Experimental and computational studies of this reaction have provided insight of kinetic mechanisms and the role of active sites. The observation of a catalyst-independent kinetic regime at relatively high concentration of Brønsted acid sites (BASs) and the nearly equivalent Diels–Alder reaction barriers of DMF with ethylene between noncatalytic and catalytic (H–Y zeolites) processes called into question the exact role of Brønsted acidity in the cycloaddition of DMF with ethylene to produce the cycloadduct intermediate.5,8 Density functional theory (DFT) calculations have shown these sites reduce the reaction barrier of the subsequent dehydration pathway from 57.6 kcal mol−1 for the uncatalyzed reaction to 19.1 kcal mol−1. The prevailing hypothesis that a BAS has a limited contribution to the cycloaddition step motivates the use of alternative BAS species (i.e., going beyond aluminum sites) as well as the identification of an alternative active site for this reaction.
Previous studies have demonstrated that Lewis acid sites (LASs) in zeolite beta (*BEA) can promote the coordination of diene (DMF) with dienophile (ethylene) to catalyze the Diels–Alder reaction. For example, comparison of the pure aluminosilicate (Al-*BEA) with Sn-*BEA revealed the latter outperforms in the Diels–Alder reaction between cyclopentadiene and p-benzoquinone.9 Another study showed that Zr-*BEA also outperforms Al-*BEA in the Diels–Alder reaction between DMF and ethylene at fixed acid site concentration.7 These observations are linked to a lower activation barrier of LAS species. Notably, ethylene–LAS binding activates the normal electron-demand mechanism, which results in the Diels–Alder reaction barrier dropping from 24.7 (uncatalyzed) to 16.9 kcal mol−1.10 Studies of these tandem reactions collectively show that both BAS and LAS species are crucial to the reaction, thus highlighting the need to design bifunctional catalysts.
Prior studies have also demonstrated that zeolite *BEA is superior to other frameworks (e.g., FAU, MFI) for this reaction.6 Examples include metal-exchanged *BEA zeolites in batch processes7 as well as a packed bed microreactor configuration employing phosphorous-decorated zeolite (P-*BEA) that can achieve exceptionally high p-xylene selectivity and DMF conversion.11 Based on these findings, we selected *BEA for the design of bifunctional catalysts. In a recent study, we showed that gallosilicate zeolites are exceptional dehydration catalysts in comparison to state-of-the-art materials.12 Introduction of Ga as a heteroatom reduces BAS strength while extra-framework Ga species provide Lewis acidity.13 The latter is attributed to Ga3+ species, which have proven to be effective for nonoxidative propane dehydrogenation.14 Moreover, we previously reported that Ga,Al-ZSM-5 zeolites containing both framework and extra-framework Ga species enhance aromatics (e.g., p-xylene) selectivity in the ethylene dehydroaromatization reaction.15
Herein, we prepare a series of Ga,Al-*BEA zeolites through one-pot and post-synthesis techniques to introduce Ga in both framework and extra-framework sites. We then evaluate these catalysts against Al-*BEA in the tandem Diels–Alder/dehydration reaction to show that Ga is an effective alternative to Sn-*BEA, Zr-*BEA, and other metal-exchanged zeolites that often require time-intensive synthesis steps, the use of crystal seeds, and/or hazardous mineralizing agents (e.g., HF) that are bypassed in this new protocol. Our findings reveal that tuning zeolite acidity by heteroatom incorporation in the crystal framework is an efficient method to optimize p-xylene production from biomass-derived DMF and ethylene.
2. Experimental
2.1. Synthesis of *BEA catalysts
A pure aluminosilicate reference sample, Al1.0-*BEA (control), was prepared from a growth mixture with a molar composition of 8 TEAOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Al2O3:50 SiO2:743 H2O using tetraethylammonium (TEA) as the organic structure-directing agent. This solution was generated by the combination of two pre-mixed solutions. In the first solution, 2.530 g TEAOH (35 wt% in water) and 0.081 g Al were mixed and stirred for 3 h. The second solution was made with 2.530 g TEAOH (35 wt%), 16.885 g H2O, and 4.510 g fumed silica. Galloaluminosilicates (Gax,Aly-*BEA) were prepared by a one-pot (direct) synthesis using a growth mixture with a molar composition of 10 TEAOH: x Ga2O3: y Al2O3: 50 SiO2: 495 H2O (x = 0.2, 0.4, 0.6; and y = 0.6) generated from the combination of two pre-mixed solutions. The first solution was prepared identical to the one above, but with the addition of gallium nitrate hydrate. For example, the Ga0.4,Al0.6-*BEA sample was prepared by mixing 4.207 g TEAOH (35 wt%) with 0.065 g Al. After stirring for 3 h, 0.468 g gallium nitrate hydrate was added and combined with the second solution containing 4.207 g TEAOH (35 wt%), 12.314 g H2O, and 6.000 g fumed silica. The mixture was vigorously stirred until it became a gel phase.
:1 Al2O3:50 SiO2:743 H2O using tetraethylammonium (TEA) as the organic structure-directing agent. This solution was generated by the combination of two pre-mixed solutions. In the first solution, 2.530 g TEAOH (35 wt% in water) and 0.081 g Al were mixed and stirred for 3 h. The second solution was made with 2.530 g TEAOH (35 wt%), 16.885 g H2O, and 4.510 g fumed silica. Galloaluminosilicates (Gax,Aly-*BEA) were prepared by a one-pot (direct) synthesis using a growth mixture with a molar composition of 10 TEAOH: x Ga2O3: y Al2O3: 50 SiO2: 495 H2O (x = 0.2, 0.4, 0.6; and y = 0.6) generated from the combination of two pre-mixed solutions. The first solution was prepared identical to the one above, but with the addition of gallium nitrate hydrate. For example, the Ga0.4,Al0.6-*BEA sample was prepared by mixing 4.207 g TEAOH (35 wt%) with 0.065 g Al. After stirring for 3 h, 0.468 g gallium nitrate hydrate was added and combined with the second solution containing 4.207 g TEAOH (35 wt%), 12.314 g H2O, and 6.000 g fumed silica. The mixture was vigorously stirred until it became a gel phase.
The following steps were then performed for all *BEA zeolite samples. The growth mixture was aged at room temperature for 24 h under continuous stirring before being transferred to a 23 mL Teflon-lined stainless-steel acid digestion bomb (Parr Instruments). Hydrothermal treatment was carried out at 140 °C under rotation (ca. 30 rpm) in a Thermo-Fisher Precision Premium 3050 Series gravity oven. The synthesis times for Al-*BEA and Gax,Aly-*BEA zeolites were 42 and 11 days, respectively. Solids were isolated from the supernatant by three cycles of centrifugation and washing with DI water, followed by drying at 50 °C. The dried samples were calcined under the flow of dried air (100 mL min−1) at 550 °C for 8 h with a ramp rate of 1 °C min−1 to generate H-form catalysts for further characterization. For comparison, Ga-impregnated Al-*BEA zeolites (Gax/Aly-*BEA with x = 0.2, 0.4, 0.6, and y = 1.0) were synthesized by an incipient wetness impregnation method (see the ESI,† for more details).
2.2. Characterization of H-form zeolites
The crystallinity of zeolite catalysts was determined by powder X-ray diffraction (XRD) using a Rigaku Smartlab X-ray diffractometer with a step size of 0.02° and Cu Kα source (40 kV, 44 mA). Textural properties were assessed from N2 physisorption isotherms using a Micromeritics 3Flex instrument at 77 K and the Brunauer–Emmett–Teller (BET) method to determine external surface area and pore volume (t-plot method). Prior to adsorption, the samples were degassed at 300 °C overnight. Scanning electron microscopy (SEM) images were obtained using a Thermo Scientific Axia ChemiSEM instrument. Energy dispersive X-ray spectroscopy (EDS) was performed on the same instrument at 15 kV and a 10 mm working distance. Elemental analysis of zeolite samples was performed by inductively coupled plasma analysis in combination with optical emission spectroscopy (ICP-OES) using an Agilent 725 instrument. Acid site density was quantified by temperature-programmed desorption of ammonia (NH3-TPD). Approximately 80 mg of catalyst was first outgassed under Ar flow (Matheson, 30 cm3 min−1) for 2 h at 550 °C with a heating ramp of 2 °C min−1. The sample was then exposed to NH3 at 150 °C until saturated, followed by flushing with Ar for 3 h at 150 °C. The ammonia desorption was monitored with a quadrupole mass spectrometer (Cirrus 3, MKS Instruments) using a heating ramp from 150 to 700 °C at a rate of 5 °C min−1.The ratio of BAS to LAS sites was estimated by Fourier transform infrared (FTIR) spectroscopy with pyridine (Py) as a probe molecule. Acid site densities were determined using extinction coefficients for BAS as 1.67 cm μmol−1 (band at 1545 cm−1) and LAS as 2.22 cm μmol−1 (band at 1455 cm−1) following reported procedures.16,17 All spectra were recorded with a Thermo Scientific Nicolet IS20 spectrometer at 2 cm−1 optical resolution. Zeolite powders were initially pressed in self-supporting discs and pre-treated in the sample cell at 500 °C for 2 h in N2. An excess of probe molecules was admitted by multiple 1.0 μL pulse injections into the FTIR sample cell held at 200 °C until the peak area reached saturation. Physisorbed molecules were subsequently removed by N2 purging. Difference spectra were obtained by subtracting the spectrum of the zeolite before probe molecule adsorption using the Thermo Scientific OMNIC series software. X-ray photoelectron spectroscopy (XPS) analysis was performed on a PHI Quantera system (physical electronics) equipped with an achromatic Al Kα X-ray source (1486.7 eV) operating at 50 W (15 kV with a spot size of 200 μm.). The pass energy of the narrow scan was set at 26 eV with a 0.1 eV step size.
2.3. Catalytic reactions
Diels–Alder/dehydration reactions were performed in a 160 mL bench-top reactor (4564 with 4848 reactor controller, Parr Instrument Company) connected with ethylene and N2 gas lines. The zeolite catalyst (600 mg) was placed in the reactor followed by DMF and n-heptane to generate a 100 mL slurry with 0.23 M DMF in n-heptane. The vessel was purged with N2 to create an oxygen-free environment. The reactor was then heated to 250 °C under constant stirring (1000 rpm) using the built-in impeller. Once reaching 250 °C (20 bar), 40 bar (partial pressure) of ethylene was charged. The temperature was maintained at 250 °C and the total pressure of the system was kept at 60 bar. Sampling of liquid aliquots was conducted at a designated reaction time. Liquid samples were filtered with a 0.22 μm filter (13 mm hydrophobic PTFE, VWR International LLC) and analyzed after mixing with n-tridecane as an internal standard by a 6890 Agilent gas chromatograph (GC) equipped with a HP-1 capillary column (19091Z-530), an autosampler (7693A, G4513A), and a flame ionization detector. Calibration of reactants and products concentration was conducted by calculating response factors using DMF, p-xylene, 2,5-hexanedione, and 1-methyl-4-propylbenzene as standard chemicals. Each GC peak was identified by comparing the retention times with standard chemicals and other product compounds.18 Concentrations of alkylated products and oligomers were estimated through lump sum of all related chemicals. Conversion of DMF, product selectivity, and p-xylene yield were calculated by the following equations: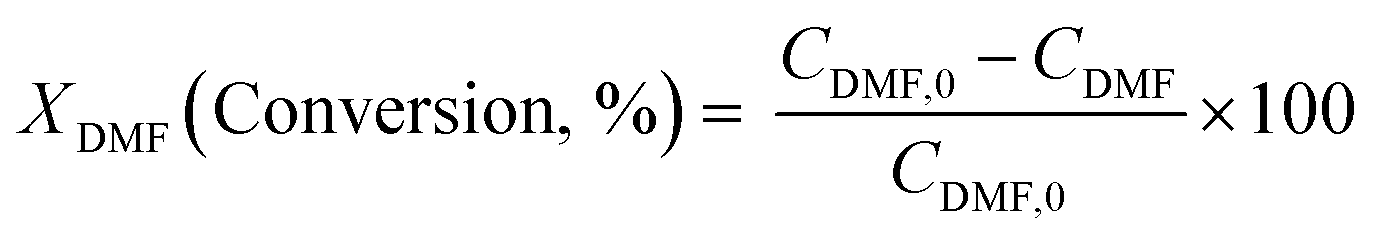 | (1) |
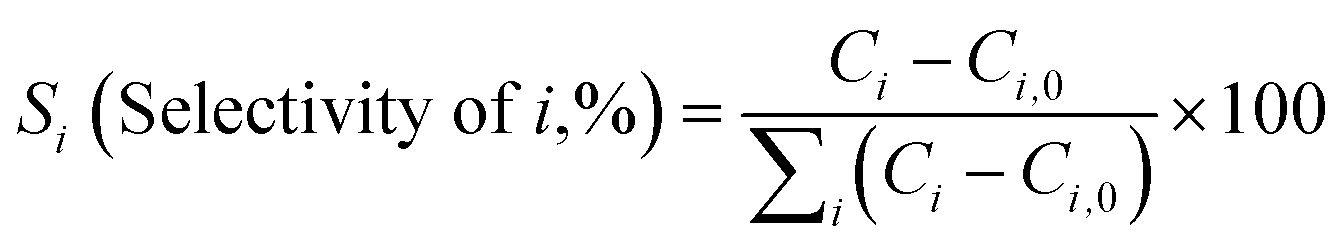 | (2) |
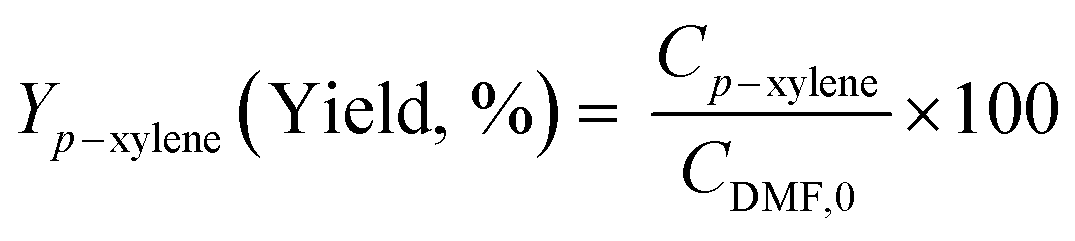 | (3) |
3. Results and discussion
3.1. Characterization of zeolite catalysts
| *BEA zeolite | Al/Gaa | S A (m2 g−1) | S ext (m2 g−1) | V micro (cm3 g−1) | Total acidity (μmol g−1)b | LAS (μmol g−1)c | BAS (μmol g−1)c | |
|---|---|---|---|---|---|---|---|---|
| ICP | EDS | |||||||
| a Solids after calcination (product yields are reported in Table S2 of the ESI† and elemental analysis is reported in Table S3 of the ESI†). b Data from NH3-TPD. c Data obtained by multiplying total acidity by the percentages of LAS and BAS from pyridine FTIR using previously reported extinction coefficients and protocols.16 | ||||||||
| Al1.0-*BEA | — | — | 608 | 60 | 0.22 | 630 | 204 | 426 |
| Ga0.6,Al0.6-*BEA | 1.0 | 1.0 | 581 | 65 | 0.21 | 625 | 328 | 297 |
| Ga0.4,Al0.6-*BEA | 1.5 | 1.7 | 626 | 79 | 0.22 | 558 | 261 | 297 |
| Ga0.2,Al0.6-*BEA | 2.8 | 3.5 | 580 | 73 | 0.21 | 533 | 263 | 270 |
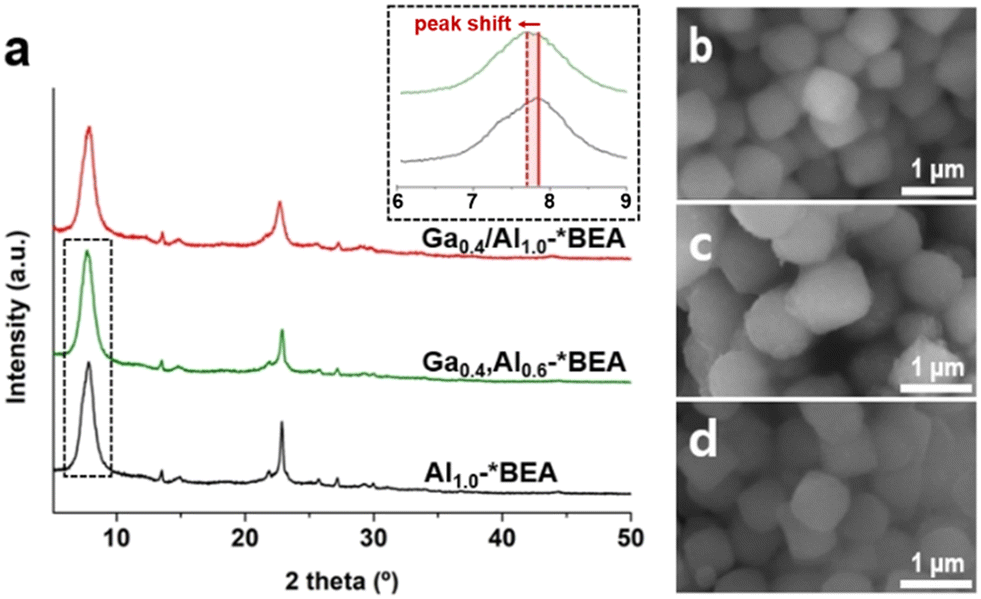 | ||
| Fig. 1 (a) Powder XRD patterns of Ga0.4/Al1.0-*BEA, Ga0.4,Al0.6-*BEA, and Al1.0-*BEA (see also Fig. S1 of the ESI† for other samples). Inset: example of the peak shift to lower 2θ with isomorphic Ga substitution. (b–d) Scanning electron microscope (SEM) images of representative (b) Ga0.4/Al1.0-*BEA, (c) Ga0.4,Al0.6-*BEA, and (d) Al1.0-*BEA samples. | ||
Many reported syntheses of *BEA zeolites in basic media (i.e., NaOH) result in crystals with spherical morphology;25 however, our synthesis protocols for Gax,Aly-*BEA and Al1.0-*BEA employ an alternative hydroxide source and result in more cubic morphologies (Fig. 1b–d) with a relatively monodisperse size distribution (Fig. S2 of the ESI†). The average particle size (diameter) of Gax,Aly-*BEA samples is ca. 900 nm, while that of Al1.0-*BEA is ca. 700 nm. The exact cause of crystal size differences is not fully understood, but does imply that the presence of Ga in the zeolite growth mixture inhibits nucleation, leading to a slightly smaller population of crystals with increased average particle size. Attempts were made to synthesize Al1.0-*BEA with slightly larger size to exactly match those of the Gax,Aly-*BEA samples by conducting a systematic parametric analysis of synthesis conditions (Table S1 and Fig. S3 of the ESI†), but we were unable to increase the average crystal size in Ga-free growth media. The impact of catalyst particle size is addressed later.
All *BEA zeolite catalysts have similar textural properties based on nitrogen adsorption/desorption measurements (Table 1 and Fig. S4 of the ESI†). The BET surface area (SA) and micropore volume (Vmicro) for samples range from 580 to 630 m2 g−1 and 0.21 to 0.22 cm3 g−1, respectively. The external surface area (Sext) for Ga,Al-*BEA samples is slightly larger (65–79 m2 g−1) than that of the control, Al1.0-*BEA (60 m2 g−1).
The acidity of all *BEA zeolites was determined using a well-established combination of two techniques: NH3-TPD and Py-FTIR.12,28 The total acidity was determined from NH3-TPD data and the relative percentages of BAS and LAS were determined from pyridine FTIR data (Table 1 and Fig. S6 of the ESI†). In general, replacing Al with Ga reduces the acidity of the catalyst. This is evident when comparing the total acid site density of Al1.0-*BEA (630 μmol g−1) to Ga0.4,Al0.6-*BEA (558 μmol g−1), which contains the same number of total heteroatoms (Al and Ga). As the Ga molar composition decreases from 0.6 to 0.2 in Gax,Aly-*BEA samples, the total acid site density reduces from 625 to 533 μmol g−1. The LAS density decreases from 328 to 261 μmol g−1 as the Ga composition is reduced from 0.6 to 0.4. Upon further reduction of the Ga composition to 0.2, the LAS density remains constant at around 263 μmol g−1.
In a previous study15 we showed that solid-state 71Ga NMR and X-ray absorption spectroscopy (XAS) were unable to resolve the fraction of framework (F) and extra-framework (EF) Ga species owing to their similar tetrahedral coordination (Fig. S7 of the ESI†). Here we use a surface-sensitive technique, X-ray photoelectron spectroscopy (XPS), as a means of estimating the percentages of each Ga species. XPS peak positions of Ga 2p3/2 in Gax,Aly-*BEA samples were detected in the range of 1119.0 to 1119.1 eV (Fig. 2). Each peak can be deconvoluted into two peaks to estimate the relative concentrations of framework Ga (1118.9–1119.1 eV) and extra-framework Ga (1120.3–1120.6 eV).12,29,30
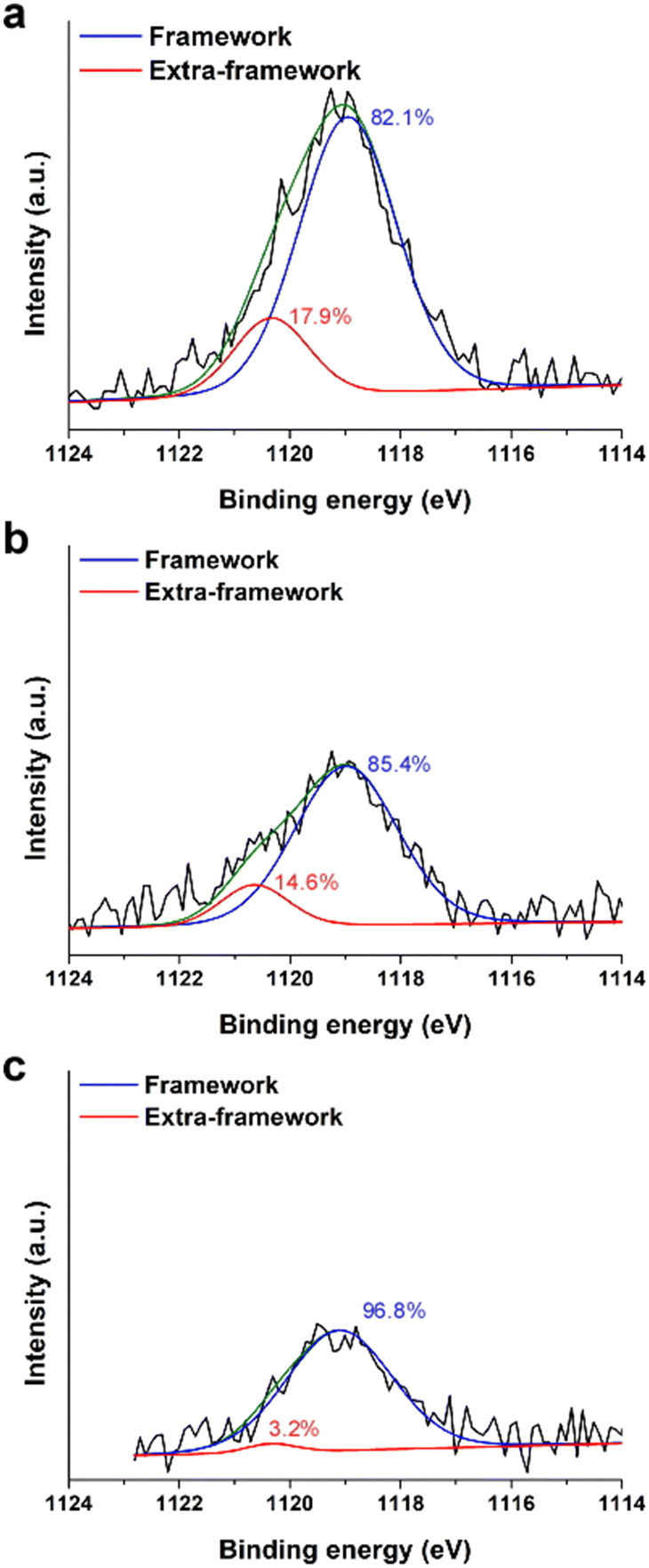 | ||
| Fig. 2 XPS spectra of Ga 2p3/2 for (a) Ga0.6,Al0.6-*BEA, (b) Ga0.4,Al0.6-*BEA, and (c) Ga0.2,Al0.6-*BEA samples. Peak positions are corrected using a carbon reference. Peak deconvolution was executed using Gaussian fits to obtain relative percentages (listed) of framework (blue) and extra-framework (red) Ga species (see also Table S5 of the ESI†). Green lines refer to fitted composite spectra. | ||
Given that XPS only probes the outermost rim (5–10 nm depth) of each sample, care should be given when trying to interpret these results as those of the bulk sample. Our findings indicate that the relative percentage of EF Ga increases with increasing Ga concentration in the Ga,Al-*BEA samples (Table S5 of the ESI†); however, we do not observe a systematic shift in BET surface area (Table 1) or loss of Vmicro, suggesting the absence of large Ga aggregates (e.g., Ga2O3 particles).
3.2. Catalytic performance
The BAS and LAS combinations from Al and Ga create a bifunctional catalyst that was assessed for the tandem Diels–Alder/dehydration reaction of DMF with ethylene (Scheme 1) in heptane solvent. All reactions were carried out in a batch reactor at 250 °C under continuous stirring. There is a monotonic increase in DMF conversion (Fig. 3a) and p-xylene selectivity (Fig. 3b) with increased reaction time. The reference Al1.0-*BEA catalyst achieves 61% DMF conversion within 24 h of reaction with a corresponding p-xylene selectivity of 76.5%. We observe an improved catalytic performance for all Gax,Aly-*BEA catalysts in terms of enhanced DMF conversion. We also observe a progressive increase in p-xylene selectivity with increased Ga content wherein the Ga0.6,Al0.6-*BEA catalyst results in the highest value of 82.2%, which is also comparable to the 80% p-xylene selectivity reported for Zr,Al-*BEA.7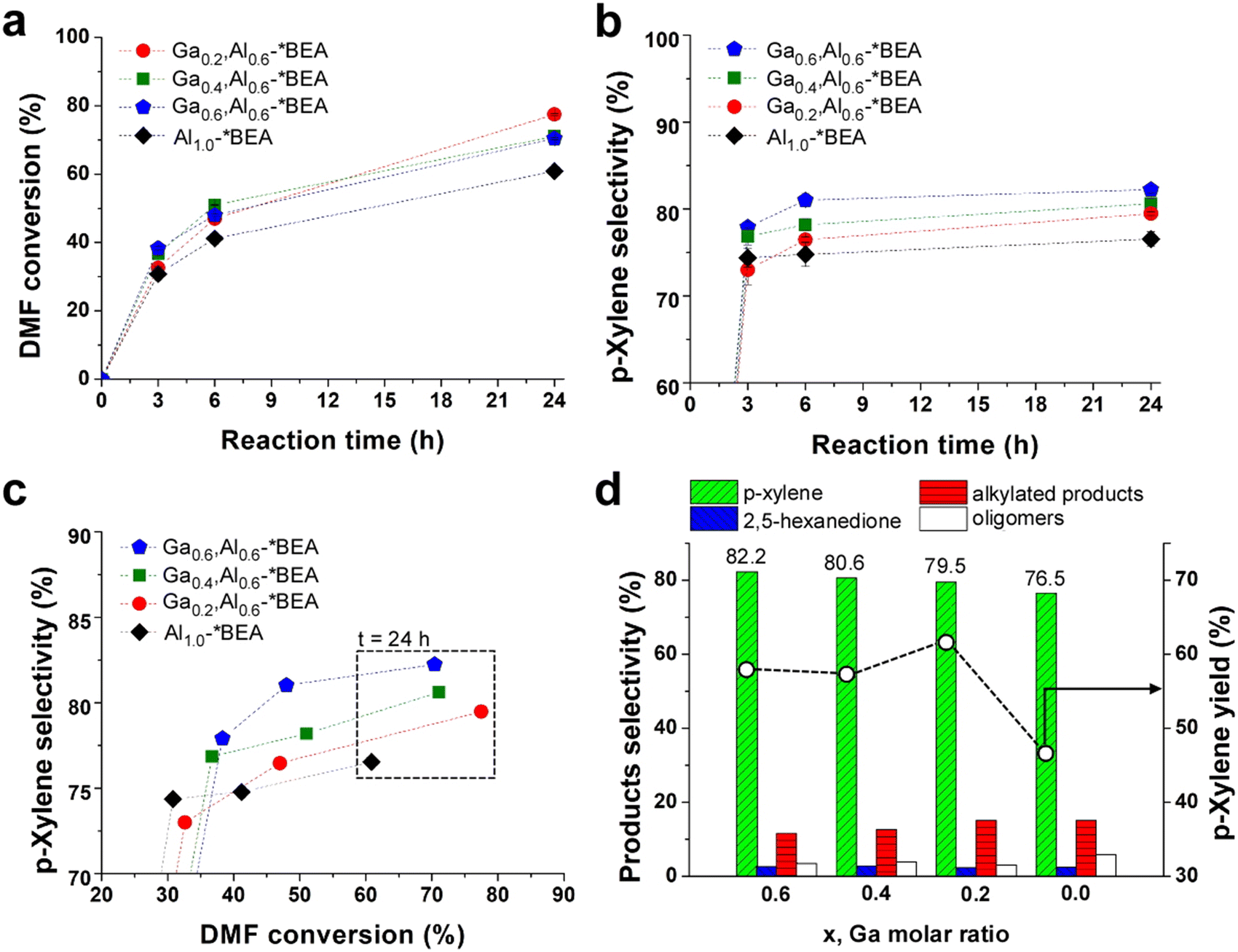 | ||
| Fig. 3 Tandem Diels–Alder/dehydration reaction performance of *BEA catalysts at 250 °C and 60 bar: (a) DMF conversion (starting from 0.23 M DMF in heptane) and (b) p-xylene selectivity vs. reaction time; (c) p-xylene selectivity vs. DMF conversion; and (d) products selectivities (histograms, left y-axis) and p-xylene yield (open circles, right y-axis) after 24 h of reaction. The x-axis lists the Ga molar ratio for each Gax,Al0.6-*BEA catalyst. Dashed lines are interpolated to guide the eye. Symbols are the average of three measurements for a single experiment. Error bars span two standard deviations (those not visible are less than the size of the symbol). This error is in good agreement with reproducibility tests that compared separate reactions in triplicate for the Al1.0-*BEA catalyst (see Fig. S10 of the ESI†). | ||
An iso-conversion plot (Fig. 3c) comparing the performance of each catalyst at the same DMF conversion shows that Ga incorporation leads to a ca. 5% increase in p-xylene selectivity. We also observed that p-xylene selectivity is proportional to the quantity of Ga, which is present in both framework and extra-framework sites within the zeolite. These acid sites are weaker than Al counterparts,31–33 which proved beneficial for alcohol dehydration reactions in our previous study of methanol and ethanol upgrading over pure gallosilicate zeolites (prepared for CHA, MFI, and MWW frameworks).12 For the tandem Diels–Alder/dehydration reaction, we show the presence of Ga impacts the distribution of undesirable products (Fig. 3d). Notably, we observe a decrease in both oligomers and alkylated products (Fig. S8 of the ESI†). The opposite is observed for 2,5-hexanedione where Ga sites initially promote its formation at early times, but the equilibrium reaction (Scheme 1) is reversed at later times as DMF is consumed, resulting in similar 2,5-hexanedione selectivity after 24 h for all zeolites tested. This time-dependent generation and consumption of 2,5-hexanedione is similar to the trend reported by Fan and coworkers for various metal-substituted *BEA zeolite catalysts.7
Distinguishing the contributions of framework and extra-framework gallium to catalyst performance is nontrivial owing to the challenges of differentiating Ga speciation by conventional characterization techniques;15 however, we prepared a separate set of Ga-impregnated catalysts, Gax/Al1.0-*BEA, where the only Ga species are extra-framework. As shown in Fig. 4a, the iso-conversion plot reveals a p-xylene selectivity for Gax/Al1.0-*BEA catalysts after 24 h of reaction that are slightly less than the Gax,Aly-*BEA catalysts (Fig. 3c), but higher than the control, Al1.0-*BEA. The conversion of Ga-impregnated samples is lower than those generated by a one-pot synthesis. This seems to indicate that framework Ga species significantly contribute to the Diels–Alder/dehydration of DMF to p-xylene. This was further validated by directly comparing catalysts on a per acid site basis using the turnover frequency (TOF), which was calculated by scaling the slope of conversion versus time on stream (for the first 3 h of reaction) by the total acidity measured by NH3-TPD, thus accounting for slight differences in sample compositions (Table S6 of the ESI†).
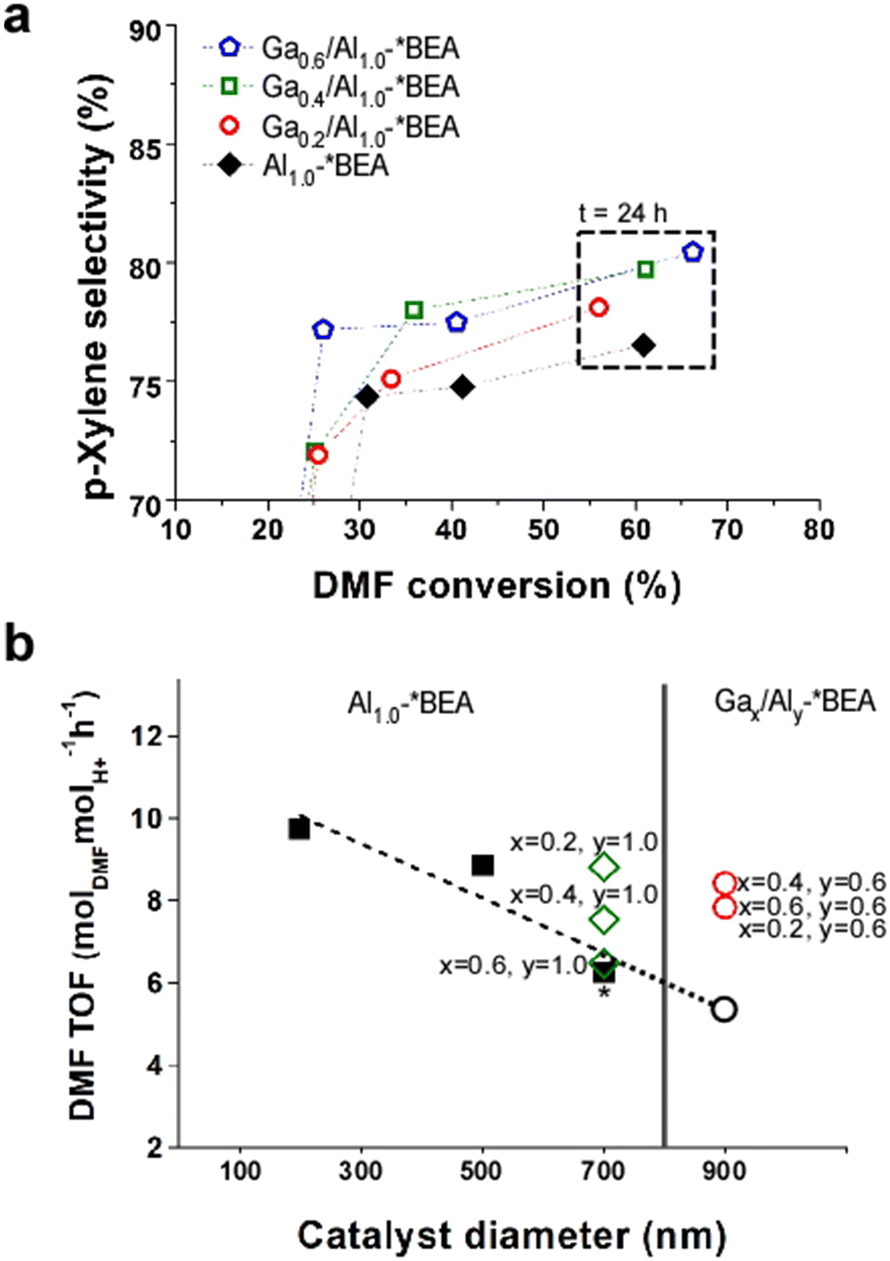 | ||
| Fig. 4 (a) Reaction performance of Ga-impregnated catalysts, Gax/Al10-*BEA, compared with the aluminosilicate control, Al1.0-*BEA (black diamonds). Data above 50% conversion correspond to 24 h of reaction time. (b) DMF turnover frequency (TOF) within the first 3 h of reaction (see Table S6 of the ESI†) for Al1.0-*BEA catalysts (closed black squares) prepared with different sizes (where the 700 nm sample used as the control is highlighted with an asterisk), and Gax,Aly-*BEA catalysts from one-pot syntheses (open red circles). The open black circle is a hypothetical Al1.0-*BEA sample of 900 nm size based on the extrapolated trendline (dashed line from linear regression). The Gax/Al10-*BEA samples prepared by Ga impregnation are shown as open green diamonds. | ||
The TOF values for Gax,Aly-*BEA catalysts (Fig. 4b, open red circles) are higher than that of the reference Al1.0-*BEA catalyst (Fig. 4b, closed black square with asterisk). The TOF values are similar for all three samples prepared by one-pot syntheses, which only differ by their Ga content. For Ga-impregnated samples (Fig. 4b, open green diamonds), TOF values are also higher than the reference and are comparable to those synthesized via the one-pot method; however, there are two key differences for Ga-impregnated catalysts. The first is that they have nearly double the Al content, and the second is the trend of reduced TOF with increasing extra-framework Ga content. The exact reason for this correlation is not fully understood, but does suggest that extra-framework Ga is less active. This is consistent with TOF calculations using total moles of both Al and Ga from elemental analysis in place of the total acidity measured by NH3-TPD (Fig. S9 of the ESI†). Under these conditions, the TOF values for the Gax,Aly-*BEA catalysts (from one-pot syntheses) are practically the same, whereas those of the Gax/Al1.0-*BEA catalysts (from post-synthesis impregnation) have much lower TOF values that fall below the control.
Another important aspect to consider when comparing different samples is the effect of particle size (i.e., diameter of zeolite *BEA crystals) on catalyst performance. There are two catalytic consequences of reduced catalyst size: (i) a kinetic effect associated with potential differences in the intrinsic activity of acid sites on exterior surfaces compared to the catalyst interior; or (ii) a mass transport effect related to the reduction of internal diffusion path length. The relative contributions of each effect for the catalysts in this study remain elusive. Mass transfer limitations can either be external (liquid-to-solid) or internal (pore diffusion).34 It is generally reported for liquid-phase reactions35,36 that larger particle size has a deleterious effect on catalytic performance. As previously mentioned, comparisons of catalysts with and without Ga were not made at constant zeolite crystal size in this study owing to the challenge of simultaneously tuning catalyst diameter and composition. Notably, the nominal aluminosilicate Al1.0-*BEA (Si/Al = 25) used as the control was synthesized with a diameter of 700 nm, whereas the Gax,Aly-*BEA catalysts (Si/(Al + Ga) = 25, when x = 0.4, y = 0.6) were prepared with sizes of ca. 900 nm. Numerous attempts were made to modify each synthesis to achieve either larger Al1.0-*BEA crystals or smaller Gax,Aly-*BEA crystals; however, these attempts proved unsuccessful. To explore the effects of size, we prepared Al1.0-*BEA samples with diameters of approximately 200 and 500 nm (see the expanded methods section in the ESI† for more details). In Fig. 4b we compare DMF turnover frequencies for different sizes of Al1.0-*BEA catalysts (black circles) and reveal enhanced activity with decreasing crystal size, consistent with reduced mass transport restrictions. If we extrapolate these trends to a higher particle size (i.e., 900 nm: Fig. 4b, open black circle), the anticipated differences in performance compared to Gax,Aly-*BEA catalysts (Fig. 4b, red squares) are even more significant than the results presented in Fig. 3c.
All Ga-impregnated samples were prepared using the Al1.0-*BEA control, and thus have the same size (700 nm). These catalysts require a higher Al content with relatively low Ga loading to achieve similar performance levels. It is unclear whether extra-framework Ga species impose diffusion limitations via partial pore blockage, although this is not evident from textural analysis (Table S4 of the ESI†), which shows no major changes in surface area or micropore volume with Ga loading. The more likely explanation is the differences in Ga speciation for materials made by a one-pot synthesis, which are apparently more active for the tandem reaction. The exact nature of these sites is unknown, but could be a different type of extra-framework species than those generated during insipient wetness impregnation. The introduction of framework Ga during the one-pot synthesis is another differentiating factor, but its relative contribution to the reaction cannot be easily differentiated from the extra-framework species. In the future, additional analysis is needed, in combination with computational studies, to better understand the active sites involved in different steps of the tandem reaction.
4. Conclusions
Tandem Diels–Alder and dehydration reactions have been suggested as a promising pathway for renewable p-xylene production. To maximize p-xylene selectivity, we developed Ga,Al-*BEA zeolites as bifunctional catalysts containing both Brønsted and Lewis acidity for this reaction. The Ga,Al-*BEA catalysts examined in this study were synthesized by both direct (one-pot) synthesis and post-synthesis incipient wetness impregnation without using fluoride media or any crystalline seeds that are critical to conventional synthesis methods used to prepare metal-substituted *BEA zeolite catalysts for this reaction. Our findings reveal that Ga,Al-*BEA catalysts provide higher DMF conversion (more than 70%) and p-xylene selectivity (more than 80%) in comparison with its aluminosilicate analogue, Al-*BEA. The presence of gallium also results in fewer byproducts, such as 2,5-hexanedione, 1-methyl-4-propyl-benzene, alkylated products, and oligomers. We show that particle size also plays a role in catalyst performance. Attempts to synthesize Ga,Al-*BEA and Al-*BEA samples with identical sizes and acid strength proved to be difficult; however, estimates of catalyst performance for Al-*BEA samples of varying size suggest that side-by-side comparisons with Ga,Al-*BEA would result in even more significant improvement than the results presented in this study, owing to the larger crystal size of Ga,Al-*BEA.Comparison of different Ga-containing zeolites revealed that the enhanced catalytic performance is attributed to both framework and extra-framework species; however, calculations of the turnover frequency reveal much higher values for samples containing framework Ga sites, suggesting the reduced acid strength of Ga Brønsted acid sites in comparison to Al sites is beneficial for this reaction, consistent with our recent discovery that Ga-zeolites are highly efficient dehydration catalysts. Our collective findings indicate that the judicious selection of heteroatoms for the design of bifunctional zeolite catalysts can be an efficient method for engineering improved tandem reactions. Moreover, we show for the first time that Ga-zeolites can be used to enhance p-xylene production from renewable resources.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support primarily from the National Science Foundation (Award DMR-2005201). Support for materials characterization was also provided by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0014468. JDR received additional support from the Welch Foundation (Award E-1794). This work was conducted in part using resources of the Shared Equipment Authority at Rice University under supervision of Dr. Bo Chen. We also thank Dr. Boris Makarenko (University of Houston) for assistance with XPS analysis. In honor of his 70th birthday, we also wish to recognize the seminal research contributions of Prof. Klavs Jensen that are a source of inspiration.References
- W. W. Kaeding, C. Chu, L. B. Young, B. Weinstein and S. A. Butter, J. Catal., 1981, 67, 159–174 CrossRef CAS.
- U. L. P. Wantanachaisaeng and K. O'Neil, Capturing Opportunities for Para-xylene Production, 2007, https://theswissbay.ch/pdf/Books/STEM/UOP-aromatics-paraxylene-capture-paper1.pdf Search PubMed.
- B. Consulting, Paraxylene Market-Global Industry Size, Share, Growth, Opportunity and Forecast, 2018–2028, 2022, https://www.blueweaveconsulting.com/report/global-paraxylene-market Search PubMed.
- Y.-C. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68–80 RSC.
- C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- C.-C. Chang, S. K. Green, C. L. Williams, P. J. Dauenhauer and W. Fan, Green Chem., 2014, 16, 585–588 RSC.
- C.-C. Chang, H. Je Cho, J. Yu, R. J. Gorte, J. Gulbinski, P. Dauenhauer and W. Fan, Green Chem., 2016, 18, 1368–1376 RSC.
- R. E. Patet, N. Nikbin, C. L. Williams, S. K. Green, C.-C. Chang, W. Fan, S. Caratzoulas, P. J. Dauenhauer and D. G. Vlachos, ACS Catal., 2015, 5, 2367–2375 CrossRef CAS.
- Á. Cantín, M. V. Gomez and A. de la Hoz, Beilstein J. Org. Chem., 2016, 12, 2181–2188 CrossRef PubMed.
- N. Nikbin, P. T. Do, S. Caratzoulas, R. F. Lobo, P. J. Dauenhauer and D. G. Vlachos, J. Catal., 2013, 297, 35–43 CrossRef CAS.
- Z. Wang, T. Goculdas, Y. W. Hsiao, W. Fan and D. G. Vlachos, Green Chem., 2024, 26, 8831–8839 RSC.
- A. Abutalib, D. Parmar, J. Kim and J. D. Rimer, J. Catal., 2024, 433, 115466 CrossRef CAS.
- L. Ni, R. Khare, R. Bermejo-Deval, R. Zhao, L. Tao, Y. Liu and J. A. Lercher, J. Am. Chem. Soc., 2022, 144, 12347–12356 CrossRef CAS PubMed.
- Y. Yuan, J. S. Lee and R. F. Lobo, J. Am. Chem. Soc., 2022, 144, 15079–15092 CrossRef CAS PubMed.
- Y. Zhou, H. Thirumalai, S. K. Smith, K. H. Whitmire, J. Liu, A. I. Frenkel, L. C. Grabow and J. D. Rimer, Angew. Chem., Int. Ed., 2020, 59, 19592–19601 CrossRef CAS PubMed.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- S. Lee and M. Choi, J. Catal., 2019, 375, 183–192 CrossRef CAS.
- P. T. M. Do, J. R. McAtee, D. A. Watson and R. F. Lobo, ACS Catal., 2013, 3, 41–46 CrossRef CAS PubMed.
- Y. Kamimura, S. Tanahashi, K. Itabashi, A. Sugawara, T. Wakihara, A. Shimojima and T. Okubo, J. Phys. Chem. C, 2011, 115, 744–750 CrossRef CAS.
- S. Han, N. Linares, T. Terlier, J. B. Hoke, J. García Martínez, Y. Li and J. D. Rimer, Angew. Chem., Int. Ed., 2022, 61, e202210434 CrossRef CAS PubMed.
- N. D. Hould, S. Senapati, H. Koller and R. F. Lobo, Top. Catal., 2012, 55, 1332–1343 CrossRef CAS.
- S. Dzwigaj, Y. Millot, J.-M. Krafft, N. Popovych and P. Kyriienko, J. Phys. Chem. C, 2013, 117, 12552–12559 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- M. A. Camblor, A. Corma and S. Valencia, Microporous Mesoporous Mater., 1998, 25, 59–74 CrossRef CAS.
- E.-H. Yuan, Colloids Surf., A, 2020, 605, 125386 CrossRef CAS.
- M. D. Oleksiak, K. Muraoka, M.-F. Hsieh, M. T. Conato, A. Shimojima, T. Okubo, W. Chaikittisilp and J. D. Rimer, Angew. Chem., Int. Ed., 2017, 56, 13366–13371 CrossRef CAS PubMed.
- H. Y. Luo, J. D. Lewis and Y. Román-Leshkov, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 663–692 CrossRef CAS PubMed.
- S. Han, K. Shilpa, A. J. Mallette, Y. Li, J. B. Hoke and J. D. Rimer, J. Cryst. Growth, 2023, 603, 126992 CrossRef CAS.
- M. Xin, E. Xing, X. Gao, Y. Wang, Y. Ouyang, G. Xu, Y. Luo and X. Shu, Ind. Eng. Chem. Res., 2019, 58, 6970–6981 CrossRef CAS.
- R. Oozeerally, J. Pillier, E. Kilic, P. B. J. Thompson, M. Walker, B. E. Griffith, J. V. Hanna and V. Degirmenci, Appl. Catal., A, 2020, 605, 117798 CrossRef CAS.
- M. S. Stave and J. B. Nicholas, J. Phys. Chem., 1995, 99, 15046–15061 CrossRef CAS.
- A. Chatterjee, T. Iwasaki, T. Ebina and A. Miyamoto, Microporous Mesoporous Mater., 1998, 21, 421–428 CrossRef CAS.
- A. J. Jones, R. T. Carr, S. I. Zones and E. Iglesia, J. Catal., 2014, 312, 58–68 CrossRef CAS.
- C. Sievers, Y. Noda, L. Qi, E. M. Albuquerque, R. M. Rioux and S. L. Scott, ACS Catal., 2016, 6, 8286–8307 CrossRef CAS.
- C. Ercan, F. M. Dautzenberg, C. Y. Yeh and H. E. Barner, Ind. Eng. Chem. Res., 1998, 37, 1724–1728 CrossRef CAS.
- S. M. Robinson, W. D. Arnold and C. H. Byers, AIChE J., 1994, 40, 2045–2054 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00362d |
| ‡ Present address: Sungmin Han – Intel Corporation, Chandler, Arizona 85226, USA. |
| This journal is © The Royal Society of Chemistry 2024 |