
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting quantum yields and circularly polarized luminescence of penta- and hexahelicenes by doping with two BN-groups†
Yannik
Appiarius
 ab,
Sandra
Míguez-Lago
c,
Pim
Puylaert
d,
Noah
Wolf
a,
Sourabh
Kumar
e,
Martin
Molkenthin
a,
Delia
Miguel
f,
Tim
Neudecker
beg,
Michal
Juríček
h,
Araceli G.
Campaña
c and
Anne
Staubitz
*ab
ab,
Sandra
Míguez-Lago
c,
Pim
Puylaert
d,
Noah
Wolf
a,
Sourabh
Kumar
e,
Martin
Molkenthin
a,
Delia
Miguel
f,
Tim
Neudecker
beg,
Michal
Juríček
h,
Araceli G.
Campaña
c and
Anne
Staubitz
*ab
aUniversity of Bremen, Institute for Organic and Analytical Chemistry, 28359 Bremen, Germany. E-mail: staubitz@uni-bremen.de
bUniversity of Bremen, MAPEX Center for Materials and Processes, 28359 Bremen, Germany
cUniversity of Granada, Department of Organic Chemistry, Unidad de Excelencia de Química, 18071 Granada, Spain
dUniversity of Bremen, Institute for Inorganic Chemistry and Crystallography, 28359 Bremen, Germany
eUniversity of Bremen, Institute for Physical and Theoretical Chemistry, 28359 Bremen, Germany
fUniversity of Granada, Department of Physical Chemistry, Unidad de Excelencia de Química, 18071 Granada, Spain
gUniversity of Bremen, Bremen Center for Computational Materials Science, 28359 Bremen, Germany
hUniversity of Zurich, Department of Chemistry, 8057 Zurich, Switzerland
First published on 25th October 2023
Abstract
The incorporation of boron–nitrogen (BN) units into polycyclic aromatic hydrocarbons (PAHs) as an isoelectronic replacement of two carbon atoms can significantly improve their optical properties, while the geometries are mostly retained. We report the first non-π-extended penta- and hexahelicenes comprising two aromatic 1,2-azaborinine rings. Comparing them with their all-carbon analogs regarding structural, spectral and (chir)optical properties allowed us to quantify the impact of the heteroatoms. In particular, BN-hexahelicene BN[6] exhibited a crystal structure congruent with its analog CC[6], but displayed a fivefold higher fluorescence quantum yield (φfl = 0.17) and an outstanding luminescence dissymmetry factor (|glum| = 1.33 × 10−2). Such an unusual magnification of both properties at the same time makes BN-helicenes suitable candidates as circularly polarized luminescence emitters for applications in materials science.
Introduction
Circularly polarized luminescence (CPL) is the differential emission of left- (IL) and right-handed (IR) circularly polarized light by chiral luminophores.1 Recently, CPL-active materials have demonstrated their immense application potential for optoelectronics,2–4 3D displays,5,6 switches for data storage,7,8 spintronics9,10 or chiral sensors.11,12Small organic molecules are particularly suitable as CPL emitters due to their structural modifiability.13 It allows the tuning of crucial optical properties like molar absorptivities (ε), fluorescence quantum yields (φfl) or emission dissymmetry factors (glum = 2 × (IL − IR)/(IL + IR)), quantifying the degree of polarization in the emission. Moreover, structure-related parameters like configurational14 and thermal15 stabilities towards decomposition are tunable, which is of high relevance for the processing into functional materials.
Among the various classes of chiral, π-conjugated molecules, helicenes are well-known for their strong optical rotation and electronic circular dichroism (ECD).16 Carbohelicenes are chiral, screw-shaped polycyclic aromatic hydrocarbons (PAHs) with n ≥ 5 ortho-fused benzenoid rings.17 Research studies have especially focused on derivatives of pentahelicene and hexahelicene because they allow chiral resolution at ambient conditions18 and gram-scale preparation by various synthetic approaches.19
For assessing the suitability of helicenes as CPL-emitting molecules, key prerequisites are high |glum| values, but also elevated ε and φfl. To combine these parameters into one, the CPL brightness factor BCPL = 0.5 × ε × φfl × |glum| has been introduced,20 allowing to compare the suitability of such luminophores for potential applications. Planar PAHs typically display high φfl values due to electronically allowed π–π* transitions, but the distorted π-planes of helicenes induce a significant decrease (φfl = 0.04 for both penta- and hexahelicene). This is a result of accelerated intersystem crossing, leading to non-emissive triplet states.21 Furthermore, the dissymmetry factors of carbohelicenes are almost exclusively below 10−2 (|glum| = 2.7 × 10−3 for penta- and 9 × 10−4 for hexahelicene).22,23 Consequently, pristine carbohelicenes (BCPL < 2 M−1 cm−1) are barely useful as CPL emitters.24
Incorporating main group elements like nitrogen,25–27 oxygen,28,29 silicon30,31 or combinations of these (e.g. B–O)32 in PAHs in general can modulate the optical properties significantly. However, this also perturbs the molecular architecture and the estimation of structural and electronic influences isolated from each other is complicated.33
Therefore, the doping with neighboring boron–nitrogen (BN) units has emerged as a popular strategy especially for planar PAHs,35–38 as it provides isoelectronic and isostructural BN-PAHs. In such compounds, the disparities in properties are almost exclusively caused by altered electronegativities of the heteroatoms,39 facilitating the comparison with the parent, all-carbon CC-PAHs.40–42
Unfortunately, reports of helical BN-derivatives have remained scarce,43–47 which especially applies to helicenes with B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N groups in the aromatic backbone.48,49 Moreover, experimental studies that compare BN-helicenes with their all-carbon counterparts are rare. To our knowledge, the work from Nowak-Król and Ingleson describes the only non-π-extended BN-helicenes so far (Fig. 1, top right).50 Besides a small structural impact, the most distinctive effect was an amplification of the radiative decay (φfl = 0.30 for the (mono)BN-pentahelicene, φfl = 0.21 for the BN-hexahelicene), compared to the all-carbon congeners, which were calculated but not synthesized. Nevertheless, this suggests the great potential of this compound class.
N groups in the aromatic backbone.48,49 Moreover, experimental studies that compare BN-helicenes with their all-carbon counterparts are rare. To our knowledge, the work from Nowak-Król and Ingleson describes the only non-π-extended BN-helicenes so far (Fig. 1, top right).50 Besides a small structural impact, the most distinctive effect was an amplification of the radiative decay (φfl = 0.30 for the (mono)BN-pentahelicene, φfl = 0.21 for the BN-hexahelicene), compared to the all-carbon congeners, which were calculated but not synthesized. Nevertheless, this suggests the great potential of this compound class.
 | ||
| Fig. 1 Top: previously reported BN-penta- and hexahelicenes. Bottom: structures studied in this work and the demonstrated benefits of such a twofold BN-substitution.34 | ||
Herein, we present the first non-π-extended penta- and hexahelicenes BN[5] and BN[6], respectively, comprising two aromatic boron–nitrogen-containing rings (Fig. 1, bottom). A multi-dimensional comparison with their all-carbon analogs CC[5] and CC[6] allowed us to gauge the exact influence of the BN-substitution on structural and (chir)optical properties. Most strikingly, the BN-substitution led to an outstanding CPL brightness of BN[6] (BCPL = 59 M−1 cm−1) while leaving the geometrical structure almost unaffected.
Results and discussion
Syntheses
BN-helicenes BN[5] and BN[6] were synthesized via transition-metal-catalyzed electrophilic cyclizations of (bis)ethynylarene precursors. These mild conditions51–53 were chosen to ensure the integrity of the moderately acidic, N-deprotected 1,2-azaborinine building blocks.Initially, electrophilic benzene- 1 and naphthalene precursors 2, equipped with two (trimethylsilyl)ethynyl substituents, were synthesized (Scheme 1).52 Subsequent Suzuki–Miyaura cross-coupling reactions with nucleophilic mesitylazaborinine 3 (ref. 54) gave TMS-alkynes 4 and 5 in yields of 72% and 75%, respectively. Under basic reaction conditions, deprotected alkynes 6 and 7 were obtained quantitatively.
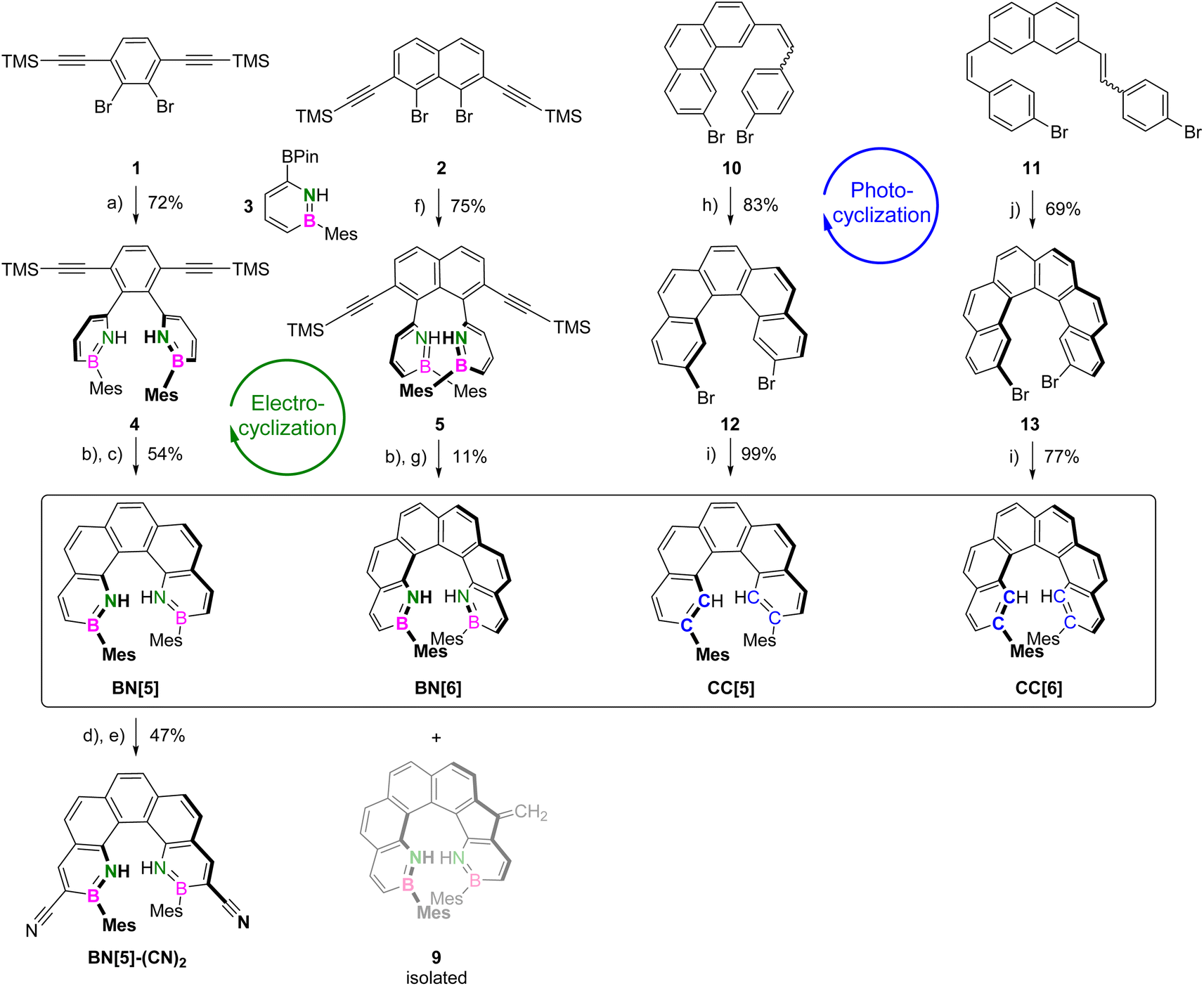 | ||
| Scheme 1 Syntheses of BN- and CC-helicenes. Reagents and conditions: (a) 3 (4.0 equiv.), potassium phosphate (3.0 equiv.), [Pd(dppf)Cl2] (5 mol%), methyl tert-butyl ether (MTBE), water, 90 °C, 2 d; (b) KOH (2.0 equiv.), MeOH, Et2O, 25 °C, 3 h; (c) 1. AuCl3 (30 mol%), mesitylene, 100 °C, 24 h; 2. Piperidine, acetonitrile, 25 °C, 1 h; (d) Bromine (2.2 equiv.), DCM, 0 °C, 2 h; (e) CuCN (10 equiv.), DMF, 170 °C, 19 h; (f) 3 (4.0 equiv.), potassium phosphate (3.0 equiv.), [Pd2(dba)3] (5 mol%), SPhos (10 mol%), THF, water, 60 °C, 17 h; g) 1. [(p-cymene)RuCl2]2 (30 mol%), AgSbF6 (30 mol%), mesitylene, 170 °C, 1 h; 2. Piperidine, acetonitrile, 25 °C, 1 h; (h) Iodine (1.1 equiv.), propylene oxide (500 equiv.), toluene, 365 nm LED, 25 °C, 30 min; (i) MesMgBr (3.0 equiv.), [Pd(dppf)Cl2] (5 mol%), 1,4-dioxane, 95 °C, 3 d; (j) Iodine (2.2 equiv.), propylene oxide (500 equiv.), toluene, 365 nm LED, 25 °C, 3 h. | ||
Our previous study of planar BN-PAHs55 showed that six-membered rings are formed almost exclusively when reacting the deprotected arylalkynes with PtCl2 at 100 °C. Transferring these conditions to BN-helicenes, however, significantly favored the competing 5-exo-dig pathway, leading to dibenzofulvene-like side-products in conversions of more than 40%. Therefore, we investigated different catalytic systems based on Pt(II), Au(I), Au(III) and Ru(II) (see ESI, Tables S1 and S2†).
In the case of BN[5], the reaction of phenylalkyne 6 with AuCl3 at 100 °C for 24 h provided the highest conversion with less than 5% endo/exo-side product 8 (see ESI, Table S1†). Derivatization of the latter with piperidine56 and chromatographic separation of the resulting Michael-type adduct eventually allowed the isolation of BN[5] in 54% yield.
On the other hand, the cyclization of naphthylalkyne 7 to BN[6] was achieved using [(p-cymene)RuCl2]2 and AgSbF6. Among all catalytic systems tested, this was the only one that favored the formation of two six-membered rings (ca. 80% of endo/endo) (see ESI, Table S2†). We assume that the reaction with Ru(II) preferably proceeds via the η1-vinylidene intermediate, assisting the nucleophilic attack at the terminal alkyne carbon.57 After heating 7 and the catalysts to 170 °C for 1 h in mesitylene and purification as for BN[5], BN[6] was obtained in a yield of 11% (Scheme 1). In contrast to the Ru(II) catalyst, the use of AuBrPPh3 and AgSbF6 allowed to selectively synthesize and isolate endo/exo-derivative 9 (see ESI, Section 2.28†).
For the syntheses of CC[5] and CC[6] (phenylethenyl)phenanthrene 10 and bis(phenylethenyl)naphthalene 11 were prepared in total yields of 52% and 18%, respectively. Mallory photocyclization reactions using UV irradiation at λ = 365 nm in the presence of iodine and propylene oxide gave dibromopenta- 12 and dibromohexahelicene 13. Subsequently, mesityl substituents were installed at positions 2,13 and 2,15, respectively, via Kumada cross-coupling reactions with mesitylmagnesium bromide. Eventually, racemic mixtures of CC[5] and CC[6] were obtained in yields of 82% and 53% over the last two steps (Scheme 1).
Introducing functional groups onto a pre-existing helicene scaffold by electrophilic aromatic substitution is highly desirable but often demanding. In a 1,2-azaborinine, the uneven charge distribution renders all carbon atoms distinct in reactivity.58 In particular, the selective electrophilic bromination at the α-carbon adjacent to boron has been shown to be a viable starting point for post-functionalizations.59–61 This methodology allowed us to selectively synthesize 3,12-dibrominated BN[5]-Br2 in 75% yield by the reaction of BN[5] with 2.2 equiv. of bromine. Subsequently, the conversion with an excess of copper(I) cyanide under Rosenmund–von Braun conditions gave BN[5]-(CN)2 in a yield of 62% (Scheme 1). In contrast, the reaction of CC[5] with bromine gave the C5,C6-dibrominated adduct.62 Upon heating it to 130 °C, rearomatization occurred and a mixture of reactant and the C5-brominated derivative was obtained. This highlights the superior selectivity of BN-PAHs in such a reaction (see ESI, Section 2.29†).
Structural analysis
Single crystals of the racemic target compounds suitable for X-ray diffraction analysis were obtained by vapor diffusion of acetonitrile, methanol, cyclohexane or n-hexane into DCM solutions of the helicenes.Comparing the bond lengths within the terminal rings (B–N: 1.426 ± 0.003 Å, C1–C2: 1.385 ± 0.003 Å, Δd ≈ 3%) showed the expected, relatively small geometric effect of the BN-substitution. Moreover, the C–C bond lengths within outer (1.34–1.38 Å) and inner helices (1.43–1.46 Å) remained almost identical, confirming the high two-dimensional isostructuralism of carbo- and BN-helicenes.63 On the other hand, the effect of BN on the spatial configurations of the pentahelicenes was remarkable (Fig. 2). Compared to CC[5] (φ = 52.4°), the torsion angle (φ) between the terminal rings (A) was only φ = 42.8° in BN[5], pointing at attracting interactions involving the NH units in close proximity (d (NH⋯N) = 2.35 Å).
 | ||
| Fig. 2 Solid-state structures as obtained from X-ray diffraction analysis. Thermal ellipsoids are shown at a 50% probability level. Hydrogen atoms (except for N–H and C1–H) are omitted for clarity. NICS(0) values for the individual rings were calculated at the MP2 (ref. 64)/cc-pVDZ65 level of theory, as implemented in the Q-Chem 6.0 program package.66C2 symmetry was assumed so that angles and distances are given as average values of both sides of the molecules. | ||
This also has implications on the molecular arrangements. While the packing structures of both compounds consist of opposite pairs of (P)- and (M)-enantiomers, there is a significant overlap of the helicene scaffolds (d = 3.59 Å between rings C) in a BN[5] crystal, hinting at the presence of π–π-interactions. Furthermore, the mesityl groups of each molecule are oriented almost parallelly (φ = 7.9°), which leads to a highly ordered packing arrangement. In CC[5], rings A and B of opposite molecules are arranged co-planarly but displaced. Also, the interplanar angle between the mesityl groups is larger (φ = 36.8°). The structural parameters of BN[5]-(CN)2 were mostly comparable with BN[5] (see ESI, Section 3.3†).
Both hexahelicenes exhibit much more similar metrics (φ = 69.1° for BN[6], φ = 66.6° for CC[6] between rings A), identical packing modes (see ESI, Fig. S10 and S14†) and very similar unit-cell parameters. The absence of additional dipolar interactions in BN[6] indicates that for hexahelicenes, the distant BN-units do not play a structure-determining role.
Besides the high planarity of the 1,2-azaborinine rings (averaged φ = 2.6° within rings A), aromaticity was clearly confirmed by nucleus-independent chemical shift (NICS) calculations (NICS(0) < −3 ppm, Fig. 2).67
Optical spectroscopy
Absorption and fluorescence spectra as well as φfl and fluorescence lifetimes (τfl) of the investigated helicenes were acquired in DCM solutions (Table 1).| Compound | λ abs , [nm] (10−3 × ε [M−1 cm−1]) | λ fl , , [nm] | φ fl | Stokes shift [cm−1] | τ fl [ns] |
f![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) f (λ [nm]) f (λ [nm]) |
|---|---|---|---|---|---|---|
| a Measurements performed in DCM solutions at c = 2.4 × 10−5 to 4.0 × 10−5 M. b Bold values represent intensity maxima. c LED, λex = 370 nm (BN-helicenes), 300 nm (carbohelicenes). d Not determinable because the 0–0 transitions were present as shoulder bands (λabs ≈ 400 nm). e Intensity averaged values because fluorescence decay consisted of more than one species (see ESI, Section 5.3 for more details). f TD-DFT at the B3LYP73–75/cc-pVDZ65 level of theory. | ||||||
| BN[5] | 283 (48.2), 363 (14.7), 383 (12.6) | 407, 428 | 0.10 | 4.7 | 0.1599 (370) | |
| CC[5] | 275 (34.2), 308 (27.0), 336 (14.8), 375 (1.0) | 409, 425 | 0.04 | 9.7e | 0.0010 (372) | |
| BN[6] | 280 (52.2), 342 (26.3), 401 (3.1), 424 (3.3) | 434, 459 | 0.17 | 540 | 7.1e | 0.0418 (400) |
| CC[6] | 266 (44.7), 318 (19.1), 390 (0.3), 414 (0.2) | 423, 444 | 0.03 | 510 | 8.7e | 0.0027 (387) |
| BN[5]-(CN)2 | 313 (28.0), 390 (14.9), 413 (16.6) | 424, 446 | 0.25 | 630 | 3.5 | 0.0053 (415) |
BN[5], CC[5], BN[6] and CC[6] revealed comparable absorption maxima (λabs = 266–283 nm) with decent molar extinction coefficients (ε = 3.4–5.2 × 104 M−1 cm−1 see ESI, Fig. S18–S22†). The absorption bands of both hexahelicenes closely resembled each other with small bathochromic shifts of BN[6] (Δλabs ≈ 10–15 nm, Fig. 3b). This suggests similar energy orderings of the differently polarized dipole transition moments.68 Most remarkably, the BN-moieties induced a substantial intensification of the least energetic 1Lb band, which is associated with fluorescence69 (BN[6]: ε = 3300 M−1 cm−1 at 424 nm, CC[6]: ε = 200 M−1 cm−1 at 414 nm).70,71 With respect to the pentahelicenes, the absorption spectra were more different in shape but both revealed shoulder bands at λabs ≈ 400 nm. The ε value of the lowest-energy transition of BN[5] was intensified by about an order of magnitude as well (Fig. 3a). This is a typical effect of BN-doping, rendering the symmetry-forbidden S0 → S1 transition of all-carbon aromatics partially allowed.72
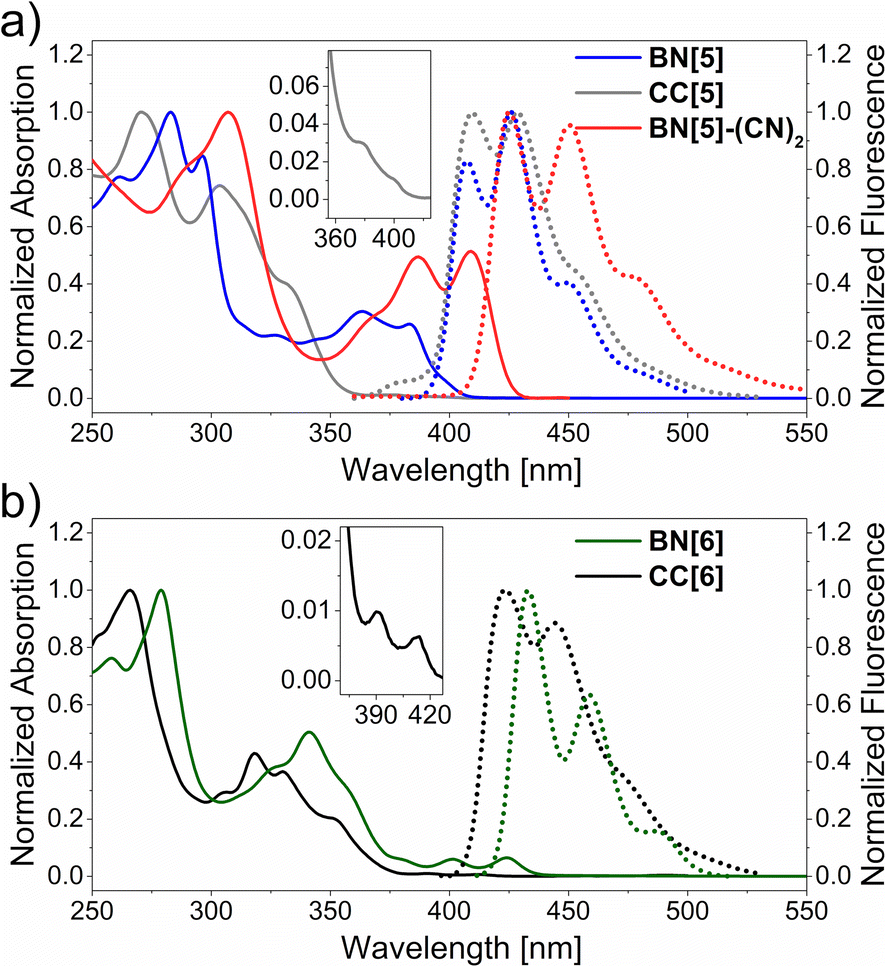 | ||
| Fig. 3 Normalized absorption (continuous lines) and fluorescence (dotted lines) spectra of pentahelicenes (a) and hexahelicenes (b) in DCM (c = 2.4 × 10−5 to 4.0 × 10−5 M). Insets show the lowest-energy absorption (shoulder) bands of the carbohelicenes. | ||
The fluorescence spectra of all five compounds consisted of at least three distinct vibronic bands, and the Stokes shifts, if determinable, were small (≈600 cm−1, Fig. 3). Compared to the very similar emission bands of the pentahelicenes (λfl ≈ 410 nm, Δλfl = 2 nm for the 0–0 bands), the difference between the hexahelicenes was slightly more pronounced (λfl = 434 nm for BN[6], λfl = 423 nm for CC[6]).
Moreover, the bathochromic effect of a twofold BN-substitution was identified by comparison with the reported (mono)BN-hexahelicene (Fig. 1, top right, λabs = 397 nm, λfl = 419 nm).50 In terms of fluorescence quantum yields, the BN-helicenes were substantially brighter (φfl = 0.10 for BN[5], φfl = 0.17 for BN[6]) than the sparsely emissive carbohelicenes (φfl = 0.04 for CC[5], φfl = 0.03 for CC[6]). Furthermore, the intensity averaged τfl of the heterohelicenes were comparably smaller than those of the studied carbohelicenes. While the difference was just 20% for the hexahelicenes, the lifetime of BN[5] (τfl = 4.7 ns) was halved compared to CC[5] (τfl = 9.7 ns). The increase of φ with a concomitant decrease of τfl for BN-helicenes can be rationalized with a larger emissive rate of these fluorophores compared to the all-carbon analogs. The influence of the mesityl-substitution on the optical features of the parent carbohelicenes (cf. hexahelicene: λabs = 410 nm, λfl = 420 nm, φfl = 0.03, τfl = 8.4 ns)76 was negligible, emphasizing that the enhancement of the photophysical properties by the BN-unit should not be limited to this particular substitution pattern.
We further investigated the structural and electronic properties of the helicenes by time-dependent density functional theory (TD-DFT) calculations at the B3LYP73–75/cc-pVDZ65 level of theory, as implemented in the Q-Chem 6.0 (ref. 66) and Gaussian 09, Rev. B.01 (ref. 77) program packages. For all helicenes, the emissive S1 states78 mostly consisted of highest occupied (HONTO) and lowest unoccupied (LUNTO) natural transition orbitals (NTOs). Moreover, the carbohelicenes revealed increased contributions of HONTO−1 and LUNTO+1 (see ESI, Section 7.4, Fig. S31†). All NTOs were delocalized over the PAH scaffolds with little to no involvement of the mesityl-groups due to their perpendicular orientations. Most noticeably, BN-substitution induced a symmetry break of the NTOs of BN[5] (Cs for HONTO, C2 for LUNTO, both are C2 symmetric for CC[5]). This feature is the cause of the higher φfl of the BN-helicenes, because the associated transition is rendered symmetry-allowed. Therefore, the related oscillator strength of BN[5] (f = 0.1599) was increased by more than two orders of magnitude compared to CC[5] (f = 0.0010). Despite the almost identically shaped NTOs of BN[6] and CC[6], the BN-helicene still revealed a tenfold increase of f. Overall, these results rationalize the experimental increase of ε in the low-energy regions.
The change of the optical properties of BN[5] upon installation of cyano-substituents was remarkable and higher than for similarly functionalized carbohelicenes:79BN[5]-(CN)2 not only revealed an intensification and a bathochromic shift of the 0–0 absorption band (Δλabs = 30 nm), but was also the most emissive compound (φfl = 0.25). In combination with the short lifetime (τfl = 3.5 ns), this indicates a particularly high fluorescence emission rate.
Chiral resolution & kinetics of racemization
The targeted helicenes were resolved into their enantiomers by high-performance liquid chromatography (HPLC), employing a chiral stationary phase (CSP). From a practical perspective, the pentahelicenes revealed a better resolution than the hexahelicenes with baseline-separated peaks (see ESI, Fig. S15 for chromatograms†). While CC[6] could not be resolved as a result of its low polarity concomitant with a poor solubility, BN[5] and BN[6] (enantiomeric excess ee = 99% for both) as well as BN[5]-(CN)2 (ee = 94%) and CC[5] (ee = 70%) were collected in their highly enantiomerically enriched forms.This enabled the determination of the half-lives (t1/2rac) and activation parameters of enantiomerization. For that purpose, each enantioenriched pentahelicene was subjected to time-course ECD measurements at 50, 60 and 70 °C. BN[5] showed a significantly lower racemization rate (t1/2rac ≈ 80 min at 60 °C) than CC[5] (t1/2rac ≈ 20 min). This translated into a Gibbs activation energy ΔG‡ (25 °C) of 25.7 kcal mol−1 for BN[5], which was higher by more than 1 kcal mol−1 compared to CC[5] (24.3 kcal mol−1, Table 2, for Eyring plots and calculations see ESI, Section 4.2†). Despite the bulkiness of the mesityl groups, the 2,13 disubstitution had a comparably small effect on the configurational stability (ΔG‡ (25 °C) = 24.1 kcal mol−1 for pentahelicene).80 In contrast to the pentahelicenes, heating an enantiopure sample of hexahelicene BN[6] to 200 °C for 14 h did not induce any racemization as analyzed by (CSP)-HPLC. Instead, higher temperatures caused a rapid decomposition.
| Compound | Kineticsb | ECDa | CPLa | |||||
|---|---|---|---|---|---|---|---|---|
| t 1/2rac [min] | ΔG‡ (25 °C) [kcal mol−1] | λ max , [nm] | Δε [M−1 cm−1] (λmax [nm]) | 103 × |gabs|g | λ max , [nm] | 103 × |glum|i | B CPL [M−1 cm−1] | |
| a Chiroptical measurements performed in DCM solutions at c = 1.3 × 10−5 to 4.0 × 10−5 M. b By time-course ECD measurements in n-heptane. t1/2rac at 60 °C. c Not measurable due to decomposition before reaching a temperature sufficient for racemization. d Not determined because chiral resolution by (CSP)-HPLC was unsuccessful. e Bold values represent intensity maxima. f Signs for (P)-enantiomers. (−) = negative CE, (+) = positive CE. g g abs = Δε/ε for the lowest energetic CE. h λ ex = 370 nm (BN-helicenes), λex = 300 nm (carbohelicenes). i g lum = 2 × (IL − IR)/(IL + IR). j B CPL = 0.5 × ε × φfl × |glum|. | ||||||||
| BN[5] | 77.6 | 25.7 | 304 (−), 373 (+), 393 (+) | 276 (304), 111 (393) | 9.9 | 423, 444 | 4.2 | 10.1 |
| CC[5] | 21.1 | 24.3 | 276 (−), 319 (+), 337 (+), 399 (−) | 152 (319), 82 (337) | 6.0 | 408, 429 | 4.4 | 3.0 |
| BN[6] | 285 (−), 344 (+), 409 (+), 431 (+) | 151 (285), 28 (431) | 10.8 | 432, 459 | 13.3 | 59.0 | ||
| CC[6] | ||||||||
| BN[5]-(CN)2 | 50.3 | 25.2 | 291 (+), 313 (−), 389 (+), 412 (+) | 187 (313), 127 (412) | 8.0 | 429 | 5.1 | 17.8 |
In order to understand the influence of BN- and mesityl-substitution on the racemization, we calculated the respective transition states and activation barriers (see ESI, Section 7.2†). Although the absolute theoretical values of ΔG‡ (T) were higher than the experimental ones, a gain in configurational stability upon BN-substitution was reproduced. We attribute this to the dipolar repulsions of the opposite NH hydrogen atoms, which are in significantly closer proximity in the transition state (d ≈ 1.5 Å for BN[5]) than in the C2-symmetric ground state (d = 2.2 Å in a crystal). In contrast, the parallelly aligned but distant mesityl groups in the transition states do not appear to play a major role.
Chiroptical spectroscopy
Electronic Circular Dichroism (ECD) measurements of the highly enriched enantiomers throughout resulted in mirror-imaged spectra (Fig. 4 and Table 2).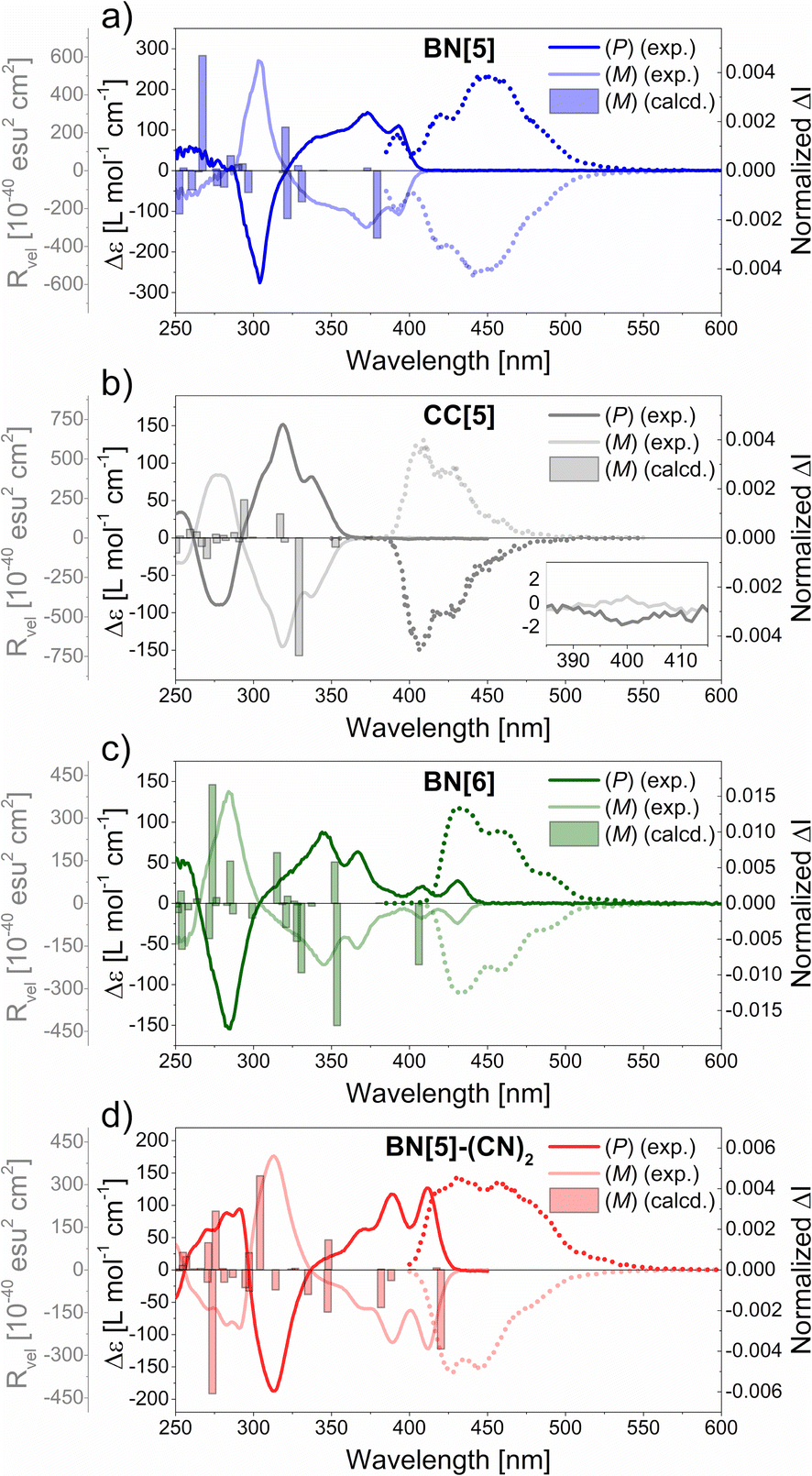 | ||
| Fig. 4 Experimental ECD (continuous lines) and CPL (dotted lines) spectra in DCM (c = 1.3 × 10−5 to 4.0 × 10−5 M) as well as computed rotatory strengths (bars outlined in grey). ΔI was normalized to the respective global maxima of |glum|. | ||
In all cases, the correlation with the computed rotatory strengths Rvel allowed us to assign that the absolute configuration of the first fraction from (CSP)-HPLC was (P), which corresponds to parent penta- and hexahelicenes (Fig. 4).69
The (P)-enantiomers of BN[5] (λ = 393 nm, Δε = +111 M−1 cm−1) and BN[5]-(CN)2 (λ = 412 nm, Δε = +127 M−1 cm−1) revealed intense, positive Cotton effects (CE) in the low-energy region. Comparably sharp, negative CE maxima were located at λ ≈ 300 nm. In contrast, the band that represents the 1La transition of (P)-CC[5] (λ = 399 nm, Δε = −1.8 M−1 cm−1) was barely visible (Fig. 4b), which closely resembles the parent pentahelicene.22 Moreover, the band for the 1Bb transition was mirrored as a positive CE at λ = 319 nm (Δε = +152 M−1 cm−1).
The ECD spectra of (P)-BN[6] and (mono)BN-hexahelicene (Fig. 1, top right)50 were similarly shaped, with one strongly negative CE (λ = 285 nm, Δε = −151 M−1 cm−1 for BN[6], λ = 245 nm, Δε = −290 M−1 cm−1 for the reported compound) and several less intense CE with opposite sign at higher wavelengths. As for the absorption and emission maxima, a twofold BN-substitution induced a significant bathochromic shift of the whole ECD spectrum (Δλ ≈ 40 nm).
The experimental absorption dissymmetry factors were typical of helicenes (|gabs| = 6–11 × 10−3, cf. 7.6 × 10−3 for pentahelicene22 and 9.2 × 10−3 for hexahelicene,23 in DCM solutions at 25 °C, see ESI, Section 5.4†). By means of TD-DFT calculations, we analyzed the contributions of electric (|μ|) and magnetic (|m|) transition dipole moments and the angle between both (θ) on absorption dissymmetries. For helicenes, the simplified expression gabs ≈ 4 × |m| × cos(θ)/|μ| applies.81 Due to the good reproduction of the experimental transition energies, we made use of the same level of theory as applied previously, although several recent studies reveal the accurate prediction of |gabs| and |glum| values by using long-range corrected functionals (see ESI, Section 7.7†).82–84
For BN[5] and BN[5]-(CN)2, strongly enhanced transition dipole moments (≈ thirtyfold increases of |m| and ≈ tenfold increases of |μ|), compared to CC[5]), were calculated for the lowest-energy transitions (see ESI, Tables S55 and S56†). However, the favorable, parallel orientations of the transition dipole moments of CC[5] (θ = 0°, cf. 114° for BN[5]) balanced out the BN-induced increase of |m|, so that |gabs| ≈ 1.0–1.3 × 10−2 for all pentahelicenes. With respect to the S0 → S1 transitions of the hexahelicenes, both |μ| and |m| of BN[6] were enhanced only by a factor of four, compared to CC[6]. Here, the more favorably aligned dipole moments result in a considerably enhanced theoretical absorption dissymmetry.
TD-DFT calculations of the geometry-optimized structures in the S1 states also allowed us to obtain the theoretical luminescence dissymmetry factors |glum| (see ESI, Table S56†). Overall, the absolute values of |μ|, |m| and θ of the S1 → S0 transition were comparable with the S0 → S1 transition, so that |glum| = 0.74–1.02 × |gabs|. Although the calculated bond lengths were very similar in both electronic states, the disparities in plane-to-plane angles were more pronounced, leading to deviating geometries (see ESI, Tables S49–53†).
Despite the previously discussed small Stokes shifts (Table 1), this could contribute to the experimental and theoretical differences between |gabs| and |glum|.
Experimental CPL spectra were obtained and normalized to the respective maxima of glum (Fig. 4 and ESI, Section 5.5†). In all cases, the sign of the CPL was identical with the sign of the lowest-energy transition in ECD. All pentahelicenes revealed comparable luminescence dissymmetry factors (|glum| = 4–5 × 10−3), which is in good agreement with the empirically observed correlation in helicenes (glum ≈ 0.61 × gabs) in the case of CC[5] and BN[5]-(CN)2.85 Due to the enhanced fluorescence quantum yields of both BN-pentahelicenes, their CPL brightness factors were considerably elevated (BCPL > 10 M−1 cm−1) compared to CC[5] (BCPL = 3.0 M−1 cm−1).
BN[6] exhibited an outstanding experimental value of |glum| = 1.33 × 10−2, which is considerably higher than of unsubstituted hexahelicene (|glum| = 9 × 10−4).23 In accordance with that, the theoretical values (|glum| = 2.3 × 10−2 for BN[6], |glum| = 0.6 × 10−2 for CC[6]) indicate a stronger impact of BN-substitution on a hexahelicene than on a pentahelicene. In particular, the much more favorable orientation of the transition dipole moments in BN[6] (135°, cf. 102° for CC[6]) is the most significant contributor to the dramatically improved theoretical |glum| value.
Compared to several reported helicenes84,86 with coordinative B←N-bonds (|glum| = 0.25–3.5 × 10−3 and BCPL < 10 M−1 cm−1), the combination of increased φfl and |glum| values led to a CPL brightness of BCPL = 59 M−1 cm−1 for BN[6]. This value is exceptionally high for a non-π-extended, monomeric helicene, and proves that BN-doping of helicenes is a concept that could foster the development of potent CPL emitting materials.
Conclusions
In summary, we have prepared and investigated the first examples of fully conjugated and non-π-extended (bis)BN-substituted penta- and hexahelicenes. Comparing them with their all-carbon congeners allowed the precise estimation of the influence of such BN-doping.As seen from the solid-state structures, the helix torsion of pentahelicene BN[5] was significantly smaller than of CC[5]. Owing to a larger spatial separation, this was not the case for BN[6], resulting in congruent structures of both hexahelicenes. The impact of BN-substitution on λabs and λfl was rather small (Δλ ≈ 10–15 nm). However, both the intensities of the low-energy absorptions as well as the φfl values were significantly elevated, peaking at 0.17 (BN[6]) and 0.25 (BN[5]-(CN)2). TD-DFT calculations allowed us to reproduce the causal, largely increased oscillator strengths of the BN-helicenes, being attributable to the reduction of symmetry. With respect to the configurational stabilities, BN[5] (t1/2rac ≈ 80 min at 60 °C) racemized four times slower than CC[5], which is possibly due to NH-repulsions in the transition state.
Mirror-imaged ECD spectra were obtained and, according to the strengthened low-energy absorptions, the Δε was considerably intensified in this region for the BN-pentahelicenes, as well. Regarding CPL, the |glum| value of BN[6] (1.33 × 10−2, cf. hexahelicene: 9 × 10−4) was unusually high, as elevated dissymmetry factors are usually associated with a loss of fluorescence efficiency.13
Taking into account that a selective dibromination of BN[5] was possible and the boron atoms may be modified with nucleophilic groups at an earlier stage of the reaction sequence,87 BN-helicenes with tailored push–pull substitution patterns could be synthesized in the future in order to explore their potential as efficient CPL emitters.
Data availability
Experimental and theoretical details supporting the statements of this article are included in the ESI.†Author contributions
Y. A. and A. S. conceived the project. Y. A. and N. W. performed the syntheses and routine analytics. Y. A. and S. M. L. performed the chiral resolutions, kinetics and chiroptical measurements, which were supervised by A. G. C. and M. J. P. P. performed the X-ray diffraction analyses. Y. A., S. M. L. and S. K. performed the DFT calculations, which were supervised by T. N. M. M. collected the quantum yields. D. M. performed the lifetime and excitation experiments. The manuscript was written by Y. A and S. M. L. All authors participated in reviewing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A.G.C. and S.M.L. acknowledge grant PID2021-127521NB-I00 funded by MCIN/AEI/10.13039/501100011033, the European Regional Development Fund (ERDF) “A way of making Europe” and Ministerio de Economía y Competitividad (BES-2016-076371). S.M.L. acknowledges the Spanish Junta de Andalucía for funding. P.P. acknowledges the Central Research Development Fund of the University of Bremen for the postdoctoral fellowship. M.J. acknowledges the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 716139) and the Swiss National Science Foundation (SNSF; no. PP00P2_198900). We thank Prof. A. J. Mota from the Department of Inorganic Chemistry from the University of Granada for performing additional DFT calculations.Notes and references
- J. Crassous, in Circularly Polarized Luminescence of Isolated Small Organic Molecules, ed. T. Mori, Springer Singapore, Singapore, 2020, pp. 53–97 Search PubMed.
- Y. Yang, R. C. da Costa, M. J. Fuchter and A. J. Campbell, Circularly polarized light detection by a chiral organic semiconductor transistor, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS.
- M. Li, S.-H. Li, D. Zhang, M. Cai, L. Duan, M.-K. Fung and C.-F. Chen, Stable Enantiomers Displaying Thermally Activated Delayed Fluorescence: Efficient OLEDs with Circularly Polarized Electroluminescence, Angew. Chem., Int. Ed., 2018, 57, 2889–2893 CrossRef CAS.
- J. Crassous, M. J. Fuchter, D. E. Freedman, N. A. Kotov, J. Moon, M. C. Beard and S. Feldmann, Materials for chiral light control, Nat. Rev. Mater., 2023, 8, 365–371 CrossRef.
- M. Schadt, Liquid Crystal Materials and Liquid Crystal Displays, Annu. Rev. Mater. Sci., 1997, 27, 305–379 CrossRef CAS.
- Y. Yang, R. C. da Costa, D.-M. Smilgies, A. J. Campbell and M. J. Fuchter, Induction of Circularly Polarized Electroluminescence from an Achiral Light-Emitting Polymer via a Chiral Small-Molecule Dopant, Adv. Mater., 2013, 25, 2624–2628 CrossRef CAS.
- N. P. M. Huck, W. F. Jager, B. de Lange and B. L. Feringa, Dynamic Control and Amplification of Molecular Chirality by Circular Polarized Light, Science, 1996, 273, 1686–1688 CrossRef CAS.
- M. Suarez and G. B. Schuster, Photoresolution of an Axially Chiral Bicyclo[3.3.0]octan-3-one: Phototriggers for a Liquid-Crystal-Based Optical Switch, J. Am. Chem. Soc., 1995, 117, 6732–6738 CrossRef CAS.
- N. Nishizawa and H. Munekata, Lateral-Type Spin-Photonics Devices: Development and Applications, Micromachines, 2021, 12, 644 CrossRef.
- R. Farshchi, M. Ramsteiner, J. Herfort, A. Tahraoui and H. T. Grahn, Optical communication of spin information between light emitting diodes, Appl. Phys. Lett., 2011, 98, 162508 CrossRef.
- G. Muller, Luminescent chiral lanthanide(iii) complexes as potential molecular probes, Dalton Trans., 2009, 9692–9707 RSC.
- C. P. Montgomery, E. J. New, D. Parker and R. D. Peacock, Enantioselective regulation of a metal complex in reversible binding to serum albumin: dynamic helicity inversion signalled by circularly polarised luminescence, Chem. Commun., 2008, 36, 4261–4263 RSC.
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Circularly Polarized Luminescence from Simple Organic Molecules, Chem.–Euro. J., 2015, 21, 13488–13500 CrossRef PubMed.
- D.-Q. He, H.-Y. Lu, M. Li and C.-F. Chen, Intense blue circularly polarized luminescence from helical aromatic esters, Chem. Commun., 2017, 53, 6093–6096 RSC.
- X.-F. Luo, H.-B. Han, Z.-P. Yan, Z.-G. Wu, J. Su, J.-W. Zou, Z.-Q. Zhu, Y.-X. Zheng and J.-L. Zuo, Multicolor Circularly Polarized Photoluminescence and Electroluminescence with 1,2-Diaminecyclohexane Enantiomers, ACS Appl. Mater. Interfaces, 2020, 12, 23172–23180 CrossRef CAS.
- M. Gingras, One hundred years of helicene chemistry. Part 1: non-stereoselective syntheses of carbohelicenes, Chem. Soc. Rev., 2013, 42, 968–1006 RSC.
- J. Crassous, I. G. Stará and I. Stary, Helicenes: Synthesis, Properties and Applications, Wiley-VCH, Weinheim, 2022 Search PubMed.
- P. Ravat, R. Hinkelmann, D. Steinebrunner, A. Prescimone, I. Bodoky and M. Juríček, Configurational Stability of [5]Helicenes, Org. Lett., 2017, 19, 3707–3710 CrossRef CAS.
- Y. Shen and C.-F. Chen, Helicenes: Synthesis and Applications, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- L. Arrico, L. Di Bari and F. Zinna, Quantifying the Overall Efficiency of Circularly Polarized Emitters, Chem.–Euro. J., 2021, 27, 2920–2934 CrossRef CAS.
- M. Sapir and E. V. Donckt, Intersystem crossing in the helicenes, Chem. Phys. Lett., 1975, 36, 108–110 CrossRef CAS.
- H. Tanaka, Y. Kato, M. Fujiki, Y. Inoue and T. Mori, Combined Experimental and Theoretical Study on Circular Dichroism and Circularly Polarized Luminescence of Configurationally Robust D3-Symmetric Triple Pentahelicene, J. Phys. Chem. A, 2018, 122, 7378–7384 CrossRef CAS.
- H. Tanaka, M. Ikenosako, Y. Kato, M. Fujiki, Y. Inoue and T. Mori, Symmetry-based rational design for boosting chiroptical responses, Commun. Chem., 2018, 1, 38 CrossRef.
- D. Göbel, S. Míguez-Lago, M. J. Ruedas-Rama, A. Orte, A. G. Campaña and M. Juríček, Circularly Polarized Luminescence of [6]Helicenes through Excited-State Intramolecular Proton Transfer, Helv. Chim. Acta, 2022, 105, e202100221 CrossRef.
- F. Chen, T. Tanaka, T. Mori and A. Osuka, Synthesis, Structures, and Optical Properties of Azahelicene Derivatives and Unexpected Formation of Azahepta[8]circulates, Chem.–Euro. J., 2018, 24, 7489–7497 CrossRef CAS.
- T. Otani, A. Tsuyuki, T. Iwachi, S. Someya, K. Tateno, H. Kawai, T. Saito, K. S. Kanyiva and T. Shibata, Facile Two-Step Synthesis of 1,10-Phenanthroline-Derived Polyaza[7]helicenes with High Fluorescence and CPL Efficiency, Angew. Chem., Int. Ed., 2017, 56, 3906–3910 CrossRef CAS.
- K. Goto, R. Yamaguchi, S. Hiroto, H. Ueno, T. Kawai and H. Shinokubo, Intermolecular Oxidative Annulation of 2-Aminoanthracenes to Diazaacenes and Aza[7]helicenes, Angew. Chem., Int. Ed., 2012, 51, 10333–10336 CrossRef CAS.
- H. Nishimura, K. Tanaka, Y. Morisaki, Y. Chujo, A. Wakamiya and Y. Murata, Oxygen-Bridged Diphenylnaphthylamine as a Scaffold for Full-Color Circularly Polarized Luminescent Materials, J. Org. Chem., 2017, 82, 5242–5249 CrossRef CAS PubMed.
- T. Matsuno, Y. Koyama, S. Hiroto, J. Kumar, T. Kawai and H. Shinokubo, Isolation of a 1,4-diketone intermediate in oxidative dimerization of 2-hydroxyanthracene and its conversion to oxahelicene, Chem. Commun., 2015, 51, 4607–4610 RSC.
- M. Murai, R. Okada, A. Nishiyama and K. Takai, Synthesis and Properties of Sila[n]helicenes via Dehydrogenative Silylation of C–H Bonds under Rhodium Catalysis, Org. Lett., 2016, 18, 4380–4383 CrossRef CAS.
- H. Oyama, K. Nakano, T. Harada, R. Kuroda, M. Naito, K. Nobusawa and K. Nozaki, Facile Synthetic Route to Highly Luminescent Sila[7]helicene, Org. Lett., 2013, 15, 2104–2107 CrossRef CAS PubMed.
- T. Katayama, S. Nakatsuka, H. Hirai, N. Yasuda, J. Kumar, T. Kawai and T. Hatakeyama, Two-Step Synthesis of Boron-Fused Double Helicenes, J. Am. Chem. Soc., 2016, 138, 5210–5213 CrossRef CAS PubMed.
- K. Dhbaibi, L. Favereau and J. Crassous, Enantioenriched Helicenes and Helicenoids Containing Main-Group Elements (B, Si, N, P), Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS.
- Formal charges at the B=N groups omitted because the aromaticity of the 1,2-azaborinine rings was verified both experimentally and theoretically Search PubMed.
- A. Abengózar, P. García-García, M. A. Fernández-Rodríguez, D. Sucunza and J. J. Vaquero, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Academic Press, 2021, vol. 135, pp. 197–259 Search PubMed.
- J. Hoffmann, B. Geffroy, E. Jaques, M. Hissler and A. Staubitz, Tuning the aggregation behaviour of BN-coronene diimides with imide substituents and their performance in devices (OLEDs and OFETs), J. Mater. Chem. C, 2021, 9, 14720–14729 RSC.
- J. Hoffmann, D. Jacquemin, M. Hissler and A. Staubitz, BN-Substituted Coronene Diimide Donor–Acceptor–Donor Triads: Photophysical, (Spectro)-Electrochemical Studies and Lewis Behavior, J. Mater. Chem. C, 2021, 9, 13926–13934 RSC.
- L. Palomino-Ruiz, S. Rodríguez-González, J. G. Fallaque, I. R. Márquez, N. Agraït, C. Díaz, E. Leary, J. M. Cuerva, A. G. Campaña, F. Martín, A. Millán and M. T. González, Single-Molecule Conductance of 1,4-Azaborine Derivatives as Models of BN-doped PAHs, Angew. Chem., Int. Ed., 2021, 60, 6609–6616 CrossRef CAS.
- Y. Appiarius and A. Staubitz, Azaborinine, Chem. Unserer Zeit, 2023, 57, 180–190 CrossRef CAS.
- S. Hashimoto, T. Ikuta, K. Shiren, S. Nakatsuka, J. Ni, M. Nakamura and T. Hatakeyama, Triplet-Energy Control of Polycyclic Aromatic Hydrocarbons by BN Replacement: Development of Ambipolar Host Materials for Phosphorescent Organic Light-Emitting Diodes, Chem. Mater., 2014, 26, 6265–6271 CrossRef CAS.
- J. S. A. Ishibashi, J. L. Marshall, A. Mazière, G. J. Lovinger, B. Li, L. N. Zakharov, A. Dargelos, A. Graciaa, A. Chrostowska and S.-Y. Liu, Two BN Isosteres of Anthracene: Synthesis and Characterization, J. Am. Chem. Soc., 2014, 136, 15414–15421 CrossRef CAS PubMed.
- Y. Appiarius, P. J. Gliese, S. A. W. Segler, P. Rusch, J. Zhang, P. J. Gates, R. Pal, L. A. Malaspina, K. Sugimoto, T. Neudecker, N. C. Bigall, S. Grabowsky, A. A. Bakulin and A. Staubitz, BN-Substitution in Dithienylpyrenes Prevents Excimer Formation in Solution and in the Solid State, J. Phys. Chem. C, 2022, 126, 4563–4576 CrossRef CAS.
- J. Full, S. P. Panchal, J. Götz, A.-M. Krause and A. Nowak-Król, Modular Synthesis of Organoboron Helically Chiral Compounds: Cutouts from Extended Helices, Angew. Chem., Int. Ed., 2021, 60, 4350–4357 CrossRef CAS PubMed.
- J.-K. Li, X.-Y. Chen, Y.-L. Guo, X.-C. Wang, A. C. H. Sue, X.-Y. Cao and X.-Y. Wang, B,N-Embedded Double Hetero[7]helicenes with Strong Chiroptical Responses in the Visible Light Region, J. Am. Chem. Soc., 2021, 143, 17958–17963 CrossRef CAS.
- F. Full, Q. Wölflick, K. Radacki, H. Braunschweig and A. Nowak-Król, Enhanced Optical Properties of Azaborole Helicenes by Lateral and Helical Extension, Chem.–Euro. J., 2022, 28, e202202280 CrossRef CAS.
- J. Guo, Z. Li, X. Tian, T. Zhang, Y. Wang and C. Dou, Diradical B/N-Doped Polycyclic Hydrocarbons, Angew. Chem., Int. Ed., 2023, 62, e202217470 CrossRef CAS.
- F. Zhang, F. Rauch, A. Swain, T. B. Marder and P. Ravat, Efficient Narrowband Circularly Polarized Light Emitters Based on 1,4-B,N-embedded Rigid Donor–Acceptor Helicenes, Angew. Chem., Int. Ed., 2023, 62, e202218965 CrossRef CAS.
- T. Hatakeyama, S. Hashimoto, T. Oba and M. Nakamura, Azaboradibenzo[6]helicene: Carrier Inversion Induced by Helical Homochirality, J. Am. Chem. Soc., 2012, 134, 19600–19603 CrossRef CAS.
- A. Abengózar, P. García-García, D. Sucunza, A. Pérez-Redondo and J. J. Vaquero, Synthesis of functionalized helical BN-benzo[c]phenanthrenes, Chem. Commun., 2018, 54, 2467–2470 RSC.
- K. Yuan, D. Volland, S. Kirschner, M. Uzelac, G. S. Nichol, A. Nowak-Król and M. J. Ingleson, Enhanced N-directed electrophilic C–H borylation generates BN–[5]- and [6]helicenes with improved photophysical properties, Chem. Sci., 2022, 13, 1136–1145 RSC.
- M. Buchta, J. Rybáček, A. Jančařík, A. A. Kudale, M. Buděšínský, J. V. Chocholoušová, J. Vacek, L. Bednárová, I. Císařová, G. J. Bodwell, I. Starý and I. G. Stará, Chimerical Pyrene-Based [7]Helicenes as Twisted Polycondensed Aromatics, Chem.–Euro. J., 2015, 21, 8910–8917 CrossRef CAS PubMed.
- L. D. M. Nicholls, M. Marx, T. Hartung, E. González-Fernández, C. Golz and M. Alcarazo, TADDOL-Derived Cationic Phosphonites: Toward an Effective Enantioselective Synthesis of [6]Helicenes via Au-Catalyzed Alkyne Hydroarylation, ACS Catal., 2018, 8, 6079–6085 CrossRef CAS.
- A. Jančařík, J. Rybáček, K. Cocq, J. Vacek Chocholoušová, J. Vacek, R. Pohl, L. Bednárová, P. Fiedler, I. Císařová, I. G. Stará and I. Starý, Rapid Access to Dibenzohelicenes and their Functionalized Derivatives, Angew. Chem., Int. Ed., 2013, 52, 9970–9975 CrossRef.
- A. W. Baggett, M. Vasiliu, B. Li, D. A. Dixon and S.-Y. Liu, Late-Stage Functionalization of 1,2-Dihydro-1,2-azaborines via Regioselective Iridium-Catalyzed C–H Borylation: The Development of a New N,N-Bidentate Ligand Scaffold, J. Am. Chem. Soc., 2015, 137, 5536–5541 CrossRef CAS.
- Y. Appiarius, T. Stauch, E. Lork, P. Rusch, N. C. Bigall and A. Staubitz, From a 1,2-Azaborinine to Large BN-PAHs via Electrophilic Cyclization: Synthesis, Characterization and Promising Optical Properties, Org. Chem. Front., 2021, 8, 10–17 RSC.
- S. Eissler, M. Kley, D. Bächle, G. Loidl, T. Meier and D. Samson, Substitution determination of Fmoc-substituted resins at different wavelengths, J. Pept. Sci., 2017, 23, 757–762 CrossRef CAS.
- C. A. Merlic and M. E. Pauly, Ruthenium-Catalyzed Cyclizations of Dienylalkynes Via Vinylidene Intermediates, J. Am. Chem. Soc., 1996, 118, 11319–11320 CrossRef CAS.
- C. R. McConnell and S.-Y. Liu, Late-stage functionalization of BN-heterocycles, Chem. Soc. Rev., 2019, 48, 3436–3453 RSC.
- L. Zi, J. Zhang, C. Li, Y. Qu, B. Zhen, X. Liu and L. Zhang, Synthesis, Properties, and Reactivity of Bis-BN Phenanthrenes: Stepwise Bromination of the Main Scaffold, Org. Lett., 2020, 22, 1499–1503 CrossRef CAS.
- A. Abengózar, I. Valencia, G. G. Otárola, D. Sucunza, P. García-García, A. Pérez-Redondo, F. Mendicuti and J. J. Vaquero, Expanding the BN-embedded PAH family: 4a-aza-12a-borachrysene, Chem. Commun., 2020, 56, 3669–3672 RSC.
- C. Zhang, BN-Phenanthrenes: Synthesis, Reactivity, and Optical Properties, Org. Lett., 2019, 21, 3476–3480 CrossRef CAS.
- M. Solà, Forty years of Clar’s aromatic π-sextet rule, Front. Chem., 2013, 1, 22 Search PubMed.
- P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Recent Advances in Azaborine Chemistry, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, A direct MP2 gradient method, Chem. Phys. Lett., 1990, 166, 275–280 CrossRef CAS.
- T. H. Dunning Jr, Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron Through Neon and Hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z.-Q. You, Y. Zhu, B. Alam, B. J. Albrecht, A. Aldossary, E. Alguire, J. H. Andersen, V. Athavale, D. Barton, K. Begam, A. Behn, N. Bellonzi, Y. A. Bernard, E. J. Berquist, H. G. A. Burton, A. Carreras, K. Carter-Fenk, R. Chakraborty, A. D. Chien, K. D. Closser, V. Cofer-Shabica, S. Dasgupta, M. de Wergifosse, J. Deng, M. Diedenhofen, H. Do, S. Ehlert, P.-T. Fang, S. Fatehi, Q. Feng, T. Friedhoff, J. Gayvert, Q. Ge, G. Gidofalvi, M. Goldey, J. Gomes, C. E. González-Espinoza, S. Gulania, A. O. Gunina, M. W. D. Hanson-Heine, P. H. P. Harbach, A. Hauser, M. F. Herbst, M. Hernández Vera, M. Hodecker, Z. C. Holden, S. Houck, X. Huang, K. Hui, B. C. Huynh, M. Ivanov, Á. Jász, H. Ji, H. Jiang, B. Kaduk, S. Kähler, K. Khistyaev, J. Kim, G. Kis, P. Klunzinger, Z. Koczor-Benda, J. H. Koh, D. Kosenkov, L. Koulias, T. Kowalczyk, C. M. Krauter, K. Kue, A. Kunitsa, T. Kus, I. Ladjánszki, A. Landau, K. V. Lawler, D. Lefrancois, S. Lehtola, R. R. Li, Y.-P. Li, J. Liang, M. Liebenthal, H.-H. Lin, Y.-S. Lin, F. Liu, K.-Y. Liu, M. Loipersberger, A. Luenser, A. Manjanath, P. Manohar, E. Mansoor, S. F. Manzer, S.-P. Mao, A. V. Marenich, T. Markovich, S. Mason, S. A. Maurer, P. F. McLaughlin, M. F. S. J. Menger, J.-M. Mewes, S. A. Mewes, P. Morgante, J. W. Mullinax, K. J. Oosterbaan, G. Paran, A. C. Paul, S. K. Paul, F. Pavošević, Z. Pei, S. Prager, E. I. Proynov, Á. R. ák, E. Ramos-Cordoba, B. Rana, A. E. Rask, A. Rettig, R. M. Richard, F. Rob, E. Rossomme, T. Scheele, M. Scheurer, M. Schneider, N. Sergueev, S. M. Sharada, W. Skomorowski, D. W. Small, C. J. Stein, Y.-C. Su, E. J. Sundstrom, Z. Tao, J. Thirman, G. J. Tornai, T. Tsuchimochi, N. M. Tubman, S. P. Veccham, O. Vydrov, J. Wenzel, J. Witte, A. Yamada, K. Yao, S. Yeganeh, S. R. Yost, A. Zech, I. Y. Zhang, X. Zhang, Y. Zhang, D. Zuev, A. Aspuru-Guzik, A. T. Bell, N. A. Besley, K. B. Bravaya, B. R. Brooks, D. Casanova, J.-D. Chai, S. Coriani, C. J. Cramer, G. Cserey, A. E. DePrince III, R. A. DiStasio Jr, A. Dreuw, B. D. Dunietz, T. R. Furlani, W. A. Goddard III, S. Hammes-Schiffer, T. Head-Gordon, W. J. Hehre, C.-P. Hsu, T.-C. Jagau, Y. Jung, A. Klamt, J. Kong, D. S. Lambrecht, W. Liang, N. J. Mayhall, C. W. McCurdy, J. B. Neaton, C. Ochsenfeld, J. A. Parkhill, R. Peverati, V. A. Rassolov, Y. Shao, L. V. Slipchenko, T. Stauch, R. P. Steele, J. E. Subotnik, A. J. W. Thom, A. Tkatchenko, D. G. Truhlar, T. Van Voorhis, T. A. Wesolowski, K. B. Whaley, H. L. Woodcock III, P. M. Zimmerman, S. Faraji, P. M. W. Gill, M. Head-Gordon, J. M. Herbert and A. I. Krylov, Software for the frontiers of quantum chemistry: An overview of developments in the Q-Chem 5 package, J. Chem. Phys., 2021, 155 Search PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- T. Mori, Chiroptical Properties of Symmetric Double, Triple, and Multiple Helicenes, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed.
- F. Furche, R. Ahlrichs, C. Wachsmann, E. Weber, A. Sobanski, F. Vögtle and S. Grimme, Circular Dichroism of Helicenes Investigated by Time-Dependent Density Functional Theory, J. Am. Chem. Soc., 2000, 122, 1717–1724 CrossRef CAS.
- Y. Nakai, T. Mori, K. Sato and Y. Inoue, Theoretical and Experimental Studies of Circular Dichroism of Mono- and Diazonia[6]helicenes, J. Phys. Chem. A, 2013, 117, 5082–5092 CrossRef CAS.
- J. B. Birks, D. J. S. Birch, E. Cordemans and E. Vander Donckt, Fluorescence of the higher helicenes, Chem. Phys. Lett., 1976, 43, 33–36 CrossRef CAS.
- A. J. V. Marwitz, M. H. Matus, L. N. Zakharov, D. A. Dixon and S.-Y. Liu, A Hybrid Organic/Inorganic Benzene, Angew. Chem., Int. Ed., 2009, 48, 973–977 CrossRef CAS.
- A. D. Becke, Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, A New Mixing of Hartree–Fock and Local Density-Functional Theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- H. Kubo, T. Hirose and K. Matsuda, Control over the Emission Properties of [5]Helicenes Based on the Symmetry and Energy Levels of Their Molecular Orbitals, Org. Lett., 2017, 19, 1776–1779 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. B.01, Wallingford CT, 2010 Search PubMed.
- M. Kasha, Characterization of electronic transitions in complex molecules, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- H. Görner, C. Stammel and J. Mattay, Excited state behaviour of pentahelicene dinitriles, J. Photochem. Photobiol., A, 1999, 120, 171–179 CrossRef.
- P. Ravat, Carbo[n]helicenes Restricted to Enantiomerize: An Insight into the Design Process of Configurationally Stable Functional Chiral PAHs, Chem.–Euro. J., 2021, 27, 3957–3967 CrossRef CAS.
- M. Cei, L. Di Bari and F. Zinna, Circularly polarized luminescence of helicenes: A data-informed insight, Chirality, 2023, 35, 192–210 CrossRef CAS PubMed.
- C. A. Guido, F. Zinna and G. Pescitelli, CPL calculations of [7]helicenes with alleged exceptional emission dissymmetry values, J. Mater. Chem. C, 2023, 11, 10474–10482 RSC.
- K. Dhbaibi, L. Abella, S. Meunier-Della-Gatta, T. Roisnel, N. Vanthuyne, B. Jamoussi, G. Pieters, B. Racine, E. Quesnel, J. Autschbach, J. Crassous and L. Favereau, Achieving high circularly polarized luminescence with push–pull helicenic systems: from rationalized design to top-emission CP-OLED applications, Chem. Sci., 2021, 12, 5522–5533 RSC.
- Z. Domínguez, R. López-Rodríguez, E. Álvarez, S. Abbate, G. Longhi, U. Pischel and A. Ros, Azabora[5]helicene Charge-Transfer Dyes Show Efficient and Spectrally Variable Circularly Polarized Luminescence, Chem.–Euro. J., 2018, 24, 12660–12668 CrossRef.
- H. Tanaka, Y. Inoue and T. Mori, Circularly Polarized Luminescence and Circular Dichroisms in Small Organic Molecules: Correlation between Excitation and Emission Dissymmetry Factors, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
- C. Shen, M. Srebro-Hooper, M. Jean, N. Vanthuyne, L. Toupet, J. A. G. Williams, A. R. Torres, A. J. Riives, G. Muller, J. Autschbach and J. Crassous, Synthesis and Chiroptical Properties of Hexa-, Octa-, and Deca-azaborahelicenes: Influence of Helicene Size and of the Number of Boron Atoms, Chem.–Euro. J., 2017, 23, 407–418 CrossRef CAS PubMed.
- A. W. Baggett and S.-Y. Liu, A Boron Protecting Group Strategy for 1,2-Azaborines, J. Am. Chem. Soc., 2017, 139, 15259–15264 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, single crystal data, HPLC chromatograms, kinetic data, photophysical data, NMR spectra and DFT calculations. CCDC 2192803, 2192802, 2192799, 2192801, 2212001 and 2212000. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02685j |
| This journal is © The Royal Society of Chemistry 2024 |