
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-entropy intermetallics: emerging inorganic materials for designing high-performance catalysts
Yuki
Nakaya
 * and
Shinya
Furukawa
*
* and
Shinya
Furukawa
*
Division of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita 565-0871, Japan. E-mail: furukawa@chem.eng.osaka-u.ac.jp
First published on 25th July 2024
Abstract
Alloy materials have been used as promising platforms to upgrade catalytic performance that cannot be achieved with conventional monometallic materials. As a result of numerous efforts, the recent progress in the field of alloy catalysis has been remarkable, and a wide range of new advanced alloys have been considered as potential electro/thermal catalysts. Among advanced alloy materials, high-entropy intermetallics are novel materials, and their excellent catalytic performance has recently been reported. High-entropy intermetallics have several advantages over disordered solid-solution high-entropy alloys, that is, greater structural/thermal stability, more facile site isolation, more precise control of electronic structures, tunability, and multifunctionality. A multidimensional compositional space is indeed limitless, but such a compositional space also provides a well-designed surface configuration because of its ordered nature. In this review, we will provide fundamental insights into high-entropy intermetallics, including thermodynamic properties, synthesis requirements, characterization techniques, roles in catalysis, and reaction examples. The comprehensive information provided in this review will highlight the great application potential of high-entropy intermetallics for catalysis, and will accelerate the development of this newly developed field.
1. Introduction
Humanity has been facing a variety of global problems, such as increasing global population, rising energy demand, and climate change.1–3 In addressing these situations, catalysis has always been a most important core research topic in the field of chemistry.4 For example, more than 80% of the chemicals produced worldwide have undergone catalysis,5 demonstrating the importance of catalysis research. Catalysis primarily aims to develop an innovative catalyst that exhibits unprecedented high catalytic activity, selectivity, and stability with great cost performance. To achieve this goal, alloy materials have been considered as promising platforms. Although alloy catalysis has a long history of nearly 100 years, which is comparable to that of catalytic chemistry itself,6 the field of alloy catalysis is advancing every day. The percentage of alloy catalysis has increased dramatically since the 1970s,6,7 highlighting the importance of alloy materials in catalytic chemistry.In the past few decades, alloy catalysts have undergone rapid advancement with the flourishing of synthesis methods, characterization technologies, analytical techniques, theoretical calculations, etc.6–8 Considerable efforts have been made, and versatile types of alloys have been synthesized, identified, and tested for catalytic reactions. Consequently, the hidden rules of alloy catalysis, such as the ensemble effect, ligand effect, and multifunctional effect, have been discovered.6,9–14 However, achieving a prospective goal of alloy catalysis, which is to arrange the atoms in a controllable way with a wide compositional space to obtain the desired catalytic properties, is significantly difficult using conventional alloy materials. For example, state-of-the-art pseudo-binary alloys and high-entropy alloys (HEAs) have recently emerged as promising catalysts because of their controllable tuning of surface environments and unlimited compositional space, respectively. However, these properties are associated with the ordered and disordered nature of alloys. If ordered alloys characterized by multicompositionality can be obtained, then the alloys would function as breakthrough catalysts because of the atomic-scale tuning of surface environments with high flexibility.
Since the Bronze Age, the addition of relatively small amounts of secondary elements to a primary element has been explored as a basic alloying strategy over millennia.15 This approach is reflected in the fact that alloys are named after their main component (e.g., ferrous alloys, titanium alloys, and nickel alloys). However, such a primary-element approach only allows for a narrow compositional space for alloys. The discovery of HEAs in 2004 (ref. 16 and 17) was a turning point in the field of alloy materials, greatly expanding the compositional space of alloys that can be studied. HEAs have emerged as a unique class of disordered alloys, comprising five or more principal metal elements at relatively high (5–35 atom%, often near equimolar) concentrations with a mixing entropy of 1.5R or higher (ΔSmix ≥ 1.5R).15,18 The greatly increased entropy promotes mixing of the immiscible elemental combinations, leading to the exploration of alloys with an unlimited compositional space that could never be achieved with traditional alloys.19–28
The earlier attempt to use HEAs in the field of catalysis was reported in 2008,29 and, particularly, the number of publications has increased vigorously in the last five years with the advancement of synthetic methods.6,30–38 Compared with materials having simple compositions, HEAs have three distinct features39,40 such as (i) an unlimited compositional space, providing a number of compositional choices, including those that have not been allowed by immiscibility; (ii) diversity of atomic configurations, providing a variety of surface environments, some of which can work as multifunctional active environments for the target reaction; and (iii) greater thermal/structural robustness, thermodynamically and kinetically improving the stability because of the increase in mixing entropy and sluggish diffusion of constituent atoms, respectively.
Consequently, HEAs function as promising platforms to achieve greater catalytic performance. However, the randomness and diversity of surface atomic arrangement often involve (1) large complexity and difficulty in ascertaining the origin of improved catalytic performance and (2) a limited number of effective atomic arrangements. Therefore, the synergistic contribution of element-specific properties is often left in a black box called the “cocktail effect” because of the disordered nature. In addition, HEAs typically have a simple disordered structure such as face-centered cubic (fcc), body-centered cubic (bcc), hexagonal close packed (hcp), or orthorhombic.15 Therefore, neither precise control of the atomic arrangement nor constructing specific and ordered active environments is possible using HEAs.
Compared with disordered alloys, intermetallics are an important and unique family of alloys that feature long-range atomic ordering with well-defined atomic positions.6,7,41–43 The constituent metals of intermetallics are separately distributed to two (or more) crystallographically distinct sites, thereby displaying well-defined surface environments suitable for clarifying or controlling structure–property relationships.5–8,44 In addition, the strong interatomic interaction often provides high structural stability. Although intermetallics have many advantages for catalysis, they lack tunability and expandability for material design because of the fixed positions and composition ratio.
Given the advantages of disordered HEAs and ordered intermetallics, high-entropy intermetallics (HEIs) are potentially efficient catalysts because of the combination of multimetallic (comprising five or more metal elements) and ordered nature.19–28 Crystallographically, HEIs can be classified as pseudo-binary alloys because HEIs have the parent intermetallic AmBn structure, in which A and/or B elements are partially substituted with other elements. Therefore, HEIs are extension of ternay/quaternary alloys of pseudo-binary alloys. Despite their promising potential, employing HEIs for catalysis has not been explored until 2019. The use of HEIs for catalysis requires downsizing from the bulk to nanoscale, and synthesizing HEIs at the nanoscale is significantly difficult. In particular, the increased number of elements and the harsh annealing conditions inevitably induce growth/agglomeration. Larger nanoparticles have a larger thermodynamic barrier of the disorder-to-order phase transition, thereby resulting in HEAs and/or phase-separated alloys.28
With the improvement of synthetic methods, the catalytic use of an HEI nanomaterial was first reported in 2020.20 To date, many reports have been published.19–28,45–48 and we will highlight the first ten reports to explain HEIs in this review paper.19–28 Moreover, all the reported HEI catalysts have shown excellent catalytic performance, surpassing those of conventional alloy catalysts. HEIs have several advantages over their HEA counterparts, including (i) greater structural/thermal stability (multicomponent), (ii) more facile site isolation (ordered structure), (iii) more precise control of electronic structures (interatomic interaction), (iv) tunability, and (v) multifunctionality, which highlight this state-of-the-art topic in chemistry. In addition, HEIs provide a number of new ordered alloys. However, the catalysis field of HEIs is in the preliminary stage, and it lacks basic information. Therefore, conducting more remarkable and deeper studies is urgently necessary to open a new paradigm in HEI catalysis. This review will provide a comprehensive understanding of the newly emerging field of HEIs with regard to thermodynamic properties, synthesis requirements, characterization techniques, roles in catalysis, and reaction examples.
2. Thermodynamics of HEI formation
In general, the synthesis of ordered structures requires a high annealing temperature of more than 600 °C to overcome the barrier of the disorder-to-order transition (Fig. 1a and b).8,19,49,50 This thermodynamic requirement is common both in conventional intermetallics and HEIs. In addition, the sluggish diffusion of component metals in multimetallic alloys requires long exposure to temperature.19 Therefore, the attempt to synthesize HEIs typically results in the bulk structure of the HEA and/or phase-separated alloys.28 Moreover, other difficulties, including the selection of constituent metals and the homogeneous distribution of metal precursors, make the synthesis of nanoscale HEIs difficult. Therefore, synthesizing HEI nanomaterials is a challenging task. In this section, we introduce the following important factors to guide the construction of HEIs.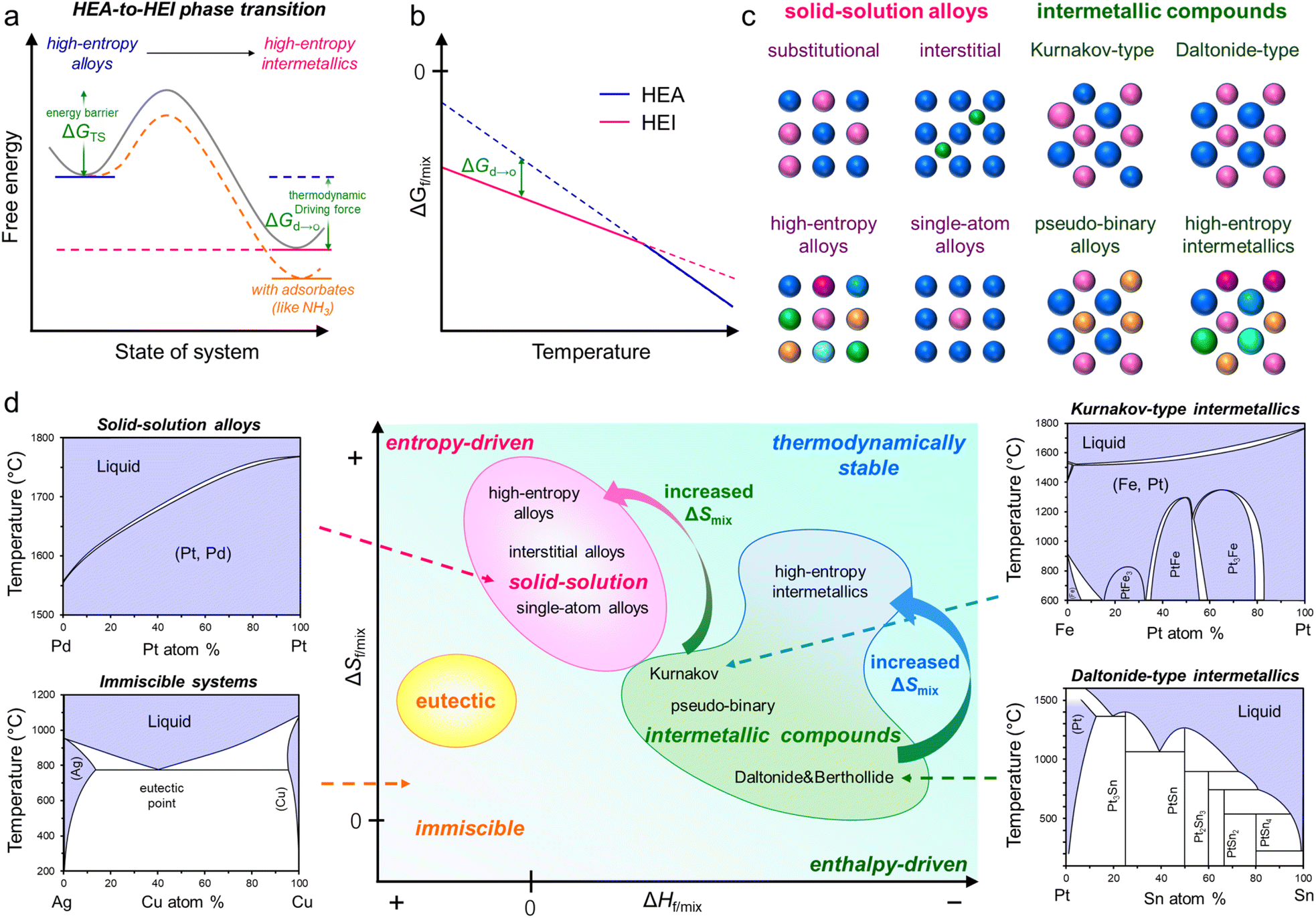 | ||
| Fig. 1 (a) Schematic illustration of the transition from a disordered HEA phase to an ordered HEI phase. (b) Gibbs free energies of the HEA and HEI structures as a function of temperature. (c) Schematic representation of various alloy structures. (d) Classification of alloy structures with regard to the thermodynamic driving force of formation. Phase diagrams of the Pd–Pt (solid solution, fully soluble), Ag–Cu (immiscible/eutectic), Fe–Pt (solid solution/Kurnakov-type intermetallic), and Pt–Sn (Daltonide-type intermetallic) systems reproduced from the AtomWork database of the National Institute for Materials Science (http://crystdb.nims.go.jp/). | ||
(1) Thermodynamics: enthalpic term vs. entropic term.
(2) Size dependence of HEI formation: surface energy.
(3) Annealing conditions: temperature/time/atmosphere.
(4) Prerequisites: proper selection of constituent metals.
2.1. Free energy of alloying
The proper design of HEIs requires a basic knowledge of thermodynamics, including solid-solution alloys, intermetallics, and HEAs (Fig. 1c and d).6,15,51,52 At present, the free energy change upon alloy formation could explain the phase diversity of various alloys (eqn (1)).| ΔGf/mix = ΔHf/mix − TΔSf/mix | (1) |
| Alloy | ΔHf (kJ mol−1) | Ref. |
|---|---|---|
a ΔHmix: mixing enthalpy for the liquid state.
b ΔHmix: mixing enthalpy for single-phase solid-solution alloys.
c Kurnakov-type intermetallics have compositional ranges on both sides of the stoichiometry, with order–disorder transition points at high temperatures.
d Berthollide-type intermetallics have compositional ranges on either side of the stoichiometry and retain their structures up to the melting points.
e Daltonide-type intermetallics have no compositions other than the stoichiometry (AmBn), therefore the m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n ratio is strictly fixed. :n ratio is strictly fixed.
|
||
| Immiscible systems | ||
| Ag0.5Cu0.5 | +3.5 | 53 |
| Fe0.5Cu0.5 | +10.5 | 54 |
| Ag0.6Ni0.4 | +22.0 | 55 |
|
||
| Solid-solution alloys | ||
| Pt0.5Co0.5 | −9.8 | 56 |
| Pt0.75Co0.25 | −9.2 | 56 |
| Cu0.5Au0.5 | −7.0 | 53 |
| Ag0.5Au0.5 | −4.2 | 53 |
| Ni0.5Fe0.5 | −3.9 | 57 |
| Pt0.5Pd0.5 | −3.6 | 58 |
| Pd0.5Ni0.5 | −0.5 | 57 |
| Pd0.5Co0.5 | 0 | 56 |
| Ni0.5Co0.5 | 0 | 56 |
|
||
| High-entropy alloys | ||
| CrFeCoNiPd | −7.0 | 59 |
| CrMnFeNiCo | −4.2 | 59 |
| VFeCoNiCu | −2.2 | 59 |
| CrFeCoNiCu0.5 | +0.5 | 59 |
| CrFeCoNiCu | +3.2 | 59 |
| CrMoFeNiCu | +4.6 | 59 |
|
||
| Kurnakov-type intermetallics | ||
| PtFe | −20.2 | 60 |
| PtCu | −16.0 | 61 |
| Pt3Fe | −15.9 | 62 |
| PtCo | −14.0 | 56 |
| Pt3Co | −12.0 | 56 |
| PtCu3 | −11.0 | 61 |
| Pt3Cu | −8.0 | 61 |
| AuCu | −8.0 | 63 |
|
||
| Berthollide-type intermetallics | ||
| PdGa | −79.4 | 64 |
| PdZn | −73.9 | 63 |
| PtZn | −65.0 | 63 |
| NiAl | −61.8 | 65 |
| PdIn | −61.4 | 66 |
| NiGa | −46.5 | 67 |
| Pt3In | −41.5 | 66 |
| Ni3Al | −41.0 | 57 |
| Ni3Sn2 | −31.8 | 68 |
| Ni3Sn | −26.3 | 68 |
| Ni3Sn4 | −25.3 | 68 |
| Ni3Ga | −25.3 | 67 |
| NiZn | −19.7 | 63 |
| Co3Mo | −5.3 | 56 |
| Co7Mo6 | −4.2 | 56 |
|
||
| Daltonide-type intermetallics | ||
| Pt3Zr | −112.9 | 62 |
| PtGe | −90.8 | 69 |
| PtSn | −74.0 | 70 |
| PtGa | −55.6 | 67 |
| Pt3Sn | −55.3 | 70 |
| PtBi2 | −49.1 | 71 |
| RhSb | −32.2 | 66 |
| Rh2Sb | −30.9 | 66 |
| PtBi | −29.5 | 71 |
|
||
| Pseudo-binary alloys | ||
| (Ni0.6Ru0.4)Al | −61.5 | 65 |
| (Ni0.4Ru0.6)Al | −63.1 | 65 |
| (Ni0.2Ru0.8)Al | −62.0 | 65 |
| (Co0.5Mn0.5)2Er | −27.2 | 72 |
Intermetallics are divided into three types, namely, Daltonide, Berthollide, and Kurnakov (footnotes in Table 1).6 A Kurnakov-type intermetallic (mostly represented by the composition formula of A3B, AB, or AB3) has a wide composition width (typically 10–20 atom%), although the crystal structure differs from that of parent metals. Although the A and B sites in the crystal are crystallographically distinct, the A(B) site is partially occupied with B(A) when the composition ratio is off-stoichiometric. Therefore, Kurnakov-type intermetallics also have a property characteristic of solid-solution alloys, that is, the (partial) random distribution of constituents undergoes a phase transition from an intermetallic to a solid-solution alloy at elevated temperatures, while the others do not. ΔHf of the Kurnakov-type is not largely negative (−8 to −16 kJ mol−1; Table 1) compared with other two types. At high temperatures, the contribution of the entropic term (−TΔSf/mix) is significantly strengthened. Therefore, the entropic term primarily dominates at elevated temperatures, and ΔGmix becomes more negative than ΔGf, allowing the phase transition to solid-solution alloys (low temperature: ΔGf < ΔGmix, high temperature: ΔGmix < ΔGf).
| ΔSmix = −R∑xiln(xi) | (2) |
| ΔSmix = −Rln(n) | (3) |
| Catalyst | Synthetic method | Temp. and pressure | Morphology and size | Parent structure/ΔHf (kJ mol−1) | Reaction | Ref. |
|---|---|---|---|---|---|---|
| Electrocatalysis | ||||||
| D15h (FeCoNi)3(TiAl) | Melt-spinning/dealloying | 1200–1500 °C/1 atm | Dendrite-like (ribbon) | Ni3Al/−41.0 (ref. 57) | HER | 20 |
| (FeCoNi)3(CuNiPd)/mNC | Ligand-assisted interfacial assembly/NH3 annealing | 750 °C/1 atm | Nanoparticle (10–50 nm) | Fe3Pd | ORR | 21 |
| PtRhFeNiCu | Impregnation/H2 reduction | 700 °C/1 atm | Nanoparticle (5–20 nm) | L12-type (A3B) | EOR | 23 |
| (PtRh)(BiSnSb) | Wet-chemistry method | 220 °C/— | Hexagonal nanoplate (average edge length ≃ 6.2 nm) | PtBi/29.6 (ref. 71) | MOR, EOR, and GOR | 22 |
| Pt(FeCoCuNi)/S–C | Surfur-anchoring method/H2 reduction | 1000 °C/1 atm | Nanoparticle (![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) = 5.1 nm) = 5.1 nm) |
PtCo/−14.0 (ref. 56) | ORR and HER | 24 |
| (PtIr)(FeCoCu)/C | Chemical co-reduction/H2 reduction | 850 °C/1 atm | Nanoparticle ( = 6 nm) |
PtFe/−20.2 (ref. 60) | ORR | 19 |
| (PtPdAu)(FeCoNiCuSn) | 5 min Joule heating/rapid cooling | ∼1100 K/1 atm | Nanoparticle ( = 5.2 nm) |
PtFe/−20.2 (ref. 60) | EOR | 28 |
|
||||||
| Thermal catalysis | ||||||
| (PtCoCu)(GeGaSn)/Ca–SiO2 | Pore-filling with freeze-dry/H2 reduction | 700 °C/1 atm | Nanoparticle ( = 2.2 nm) |
PtGe/−90.8 (ref. 69) | PDH | 25 |
| (PtCoNi)(InGaSn)/CeO2 | Impregnation/H2 reduction | 600 °C/1 atm | Nanoparticle (∼4 nm) | PtSn/−74.0 (ref. 70) | CO2-ODP | 27 |
| NiFeCuGaGe/SiO2 | Pore-filling with freeze-dry/H2 reduction | 700 °C/1 atm | Nanoparticle ( = 4.9 nm) |
NiGa/−46.5 (ref. 67) | Acetylene semihydrogenation | 26 |
Hu et al. performed Monte Carlo (MC) simulations for the quinary Pt(Fe0.7Co0.1Ni0.1Cu0.1) nanoparticle at 1100 K.28 A random HEA structure was made as an initial state, in which the constituent atoms gradually diffused to form the L10-type HEI phase. During this process, the formation enthalpy decreased significantly because of the strong interaction via the spin–orbital coupling and hybridization between non-noble metal 3d and noble metal 5d states. In addition, the more negative enthalpy results in a lower Gibbs free energy of the HEI than that of the HEA over a wide temperature range (∼273 to 1500 K), indicating that the phase transition from HEA to HEI is thermodynamically favorable (ΔGf < ΔGmix, Fig. 1b). These results demonstrate that the combination of the enthalpic and entropic effects in HEIs significantly improves the thermodynamic stability.
2.2. Size dependence of HEI formation
With regard to thermodynamics, the size of multimetallic alloys plays an important role in the synthesis of HEIs. A report by Hu et al. is a good example to understand the size dependence of the multi-elemental disorder-to-order phase transition.28 They synthesized differently sized PtFe0.7Co0.1Ni0.1Cu0.1 by changing the heating/cooling conditions (Fig. 2a). High-angle annular dark-field scanning transmission electron microscopy with energy-dispersive X-ray spectroscopy (HAADF-STEM-EDX) revealed that the 5 nm alloy had a single-phase HEI structure, whereas nanoparticles with a size of 20 and 150 nm had partially ordered (HEI + HEA) and phase-separated structures, respectively (Fig. 2b). Fig. 2c shows the long-range ordering (LRO) parameter (defined using eqn (4)) as a function of nanoparticle size.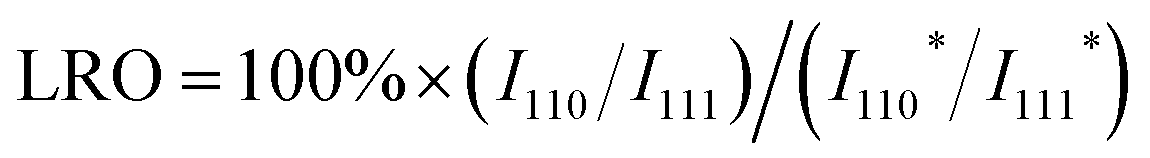 | (4) |
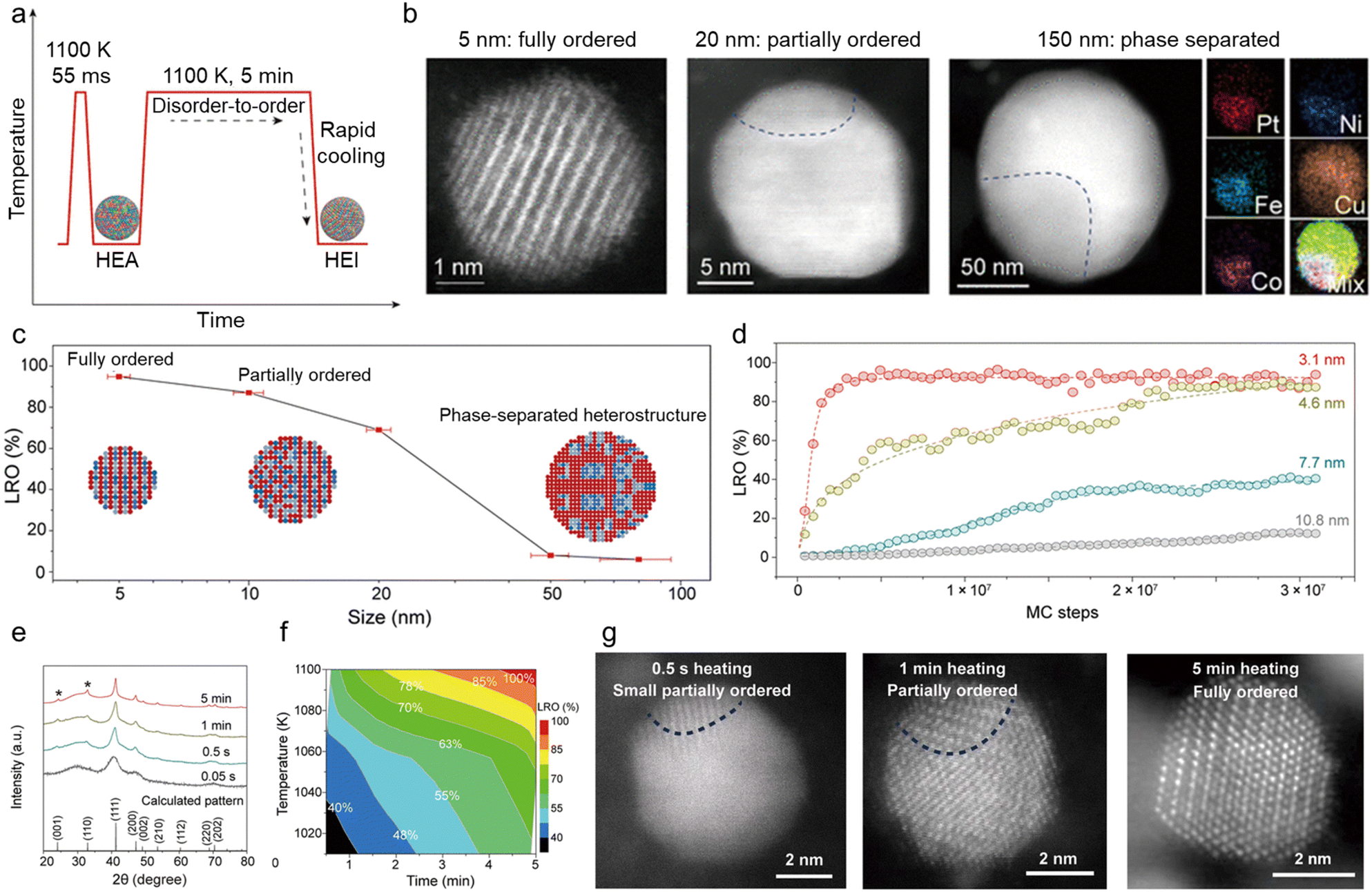 | ||
| Fig. 2 (a) The formation of HEI nanoparticles in the Joule-heating process, with regard to temperature evolution as a function of the heating time. (b) HAADF-STEM images of nanoparticles of increasing size (∼5, 20, and 150 nm), in which the smallest nanoparticle features an ordered structure, whereas the larger particles become increasingly disordered, corresponding to the schematics shown in (c). EDX mapping of the 150 nm particle shows its phase-separated structure. (c) The LRO as a function of the quinary HEI particle size. The insets schematically show the microstructural evolution as a function of the particle size. (d) The simulated LROs of differently sized nanoparticles (3.1, 4.6, 7.7, and 10.8 nm) as a function of MC simulation steps. (e) XRD patterns of Pt(Fe0.7Co0.1Ni0.1Cu0.1) synthesized at different heating times at ∼1100 K and the virtual XRD pattern of the fully ordered Pt(Fe0.7Co0.1Ni0.1Cu0.1) HEI computed using the virtual XRD analysis method. The asterisk (*) represents the superlattice peaks of the intermetallic. (f) The time and temperature dependence of the LRO of Pt(Fe0.7Co0.1Ni0.1Cu0.1). (g) HAADF-STEM images showing the evolution of nanoparticles held at ∼1100 K via Joule-heating for different time durations (0.5 s, 1.0 min, and 5 min). The Pt(Fe0.7Co0.1Ni0.1Cu0.1) sample obtained by heating for 0.5 s, 1 min, and 5 min showed a small partially ordered structure, slightly more but still partial ordering, and a fully ordered structure. Reproduced with permission from ref. 28. Copyright 2022 American Associate for the Advancement of Science. | ||
2.3. Annealing conditions
In addition to the abovementioned factors, annealing conditions, including the (i) temperature, (ii) time duration, and (iii) atmosphere, are important factors that must be considered in the construction of HEIs. It should be noted that factors (i) and (ii) are categorized as kinetics, while factor (iii) is categorized as thermodynamics. The L10-type Pt(Fe0.7Co0.1Ni0.1Cu0.1)/C system is a good example to understand the mechanism by which the annealing temperature (T) and time duration (t) play important roles in the construction of ordered HEI structures.28 First, disordered PtFe0.7Co0.1Ni0.1Cu0.1 HEA nanoparticles were synthesized by heating the metal precursors for tens of milliseconds at ∼1100 K to mix the different elements without phase separation. Then, the as-synthesized HEA nanoparticles were rapidly reheated to 1100 K for an additional ∼5 min (Fig. 2a). Fig. 2e and f show the X-ray diffraction (XRD) patterns of the sample at different heating times at ∼1100 K and the effects of heating temperature and time on LRO, respectively (see Fig. 2g for the HAADF-STEM images). Annealing at a high temperature for a long time promoted the gradual phase transition from disordered to ordered structures.A similar result was reported for the Pt(FeCoCuNi)/S-doped carbon (S–C) system (PtCo-type HEI).24 The XRD pattern of the sample annealed at 500 °C showed the disordered fcc phase, and the fcc HEA structure can be formed even at T = 170 °C for t = 5 min. These results demonstrated a low thermodynamic barrier for the formation of a fcc HEA structure. By contrast, the XRD pattern of the samples annealed at T ≥ 600 °C showed the superlattice peaks of the ordered face-centered tetragonal PtCo-phase, including (001) at 24.0° and (110) at 32.8°. In quantitatively estimating the degree of ordering, the ratio of the (110) peak area to the combined area of the (111), (200), and (002) peaks, denoted as S(110)/(S(111) + S(200) + S(002)), was used. When T was increased from 600 °C to 900 °C for t = 25 min, the ordering degree increased from 35% to 76%. In addition, when t was extended from 25 to 120 min at T = 700 °C, the ordering degree also increased from 52% to 65%. These results demonstrated that the control of the annealing temperature and time duration is of great importance for the synthesis of fully ordered HEI structures.
In addition, the atmosphere plays an important role in the disorder-to-order transition. Zhu et al. investigated the effect of annealing atmosphere on the nanoparticulate FeCoNiCuPd system.21 Despite the high annealing temperature of 750 °C, the N2 atmosphere resulted in a typical fcc phase. By contrast, the use of NH3 resulted in the formation of the ordered Fe3Pd-type HEI structure. Density functional theory (DFT) calculations showed that the adsorption energies of N2 and NH3 were −0.11 and −0.32 eV on the HEI, while −0.44 and −0.11 eV on the HEA, respectively. Therefore, NH3 prefers to be adsorbed on the HEI surface, whereas N2 prefers to be adsorbed on the HEA surface. Moreover, molecules that adsorb more strongly on HEIs lower the thermodynamic barrier of the disorder-to-order transition (Fig. 2a), thereby promoting HEI formation. These fundamental insights into annealing conditions will provide an important impetus for the synthesis of new HEI nanomaterials.
2.4. Selection of elements
So far, we have explained how to overcome the thermodynamic barrier for the construction of HEIs. However, prerequisites should be considered to achieve this goal, including the proper selection of constituent elements and the homogeneous distribution of metal precursors. Compared with HEAs, HEIs have specific structures derived from the parent intermetallics (AmBn). When the electronegativity of a secondary metal is largely different from that of the primary metal, a pair of the elements tends to form the intermetallic to maximize charge redistribution rather than to increase the disorder to obtain the entropic stabilization. If the electronic properties of the added elements are largely different from those of A and B, then the degree of charge redistribution is decreased. Therefore, enthalpic stabilization may no longer be greater than the alloy system, and entropic stabilization is preferred, thereby leading to the formation of HEAs. Therefore, the additional elements should have electronic properties close to those of either A or B to maintain the parent ordered structure.3. Synthetic methods for HEI nanomaterials
Controlled synthesis of HEI nanomaterials is the basic requirement for the research of HEIs in heterogeneous catalysis, which raises the bar of this field. Here, we introduce the representative synthetic methods reported in the literature (Table 2). In 2020, Jia et al. reported the catalytic properties of HEIs for the first time.20 They synthesized the (FeCoNi)3(TiAl) HEI catalyst by combining the melt-spinning technique and de-alloying process. They first prepared a parent alloy ingot whose atomic composition was well designed with homogeneous distribution by arc-melting, then melted it again at high temperatures of 1200–1500 °C under an Ar atmosphere, and the molten metal alloy was ejected onto a rotating Cu wheel surface. This unique process resulted in as-spun HEI ribbons with a nominal thickness of 30–40 μm. The obtained ribbons were cut into segments and then dealloyed by a chemical extraction process using 1.0 M HCl. As a result, they obtained the L12-type (FeCoNi)3(TiAl) HEI with a dendritic surface morphology. In 2022, Cui et al. synthesized the (PtPdAu)(FeCoNiCuSn) HEI by Joule heating on a carbonized wood substrate.28 Eight different types of metal chlorides were deposited on a carbonized wood substrate using EtOH as a solvent. Then, the as-deposited metal precursors were heated by Joule heating using electrical pulses. The reaction temperature can be controlled with an electric current. Although this unique process can endow the octonary (PtPdAu)(FeCoNiCuSn) nanoparticles with a size of 4–5 nm, this requires special equipment setup and can only be applied to conductive supports. Several synthetic methods, such as the wet-chemistry, chemical co-reduction, and impregnation methods have also been reported as simpler methods. Chen et al. synthesized (PtRh)(BiSnSb) HEI nanoplates by the wet-chemistry method.22 A mixture of metal precursors, ascorbic acid, cetyltrimethyl ammonium bromide, octadecene, and oleylamine was heated at 220 °C under magnetic stirring, forming hexagonal nanoplates with an average edge length of 6.2 nm. Feng et al. synthesized (PtIr)(FeCoCu) HEI nanoparticles with a size of 6 nm by chemical co-reduction.19 Under an Ar atmosphere, a mixed solution of metal precursors and carbon powders was treated with NaBH4 and collected by centrifugation. The obtained product was washed and dried under Ar protection, and then annealed at 850 °C under flowing H2/Ar. A disordered phase can be obtained instead of the ordered phase when it was annealed at 400 °C. Nakaya and Furukawa et al. synthesized HEI nanoparticles supported on a SiO2 support by the pore-filling freeze-drying method.25,26 First, the metal precursors were added dropwise to the SiO2 support with a minimum solution volume to fill the pores of SiO2. The obtained mixture was quickly frozen using liquid nitrogen and dried in a vacuum at −5 °C. The obtained powder was dried at 90 °C overnight and calcined at 400 °C, followed by reduction at 700 °C with flowing hydrogen. Importantly, the combination of the pore-filling and freeze-drying allows the homogeneous deposition of the metal precursors on the support, resulting in the successful synthesis of (PtCoCu)(GeGaSn)25 and NiFeCuGaGe26 HEI nanoparticles. The preparation of HEIs using a conventional impregnation method with H2 reduction has also been reported by Furukawa et al.27 They synthesized (PtCoNi)(InGaSn) HEI nanoparticles on a CeO2 support. Since, this research field has been growing, various effective synthetic methods will be developed in the near future.4. Roles of HEIs in catalysis
In this chapter, we briefly outline the roles of HEIs in catalysis. Geometric and electronic effects are the typical factors that can be considered to explain the origin of high performance of alloy catalysts. Although these effects are often discussed independently of one another, conducting a separate discussion of these effects is generally difficult. However, this may be possible if one selects appropriate systems and carefully discusses the details. In this chapter, we introduce the representative roles of HEIs in catalysis, that is, (1) geometric site isolation, (2) fine-tuning of the electronic structure, and (3) multifunctional effects.4.1. Geometric effects: site isolation
The atomic-scale control of surface ensembles is a challenging but an attractive approach to achieve an unprecedented catalytic performance. One of the major advantages of HEIs is the construction of metallic single-atom (M1) sites. In recent years, M1 sites embedded in alloy materials have attracted attention as promising active sites for a variety of reactions.25,75–79 However, the complete isolation of active sites requires a large excess (>20 equiv.) of counterpart metals in disordered alloy structures (e.g., single-atom alloys (SAAs) and HEAs).6,25 In addition, the poor thermal stability of the parent alloy materials leads to severe sintering at high operation temperatures.77 Therefore, the facile construction of thermally stable M1 sites is highly desired. Compared with disordered alloys, intermetallics are promising architectures because of their well-defined ordered structures, which result in small surface active metal–metal coordinations. However, even equimolar intermetallics typically do not provide isolated sites by themselves because of the crystallographically determined surface configurations. The selective substitution of crystallographically distinct sites allows the further dilution of such coordinations while maintaining the thermal stability derived from the parent ordered structures. In addition, the combination of the significantly negative formation enthalpy of the parent materials and the increased mixing entropy in HEIs can dramatically improve the structural stability.6 Therefore, the isolation of active metals using HEIs is highly attractive. For example, Nakaya et al. reported the construction of isolated Pt sites on the PtGe-type HEI.25 As shown in Fig. 3, the (020) surface of the PtGe intermetallic (FeAs-type structure, space group: Pnma) provides one-dimensionally aligned Pt columns, where the surface Pt–Pt coordination number is only two. The Pt and Ge sites were selectively substituted by much less active Co/Cu and inert Ga/Sn, respectively. Detailed structural analysis revealed that only isolated Pt (Pt1) sites were exposed on the surface of the HEI (Pt fraction: 0.25). Pt1@(PtCoCu)(GeGaSn) acted as thermally stable active sites, providing significantly high selectivity and durability for propane dehydrogenation. Based on the segregation energy obtained by DFT calculations, the HEI structures with Pt1 sites were electronically more favorable than those with Pt–Pt coordinations. As control experiments, the quaternary (PtCoCu)Ge and quinary HEA catalysts were also synthesized. However, the (PtCoCu)Ge and HEA catalysts suffered from low thermal stability and coke deposition (induced by Pt–Pt ensembles), respectively. These results indicated that the multimetallization of Ge sites by Ga/Sn improved the thermal stability because of the increased mixing entropy and the use of the HEI achieved more efficient site isolation. DFT calculations showed that the undesired side reactions on the HEI surface were effectively suppressed by facile propylene desorption. The density of states (DOS) projected onto the d-orbitals of the surface Pt atoms verified the absence of a significant difference in the d-band shape and center between PtGe and the HEI. Therefore, the site isolation effect primarily causes the superior catalytic performance. However, the Pt1 site on Pt–Cu SAA (Pt1@Cu) possessed the d-band center (εd) much closer to the Fermi level, indicating the potentially low selectivity for PDH. The experimental and theoretical results supported the inferior selectivity of Pt1@Cu for PDH. Therefore, although the ligand effect is not involved in this multimetallization, the electronic properties of the parent materials are significantly important. A similar site isolation methodology has also been reported for other catalytic systems to achieve a remarkable catalytic performance.26,27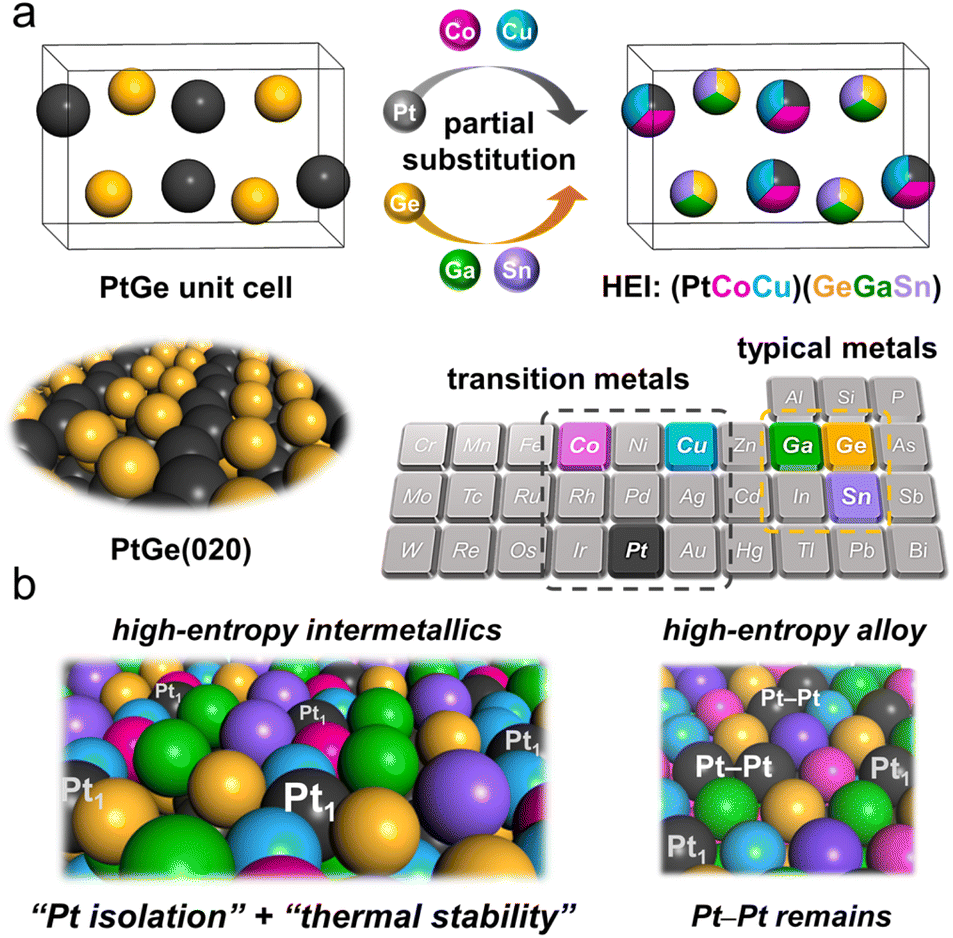 | ||
| Fig. 3 Catalyst design concept for thermally stable isolated Pt sites using the HEI. (a) Pt and Ge sites in the intermetallic PtGe are partially substituted by Co/Cu and Ge/Sn, respectively, resulting in the formation of the PtGe-type (PtCoCu)(GeGaSn). (b) Illustrations of the (020) surface of the (PtCoCu)(GeGaSn) HEI (left) and the (111) surface of the Pt-based senary HEA (right). | ||
4.2. Electronic effects: fine-tuning
The other advantage of HEIs is the fine-tuning of the electronic structure caused by the combination of the ordered and multimetallic nature, which alters the adsorption properties and resulting catalysis. However, in general, the geometric and electronic effects interfere with each other. Therefore, extracting the electronic effect while excluding the geometric effect is difficult. The catalytic system with small atoms/molecules is the appropriate reaction to minimize the geometric effect and to outline the electronic effect of HEIs. Here, we introduce the fine-tuning of the electronic structure using the oxygen reduction reaction (ORR: 1/2O2 + 2H+ + 2e− → H2O) as an example. The ORR is a desirable reaction, and its sluggish kinetics must be boosted (see Section 5.2.2 for details on the ORR). A volcano plot (Sabatier principle) is observed between the oxygen binding energy and the ORR activity.6,80–83 In addition, the oxygen binding energy is correlated with the d-band center.6,80–83 Therefore, the d-band center is the commonly used descriptor to predict/interpret the ORR activity. Based on the predictions, the ORR activity reaches the maximum when the d-band center of active metals is downshifted by approximately 0.32 eV than that of pure Pt(111).80,81 Therefore, tremendous efforts have been devoted to tune the d-band center close to the optimum value by alloying. However, the fine-tuning of the electronic structure using conventional solid-solution alloys, intermetallics, and even HEAs is highly difficult.40,82,83Here, the unique report by Feng et al. provides a comprehensive understanding of the effects of ordered and multi-element properties on the electronic features of HEIs.19 Low-index facets are thermodynamically stable, but they have inferior ORR activities compared with high-index facets. However, they succeeded in transforming the low active (001) facet into a highly active surface using the PtFe-type (PtIr)(FeCoCu) HEI. As shown in Fig. 4a, the εd value of the Pt(001) was upshifted by 0.09 eV from that of Pt(111), which was away from the optimum value. Therefore, the ORR activity on Pt(001) is inferior to that on Pt(111) because of the strong oxygen binding energy. For HEA(111), HEA(001), and HEI(111), the downshifts of εd were 0.37, 0.17, and 0.28 eV, respectively (Fig. 4b and c). By contrast, εd of HEI(001) was downshifted by 0.31 eV, which is closest to the optimal value (0.32 eV, Fig. 4c). The larger downshift of HEI(001) than that of HEI(111) can be explained by the spatial configuration of the nearest neighbors of the surface Pt (Fig. 4d). The dyx, dyz, and dxz orbitals directly face four non-noble and four noble metal atoms (eight in total) for Pt at the HEI(001) surface, whereas the orbitals directly face two non-noble and one noble metal atoms (three in total) for Pt at the HEI(111) surface. Therefore, the (001) surface of the HEI can be tuned more directly by other metal atoms than the (111) surface. Consequently, the (001) surface of the (PtIr)(FeCoCu) HEI exhibited an outstanding ORR activity because of optimized εd of Pt. Therefore, HEIs are promising architectures for fine-tuning of the electronic structure because of the combination of ordered and multimetallic nature.
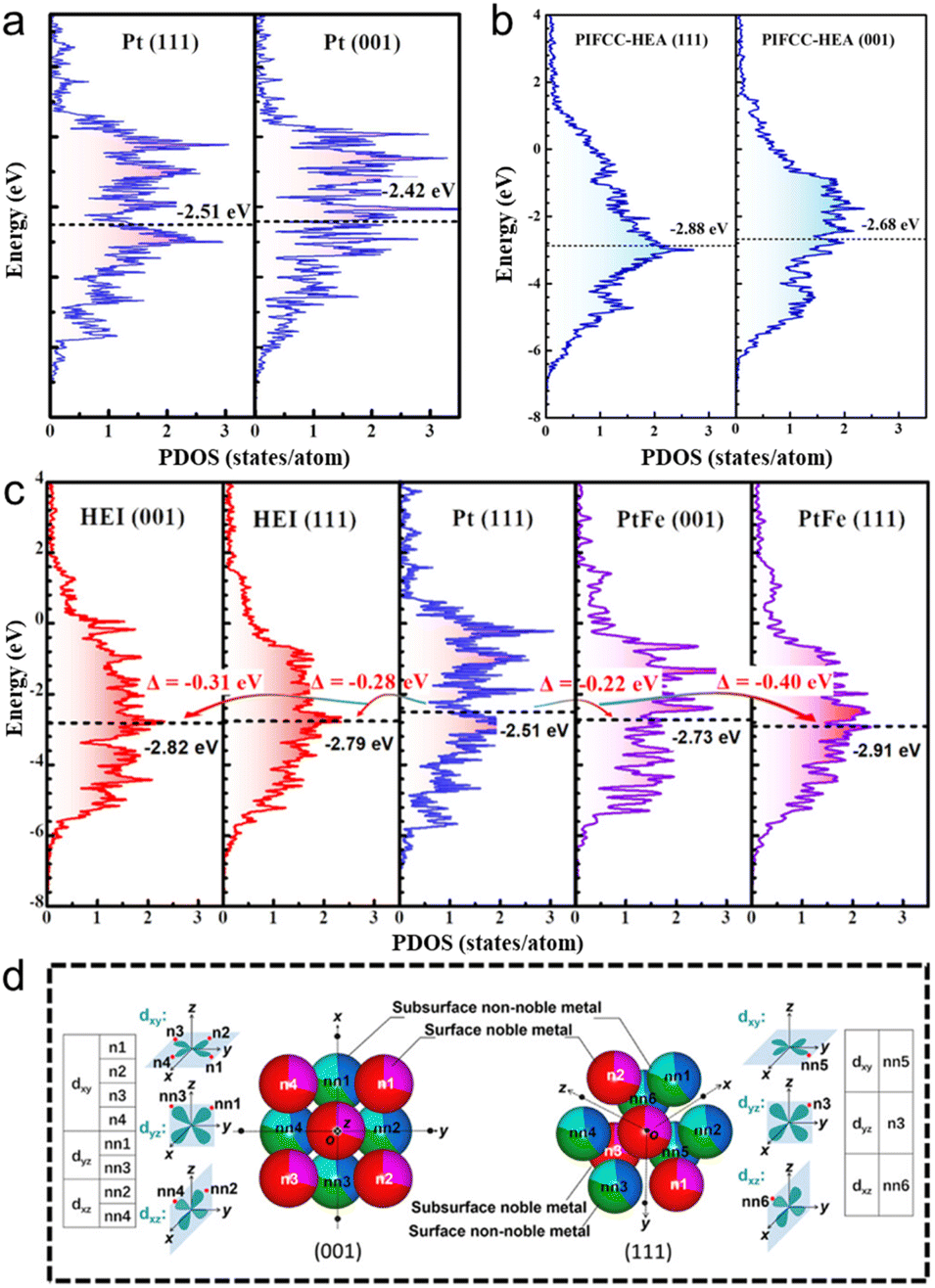 | ||
| Fig. 4 DOS projected onto the d-orbitals of the (111) and (001) surfaces of (a) pure Pt and (b) PtIrFeCoCu HEA. (c) DOS projected onto the d-orbitals of the surface Pt atoms in the (PtIr)(FeCoCu)-HEI(001), HEI(111), Pt(111), PtFe(001), and PtFe(111) surfaces. (d) Spatial configurations of the nearest neighbors of the surface Pt in the HEI(001) and HEI(111) surfaces. Reproduced with permission from ref. 19. Copyright 2023 American Chemical Society. | ||
4.3. Multifunctional effects: synergetic contribution of element-specific functions
Another characteristic of HEIs is the provision of multifunctional effects, which are neither geometric nor electronic effects. In general, the geometric and electronic effects are specified when the counterpart metals are catalytically less active or inert for the reaction. Therefore, only the change in the catalytic/adsorptive characteristic of the main active metals is included in the main discussion. By contrast, multifunctional effects occur when the counterpart metals, which contribute positively and actively to catalysis, are incorporated in the alloy. The synergetic contributions of the element-specific functions of the main and counterpart metals can effectively catalyze the complex catalysis, which requires catalyzing different substrates/intermediates. These multifunctional effects have been observed in the alloy materials including the random (e.g., solid-solution alloys and HEAs) and ordered (e.g., binary and pseudo-binary intermetallics) alloys.6,84–88 Multifunctional effects have also been reported in HEIs.24,27 The PtSn-type (PtCoNi)(SnInGa) HEI system reported by Furukawa et al.27 is a representative example of the multifunctional effect using a HEI (Fig. 5). For the oxidative dehydrogenation of propane using CO2 as an oxidant (CO2-ODP; C3H8 + CO2 → C3H6 + CO + H2O), in addition to the dehydrogenation ability, the CO2 activation ability is also necessary to complete the reaction. Therefore, constructing a multifunctional architecture is necessary. In this context, the Pt and Sn sites of the PtSn intermetallic were multimetallized to form the (PtCoNi)(SnInGa) HEI. The introduction of Co/Ni can improve the CO2 activation ability. Consequently, the synthesized (PtCoNi)(SnInGa)/CeO2 HEI catalyst exhibited significantly high specific activity for propylene and long catalyst lifetime in CO2-ODP at 600 °C.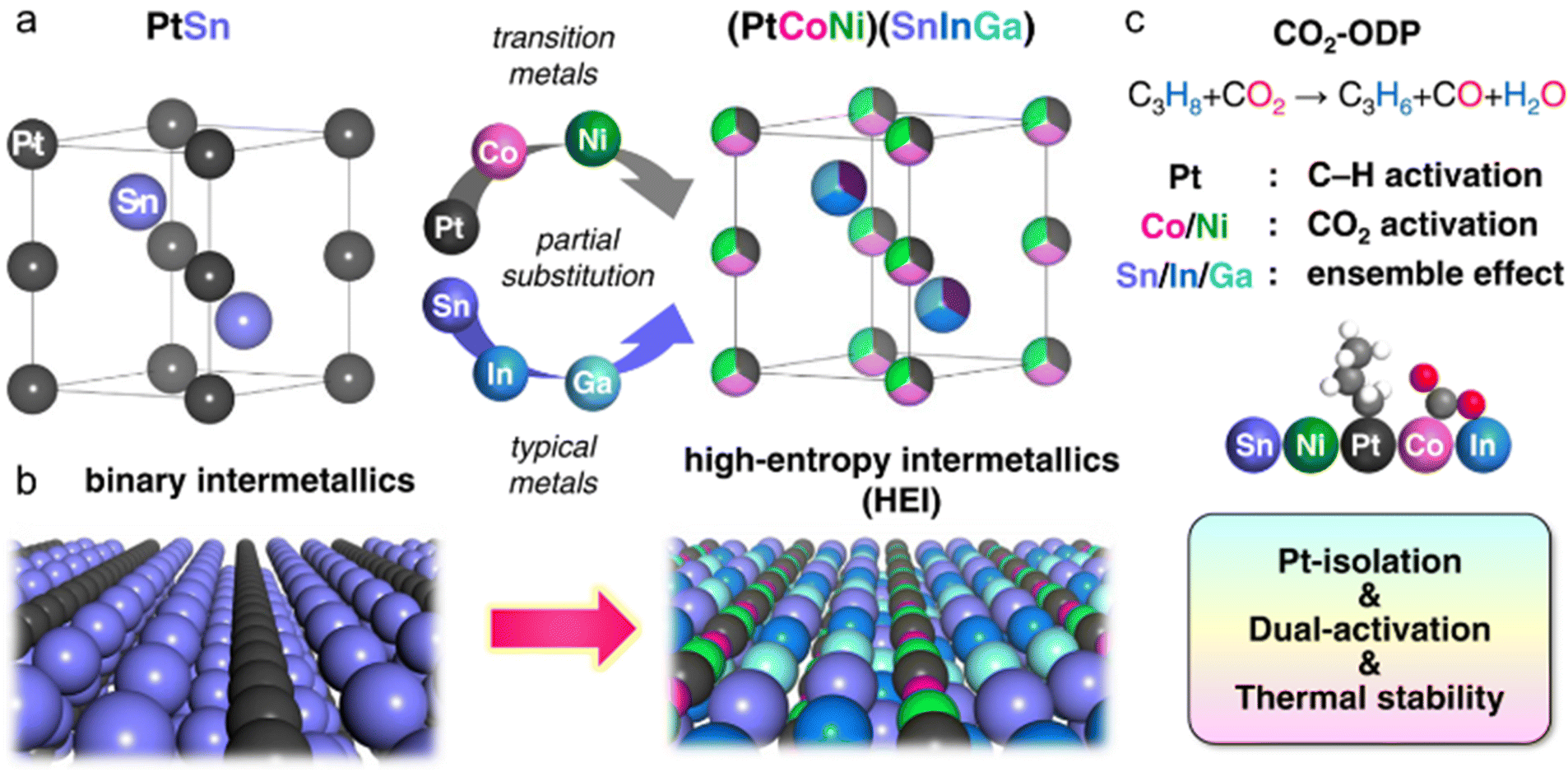 | ||
| Fig. 5 (a) Catalyst design concept based on the HEI. Pt and Sn sites in intermetallic PtSn (hexagonal NiAs-type structure, space group: P63/mmc) are partially substituted by Co/Ni and In/Ga, respectively, to form a PtSn-type HEI (PtCoNi) (SnInGa). (b) Atomic arrangement of the most stable (110) surfaces of PtSn (left) and HEI (PtCoNi)(SnInGa) (right). (c) The role of each metal and the effect of multimetallization on the catalysis of CO2-ODP. | ||
4.4. Mixing effects
As the number of constituent metals increases, the physical and/or chemical properties of alloys are often improved.6 In recent years, the “mixing effect” has been proposed as one of the positive roles played by multimetallic alloys in catalysis. Although the mixing effect itself does not directly affect the adsorption and/or catalytic behavior, it modifies the alloying degree and/or the stability of alloy phases by multlimetallization. An example is the enhancement in the mutual solubility of the constituent metals. Even when the two constituent metals have a miscibility gap or low miscibility, the degree of alloying can be enhanced by increasing the number of constituent metals due to the increase in ΔSmix. This phenomenon is also known as the entropy effect (Section 2.1 for details). Another example is the improvement of structural and/or chemical stability. The ΔG value for alloy formation becomes more negative owing to the contribution of larger ΔSmix, which makes the alloy thermodynamically more stable. In addition, the atomic diffusion rate in the crystal lattice decreases due to the large difference in the atomic radii of the constituent metals (sluggish diffusion effect), which enhances the robustness of the alloy phase. This effect also enhances the anti-sintering properties of alloy nanoparticles. Therefore, the multimetallization can increase the stability of alloys thermodynamically and kinetically, thus improving the catalyst durability and robustness even under harsh conditions (see Chapter 6 for some examples). Notably, however, the true picture of the entropy effect remains unclear. Developing further experimental and theoretical rationale is expected for a clearer understanding of this chemistry.5. Identification of HEI structures
In this section, we explain the detailed analytical method used to determine the formation of HEIs. The priority of measurements is first XRD and HAADF-STEM-EDX and then X-ray absorption fine structure (XAFS) if the aforementioned methods do not provide sufficient evidence. The combination of each analysis allows for more detailed structure determination.5.1. XRD
Laboratory or synchrotron XRD is a powerful technology to prove the ordered phase. Fig. 6a shows the laboratory XRD patterns of the PtRhFeNiCu HEA and HEI catalysts.23 The HEA showed a typical Pt-like fcc feature. By contrast, the XRD pattern of the HEI showed the (100) and (110) characteristic diffraction peaks of an ordered L12 structure (A3B-type). In addition, the diffraction angles were shifted to higher angles, revealing the larger lattice shrinkage in the HEI than in the HEA. Fig. 6b shows the synchrotron XRD patterns of PtGe-type (PtCoCu)(GeGaSn) HEI nanoparticles.25 The synthesized HEI showed a PtGe-type diffraction pattern, unlike fcc-, bcc-, or hcp-HEAs. The diffraction angles were largely shifted to higher angles because of lattice shrinkage (5.6%). The theoretical lattice shrinkage was calculated to be 5.4%, which was consistent with the experimental result. Notably, the intensity of the 011 diffraction for the PtGe-type HEI was low. Upon the incorporation of differently sized elements, the crystal order decreases because of lattice distortion. This decrease in order is more pronounced at longer periods, thereby lowering the relative diffraction intensity at lower angles. Such a decrease in diffraction intensity has also been reported in HEI and fcc HEA systems.89–91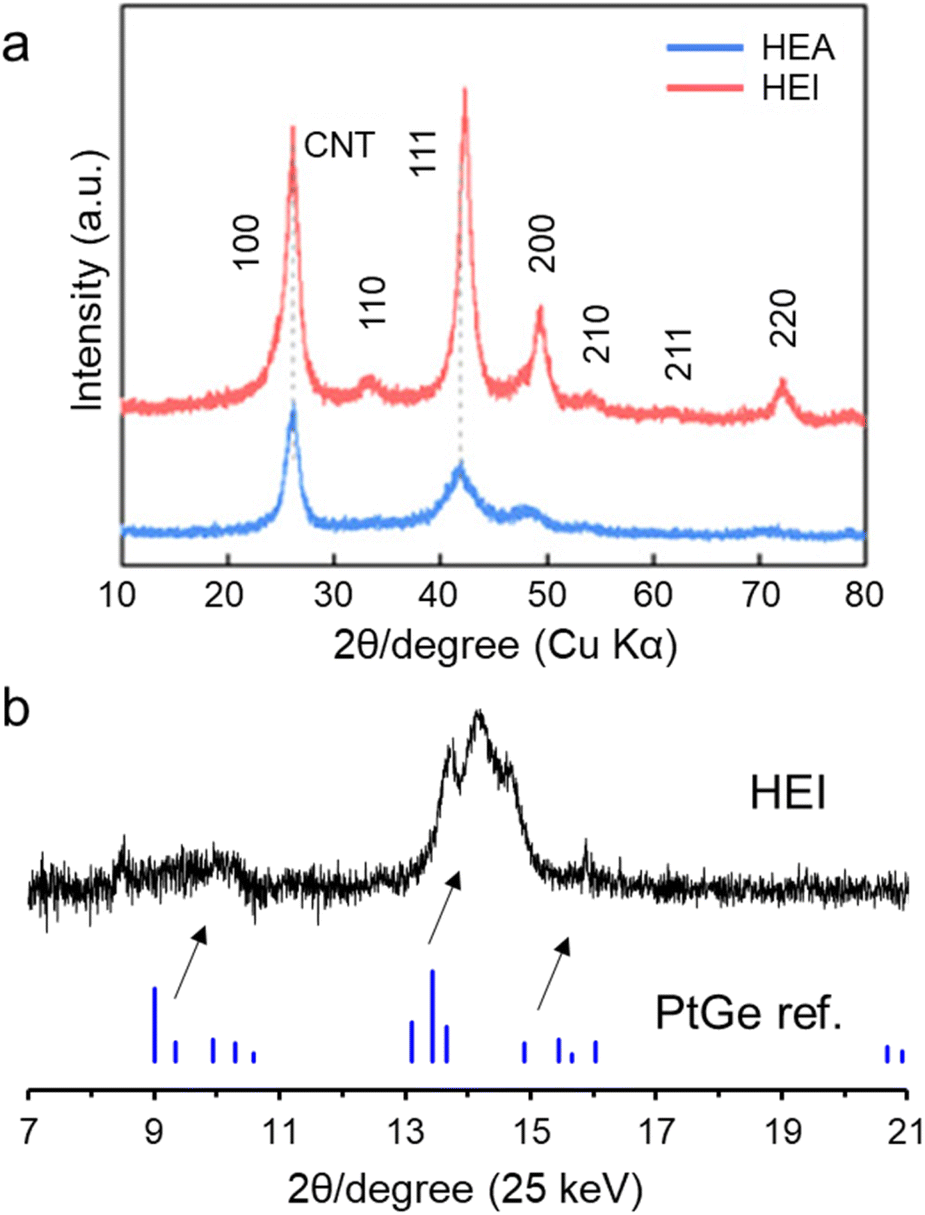 | ||
| Fig. 6 (a) XRD patterns of the PtRhFeNiCu HEA and HEI catalysts.23 (b) Synchrotron XRD pattern of the (PtCoCu)(GeGaSn)/Ca–SiO2 catalyst. | ||
5.2. HAADF-STEM-EDX
HAADF-STEM with EDX spectroscopy is also a powerful technology for identifying the formation of HEIs. In this section, we use the (FeCoNi)3(NiCuPd) system as an example to explain how the HEI structure can be identified using HAADF-STEM-EDX.21 Wang et al. synthesized the nanoparticulate (FeCoNi)3(NiCuPd) HEI by annealing under an NH3 atmosphere. Elemental maps of a single nanoparticle revealed the homogeneous distribution of Fe, Co, Ni, Cu, and Pd. Given the different average atomic numbers, the HAADF-STEM image distinguished two types of atomic columns, corresponding to the projection of the ordered L12 structure (Fe3Pd phase) along the [100] direction (Fig. 7a). The corresponding fast Fourier transform (FFT) pattern showed (0![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and (00) reflections in the reciprocal space (Fig. 7b), confirming the L12 structure. The L12 structure was further observed along the [110] direction (Fig. 7c and d). As shown in Fig. 7e, FeCo, and CuPd occupied the face-centered (A) and vertex (B) sites of the L12 A3B structure, respectively. By contrast, Ni was randomly distributed at both sites. These results indicated the formation of the (FeCoNi)3(NiCuPd) HEI. The use of HAADF-STEM-EDX and FFT patterns is of great importance for the identification of HEI structures. However, notably, HAADF-STEM-EDX analysis only provides the local information and not the whole information. Therefore, the conclusion should be drawn in combination with other measurements such as XRD.
) and (00) reflections in the reciprocal space (Fig. 7b), confirming the L12 structure. The L12 structure was further observed along the [110] direction (Fig. 7c and d). As shown in Fig. 7e, FeCo, and CuPd occupied the face-centered (A) and vertex (B) sites of the L12 A3B structure, respectively. By contrast, Ni was randomly distributed at both sites. These results indicated the formation of the (FeCoNi)3(NiCuPd) HEI. The use of HAADF-STEM-EDX and FFT patterns is of great importance for the identification of HEI structures. However, notably, HAADF-STEM-EDX analysis only provides the local information and not the whole information. Therefore, the conclusion should be drawn in combination with other measurements such as XRD.
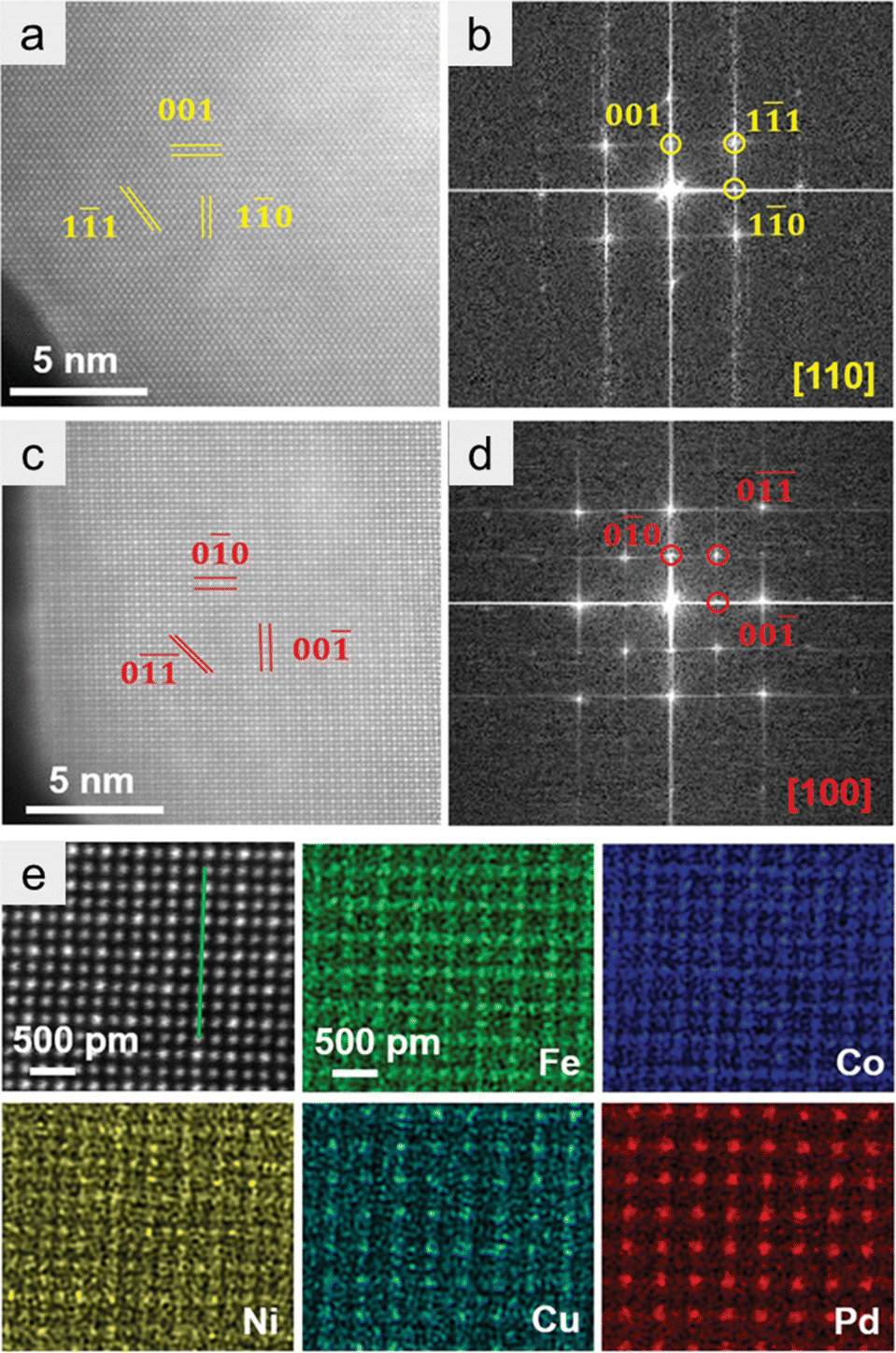 | ||
| Fig. 7 (a and b) Atom-resolved HAADF-STEM image and the corresponding FFT pattern along the [100] zone axis. (c and d) Atom-resolved HAADF-STEM image and the corresponding FFT pattern along the [110] zone axis. (e) Atom-resolved HAADF-STEM image and corresponding EDS maps. Reproduced with permission from ref. 21. Copyright 2022 Wiley-VCH. | ||
5.3. XAFS
As explained in the previous sections, the combination of XRD and HAADF-STEM-EDX is of great importance for the identification of HEIs. However, for extremely small nanoparticles (<3 nm), determining the exact location of each element by elemental mapping is difficult. In addition, FFT analysis cannot be applied to the image of a narrow periodic repeating structure. In such cases, further structural analysis by XAFS plays a crucial role. A good example to understand the fundamental insights for XAFS analysis of HEIs is a PtGe-type (PtCoCu)(GeGaSn) HEI (average size = 2.2 nm).25 Synchrotron XRD and HAADF-STEM-EDX revealed the PtGe-type structure and homogeneous distribution of all constituent elements in a nanoparticle, respectively (Fig. 6b and 8a). Quantitative analysis of some nanoparticles elucidated the unity atomic ratios of (Pt + Co + Cu)/(Ge + Ga + Sn). However, quantitative analysis does not provide the information for selective substitutions. XAFS analysis was performed for Pt LII-, Co K-, Cu K-, Ge K-, Ga K-, and Sn K- edges to study the coordination environment of the atoms in the HEI. As shown in Fig. 8b, the HEI presented a Pt LII-edge XANES characteristic that is similar to those of bulk and nanoparticulate PtGe intermetallics but different from that of nanoparticulate Pt. The raw extended XAFS (EXAFS) oscillation also showed a similar tendency (Fig. 8c). In addition, the EXAFS oscillation did not coincide with those of Pt0.5Co0.5, Pt0.5Cu0.5, PtGa, and PtSn, indicating that the PtGe-type structure was retained even after multimetallization. Compared with PtGe, the HEI displayed two peaks at 2.0 and 2.5 Å (Fig. 8d), which could be assigned to Pt–Ge(Ga) and Pt–Sn scatterings, respectively, demonstrating the incorporation of Sn into the Ge site. EXAFS curve-fitting analysis allowed the assignment of all the associated transition-typical metal scatterings, providing comprehensive support for the formation of the (PtCoCu)(GeGaSn) HEI structure. They also synthesized the quinary multimetallic alloy using PtGa as the parent structure. However, structural analysis revealed the formation of the disordered HEA. ΔHf of PtGe (−90.8 kJ mol−1)69 is more negative than that of solid-solution alloys (Pt0.5Co0.5: −9.8 kJ mol−1)56 and intermetallics (PtGa: −55.6 kJ mol−1,67 PtSn: −74.0 kJ mol−1).70 Therefore, the large contribution of the enthalpic effect is an important driving force to maintain the parent ordered structure (see Section 2.1 for the detailed discussion on thermodynamics). The determination of the HEI structure by XAFS analysis has also been successful in other systems,23,26–28 providing useful structural information. Notably, the XAFS spectra often overlap with one another, therefore, the constituent elements and the choice of their edges for XAFS should be considered before performing the XAFS measurement.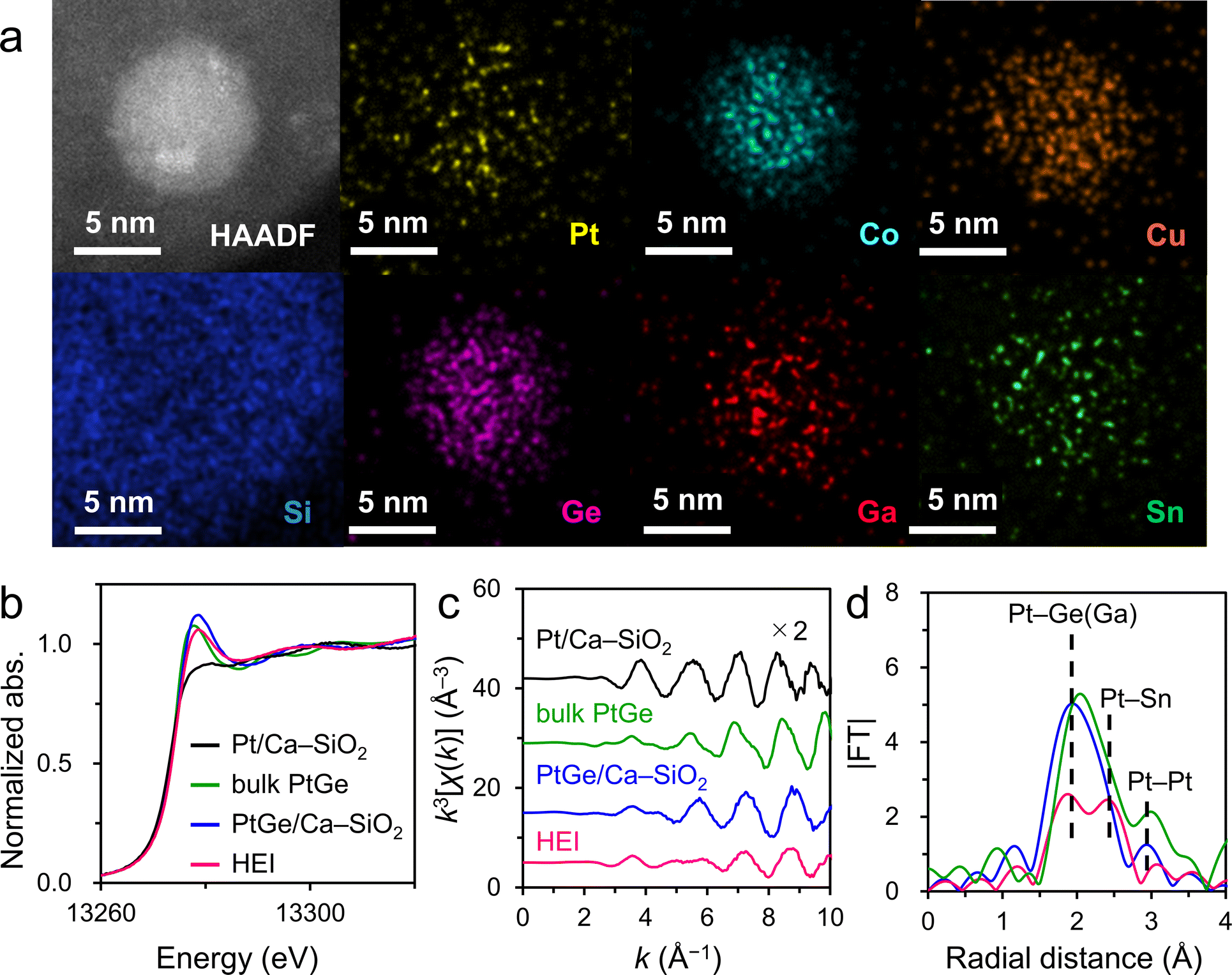 | ||
| Fig. 8 (a) HAADF-STEM image and the corresponding elemental maps for a single nanoparticle in the (PtCoCu)(GeGaSn)/Ca–SiO2 catalyst. Pt LII-edge (b) XANES, (c) k3-weighted EXAFS oscillations, and (d) Fourier-transformed EXAFS of Pt/Ca–SiO2, bulk PtGe, PtGe/Ca–SiO2, and HEI(0.25). | ||
6. Catalysis of HEIs
To date, 10 reports have been found on the application of HEIs, including three in thermal catalysis25–27 and seven in electrocatalysis.19–24,28 Among these reports, HEIs have been found to have a unique catalytic performance that dramatically exceeds those of conventional catalysts. In guiding the development of new HEI catalysts, the HEI for each reaction is presented in this chapter. The mechanism of enhancing the catalytic performance would provide fundamental insights into a more rational catalyst design based on HEIs.6.1. Thermal catalysis
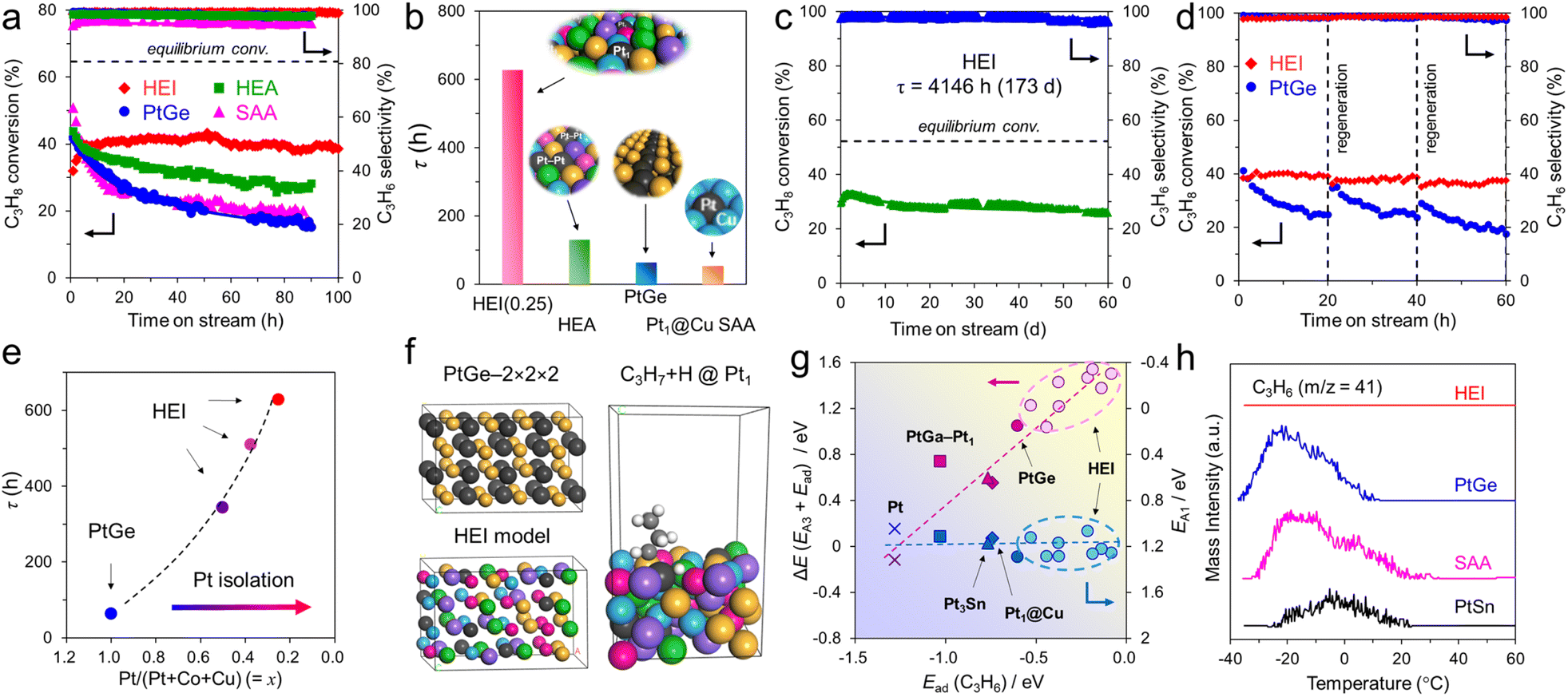 | ||
| Fig. 9 Catalytic performance of the HEI in PDH and DFT calculations. (a) Catalytic performance of PtGe, HEI(0.25), SAA, and HEA in PDH at 600 °C without co-feeding H2. (b) Summarized mean catalyst life (τ = kd−1) of HEI(0.25) and control catalysts in PDH without co-feeding H2. (c) Long-term stability test of HEI(0.25) in PDH at 600 °C with co-feeding H2. (d) Reusability of PtGe and HEI(0.25) in PDH at 600 °C without co-feeding H2 after repeated regeneration processes. (e) Relationship between the mean catalyst life (τ = kd−1) and the degree of Pt isolation represented by x as the Pt/(Pt+ Co + Cu) molar ratio in PtGe and the HEI. (f) Model structure of the HEI (left bottom) derived from PtGe (left top) for DFT calculations. An example of the HEI slab model for PDH: C3H7 + H at a Pt1 site (right). (g) Relationship between Ead(C3H6) and ΔE or EA1 for various Pt-based surfaces. (h) C3H6-TPD for Pt-based catalysts. | ||
The use of the HEI catalyst has also been reported for the oxidative dehydrogenation of propane using CO2 as a soft oxidant (CO2-ODP; C3H8 + CO2 → C3H6 + CO + H2O).27 This reaction not only produces propylene but can also convert CO2 to a value-added chemical, making it an attractive alternative to PDH. In addition to the dehydrogenation capability with high propylene selectivity, the CO2 activation capability is also required to cycle this reaction. Therefore, a multifunctional alloy architecture is necessary. Furukawa et al. synthesized the (PtCoNi)(SnInGa)/CeO2 HEI catalyst and reported its remarkable catalytic performance.27 They selected the PtSn structure, which is a well-known selective catalyst in PDH, because of its significantly negative ΔHf value (−74.0 kJ mol−1).70 The Pt and Sn sites of PtSn were selectively substituted with Co/Ni and In/Ga to improve CO2 activation ability and thermal stability, respectively. Notably, most of the Ga species were oxidized even after H2 reduction. Considering the order of reduction potentials (Ga3+ + 3e− → Ga(0): −0.55 V vs. SHE < CeO2 + e− → Ce2O3: −0.36 V < In3+ + 3e− → In(0): −0.34 V), the reduced Ga species would be re-oxidized by the CeO2 lattice oxygen because of its oxophilic nature. Therefore, only a small fraction of Ga participated in alloying. The constructed (PtCoNi)(SnInGa)/CeO2 HEI catalyst exhibited a higher catalytic activity, C3H6 selectivity, long-term stability, and CO2 utilization efficiency at 600 °C than PtSn/CeO2, Pt–Co–In/CeO2, and previously reported catalysts (Fig. 10a–c). The control experiments with binary alloy catalysts (Pt–M/CeO2, M = Co, Ni, Sn, In, and Ga) revealed that the incorporation of Ni/Co is the key to improve the CO2 activation ability. In addition, typical metals were necessary to inhibit the dry reforming of propane, resulting in high propylene selectivity. A mechanistic study was performed to determine the origin of the enhanced catalysis. The apparent activation energies for propane dehydrogenation were as follows: 128.0 kJ mol−1 (HEI), 138.3 kJ mol−1 (Pt–Co–In), and 143.9 kJ mol−1 (PtSn). A similar trend was also observed for the apparent activation energies for CO2 reduction (HEI: 102.5 kJ mol−1, Pt–Co–In: 130.3 kJ mol−1, and PtSn: 141.5 kJ mol−1). Therefore, the activation of propane and CO2 was kinetically accelerated in the HEI structure. DFT calculations were performed to investigate the role of the HEI phase. In modeling the HEI structure, the Pt and Sn sites in the PtSn (2 × 2 × 2) supercell were partially and randomly replaced with Ni/Co and In, respectively (Fig. 10d and e). The adsorption (Ead) and C–H activation energies (EA) of propylene were calculated using eight different adsorption sites and conformations (Fig. 10f). Ead and EA have narrow ranges (Ead: −51 to −35 kJ mol−1, EA: 137 to 147 kJ mol−1). ΔE (= EA + Ead = EA − Ed (Ed: propylene desorption energy)), which is the well-known descriptor for selectivity, of the HEI (98.9 kJ mol−1) was higher than that of Pt–Co–In (45.7 kJ mol−1). This result indicated that propylene desorption was significantly accelerated because of the effective inhibition of the 3rd C–H cleavage upon Pt isolation. In addition, EA values of propane to propylene and CO2 reduction were also calculated (Fig. 10g and h). The EA value for propane dehydrogenation and CO2 activation was decreased after multimetallization. Notably, the trends obtained from DFT calculations were consistent with the experimental results. Based on the d-DOS of surface transition metals, the HEI showed an intense peak near the Fermi level because of the 3d state of Ni and Co. Therefore, the upshift of the d-band center by the incorporation of Ni and Co is the key for the enhanced activity in propane dehydrogenation and CO2 reduction.
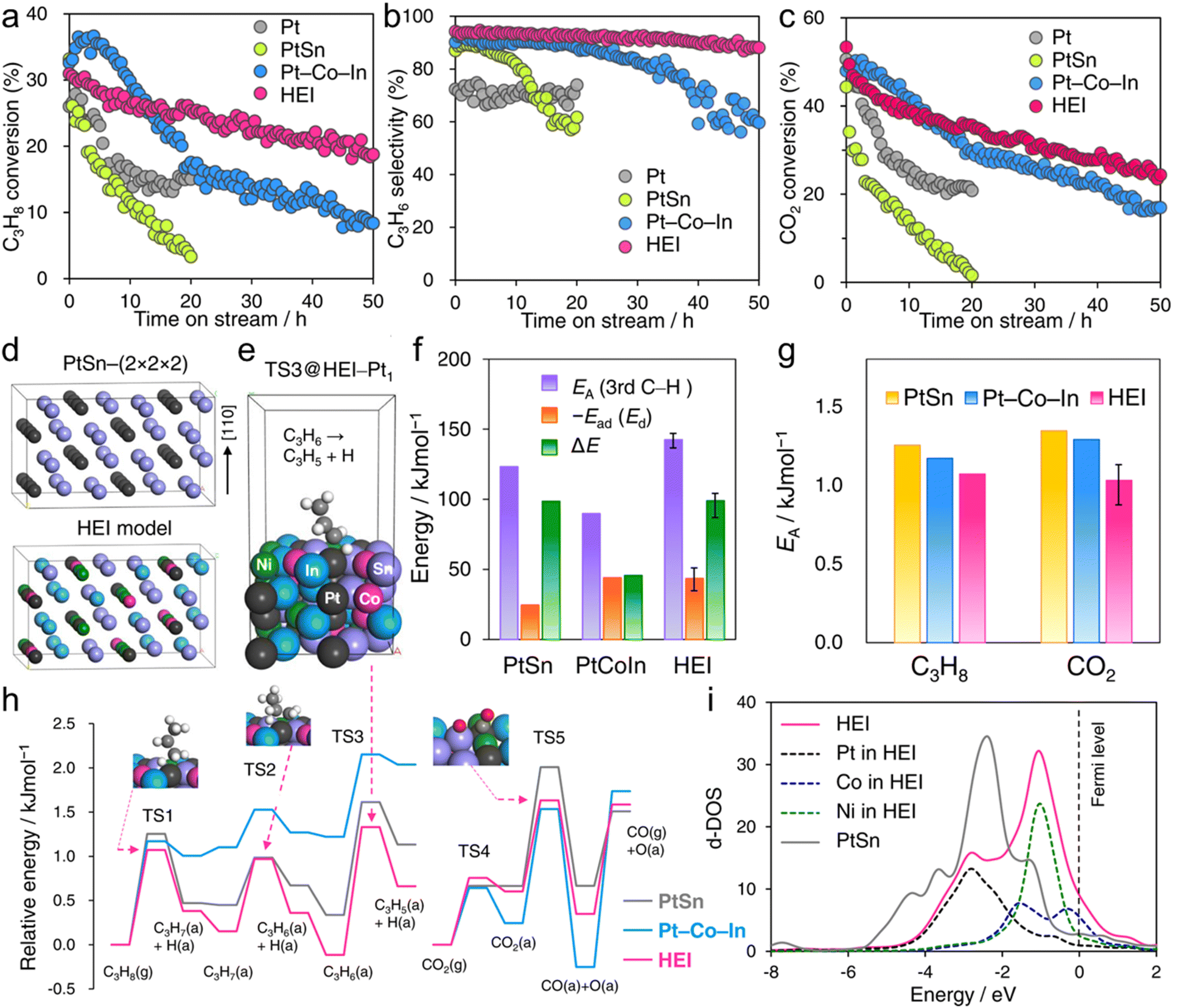 | ||
| Fig. 10 Time course of (a) C3H8 conversion, (b) C3H6 selectivity in hydrocarbons, and (c) CO2 conversion in the CO2-ODP at 600 °C. (d) Model structures of PtSn and the PtSn-based HEI for DFT calculations. (e) An example of the HEI slab model for CO2-ODP for overdehydrogenation: C3H6 → C3H5 +H at a Pt1-HEI site on the (110) surface (HEI(001):B3). (f) Comparison of EA of the third C–H activation (overdehydrogenation of C3H6), −Ead (Ed) of C3H6, and ΔE (= EA + Ead) on the surface of PtSn, Pt–Co–In, and the HEI (average). (g) EA of propane dehydrogenation and CO2 activation. For propane dehydrogenation, EA of the first C–H scission was shown. (h) Energy diagrams of propane dehydrogenation and CO2 reduction on the surface of PtSn, Pt–Co–In and the HEI. (i) d-DOS of the surface transition metals on HEI(004):B and PtSn(110). | ||
:H2:He (=1:10:39) stream, the synthesized (NiFeCu)(GaGe)/SiO2 HEI retained high ethylene selectivity (93–98%) even after reaching 100% conversion of acetylene, whereas NiGa showed a significant drop in selectivity after reaching 100% conversion (Fig. 11b and c). The catalytic activity of the HEI with approximately one/sixfold Ni amount was comparable to that of NiGa, indicating the improved catalytic activity. In addition, the HEI showed higher durability than those of binary NiGa, quaternary NiCuGaGe, NiFeGaGe, and NiFeCuGa catalysts (Fig. 11d). These results indicated the improvement of activity, selectivity, and durability upon multimetallization. Kinetic studies revealed the following apparent activation energy and reaction orders for HEI and NiGa:
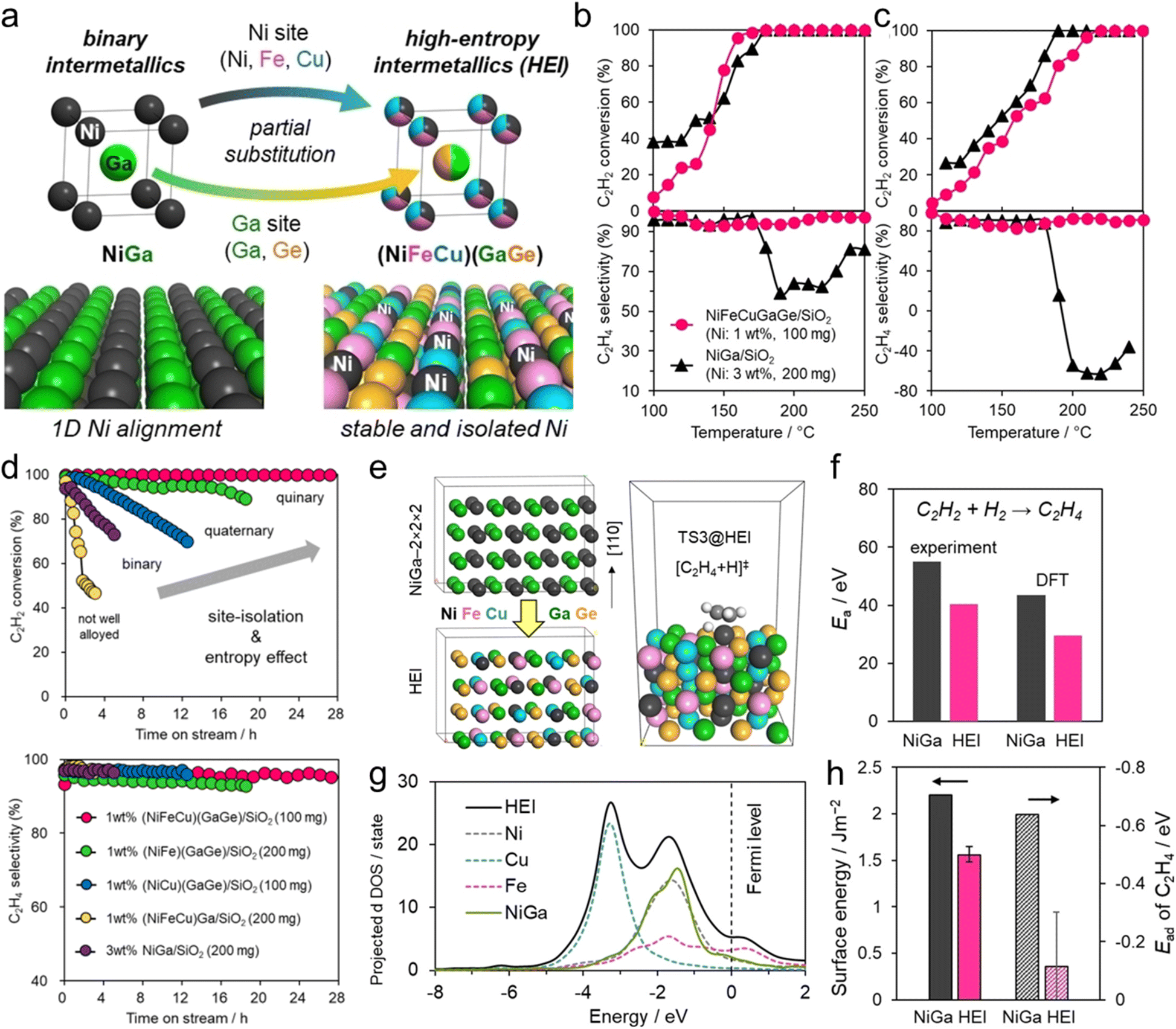 | ||
| Fig. 11 (a) Multimetallization of NiGa to (NiFeCu)(GaGe) HEI and the (110) planes of NiGa and HEI. Catalytic performance of NiGa/SiO2 and NiFeCuGaGe/SiO2 (HEI) in acetylene semihydrogenation. C2H2 conversion and C2H4 selectivity in the (b) absence (C2H2:H2:He = 1:10:39 mL min−1) and (c) presence of excess ethylene (C2H2:H2:C2H4:He = 1:10:10:39 mL min−1). (d) Time-course of acetylene conversion and ethylene selectivity in long-term stability tests. (e) Optimized bulk structures of the parent NiGa supercell and the HEI model, and an example of a TS image: hydrogen attack on C2H2 on the HEI. (f) Activation energy of acetylene hydrogenation. (g) DOS profiles projected on d-orbitals. The dotted line shows the partial d-DOS of the HEI projected onto each transition metal. (h) Surface energy and adsorption energy of ethylene on Ni sites. | ||
(1) Apparent activation energy: 40.4 kJ mol−1 (HEI) and 55.1 kJ mol−1 (NiGa)
(2) Reaction order for acetylene: −0.13 (HEI) and −0.91 (NiGa)
(3) Reaction order for hydrogen: 0.80 (HEI) and 2.27 (NiGa).
These results indicated that the increase in catalytic activity by multimetallization was attributed to the following two factors: (1) decreased energy barrier and (2) increased hydrogen supply (i.e., appearance of vacant active sites) to the surface. DFT calculations were performed on the (110) planes of the HEI and NiGa to explore the origin of the high catalytic performance (Fig. 11e). The HEI showed a lower activation barrier for acetylene hydrogenation than NiGa (Fig. 11f). In addition, the adsorption energy of ethylene was reduced dramatically after multimetallization. Notably, the DOS projected onto the d-orbitals verified that the electronic structure of Ni did not change upon multimetallization, indicating the negligible contribution of the ligand effect (Fig. 11g). By contrast, the surface energy of the (110) plane showed a dramatic decrease in various models, which was related to the decrease in the adsorption energy of ethylene (Fig. 11h). Therefore, the enhanced ethylene desorption was attributed to the decreased surface energy upon multimetallization. These results were consistent with the enhanced activity drawn from the experimental results. In addition, the free energy barrier for the overhydrogenation of ethylene increased upon multimetallization, proving the update of ethylene selectivity without sacrificing the catalytic activity. Consequently, the origin of the high catalytic performance was proven by DFT calculations. Therefore, the ligand effect has no significant contribution to this system.
6.2. Electrocatalysis
Wang et al. synthesized disordered, partially ordered, and highly ordered (PtCo-phase) Pt(FeCoCuNi) nanocrystals and tested their HER performance in 1 M KOH aqueous solution.24 The highly ordered Pt(FeCoCuNi) nanocrystal required an overpotential of only 20 mV vs. the reversible hydrogen electrode (RHE) to deliver 10 mA cm−2, whereas the partially ordered and disordered counterparts and the commercial Pt/C showed overpotential of 32, 47, and 38 mV (Fig. 12a and b). The Tafel slope of the highly ordered Pt(FeCoCuNi) was 31 mV dec−1, which was smaller than those of the partially ordered (39 mV dec−1) and disordered Pt(FeCoCuNi) (56 mV dec−1) and Pt/C (54 mV dec−1) (Fig. 12a and c). The mass activity of the highly ordered Pt(FeCoCuNi) at a potential of −0.2 V vs. RHE was 71.9 A mgPt−1, which was 2.1, 3.0, and 5.6 times higher than those of the partially ordered (34.8 A mgPt−1) and disordered counterparts (23.8 A mgPt−1) and Pt/C (12.9 A mgPt−1), respectively (Fig. 12d). Therefore, the ordering degree of multimetallic alloys plays an important role in facilitating the alkaline HER activity and the HEI is the active structure. A durability test of more than 50 h of chronopotentiometry at a current density of 10 mA cm−2 in 1 M KOH electrolyte was also performed (Fig. 12e). The stability result was in the following order: commercial Pt/C ≪ disordered < partially ordered < highly ordered Pt4FeCoCuNi. The same stability trend was also observed for linear sweep voltammetry of 10000 cyclic voltammetry cycles, demonstrating the critical role of the ordered structure (Fig. 12f).
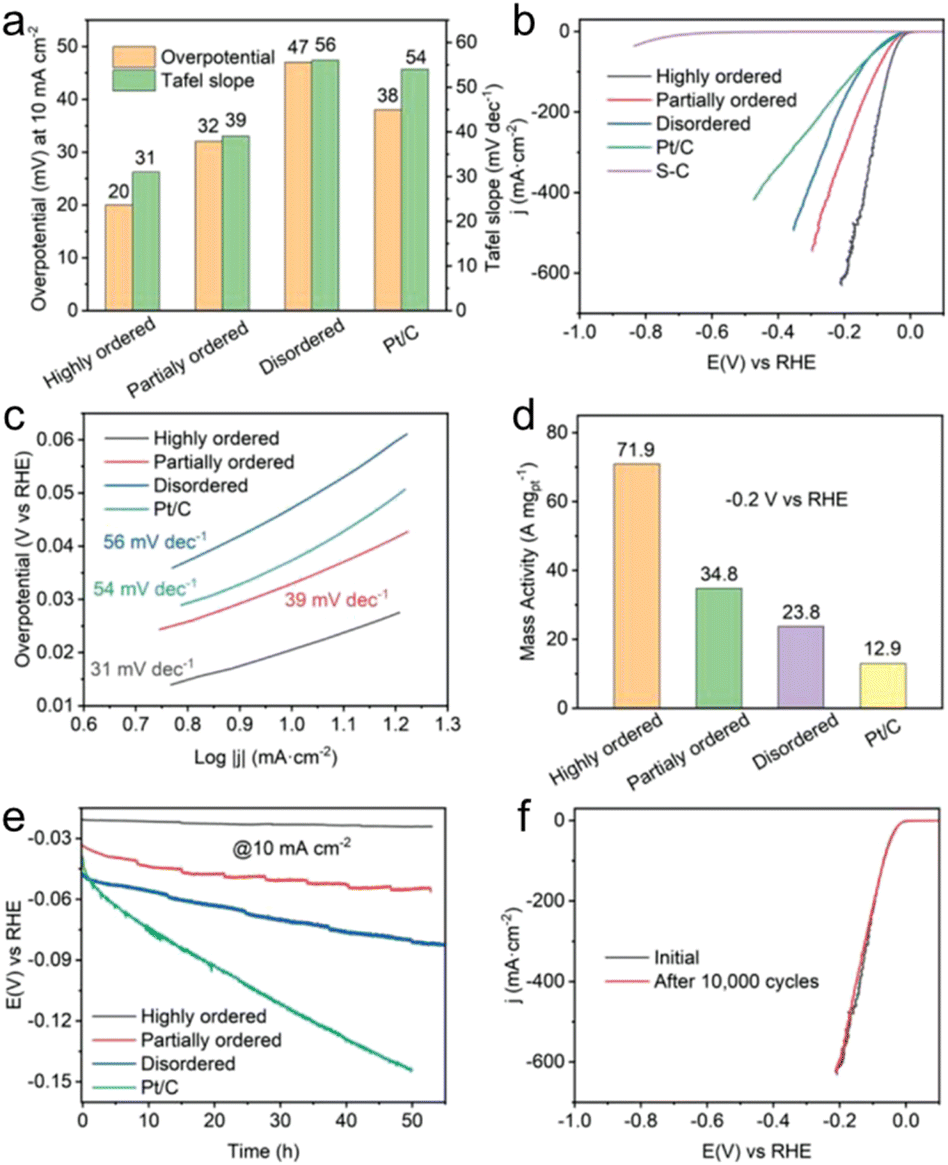 | ||
| Fig. 12 Alkaline HER performance and characterization after durability tests. (a) Comparison of overpotentials at 10 mA cm−2 and Tafel slopes among Pt(FeCoCuNi) catalysts with different ordering degrees and commercial Pt/C. (b) HER linear sweep voltammetry curves and (c) the corresponding Tafel slopes of highly ordered, partially ordered, disordered Pt(FeCoCuNi), bare S-doped carbon, and commercial Pt/C in 1 M KOH. (d) Comparison of HER mass activities for Pt(FeCoCuNi) with different ordering degrees and commercial Pt/C at −0.2 V vs. RHE. (e) Chronopotentiometry curves of highly ordered, partially ordered, and disordered Pt(FeCoCuNi) catalysts recorded at a constant cathodic current density of 10 mA cm−2. (f) HER polarization curves of the highly ordered Pt(FeCoCuNi) catalyst before and after 10000 CV cycles. Reproduced with permission from ref. 21. Copyright 2022 Wiley-VCH. | ||
Jia et al. synthesized the L12-type (FeCoNi)3(TiAl) HEI with a dendritic surface morphology and tested its catalytic performance for the HER in 1.0 M KOH aqueous solution.20 This catalyst exhibited significantly lower overpotential (88.2 mV at 10 mA cm−2) and Tafel slope (40.1 mV dec−1) than the Pt sheet (145 mV, 79.3 mV dec−1). The HER activity achieved in this non-noble metal-based HEI was comparable to the top-level performance of noble metal-based electrocatalysts, highlighting the excellent cost performance. The 40 h durability test at a current density of 100 mA cm−2 showed no significant overpotential change. Inductively coupled plasma-optical emission spectrometry of the spent catalyst showed that only a small amount of Al was leached out (0.276 mmol L−1), whereas the dissolution of Fe, Co, Ni, and Ti was below the detection limit. DFT calculations were also performed on the (111) and (100) surfaces of (FeCoNi)3(TiAl). The Ti atom had a preferential adsorption energy for H2O compared with other elements because of the strong charge transfer from Ti to H2O, which promoted H2O splitting. In addition, all the active sites that coordinated with one single-Al atom showed moderate adsorption energy for H, indicating balanced H adsorption and accelerated H2 desorption. Partial DOS revealed that the d-band center of the d-orbital was significantly downshifted relative to the Fermi level (Ef) for the single-Al coordinated active site, which weakened the adsorption of H(ad) because of the more filled anti-bonding electronic states. The scenario of active sites on an Al-rich surface was also confirmed. The active sites that coordinated with dual-Al atoms could not stably adsorb hydrogen, resulting in decreased H2 evolution ability. The ordered HEI structure can avoid Al-rich surfaces compared with the conventional disordered HEA structure. Therefore, the outstanding HER performance was due to the fine-tuning of the electronic structure by combining the chemical synergistic and structural site isolation effects. However, X-ray photoelectron spectroscopy (XPS) measurement confirmed that the surface was mostly oxidized to oxides and hydroxides, indicating that only a small fraction of zero-valent metals remained on the surface. Therefore, the conclusions drawn from the assumption that the surface is in the metallic state are not adequate as explanations, and it needs to be clarified what kind of surface structure actually contributes to catalysis.
Zhu et al. applied the nanoparticulate (FeCoNi)3(NiCuPd) HEI with an L12 phase supported on 2D nitrogen-rich mesoporous carbon nanosheets (mNC), and reported its excellent catalytic performance for the ORR in alkaline media (0.1 M KOH).21 The synthesized HEI achieved a large half-wave potential (0.90 eV), a significantly positive shift of ≈30 and ≈60 mV relative to the commercial Pt/C, and corresponding disordered quinary FeCoNiCuPd HEA. Among them, the HEI exhibited the lowest Tafel slope (55 mV dec−1) (Pt/C: 105 mV dec−1, HEA: 67 mV dec−1). The calculated mass activity of the HEI at 0.9 V was 2.037 A mgPd−1, which was 10.0 and 12.0 times larger than those of Pt/C (0.204 A mgPd−1) and the HEA (0.169 A mgPd−1), respectively. The comprehensive ORR performance of the HEI was superior to those of the recently reported HEA electrocatalysts. The Koutecky–Levich plots for the HEI indicated that the number of electrons transferred (n) was estimated to be ≈4.0 at 0.3–0.5 V, indicating that O2 was directly converted to H2O. Durability was evaluated by accelerated durability experiments between 0.2 and 1.1 V (vs. RHE) in 0.1 M KOH solution. After 10000 cycles, the HEI resulted in only 0.01 V decay and retained the HEI structure. In addition, chronoamperometric measurement showed that the HEI catalyst retained 95.4% of its initial current density after 18 h of continuous testing, which is better than that of Pt/C (retained only 71.0%). These durability tests confirmed the superior durability of the HEI. DFT calculations revealed that the HEI structure had a more robust structural configuration than that of the HEA structure, indicating the greater durability of the HEI structure for electrocatalysis.
The ORR of the Pt(FeCoCuNi) HEI catalyst, which exhibited a high HER performance, was also tested in 0.1 M HClO4.24 Pt(FeCoCuNi) HEI exhibited a large half-wave potential of 0.943 V, which was higher than that of the partially ordered and disordered counterparts (0.927 and 0.910 V, respectively). The mass activity at 0.9 V vs. RHE was 3.78 A mgPd−1, which was 2.1-, 4.4-, and 18.9-fold greater than those of the partially ordered and disordered counterparts and Pt/C, respectively. The HEI exhibited the lowest Tafel slope (51.5 mV dec−1) compared with its partially ordered (57.3 mV dec−1) and disordered counterparts (64.1 mV dec−1) and Pt/C (77.9 mV dec−1). The distinct ORR activity was correlated with the degree of ordering. The accelerated degradation test for 30000 cycles between +0.5 and 1.1 V vs. RHE revealed that E1/2 of the highly ordered HEI was negatively shifted by 7 mV, which was smaller than 13 and 28 mV of the partially ordered and disordered counterparts. Furthermore, the HEI retained 74% of the initial mass activity, exceeding the partially ordered (56%) and disordered (38%) counterparts. These results indicated that the HEI has more robust stability than the HEA. However, structural analysis of the spent HEI catalyst revealed the core/shell architecture with a Pt-rich shell because of the leaching and dissolution of non-noble metals under acidic ORR conditions.
In 2023, Feng et al. synthesized the PtIrFeCoCu HEA and (PtIr)(FeCoCu) HEI by controlling the annealing temperatures and tested their catalytic performance for the ORR in N2-purged 0.1 M HClO4 solution.19 Based on the structural analysis, the HEI possessed a PtFe-type ordered structure, in which Ir and Fe/Cu partially substituted the Pt and Co sites, respectively (Fig. 13a and b). By contrast, the HEA counterpart had a disordered solid-solution structure. The mass activity at 0.9 V was 1.29 A mgnoble metal−1, outperforming the HEA counterpart (0.81 A mgnoble metal−1) and commercial Pt/C (0.098 A mgnoble metal−1, Fig. 13c). When the potential was reduced to 0.85 V, the HEI exhibited a mass activity of 7.14 A mgnoble metal−1, which was 2.4 and 20.4 times higher than those of HEA (3.03 A mgnoble metal−1) and Pt/C (0.35 A mgnoble metal−1), respectively. The HEI presented a higher half-wave potential (E1/2, 0.894 V) than that of the commercial Pt/C (0.853 V). Tafel plots identified the lower Tafel slope of the HEI (63.5 mV dec−1) than that of Pt/C (75.6 mV dec−1). In addition, as the ordering degrees of the HEI were increased by elevating the annealing temperature, the ORR activity was gradually elevated. These results indicated the remarkable ORR performance of the HEI. Durability tests were also evaluated. After a 60 h constant test in 0.1 M HClO4, the HEI retained 100% of the initial current density, whereas Pt/C retained only 64.4% after 10 h test (Fig. 13d). Furthermore, E1/2 of the HEI was degraded by only 9 mV after the accelerated degradation test for 60000 cycles between +0.6 and 1.1 V vs. RHE, indicating its superiority to Pt/C (loss of 49 mV) and other reported catalysts (Fig. 13e and f). Structural analysis of the spent HEI catalyst confirmed no apparent change, indicating structural stability. Theoretical calculations were performed to obtain further insights into the high catalytic activity and structural robustness of the HEI. The dissolution energy calculation revealed that the outstanding stability of the HEI is due to the high-entropy stabilization effect, which leads to the positive dissolution energy (>1 eV) of the five elements (Fig. 13g). Furthermore, the dynamic processes of the rate-determining step on Pt(111), HEI(111), Pt(001), and HEI(011) surfaces were calculated (Fig. 13h). Two main mechanisms were utilized, namely, (1) O2 hydrogenation and (2) O2 dissociation (Fig. 13i). The ORR would favorably proceed with O2 hydrogenation on Pt(111), but the ORR would proceed with O2 dissociation on Pt(001) after O2 is adsorbed on the surface. Theoretical calculations showed that HEI(111) and Pt(111) preferred the former mechanism with the rate-determining step of O* + H* → OH*, whereas HEI(001) and Pt(001) preferred the latter mechanism with the rate-determining step of OH* + H* → H2O. The calculated activation barriers for the rate-determining steps were in the following order: HEI(001) < HEI(111) < Pt(111) < (001). As previously reported, Pt(001) showed a lower ORR activity than Pt(111),110 whereas HEI(001) showed a higher ORR activity than HEI(111) (Fig. 13i). The d-band center of HEI(001) was shifted by 0.31 eV relative to that of pure Pt(111), which was closest to the optimum value (0.32 eV).80,81 Consequently, the optimized decrease in the d-band center relieves the overbinding of surface oxygen, thereby boosting the activity for the ORR. Details of the electronic structure are described in Section 3.2. In situ electrochemical Raman spectra confirmed the presence of OH* and OOH*, which are important intermediate species for the ORR occurring on HEI(001) and HEI(111), respectively. The stronger peak of OH* than that of OOH* indicated that the HEI(001) surface is the main active site for a faster ORR. Consequently, they succeeded in transforming a conventional low activity facet into a highly active facet using the HEI.
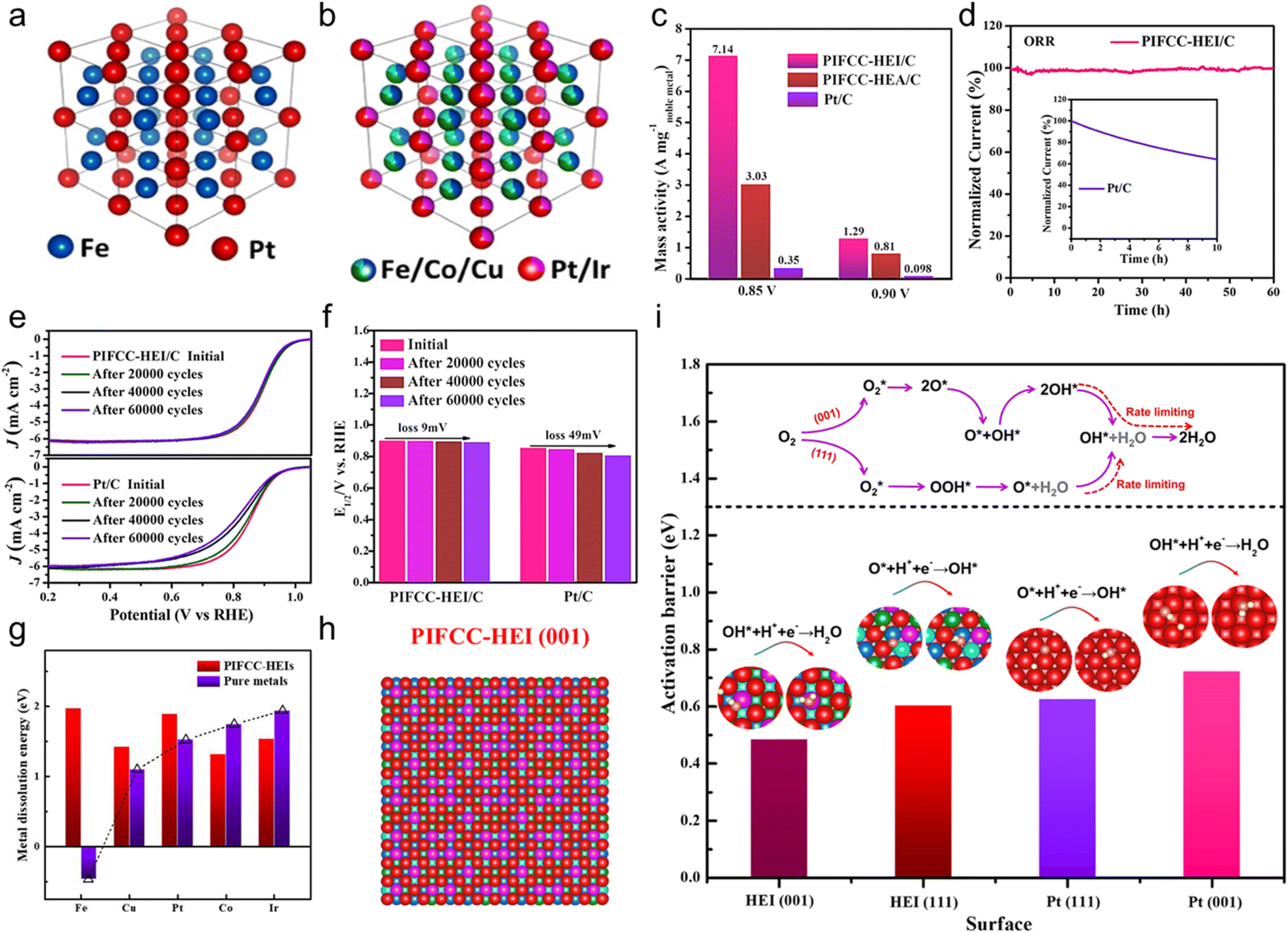 | ||
| Fig. 13 Simulated crystal structure of the (a) (PtIr)(FeCoCu) HEI and (b) PtFe intermetallic. (c) Mass activity comparisons of HEI/C and commercial Pt/C at 0.85 and 0.9 V vs. RHE, normalized by noble metal loading. (d) Stability test comparison of HEI/C and commercial Pt/C through the constant applied potential for 60 h. (e) ORR polarization curves of HEI/C and commercial Pt/C before and after cyclic voltammetry. (f) Quantitative comparisons of the half-wave potential of HEI/C and commercial Pt/C before and after cyclic voltammetry. (g) Comparison of the metal dissolution energy of the HEI and pure metals. (h) Top view of the HEI(001) surface model. Red, purple, navy blue, green, and cyan refer to Pt, Ir, Fe, Co, and Cu atoms, respectively. (i) Activation barriers for the rate-determining step of the ORR on the PIFCC-HEI (001) surface, PIFCC-HEI (111) surface, Pt (111) surface, and Pt (001) surface. Reproduced with permission from ref. 19. Copyright 2023 American Chemical Society. | ||
In 2022, Chen et al. reported a (PtRh)(BiSnSb) HEI nanoplate as a highly efficient methanol oxidation catalyst. Pt/Rh and Bi/Sn/Sb occupied the Pt and Bi columns, respectively, of the hcp PtBi structure (Fig. 14a and b).22 The similar structural configurations (hcp) of PtBi, RhBi, PtSn, and PtSb allow the substitution of Pt and Bi sites by Rh and Sb/Sn, respectively. The elemental mapping and simulation revealed that the atomic percentages of Rh and Bi are relatively higher in the central parts, and the atomic percentages of Pt, Sn, and Sb are relatively higher near the edges (Fig. 14c and d). For comparison, Pt(BiSnSb) and PtBi nanoplates were also synthesized. XPS measurement showed that the (PtRh)(BiSnSb) HEI has the lowest Pt 4f binding energy compared with Pt(BiSnSb), PtBi, and Pt/C (Fig. 14e). Therefore, the incorporation of Rh improved the electron transfer efficiency. Prior to electrocatalysis, (PtRh)(BiSnSb), Pt(BiSnSb), and PtBi nanoplates were dispersed on commercial carbon, and then used for the MOR in an Ar-saturated 1.0 M KOH + 1.0 M MeOH aqueous solution. (PtRh)(BiSnSb) exhibited a higher MOR mass activity of 19.5 A mg−1Pt+Rh than those of Pt(BiSnSb), PtBi, Pt/C, and previously reported catalysts. In addition, (PtRh)(BiSnSb) achieved excellent EOR and GOR mass activities (15.6 A mg−1Pt+Rh and 7.5 A mg−1Pt+Rh, respectively), which were higher than those of Pt(BiSnSb) and Pt/C. These results indicated the synergistic effect of five metals in accelerating AORs. DFT calculations indicated the optimization of the d-band center upon the introduction of Rh. Consequently, CH3OH and CO2 were strongly adsorbed on (PtRh)(BiSnSb), whereas CO adsorption was significantly suppressed. Consequently, (PtRh)(BiSnSb) preferred the CO2 pathway with a rate-determining step of 0.41 eV compared to the CO pathway with a rate-determining step of 0.77 eV. By contrast, Pt(BiSnSb) showed larger energy barriers than (PtRh)(BiSnSb) and stronger preference for the CO pathway. The durability of the MOR was also tested. After 5000 cyclic voltammetry cycles, (PtRh)(BiSnSb), Pt(BiSnSb), and Pt/C lost 29.8%, 37.3%, and 76.9% of their initial activities, respectively (Fig. 14f). Therefore, the HEI architecture is of great importance to improve activity and durability. However, EDX mapping confirmed the leaching of Bi/Sn/Sb compositions in the (PtRh)(BiSnSb) and Pt(BiSnSb) nanoplates, which results in catalyst degradation (Fig. 14g). Therefore, no matter how excellent the structural stability of the HEI is, it is difficult to completely alter the element-specific properties. Therefore, it is important to carefully select the constituent elements.
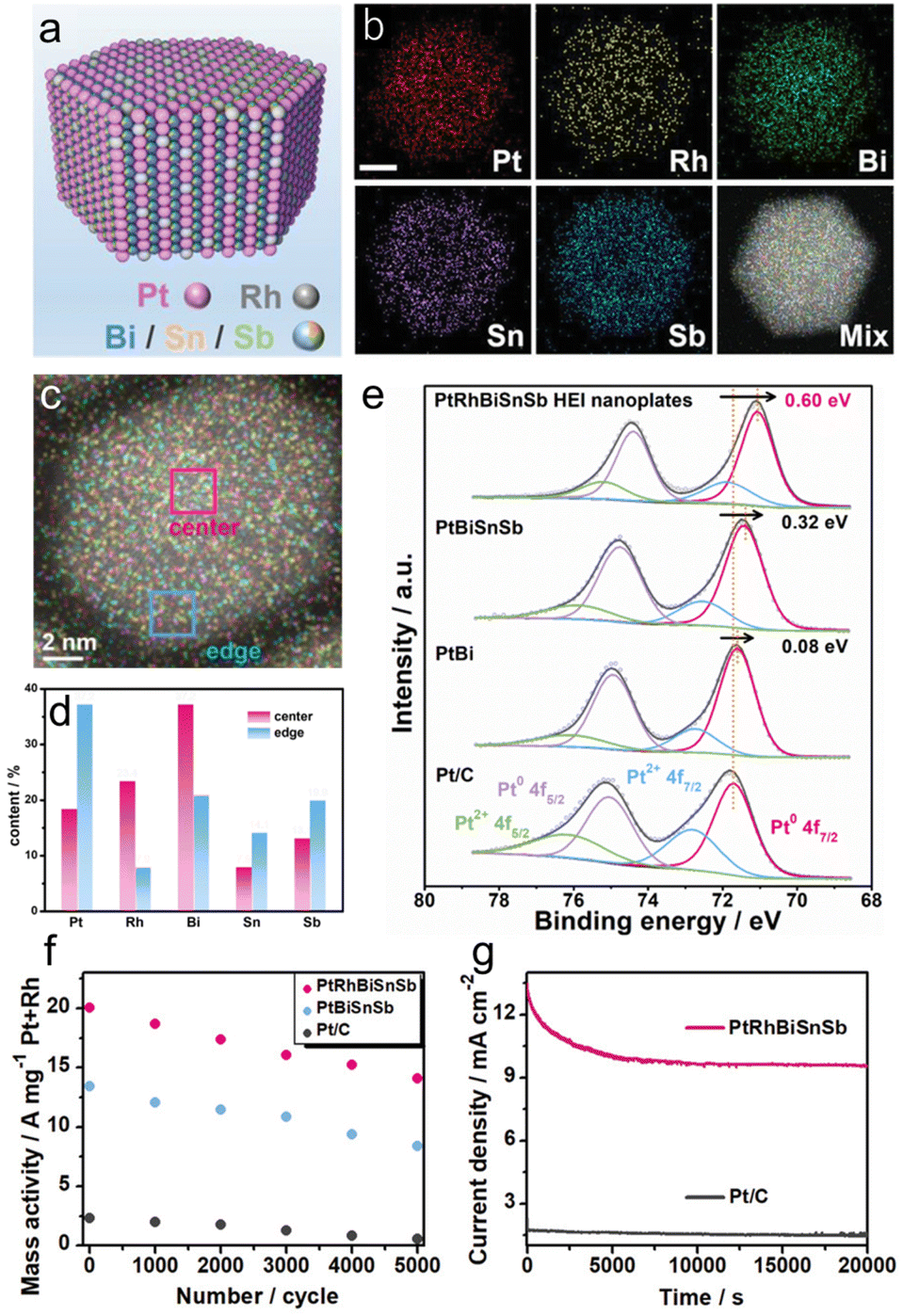 | ||
| Fig. 14 (a) The schematic illustration shows the atomic arrangement of Pt/Rh/Bi/Sn/Sb atoms on the surface of the (PtRh)(BiSnSb) HEI nanoplate. (b) TEM image. (c) EDX mapping image of a nanoplate. (d) Atomic ratios of Pt/Rh/Bi/Sn/Sb at different positions on the nanoplate obtained from the EDX results. (e) The Pt 4f XPS spectra of the (PtRh)(BiSnSb) HEI nanoplates, Pt(BiSnSb) nanoplates, PtBi nanoplates, and Pt/C. (f) The changes in mass activities of different catalysts before and after long-term potential cycling. (g) Long-term chronoamperometry for the MOR of different catalysts recorded at 0.7 V versus RHE. Reproduced with permission from ref. 22. Copyright 2022 Wiley-VCH. | ||
Another example of the EOR is the L12-type PtRhFeNiCu HEI system reported by Wang et al.23 The mass activity of the HEI in N2-saturated 0.1 M HClO4 + 0.2 M EtOH aqueous solution was 0.914 A mgPt−1, which was higher than those of the HEA counterpart (0.578 A mgPt−1) and Pt/C (0.202 A mgPt−1) catalysts. As a control experiment, the electrocatalytic activity of Rh/C was also evaluated under the same conditions, which showed a lower activity. During the electrochemical test, three peaks were observed, that is, (1) backward peak (Ib), (2) first peak (If1), and (3) second peak (If2). If1 represents the oxidation of EtOH to acetaldehyde, acetic acid, and CO2, whereas If2 represents the formation of acetic acid. Therefore, the ratio of If1/If2 is used to test the C–C bond cleavage ability. In addition, the ratio of If1/Ib is used to indicate the CO intermediate tolerance. As expected, the HEI catalyst exhibited the highest If1/If2 and If1/Ib values, indicating its strong C–C bond breaking ability and CO intermediate tolerance compared to the HEA counterpart and Pt/C catalysts. Based on the CO stripping experiment, the HEI showed the lowest onset potential for CO removal, proving CO tolerance. Hu et al. reported that the octonary (Pt0.8Pd0.1Au0.1)(Fe0.6Co0.1Ni0.1Cu0.1Sn0.1) HEI exhibited improved EOR activity, which was 8 and 12 times higher than that of the binary PtFe intermetallic and commercial Pt/C catalysts respectively.28 However, the report focused on the synthesis of small HEI nanoparticles, which lacked information about the catalytic performance.
7. Concluding remarks and future directions
The design of alloy catalysts has entered a new era, and a variety of alloy materials have been reported as promising catalyst architectures. Alloy catalysis aims to manipulate the metal atoms in a controllable way with a wide compositional space, thereby achieving the desired catalytic properties. In the past few years, the development of HEI catalysts has been observed, which integrates metal species with controlled atomicity and wide compositional space. The emergence of HEIs demonstrates that we are moving toward the cherished goal in alloy catalysis. In this review, we have comprehensively summarized the information on the state-of-the-art field of HEIs with regard to thermodynamic properties, synthesis requirements, characterization techniques, roles in catalysis, and reaction examples. We hope that this review will provide the direction for the development of innovative catalysts based on state-of-the-art HEIs. Practical application of the developed materials is also an important for the future in the catalysis research. For example, long-term stability under harsh reaction conditions or large-scale production and supply of catalyst materials should be the key requirement for this goal. For the former, some papers have already focused on the long-term stability under the reaction conditions. However, for successful application under practical conditions, further understanding of the structural/compositional changes of HEIs at the atomic level is necessary. For the latter, special care, techniques, and apparatus have been needed for the synthesis of HEI catalysts with high homogeneity and size control even in lab-scale production. Therefore, establishing an easy, simple, and cost-effective procedure that can be transferred to an industrial large scale is highly desired. We hope that this review will be a hint for researchers to overcome these challenges in the near future.Author contributions
Y. N. and S. F. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by JSPS KAKENHI (Grants 20H02517, 21J20594, 22KJ0016, and 23H00308).Notes and references
-
S. Venkata Mohan and A. Pandey, Biomass, Biofuels, Biochem. Biohydrogen, 2nd edn, 2019, vol. 305, pp. 1–23 Search PubMed
.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS PubMed
- Y. Nakaya and S. Furukawa, Chem. Rev., 2023, 123, 5859–5947 CrossRef CAS PubMed
- S. Furukawa and T. Komatsu, ACS Catal., 2017, 7, 735–765 CrossRef CAS
- Y. Yan, J. S. Du, K. D. Gilroy, D. Yang, Y. Xia and H. Zhang, Adv. Mater., 2017, 29, 1605997 CrossRef PubMed
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS PubMed
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed
- S. Mitchell and J. Pérez-Ramírez, Nat. Rev. Mater., 2021, 6, 969–985 CrossRef
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS PubMed
- T. Zhang, A. G. Walsh, J. Yu and P. Zhang, Chem. Soc. Rev., 2021, 50, 569–588 RSC
- E. P. George, D. Raabe and R. O. Ritchie, Nat. Rev. Mater., 2019, 4, 515–534 CrossRef CAS
- J. W. Yeh, S. K. Chen, S. J. Lin, J. Y. Gan, T. S. Chin, T. T. Shun, C. H. Tsau and S. Y. Chang, Adv. Eng. Mater., 2004, 6, 299–303 CrossRef CAS
- B. Cantor, I. T. H. Chang, P. Knight and A. J. B. Vincent, Mater. Sci. Eng., A, 2004, 375–377, 213–218 CrossRef
- B. Wang, Y. Yao, X. Yu, C. Wang, C. Wu and Z. Zou, J. Mater. Chem. A, 2021, 9, 19410–19438 RSC
- G. Feng, F. Ning, Y. Pan, T. Chen, J. Song, Y. Wang, R. Zou, D. Su and D. Xia, J. Am. Chem. Soc., 2023, 145, 11140–11150 CrossRef CAS PubMed
- Z. Jia, T. Yang, L. Sun, Y. Zhao, W. Li, J. Luan, F. Lyu, L. C. Zhang, J. J. Kruzic, J. J. Kai, J. C. Huang, J. Lu and C. T. Liu, Adv. Mater., 2020, 32, 2000385 CrossRef CAS PubMed
- G. Zhu, Y. Jiang, H. Yang, H. Wang, Y. Fang, L. Wang, M. Xie, P. Qiu and W. Luo, Adv. Mater., 2022, 34, 2110128 CrossRef CAS PubMed
- W. Chen, S. Luo, M. Sun, X. Wu, Y. Zhou, Y. Liao, M. Tang, X. Fan, B. Huang and Z. Quan, Adv. Mater., 2022, 34, 2206276 CrossRef CAS PubMed
- D. Wang, Z. Chen, Y. Wu, Y. Huang, L. Tao, J. Chen, C. Dong, C. V. Singh and S. Wang, SmartMat, 2023, 4, 1–11 Search PubMed
- Y. Wang, N. Gong, H. Liu, W. Ma, K. Hippalgaonkar, Z. Liu and Y. Huang, Adv. Mater., 2023, 35, 2302067 CrossRef CAS PubMed
- Y. Nakaya, E. Hayashida, H. Asakura, S. Takakusagi, S. Yasumura, K. Shimizu and S. Furukawa, J. Am. Chem. Soc., 2022, 144, 15944–15953 CrossRef CAS PubMed
- J. Ma, F. Xing, Y. Nakaya, K. Shimizu and S. Furukawa, Angew. Chem., Int. Ed., 2022, e202200889 CAS
- F. Xing, J. Ma, K. Shimizu and S. Furukawa, Nat. Commun., 2022, 13, 5065 CrossRef CAS PubMed
- M. Cui, C. Yang, S. Hwang, M. Yang, S. Overa, Q. Dong, Y. Yao, A. H. Brozena, D. A. Cullen, M. Chi, T. F. Blum, D. Morris, Z. Finfrock, X. Wang, P. Zhang, V. G. Goncharov, X. Guo, J. Luo, Y. Mo, F. Jiao and L. Hu, Sci. Adv., 2022, 8, eabm4322 CrossRef CAS PubMed
- C. F. Tsai, P. W. Wu, P. Lin, C. G. Chao and K. Y. Yeh, Jpn. J. Appl. Phys., 2008, 47, 5755–5761 CrossRef CAS
- H. Li, Y. Han, H. Zhao, W. Qi, D. Zhang, Y. Yu, W. Cai, S. Li, J. Lai, B. Huang and L. Wang, Nat. Commun., 2020, 11, 5432 CrossRef PubMed
- Y. Lu, K. Huang, X. Cao, L. Zhang, T. Wang, D. Peng, B. Zhang, Z. Liu, J. Wu, Y. Zhang, C. Chen and Y. Huang, Adv. Funct. Mater., 2022, 32, 2110645 CrossRef CAS
- K. Mori, N. Hashimoto, N. Kamiuchi, H. Yoshida, H. Kobayashi and H. Yamashita, Nat. Commun., 2021, 12, 3884 CrossRef CAS PubMed
- S. Gao, S. Hao, Z. Huang, Y. Yuan, S. Han, L. Lei, X. Zhang, R. Shahbazian-Yassar and J. Lu, Nat. Commun., 2020, 11, 2016 CrossRef CAS PubMed
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 13833–13838 CrossRef CAS PubMed
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, I. Gueye, O. Seo, J. Kim, S. Hiroi, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, Chem. Sci., 2020, 11, 12731–12736 RSC
- X. Wang, Q. Dong, H. Qiao, Z. Huang, M. T. Saray, G. Zhong, Z. Lin, M. Cui, A. Brozena, M. Hong, Q. Xia, J. Gao, G. Chen, R. Shahbazian-Yassar, D. Wang and L. Hu, Adv. Mater., 2020, 32, 2002853 CrossRef CAS PubMed
- P. Xie, Y. Yao, Z. Huang, Z. Liu, J. Zhang, T. Li, G. Wang, R. Shahbazian-Yassar, L. Hu and C. Wang, Nat. Commun., 2019, 10, 4011 CrossRef PubMed
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu, M. Rehwoldt, D. Yu, M. R. Zachariah, C. Wang, R. Shahbazian-Yassar, J. Li and L. Hu, Science, 2018, 359, 1489–1494 CrossRef CAS PubMed
- Y. Yao, Q. Dong, A. Brozena, J. Luo, J. Miao, M. Chi, C. Wang, I. G. Kevrekidis, Z. J. Ren, J. Greeley, G. Wang, A. Anapolsky and L. Hu, Science, 2022, 376, eabn3103 CrossRef CAS PubMed
- Y. Xin, S. Li, Y. Qian, W. Zhu, H. Yuan, P. Jiang, R. Guo and L. Wang, ACS Catal., 2020, 10, 11280–11306 CrossRef CAS
- V. S. Marakatti and S. C. Peter, Prog. Solid State Chem., 2018, 52, 1–30 CrossRef CAS
- S. Furukawa, T. Komatsu and K. I. Shimizu, J. Mater. Chem. A, 2020, 8, 15620–15645 RSC
- X. Ji, K. T. Lee, R. Holden, L. Zhang, J. Zhang, G. A. Botton, M. Couillard and L. F. Nazar, Nat. Chem., 2010, 2, 286–293 CrossRef CAS PubMed
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. Disalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed
- T. Chen, C. Qiu, X. Zhang, H. Wang, J. Song, K. Zhang, T. Yang, Y. Zuo, Y. Yang, C. Gao, W. Xiao, Z. Jiang, Y. Wang, Y. Xiang and D. Xia, J. Am. Chem. Soc., 2024, 146, 1174–1184 CrossRef CAS PubMed
- T. Shen, D. Xiao, Z. Deng, S. Wang, L. An, M. Song, Q. Zhang, T. Zhao, M. Gong and D. Wang, Angew. Chem., Int. Ed., 2024, e202403260 CAS
- S. Zheng, J. Hu, R. Feng, J. Xu, Y. Yu, L. Li, W. Liu, W. Zhang, F. Huo and F. Saleem, Small Struct., 2024, 2300537 CrossRef CAS
- Y. Wang, X. Y. Zhang, H. He, J. J. Chen and B. Liu, Adv. Energy Mater., 2024, 14, 1–10 Search PubMed
- Z. Cui, H. Chen, M. Zhao, D. Marshall, Y. Yu, H. Abruña and F. J. Disalvo, J. Am. Chem. Soc., 2014, 136, 10206–10209 CrossRef CAS PubMed
- J. Liang, F. Ma, S. Hwang, X. Wang, J. Sokolowski, Q. Li, G. Wu and D. Su, Joule, 2019, 3, 956–991 CrossRef CAS
- D. B. Miracle, Mater. Sci. Technol., 2015, 31, 1142–1147 CrossRef CAS
- F. Lin, M. Li, L. Zeng, M. Luo and S. Guo, Chem. Rev., 2023, 123, 12507–12593 CrossRef CAS PubMed
- K. Fitzner, Q. Guo, J. Wang and O. J. Kleppa, J. Alloys Compd., 1999, 291, 190–200 CrossRef CAS
- T. Fang, L. Wang, C. X. Peng and Y. Qi, J. Phys.: Condens.Matter, 2012, 24, 505103 CrossRef CAS PubMed
- J. H. He, H. W. Sheng and E. Ma, Appl. Phys. Lett., 2001, 78, 1343–1345 CrossRef CAS
- A. K. Niessen, A. R. Miedema, F. R. de Boer and R. Boom, Physica B+C, 1988, 151, 401–432 CrossRef CAS
- A. K. Niessen and A. R. Miedema, Phys. B, 1988, 152, 303–346 CrossRef CAS
- J. B. Darby and K. M. Myles, Metall. Trans., 1972, 3, 653–657 CrossRef CAS
- Y. F. Ye, Q. Wang, J. Lu, C. T. Liu and Y. Yang, Mater. Today, 2016, 19, 349–362 CrossRef CAS
-
A. Olga Hermine and Kubik, PhD thesis, University of London, 1966
- T. Abe, B. Sundman and H. Onodera, J. Phase Equilib. Diffus., 2006, 27, 5–13 CrossRef CAS
- J. W. Richards, J. Franklin Inst., 1891, 131, 283–298 Search PubMed
- Y. Hsiao, Y. A. Chang and H. Ipser, J. Electrochem. Soc., 1977, 124, 1235 CrossRef CAS
- J. Gröbner, R. Wenzel, G. G. Fischer and R. Schmid-Fetzer, J. Phase Equilib., 1999, 20, 615–625 CrossRef
- H.-N. Su and P. Nash, J. Alloys Compd., 2005, 403, 217–222 CrossRef CAS
- W. E. Liu and S. E. Mohney, Mater. Sci. Eng., B, 2003, 103, 189–201 CrossRef
- W. E. Liu and S. E. Mohney, J. Electron. Mater., 2003, 32, 1090–1099 CrossRef CAS
- E. Diagram, Bull. Alloy Phase Diagrams, 1985, 6, 350–359 CrossRef
- W. G. Jung and O. J. Kleppa, J. Alloys Compd., 1991, 176, 301–308 CrossRef CAS
- P. Anres, M. Gaune-Escard, J. P. Bros and E. Hayer, J. Alloys Compd., 1998, 280, 158–167 CrossRef CAS
- T. A. Stolyarova, M. V. Voronin, E. G. Osadchii and E. A. Brichkina, Geochem. Int., 2015, 53, 845–847 CrossRef CAS
- Y. G. Kim, U. I. Chung and J. Y. Lee, Acta Metall. Mater., 1992, 40, 1497–1503 CrossRef CAS
- F. Otto, A. Dlouhý, K. G. Pradeep, M. Kuběnová, D. Raabe, G. Eggeler and E. P. George, Acta Mater., 2016, 112, 40–52 CrossRef CAS
- F. Huang and J. F. Banfield, J. Am. Chem. Soc., 2005, 127, 4523–4529 CrossRef CAS PubMed
- Y. Nakaya, F. Xing, H. Ham, K. Shimizu and S. Furukawa, Angew. Chem., Int. Ed., 2021, 60, 19715–19719 CrossRef CAS PubMed
- Y. Nakaya, J. Hirayama, S. Yamazoe, K. Shimizu and S. Furukawa, Nat. Commun., 2020, 11, 2838 CrossRef CAS PubMed
- G. Sun, Z. J. Zhao, R. Mu, S. Zha, L. Li, S. Chen, K. Zang, J. Luo, Z. Li, S. C. Purdy, A. J. Kropf, J. T. Miller, L. Zeng and J. Gong, Nat. Commun., 2018, 9, 4454 CrossRef PubMed
- Y. Ren, Y. Tang, L. Zhang, X. Liu, L. Li, S. Miao, D. Sheng Su, A. Wang, J. Li and T. Zhang, Nat. Commun., 2019, 10, 4500 CrossRef PubMed
- Q. Sun, N. Wang, Q. Fan, L. Zeng, A. Mayoral, S. Miao, R. Yang, Z. Jiang, W. Zhou, J. Zhang, T. Zhang, J. Xu, P. Zhang, J. Cheng, D. C. Yang, R. Jia, L. Li, Q. Zhang, Y. Wang, O. Terasaki and J. Yu, Angew. Chem., Int. Ed., 2020, 59, 19450–19459 CrossRef CAS PubMed
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed
- Y. J. Wang, W. Long, L. Wang, R. Yuan, A. Ignaszak, B. Fang and D. P. Wilkinson, Energy Environ. Sci., 2018, 11, 258–275 RSC
- I. E. L. Stephens, A. S. Bondarenko, U. Grønbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744–6762 RSC
- H. Xu, Q. Fu, X. Guo and X. Bao, ChemCatChem, 2012, 4, 1645–1652 CrossRef CAS
- H. Zhang, X. Liu, N. Zhang, J. Zheng, Y. Zheng, Y. Li, C. J. Zhong and B. H. Chen, Appl. Catal., B, 2016, 180, 237–245 CrossRef CAS
- J. Jeon, K. Kon, T. Toyao, K. Shimizu and S. Furukawa, Chem. Sci., 2019, 10, 4148–4162 RSC
- F. Xing, Y. Nakaya, S. Yasumura, K. Shimizu and S. Furukawa, Nat. Catal., 2022, 5, 55–65 CrossRef CAS
- Y. Nakaya, M. Miyazaki, S. Yamazoe, K. Shimizu and S. Furukawa, ACS Catal., 2020, 10, 5163–5172 CrossRef CAS
- J. Y. He, H. Wang, H. L. Huang, X. D. Xu, M. W. Chen, Y. Wu, X. J. Liu, T. G. Nieh, K. An and Z. P. Lu, Acta Mater., 2016, 102, 187–196 CrossRef CAS
- J. Joseph, N. Stanford, P. Hodgson and D. M. Fabijanic, J. Alloys Compd., 2017, 726, 885–895 CrossRef CAS
- R. J. Vikram, B. S. Murty, D. Fabijanic and S. Suwas, J. Alloys Compd., 2020, 827, 154034 CrossRef CAS
- Y. Nakaya, E. Hayashida, H. Asakura, K. Shimizu and S. Furukawa, ChemRxive, 2021, preprint, DOI:10.26434/chemrxiv-2021-7pz5m.
- S. Chen, Z. Zhao, S. Chen, Z. Zhao, R. Mu, X. Chang, J. Luo, S. C. Purdy, A. J. Kropf, G. Sun, C. Pei, J. T. Miller, X. Zhou, E. Vovk, Y. Yang and J. Gong, Chem, 2021, 7, 1–19 Search PubMed
- Y. Nakaya and S. Furukawa, ChemPlusChem, 2022, 87, e202100560 CrossRef CAS PubMed
- A. Borodziński and G. C. Bond, Catal. Rev.: Sci. Eng., 2006, 48, 91–144 CrossRef
- A. Borodziński and G. C. Bond, Catal. Rev.: Sci. Eng., 2008, 50, 379–469 CrossRef
- R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Science, 2008, 320, 1320–1322 CrossRef CAS PubMed
- C. S. Spanjers, J. T. Held, M. J. Jones, D. D. Stanley, R. S. Sim, M. J. Janik and R. M. Rioux, J. Catal., 2014, 316, 164–173 CrossRef CAS
- C. S. Spanjers, R. S. Sim, N. P. Sturgis, B. Kabius and R. M. Rioux, ACS Catal., 2015, 5, 3304–3315 CrossRef CAS
- Y. Niu, X. Huang, Y. Wang, M. Xu, J. Chen, S. Xu, M. G. Willinger, W. Zhang, M. Wei and B. Zhang, Nat. Commun., 2020, 11, 3324 CrossRef CAS PubMed
- Y. Wang, B. Liu, X. Lan and T. Wang, ACS Catal., 2021, 11, 10257–10266 CrossRef CAS
- M. Hu, W. Yang, S. Liu, W. Zhu, Y. Li, B. Hu, Z. Chen, R. Shen, W. C. Cheong, Y. Wang, K. Zhou, Q. Peng, C. Chen and Y. Li, Chem. Sci., 2019, 10, 614–619 RSC
- Y. Cao, H. Zhang, S. Ji, Z. Sui, Z. Jiang, D. Wang, F. Zaera, X. Zhou, X. Duan and Y. Li, Angew. Chem., 2020, 132, 11744–11749 CrossRef
- M. Armbrüster, K. Kovnir, M. Friedrich, D. Teschner, G. Wowsnick, M. Hahne, P. Gille, L. Szentmiklósi, M. Feuerbacher, M. Heggen, F. Girgsdies, D. Rosenthal, R. Schlögl and Y. Grin, Nat. Mater., 2012, 11, 690–693 CrossRef PubMed
- L. Zhang, K. Doyle-Davis and X. Sun, Energy Environ. Sci., 2019, 12, 492–517 RSC
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef PubMed
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed
- Z. F. Huang, J. Wang, Y. Peng, C. Y. Jung, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7, 1700544 CrossRef
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed
- J. T. L. Gamler, H. M. Ashberry, S. E. Skrabalak and K. M. Koczkur, Adv. Mater., 2018, 30, 1801563 CrossRef PubMed
- R. Rizo and B. Roldan Cuenya, ACS Energy Lett., 2019, 4, 1484–1495 CrossRef CAS PubMed
- M. Li, Z. Zhao, W. Zhang, M. Luo, L. Tao, Y. Sun, Z. Xia, Y. Chao, K. Yin, Q. Zhang, L. Gu, W. Yang, Y. Yu, G. Lu and S. Guo, Adv. Mater., 2021, 33, 2103762 CrossRef CAS PubMed
- J. Zhang, M. Yuan, T. Zhao, W. Wang, H. Huang, K. Cui, Z. Liu, S. Li, Z. Li and G. Zhang, J. Mater. Chem. A, 2021, 9, 20676–20684 RSC
| This journal is © The Royal Society of Chemistry 2024 |