
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonitoring the synthesis of neutral lipids in lipid droplets of living human cancer cells using two-color infrared photothermal microscopy†
Chanjong
Park
 ab,
Jong Min
Lim‡
*a,
Seok-Cheol
Hong
ac and
Minhaeng
Cho
*ab
ab,
Jong Min
Lim‡
*a,
Seok-Cheol
Hong
ac and
Minhaeng
Cho
*ab
aCenter for Molecular Spectroscopy and Dynamics, Institute for Basic Science, Seoul, 02841 Korea. E-mail: mcho@korea.ac.kr
bDepartment of Chemistry, Korea University, Seoul, 02841 Korea
cDepartment of Physics, Korea University, Seoul, 02841 Korea
First published on 1st December 2023
Abstract
There has been growing interest in the functions of lipid droplets (LDs) due to recent discoveries regarding their diverse roles. These functions encompass lipid metabolism, regulation of lipotoxicity, and signaling pathways that extend beyond their traditional role in energy storage. Consequently, there is a need to examine the molecular dynamics of LDs at the subcellular level. Two-color infrared photothermal microscopy (2C-IPM) has proven to be a valuable tool for elucidating the molecular dynamics occurring in LDs with sub-micrometer spatial resolution and molecular specificity. In this study, we employed the 2C-IPM to investigate the molecular dynamics of LDs in both fixed and living human cancer cells (U2OS cells) using the isotope labeling method. We investigated the synthesis of neutral lipids occurring in individual LDs over time after exposing the cells to excess saturated fatty acids while simultaneously comparing inherent lipid contents in LDs. We anticipate that these research findings will reveal new opportunities for studying lesser-known biological processes within LDs and other subcellular organelles.
Introduction
Lipid droplets (LDs) are ubiquitous organelles in eukaryotic cells, consisting of a neutral lipid core surrounded by a phospholipid monolayer. Although LDs were long thought to serve as a storage site for neutral lipids, it has been revealed that they are pivotal players extending beyond energy homeostasis, including vitamin metabolism,1 lipotoxicity regulation,2 and cell signaling.3 Excessive enlargement or accumulation of LDs could result in health problems. Certain rare genetic disorders associated with LD metabolism lead to a systemic increase in the size and number of these cytosolic inclusions.4 Many human pathologies, including obesity, diabetes, and cardiovascular diseases, are often linked to the appropriate size and quantity of LDs. Maintaining the balance in LD size and number is crucial for overall health. Understanding LD dynamics is, thus, a prerequisite to comprehending the intricate functions of LDs and their far-reaching implications for an array of physiological and cellular processes.However, investigating LDs, particularly their dynamic behavior, presents challenges and complexities. These dynamic entities exhibit a remarkable capacity to fluctuate in size, number, and location within cells, which is determined by various physiological and environmental factors. This intrinsic dynamism complicates the task of capturing their behavior within a specific observational time. The broad complexity of lipid profiles is also an issue in analyzing the lipid composition of LDs. Fluorescence imaging, commonly employed for LD investigation, has certain limitations like cytotoxicity, photobleaching, and the dearth of acyl-specific labeling techniques to target neutral lipids directly.5 Lipids conjugated with a fluorophore may introduce potential interference to metabolic functionality.6 Even advanced imaging modalities such as confocal and super-resolution microscopy still pose challenges due to the labeling characteristics. Thus, developing novel methods for enabling label-free real-time monitoring of chemical compositions of LDs without phototoxicity has emerged as an imperative pursuit.
IR photothermal microscopy (IPM) has undergone significant methodological developments over the past decade7–24 and has been considered a powerful tool to investigate molecular dynamics in living systems without exogenous labels.25 This IR microscopic technique accomplished an enhanced spatial resolution of sub-micrometer level and chemical information by probing the IR pulse-induced transient photothermal dynamics with a non-resonant visible light source. These exceptional attributes of IPM facilitate subcellular-level exploration of the intracellular structures. Moreover, it should be noted that the non-phototoxic characteristic of IPM also allows a long-time evolution of living systems covering the lifecycle of cells. Dark-field scattering-based IR photothermal imaging (IPI), a representative among various IPM branches, has been widely applied to investigate molecular information in biological specimens9,16,20,22,26,27 as well as materials.28–31 Its potential for studying the dynamics of biomolecules in living organisms was demonstrated with the visualization of LDs in living cells by targeting the ester group of lipids.9 Our group has also harnessed IPM to investigate protein dynamics of cellular cytoplasm and protein-rich vesicle transport in living cells.26
Thus, in this study, we demonstrate the utility of the IPI technique for investigating lipid dynamics in human cancer cells (U2OS) and human hepatocellular carcinoma cells (Huh-7), specifically focusing on triacylglycerol, the neutral lipid in LDs. Fig. 1A illustrates the process of neutral lipid synthesis from free long-chain fatty acids (FAs) in a living cell. Cells primarily acquire FAs through two sources: (1) de novo lipogenesis, the conversion of glucose into FAs, and (2) direct uptake from plasma. The latter, particularly interesting to us, occurs either through fatty acid transport proteins or simple diffusion across cell membranes.32 In the cell cytoplasm, free FAs are conjugated with coenzyme A (CoA) to form acyl-CoA in a process known as activation. This is facilitated by an enzyme, long-chain acyl-CoA synthetase (ACSL).33 Once activated, the fatty acids (now in the form of acyl-CoA) serve as precursors in the synthesis of membrane and neutral lipids. This includes the production of lysophosphatidic acid, phosphatidic acid, diacylglycerol (DAG), and triacylglycerol (TAG), all catalyzed by various lipid biosynthetic enzymes.34 These lipids play critical roles in cellular processes such as membrane formation, energy storage, and signaling pathways. Notably, TAG, a major metabolite for energy storage, is synthesized through the enzymatic combination of acyl-CoA and DAG by diglyceride acyltransferase (DGAT). Subsequently, TAG accumulates in LDs, serving a crucial role in storing excess energy as neutral lipids within the cell.
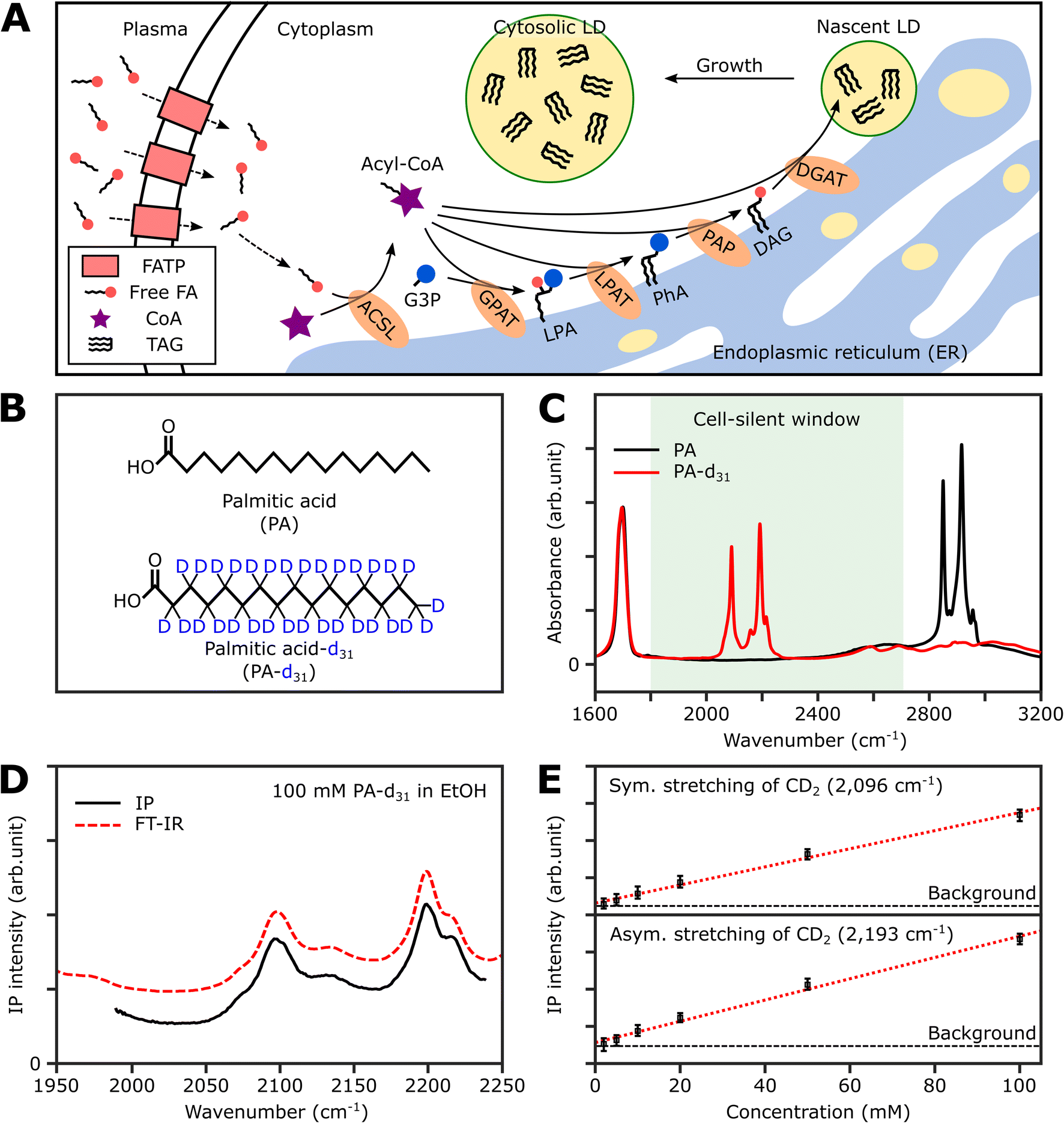 | ||
| Fig. 1 Deuterated palmitic acid is a tracer for neutral lipid synthesis. (A) Schematic illustration of neutral lipid synthesis using dietary long-chain fatty acid in a mammalian cell. FATP, fatty acid transport protein; G3P, glycerol-3-phosphate; GPAT, glycerol-3-phosphate acyltransferase; LPA, lysophosphatidic acid; LPAT, lysophosphatidate acyltransferase; PhA, phosphatidic acid; PAP, phosphatidic acid phosphatase. (B) Chemical structures of PA and PA-d31. In the structure of PA, all hydrogen atoms bonded to carbons are omitted. (C) FT-IR spectra of PA and PA-d31. The FT-IR spectra were obtained for powder samples in an attenuated total reflection geometry. (D) IP spectra and FT-IR spectra of 100 mM PA-d31 solution in ethanol. The offset is for clarity. Each data point in the IP spectra was obtained by 50 ms averaging and normalization to the intensity of the IR excitation pulse. Stepsize, 1 cm−1. (E) Linear dependence of IP responses on PA-d31 concentration at 2096 cm−1 and 2193 cm−1. The detection limit of our IPM for PA-d31 was estimated to be about 2 mM. The IP intensities were obtained by averaging over 100 ms, and the error bars represent the range between the maximum and minimum values of the IP signal intensity. | ||
In response to excess free FAs, cells increase TAG synthesis as a protective mechanism against lipotoxicity. This heightened TAG synthesis leads to an enhanced accumulation of neutral lipids within LDs. By storing excessive FAs in the form of TAG in LDs, cells can mitigate the detrimental effects of lipotoxicity, ensuring the proper regulation of lipid metabolism and maintaining cellular homeostasis.2 Our study employs the two-color IPI technique to investigate lipid dynamics in LDs. In this technique, two different IR contrasts, originating from different molecular vibrations, are investigated simultaneously using modulation-frequency multiplexing and dual-frequency lock-in detection.21 It is efficient for monitoring or tracking the trajectories of LDs in living cells based on the inherent IR absorption of target. It provides precise IR absorption information about their neutral lipid content with superior spatial and spectral resolutions and allows for distinguishing intracellular LDs from other organelles. Specifically, we focus on analyzing cells that have been supplied with exogenous fatty acids. This approach enables a quantitative assessment of neutral lipid contents in LDs at various time points following the addition of exogenous fatty acids with distinct compositions.
Furthermore, we selectively measured the IP response of neutral lipids synthesized from deuterated fatty acids. Despite the advantages of IPM, its practical application for investigating lipids inside LDs has been restricted by a broad IR spectral overlap between various lipids due to their structural similarity. Isotope labeling, which involves replacing one or more atoms in target molecules with its non-radioactive isotope having a different mass, could address this limitation. This strategy shifts vibrational frequencies into distinct regions, enabling discrimination between inherent and labeled molecules without substantially altering chemical properties. Deuterium labeling, with its IR absorption frequencies placed in the cell-silent spectral region (1800 cm−1 to 2600 cm−1), finds substantial favor in biological research.35,36 Devoid of significant interference from endogenous cellular components, deuterium-labeled biomolecules can be tracked, thus facilitating the investigation of specific metabolite production via the incorporation of exogenous nutrients.
Quantification of endogenous and newly synthesized lipids was achieved through analysis of IP responses corresponding to CH2 and CD2 stretching vibrations of LDs. This facilitated quantitative evaluation of lipid composition shifts within individual LDs over the exposure time to excess fatty acids. We also achieved real-time monitoring of the lipid synthesis process within individual LDs of living cells. Tracking two lipid species, distinguished by CD2 and CH2 groups, during neutral lipid synthesis from deuterated fatty acids afforded insights into dynamic lipid composition changes, thus offering invaluable real-time perspectives. This pioneering study not only underscores the potential of two-color IPM as a valuable tool in LD research but also introduces a robust approach for probing intricate molecular dynamics within living cells.
Materials and methods
Two-color IR photothermal microscope
The two-color IPM technique utilizes a pair of pulsed IR lasers and a continuous wave (CW) visible laser to examine two distinct IR-absorbing vibrational modes.22 This approach enables microscopic investigations with chemical contrasts, offering a spatial resolution of a few hundred nanometers. In this study, we opted for an IR laser source spanning from 1980 cm−1 to 2239 cm−1. The rationale behind this choice is to focus on a specific molecular vibration within the cell-silent spectral window – namely, the C–D stretching vibration. Another IR spectral window of our two-color IPM covers the range from 2740 cm−1 to 2970 cm−1. These spectral ranges facilitate the concurrent measurement of absorbance for both C–D and C–H stretching vibrations. Further details about the experimental setup can be found in ESI Note 1.†Cell culture
U2OS cell line (human bone osteosarcoma epithelial cells) and Huh-7 cell line (human hepatocellular carcinoma cells) were supplied from Korean Cell Line Bank (Seoul, Korea) and cultured in DMEM (Welgene, LM 001-17) supplemented with penicillin (100 U mL−1), streptomycin (100 μg mL−1), 10% heat-inactivated fetal bovine serum (Welgene, S1010-01) at 37 °C and pH 7.4 (5% CO2). For IP measurements, the cells were subcultured with a cell density of 100 cells per mm2 in μ-Slide I Luer (ibidi, #80181), whose bottom is a borosilicate glass (170 μm in thickness) coated with 0.2 mg mL−1 poly-D-lysine (Sigma-Aldrich, P7886). To obtain IP spectra of individual LDs, cells were fixed using a fixation buffer containing 4% paraformaldehyde (Biolegend, #420801). The cells were treated with the fixation buffer for 10 minutes and then washed three times with phosphate-buffered saline (Welgene, LB 004-02).Preparation of fatty acid-BSA conjugate solution
In the present work, we utilized palmitic acid (PA) and its deuterated analogue (PA-d31) to demonstrate the capability of IPM in monitoring the dynamic organization of neutral lipids in LDs. Due to the hydrophobic nature of PA and PA-d31 stemming from their long aliphatic chains, it was necessary to associate them with water-soluble protein carriers before being administered to cells.Bovine serum albumin (BSA) is commonly used as a protein vehicle for facilitating the delivery of these hydrophobic compounds into cells. This approach enables efficient cellular uptake and allows for investigating their effects.37 The protocol for conjugating the PAs and BSA is outlined as follows. PA (Sigma-Aldrich, P5585) and PA-d31 (Sigma-Aldrich, 366897) were first prepared in ethanol (Sigma-Aldrich, 51976) at a concentration of 100 mM. These solutions were subsequently diluted to 5 mM using a 10% BSA solution (Sigma-Aldrich, A1595). The resulting mixture was then subjected to sonication for 15 minutes, followed by an incubation at 50 °C and continuous shaking for an additional 15 minutes. These steps were taken to ensure proper dispersion and conjugation of the hydrophobic compounds with the BSA protein.
After the sonication and heating processes, the solutions were filtered through a 0.2 μm polyethersulfone (PES) filter to separate and collect the well-dispersed fraction. The final concentrations in these stock solutions were 5 mM PA or PA-d31, 10% BSA (equivalent to 1.5 mM BSA), and 5% ethanol. Further dilution was performed using a growth medium (DMEM supplemented with 10% FBS) to achieve the desired concentration of fatty acids (250 μM in this study), appropriate for cellular administration in in vitro experiments.
Fluorescence imaging
Nile Red (Sigma-Aldrich, 19123) was employed to label cellular LDs. The powder was initially dissolved in DMSO to create a stock solution with a concentration of 1 mM. A staining solution was prepared by adding the stock solution to the cell medium, resulting in a final concentration of 500 nM. Before imaging, the cells in μ-Slide I Luer were stained with 200 μL of the staining solution for 30 minutes and then observed under a microscope (Olympus, IX 73) coupled with a 40X objective lens (Olympus, LUCPlanFL N, 0.60NA). The excitation and emission filters used pass the range of wavelength from 464 nm to 495 nm and from 516 nm to 559 nm, respectively.Results
Strategy for IR photothermal imaging of lipid synthesis
To selectively quantify newly synthesized neutral lipids, we considered various metabolites and identified PA-d31 as a suitable candidate due to its distinct characteristics from the naturally occurring lipids in cells. PA, a prevalent FA in living organisms, constitutes a major component of LDs (in the form of TAG) and cell membranes (as phospholipids).38Fig. 1B displays the chemical structures of both PA and PA-d31. PA, composed exclusively of C–H chemical bonds, exhibits strong IR absorption peaks at 2850 cm−1 and 2916 cm−1, corresponding to the symmetric and antisymmetric CH2 stretching modes, respectively (Fig. 1C).The substitution of every aliphatic hydrogen in PA with deuterium causes the IR absorption to shift to a frequency region without cell interference (with absorption peaks at 2089 cm−1 and 2191 cm−1, indicated by the red line in Fig. 1C). This alteration enables the differentiation of neutral lipids derived from PA-d31, allowing for precise identification.
Spectral information and detection sensitivity
We assessed the IP response of our setup using PA-d31, the target molecule for monitoring neutral lipid synthesis. We measured the IP spectrum of a 100 mM PA-d31 solution in ethanol and normalized the obtained data by dividing the signal intensity at each IR frequency by the corresponding output intensity of the IR excitation lasers. As depicted in Fig. 1D, the intensity-normalized IP spectrum is in excellent agreement with the corresponding FT-IR spectrum.We then examined the concentration-dependent IP response at the absorption peaks of the symmetric and antisymmetric CD2 stretch modes at 2096 cm−1 and 2193 cm−1, respectively (Fig. 1E and S2†). The IP responses at these frequencies exhibited linear proportionality to the concentration of the PA-d31 solution, fitting well into linear regression lines (red dotted lines in Fig. 1E). Our instrument's capability to detect PA-d31 in ethanol was demonstrated down to a concentration of 2 mM, demonstrating the detection limit of our setup.
IP measurement of LDs in U2OS cell
To demonstrate the feasibility of investigating LDs using two-color IPM, we conducted two-color IP measurements of LDs in normal U2OS cells, targeting the CH2 vibration in lipids. Fig. 2A presents both the single-color and two-color IP images of fixed U2OS cells.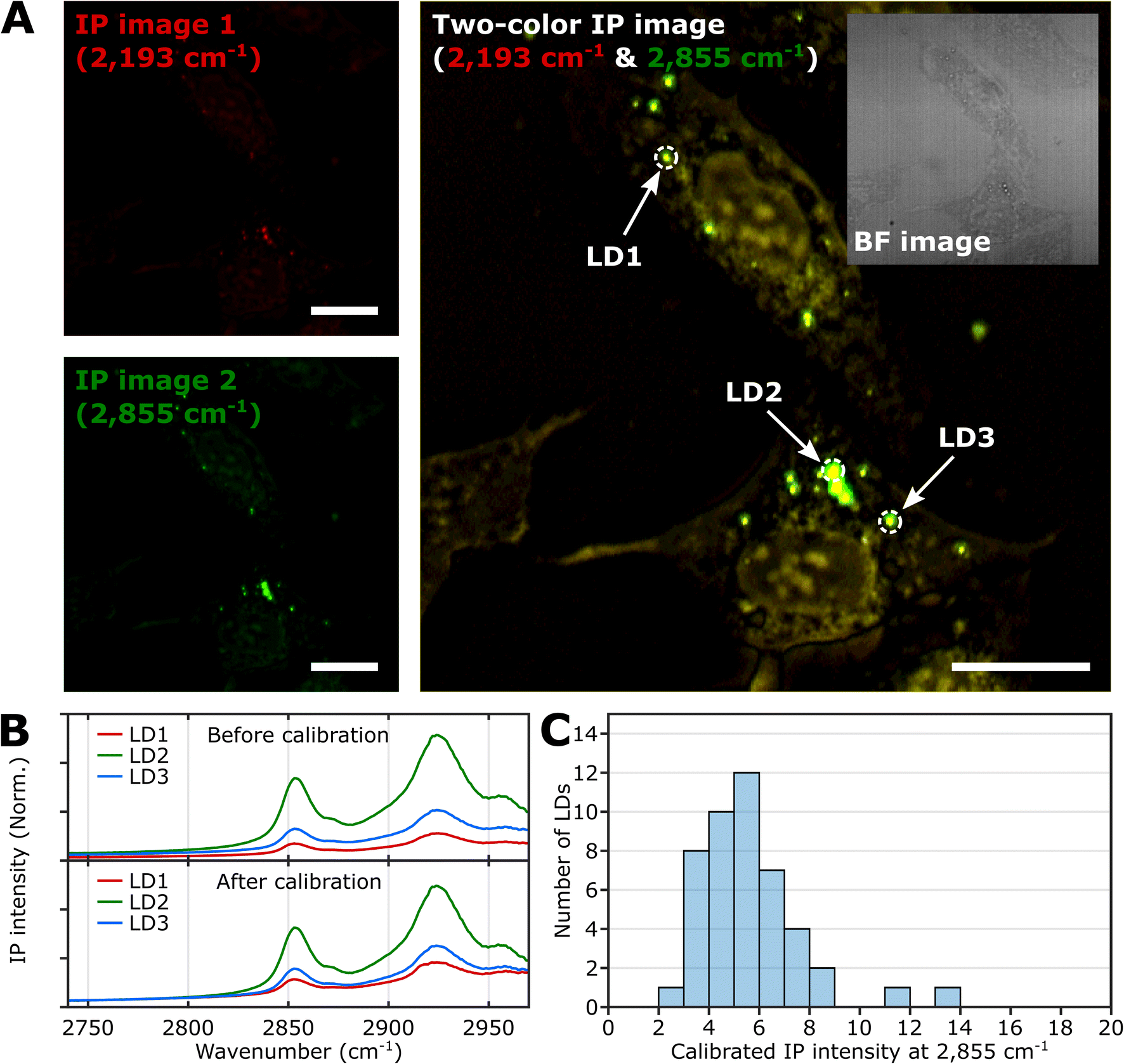 | ||
| Fig. 2 IPI measurement of LDs in U2OS cells. (A) Single-color (left) and two-color (right) IP images at 2193 cm−1 and 2855 cm−1. The two-color IP image was constructed by superimposing IP images simultaneously obtained with IR excitations of 2855 cm−1 (45 kHz) and 2193 cm−1 (50 kHz), which are marked by false green and red colors, respectively. The IPI contrasts were adjusted to reveal the cellular structures. The inset image is the bright-field image of the corresponding region. Cutoff frequency, 70 Hz. Step size, 200 nm. Pixel dwell time, 2 ms. Probe power, 15 mW. IR excitation power, 0.2 mW (at 2193 cm−1) and 0.1 mW (at 2855 cm−1). Scale bars, 20 μm. (B) IP spectra of LDs before and after background calibration. Three LDs are indicated in Fig. 2A. Each data point in the IP spectra was obtained by 50 ms averaging and normalization to the intensity of the IR excitation pulse. The spectra on the bottom were calibrated by matching the background signal level in the off-resonant spectral region (2740 cm−1 to 2760 cm−1). Stepsize, 1 cm−1. (C) Histogram for IP intensity at 2855 cm−1 of LDs. We obtained IP spectra of 47 LDs within U2OS cells from seven different regions, and the histogram shows the distribution of IP intensities at 2855 cm−1 of the calibrated IP spectra. | ||
For generating the two IP images, IR excitations at 2193 cm−1 and 2855 cm−1 were employed. These IR excitations were applied with repetition rates of 50 kHz and 45 kHz, respectively. The IPI contrast at 2855 cm−1 corresponds to the spatial distribution of molecular species containing CH2 groups, primarily lipids. Meanwhile, the contrast observed at 2193 cm−1 represents the background IP signal. Notably, a few round-shaped organelles were discernible in both the single-color IP images. However, the IPI contrast at 2193 cm−1 is negligible compared to that at 2855 cm−1. It should be noted that the IR absorption by the background water affects the IPI contrast under investigation.20,22 To effectively distinguish the LD signal from the undesired water-related signal in the IPI measurements, we adjusted the power of both IR excitations. This adjustment was made in order to match the IP intensities at the background region of the two IP images obtained at 2193 cm−1 and 2855 cm−1, respectively (see ESI Note 3†).
As depicted in Fig. 2B, the IP spectra of these round-shaped organelles exhibit two absorption peaks at 2855 cm−1 and 2924 cm−1, indicating a considerable presence of molecular species containing CH2 groups within the structures. These organelles are likely to be LDs, as they have a substantial amount of TAGs bearing long-chain methylene bridges (–CH2–). These methylene bridges are highly responsive to the IR beam at 2855 cm−1, which explains the pronounced IPI contrast observed in the IP images.
Conversely, there is an absence of observed IR absorption peaks linked to CD stretching vibrations in the range of 2030 to 2230 cm−1 (Fig. S4B†). This finding supports the conclusion that there is an abundant presence of CH2 groups within the LDs, thereby substantiating their identification in our study.
While we have demonstrated the capability of IPM to measure the spatial distribution and spectra of LDs in cells, there is still an issue to address in the context of IPI measurements of LDs. The inherent variability in the size and shape of LDs can potentially alter the beam path of the visible probe as it traverses through the specimen en route to the detector. This phenomenon can consequently influence the measured IP signal.
As shown in the spectra (upper panel in Fig. 2B), the IP responses of LDs exhibit considerable IP intensity, even at wavelengths not directly connected to the CH vibration peak. To remove this background signal, we derived calibration constants from each IP spectrum. These constants give all IP spectra the same background baseline in the wavenumber range of 2740 to 2760 cm−1. Subsequently, we divided each IP spectrum by the corresponding constant.
The background-corrected IP spectra exhibit an excellent match in the non-resonant spectral region below 2800 cm−1 (Fig. 2B, bottom) and across other spectral regions spanning from 1989 cm−1 to 2239 cm−1 (Fig. S4†). This correction procedure ensures consistency and reliability in our IPI measurements, providing a more precise and meaningful data analysis. Thorough theoretical details for the background correction method are presented and discussed in ESI Note 4.†
Subsequently, we obtained the distribution of LDs based on their IP intensity, which is anticipated to correlate closely with their size (Fig. 2C). The IP intensity distribution from LDs was positively skewed, consistent with previous experiments that unveiled the prevalence of small LDs, ranging from 400 to 600 nm, within U2OS cells under standard growth conditions.39 For the comprehensive set of IP images and spectra of LDs employed in Fig. 2C, refer to ESI Note 5.†
Excess fatty acid-induced lipid synthesis
Three groups of U2OS cells were cultured under different palmitate conditions (250 μM PA, 250 μM PA-d31, and standard growth medium) for 24 hours and subsequently fixed. These specimens were then subjected to two-color IPI measurements using IR excitations at 2193 cm−1 (50 kHz) and 2855 cm−1 (45 kHz). This approach enables the visualization of spatial distributions of lipid components involving C–D and C–H vibrations, respectively.Fig. 3A shows the merged two-color IP images taken at 2193 cm−1 and 2855 cm−1, with a consistent color scheme akin to Fig. 2A. A noticeable change in cell morphology was observed in the FA-treated cells, characterized by a considerable emergence of round-shaped organelles, distinct from the control group. The observed increase in the number of LDs in both groups exposed to an excess amount of PA and PA-d31 suggests a potential accumulation of newly synthesized neutral lipids derived from the exogenous PA or PA-d31.
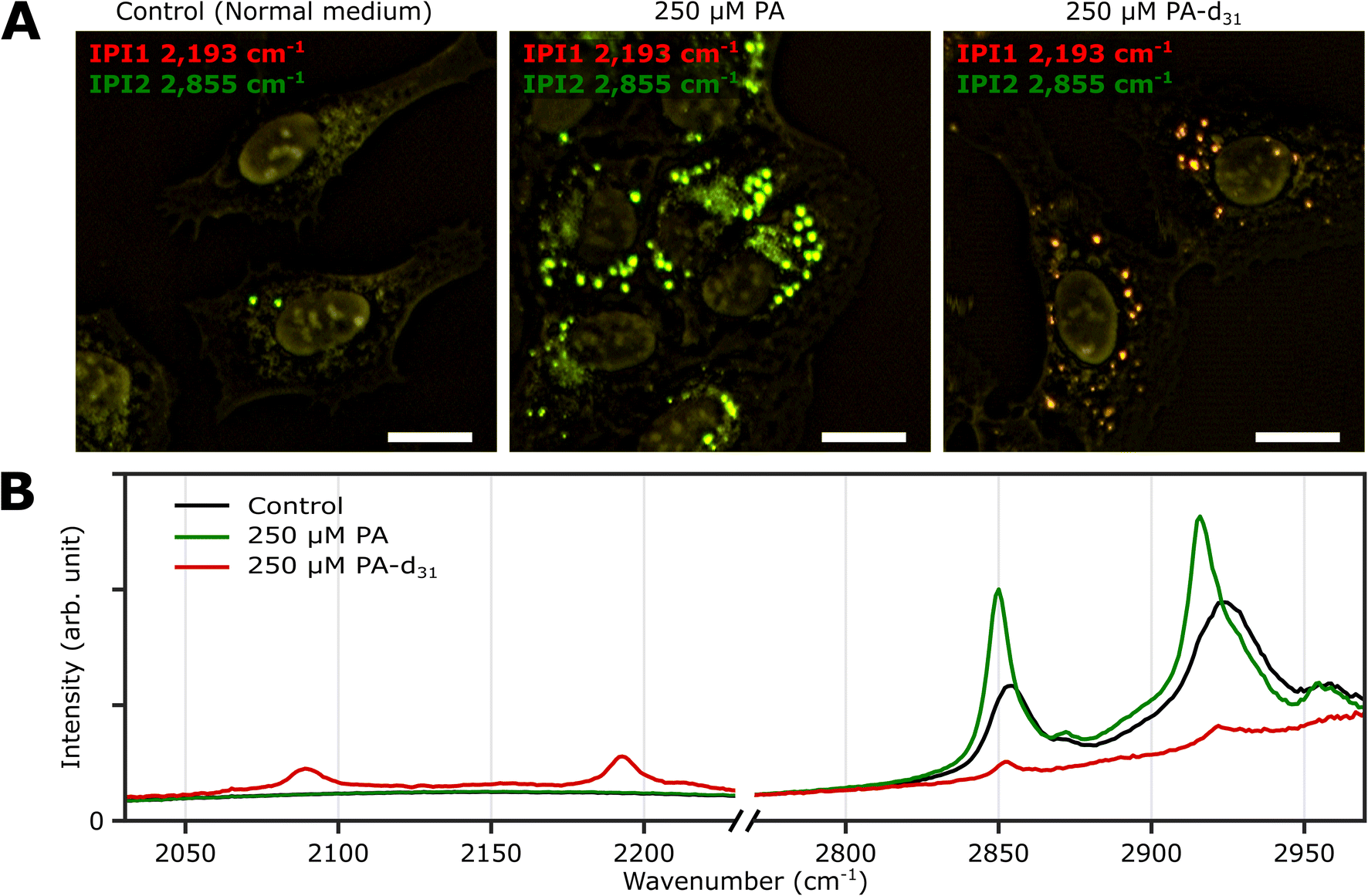 | ||
| Fig. 3 Two-color IPI measurements of U2OS cells cultured exposed to PA and PA-d31. (A) Two-color IP images of fixed U2OS cells cultured in different growth media (250 μM PA, 250 μM PA-d31, and standard medium) for 24 hours. The green and red false colors indicate IPI contrasts of 2855 cm−1 and 2193 cm−1, respectively. Cutoff frequency, 70 Hz. Step size, 200 nm. Pixel dwell time, 2 ms. Probe power, 15 mW. IR excitation power, 0.2 mW (at 2193 cm−1) and 0.1 mW (at 2855 cm−1). Scale bar, 20 μm. (B) Representative IP spectra of LDs observed in Fig. 3A at two different spectral regions. Each data point in the IP spectra was obtained by 50 ms averaging and normalization to the intensity of the IR excitation pulse. Stepsize, 1 cm−1. | ||
The IPI contrast at 2855 cm−1, representing normal (non-deuterated) neutral lipids, exhibited significant intensity across all groups. However, the IPI contrast at 2193 cm−1 manifested only within the U2OS cells exposed to PA-d31 (Fig. 3B). This finding strongly implies that the uptake of excessive PA-d31 triggered the active synthesis of neutral lipids within the cells, resulting in their subsequent accumulation within LDs. Moreover, it indicates that the IR response at 2193 cm−1 is specific to the presence of PA-d31, which can serve as a chemical marker for newly synthesized neutral lipids originating from exogenous PA-d31.
We found a notable difference in IR absorption peaks related to CH2 stretching vibrations between PA-treated and untreated cells (green and black lines in Fig. 3B). Although LDs in both cell groups cultured in PA and normal medium showed strong IR absorption peaks between 2800 cm−1 and 2970 cm−1, the peak positions in the IP spectra appeared to be quite different. This variance arises from differences in the molecular composition within LDs. TAG consists of three acyl chains, and their chain length and degree of saturation can be diverse.40 This heterogeneity in acyl groups of TAG contributes to the broadening of spectral peaks related to CH2 vibrations in the IP spectrum of normal (PA-untreated) cells (black line). In contrast, the spectrum of PA-treated cells (green line) predominantly reflects acyl groups constituted by PA.
These outcomes strongly support the efficacy of our IPI technique in discriminating neutral lipids synthesized using exogenous PA labeled with a distinctive vibrational tag (C–D vibration in this case). This capability, in turn, facilitates the analysis of lipid dynamics within LDs, effectively distinguishing them from endogenous lipids, a feat not achievable with traditional fluorescent dyes used for LD labeling, such as Bodipy and Nile Red. These dyes only reveal the hydrophobic environment inside LDs and cannot differentiate between neutral lipids composed of PA and PA-d31, unlike the IPI technique (Fig. S6†). Consistent results were replicated in experiments using the Huh-7 cell line (refer to ESI Note 7†), indicating the feasibility of investigating neutral lipid synthesis inside cellular LDs with 2C-IPM across various cell types.
Investigation of neutral lipid synthesis over time
To investigate the time-dependent dynamics of neutral lipids within LDs, we acquired IP images and spectra of LDs at different time points following the administration of 250 μM PA-d31. In Fig. 4A, we present two-color IP images of fixed U2OS cells that were treated with PA-d31 for specified time intervals. The color scheme employed is the same as in the previous two-color IP images (red: 2193 cm−1, green: 2855 cm−1).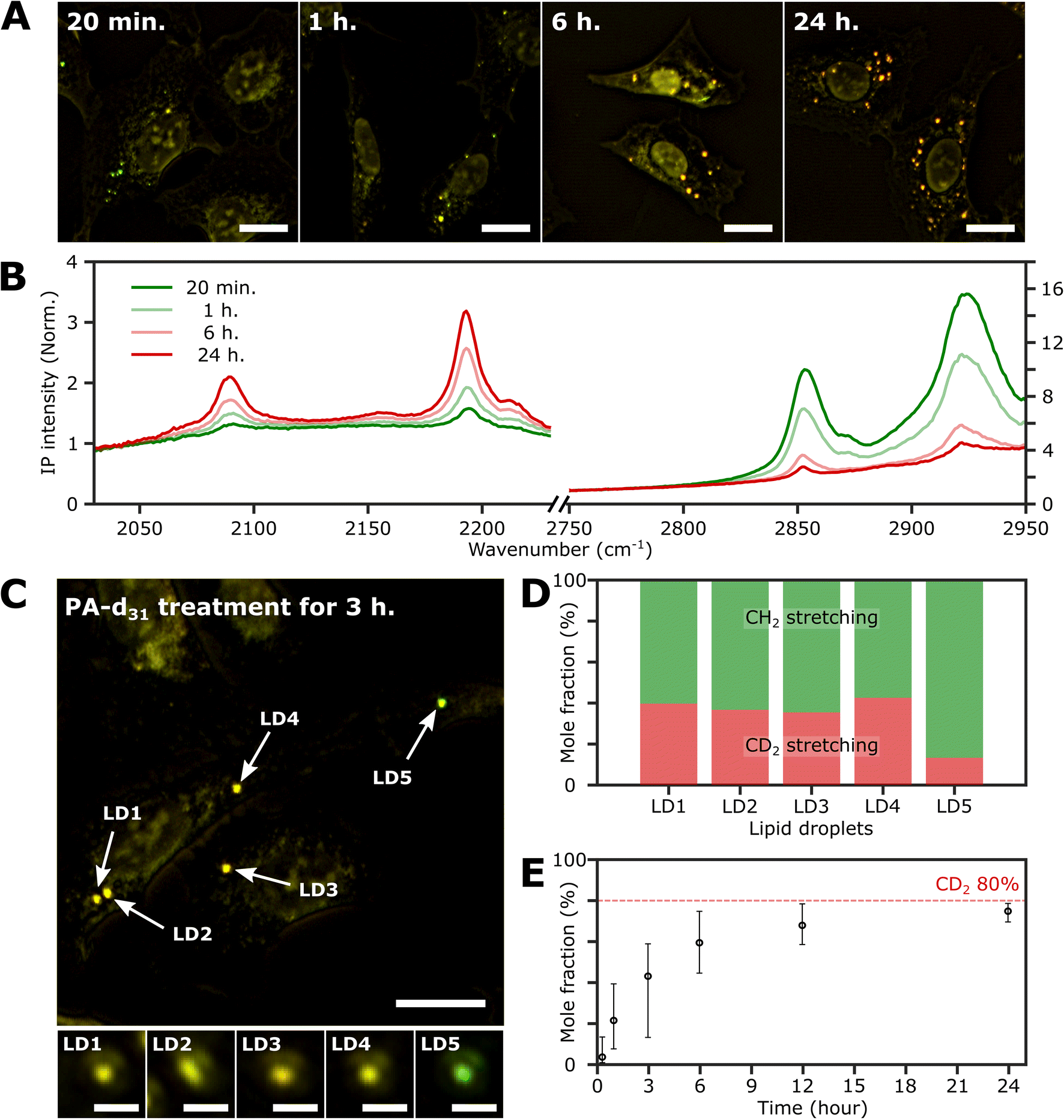 | ||
| Fig. 4 Investigation of the LDs with time. (A) Two-color (excitation: 2193 cm−1 and 2855 cm−1) IP images of fixed U2OS cells at different time points after PA-d31 administration. PA-d31 treatment times were indicated in the upper left corner of each image. The color scheme is the same as in Fig. 2A. Cutoff frequency, 70 Hz. Step size, 200 nm. Pixel dwell time, 2 ms. Probe power, 15 mW. IR excitation power, 0.2 mW (at 2193 cm−1) and 0.1 mW (at 2855 cm−1). Scale bars, 20 μm. (B) IP spectra of LDs at time points (20 min, 1 h, 6 h, and 24 h) after PA-d31 administration. Each data point in the IP spectra was obtained by 50 ms averaging and normalization to the intensity of the IR excitation pulse. Stepsize, 1 cm−1. The y-axis scales in the two regions (2030 cm−1 to 2230 cm−1 and 2750 cm−1 to 2950 cm−1) are adjusted differently to enhance clarity. (C) Two-color IP images of fixed U2OS cells treated with PA-d31 for 3 hours. The experimental condition is the same as that of Fig. 4A. Scale bars, 20 μm. The figures below depict magnified images of five LDs with 2 μm scale bars. (D) Mole fractions of CD2 and CH2 groups from neutral lipids of Fig. 4C. Two wavelengths of 2193 cm−1 and 2855 cm−1 were chosen to calculate the mole fraction. (E) Temporal change in mole fraction. The mole fraction of 10 different LDs was averaged at each time point. The error bars represent the upper and lower bounds of the mole fraction, corresponding to the maximum and minimum values, respectively. | ||
With an exposure time of 20 minutes, the LDs showed a strong IP response to the 2855 cm−1 IR beam while displaying a weak signal at 2193 cm−1. This observation suggests a notable abundance of endogenous lipids featuring methylene groups in LDs in comparison to the neutral lipids synthesized from PA-d31 during the early stages. After 1 hour of exposure to PA-d31, the two-color IP image displayed numerous LDs in yellow hues, indicating comparable IP intensities at both 2193 cm−1 and 2855 cm−1. This yellow appearance implies a balanced presence of endogenous and newly synthesized lipids from PA-d31 within the LDs during this specific time frame. After 6 hours or more, the LDs appeared reddish-orange in the image, signifying a predominant IP response at 2193 cm−1 over 2855 cm−1.
Fig. 4B displays the IP spectra of LDs at distinct time points subsequent to the administration of PA-d31 (20 min, 1 h, 6 h, and 24 h). LDs of comparable size were carefully chosen to account for any size-related impact on the IP signal. Our IPM was capable of detecting discernible IR absorption peaks corresponding to neutral lipids composed of PA-d31, even within cells treated for as brief as 20 minutes. The absorbances of CD2 vibrations (2096 cm−1 and 2193 cm−1) increased with increasing exposure time, indicating an accumulation of newly synthesized lipids from PA-d31 inside the LDs over time.
On the other hand, the absorption peaks related to CH2 vibrations (2855 cm−1 and 2923 cm−1) showed a decreasing pattern, indicating a decrease in the relative content of endogenous lipids within the LDs as the exposure time to PA-d31 increased. However, it is essential to note that the trends observed in the IP spectra do not consistently align, as LDs in heterogeneous environments might synthesize neutral lipids at varying rates and possess different sizes. The mole fraction of endogenous and newly synthesized neutral lipids within each LD can be a quantitative measure for comparing LD dynamics, regardless of their size. We have derived an equation for calculating the mole fraction of lipid species composed of CD2, denoted as χCD2,
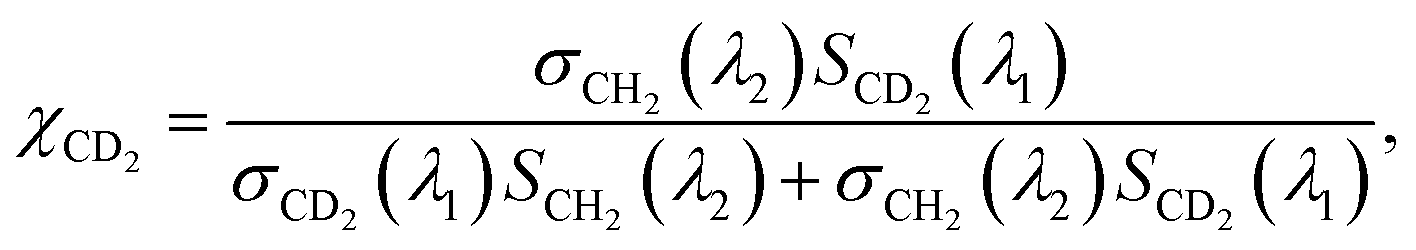 | (1) |
Fig. 4C depicts the two-color IP image of U2OS cells subjected to a 3 hour treatment with PA-d31, wherein five distinct LDs were observed and subsequently numbered for identification. To facilitate direct comparison of their IP signals, magnified IP images of these five LDs, each covering a 4 μm × 4 μm area, are presented in the lower panels of Fig. 4C. Notably, LD5 exhibited a discernibly different color (yellowish green) in contrast to the other LDs (yellow), indicating distinct IP responses to the IR excitations.
The mole fraction of CD2 lipid species was calculated for each of these five LDs, revealing a noteworthy dissimilarity in the mole fraction of CD2 lipid species between LD5 and the other LDs (Fig. 4D). This observation indicates the presence of heterogeneity in lipid synthesis rates among these LDs. The mole fraction of CD2 lipid species within LDs demonstrated a progressive increase, reaching 70–80% with a prolonged PA-d31 treatment time (Fig. 4E and S9†). This significant increase indicates a substantial accumulation of neutral lipids originating from PA-d31 within the LDs.
Monitoring neutral lipid synthesis in living cells
An important advantage of the two-color IPI technique is its capability to simultaneously monitor two distinct molecular species within a living cell, as demonstrated in our previous works.22 To investigate lipid dynamics, specifically focusing on the synthesis of neutral lipids within individual LDs, we conducted a time-lapse two-color IPI on living U2OS cells treated with 250 μM PA-d31. Every 10 min, we simultaneously captured two-color IP images at 2193 cm−1 (50 kHz) and 2855 cm−1 (45 kHz) (Fig. 5A, S10, and Movie 1†).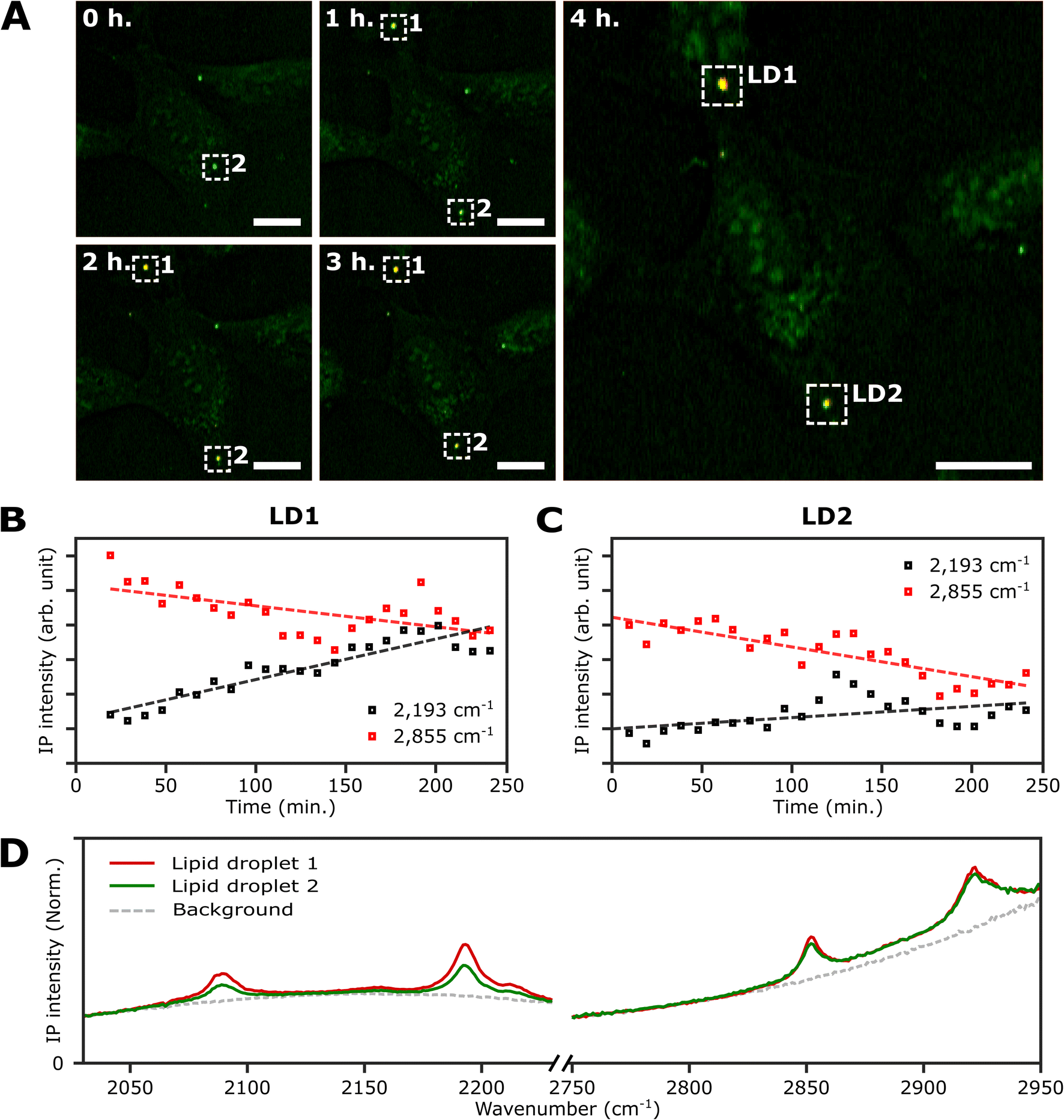 | ||
| Fig. 5 Real-time monitoring of neutral lipid synthesis in living U2OS cells. (A) Time-lapse two-color (excitation: 2193 cm−1 and 2855 cm−1) IP images of a live U2OS cell. The two-color IP images were obtained every 10 minutes for 4 hours. The color scheme is the same as in Fig. 3. Cutoff frequency, 70 Hz. Step size, 400 nm. Pixel dwell time, 2 ms. Probe power, 15 mW. IR excitation power, 0.2 mW (at 2193 cm−1) and 0.1 mW (at 2855 cm−1). Scale bars, 20 μm. (B and C) Temporal IP intensity profiles of LDs in living U2OS cells. The maximal intensities of 2193 cm−1 and 2855 cm−1 of LD1 and 2 in Fig. 5A are plotted with time, indicated as white dotted boxes. Linear lines fitted to the temporal profiles of each IP intensity are displayed as dotted lines to show their respective trends. (D) IP spectra of the two LDs. Each data point in the IP spectra was obtained by 50 ms averaging and normalization to the intensity of the IR excitation pulse. Step size, 1 cm−1. | ||
In the early stages of the time-lapse observation, all LDs exhibited a strong IP response at 2855 cm−1 while showing a minimal response at 2193 cm−1. We noted that the IP signal intensities at 2193 cm−1, measured at both LDs located within the white dotted boxes in Fig. 5A, exhibited a gradual increase over time (Fig. 5B and C). The IP signal intensity of LD1 was analysed after 20 minutes (Fig. S10†). The observed increase in IP intensities at 2193 cm−1 is believed to result from the conversion of exogenous PA-d31 into neutral lipids, TAG, within the LDs. After completing the time-lapse measurement for 4 hours, the specimen was promptly fixed to acquire the IP spectra of the LDs. Fig. S11† presents the two-color IP images measured within the same region of the fixed specimen, exhibiting cellular morphology closely resembling the last set of time-lapse images.
By analyzing the IP spectra from the two LDs observed in the IP image, distinctive IR absorptions at 2096 cm−1 and 2193 cm−1, as well as 2855 cm−1 and 2923 cm−1, were revealed (Fig. 5D). Based on the presence of IR absorption peaks at 2096 cm−1 and 2193 cm−1, we deduced that the temporal intensity augmentation observed at 2193 cm−1 during the time-lapse two-color IPI study signifies the process of neutral lipid synthesis within the LDs.
Discussion
Fluorescent labeling has been considered a high-priority method for studying LD dynamics over the past decades.41,42 Although the structure and function of LDs have been extensively studied, in particular, with the help of super-resolution techniques, molecular dynamics occurring in LDs cannot be easily investigated using fluorescent dyes such as Bodipy and Nile Red because they can only report the hydrophobicity in LDs. In the present work, we demonstrated that the two-color IPI technique is feasible to monitor neutral lipid synthesis occurring in living cells as well as quantitatively analyze the fractions of endogenous and newly synthesized neutral lipids inside individual LDs. It shows a sufficient potential of IPM to supplement or surpass fluorescence microscopy in studying LD dynamics by providing spatially-resolved molecular information based on IR spectroscopy.Although relatively slow molecular dynamics of lipid metabolism (i.e., neutral lipid synthesis) was considered in the present work, IPM could also be applied to investigate faster molecular dynamics. Due to its sample-scanning modality, the acquisition time for an IP image depends on the size of the region of interest, more precisely, the number of pixels forming an image. In our case, IPI measurements could be performed even with a pixel dwell time of 500 μs, restricted by the physical limitation of the sample scanner, while maintaining a considerable signal-to-noise ratio (Fig. S12†). It only takes 5 seconds to obtain an image of 100 by 100 pixels, allowing to monitor faster molecular dynamics occurring in living systems. We emphasize again that 10 minutes of measurement interval used here does not mean the instrumental limitation in imaging speed. Since neutral lipids inside LDs were detected with our IPM after at least 20 minutes of exposure to PA-d31 (Fig. 4B), the parameter was deemed sufficient to investigate neutral lipid synthesis in LDs.
In the present work, newly synthesized and endogenous lipids inside LDs were analyzed by measuring the IR absorbance of CD2 and CH2 stretching vibrations (2193 cm−1 and 2855 cm−1, respectively) at fixed specimens as well as living cells. However, the current detection sensitivity of 2 mM for PA-d31 is insufficient for IPM to investigate other lipid dynamics, especially related to the endoplasmic reticulum, where other metabolic conversions of acyl-CoA to membrane lipids were known to occur.37 Despite the abundance of C–D chemical bonds per PA-d31 molecule (31 of C–D chemical bonds), the detection sensitivity of IPM for PA-d31 was only comparable to those for other chemical compounds such as ascorbic acid and N-methylacetamide (NMA) having a single chemical bond per molecule, which were estimated to 2 and 5.1 mM, respectively.26 It is mainly due to the fact that the molar extinction coefficient of C–D stretching vibration is lower than those of carbonyl and amide I vibrations in the ascorbic acid and NMA, respectively. A change in the IR label is expected to enhance the detection sensitivity. Azido group (N3-) could be a representative candidate since its asymmetric stretch mode has a much larger molar extinction coefficient than the C–D stretch mode (2 orders of magnitude larger), and the corresponding IR absorption peak resides in the cell-silent spectral region.43 Palmitic acid labeled with the azido group was reported to be incorporated in cellular metabolism in a HEK293T cell.44
Conclusion
In this work, we have demonstrated that two-color IPI could be applied to investigate lipid dynamics occurring in individual LDs focusing on neutral lipids such as TAG composed of CD2 and CH2 chemical bonds. Two-color IPI could distinguish different lipid species (endogenous and newly synthesized) of individual LDs and measure their molecular fraction in time after the onset of lipotoxic condition (exposed to excess PA-d31) based on the measurement of IP responses from CD2 and CH2 vibrations. A noteworthy aspect of our study is that we monitored the synthesis of neutral lipids and their accumulation in LDs in fixed and living human cells. For the first time, lipid synthesis in living cells was monitored using IR photothermal microscopy. We anticipate that this technique could provide unprecedented access to cytological investigations and new insights into LD dynamics and the synthesis in living cells.Data availability
The authors declare that all data supporting the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Conceptualization: MC, CP. Data curation: CP. Formal analysis: CP. Funding acquisition: MC. Investigation: CP, JML, MC. Methodology: CP, JML, MC. Project administration: MC. Resources: MC. Supervision: MC, SCH. Validation: CP, JML, SCH, MC. Visualization: CP. Writing – original draft: CP, MC. Writing – review & editing: CP, JML, SCH, MC.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Institute for Basic Science (IBS-R023-D1) and in part by the Global Research and Development Center Program (2018K1A4A3A01064272) through the NRF of Korea (S.-C. H.). The authors sincerely thank Professor Seung-Hoi Koo for discussing the biological importance of LDs.References
- W. S. Blaner, Y. Li, P. J. Brun, J. J. Yuen, S. A. Lee and R. D. Clugston, Subcell. Biochem., 2016, 81, 95–125 CrossRef CAS PubMed.
- T. B. Nguyen, S. M. Louie, J. R. Daniele, Q. Tran, A. Dillin, R. Zoncu, D. K. Nomura and J. A. Olzmann, Dev. Cell, 2017, 42, 9–21 CrossRef CAS PubMed.
- P. T. Bozza, I. Bakker-Abreu, R. A. Navarro-Xavier and C. Bandeira-Melo, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2011, 85, 205–213 CrossRef CAS PubMed.
- S. Missaglia, R. A. Coleman, A. Mordente and D. Tavian, Cells, 2019, 8, 187 CrossRef PubMed.
- J. Zhang, J. Nie, H. Sun, J. Li, J. P. Andersen and Y. Shi, Mol. Metab., 2022, 61, 101511 CrossRef CAS PubMed.
- C. Paulusma, R. Houwen and P. Williamson, Nat. Med., 2011, 17, 413 CrossRef CAS PubMed.
- E. S. Lee and J. Y. Lee, Appl. Phys. Lett., 2009, 94, 261101 CrossRef.
- R. Furstenberg, C. A. Kendziora, M. R. Papantonakis, V. Nguyen and R. A. McGill, Proc. SPIE, 2012, 8374, 837411 CrossRef.
- D. Zhang, C. Li, C. Zhang, M. N. Slipchenko, G. Eakins and J. X. Cheng, Sci. Adv., 2016, 2, e1600521 CrossRef PubMed.
- Z. Li, K. Aleshire, M. Kuno and G. V. Hartland, J. Phys. Chem. B, 2017, 121, 8838–8846 CrossRef CAS PubMed.
- Y. Bai, D. Zhang, C. Li, C. Liu and J. X. Cheng, J. Phys. Chem. B, 2017, 121, 10249–10255 CrossRef CAS PubMed.
- K. Toda, M. Tamamitsu, Y. Nagashima, R. Horisaki and T. Ideguchi, Sci. Rep., 2019, 9, 9957 CrossRef PubMed.
- Y. Bai, D. Zhang, L. Lan, Y. Huang, K. Maize, A. Shakouri and J. X. Cheng, Sci. Adv., 2019, 5, eaav7127 CrossRef CAS PubMed.
- D. Zhang, L. Lan, Y. Bai, H. Majeed, M. E. Kandel, G. Popescu and J. X. Cheng, Light: Sci. Appl., 2019, 8, 116 CrossRef CAS PubMed.
- M. Schnell, S. Mittal, K. Falahkheirkhah, A. Mittal, K. Yeh, S. Kenkel, A. Kajdacsy-Balla, P. S. Carney and R. Bhargava, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 3388–3396 CrossRef CAS PubMed.
- M. Tamamitsu, K. Toda, H. Shimada, T. Honda, M. Takarada, K. Okabe, Y. Nagashima, R. Horisaki and T. Ideguchi, Optica, 2020, 7, 359–366 CrossRef CAS.
- P. D. Samolis, D. Langley, B. M. O'Reilly, Z. Oo, G. Hilzenrat, S. Erramilli, A. E. Sgro, S. McArthur and M. Y. Sander, Biomed. Opt. Express, 2021, 12, 303–319 CrossRef CAS PubMed.
- Y. Zhang, C. Yurdakul, A. J. Devaux, L. Wang, X. G. Xu, J. H. Connor, M. S. Ünlü and J. X. Cheng, Anal. Chem., 2021, 93, 4100–4107 CrossRef CAS PubMed.
- S. Zhang, K. Kniazev, I. M. Pavlovetc, S. Zhang, R. L. Stevenson and M. Kuno, J. Chem. Phys., 2021, 155, 214202 CrossRef CAS PubMed.
- J. Yin, L. Lan, Y. Zhang, H. Ni, Y. Tan, M. Zhang, Y. Bai and J. X. Cheng, Nat. Commun., 2021, 12, 7097 CrossRef CAS PubMed.
- E. M. Paiva and F. M. Schmidt, Anal. Chem., 2022, 94, 14242–14250 CrossRef CAS PubMed.
- C. Park, J. M. Lim, S. C. Hong and M. Cho, Analyst, 2023, 148, 2395–2402 RSC.
- K. M. Tracy, M. V. Barich, C. L. Carver, B. M. Luther and A. T. Krummel, J. Phys. Chem. Lett., 2016, 7, 4865–4870 CrossRef CAS PubMed.
- A. L. Serrano, A. Ghosh, J. S. Ostrander and M. T. Zanni, Opt. Express, 2015, 23, 17815–17827 CrossRef CAS PubMed.
- S. Adhikari, P. Spaeth, A. Kar, M. D. Baaske, S. Khatua and M. Orrit, ACS Nano, 2020, 14, 16414–16445 CrossRef CAS PubMed.
- J. M. Lim, C. Park, J. S. Park, C. Kim, B. Chon and M. Cho, J. Phys. Chem. Lett., 2019, 10, 2857–2861 CrossRef CAS PubMed.
- A. Spadea, J. Denbigh, M. J. Lawrence, M. Kansiz and P. Gardner, Anal. Chem., 2021, 93, 3938–3950 CrossRef CAS PubMed.
- C. Li, D. Zhang, M. N. Slipchenko and J. X. Cheng, Anal. Chem., 2017, 89, 4863–4867 CrossRef CAS PubMed.
- R. Chatterjee, I. M. Pavlovetc, K. Aleshire, G. V. Hartland and M. Kuno, ACS Energy Lett., 2018, 3, 469–475 CrossRef CAS.
- S. Mondal, J. Kang, K. Park, J. M. Lim, J. H. Ha, K. Kwak and M. Cho, J. Phys. Chem. Lett., 2021, 12, 9275–9282 CrossRef CAS PubMed.
- J. S. Böke, J. Popp and C. Kraft, Sci. Rep., 2022, 12, 18785 CrossRef PubMed.
- X. Su and N. A. Abumrad, Trends Endocrinol. Metab., 2009, 20, 72–77 CrossRef CAS PubMed.
- E. Soupene and F. A. Kuypers, Exp. Biol. Med., 2008, 233, 507–521 CrossRef CAS PubMed.
- P. Fagone and S. Jackowski, J. Lipid Res., 2009, 50, S311–S316 CrossRef PubMed.
- L. Shi, X. Liu, L. Shi, H. T. Stinson, J. Rowlette, L. J. Kahl, C. R. Evans, C. Zheng, L. E. P. Dietrich and W. Min, Nat. Methods, 2020, 17, 844–851 CrossRef CAS PubMed.
- X. Liu, L. Shi, L. Shi, M. Wei, Z. Zhao and W. Min, Adv. Sci., 2022, 9, 2105437 CrossRef CAS PubMed.
- K. D. Ono-Moore, M. L. Blackburn and S. H. Adams, Am. J. Physiol.: Endocrinol. Metab., 2018, 315, E780–E794 CrossRef CAS PubMed.
- G. Carta, E. Murru, S. Banni and C. Manca, Front. Physiol., 2017, 8, 902 CrossRef PubMed.
- T. Exner, C. A. Beretta, Q. Gao, C. Afting, I. Romero-Brey, R. Bartenschlager, L. Fehring, M. Poppelreuther and J. Füllekrug, J. Lipid Res., 2019, 60, 1333–1344 CrossRef CAS PubMed.
- V. Ruiz-Gutierrez, E. Montero and J. Villar, J. Chromatogr., 1992, 581, 171–178 CrossRef CAS PubMed.
- P. Greenspan, E. P. Mayer and S. D. Fowler, J. Cell Biol., 1985, 100, 965–973 CrossRef CAS PubMed.
- L. L. Listenberger, A. M. Studer, D. A. Brown and N. E. Wolins, Curr. Protoc. Cell Biol., 2016, 71, 4.31.1–4.31.14 Search PubMed.
- J. Ma, I. M. Pazos, W. Zhang, R. M. Culik and F. Gai, Annu. Rev. Phys. Chem., 2015, 66, 357–377 CrossRef CAS PubMed.
- J. Greaves, K. R. Munro, S. C. Davidson, M. Riviere, J. Wojno, T. K. Smith, N. C. Tomkinson and L. H. Chamberlain, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E1365–E1374 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04705a |
| ‡ Present address: Department of Chemistry, Kyungpook National University, Daegu 41566, Korea. |
| This journal is © The Royal Society of Chemistry 2024 |